DOI:
10.1039/C9SC03790J
(Edge Article)
Chem. Sci., 2019,
10, 10716-10722
Synthesis of bicyclo[3.1.0]hexanes by (3 + 2) annulation of cyclopropenes with aminocyclopropanes†
Received
31st July 2019
, Accepted 7th October 2019
First published on 8th October 2019
Abstract
We report the convergent synthesis of bicyclo[3.1.0]hexanes possessing an all-carbon quaternary center via a (3 + 2) annulation of cyclopropenes with cyclopropylanilines. Using an organic or an iridium photoredox catalyst and blue LED irradiation, good yields were obtained for a broad range of cyclopropene and cyclopropylaniline derivatives. The reaction was highly diastereoselective when using difluorocyclopropenes together with a removable substituent on the cyclopropylaniline, giving access to important building blocks for medicinal chemistry. With efficient methods existing for the synthesis of both reaction partners, our method grants a fast access to highly valuable bicyclic scaffolds with three contiguous stereocenters.
1. Introduction
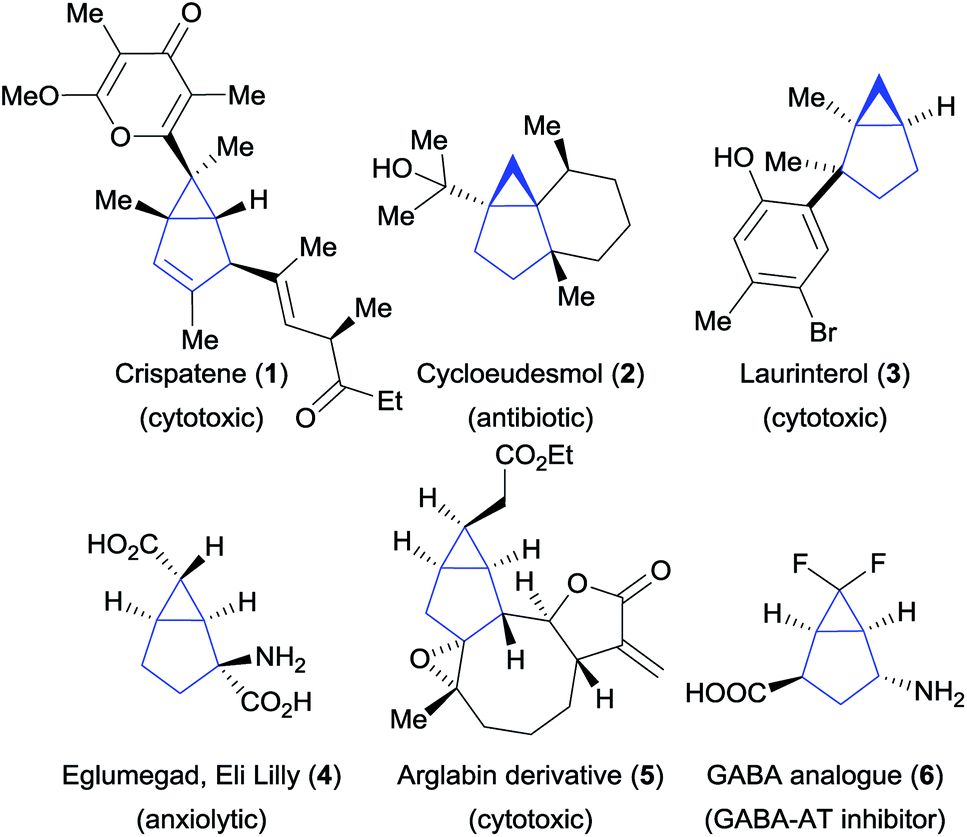
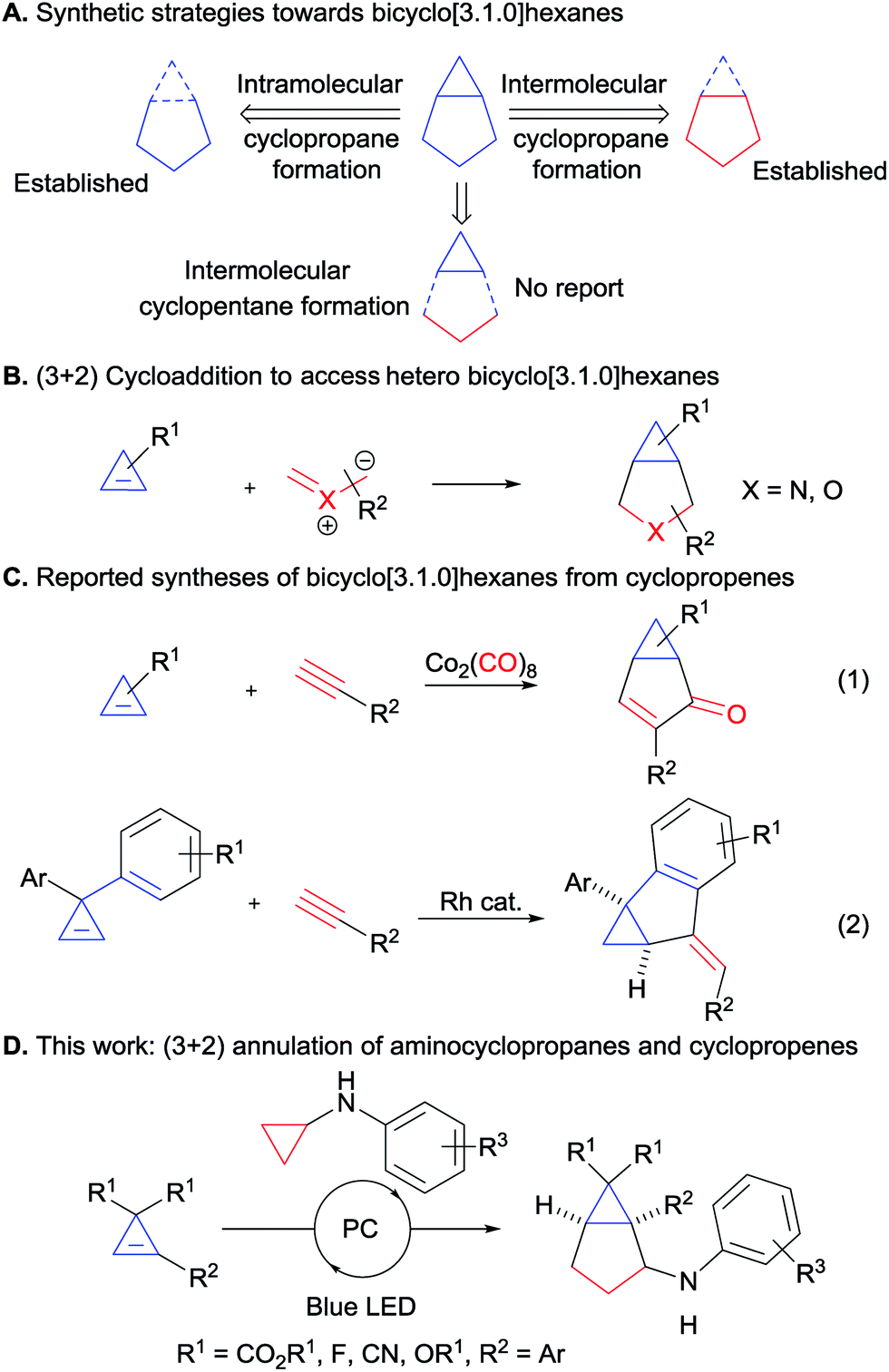
Bicyclo[3.1.0]hexanes are prevalent scaffolds in natural products and synthetic bioactive compounds (Fig. 1).1 For instance, crispatene (1) as well as the two sesquiterpenes cycloeudesmol (2) and laurinterol (3), were isolated from marine sources and exhibit potent bioactivities.2 Eli Lilly's glutamate derivative 4 and the arglabin derivative 5 are examples of promising synthetic drugs for the treatment of psychiatric disorders3 and cancer,4 respectively. Fluorinated analogues such as 6![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 5 are increasingly important in medicinal chemistry.6 In addition, the high ring strain of these bicyclic scaffolds make them valuable synthetic intermediates,7 but at the same time makes their synthesis challenging. Various methods for their preparation have been developed, relying for the most part on the construction of the three-membered ring (Scheme 1A). This approach has led to the development of efficient intramolecular cyclization, cyclopropanation and transannular reactions.8 However, these methods often require sophisticated and pre-functionalized starting materials. Intermolecular processes have been mostly limited to cyclopropanation reactions based on the use of carbenes and metallocarbenes.9 Surprisingly, the convergent synthesis of bicyclo[3.1.0]hexanes via a (3 + 2) annulation process to build the five-membered ring has not been reported so far.
