
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Functionalized silica nanoplatform as a bimodal contrast agent for MRI and optical imaging†
Sarah
Garifo
 a,
Dimitri
Stanicki
a,
Sébastien
Boutry
b,
Lionel
Larbanoix
b,
Indiana
Ternad
a,
Robert N.
Muller
ab and
Sophie
Laurent
*ab
a,
Dimitri
Stanicki
a,
Sébastien
Boutry
b,
Lionel
Larbanoix
b,
Indiana
Ternad
a,
Robert N.
Muller
ab and
Sophie
Laurent
*ab
aGeneral, Organic and Biomedical Chemistry Unit, NMR and Molecular Imaging Laboratory, University of Mons, 19 avenue Maistriau, 7000 Mons, Belgium. E-mail: sophie.laurent@umons.ac.be; Tel: +32-(0)65-37-35-25
bCenter for Microscopy and Molecular Imaging (CMMI), 8 rue Adrienne Bolland, 6041 Gosselies, Belgium
First published on 20th September 2021
Abstract
The preparation of an efficient bimodal single probe for magnetic resonance (MRI) and optical imaging (OI) is reported. Paramagnetic properties have been obtained by the non-covalent encapsulation of the clinically used Gd3+ chelate (i.e., Gd-HP-DO3A) within silica nanoparticles through a water-in-oil microemulsion process. To ensure colloidal stability, the surface of the particles was modified by means of treatment using PEG-silane, and further functionalized photochemically using a diazirine linker bearing carboxylic functions. Optical properties were obtained by the covalent grafting of a near-infrared emitting probe (NIR) on the resulting surface. The confinement of Gd complexes within the permeable matrix resulted in a significant increase in longitudinal relaxivities (>500% at 20 MHz) in comparison with the relaxivities of free chelate, while the post-functionalization process of PEG with fluorescent compounds appeared promising for the derivatization procedure. Several physico-chemical properties attested to the efficient surface modification and confirmed covalent grafting. Preliminary imaging experiments complete this study and confirm the potential of the presented system for preclinical imaging experiments.
Introduction
For several decades, magnetic resonance imaging (MRI) has been used as a reference tool for medical diagnosis by allowing the tridimensional visualization of various organs and tissues noninvasively and with remarkable spatial resolution (at the submillimeter level for the most efficient devices).1,2 The intensity of the signal depends on the intrinsic parameters, namely, the local proton concentration as well as their longitudinal (R1) and transverse (R2) relaxation rates. Depending on the environment and their dynamics, the relaxation rates will change according to the nature of the tissue. The specificity of an MRI exam can be increased by injecting a contrast agent (CA), which will act as a nuclear relaxation booster.3 Typically, paramagnetic ions are able to enhance both R1 and R2 by catalyzing the return of water protons to the ground state, thus improving the image contrast where the ions are located. It is estimated that up to 40% of clinical MR examinations are performed by using paramagnetic chelates.4,5 However, as a consequence of the poor sensitivity of MRI, a high local concentration of contrast agent is needed to achieve visualizable contrast enhancement.6 To improve the relaxivity of a paramagnetic substance, a conventional strategy is to increase the molecular weight of the structure to increase the rotational correlation time value (i.e., τR).7,8 If the initial strategies implied the development of low molecular weight macrocycles that are able to interact with plasma proteins (e.g., albumin) in vivo,9,10 then current strategies tend towards the design of macromolecular systems resulting either from covalent grafting of a paramagnetic complex onto a polymeric support (e.g., dendrimers),6,11–14 or from the noncovalent confinement of the complex within porous and water-permeable supports,15–18 (e.g., apoferritin).16,17 Among the different developed systems, those resulting from the combination of a paramagnetic substance and inorganic nanoparticles have been extensively studied and have highlighted promising platforms that are able to deliver a large number of relaxophores at the site of interest.11,19–21 More specifically, silica NPs (SiO2-NPs) appear to be particularly interesting supports because they offer many advantages over other nanosystems, such as tunable and well-defined structures in terms of size, morphology and porosity.11,22 In addition, their biocompatibility,12,23,24 ease of surface modification,25–31 and possibility of entrapping molecules in their cores render SiO2-NPs particularly valuable materials for biomedical purposes.11,25,32–36 If most of the systems using silica as a matrix proposed the covalent grafting of Gd-chelates onto the surface of porous particles, the use of amorphous silica for the noncovalent confinement of Gd complexes has been poorly studied despite the fact that such a strategy frees up the surface for other molecules of interest (e.g., peptides, antibodies, siRNA, …).For this study, we proposed to encapsulate the clinically used Gd-HP-DO3A inside the silica matrix. To ensure aqueous colloidal stability, the silica surfaces were modified by PEGylated silane chains. To introduce functional groups of interest, we decided to explore a non-conventional strategy which implies the use of a carboxylated photoactivable moiety (i.e., 3-phenyl-3-(trifluoromethyl)diazirine) in solution. By proceeding this way, we see an efficient way to anchor ligands exclusively on the external soluble parts of particles. Because of its complementarity with MRI, we chose to focus our efforts on the development of an optical imaging probe, by grafting a near-infrared luminescent dye (NIR-dye) onto the surface of the resulting nano-objects. Such modification allowed to design a prototype for multimodal imaging as a proof of concept for functionalization procedure. The performance of the as-prepared nanoplatform as a potential probe was evaluated according to physico-chemical parameters and multimodal imaging experiments.
Experimental section
Materials
Hexan-1-ol (99%), Triton™ X-100, ammonium hydroxide (30–33%), hydrochloric acid (37%), sulfuric acid (95%), gadolinium atomic absorption standard, acetone (97%), sodium phosphate monobasic monohydrate (NaH2PO4·H2O, 98%) and sodium phosphate dibasic (Na2HPO4, 99%) were purchased from Sigma-Aldrich (Belgium). Diethyl ether (99%) was obtained from Chem-Lab (Belgium). Tetraethoxysilane (TEOS, 99.9%) was obtained from Fluka (Belgium). Cyclohexane (99.9%) and acetonitrile (99.8%) were provided from VWR (Belgium). 3-[Methoxy(polyethylenec oxide)propyl] (triethoxysilylpropyl)carbamate (PEG, 2000 g mol−1) was purchased from Biochempeg Scientific (USA). 4-(3-Diazirin-3-yl)benzoic acid (3H-DiazBA, 97%) was obtained from TCI Chemicals (Belgium). N-(3-Dimethylaminopropyl)-N′-ethylcarbodiimide hydrochloride (EDC·HCl) was purchased from Roth. ProHance® (Gd-HP-DO3A) was purchased from Altana Pharma (Belgium). All the materials mentioned above were used directly, without any further treatment. Stirred cells and membranes for ultrafiltration (MWCO = 100 kDa) were purchased from Merck Millipore (USA) and Metrohm (Belgium). Membranes Spectre/Por® (MWCO = 12–14 kDa) for dialysis were acquired from VWR (Belgium).Preparation of empty silica nanoparticles as a control sample (PEG-coated SiO2-NPs)
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1, v/v), washed five times with acetone, and finally resuspended in DI water (5 mL). The resulting suspension was dialyzed (MWCO = 12–14 kDa) at 35 °C against DI water to remove physisorbed species. After the purification steps, the suspension was centrifuged (2700g, 5 minutes) to remove any agglomerates and the supernatant was carefully separated and characterized. 1H NMR (500 MHz, D2O) δ (ppm): [3.7–3.5] (m); 3.4 (s, 3H); 1.6 (q, 2H, J = 7.7 Hz); 0.6 (t, 2H, J = 7.9 Hz).
:1, v/v), washed five times with acetone, and finally resuspended in DI water (5 mL). The resulting suspension was dialyzed (MWCO = 12–14 kDa) at 35 °C against DI water to remove physisorbed species. After the purification steps, the suspension was centrifuged (2700g, 5 minutes) to remove any agglomerates and the supernatant was carefully separated and characterized. 1H NMR (500 MHz, D2O) δ (ppm): [3.7–3.5] (m); 3.4 (s, 3H); 1.6 (q, 2H, J = 7.7 Hz); 0.6 (t, 2H, J = 7.9 Hz).
Surface functionalization with carboxylic groups through photochemistry process (functionalized PEG-coated-SiO2-NPs)
Carboxylic functions were introduced onto PEG-coated SiO2 nanoparticles by mean of a photochemical treatment in the presence of a carboxylated diazirine system. The UV-sensitive 4-(3-diazirin-3-yl)benzoic acid (3H-DiazBA) crosslinker was solubilized in acetonitrile and the same volume of nanoparticle suspension (1%wt/v) was added to obtain a composition of acetonitrile/DI water (1:1, v/v) using 20 μmol of 3H-DiazBA per milliliter of PEGylated particles suspension. The homogeneous mixture was then irradiated with ultra-violet light (365 nm) at a distance of 3 cm from the light source. Irradiance experiments were performed as a function of time under ambient atmosphere and continuous magnetic stirring. Hereafter, the samples treated with 3H-DiazBA were then dialyzed against deionized water (MWCO = 12–14 kDa). The concentration of carboxylated groups onto fully PEG-coated SiO2-NPs was determined spectroscopically by following the UV absorbance peak of the linker (233 nm). The nanoparticle density was obtained gravimetrically after lyophilizing of the suspension followed by a drying step at 80 °C during 24 hours. 1H NMR (500 MHz, D2O) δ (ppm): 7.8 (d, J = 8.1 Hz); 7.4 (d, J = 8.1 Hz); [3.7–3.5] (m); 3.4 (s, 3H); 1.6 (q, J = 7.7 Hz); 0.6 (t, J = 7.9 Hz); 19F NMR (470 MHz, D2O) δ (ppm): 72.
Synthesis of paramagnetic silica nanoparticles (PEG-coated SiO2-Gd-NPs)
Stable paramagnetic SiO2-NPs were obtained by replacing water by a solution of Gd-HP-DO3A (1 mL) in the above-described protocol. To achieve optimal efficiency, three samples were prepared using 25, 50 and 100 mM Gd-HP-DO3A solution in deionized water. Optimizations were performed using three replicates in each condition.Preparation of multimodal (fluorescent and paramagnetic) particles by the insertion of NIR-dyes onto functionalized PEG-coated SiO2-Gd-NPs (NIR-dye PEG-coated SiO2-Gd-NPs)
The entire synthetic process for the preparation of such particles is summarized in Scheme 1.
