
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceDevelopment of a hydrolysis-based small-molecule hydrogen selenide (H2Se) donor†
Turner D.
Newton
 and
Michael D.
Pluth
*
and
Michael D.
Pluth
*
Department of Chemistry and Biochemistry, Materials Science Institute, Institute of Molecular Biology, University of Oregon, Eugene, OR 97403, USA. E-mail: pluth@uoregon.edu
First published on 11th October 2019
Abstract
Selenium is essential to human physiology and has recently shown potential in the treatment of common pathophysiological conditions ranging from arsenic poisoning to cancer. Although the precise metabolic and chemical pathways of selenium incorporation into biomolecules remain somewhat unclear, many such pathways proceed through hydrogen selenide (H2Se/HSe−) formation. Despite this importance, well-characterized chemistry that enables H2Se release under controlled conditions remains lacking. Motivated by this need, we report here the development of a hydrolysis-based H2Se donor (TDN1042). Utilizing 31P and 77Se NMR experiments, we demonstrate the pH dependence of H2Se release and characterize observed reaction intermediates during the hydrolysis mechanism. Finally, we confirm H2Se release using electrophilic trapping reagents, which not only demonstrates the fidelity of this donor platform but also provides an efficient method for investigating future H2Se donor motifs. Taken together, this work provides an early example of an H2Se donor that functions through a well-defined and characterized mechanism.
Introduction
Selenium is often regarded as a toxic metalloid, but it is also an essential bioinorganic dietary micronutrient.1–3 For example, geographic regions with selenium-deficient soil display unusually high occurrences of conditions including Keshan and Kashin-Beck diseases in the population, which are both tied to a dietary scarcity of selenium.4,5 Dietary sources of selenium are typically selenomethionine (SeMet) and selenate salts (SeO42−), which must pass through complex metabolic pathways prior to incorporation into selenium-containing biomolecules.6 In the body, selenium exerts function primarily as selenocysteine, often referred to as the 21st amino acid, which is incorporated into and gives rise to the often unique reactivity of selenoproteins.7Twenty-five selenoproteins have been identified in humans and fall into several main categories. These categories include