5 are increasingly important in medicinal chemistry.6 In addition, the high ring strain of these bicyclic scaffolds make them valuable synthetic intermediates,7 but at the same time makes their synthesis challenging. Various methods for their preparation have been developed, relying for the most part on the construction of the three-membered ring (Scheme 1A). This approach has led to the development of efficient intramolecular cyclization, cyclopropanation and transannular reactions.8 However, these methods often require sophisticated and pre-functionalized starting materials. Intermolecular processes have been mostly limited to cyclopropanation reactions based on the use of carbenes and metallocarbenes.9 Surprisingly, the convergent synthesis of bicyclo[3.1.0]hexanes via a (3 + 2) annulation process to build the five-membered ring has not been reported so far.
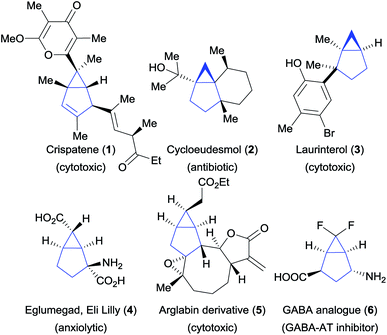 |
| | Fig. 1 Natural products and synthetic bioactive compounds containing the bicyclo[3.1.0]hexane scaffold. | |
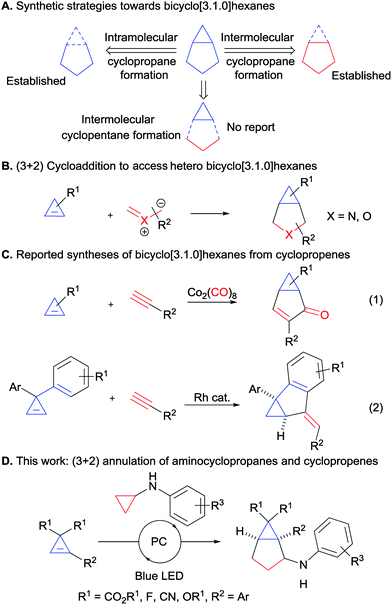 |
| | Scheme 1 Synthesis of bicyclo[3.1.0]hexanes. (A) Synthetic strategies towards bicyclo[3.1.0]hexanes. (B) (3 + 2) cycloaddition to access hetero bicyclo[3.1.0]hexanes from cyclopropenes. (C) Reported syntheses of bicyclo[3.1.0]hexanes from cyclopropenes. (D) This work: (3 + 2) annulation of aminocyclopropanes and cyclopropenes). | |
Cyclopropenes appeared to us as ideal two-carbon partners for this unprecedented approach. With an increasing number of methods for their synthesis combined with high reactivity, they have emerged as key building blocks in organic synthesis.10 In particular, the synthesis of diester and difluoro-substituted cyclopropenes is now well-established.11 The latter would give access to important fluorinated derivatives such as GABA analogue 6 (Fig. 1). Annulations involving cyclopropenes have been highly successful in the past, especially in the case of (4 + 2) processes leading to 6-membered rings.12 In contrast, (3 + 2) annulations have been limited to the formation of heterocycles, such as tetrahydrofuran or pyrrolidine derivatives, starting from carbonyl or imine ylides (Scheme 1B).13 To date, the only reports for the synthesis of [3.1.0] carbocycles starting from cyclopropenes are Pauson–Khand reactions14a,b or a rhodium-catalyzed annulation of diarylcyclopropenes with alkynes (Scheme 1C, eqn (1) and (2)).14c
Based on our interest in the development of annulation methodologies exploiting ring strain,15 we believed that activated cyclopropanes could function as ideal three carbon partners in a (3 + 2) annulation with cyclopropenes for the synthesis of bicyclo[3.1.0]hexanes. We report herein the successful implementation of this strategy via an unprecedented photoredox-mediated (3 + 2) annulation between diester-substituted cyclopropenes and aminocyclopropanes, which displayed a broad scope and proceeded under mild conditions. Moreover, a diastereoselective version could be developed with difluorocyclopropenes for accessing important building blocks for synthetic and medicinal chemistry.
2. Results and discussions
Discovery and optimization of the (3 + 2) annulation
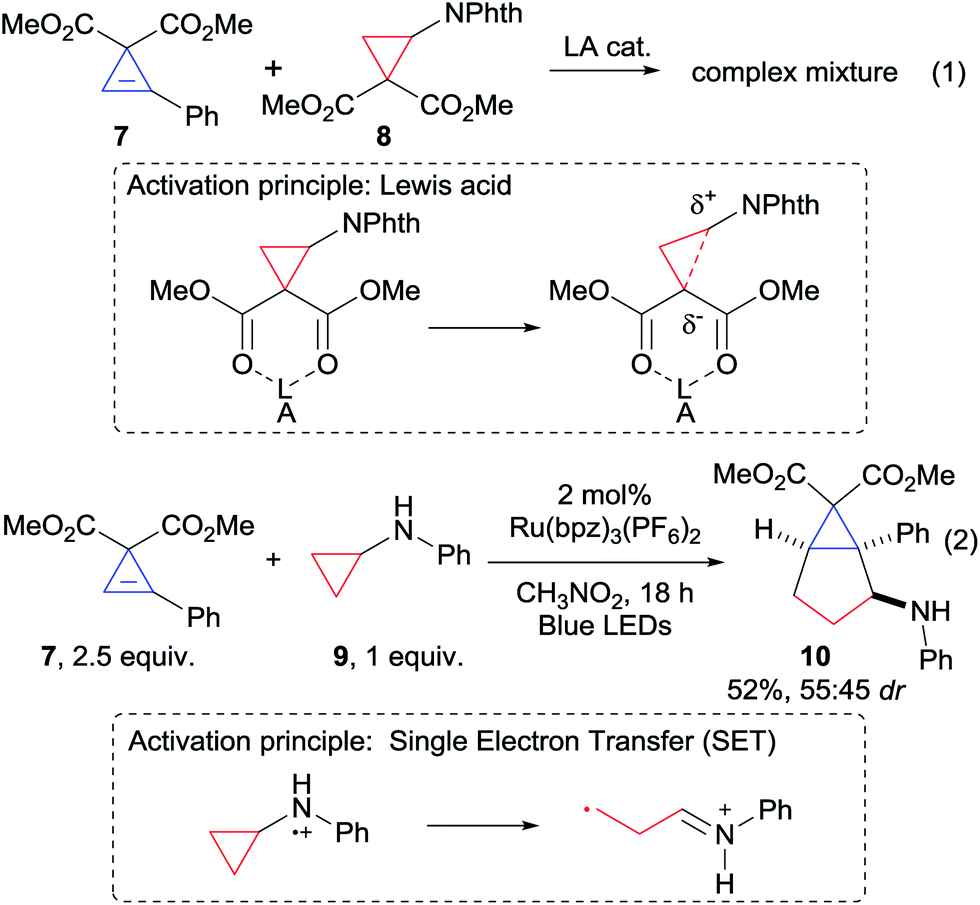
Based on our expertise with the Lewis acid activation of donor–acceptor aminocyclopropanes,15 we examined the reaction of the easily accessible aryl-substituted cyclopropene 7 with cyclopropane 8 (Scheme 2, eqn (1)). However, a complex mixture of products was obtained.16 We speculated that Lewis acid activation of cyclopropane 8 was not possible in presence of the sensitive and possibly chelating cyclopropene 7. Therefore, we decided to turn to another mode of activation based on single electron transfer (SET) and radical intermediates (Scheme 2, eqn (2)). To the best of our knowledge, only two examples of (4 + 2) annulations based on light-initiated radical processes have been reported with cyclopropenes.12a,f In this context, the activation of simple donor aminocyclopropanes via oxidation with stoichiometric reagents, metal catalysts or photoredox catalysts has attracted strong interest, and the resulting radicals have been reported to participate in annulation reaction with olefins or alkynes.17 Nevertheless, reactions with cyclopropenes were never reported. Gratifyingly, when Zheng photoredox conditions17c were examined using aminocyclopropane 9, the desired annulation product 10 could be obtained in 52% as a mixture of diastereoisomers (Scheme 2, eqn (2)). We decided therefore to optimize this result (Table 1, entry 1).