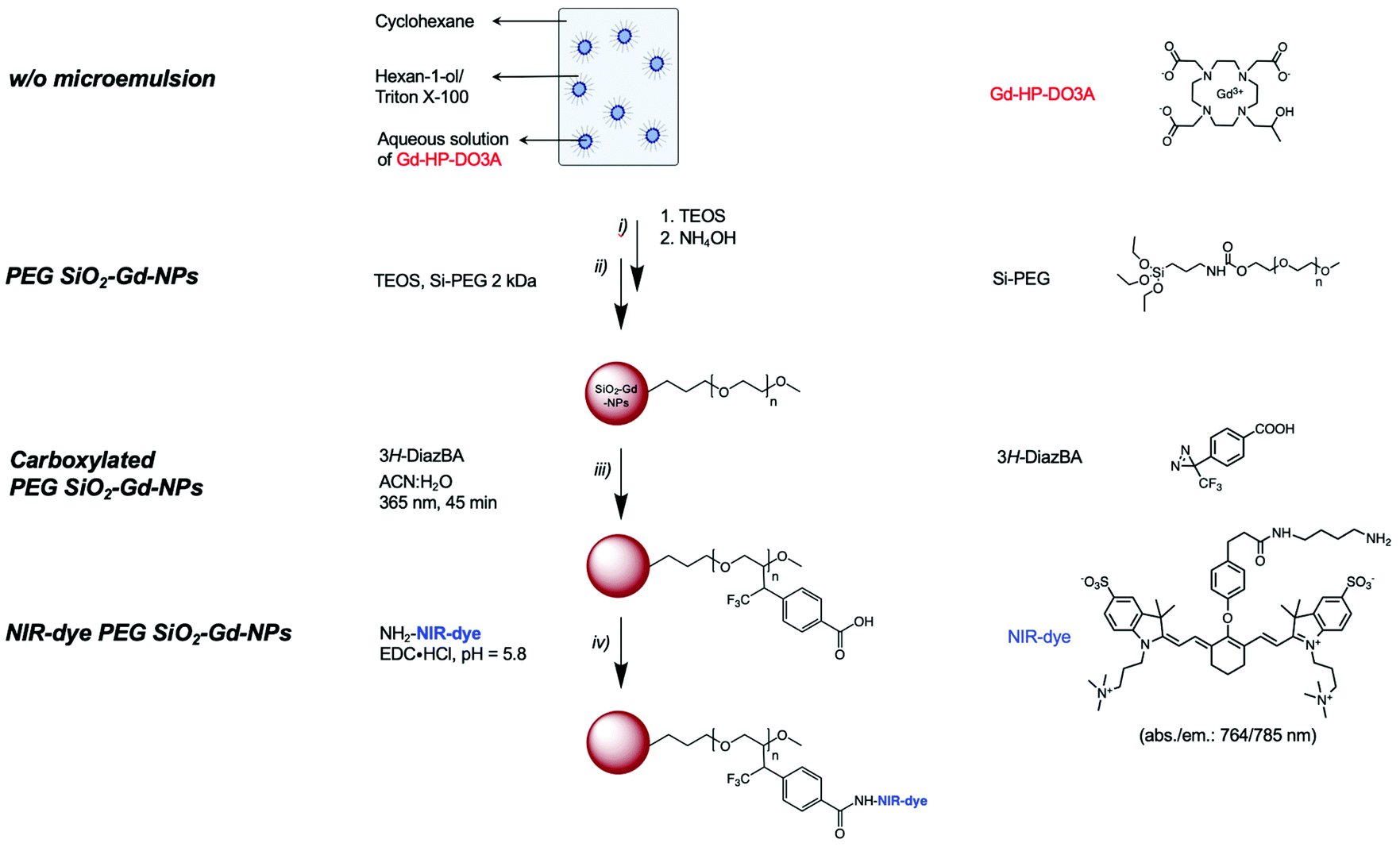 | ||
| Scheme 1 Schematic synthetic route leading to the proposed multimodal nanoplatform (NIR-dye PEG-coated SiO2-Gd-NPs). Step (i): Formation of the w/o microemulsion with encapsulation process during SiO2-NPs formation; step (ii): hydrolysis and condensation of the silica precursor into aqueous nanoreactors and PEGylation of the nanospheres; step (iii): functionalization through UV-sensitive carboxylated diazirine linker insertion and step (iv): grafting of compounds of interest (fluorescent molecule). | ||
Instrumentation
The specific surface area of empty SiO2-NP and SiO2-Gd-NP (25 mM in w/o) samples were measured at −196 °C by the nitrogen adsorption and desorption isotherms using a surface area analyzer (Micromeritics Gemini 2400 surface analyzer, Brussels, Belgium). Freeze-drying silica nanoparticles were first degassed at 150 °C for 48 h under argon gas flow before analysis. The surface areas were determined according to the Brunauer–Emmett–Teller (BET) and Barrett–Joyner–Halenda (BJH) procedures from the nitrogen adsorption sections of the resulting isotherms.
Thermogravimetric analyses (TGA) were performed on a TA Q500 device (TA Instruments, New Castle, United States) in an ambient atmosphere. Suspensions were dried, and the as-obtained powders (10 mg) were disposed of in a Platine crucible. Samples were first heated at 120 °C for 10 minutes at a temperature elevation rate of 10 °C per minute with an air flow of 25 mL min−1 to prevent solvent residues. Then, a temperature of 600 °C was reached (heating rate of 10 °C min−1) and maintained for 5 minutes. The mass loss was monitored over the process. The number of PEG chains (nPEG) anchored onto the silica surface per particle has been estimated through thermogravimetric analysis using the following equations as referred to in previously published references.40,41 Since the organic-to-inorganic ratio is measured from weight loss on the experimental TGA curve and the molecular weight of the coating molecule (MPEGw) is known as shown below (eqn. (1))
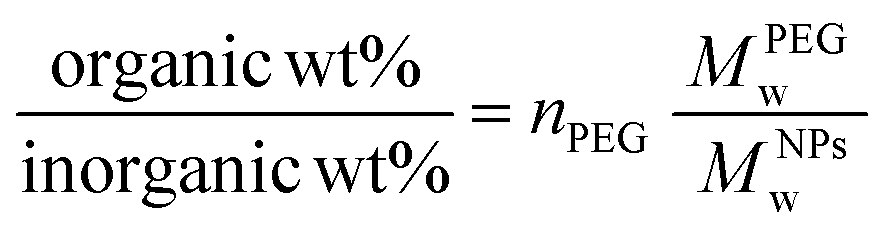 | (1) |
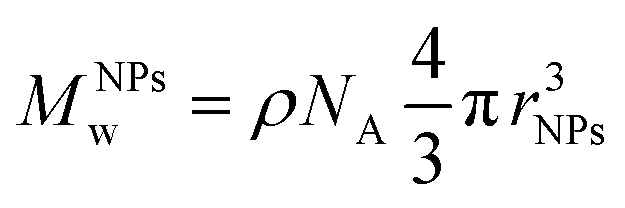 | (2) |
Finally, the PEG chain density onto a single particle (DPEG [chain per mm2]) was estimated as (eqn (3))
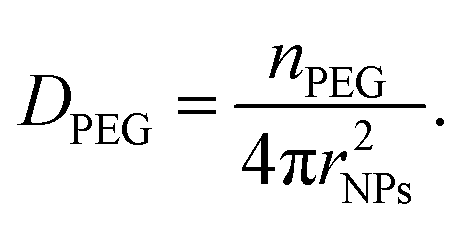 | (3) |
Proton and fluorine NMR spectra were acquired using a 500 MHz Bruker Avance II instrument (Bruker, Germany) operating at 500.13 MHz (1H) and 470.59 MHz (19F). NMR samples were obtained by mixing 540 μl of concentrated NP suspension and 60 μL of deuterated solvent. The spectra were recorded at 25 °C in H2O/D2O (10%) using water 1H NMR peak suppression sequence. Chemical shifts (δ) are presented in parts per million (ppm) referenced to D2O (4.79 ppm, 1H NMR) as the internal reference. Coupling constants (J in Hz) on 1H NMR are given in hertz and the resonance multiplicity is described as s (singlet), d (doublet), t (triplet), q (quintuplet), m (multiplet).
NMR self-diffusion measurements as diffusion ordered spectroscopy (DOSY-NMR) experiments were conducted at 25 °C on a 600 MHz Bruker Avance spectrometer (Bruker, Germany) equipped with a field gradient probe using a 2D STE-LED (stimulated echo-longitudinal eddy current delay) sequence with a gradient pulse length (δ) of 4 ms, a diffusion time (Δ) of 500 ms and gradients ranging from 2% to 95%. The translational diffusion coefficients (D in cm2 s−1) were obtained using the following relation,42 (eqn (4))
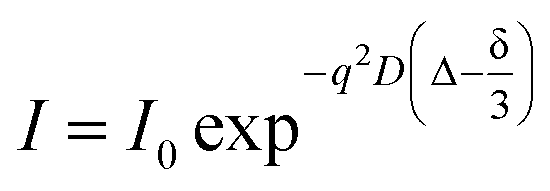 | (4) |
Spectroscopic measurements were recorded at room temperature. The crosslinker molecule was quantified by UV absorbance measurements (230 nm) after solubilizing various concentration of 3H-DiazBA for establishing a linear calibration curve (coefficient of determination, R2 > 0.99). The absorbance of the NP samples treated with the linker in CH3CN/H2O (1:1, v/v) was compared to determine their concentration. Absorbance properties were characterized by a Lambda 35 UV-visible spectrometer (PerkinElmer, Wellesley, MA) with a scanning rate set at 480 nm min−1. Silica contribution was subtracted from the absorbance background. Following the similar procedure described in the quantification of the linker, the content of grafted dyes was reached (764 nm). Emission spectra in the NIR range were recorded on a PerkinElmer LS 55 spectrofluorimeter equipped with a R928 photomultiplicator (PerkinElmer, USA). The excitation and emission slits were fixed to 10 nm and the scanning speed was set to 100 nm min−1.
Infrared (IR) spectra were recorded on a PerkinElmer FTIR Spectrum 100 (Manchester, UK).
The total gadolinium ion concentration of each sample was determined by inductively coupled plasma-atomic emission spectroscopy (ICP-AES) analysis under argon plasma on a Varian Liberty Series II instrument (Varian Inc., Palo Alto, USA). Briefly, the samples (500 μL) were digested in a mixture of H2SO4 (95%; 300 μL)/HCl (37%; 300 μL) and exposed to microwave radiation using a Milestone MLS 1200 Mega microwave laboratory station (Analis, Belgium). The volumes of the resulting acid-treated samples were adjusted to 5 mL using deionized water. Prior to quantification, standards were prepared from Gd3+ single element standard (1001 μg mL−1) on diluted H2SO4/HCl solution. Calibration curves (R2 > 0.99) using different concentration of ions standards were used to determine the gadolinium ion concentration of our samples for ICP.
Relaxometric properties including longitudinal (R1) and transversal (R2) relaxation rates of the aqueous suspension of each sample were studied adjusting the temperature to 37 °C (±0.1 °C). Both R1 and R2 were measured at 20 MHz (0.47 T) and 60 MHz (1.41 T) on Bruker Minispec mq-20 and mq-60 analyzers (Karlsruhe, Germany), respectively. Nuclear magnetic resonance dispersion (NMRD) profiles were recorded using a fast field cycling Spinmaster relaxometer (STELAR, Mede, Italy) measuring longitudinal relaxation rate (R1) over the range of proton Larmor resonance frequencies from 0.01 to 40 MHz (0.24 mT to 1 T).
As silica nanoparticles contribute to the water relaxation rate at low field, NMRD profile of empty non-paramagnetic SiO2-NPs was recorded, fitted and the diamagnetic contributions were subtracted from the recorded profile obtained for paramagnetic SiO2-NPs. Specific parameters were fixed (q = 1; r = 0.31 nm; τM = 500 ns). Approximated parameters (including diffusion coefficients, DNMRD) were determined by fitting these NMRD curves numerically with the MINUIT program. Whereas the theoretical adjustment of the profile of conventional free Gd-HP-DO3A was fitted according to the conventional Solomon–Bloembergen–Morgan relaxation model. R1, R2 values and longitudinal relaxation rates curves were normalized per paramagnetic gadolinium ion concentration. The relaxivity as defined as the enhancement rates of water protons per 1 millimolar per L of the paramagnetic center (ri in mM−1 s−1) was determined through the following relation (eqn (5))
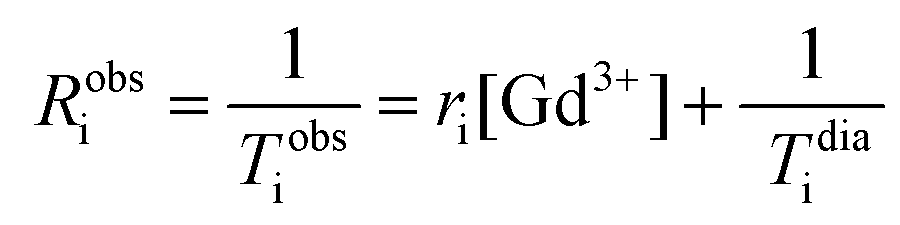 | (5) |
000 ms) was acquired using different TEs (210 and 70 ms), NEX 1, MTX 160 × 96, FOV = 4.00 × 2.50 cm, slice thickness = 1.25 × 1.25 mm. At 9.4 1 Tesla: RARE method (TR = 300 ms, TE = 12.0 ms, NEX = 4, MTX = 192 × 128, FOV = 4.00 × 2.50 cm, slice thickness = 1.25 × 1.25 mm); MSME T2-weighted sequence (TR = 10000 ms) was acquired using different TEs (85.5 ms), NEX 1, MTX 160 × 96, FOV = 4.00 × 2.50 cm, slice thickness = 1.25 × 1.25 mm. Optical phantoms were acquired with a Photon Imager Optima (Biospace Lab, Paris, France) after excitation at 712 nm for a 797 nm emission and data were analyzed using M3Vision software (Biospace Lab) by drawing ROI around each well of the plate. The fluorescent signal was expressed as photons per second per square centimeter per steradian (ph per s per cm2 per sr). Photoacoustic imaging, as multispectral optoacoustic tomography (MSOT), experiments were carried out on a InVision 256-TF instrument (iThera Medical GmbH, Munich, Germany). Samples were excited from 660 to 930 nm (5 nm steps) and data were analyzed using viewMSOT 4.0 software (iThera Medical). Optoacoustic signals were extracted as MSOT arbitrary unit (a.u.). All phantom images were recorded at several concentrations in order to compare the contrast generated by our agent.