glutathione peroxidases (Gpx), which scavenge harmful peroxide species, thioredoxin reductases (TrxR), which regulate thiol-disulfide redox homeostasis, iodothyronine deiodinases (DIOs), which regulate thyroid hormone equilibria, and specialized selenoproteins that exhibit alternative functions, such as protein folding and selenium transport.3 Selenium-deficient environments often result in the preferential expression of these proteins, whereas selenium-rich environments result in the upregulation of selenium excretion pathways to mitigate selenium toxicity.8 Many of these pathways are hypothesized to proceed through the intermediate formation of hydrogen selenide (H2Se/HSe−), which is an important yet elusive small biomolecule of interest (Fig. 1).9
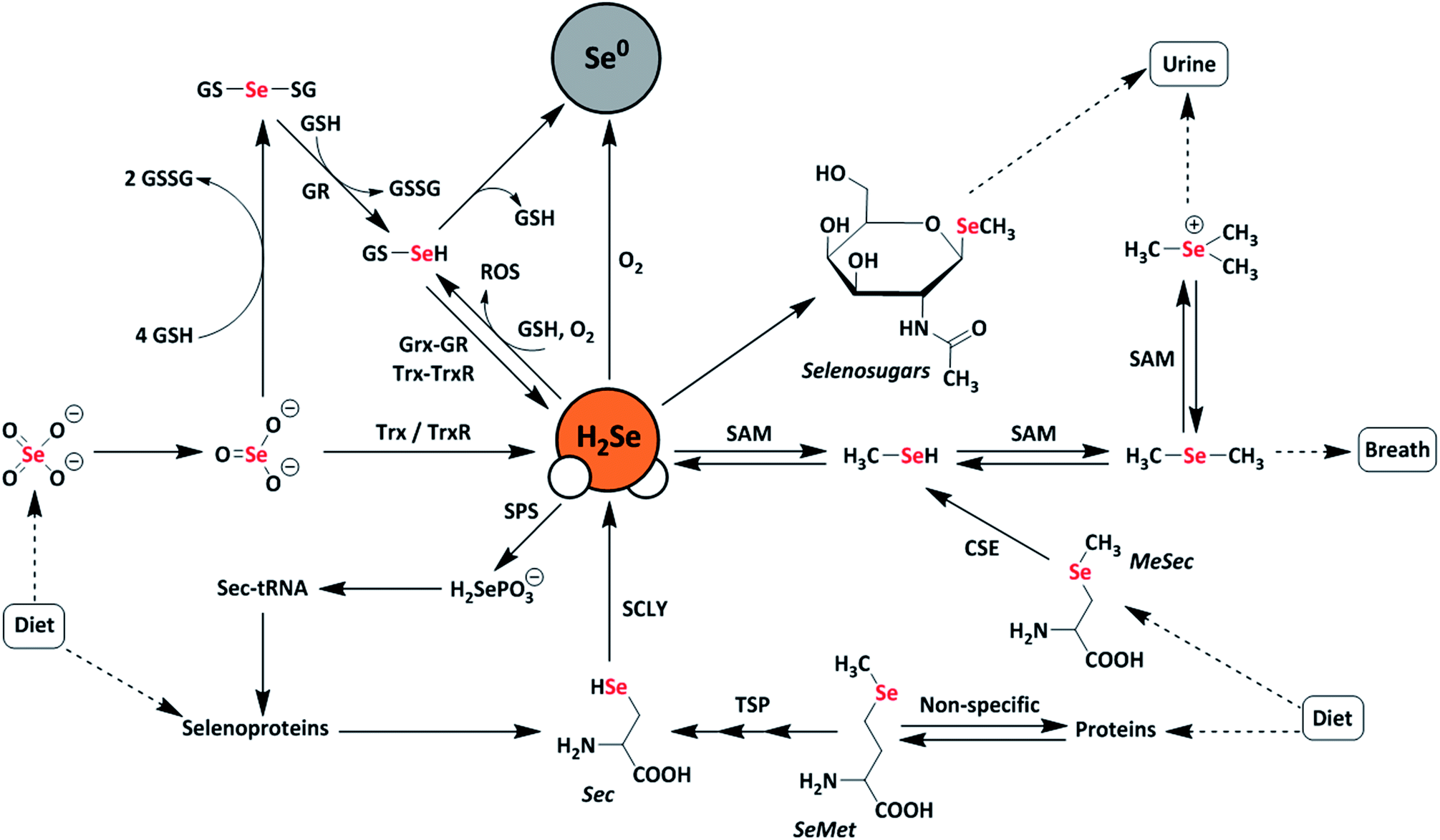 | ||
| Fig. 1 Schematic representation of metabolic pathways of dietary selenium compounds including various reactive selenium species. Selenate (SeO42−), selenite (SeO32−), selenophosphate (H2SePO3−), selenocysteine (Sec), selenomethionine (SeMet), methylselenocysteine (MeSec), thioredoxin (Trx), thioredoxin reductase (TrxR), glutaredoxin (Grx), glutathione reductase (GR), glutathione (GSH), diglutathione (GSSG), selenodiglutathione (GSSeSG), glutathioselenol (GSSeH), selenophosphate synthetase (SPS), selenocysteine lyase (SCLY), S-adenosylmethionine (SAM), transsulfuration pathway (TSP), elemental selenium (Se0), and reactive oxygen species (ROS). | ||
A common approach to increase the bioavailability of selenium is to use exogenous synthetic selenium-containing small molecules. For example, the organoselenium compound ebselen mimics the behavior of glutathione peroxidase and exhibits cytoprotective, anti-inflammatory, and antioxidant effects.10,11 Similarly, the glutathione-mediated reduction of selenite (SeO32−) to elemental selenium is thought to proceed through a selenodiglutathione (GS-Se-SG) intermediate en-route to a selenopersulfide (GS-SeH), which subsequently either decomposes to GSH and Se0 or is converted to H2Se through both enzymatic and non-enzymatic pathways.12,13 More recently, the hydrolysis of phthalic selenoanhydride was used to generate reactive selenium species (RSeS) to examine the differential synergies of these compounds with H2S and GSH in radical scavenging.14 In this investigation, H2Se release was proposed during hydrolysis but was not observed directly in the experiments.