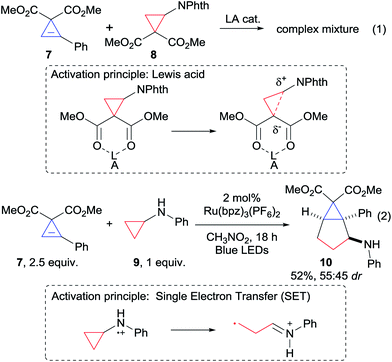 |
| | Scheme 2 First attempts of (3 + 2) annulation. | |
Table 1 Optimization of the (3 + 2) annulation
|
|
| Entry |
X
|
Y
|
Photocat. (mol%) |
[M] |
Yielda |
|
Isolated on 0.1 mmol scale.
Isolated yield on 0.3 mmol scale.
|
| 1 |
2.5 |
1.0 |
Ru(bpz)3(PF6)2 (2) |
0.1 M |
52% |
| 2 |
1.0 |
1.5 |
Ru(bpz)3(PF6)2 (2) |
0.1 M |
53% |
| 3 |
1.0 |
1.5 |
4CzIPN (5) |
0.1 M |
60% |
| 4 |
1.0 |
1.5 |
4CzIPN (5) |
0.4 M |
82% |
| 5 |
1.0 |
1.8 |
4CzIPN (5) |
0.4 M |
86% |
| 6 |
1.0 |
1.8 |
4DPAIPN (5) |
0.4 M |
89% (87%)b |
Eighteen hours of reaction were required for the full conversion of cyclopropylaniline 9. Cyclopropene 7 was stable under these reaction conditions. However degassing the solvent was required to limit degradation of cyclopropylaniline 9. Consistent with previous reports,17c,d nitromethane was superior to all other solvents. No improvement was observed when varying catalyst, concentration and stoichiometry (see ESI for more details†). A drawback of Zheng's optimized conditions is the use of a large excess of olefin (typically 5 equivalents). Cyclopropylaniline 9 being synthetically more accessible,18 we attempted to use it in excess and obtained a similar yield (Table 1, entry 2). We then examined the organic dye 2,4,5,6-tetra(9H-carbazol-9-yl)isophthalonitrile (4CzIPN), as photoredox catalyst. 4CzIPN has a close oxidation potential compared to the ruthenium catalyst, but it is a better reductant (E1/2 (*P/P−) = +1.35 and E1/2 (P/P−) = −1.21 vs. +1.45 and −0.80 for Ru(bpz)3(PF6)2).19,20 Its use led to a slightly improved yield of 60% (Table 1, entry 3). To the best of our knowledge, this is the first report on the use of an organic dye for the photoredox-based activation of aminocyclopropanes. Increasing the concentration to 0.4 M, 10 could be obtained in 82% (Table 1, entry 4). Using a higher concentration was not possible due to the low solubility of the catalyst. Eventually, using 1.8 equiv. of cyclopropylaniline 9 led to 86% yield of product 10 (Table 1, entry 5). However, 4CzIPN gave lower yields for other cyclopropene and cyclopropylaniline derivatives. We found that the less oxidizing, but stronger reducing, 2,4,5,6-tetrakis(diphenylamino)isophthalonitrile (4DPAIPN, E1/2 (*P/P−) = +0.90 and E1/2 (P/P−) = −1.65)21 was a more general catalyst. This dye was first reported by Zhang and Luo,19a but Zeitler and co-workers later showed that the dye used in their work had another structure.22 Our group was the first to access the originally proposed structure.21 In case of 7 and 9, 10 was obtained in similar yield using 4DPAIPN, and the reaction could be easily scaled up to 0.3 mmol (Table 1, entry 6). Importantly, the yield obtained is much higher than the previously reported one in radical-based annulation of cyclopropenes (16–51%),12f demonstrating that this strategy can be highly efficient. Product 10 was obtained as a nearly 1:1 mixture of diastereoisomers, which could be isolated in pure form after column chromatography.23
Scope of the (3 + 2) annulation
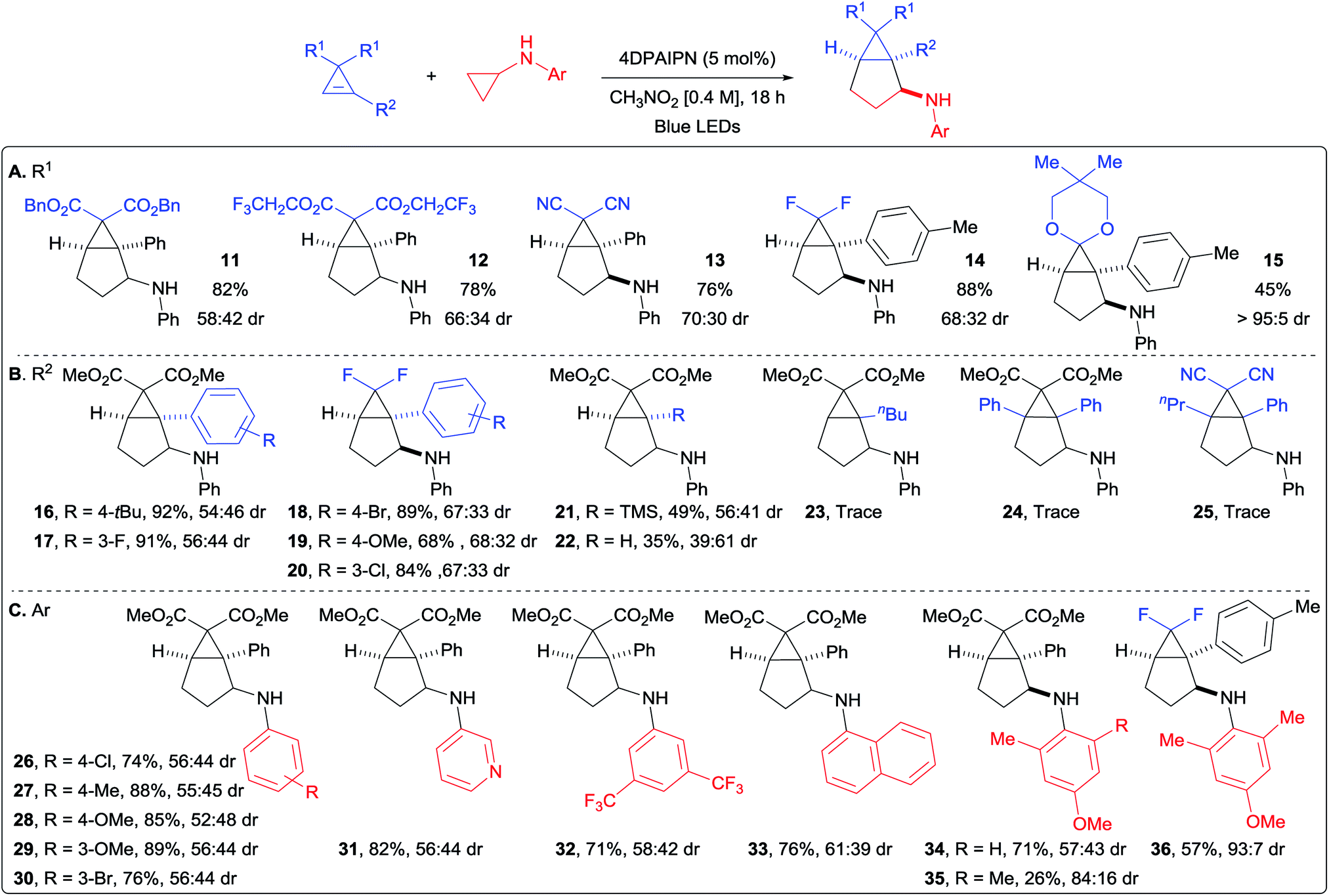