Due to stronger contrast effect at low field, main MRI experiments were carried out at 1 T, with an additional pre-injection and day 1 post-injection imaging session at 9.4 T. Signal enhancement at this higher field was nevertheless illustrated in some preliminary experiments after i.v. injection of a double dose (8.6 μmol Gd per kg) compared to 1 T MRI experiments, to compensate for the expected loss of contrast effect. This dose was fractionated in four sub-doses contained in roughly equal volumes (in fact corresponding to an injection of the total volume (200 μL) in four progressive steps, with an image acquisition after each step) to get basic visual information about dose-related signal enhancement properties at high pre-clinical field in one mouse. Two other mice underwent a follow-up of signal enhancement in first hours after injection of this double dose (total n = 3). Parameters at 9.4 T (RARE T1-weighted: TR = 450 ms, TE = 8.1 ms, NEX = 6, MTX = 256 × 320, FOV = 3.30 × 4.50 cm, slice thickness = 1.00 mm). Additional sequences were made with the aim of detecting a nanoparticle-induced negative contrast (T2-weighted), focused on liver T2 value (before and at day 1 after experiments at 1 T as above mentioned) and in renal pelvis, facilitating negative enhancement depiction due to a background initially showing hypersignal on the T2-W image (in the first hours post-double dose injection follow-up), also considering the fact that T2 effects are stronger at higher field (shorter T2). Other parameters at 9.4 T (RARE T2-weighted: TR = 4270 ms, TE = 75 ms, NEX = 5, MTX = 192 × 256, FOV = 3.30 × 4.50, slice thickness = 1.00 mm; MSME (multi-slice-multi-echo): TR = 2003 ms, TE = 7.9 ms (×24, thus up to 189.8 ms), NEX = 2, MTX = 192 × 256, FOV = 3.30 × 4.50 cm, slice thickness = 1.00 mm).
External references were prepared in flexible tubes (polyethylene 1.14 mm internal diameter), containing Dotarem (Guerbet, 500 mM Gd in stock solution) diluted in water (800×; thus 0.625 mM Gd), and then mixed with Deuterium oxide (D2O) for decreasing the number of unpaired protons (able to respond to RF pulse). One volume diluted Dotarem + 1 (9.4 T experiments) or 2 (1 T experiments) volumes of D2O. Images were collected and analyzed using the ParaVision software (Bruker), containing the Image Display tool, allowing for quantifications (signal intensity, relaxation time (depending on the sequence)) by drawing of regions of interest (ROI) at adequately selected place on the image. ROI were placed in the liver, renal pelvis and inferior vena cava, as identified on image slices. Positive signal enhancement was calculated from signal intensity measurements in specific ROIs divided by a signal intensity value measured in the external reference (“signal/external ref” ratio). Enhancement was then represented as an evolution in percentage calculated from the pre-contrast “signal/external ref” ratio.
Results and discussion
Surface study: preparation of stable PEG-coated SiO2-NPs
As depicted in Scheme 1, the preparation of the targeted systems implies:i. The encapsulation of paramagnetic chelates within a silica matrix;
ii. The PEGylation of the resulting objects;
iii. The introduction of functional group by mean of photochemical treatment in the presence of a diazirine clip;
iv. The covalent grafting of a NIR-dye onto the surface of the carboxylated particles.
In order to facilitate further physico-chemical characterizations, we decided to optimize the different surface modification steps (i.e., PEGylation and the “clipping” step) by using “empty” non-paramagnetic NPs.
Regarding the synthesis of SiO2-NPs, sol–gel methods using tetraethyl orthosilicate (TEOS) as a precursor are often preferred due to the ability to use mild experimental conditions and the superior control they provide concerning the properties of the resulting objects.43 Considering the well-described mechanism of the hydrolysis/condensation of TEOS units, it is obvious that experimental parameters such as pH, concentration, temperature or the type of solvent will have a significant influence on the hydrolysis/condensation kinetics and will impact the final characteristics of the resulting particles.43–45 Bearing these considerations in mind, we have chosen to carry out the reaction in water-in-oil (w/o) microemulsion type systems according to a well-described procedure.46–49
The general process begins with the formation of a reverse w/o microemulsion by mixing cyclohexane/hexanol/Triton X-100/water in a 9/2/2/1 volume ratio. After the formation of the microemulsion, TEOS is added to the mixture and then hydrolyzed by alkalizing the medium through the addition of ammonia. Several studies have described the excellent properties obtained by using a PEG-based coating, as a consequence of its hydrophilicity, biocompatibility and stealth properties conferred to the bearing nanosystem.50–52 Indeed, it is already well documented that a PEG coating, which helps to reduce nonspecific blood protein adsorption, acts as a protective layer for phagocyte adhesion.51,53,54 Consequently, the surface of the nanoparticles was treated exclusively with PEG-silane. A chain length of 44 monomer units (PEG 2 kDa) was considered for the development of the targeted multimodal platform. At the end of the reaction, the SiO2-NPs were isolated by precipitation in an acetone/ether mixture, washed several times with acetone and finally redispersed in demineralized water before dialysis and characterization.
Good control over the size distribution was confirmed by transmission electron microscopy (TEM) analysis, for which uniform spherical objects with a mean diameter DTEM = 28.26 ± 3.18 nm (PDITEM: 1.04) were observed (ESI, Fig. S1†). The stability and hydrodynamic diameter were evaluated by PCS. These measurements confirmed the production of well-dispersed nanostructures characterized by a single, narrow population displaying a mean hydrodynamic diameter close to 110 nm (Fig. S2;†DDLSH: 114 nm; PDIDLS: 0.17).
NMR (Fig. 3a) analysis were used to follow surface modification. First, the grafting of PEG onto the surface was assessed by the presence of the protons expected signals in NMR experiment. Thus, in addition to the presence of a signal at 3.6 ppm corresponding to the resonance of the methylene protons of the PEG unit, we noted the presence of signals at 0.6 ppm and 1.6 ppm attributable to the protons of the methylene groups at α and β positions of the silica atom, respectively. To determine the nature of this grafting procedure (adsorption or covalent), a diffusion ordered spectroscopy (DOSY) sequence was used. The polymer, whether free-moving or restricted by anchoring to a nanoscale support, was characterized by different translational movements, which were characterized by self-diffusion coefficients (D [m2 s−1]) at a given temperature. In this particular case, the characteristic signals of PEG were compared to distinguish them according to their molecular translational diffusion coefficient value (Fig. 1). According to the interpretation of the DOSY spectrum, it appeared that dialyzing the sample at a slightly higher temperature (i.e., 35 °C vs. room temperature) is enough to efficiently remove all physisorbed species. It should be noted that any purification methods performed at room temperature failed to remove free PEG, as indicated by the bimodal DOSY profiles, and percentages as high as 80% of ungrafted PEG were estimated. After thermal treatment, the fitting appeared linear with an estimated diffusion coefficient value of 4.1 × 10−7 cm2 s−1.
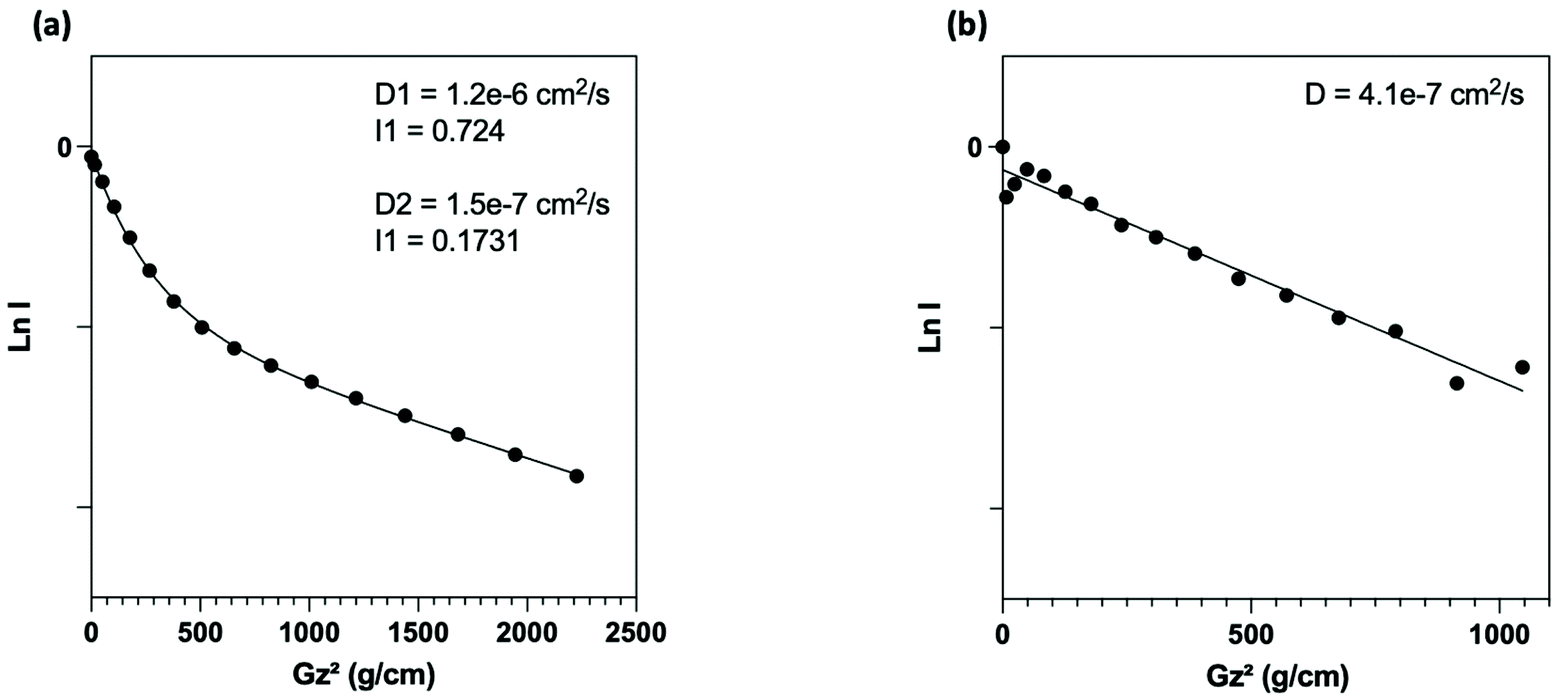 | ||
| Fig. 1 DOSY-NMR experiments. Semilogarithmic plot of echo intensity vs. the square of the gradient strength recorded on PEGylated SiO2-NP systems after dialyzing the samples (a) at room temperature; (b) at 35 °C. | ||
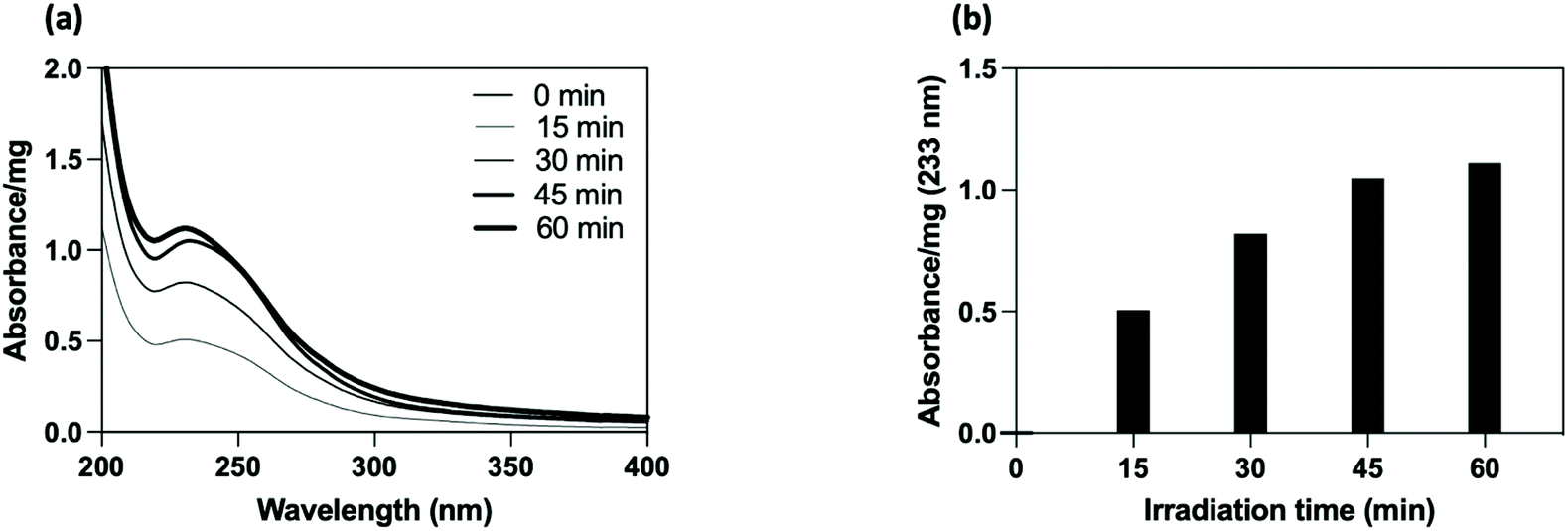 | ||
| Fig. 2 UV irradiation time study. (a) UV spectra after purification and (b) plot result of the linker absorbance (233 nm) on PEG-coated SiO2-NPs normalized per mg of particles for different time of exposure. | ||