Interest in developing chemical tools for investigating H2Se and related RSeS has grown in the last few years, with new investigations into the molecular recognition of HSe− in synthetic host-guest systems15 and with the advent of first-generation fluorescent probes for H2Se detection.16,17 In part, these investigations are motivated by potential roles of selenides in treating conditions ranging from arsenic poisoning, in which toxic arsenic species can react with H2Se to form readily-excreted products,18,19 to cancer, in which H2Se can induce oxidative stress under normoxic conditions or reductive stress under hypoxic conditions in HepG2 cells, resulting in HMGB1 protein damage and ultimately apoptosis.20 At physiological pH, almost all H2Se exists as HSe− due to the acidity of the diprotic form of H2Se (pKa = 3.9). The high redox activity and high nucleophilicity of H2Se/HSe−, when coupled with the relatively low biological selenium content (∼0.2 mg kg−1 in humans), make investigations into the biological roles of H2Se difficult.21,22 Building from these past results and increased interest in biorelevant small RSeS, we viewed that well-characterized, synthetic small molecules that release H2Se directly and under controlled conditions would provide a much-needed chemical tool for expanding research related to the chemical biology of selenium. Here we report the development and characterization of a hydrolysis-based small-molecule H2Se donor and provide insights into the reaction mechanism and methods for direct H2Se trapping.
Results and discussion
Drawing parallels to biological organosulfur chemistry, the last fifteen years have witnessed a surge in research related to hydrogen sulfide (H2S/HS−) as an important reactive sulfur species and gasotransmitter.23 Substantial efforts have focused on the development of small-molecule H2S donors for delivery to biological environments.24–26 Although the structure and complexity of such systems have evolved significantly, an early and broadly-used example of such donors is the hydrolysis-activated donor GYY4137, which relies on the hydrolytic cleavage of P![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) S bonds to generate H2S.27 GYY4137 has been used in >200 publications to date (Web of Science) and exhibits anti-inflammatory, vasorelaxant, and anti-cancer as well as other effects in different biological models27–29 with diverse applications ranging from medicinal to agricultural science.30,31 Motivated by the broad utility of this approach to access H2S donor motifs, we sought to use similar chemistry to generate well-defined H2Se donors that are activated by PSe bond hydrolysis. To prepare such a donor, we treated Woollins' reagent with an excess of morpholine, drawing parallels to the synthesis of GYY4137, to generate TDN1042 in moderate yield (Fig. 2a). The resultant product was characterized by 1H, 13C{1H}, 31P, and 77Se NMR spectroscopy (Fig. S1–S4†). Single crystals suitable for X-ray diffraction were grown by layering hexane onto a solution of TDN1042 in CH2Cl2, which confirmed the molecular structure (Fig. 2b).
S bonds to generate H2S.27 GYY4137 has been used in >200 publications to date (Web of Science) and exhibits anti-inflammatory, vasorelaxant, and anti-cancer as well as other effects in different biological models27–29 with diverse applications ranging from medicinal to agricultural science.30,31 Motivated by the broad utility of this approach to access H2S donor motifs, we sought to use similar chemistry to generate well-defined H2Se donors that are activated by PSe bond hydrolysis. To prepare such a donor, we treated Woollins' reagent with an excess of morpholine, drawing parallels to the synthesis of GYY4137, to generate TDN1042 in moderate yield (Fig. 2a). The resultant product was characterized by 1H, 13C{1H}, 31P, and 77Se NMR spectroscopy (Fig. S1–S4†). Single crystals suitable for X-ray diffraction were grown by layering hexane onto a solution of TDN1042 in CH2Cl2, which confirmed the molecular structure (Fig. 2b).