With optimized conditions in hand, we started to vary the substituents on the cyclopropene (Scheme 3A). We found that different esters were suitable for the transformation (products 11 and 12). Both dicyano- and difluoro-cyclopropenes furnished the corresponding cycloadducts 13 and 14 in high yields and improved diastereoselectivity. An acetal protected cyclopropenone could also be used as partner in the (3 + 2) cycloaddition, leading to compound 15 in 45% yield as a single diastereoisomer. Different substituents on the olefin were then investigated (Scheme 3B). With aryl groups, both electron-withdrawing and -donating groups were well tolerated in meta and para positions (products 16–20). A notable decrease in yield was only observed with the strongly donating para-methoxy group (19). Interestingly, the TMS- and un-substituted cyclopropenes afforded the corresponding bicyclo[3.1.0]hexanes 21 and 22 in moderate yields. This decrease in yield is most likely due to the lower stability of the cyclopropyl radical. Alkyl substituted and disubstituted cyclopropenes could not be used, and only trace amount of the desired products 23, 24 and 25 were observed. We then turned our attention towards the aromatic ring of the cyclopropylanilines, bearing electronically and sterically diverse substituents (Scheme 3C).24 Both electron-withdrawing and -donating groups were compatible in para and meta positions, delivering the expected products 26–30 in good to excellent yields. The reaction was also applicable to heterocyclic substituents, as well as disubstituted aryl groups on the cyclopropylamine without any significant impact on the yields of products 31 and 32. The sterically demanding naphthyl substituent led also to a high yield of product 33. When considering that para-methoxyphenyl (PMP) can be cleaved under oxidative conditions, we decided to investigate more sterically hindered derivatives to see if the diastereoselectivity could be improved. Adding one methyl in ortho position did affect neither the yield nor the stereochemical outcome (product 34). Having two methyl groups in ortho position decreased the yield, but increased significantly the diastereoselectivity in the formation of 35. When considering the importance of fluorinated compounds in medicinal chemistry,6 we then turned to difluorocyclopropenes. With N-cyclopropyl-4-methoxy-2,6-dimethylaniline (37), we could isolate the corresponding product 36 in 57% yield with an improved diastereomeric ratio of 93:7. Considering the importance of fluorinated derivatives, we decide to re-optimize this transformation (see ESI for details†) and explore its scope.
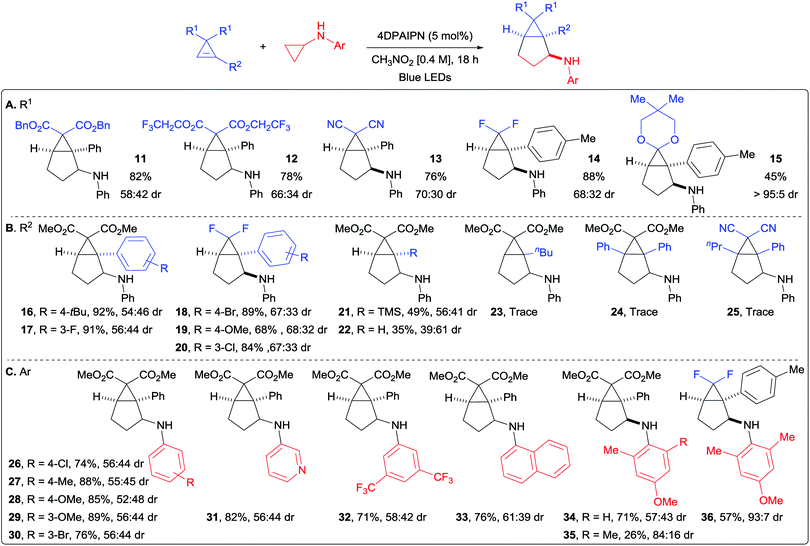 |
| | Scheme 3 Scope of the (3 + 2) annulation. Reaction conditions: cyclopropene (1.0 equiv., 0.3 mmol), 4DPAIPN (5 mol%) and cyclopropylaniline (1.8 equiv.) in CH3NO2 (0.4 M) at rt. Isolated yields are given. dr determined by 1H NMR of the crude products. (A) Scope of substituents R1 on cyclopropene. (B) Scope of substituents R2 on cyclopropene. (C) Scope of aryl substituents on cyclopropylamine. | |