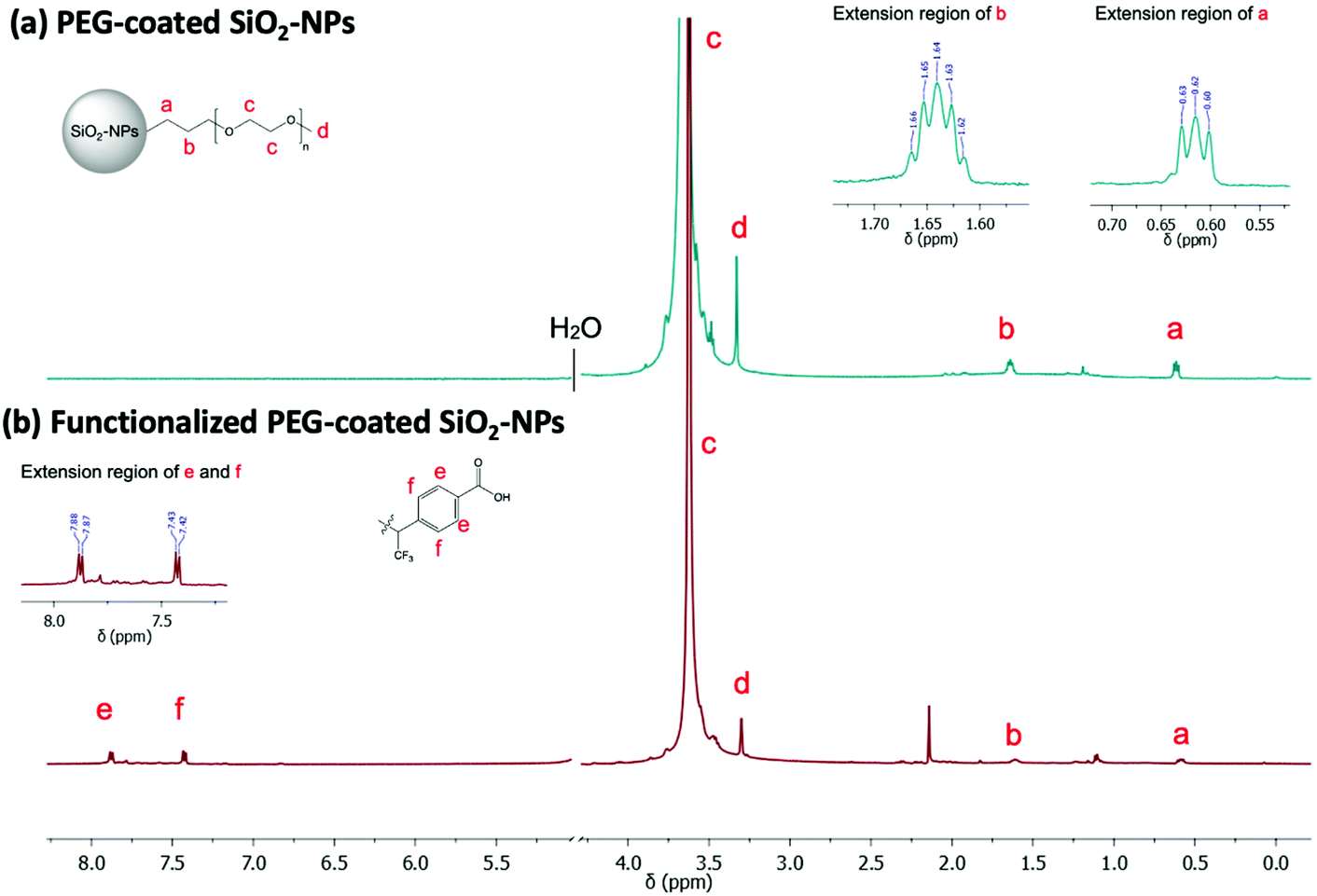 | ||
| Fig. 3 Stacked 1H spectra of empty PEG-coated silica nanoparticles (D2O, 500 MHz). 1H NMR spectrum (a) before and (b) after photografting (365 nm, 45 min) and purification step. NMR peak at 2.2 ppm (bottom spectrum) arises from acetone. | ||
Thermogravimetric analyses (TGAs; ESI, Fig. S3†) completed the characterization by allowing a rough estimation of the PEG grafting densities at approximately 0.5 PEG per nm2.
Surface functionalization with carboxylic groups
The PEG-coated SiO2-NPs were functionalized by a photochemical treatment using a carboxylated UV-sensitive crosslinker. The principle was to take advantage of a bi-reactive linker which owns an UV light activable function (diazirine) on one side and a reactive group (carboxylic function) on the other one. Under appropriate light, the linker leads to photogenerated carbene species which randomly insert on compound located at its close vicinity (i.e., in C–H bonds from the polymer coating chain). In the present study, the grafting procedure implied the use of 4-[3-(trifluoromethyl)-3H-diazirin-3-yl]benzoic acid (3H-DiazBA) linker with a concentration of 2 μmol of 3H-DiazBA per milligram of PEGylated particles, and activated by irradiating the mixture at 365 nm at room temperature and directly in liquid media.55–57 The introduction of the linker on the particles was followed by UV spectroscopy thanks to appearance of a characteristic absorption peak at 233 nm (Fig. 2a). As shown by the irradiation time study, efficient grafting were reached within less than an hour (45 min, 95% in comparison to 60 min) (Fig. 2b). To assess the covalent linkage and the efficacy of the purification steps, we used a non-irradiated sample as negative control (i.e., a particles/diazirine mixture that suffered all the experimental steps, except the irradiation). Hence, we attested the covalent insertion of the linker on PEG chain and showed success to eliminate ungrafted linker by dialysis.Using these conditions, we incorporated 110 nmol of covalently grafted linker per mg of PEGylated particles for further post-functionalization experiment.
The linker insertion was confirmed by FTIR and 1H/19F NMR spectroscopy. Using FTIR, we could notice, among other, the appearance of a band around 1710 cm−1 attributable to C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O vibrations of the introduced carboxylic functions (Fig. S4†). In the 1H NMR spectra, we observed two major doublets in the aromatic region (Fig. 3b) which are characteristic of the benzyl ring of the linker. Additional doublet signals were observed in the aromatic region as a result of the random distribution of the linker along the PEG chains. Other signal of interest may be confused with the PEG massif from 3.8 to 3.3 ppm as well as with the suspension solvent (H2O/D2O 10%). By integrating one of the aromatic doublets (7.8–7.4 ppm, 2H), and comparing its intensity to singlet of the PEG-methoxy group (3.6 ppm, 3H), we could estimate a ratio of one linker per PEG chain. Finally, the corresponding 19F NMR spectrum showed an intense peak at 72 ppm which may correspond to the three chemically and magnetically equivalents fluorine atoms of the linker (result not shown).
O vibrations of the introduced carboxylic functions (Fig. S4†). In the 1H NMR spectra, we observed two major doublets in the aromatic region (Fig. 3b) which are characteristic of the benzyl ring of the linker. Additional doublet signals were observed in the aromatic region as a result of the random distribution of the linker along the PEG chains. Other signal of interest may be confused with the PEG massif from 3.8 to 3.3 ppm as well as with the suspension solvent (H2O/D2O 10%). By integrating one of the aromatic doublets (7.8–7.4 ppm, 2H), and comparing its intensity to singlet of the PEG-methoxy group (3.6 ppm, 3H), we could estimate a ratio of one linker per PEG chain. Finally, the corresponding 19F NMR spectrum showed an intense peak at 72 ppm which may correspond to the three chemically and magnetically equivalents fluorine atoms of the linker (result not shown).
Encapsulation study: preparation of paramagnetic nanostructures (PEG-coated SiO2-Gd-NPs)
In addition to providing better control over the size of nanoparticles, water-in-oil microemulsion is particularly effective for the encapsulation of water-soluble molecules within the silica matrix.48,58 While the encapsulation of fluorescent products has already been frequently studied,25,32–35 gadolinium-based complexes are much less referenced, as this type of compound is preferentially grafted on the surface of the matrix. If this process is highly effective in improving the relaxivities of the complex, it should be emphasized that a modification of the ligand structure is often requested to introduce the needed functional group to allow coupling, which can lead to a decreased stability of the complex. In contrast, the confinement of paramagnetic complexes within the silica matrix does not require any structural modifications and allows the use of clinically used complexes. Because of its commercial availability, great stability, and efficiency as a contrast agent, we chose to encapsulate Gd-HP-DO3A.To do so, water was replaced by an aqueous solution of the contrast agent during the emulsion step. To reach the best relaxometric properties, the initial concentration of gadolinium was varied from 25 mM, 50 mM to 100 mM. Each experiment was performed in triplicate, and each sample was characterized by PCS, TEM and relaxometry. Whatever the conditions, stable colloidal suspensions were obtained as indicated by PCS. Strikingly, an increase in the mean hydrodynamic diameter was observed when increasing the initial Gd-chelate concentration (Fig. 4a and b). TEM images (Fig. 4c–e) confirmed this trend by showing particles sizes ranging from 16 nm to 26 nm (DTEM = 15.9 ± 2.8 nm; PDITEM = 1.1; DTEM = 18.1 ± 3.1 nm; PDITEM = 1.1; DTEM = 25.6 ± 5.7 nm; PDITEM = 1.2). It may appear difficult to explain the reason of such size differences since microemulsion are dynamic systems. However, one must remember that the several experimental parameters such as the intra-micellar pH or the ionic strength have a strong influence on the hydrolysis and condensation of TEOS units, thus on the final particles properties.48
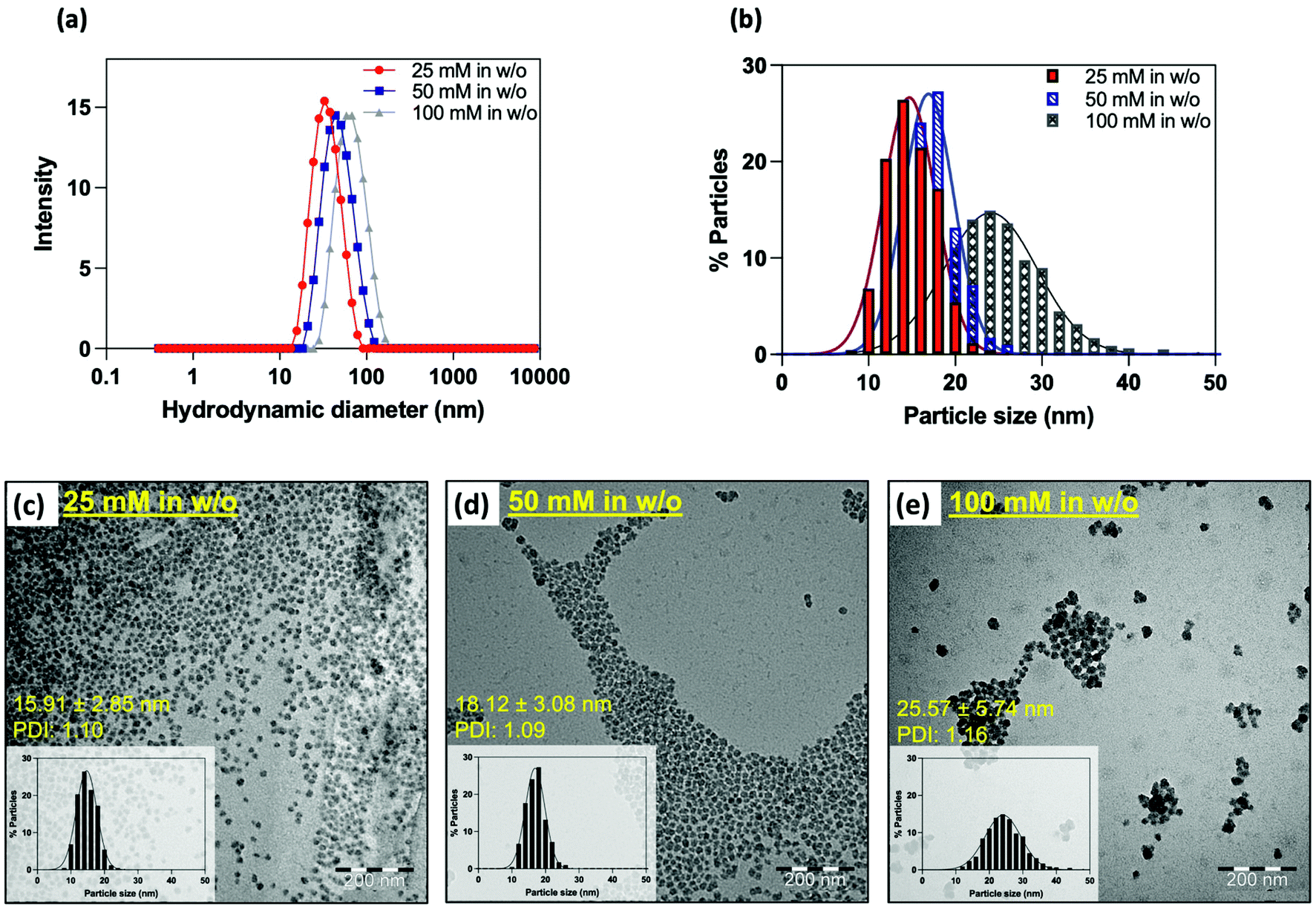 | ||
| Fig. 4 Comparison of the NP size obtained as a result of the different encapsulation study. (a) Hydrodynamic size distributions (DLS, intensity-weighted) of NP16/18/25 nmGd particles in aqueous suspension. (b) Histogram of particle size distribution from TEM analysis of the synthetized NP16/18/25 nmGd. The mean particles diameter from statistical analysis of NP16 nmGd (25 mM in w/o) was 15.91 ± 2.85 nm, TEM polydispersity index (PDITEM): 1.10; DTEM of NP18 nmGd (50 mM in w/o) = 18.12 ± 3.08 nm (PDITEM = 1.09); DTEM of NP25 nmGd (100 mM in w/o) = 25.57 ± 5.74 nm (PDITEM = 1.16). TEM images (scale bar: 200 nm) of the corresponding PEG-coated SiO2-Gd-NPs obtained with (c) 25 mM in w/o, (d) 50 mM in w/o and (e) 100 mM in w/o (scale bare: 200 nm). Insets show the TEM size distributions determined by statistical analysis. Average DTEM diameters were estimated over multiple TEM images recorded for each triplicate. | ||
The proton longitudinal relaxivities of the different samples were determined by means of the acquisition of their NMRD profiles measured at 37 °C on a per millimolar of Gd3+ basis (Fig. 5). First, it should be noted that all the prepared samples showed a significant increase in their r1 values when compared to those of free Gd-HP-DO3A in all frequency ranges. Especially at approximately 20 MHz, these values are approximately 5 to 10 times higher than the value observed for the free complex and are 3 to 5-fold higher at 60 MHz. The shape of the overall curves alongside the appearance of a bump above 10 MHz suggest effective encapsulation within the matrix as well as efficient water exchange between the inner-particle water molecule and the outer-particle water.