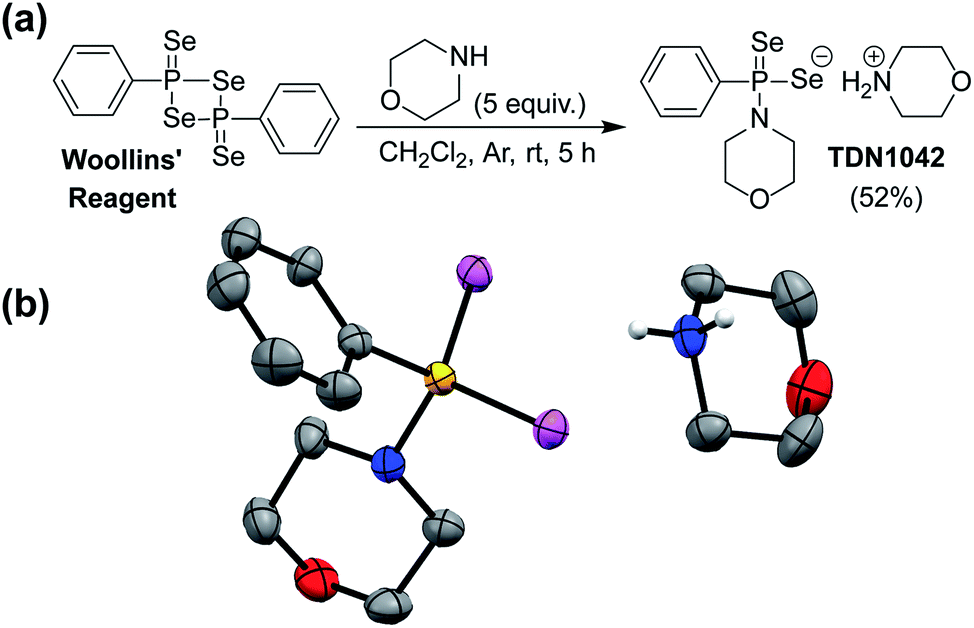 | ||
| Fig. 2 (a) Synthesis of TDN1042 from Woollins' reagent. (b) ORTEP diagram (50% ellipsoids) of TDN1042. Hydrogen atoms, except those on the morpholinium nitrogen, are omitted for clarity. | ||
With TDN1042 in hand, we next evaluated its reaction chemistry by NMR spectroscopy. Initial studies using 31P NMR spectroscopy in wet DMSO-d6 revealed the clean conversion of TDN1042 to phenylphosphonic acid (PPA) as expected (Fig. 3a). We next monitored the hydrolysis in buffered aqueous solutions using quantitative 31P NMR spectroscopy with triethylphosphate (TEP) as an internal integration standard. In these experiments, we also observed clean conversion of TDN1042 (δ(31P) = 61 ppm) to the expected PPA hydrolysis product (δ(31P) = 12 ppm). To determine the effect of pH on this reaction, we measured the rate of hydrolysis of TDN1042 (10 mM) in citrate buffer (50 mM) ranging from pH 3.0 to pH 6.0 in flame-sealed NMR tubes at ambient temperature (Fig. 3b). The resulting hydrolysis data (Fig. 3c) revealed an increase in rate at more acidic pH values, which is consistent with the expected hydrolysis mechanism. A similar pH dependence was observed for GYY4137 in a previous report,27 although the experimental conditions and methods used to monitor rates and product conversions are too dissimilar to those used here for TDN1042 to make direct quantitative comparisons. This similarity in pH dependences does, however, suggest that TDN1042 could find utility in biological contexts much like GYY4137.
 | ||
| Fig. 3 (a) Proposed hydrolysis mechanism of TDN1042 resulting in H2Se release. (b) 31P NMR spectra during the hydrolysis of TDN1042 showing the consumption of TDN1042 (61 ppm) and generation of PPA (12 ppm). (c) pH dependence of TDN1042 during the hydrolysis experiments. (d) pH dependence of PPA formation during the hydrolysis experiments. General conditions: 10 mM TDN1042 in 50 mM citrate buffer ranging from pH 3.0 to pH 6.0 at room temperature. | ||