Diastereoselective (3 + 2) annulation with difluorocyclopropenes
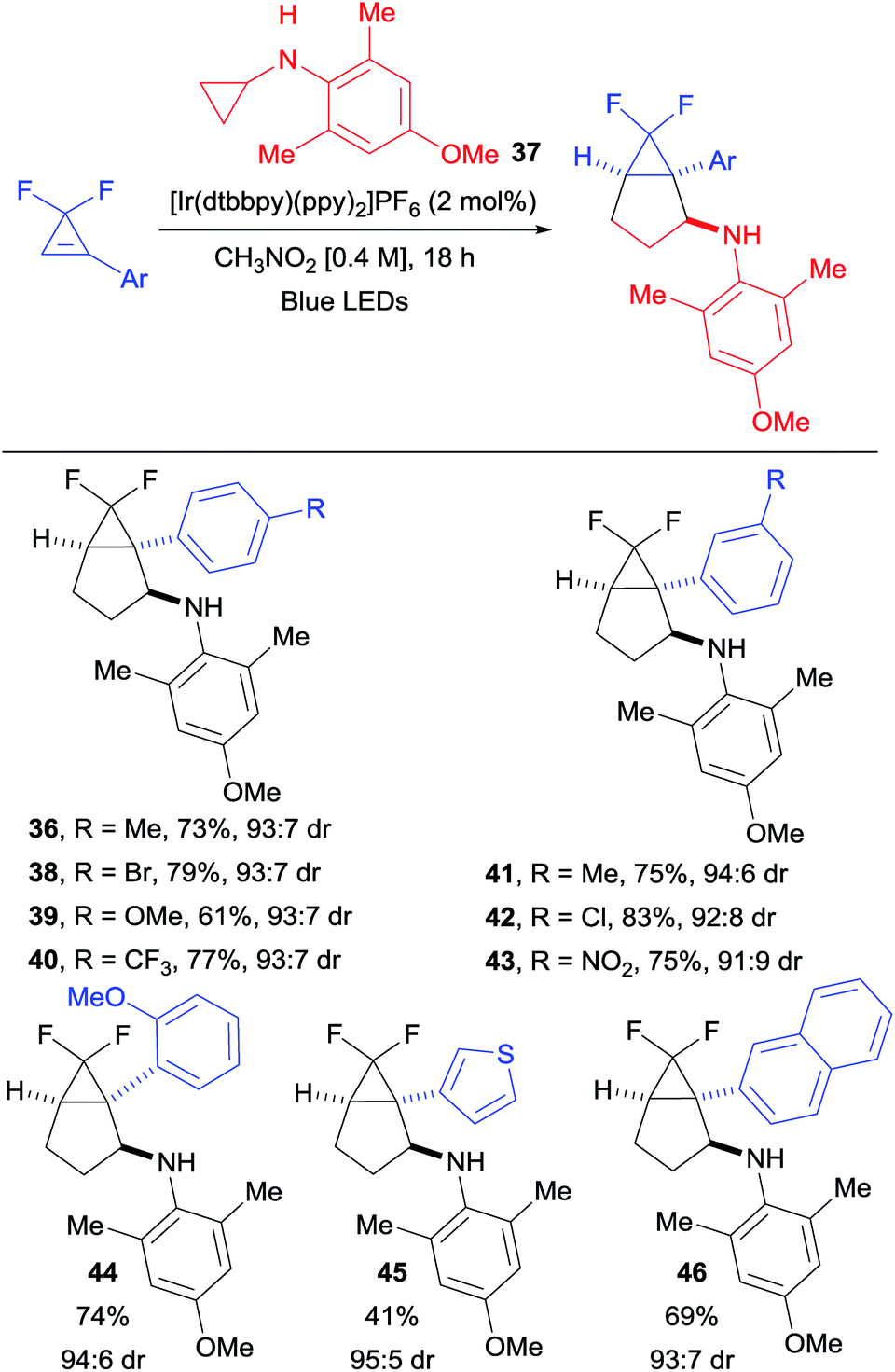
We found that employing the less oxidizing [Ir(dtbbpy)(ppy)2]PF6 photocatalyst (E1/2 (*P/P−) = +0.66 and E1/2 (P/P−) = −1.51),20 combined with a larger excess of cyclopropylaniline 37 (easily synthesized in one step from cheap and commercially available starting materials), allowed us to isolate 36 in a satisfying 73% yield with the same diastereoselectivity (Scheme 4). Under these conditions, difluorocyclopropenes bearing electron-withdrawing, as well as electron-donating groups in meta and para positions delivered the corresponding products 38–43 in good yields and diastereoselectivity. A slight decrease in yield was only observed when a strongly donating para-methoxy substituent was introduced (product 39). Ortho substitution was also tolerated as illustrated with cyclopropane 44. A thienyl-substituted difluorocyclopropene was compatible with our conditions, affording the corresponding bicyclic product 45 in moderate yield. A bulky 2-naphthyl substituent was also tolerated in the annulation, delivering 46 in a 69% yield.
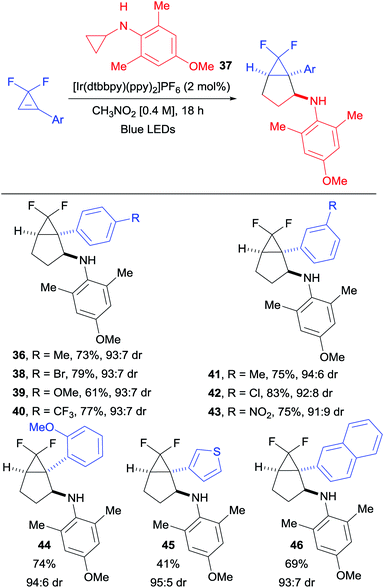 |
| | Scheme 4 Diastereoselective (3 + 2) cycloaddition with difluoro-cyclopropenes. Reaction conditions: difluorocyclopropene (1.0 equiv., 0.30 mmol), [Ir(dtbbpy)(ppy)2]PF6 (5 mol%) and N-cyclopropyl-4-methoxy-2,6-dimethylaniline (37) (2.5 equiv.) in CH3NO2 (0.4 M) at rt. Isolated yields are given. The dr was determined by 19F NMR spectroscopy of the crude products. | |
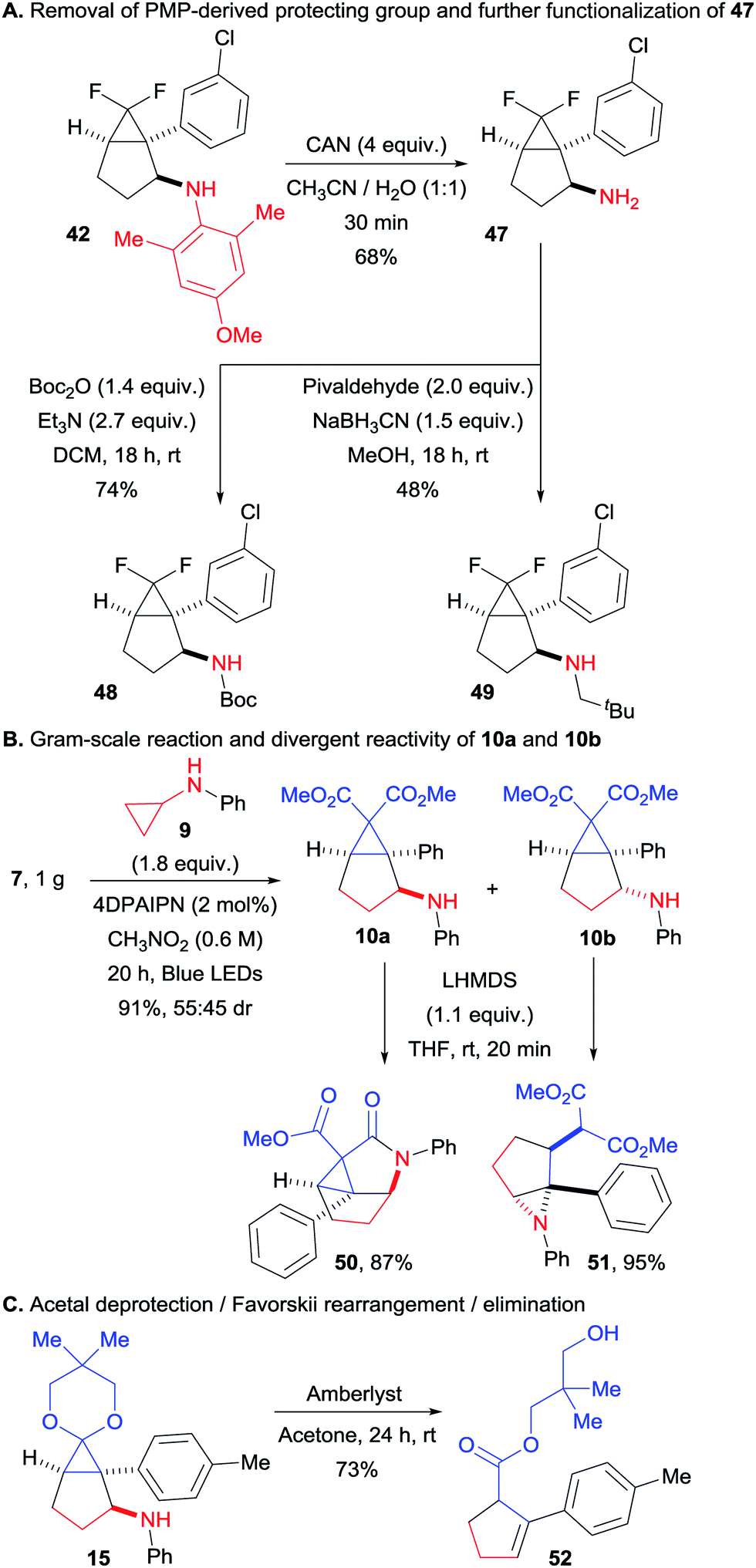
We then turned our attention to synthetically relevant transformations of the obtained bicyclo[3.1.0]hexanes. Starting with compound 42, we could easily remove the bulky PMP group (Scheme 5A). This transformation proceeded smoothly in only 30 minutes at room temperature, delivering the free amine 47. The latter could be easily further functionalized without any alteration of the bicyclic structure as illustrated by products 48 and 49, obtained via Boc protection and reductive amination with pivaldehyde respectively. In order to assess the preparative value of the transformation, we carried a gram scale experiment with compounds 7 and 9 (Scheme 5B). We found that at this scale a lower catalyst loading (2 instead of 5 mol%) along with higher concentration (0.6 instead of 0.4 M) allowed to get the desired product in a very good yield of 91%, with the same diastereomeric ratio of 55:45. The two diastereoisomers of this transformation could be separated by column chromatography. In presence of a strong base, 10a delivered the tricyclic compound 50 by amide bond formation, facilitated by the close spatial proximity of the aniline and one of the ester groups. In contrast, 10b was converted to aziridine 51via attack of the nitrogen atom on the donor–acceptor cyclopropane. Finally, we submitted tricyclic compound 15 to acidic conditions, which resulted in acetal deprotection, followed by a Favorskii-like rearrangement and a retro-Michael addition of the aniline group to give cyclopentene 52 in 73% yield (Scheme 5C).