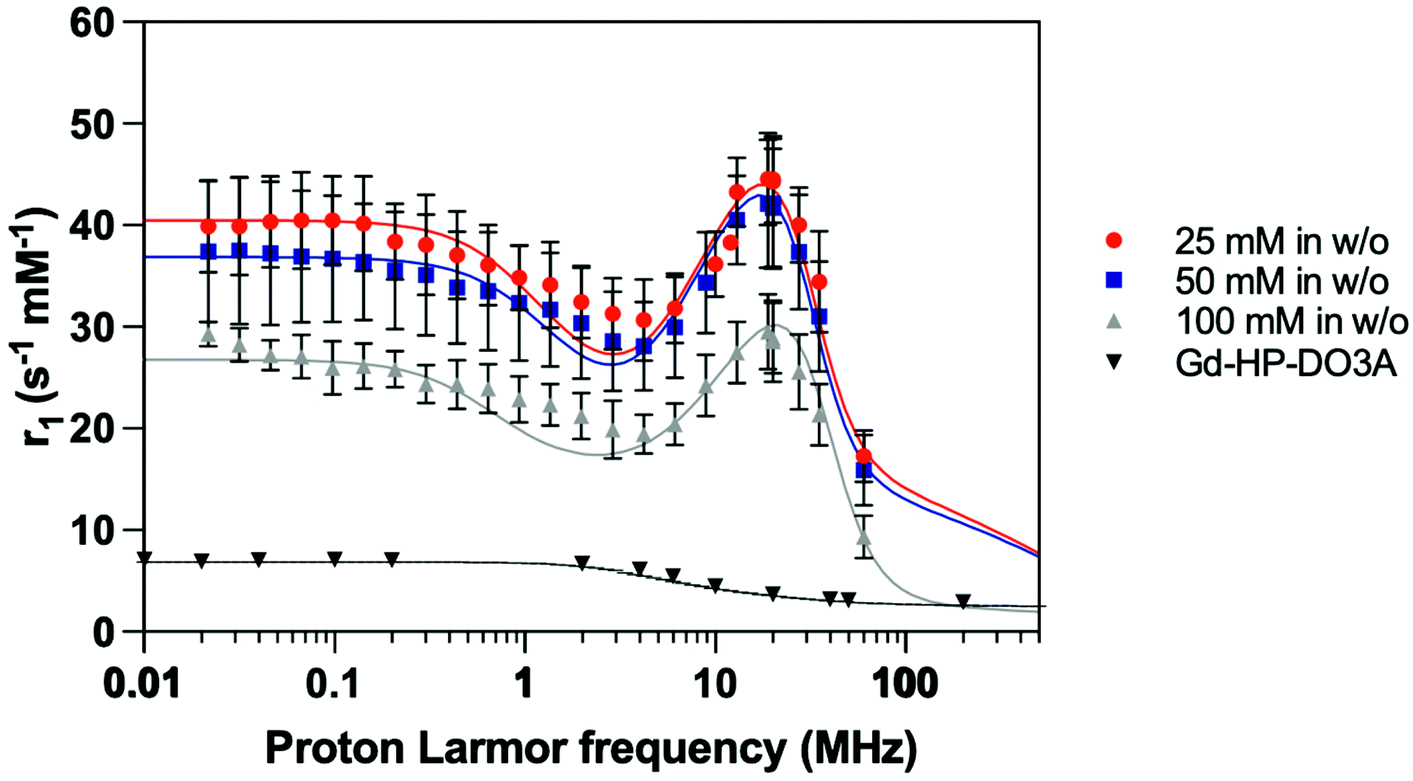 | ||
| Fig. 5 NMRD profiles (T = 37 °C) obtained for the paramagnetic complexes entrapped on PEG-coated SiO2-Gd-NPs (16, 18 and 25 nm) as a result of [Gd-HP-DO3A] in w/o procedure (triangles, squares and squares on edge, respectively) and free Gd-HP-DO3A complex (circles) in aqueous solution. Longitudinal (r1) relaxometric measurements plots are normalized per paramagnetic gadolinium ion concentration (s−1 mM−1). | ||
A decrease in the r1 values when increasing the size of the particle was observed as described elsewhere.36 Since the Gd3+ concentration was determined by ICP-AES for each sample, an estimation of the amount of Gd-HP-DO3A chelates entrapped inside a single silica particle was approximated assuming that SiO2-NP cores are monodisperse (ESI†). Herein, smaller NP16 nmGd systems carried out about ∼80 complexes within their structure, whereas NP18 nmGd contained about more than ∼130 and larger NP25 nmGd about ∼300. Thus, the r1 increase may be attributed to the smaller particle size observed when incorporating a lower initial chelate concentration. Considering that the observed boost in relaxivity is the consequence of an increase in the rotational correlation time (inner-sphere effect), but also of an increase in the diffusion correlation time for water molecules (outer-sphere effect), which are confined and forced to interact for a longer period with the Gd complex adsorbed to the inner pore surface,59 one may suggest that when decreasing the particle size, the chelates located on the outer particle shell contribute to the relaxation to a higher extent in comparison with the contribution of complexes embedded more deeply (Table 1). The coefficients of diffusion approximated from the fitting of NMRD profiles (Table 1; DNMRD) confirm this trend by showing a significant increase (approx. 7 times) when comparing NP16 nmGd and NP25 nmGd.
| Samples (conditions during encapsulation) | Hydrodynamic diameter (intensity-weighted distribution) | Particle diameter | Relaxometric measurements (T = 37 °C) (s−1 mM−1) | D NMRD (cm2 s−1) | ||||
|---|---|---|---|---|---|---|---|---|
| B 0 = 0.47 T | B 0 = 1.41 T | |||||||
| D DLSH (nm) | D TEM (nm) | PDI | r 1 para | r 2 para | r 1 para | r 2 para | ||
| 25 mM | 33.4 ± 1.2 | 15.9 ± 2.8 | 1.1 | 44.9 ± 2.4 | 61.1 ± 6.2 | 17.3 ± 2.5 | 127 ± 3 | 2.5 ± 0.1 × 10−6 |
| 50 mM | 39.1 ± 4.6 | 18.1 ± 3.1 | 1.1 | 42.2 ± 3.9 | 58.8 ± 11 | 15.9 ± 3.5 | 105 ± 3 | 2.8 ± 0.2 × 10−6 |
| 100 mM | 66.9 ± 4.7 | 25.6 ± 5.7 | 1.2 | 28.9 ± 3.4 | 43.8 ± 3.2 | 9.3 ± 2.1 | 95 ± 4 | 1.9 ± 0.04 × 10−5 |
| Gd-HP-DO3A | — | — | — | 3.6 | — | 3.0 | — | 3.3 × 10−3 |
The observations suggested efficient physico-chemical parameters of the synthesized PEG-coated SiO2-Gd-NPs systems using [Gd-HP-DO3A] = 25 mM in the encapsulation step. As a result, monodisperse and smaller NPs with a mean SiO2-Gd-NPs particles size of 15.9 ± 2.8 nm (PDITEM: 1.1). From the analysis of the relaxometric properties, it appears that the proposed system shows similar or even better longitudinal relaxivities than systems decorated by Gd-chelates on their outer-shell.25
The nitrogen adsorption and desorption isotherms are shown in Fig. SI 5.† Even if the SiO2-NPs produced by reverse microemulsion are supposed to be solid and not porous, the shape of the obtained isotherms suggests the formation of a mesoporous material.60 The w/o encapsulation procedure leads thus to spherical nanomaterial characterized by small pores (<2 nm) which enable the diffusion of water molecules while restricting the diffusion of the chelates outside the matrix.
Preparation and characterizations of the proposed multimodal nanoplatform (NIR-dye PEG-coated SiO2-Gd-NPs)
With respect to MRI, optical imaging (OI) appears to be a complementary technique characterized by its high sensitivity. Combining MRI and OI modalities within the same probe offers double tracking possibilities that are particularly useful for preclinical imaging studies. Near infrared absorbing/emitting chromophores (650–950 nm) are good candidates for in vivo imaging because of their minimal tissue absorbance and poor signal-to-noise ratio due to tissue autofluorescence. For this work, we were interested to work with an analog of indocyanine green (i.e., the zwitterionic fluorophore indocyanine,37–39Scheme 1), which is characterized by its remarkable physico-chemical properties (water solubility, high quantum yield, etc.) and great photostability.To reach the desired platform, paramagnetic PEG-coated SiO2-Gd-NPs were prepared according to the conditions previously optimized. Briefly, Gd-HP-DO3A ([Gd3+] = 25 mM in w/o) was directly introduced during the microemulsion step, followed by the PEGylation. After purification, the PEG corona was modified by mean of a photochemical process using carboxylated diazirine. The targeted fluorescent properties were obtained by treating the resulting particles with the NIR-dye and EDC in Sorensen buffer. A similar sample without EDC was prepared in parallel in order to assess the efficiency of the grafting conditions and the purification process. After isolation and purification of the resulting suspension, the bimodal SiO2-NPs were characterized with respect to stability, morphology, relaxometry and spectroscopic properties. DLS measurements were performed before and after functionalization of the silica nanoparticles and assessed the stability of the suspensions in aqueous and physiological media (Fig. S6†). On the contrary to the control sample, the particles treated with EDC showed a strong absorption/emission signal after modification with the NIR-dye (Fig. S7a†). By absorption spectroscopy, we approximated a concentration of approximately 4 nmol mg−1 of particles which correspond to the functionalization yield (relative to the quantified carboxylic functions) of 15% (∼11 dyes per particle). Optical imaging analysis of the platform confirmed its detectability at concentrations as low as 5 nM in FLI after excitation at 687 nm (Fig. S7b and c†). The number of counted photons per unit of time per unit of area while taking into account the spherical field view of the camera (intensity; photons per s per cm2 per sr) were plotted as a function of scanning wavelength in Fig. S7c.†
As expected according to previous physico-chemical characterizations, phantoms MRI experiments revealed a strong increase (T1-W) or decrease (T2-W) in the signal at high magnetic fields (1 T and at 9.4 Tesla) in-per Gd3+ in comparison to water (Fig. 6) and to free Gd-HP-DO3A (Fig. S8†).