Having established that the hydrolysis of TDN1042 results in PPA formation, we next sought to confirm H2Se release directly. To monitor H2Se release, our goal was to trap H2Se directly rather than use fluorogenic probes in case a reactive intermediate en-route to H2Se release resulted in the activation of such systems. To accomplish this labeling, we used benzyl bromide (BnBr) as an electrophilic trapping agent and monitored the reaction by 31P and 77Se NMR spectroscopy (Fig. 4a). On the basis of the proposed release mechanism of H2Se from TDN1042, we expected that, akin to initial protonation, BnBr would initially alkylate the PSe moiety, which would activate TDN1042 toward hydrolysis to release benzyl selenol (BnSeH) with subsequent alkylation by BnBr to generate dibenzyl selenide (Bn2Se). By monitoring the reaction by both 31P and 77Se NMR spectroscopy, we observed immediate formation of an intermediate (1) upon addition of BnBr and H2O to a solution of TDN1042 in DMSO-d6 (Fig. 4b and c). Intermediate 1 exhibited a singlet in the 31P spectrum (δ = 69 ppm) with two sets of selenium satellites with different coupling constants (JP–Se = 786 Hz, JP–Se = 401 Hz). This coupling pattern is consistent with inequivalent selenium environments in 1 and is in contrast to the single set of selenium satellites seen in TDN1042 (δ(31P) = 61 ppm, 1JP–Se = 671 Hz). Furthermore, the 77Se NMR spectrum of 1 revealed both a doublet (δ = −129 ppm, 1JP–Se = 786 Hz) and a doublet of triplets (δ = 354 ppm, 1JP–Se = 401 Hz, 2JSe–H = 11 Hz), which correspond to the PSe and P–SeCH2Ph moieties, respectively (Fig. 4e). As the reaction proceeded, the intensity of the δ = −129 and 354 ppm peaks decreased, with concomitant formation of PPA (δ(31P) = 12 ppm) in the 31P NMR spectrum. The 77Se NMR spectrum revealed two Se-containing products, with a triplet at δ(77Se) = 394 ppm and a pentet at δ(77Se) = 330 ppm. The 330 ppm resonance corresponds to Bn2Se, in which the selenium signal is split by two sets of benzylic protons. The 394 ppm resonance corresponds to dibenzyl diselenide (Bn2Se2), which was confirmed by comparison with an authentic Bn2Se2 sample (Fig. S13†). Formation of the diselenide is likely due to the auto-oxidation of BnSeH, which as has been observed previously.32 Taken together, these alkylation experiments support the mechanism of H2Se release and provide mechanistic insights into the hydrolysis mechanism.