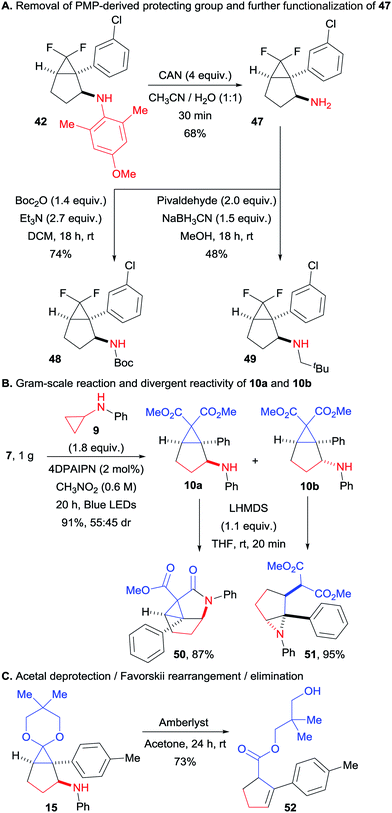 |
| | Scheme 5 Gram-scale (3 + 2) annulation and product modifications. (A) Removal of PMP-derived protecting group and further functionalization of 47. (B) Gram-scale reaction and divergent reactivity of 10a and 10b. (C) Acetal deprotection/Favorskii rearrangement/elimination. | |
3. Conclusions
In conclusion, we have developed the first (3 + 2) annulation of cyclopropenes and cyclopropanes, providing a complementary and convergent strategy towards substituted bicyclo[3.1.0]hexanes having three contiguous stereocenters, including an all-carbon benzylic quaternary center. Based on an organocatalyzed photoredox-mediated ring-opening of aminocyclopropanes, the transformation was high yielding and broadly applicable, demonstrating that radical-based strategies can be highly efficient for annulation reactions of cyclopropenes. High diastereoselectivity could be achieved by combining a bulky cyclopropylaniline with difluorocyclopropenes. Our work constitutes a new convergent strategy for the synthesis of important carbocyclic building blocks in synthetic and medicinal chemistry.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
We thank the Swiss National Science Foundation (Grant No. 200021_165788) and EPFL for financial support. We thank Dr R. Scopelliti and Dr F. F. Tirani from ISIC at EPFL for X-ray analysis. Dr Franck Le Vaillant and Marion Garreau from LCSO are acknowledged for their help through meaningful discussions.
Notes and references
-
(a) H. S. Kim, M. Ohno, B. Xu, H. O. Kim, Y. S. Choi, X. D. Ji, S. Maddileti, V. E. Marquez, T. K. Harden and K. A. Jacobson, J. Med. Chem., 2003, 46, 4974 CrossRef CAS PubMed
 ;
(b) J. Parks, T. Gyeltshen, V. Prachyawarakorn, C. Mahidol, S. Ruchirawat and P. Kittakoop, J. Nat. Prod., 2010, 73, 992 CrossRef CAS PubMed ;
(c) P. D. Boatman, B. Lauring, T. O. Schrader, M. Kasem, B. R. Johnson, P. Skinner, J. K. Jung, J. Xu, M. C. Cherrier, P. J. Webb, G. Semple, C. R. Sage, J. Knudsen, R. Chen, W. L. Luo, L. Caro, J. Cote, E. Lai, J. Wagner, A. K. Taggart, E. Carballo-Jane, M. Hammond, S. L. Colletti, J. R. Tata, D. T. Connolly, M. G. Waters and J. G. Richman, J. Med. Chem., 2012, 55, 3644 CrossRef CAS PubMed ;
(d) M. L. Liu, Y. H. Duan, Y. L. Hou, C. Li, H. Gao, Y. Dai and X. S. Yao, Org. Lett., 2013, 15, 1000 CrossRef CAS PubMed .
;
(b) J. Parks, T. Gyeltshen, V. Prachyawarakorn, C. Mahidol, S. Ruchirawat and P. Kittakoop, J. Nat. Prod., 2010, 73, 992 CrossRef CAS PubMed ;
(c) P. D. Boatman, B. Lauring, T. O. Schrader, M. Kasem, B. R. Johnson, P. Skinner, J. K. Jung, J. Xu, M. C. Cherrier, P. J. Webb, G. Semple, C. R. Sage, J. Knudsen, R. Chen, W. L. Luo, L. Caro, J. Cote, E. Lai, J. Wagner, A. K. Taggart, E. Carballo-Jane, M. Hammond, S. L. Colletti, J. R. Tata, D. T. Connolly, M. G. Waters and J. G. Richman, J. Med. Chem., 2012, 55, 3644 CrossRef CAS PubMed ;
(d) M. L. Liu, Y. H. Duan, Y. L. Hou, C. Li, H. Gao, Y. Dai and X. S. Yao, Org. Lett., 2013, 15, 1000 CrossRef CAS PubMed .
-
(a) W. Fenical and J. J. Sims, Tetrahedron Lett., 1974, 15, 1137 CrossRef ;
(b) C. Ireland and J. Faulkner, Tetrahedron, 1981, 37, 233 CrossRef ;
(c) S. Garcia-Davis, E. Viveros-Valdez, A. R. Diaz-Marrero, J. J. Fernández, D. Valencia-Mercado, O. Esquivel-Hernández, P. Carranza-Rosales, I. E. Carranza-Torres and N. E. Guzman-Delgado, Mar. Drugs, 2019, 17, 201 CrossRef CAS PubMed .
- J. A. Monn, M. J. Valli, S. M. Massey, R. A. Wright, C. R. Salhoff, B. G. Johnson, T. Howe, C. A. Alt, G. A. Rhodes, R. L. Robey, K. R. Griffey, J. P. Tizzano, M. J. Kallman, D. R. Helton and D. D. Schoepp, J. Med. Chem., 1997, 40, 528 CrossRef CAS PubMed .
- R. Csuk, A. Heinold, B. Siewert, S. Schwarz, A. Barthel, R. Kluge and D. Ströhl, Arch. Pharm., 2012, 345, 215 CrossRef CAS PubMed .
- Z. Wang and R. B. Silverman, Bioorg. Med. Chem., 2006, 14, 2242 CrossRef CAS PubMed .