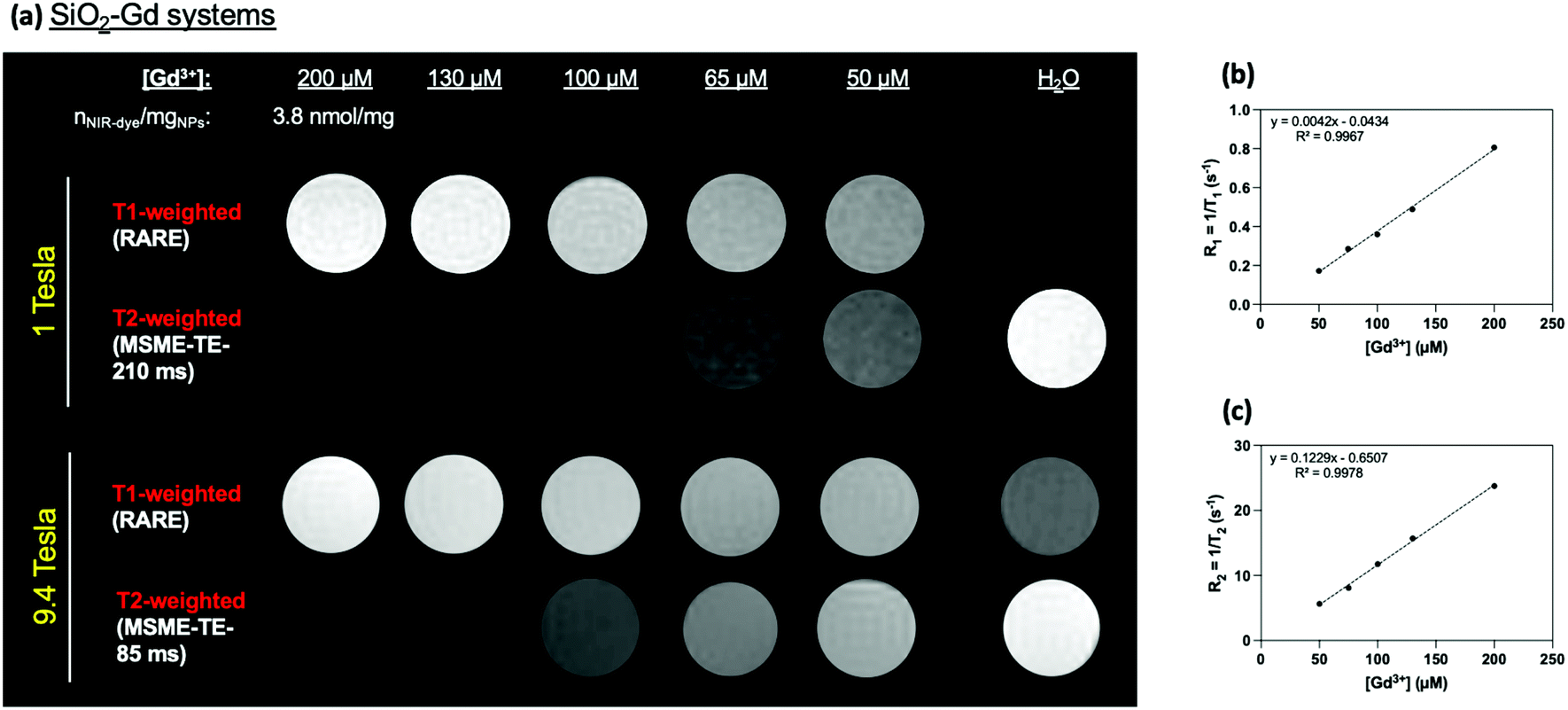 | ||
| Fig. 6 (a) T1- and T2-weighted phantom MR images recorded at 1 T (42 MHz) and 9.4 T (400 MHz) of the synthesized MRI/OI multimodal silica probe from left to right, of a suspension of 200 μM of gadolinium (11 mg of particles per mL; 46 μM of SiO2-NPs-anchored NIR-dye) and diluted samples. The images on the far right correspond to a phantom MR image of water (T1-W, RARE sequence and T2-W, MSME). Relaxation rates values showing the quantitative evolution of Gd3+ concentration-dependance using (b) T1-W and (c) T2-W (9.4 T). | ||
Small animals MRI and optical imaging
In vivo evaluations were performed using preclinical MRI scanners (1 T and 9.4 T) and FLI devices. For biodistribution studies, this version of NIR-dye PEG-coated SiO2-Gd-NPs was injected intravenously (i.v.) in the tail vein of healthy mice.Short-term biodistribution studies were performed by FLI for up to three weeks after i.v. injection with NIR-dye PEG-SiO2-Gd-NPs (0.9% NaCl) (Fig. S9†). FLI (phantoms, in vivo and ex vivo) experiments on the SiO2-NPs were carried out under excitation at 712 nm, and the fluorescence was collected at 797 nm. Measurement of signal intensity in the neck area were used as reference for blood circulating NIR-dye PEG-coated SiO2-Gd-NPs. Following the i.v. administration, the whole body of mice become fluorescent, and signal was increased up to 100% at 5 min, the first measured timing (Fig. 7a). Then it started to decrease progressively to only reach the pre-i.v. level at the later timepoints, ±25% at 1 day, ±7% at 7 days ± 2% at 21 days. This indicates a long persistence life of our NPs into the blood stream. FLI images showed a fast and strong accumulation into the bladder from the first minutes that followed the injection to 30 min after (signal increased of 600%). Then mice were allowed to wake up, and the miction emptied the bladder to a far lower level. Kidneys were also highlighted after injection and the signal was maximal at the first imaging time (i.e., 5 min) with an increase of signal intensity up to 150%. The residual signal significantly decreased of 80% after 24 h and reached the basal level (i.e., the neck level). From this timing, kidneys could not be distinguished anymore on images. Signal decreased followed the same speed as the rest of the body (Fig. 7a). Bladder and kidneys observations suggest a fast clearance of a fraction of NP by the urinary tract. The liver was also highlighted from the first timing, but the signal decreased, as compared to kidneys and the neck, much slowly. At later timings (1 day and later), the liver is still well highlighted and can be detected to 7 days and more.
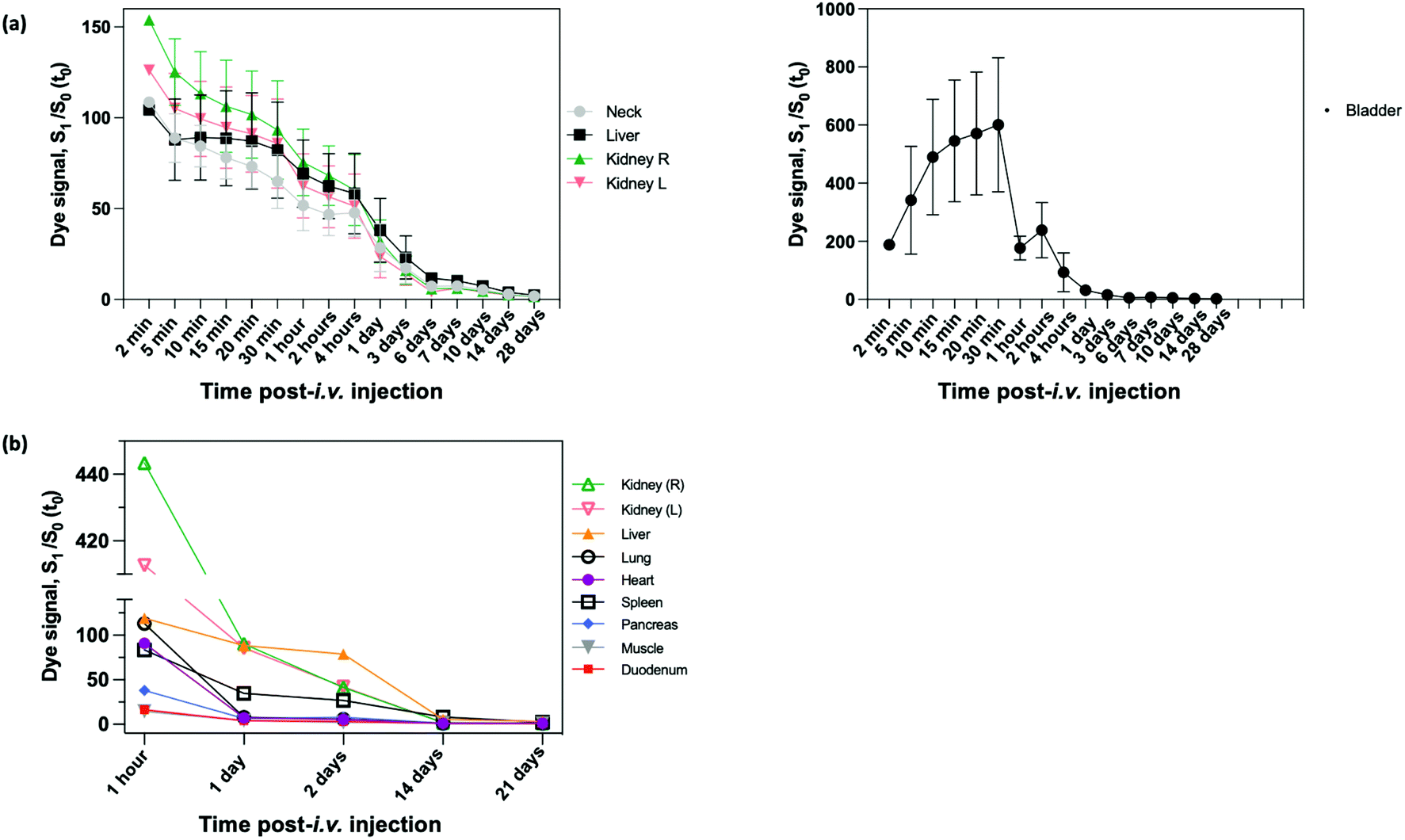 | ||
| Fig. 7 (a) Evolution of the in vivo fluorescence intensity in neck, liver, kidneys over time from pre-i.v., to 28 days post-injection (n = 3). (b) Ex vivo FLI signal decrease for 1 hour to 21 days after the injection (n = 3). | ||
At different timing after injection (1 h, 1, 2, 14 and 21 days), mice were sacrificed after the study, and different organs of interest were collected and then directly visualized by ex vivo FLI. Signal was compared to organs coming from a non-injected mouse and expressed as ratio. One hour after injection, kidneys are very strongly fluorescent with an increase of fluorescence intensities (more than 400 higher), while spleen, lungs, heart, liver showed a strong and similar fluorescence intensities (±100-fold increase) (Fig. 7b). Pancreas showed a moderate fluorescent intensity (40-fold increase), and duodenum and muscle the weaker ones (±15-fold increase). At 1 day, fluorescence intensities of heart, lungs, pancreas, duodenum and muscle decreased to reach similar values comprised between 5 to 10-fold. Interestingly, fluorescence intensities of kidneys dropped strongly to ±90-fold, while the liver on decreased only a little to reach also 90-fold. Fluorescence intensity of spleen decreased also more slowly than most of organs and reach 35-fold. Two days after injection, fluorescence intensity of kidneys continued to fall down, while the liver and spleen were stable. At 2 weeks, most organs (heart, lungs, muscle, duodenum, pancreas and kidneys) reached the control level, while the fluorescence signal of liver is still 5× higher, and interestingly the signal of spleen 8-fold time higher. Finally, spleen and liver are still weakly fluorescent (ratio of ±3) 3 weeks after the injection. Taking together, these ex vitro results confirmed in vivo observation. They showed the intense passage of NPs through kidneys just after injection, and a liver and spleen accumulation a later timepoints. As compared to muscle as reference organ, there is no specific accumulation into duodenum, lungs, heart, pancreas. Consequently, it seems that a fraction of NPs is cleared by renal clearance path in the minutes/hour following injection, and then the persistence fraction of NP accumulated into liver and spleen. As there is no signal in pancreas and duodenum, the biliary pathway could be excluded as excretion route.
Weeks after the injection, animals were sacrificed after the study and different organs of interest were collected in accordance to previously described ethical protocol of the CMMI. These include muscle, lung, heart, duodenum, spleen, pancreas, gall bladder, liver, adrenal gland (right and left) and kidney (right and left) were visualized by FLI (Fig. S10†). In the spleen from day 14 to day 21 a decrease of 75% was observed.
Vascular enhancement in the 3 first post-injection hours was monitored with MRI on inferior vena cava in SKH mice and in main organs implied in nanoparticles elimination and possible storage/metabolization (kidney, liver). Despite a low Gd dose (4.3 μmol Gd per kg) regarding usually injected contrast agents (tens of μmol per kg), satisfying MR T1 (positive) signal enhancement was measured in vena cava and represented as the evolution of signal intensity ratio between vena cava and an external reference, in percentage regarding the pre-injection value. Signal increase remained nonsignificantly fluctuating during the 3 h observation, thus stable around a 150% value. No more enhancement could be measured at day 1 after injection, suggesting that nanoparticles underwent restricted distribution (mainly vascular as could have been expected from their size) and apparently slow elimination from this compartment but significant enough to reduce their concentration below a detection threshold in MRI 24 h post-injection (Fig. 8a and b).