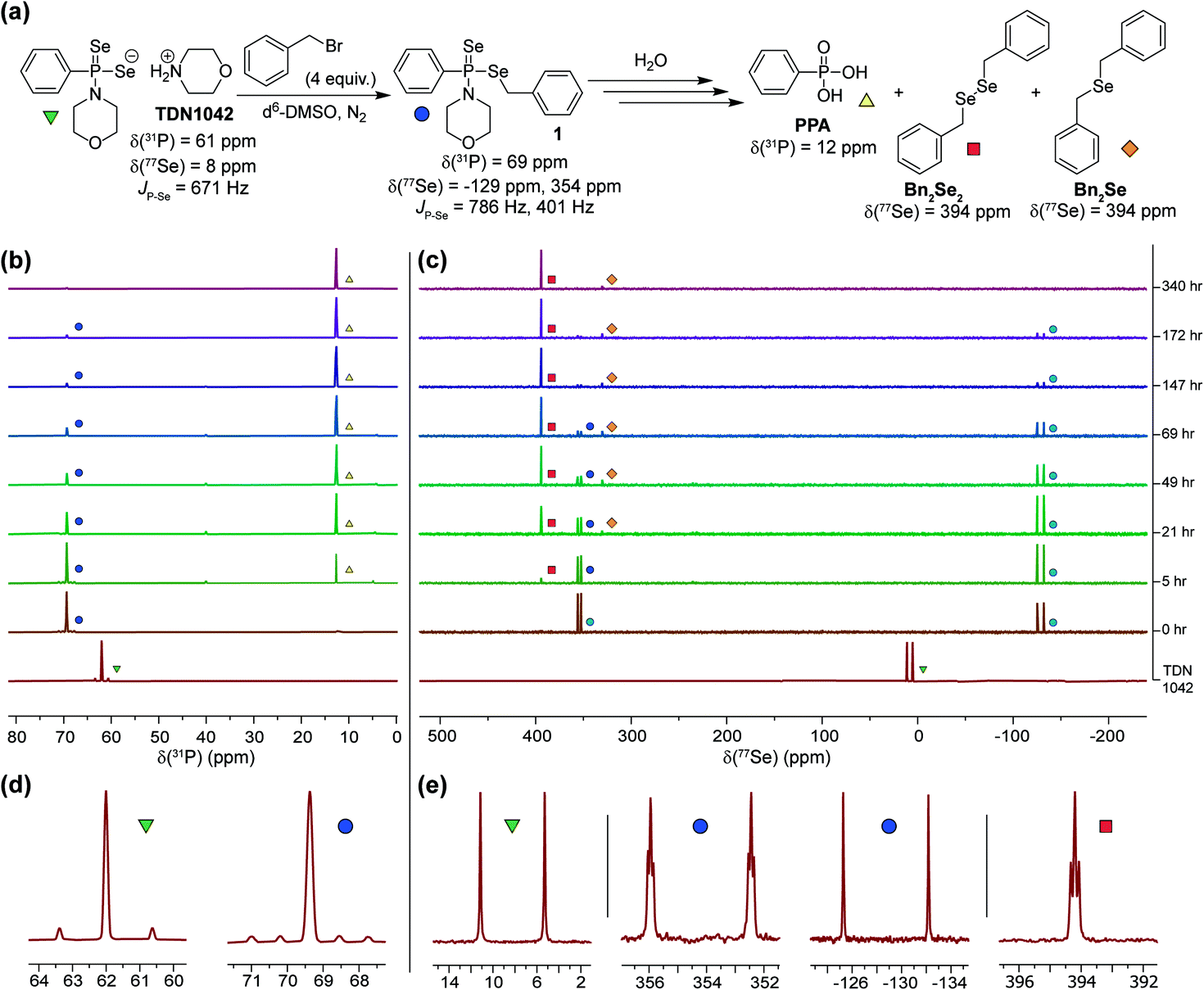 | ||
| Fig. 4 (a) Proposed hydrolysis and trapping pathways. See Scheme S3† for a more detailed mechanism. (b) 31P NMR spectra during alkylation and hydrolysis. (c) 77Se NMR spectra during alkylation and hydrolysis. (d and e) Expanded regions of the 31P and 77Se NMR spectra highlight the observed coupling patterns. | ||
To definitively establish H2Se release, we next performed experiments in which the electrophilic trapping agent was separate from the donor. For this investigation, we used 2,4-dinitrofluorobenzene (FDNB) as an electrophilic labeling reagent to trap the H2Se released and volatilized into the headspace of the reaction apparatus.33,34 In these experiments, a vial containing an aqueous solution of TDN1042 was acidified with HCl and sparged with N2 to help volatilize any H2Se into the headspace, which was subsequently bubbled through a trapping solution containing a large excess of FDNB (Fig. 5a). A final solution containing AgNO3 was used to scavenge any unreacted H2Se. Using HPLC, we observed formation of both di(2,4-dinitrophenyl) selenide ((DNP)2Se) and the corresponding diselenide ((DNP)2Se2) in the trapping solution (Fig. 5b and S14†), which is consistent with directly trapping H2Se as well as the auto-oxidation process. The identity of the observed products was confirmed by comparison to authentic samples of (DNP)2Se and (DNP)2Se2 synthesized according to published procedures (Fig. S18†).35 Taken together, these results confirm that TDN1042 releases H2Se directly.
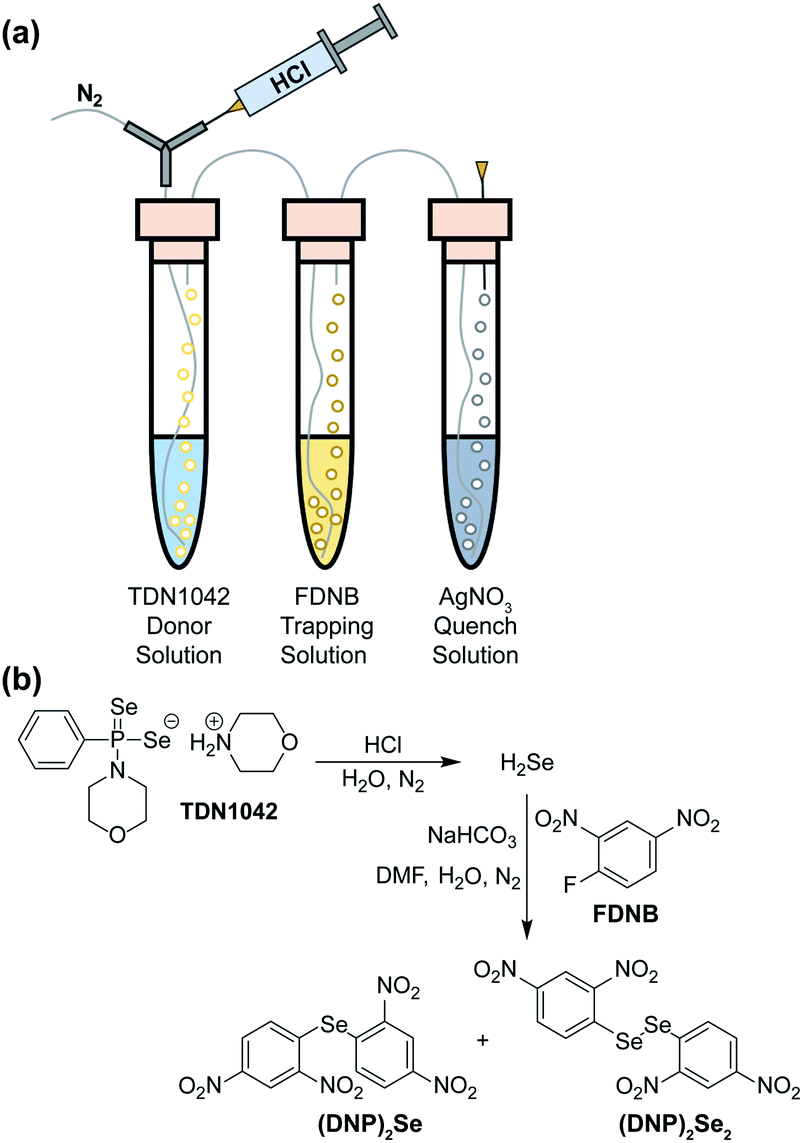 | ||
| Fig. 5 (a) Experimental setup for volatilization and trapping of H2Se with FDNB. (b) General reaction pathways for (DNP)2Se and (DNP)2Se2 formation. | ||
Conclusions