-
(a) K. Müller, C. Faeh and F. Diederich, Science, 2007, 317, 1881 CrossRef PubMed ;
(b) S. Purser, P. R. Moore, S. Swallow and V. Gouverneur, Chem. Soc. Rev., 2008, 37, 320 RSC ;
(c) E. P. Gillis, K. J. Eastman, M. D. Hill, D. J. Donnelly and N. A. Meanwell, J. Med. Chem., 2015, 58, 8315 CrossRef CAS PubMed ;
(d) N. A. Meanwell, J. Med. Chem., 2018, 61, 5822 CrossRef CAS PubMed ;
(e) A. Pons, T. Poisson, X. Pannecoucke, A. B. Charette and P. Jubault, Synthesis, 2016, 48, 4060 CrossRef CAS .
-
(a) R. Sarpong, J. T. Su and B. M. Stoltz, J. Am. Chem. Soc., 2003, 125, 13624 CrossRef CAS PubMed ;
(b) H.-U. Reissig and R. Zimmer, Chem. Rev., 2003, 103, 1151 CrossRef CAS PubMed ;
(c) B. Bajtos, M. Yu, H. Zhao and B. L. Pagenkopf, J. Am. Chem. Soc., 2007, 129, 9631 CrossRef CAS PubMed ;
(d) T. Sawada and M. Nakada, Org. Lett., 2013, 15, 1004 CrossRef CAS PubMed ;
(e) H. K. Grover, M. R. Emmett and M. A. Kerr, Org. Biomol. Chem., 2015, 13, 655 RSC ;
(f) Y. Ochi, S. Yokoshima and T. Fukuyama, Synthesis, 2017, 49, 96 CAS ; for a general review on reaction of activated cyclopropanes, see: ;
(g) T. F. Schneider, J. Kaschel and D. B. Werz, Angew. Chem., Int. Ed., 2014, 53, 5504 CrossRef CAS PubMed .
- For selected recent examples, see:
(a) J.-J. Feng and J. Zhang, J. Am. Chem. Soc., 2011, 133, 7304 CrossRef CAS PubMed ;
(b) T. Miura, N. D. Yadav, H. Iwasaki, M. Ozeki, N. Kojima and M. Yamashita, Org. Lett., 2012, 14, 6048 CrossRef CAS PubMed ;
(c) C. H. Oh, L. Piao and J. H. Kim, Synthesis, 2013, 45, 174 CrossRef CAS ;
(d) D. Qian, H. Hu, F. Liu, B. Tang, W. Ye, Y. Wang and J. Zhang, Angew. Chem., Int. Ed., 2014, 53, 13751 CrossRef CAS PubMed ;
(e) K. Masutomi, K. Noguchi and K. Tanaka, J. Am. Chem. Soc., 2014, 136, 7627 CrossRef CAS PubMed ;
(f) Y. Yang, J. Li, B. Du, C. Yuan, B. Liu and S. Qin, Chem. Commun., 2015, 51, 6179 RSC ;
(g) C. Luo, Z. Wang and Y. Huang, Nat. Commun., 2015, 6, 10041 CrossRef PubMed ;
(h) G. Xu and P. Renaud, Angew. Chem., Int. Ed., 2016, 55, 3657 CrossRef CAS PubMed ;
(i) P. T. Bohan and F. Dean Toste, J. Am. Chem. Soc., 2017, 139, 11016 CrossRef CAS PubMed ;
(j) Z. Z. Zhang, Y. Q. Han, B. B. Zhan, S. Wang and B. F. Shi, Angew. Chem., Int. Ed., 2017, 56, 13145 CrossRef CAS PubMed ;
(k) L. Ye, Q. S. Gu, Y. Tian, X. Meng, G. C. Chen and X. Y. Liu, Nat. Commun., 2018, 9, 227 CrossRef PubMed ;
(l) I. Riano, U. Uria, E. Reyes, L. Carrillo and J. L. Vicario, J. Org. Chem., 2018, 83, 4180 CrossRef CAS PubMed ;
(m) A. López-Rodríguez, G. Domínguez and J. Pérez-Castells, J. Org. Chem., 2019, 84, 924 CrossRef PubMed .
- Selected examples:
(a) A. Ebinger, T. Heinz, G. Umbricht and A. Pfaltz, Tetrahedron, 1998, 54, 10469 CrossRef CAS ;
(b) Virender, S. L. Jain and B. Sain, Tetrahedron Lett., 2005, 46, 37 CrossRef CAS ;
(c) D. Marcoux, S. Azzi and A. B. Charette, J. Am. Chem. Soc., 2009, 131, 6970 CrossRef CAS PubMed ;
(d) M. L. Rosenberg, A. Krivokapic and M. Tilset, Org. Lett., 2009, 11, 547 CrossRef PubMed ;
(e) H. Xiong, H. Xu, S. Liao, Z. Xie and Y. Tang, J. Am. Chem. Soc., 2013, 135, 7851 CrossRef CAS PubMed ;
(f) M. J. González, J. González, L. A. Lõpez and R. Vicente, Angew. Chem., Int. Ed., 2015, 54, 12139 CrossRef PubMed ;
(g) R. M. Bychek, V. V. Levterov, I. V. Sadkova, A. A. Tolmachev and P. K. Mykhailiuk, Chem.–Eur. J., 2018, 24, 12291 CrossRef CAS PubMed ;
(h) J. Werth and C. Uyeda, Angew. Chem., Int. Ed., 2018, 57, 13902 CrossRef CAS PubMed .
-
(a) M. Rubin, M. Rubina and V. Gevorgyan, Chem. Rev., 2007, 107, 3117 CrossRef CAS PubMed ;
(b) I. Marek, S. Simaan and A. Masarwa, Angew. Chem., Int. Ed., 2007, 46, 7364 CrossRef CAS PubMed ;
(c) Z. B. Zhu, Y. Wei and M. Shi, Chem. Soc. Rev., 2011, 40, 5534 RSC ;
(d) L. Dian and I. Marek, Chem. Rev., 2018, 118, 8415 CrossRef CAS PubMed .
-
(a) Y. Bessard and M. Schlosser, Tetrahedron, 1991, 47, 7323 CrossRef CAS ;
(b) M. Rubin and V. Gevorgyan, Synthesis, 2004, 2004, 796 CrossRef ;
(c) X. C. Hang, W. P. Gu, Q. Y. Chen and J. C. Xiao, Tetrahedron, 2009, 65, 6320 CrossRef CAS ;
(d) K. Oshiro, Y. Morimoto and H. Amii, Synthesis, 2010, 2010, 2080 CrossRef ;
(e) F. Wang, T. Luo, J. Hu, Y. Wang, H. S. Krishnan, P. V. Jog, S. K. Ganesh, G. K. S. Prakash and G. A. Olah, Angew. Chem., Int. Ed., 2011, 50, 7153 CrossRef CAS PubMed ;
(f) L. Li, F. Wang, C. Ni and J. Hu, Angew. Chem., Int. Ed., 2013, 52, 12390 CrossRef CAS PubMed ;
(g) P. Rullière, P. Cyr and A. B. Charette, Org. Lett., 2016, 18, 1988 CrossRef PubMed .
- Selected examples:
(a) A. Padwa, G. D. Kennedy and M. W. Wannamaker, J. Org. Chem., 1985, 50, 5334 CrossRef CAS ;
(b) M. Sodupe, R. Rios, V. Branchadell, T. Nicholas, A. Oliva and J. J. Dannenberg, J. Am. Chem. Soc., 1997, 119, 4232 CrossRef CAS ;
(c) J. D. Xidos, T. L. Gosse, E. D. Burke, R. A. Poirier and D. J. Burnell, J. Am. Chem. Soc., 2001, 123, 5482 CrossRef CAS PubMed ;
(d) J. R. Henderson, M. Parvez and B. A. Keay, Org. Lett., 2007, 9, 5167 CrossRef CAS PubMed ;
(e) L. A. Fisher, N. J. Smith and J. M. Fox, J. Org. Chem., 2013, 78, 3342 CrossRef CAS PubMed ;
(f) N. S. Dange, A. Hussain Jatoi, F. Robert and Y. Landais, Org. Lett., 2017, 19, 3652 CrossRef CAS PubMed ;
(g) B. J. Levandowski and K. N. Houk, J. Am. Chem. Soc., 2016, 138, 16731 CrossRef CAS PubMed ;
(h) N. Semakul, K. E. Jackson, R. S. Paton and T. Rovis, Chem. Sci., 2017, 8, 1015 RSC ;
(i) B. Oller-Salvia, G. Kym and J. W. Chin, Angew. Chem., Int. Ed., 2018, 57, 2831 CrossRef CAS PubMed .