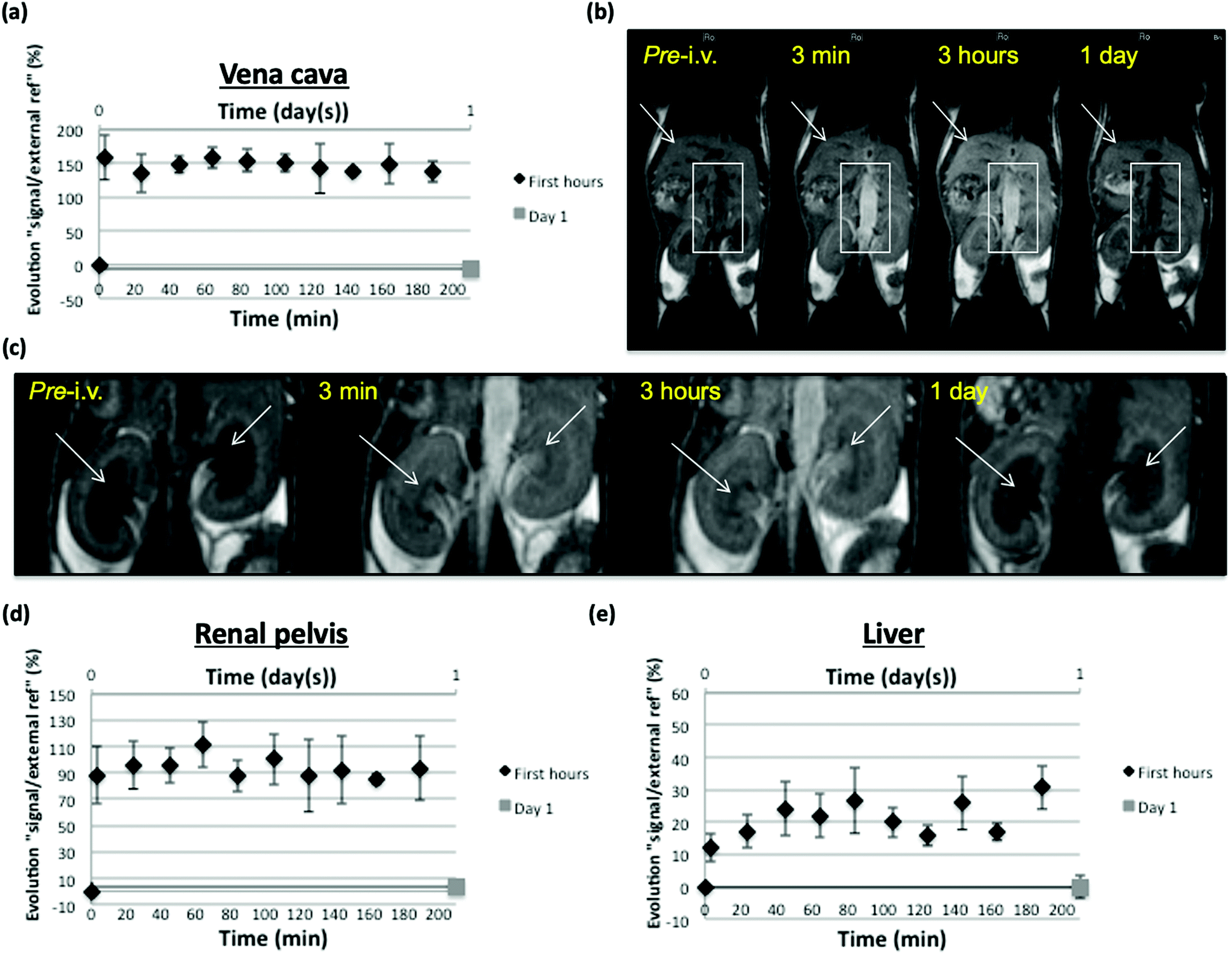 | ||
| Fig. 8 Analysis of positive signal enhancement at 1 T and representative T1-weighted MR images before and during 3 first hours (black sign) + 1 day after (gray sign) i.v. injection of NIR-dye PEG-SiO2-Gd-NPs (4.3 μmol kg−1; signal enhancement data are normalized as a calculated ratio between respective signals in organs or vessel specific ROIs and in an external Gd-containing reference and represented as a percentage of the pre-injection value). (a) and (b) concern vena cava (surrounded by a white square); (c) and (d) concern renal pelvis (white arrow points at the renal pelvis); (e) and (b) concern the liver (white arrow points at the liver on (b)). Mean values ± std deviation of the mean are represented on graphs (n = 3). | ||
Signal measurements were also focused on kidney pelvis as a clue of their renal filtration and elimination. Represented as a value calculated the same way as from vena cava measurements, kidney pelvis enhancement was evident and remained around 90% during the 3 hours of observation, suggesting ongoing renal elimination, allowed by nanoparticle properties (size, charge, shape) compatible with renal filtration of at least a part of the injected dose.60 Again, the calculated value did not reflect any MRI detectable pelvis enhancement at day 1 post-injection, suggesting that most of kidney elimination of nanoparticles took place in this period of time, possibly due to their restricted distribution allowing them to mostly remain directly clearable from the blood vessels (Fig. 8c and d). As an important organ for distribution and clearance of nanoparticles, signal measurements were also focused on the liver. Liver is indeed a well perfused organ that has permeable vascularization (sinusoids), which is important for distribution aspects, and has cell populations (Kupffer cells, hepatocytes) that will allow uptake and possible storage/metabolization of the nanoparticles.61 At least, the calculated value reflecting signal enhancement in liver showed an increase that fluctuates (maybe due to respiratory motion in this part of the body, that influences the measured signal) roughly between 15 and 30% regarding pre-injection value. This enhancement appeared to be simultaneous to the vascular enhancement observed during 3 hours, suggesting that it was related to the nanoparticle content of blood and thus to the local distribution of this compound due to liver perfusion and vessel permeability. At day one, calculated enhancement did not suggest any MRI detectable liver retention of nanoparticles (Fig. 8e). It would have been expected that a portion of the nanoparticle dose remain in liver sinusoids, possibly taken up by Kupffer cells and/or hepatocytes, but the amount of compound may be too low to induce sufficient contrast in the T1-weighted imaging conditions used (indeed, the PEG coating is expected to reduce such capture phenomena).62
It may also be that the MRI contrast effect of nanoparticles is decreased when stored and/or metabolized in the liver but it has to be further investigated. Negative (T2) contrast could have been expected to be stronger at 9.4 T, especially when nanoparticles are accumulated (as it was observed in the hypersignal background of renal pelvis using double dose, see further in Fig. S11b and c,† but no T2 value decrease could be showed in the liver from this higher field at day 1 post-injection using MSME T2-W sequence (Fig. S11a†).
As preliminary observation, progressive signal increase at day 0 was recorded in vena cava on one test mouse by injection of 200 μL of compound (total dose around 8.6 μmol Gd per kg), 50 per 50 μL (1/4, 1/2, 3/4, total); each injection step separated by one image acquisition (5 min 24 s), meaning that the whole dose was injected in a total of approximately 20 min. Data up to 80 min after injection of the total dose were compiled for 3 mice and, apart from fluctuations, signal enhancement in vena cava (as reported to an external reference and expressed as a percentage), remained quite stable as observed at 1 T, and reached a max mean value situated around 90%, which appeared to be evident but inferior to the enhancement value reached with half the dose at 1 T, underlining the expected lesser positive (T1) effect of the Gd-based nanoparticles at higher field (Fig. S11d–f†). The above-mentioned signal decrease due to kidney elimination (more evident in renal pelvis) appeared to be progressively taking place in T2-weighted MRI (RARE T2-W), probably reflecting accumulation of nanoparticles in first hours post-injection, being subsequently excreted via the ureter to the bladder (Fig. S11b and c†) as observed in optical imaging experiments.
From observations performed at 1 T and 9.4 T with NPs after i.v. injection and from their relaxometric properties at those fields, it may be suggested that in vivo experiments seeking T1 (positive) contrast should be performed at 1 T with an adequate dose; that can indeed be relatively low (4.3 μmol Gd per kg) compared to usual Gd chelates (around 100 μmol Gd per kg). Their T2 effect is stronger at 9.4 T and negative contrast should thus more evidently take place (maybe partially masking positive effect) than at 1 T. However, tissues also have shorter T2 at higher field strength, which narrows possibility of T2 contrast, especially in the liver where NP accumulation can usually be expected. NP-induced T2 contrast was indeed only depicted in long T2 area; the renal pelvis. Conditions for T2 contrast appear to be a compromise between injected dose, amount of accumulated NPs and basal relaxometric properties of the tissue where they (momentarily) accumulate. It has to be noticed that performances (signal/noise ratio, resolution) are lower on the ICON (1 T) regarding the Biospec (9.4 T), and that may also influence the selection of one or the other scanner for in vivo experiments with those NPs depending on requested image precision.
Conclusions
In this study, the noncovalent incorporation of Gd-HP-DO3A chelates into PEG-coated silica nanoparticles through a w/o microemulsion procedure was investigated. By doing so, stable suspensions exhibiting significantly improved relaxivity values were reached. Even if the mechanism is not fully understood, it is likely that such improvement results from the combination of inner-sphere (increase of the rotational correlation time) and outer-sphere (increase of the translational correlation time within the permeable matrix) components. In a second step, we demonstrated the feasibility of preparing stable functionalized PEG-coated SiO2-NPs through the covalent insertion of a carboxylic aryl diazirine photolinker on the outer coating corona and its coupling with NIR-dyes was then performed. The as-proposed nanosized platform exhibited remarkable fluorescent properties within the NIR window as well as promising high per-gadolinium r1 relaxivity, demonstrating the feasibility of using such platform for optical and MR imaging. The strong brightening signal using classical T1-weighted sequences were notable, even at 9.4 T. If it is clear that early diagnosis of molecular events studies using such platform is of great interest, further studies will focus on the development of surfaces that can be post-derivatized for molecular imaging. Interestingly, as a complementary method for fluorescent probes, optoacoustic tomography (MSOT) as an emerging preclinic imaging modality show promising interest (i.e., non-invasive, high sensitivity, penetration depth in tissue up to several centimeters) for further molecular imaging application. On the basis of optical and acoustic properties of the NIR-dye, some preliminary recorded phantoms experiments completed the study (Fig. S12†).Author contributions
Conceptualization, S.G., D.S. and S.L. Formal Analysis, S.G., D.S., I.T., S.B., L.L and S.L. Investigation, S.G., D.S., I.T., S.B., L.L. Methodology, S.G., D.S., and S.L. Project administration, R.N.M. and S.L. Funding acquisition, R.N.M. and S.L. Supervision, D.S., R.N.M. and S.L. Writing – original draft, S.G., D.S., S.B. and L.L. Writing – review & editing, S.G., D.S., S.B., L.L., I.T., R.N.M. and S.L.Conflicts of interest
The authors declare no conflict of interest.Acknowledgements
The authors thank the Center for Microscopy and Molecular Imaging (CMMI, supported by the European Regional Development Fund and the Walloon Region). The authors would like to acknowledge the unit of Therapeutic Chemistry and Pharmacognosy (Pr. P. Duez; UMONS) for the access to ICP-AES instrument and Mr C. Palmieri and Ms. G. Ayata for their investment and participation in the ICP characterizations. The authors are also grateful to the laboratory of Polymeric and Composite Materials (Pr. J.-M. Raquez; UMONS) and Dr S. Benali for the use of TGA equipment. Ms. C. Pierart is acknowledged for the record of NMRD profiles. Dr. C. Henoumont is thanked for relaxometric and DOSY experiments and for her great support in interpretations of the results. Mr T. Vangijzegem is acknowledged for his help in multiple analysis and for microscopy and TGA experiments. Authors warmly thank Prof. P.-C. Soleil and Prof. S. Meunier (Haute École Louvain en Hainaut, HELHa) for BET characterizations. This work was supported by the financial support of the Fond National de la Recherche Scientifique (FNRS), the ARC Programs of the French Community of Belgium, COST actions and the Walloon Region (Prother-Wal and Interreg projects). Authors thank the Center for Microscopy and Molecular Imaging (CMMI, supported by European Regional Development Fund and Wallonia). This project has also received funding from European Union's Horizon 2020 Research and Innovation Programme (grant agreement no. 863099).Notes and references
- S. Laurent, C. Henoumont, D. Stanicki, S. Boutry, E. Lipani, S. Belaid, R. N. Muller and L. Vander Elst, MRI Contrast Agents: From Molecules to Particles, 2017 Search PubMed.
- D. W. McRobbie, MRI from picture to proton, Cambridge University Press, Cambridge, UK, New York, 2006 Search PubMed.