Here we report the development and characterization of the hydrolysis-based H2Se donor TDN1042. Using multinuclear NMR experiments, we monitored the reaction pathway for H2Se release and confirmed H2Se generation using different electrophilic trapping methods. We anticipate that this well-characterized H2Se donor will find utility in biological investigations into the roles of H2Se and related reactive selenium species in the future and will facilitate the development of chemical tools for further investigating H2Se in chemical biology.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This research was supported by the NSF (CHE-1454747) and Dreyfus Foundation. NMR capabilities in the UO CAMCOR facility are supported by the NSF (CHE-1427987, CHE-1625529). We would like to thank Dr Samantha N. MacMillan from the X-ray diffraction facility at Cornell University for crystal structure determination.Notes and references
- R. C. McKenzie, T. S. Rafferty and G. J. Beckett, Immunol. Today, 1998, 19, 342–345 CrossRef CAS PubMed.
- L. Schomburg, U. Schweizer and J. Kohrle, Cell. Mol. Life Sci., 2004, 61, 1988–1995 CrossRef CAS PubMed.
- H. J. Reich and R. J. Hondal, ACS Chem. Biol., 2016, 11, 821–841 CrossRef CAS PubMed.
- J. S. Chen, Asia Pac, J. Clin. Nutr., 2012, 21, 320–326 Search PubMed.
- P. Sudre and F. Mathieu, Int. Orthop., 2001, 25, 175–179 CrossRef CAS PubMed.
- L. V. Papp, J. Lu, A. Holmgren and K. K. Khanna, Antioxid. Redox Signaling, 2007, 9, 775–806 CrossRef CAS PubMed.
- D. L. Hatfield, P. A. Tsuji, B. A. Carlson and V. N. Gladyshev, Trends Biochem. Sci., 2014, 39, 112–120 CrossRef CAS PubMed.
- R. F. Burk and K. E. Hill, Annu. Rev. Nutr., 2015, 35, 109–134 CrossRef CAS PubMed.
- K. A. Cupp-Sutton and M. T. Ashby, Antioxidants, 2016, 5, 42–59 CrossRef PubMed.
- T. Schewe, General Pharmacology: The Vascular System, 1995, 26, 1153–1169 CrossRef CAS PubMed.
- J. L. Wedding, B. Lai, S. Vogt and H. H. Harris, Biochim. Biophys. Acta, 2018, 1862, 2393–2404 CrossRef CAS PubMed.
- H. S. Hsieh and H. E. Ganther, Biochemistry, 1975, 14, 1632–1636 CrossRef CAS PubMed.
- J. Kessi and K. W. Hanselmann, J. Biol. Chem., 2004, 279, 50662–50669 CrossRef CAS PubMed.