- Selected examples:
(a) A. P. Molchanov, V. V. Diev, J. Kopf and R. R. Kostikov, Russ. J. Org. Chem., 2004, 40, 431 CrossRef CAS ;
(b) V. V. Diev, R. R. Kostikov, R. Gleiter and A. P. Molchanov, J. Org. Chem., 2006, 71, 4066 CrossRef CAS PubMed ;
(c) V. V. Diev, O. N. Stetsenko, T. Q. Tung, R. R. Kostikov and A. P. Molchanov, J. Org. Chem., 2008, 73, 2396 CrossRef CAS PubMed ;
(d) A. S. Filatov, N. A. Knyazev, A. P. Molchanov, T. L. Panikorovsky, R. R. Kostikov, A. G. Larina, V. M. Boitsov and A. V. Stepakov, J. Org. Chem., 2017, 82, 959 CrossRef CAS PubMed ;
(e) A. S. Filatov, N. A. Knyazev, M. N. Ryazantsev, V. V. Suslonov, A. G. Larina, A. P. Molchanov, R. R. Kostikov, V. M. Boitsov and A. V. Stepakov, Org. Chem. Front., 2018, 5, 595 RSC ;
(f) A. S. Filatov, S. Wang, O. V. Khoroshilova, S. V. Lozovskiy, A. G. Larina, V. M. Boitsov and A. V. Stepakov, J. Org. Chem., 2019, 84, 7017 CrossRef CAS PubMed ;
(g) A. Deangelis, M. T. Taylor and J. M. Fox, J. Am. Chem. Soc., 2009, 131, 1101 CrossRef CAS PubMed ;
(h) H. L. Teng, Y. Luo, M. Nishiura and Z. Hou, J. Am. Chem. Soc., 2017, 139, 16506 CrossRef CAS PubMed ;
(i) H. Deng, W. L. Yang, F. Tian, W. Tang and W. P. Deng, Org. Lett., 2018, 20, 4121 CrossRef CAS PubMed ;
(j) Y. Yuan, Z.-J. Zheng, F. Ye, J.-H. Ma, Z. Xu, X.-F. Bai, L. Li and L.-W. Xu, Org. Chem. Front., 2018, 5, 2759 RSC .
-
(a) I. Marchueta, X. Verdaguer, A. Moyano, M. A. Pericàs and A. Riera, Org. Lett., 2001, 3, 3193 CrossRef CAS PubMed ;
(b) M. K. Pallerla and J. M. Fox, Org. Lett., 2005, 7, 3593 CrossRef CAS PubMed ;
(c) T. Sawano, M. Hashizume, S. Nishimoto, K. Ou and T. Nishimura, Org. Lett., 2015, 17, 2630 CrossRef CAS PubMed .
-
(a) F. de Nanteuil and J. Waser, Angew. Chem., Int. Ed., 2011, 50, 12075 CrossRef CAS ;
(b) F. Benfatti, F. de Nanteuil and J. Waser, Chem.–Eur. J., 2012, 18, 4844 CrossRef CAS PubMed ;
(c) F. Benfatti, F. de Nanteuil and J. Waser, Org. Lett., 2012, 14, 386 CrossRef CAS PubMed ;
(d) F. de Nanteuil, F. De Simone, R. Frei, F. Benfatti, E. Serrano and J. Waser, Chem. Commun., 2014, 50, 10912 RSC ;
(e) F. de Nanteuil, E. Serrano, D. Perrotta and J. Waser, J. Am. Chem. Soc., 2014, 136, 6239 CrossRef CAS PubMed ;
(f) S. Racine, F. de Nanteuil, E. Serrano and J. Waser, Angew. Chem., Int. Ed., 2014, 53, 8484 CrossRef CAS PubMed ;
(g) E. Serrano, F. de Nanteuil and J. Waser, Synlett, 2014, 25, 2285 CrossRef CAS ;
(h) S. Racine, B. Hegedüs, R. Scopelliti and J. Waser, Chem.–Eur. J., 2016, 22, 11997 CrossRef CAS ;
(i) S. Racine, J. Vuilleumier and J. Waser, Isr. J. Chem., 2016, 56, 566 CrossRef CAS ;
(j) J. Preindl, S. Chakrabarty and J. Waser, Chem. Sci., 2017, 8, 7112 RSC .
- See ESI† for details of the tested reaction conditions.
-
(a) J. D. Ha, J. W. Lee, S. C. Blackstock and J. K. Cha, J. Org. Chem., 1998, 63, 8510 CrossRef CAS ;
(b) C. Madelaine, Y. Six and O. Buriez, Angew. Chem., Int. Ed., 2007, 46, 8046 CrossRef CAS PubMed ;
(c) S. Maity, M. Zhu, R. S. Shinabery and N. Zheng, Angew. Chem., Int. Ed., 2012, 51, 222 CrossRef CAS PubMed ;
(d) T. H. Nguyen, S. Maity and N. Zheng, Beilstein J.
Org. Chem., 2014, 10, 975 CrossRef PubMed ;
(e) T. H. Nguyen, S. A. Morris and N. Zheng, Adv. Synth. Catal., 2014, 356, 2831 CrossRef CAS PubMed ;
(f) S. A. Morris, J. Wang and N. Zheng, Acc. Chem. Res., 2016, 49, 1957 CrossRef CAS PubMed ;
(g) Y. Kuang, Y. Ning, J. Zhu and Y. Wang, Org. Lett., 2018, 20, 2693 CrossRef CAS PubMed ;
(h) W. Liu, Y. Kuang, Z. Wang, J. Zhu and Y. Wang, Beilstein J. Org. Chem., 2019, 15, 542 CrossRef CAS PubMed .
- W. Cui and R. N. Loeppky, Tetrahedron, 2001, 57, 2953 CrossRef CAS .
-
(a) J. Luo and J. Zhang, ACS Catal., 2016, 6, 873 CrossRef CAS ;
(b) F. Le Vaillant, M. Garreau, S. Nicolai, G. Gryn'Ova, C. Corminboeuf and J. Waser, Chem. Sci., 2018, 9, 5883 RSC .
- C. K. Prier, D. A. Rankic and D. W. C. MacMillan, Chem. Rev., 2013, 113, 5322 CrossRef CAS PubMed .
- M. Garreau, F. Le Vaillant and J. Waser, Angew. Chem., Int. Ed., 2019, 58, 8182 CrossRef CAS PubMed .
- E. Speckmeier, T. G. Fischer and K. Zeitler, J. Am. Chem. Soc., 2018, 140, 15353 CrossRef CAS PubMed .
- The stereochemistry of the minor diastereoisomer was assigned by analogy with compound 16b, for which a crystal structure could be obtained by X-ray diffraction (CCDC number: 1938577†). The proposed structure of the major diastereoisomer is supported by the easy conversion of 10a to 50 (Scheme 5B). The other compounds were assigned based on analogy of the NMR spectra.
- When commercially available 1-bromo-4-cyclopropoxybenzene was used as substrate, no conversion was observed.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 1938577. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c9sc03790j |
|
| This journal is © The Royal Society of Chemistry 2019 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence *
*