- P. Caravan, J. J. Ellison, T. J. McMurry and R. B. Lauffer, Chem. Rev., 1999, 99, 2293–2352 CrossRef CAS PubMed.
- V. M. Runge, Invest. Radiol., 2016, 51, 273–279 CrossRef CAS PubMed.
- V. C. Pierre, M. J. Allen and P. Caravan, J. Biol. Inorg. Chem., 2014, 19, 127–131 CrossRef CAS PubMed.
- E. Blumfield, D. W. Swenson, R. S. Iyer and A. L. Stanescu, Pediatr. Radiol., 2019, 49, 448–457 CrossRef PubMed.
- P. Caravan, C. T. Farrar, L. Frullano and R. Uppal, Contrast Media Mol. Imaging, 2009, 4, 89–100 CrossRef CAS PubMed.
- Cancer therapeutics and imaging: molecular and cellular engineering and nanobiomedicine, ed. K. Rege and S. Goklany, World Scientific, New Jersey, 2017 Search PubMed.
- P. Caravan, Acc. Chem. Res., 2009, 42, 851–862 CrossRef CAS PubMed.
- E. Boros and P. Caravan, J. Med. Chem., 2013, 56, 1782–1786 CrossRef CAS PubMed.
- L. Tang and J. Cheng, Nano Today, 2013, 8, 290–312 CrossRef CAS PubMed.
- S. Gündüz, T. Savić, Đ. Toljić and G. Angelovski, JoVE, 2016, 54776–54786 Search PubMed.
- J. Pellico, C. M. Ellis and J. J. Davis, Contrast Media Mol. Imaging, 2019, 2019, 1–13 CrossRef PubMed.
- W. Zhang, L. Liu, H. Chen, K. Hu, I. Delahunty, S. Gao and J. Xie, Theranostics, 2018, 8, 2521–2548 CrossRef CAS PubMed.
- N. Kamaly and A. D. Miller, Int. J. Mol. Sci., 2010, 11, 1759–1776 CrossRef CAS PubMed.
- S. Aime, C. Cabella, S. Colombatto, S. Geninatti Crich, E. Gianolio and F. Maggioni, J. Magn. Reson., 2002, 16, 394–406 Search PubMed.
- J. C. Cutrin, S. G. Crich, D. Burghelea, W. Dastrù and S. Aime, Mol. Pharm., 2013, 10, 2079–2085 CrossRef CAS PubMed.
- Z. Cheng, A. Al Zaki, I. W. Jones, H. K. Hall, C. A. Aspinwall and A. Tsourkas, Chem. Commun., 2014, 50, 2502–2505 RSC.
- O. M. Koo, I. Rubinstein and H. Onyuksel, Nanomedicine, 2005, 1, 193–212 CrossRef CAS PubMed.
- D. Pan, G. M. Lanza, S. A. Wickline and S. D. Caruthers, Eur. J. Radiol., 2009, 70, 274–285 CrossRef PubMed.
- D. Zhu, F. Liu, L. Ma, D. Liu and Z. Wang, IJMS, 2013, 14, 10591–10607 CrossRef PubMed.
- M. Kaasalainen, V. Aseyev, E. von Haartman, D. Ş. Karaman, E. Mäkilä, H. Tenhu, J. Rosenholm and J. Salonen, Nanoscale Res. Lett., 2017, 12, 74–84 CrossRef PubMed.
- T. Yu, A. Malugin and H. Ghandehari, ACS Nano, 2011, 5, 5717–5728 CrossRef CAS PubMed.
- V. Balakrishnan, H. A. Ab Wab, K. Abdul Razak and S. Shamsuddin, J. Nanomater., 2013, 2013, 1–8 CrossRef.
- E. Lipani, S. Laurent, M. Surin, L. Vander Elst, P. Leclère and R. N. Muller, Langmuir, 2013, 29, 3419–3427 CrossRef CAS PubMed.
- F. Carniato, M. Muñoz-Úbeda, L. Tei and M. Botta, Dalton Trans., 2015, 44, 17927–17931 RSC.
- C. Guo, J. Hu, A. Bains, D. Pan, K. Luo, N. Li and Z. Gu, J. Mater. Chem. B, 2016, 4, 2322–2331 RSC.
- F. Carniato, L. Tei, J. Martinelli and M. Botta, Eur. J. Inorg. Chem., 2018, 8 Search PubMed.
- F. Carniato, L. Tei and M. Botta, Eur. J. Inorg. Chem., 2018, 2018, 4936–4954 CrossRef CAS.
- P. Mathieu, M. Chalet, M. M. Clain, L. Teulon, E. Benoist, N. Leygue, C. Picard, S. Boutry, S. Laurent, D. Stanicki, C. Hénoumont, F. Novio, J. Lorenzo, D. Montpeyó, D. Ciuculescu-Pradines and C. Amiens, New J. Chem., 2020, 44, 18031–18047 RSC.
- S. L. C. Pinho, H. Faneca, C. F. G. C. Geraldes, J. Rocha, L. D. Carlos and M.-H. Delville, Eur. J. Inorg. Chem., 2012, 2012, 2828–2837 CrossRef CAS.
- R. P. Bagwe, C. Yang, L. R. Hilliard and W. Tan, Langmuir, 2004, 20, 8336–8342 CrossRef CAS PubMed.
- W. J. Rieter, J. S. Kim, K. M. L. Taylor, H. An, W. Lin, T. Tarrant and W. Lin, Angew. Chem., Int. Ed., 2007, 46, 3680–3682 CrossRef CAS PubMed.
- S. Santra, R. P. Bagwe, D. Dutta, J. T. Stanley, G. A. Walter, W. Tan, B. M. Moudgil and R. A. Mericle, Adv. Mater., 2005, 17, 2165–2169 CrossRef CAS.
- X. He, X. Wu, K. Wang, B. Shi and L. Hai, Biomaterials, 2009, 30, 5601–5609 CrossRef CAS PubMed.
- N. Wartenberg, P. Fries, O. Raccurt, A. Guillermo, D. Imbert and M. Mazzanti, Chem. – Eur. J., 2013, 19, 6980–6983 CrossRef CAS PubMed.
- H. Hyun, M. W. Bordo, K. Nasr, D. Feith, J. H. Lee, S. H. Kim, Y. Ashitate, L. A. Moffitt, M. Rosenberg, M. Henary, H. S. Choi and J. V. Frangioni, Contrast Media Mol. Imaging, 2012, 7, 516–524 CrossRef CAS PubMed.
- H. S. Choi, S. L. Gibbs, J. H. Lee, S. H. Kim, Y. Ashitate, F. Liu, H. Hyun, G. Park, Y. Xie, S. Bae, M. Henary and J. V. Frangioni, Nat. Biotechnol., 2013, 31, 148–153 CrossRef CAS PubMed.
- D. Stanicki, L. Larbanoix, S. Boutry, T. Vangijzegem, I. Ternad, S. Garifo, R. N. Muller and S. Laurent, J. Mater. Chem. B, 2021, 9, 5055–5068 RSC.
- A. Hannecart, D. Stanicki, L. Vander Elst, R. N. Muller, S. Lecommandoux, J. Thévenot, C. Bonduelle, A. Trotier, P. Massot, S. Miraux, O. Sandre and S. Laurent, Nanoscale, 2015, 7, 3754–3767 RSC.
- T. Vangijzegem, D. Stanicki, S. Boutry, Q. Paternoster, L. Vander Elst, R. N. Muller and S. Laurent, Nanotechnology, 2018, 29, 265103–265117 CrossRef CAS PubMed.
- R. Colsenet, O. Söderman and F. Mariette, Macromolecules, 2006, 39, 1053–1059 CrossRef CAS.
- C. J. Brinker, J. Non-Cryst. Solids, 1988, 100, 31–50 CrossRef CAS.
- E. A. Hauser, Clays Clay Miner., 1955, 4, 45–53 CrossRef.
- S. Chatterji, Cem. Concr. Compos., 2005, 27, 788–795 CrossRef CAS.
- C.-L. Chang and H. S. Fogler, AIChE J., 1996, 42, 3153–3163 CrossRef CAS.
- M. A. López-Quintela, C. Tojo, M. C. Blanco, L. García Rio and J. R. Leis, Curr. Opin. Colloid Interface Sci., 2004, 9, 264–278 CrossRef.
- K. S. Finnie, J. R. Bartlett, C. J. A. Barbé and L. Kong, Langmuir, 2007, 23, 3017–3024 CrossRef CAS PubMed.
- C. Aubery, C. Solans, S. Prevost, M. Gradzielski and M. Sanchez-Dominguez, Langmuir, 2013, 29, 1779–1789 CrossRef CAS PubMed.
- R. Michel, S. Pasche, M. Textor and D. G. Castner, Langmuir, 2005, 21, 12327–12332 CrossRef CAS PubMed.
- P. Charles, V. Stubbs, C. Soto, B. Martin, B. White and C. Taitt, Sensors, 2009, 9, 645–655 CrossRef CAS PubMed.
- S. M. S. Björkegren, L. Nordstierna, A. Törncrona, M. E. Persson and A. E. C. Palmqvist, J. Colloid Interface Sci., 2015, 452, 215–223 CrossRef PubMed.
- C. Bernhard, S. J. Roeters, J. Franz, T. Weidner, M. Bonn and G. Gonella, Phys. Chem. Chem. Phys., 2017, 19, 28182–28188 RSC.
- V. Cauda, A. Schlossbauer and T. Bein, Microporous Mesoporous Mater., 2010, 132, 60–71 CrossRef CAS.
- V. Pourcelle, C. S. Le Duff, H. Freichels, C. Jérôme and J. Marchand-Brynaert, J. Fluorine Chem, 2012, 140, 62–69 CrossRef CAS.
- V. Pourcelle, S. Laurent, A. Welle, N. Vriamont, D. Stanicki, L. Vander Elst, R. N. Muller and J. Marchand-Brynaert, Bioconjugate Chem., 2015, 26, 822–829 CrossRef CAS PubMed.
- L. Dubinsky, B. P. Krom and M. M. Meijler, Bioorg. Med. Chem., 2012, 20, 554–570 CrossRef CAS PubMed.
- S. Liang, C. L. John, S. Xu, J. Chen, Y. Jin, Q. Yuan, W. Tan and J. X. Zhao, in Advanced Fluorescence Reporters in Chemistry and Biology II, ed. A. P. Demchenko, Springer Berlin Heidelberg, Berlin, Heidelberg, 2010, vol. 9, pp. 229–251 Search PubMed.
- R. Sethi, J. S. Ananta, C. Karmonik, M. Zhong, S. H. Fung, X. Liu, K. Li, M. Ferrari, L. J. Wilson and P. Decuzzi, Contrast Media Mol. Imaging, 2012, 7, 501–508 CrossRef CAS PubMed.
- S. Swar, V. Máková and I. Stibor, Materials, 2019, 12, 3202 CrossRef CAS PubMed.
- W. Poon, Y. N. Zhang, B. Ouyang, B. R. Kingston, J. L. Y. Wu, S. Wilhelm and W. C. W. Chan, ACS Nano, 2019, 13, 5785–5798 CrossRef CAS PubMed.
- H. Arami, A. Khandhar, D. Liggitt and K. M. Krishnan, Chem. Soc. Rev., 2015, 44, 8576–8607 RSC.
Footnote |
| † Electronic supplementary information (ESI) available: Fig. S1. Additional TEM images and histograms of empty SiO2-NPs through network formation. Fig. S2: Stability measurements of PEG-coated SiO2-NPs by DLS. Fig. S3: Thermogravimetric curve of empty PEG-coated SiO2-NPs systems. Fig. S4: Surface modification: FTIR spectra of SiO2-NPs, PEG-coated SiO2-NPs and functionalized PEG SiO2-NPs. Eqn (S1)–(S4). Number of Gd3+ complexes embedded inside silica NPs. Table S1: Number of entrapped complexes inside SiO2-Gd-NPs. Fig. S5: Stability measurements of functionalized PEG-coated SiO2-Gd-NPs by DLS. Fig. S6: NIR luminescent properties of the multimodal platform: spectroscopy and imaging. Fig. S7: Phantoms MRI of SiO2-Gd-NPs vs. free Gd-HP-DO3A vs. water. Fig. S8: BET characterizations. Fig. S9: In vivo FLI. Fig. S10: Ex vivo FLI of organs of interest (after 14 days). Fig. S11: In vivo MRI examination at 9.4 T. Fig. S12: Photoacoustic imaging experiments: PA spectra recorded on phantom images of NIR-dye PEG-coated SiO2-Gd-NPs at various concentrations and evolution of MSOT signal (770 nm). See DOI: 10.1039/d1nr04972k |
| This journal is © The Royal Society of Chemistry 2021 |