- A. Kharma, A. Misak, M. Grman, V. Brezova, L. Kurakova, P. Barath, C. Jacob, M. Chovanec, K. Ondrias and E. Domínguez-Álvarez, New J. Chem., 2019, 43, 11771–11783 RSC.
- H. A. Fargher, N. Lau, L. N. Zakharov, M. M. Haley, D. W. Johnson and M. D. Pluth, Chem. Sci., 2019, 10, 67–72 RSC.
- F. P. Kong, L. H. Ge, X. H. Pan, K. H. Xu, X. J. Liu and B. Tang, Chem. Sci., 2016, 7, 1051–1056 RSC.
- F. P. Kong, Y. H. Zhao, Z. Y. Liang, X. J. Liu, X. H. Pan, D. R. Luan, K. H. Xu and B. Tang, Anal. Chem., 2017, 89, 688–693 CrossRef CAS PubMed.
- H. J. Sun, B. Rathinasabapathi, B. Wu, J. Luo, L. P. Pu and L. Q. Ma, Environ. Int., 2014, 69, 148–158 CrossRef CAS PubMed.
- I. Zwolak, Biol. Trace Elem. Res., 2019, 1–20 Search PubMed.
- X. H. Pan, X. X. Song, C. Wang, T. T. Cheng, D. R. Luan, K. H. Xu and B. Tang, Theranostics, 2019, 9, 1794–1808 CrossRef CAS PubMed.
- H. A. Schroeder, D. V. Frost and J. J. Balassa, J. Chronic Dis., 1970, 23, 227–243 CrossRef CAS PubMed.
- M. Navarro-Alarcon and C. Cabrera-Vique, Sci. Total Environ., 2008, 400, 115–141 CrossRef CAS PubMed.
- R. Wang, FASEB J., 2002, 16, 1792–1798 CrossRef CAS PubMed.
- S. D. Zanatta, B. Jarrott and S. J. Williams, Aust. J. Chem., 2010, 63, 946–957 CrossRef CAS.
- M. M. Cerda, T. D. Newton, Y. Zhao, B. K. Collins, C. H. Hendon and M. D. Pluth, Chem. Sci., 2019, 10, 1773–1779 RSC.
- C. M. Levinn, A. K. Steiger and M. D. Pluth, ACS Chem. Biol., 2019, 14, 170–175 CrossRef CAS PubMed.
- L. Li, M. Whiteman, Y. Y. Guan, K. L. Neo, Y. Cheng, S. W. Lee, Y. Zhao, R. Baskar, C. H. Tan and P. K. Moore, Circulation, 2008, 117, 2351–2360 CrossRef CAS PubMed.
- L. Li, M. Salto-Tellez, C. H. Tan, M. Whiteman and P. K. Moore, Free Radical Biol. Med., 2009, 47, 103–113 CrossRef CAS PubMed.
- Z. W. Lee, X. Y. Teo, E. Y. W. Tay, C. H. Tan, T. Hagen, P. K. Moore and L. W. Deng, Br. J. Pharmacol., 2014, 171, 4322–4336 CrossRef CAS PubMed.
- G. L. Meng, J. Wang, Y. J. Xiao, W. L. Bai, L. P. Xie, L. Y. Shan, P. K. Moore and Y. Ji, J. Biomed. Res., 2015, 29, 203–213 Search PubMed.
- J. M. Carter, E. M. Brown, J. P. Grace, A. K. Salem, E. E. Irish and N. B. Bowden, PLoS One, 2018, 13, e0208732 CrossRef PubMed.
- A. R. M. de Oliveira, L. Piovan, F. Simonelli, A. Barison, M. D. C. Santos and M. B. M. de Mello, J. Organomet. Chem., 2016, 806, 54–59 CrossRef.
- H. E. Ganther and R. J. Kraus, Anal. Biochem., 1984, 138, 396–403 CrossRef CAS PubMed.
- H. E. Ganther and R. J. Kraus, Methods Enzymol., 1987, 143, 32–38 CAS.
- D. F. Twiss, J. Chem. Soc., 1914, 105, 1672–1678 RSC.
Footnote |
| † Electronic supplementary information (ESI) available: Synthetic details, NMR spectra, HPLC experiments, crystallographic information. CCDC 1953366. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c9sc04616j |
| This journal is © The Royal Society of Chemistry 2019 |