
DePEGylation strategies to increase cancer nanomedicine efficacy
Li
Kong
,
Frederick
Campbell
and
Alexander
Kros
 *
*
Leiden Institute of Chemistry – Supramolecular and Biomaterial Chemistry, Leiden University, Einsteinweg 55, 2333CC Leiden, The Netherlands. E-mail: a.kros@chem.leidenuniv.nl
First published on 4th December 2018
Abstract
To maximize drug targeting to solid tumors, cancer nanomedicines with prolonged circulation times are required. To this end, poly(ethylene glycol) (PEG) has been widely used as a steric shield of nanomedicine surfaces to minimize serum protein absorption (opsonisation) and subsequent recognition and clearance by cells of the mononuclear phagocyte system (MPS). However, PEG also inhibits interactions of nanomedicines with target cancer cells, limiting the effective drug dose that can be reached within the target tumor. To overcome this dilemma, nanomedicines with stimuli-responsive cleavable PEG functionality have been developed. These benefit from both long circulation lifetimes en route to the targeted tumor as well as efficient drug delivery to target cancer cells. In this review, various stimuli-responsive strategies to dePEGylate nanomedicines within the tumor microenvironment will be critically reviewed.
 Left to right: Frederick Campbell, Li Kong and Alexander Kros | Li Kong obtained her BSc and MSc in the School of Chemistry and Chemical Engineering from Shandong University, under the supervision of Prof. Dr Jingcheng Hao and Prof. Dr Aiyou Hao. In 2018, She received her PhD degree under the supervision of Dr Frederick Campbell and Prof. Dr Alexander Kros in Leiden University. Currently, she is a postdoctoral researcher in the group of Prof. Alexander Kros. Her main research interests focus on the photo-activated drug delivery systems and their application in vitro and in vivo. |
Frederick Campbell is a postdoctoral researcher within the Supramolecular and Biomaterials Chemistry group, Leiden Institute of Chemistry. He received his PhD degree from the University of Leeds, UK in 2009 before conducted postdoctoral research at University College London, UK. He moved to Leiden on a EU Marie Curie Fellowship in 2013. His research interests lie in understanding the fundamental behavior of nanoparticles in vivo, using the embryonic zebrafish as animal model, to elucidate more effective and simpler nanomedicines. |
Alexander Kros is professor of Supramolecular & Biomaterials Chemistry at the Leiden Institute of Chemistry. He received his PhD degree under supervision of Prof. Nolte from the Radboud University, Nijmegen, the Netherlands. After a postdoctoral position in the group of Prof. Tirrell at Caltech he obtained a position at Leiden University. His research interests are synthetic membrane fusion systems, targeted drug delivery systems and using the zebrafish model to make the translation of nanomedicines from lab to clinic more efficient. |
1. Introduction
In the treatment of cancer, the main challenge is how to deliver cytotoxic drugs to cancer cells while minimizing off-target toxicity in healthy cells and tissue. Patients currently undergoing cancer chemotherapy will typically experience debilitating side effects1 (e.g. impaired immune system, nausea, cardiomyopathy, hair loss), and in many cases, the cumulative lifetime dose of an anti-cancer drug (e.g. doxorubicin; 550 mg kg−1) must be limited, irrespective of therapeutic success, to avoid permanent bodily damage.2 Efforts have therefore been made to develop nanomedicines capable of delivering drugs specifically to cancer cells.3Over the past 30 years, two clinically effective targeted cancer therapies have emerged: antibody–drug conjugates (ADCs) and nanoparticle-based systems. Currently, 4 ADCs and 7 distinct nanoparticle-based drug delivery systems (DDS), targeted against a variety of human cancers, have received market approval.4,5 For ADCs, active targeting of cancer cells is achieved through antibody recognition of (over-)expressed receptors (tumor-associated antigens).6 Once bound, ADCs are endocytosed, the conjugated drug released and the cell destroyed. Although effective, ADCs are costly to manufacture, can elicit adverse immunogenic responses (limiting repeat dosing) and are largely restricted to the delivery of small molecule (and serum stable) drugs.7 In the case of nanoparticle-based DDS, drugs are encapsulated within a self-assembled nanoparticle, hidden and protected from the in vivo environment. Pharmacokinetic (PK) profiles are dictated by the nanoparticle and, in theory, it is possible to deliver almost any therapeutic cargos, from small molecule drugs to plasmid DNA, to target cells and tissue within the body. An enormous variety of nanoparticle-based DDS have been reported, however the most widely investigated, and the majority approved for clinical application, are liposomes.8 In the targeted treatment of cancer, all clinically approved nanoparticle-based nanomedicines are liposomes designed to passively target tumors via the enhanced permeability and retention (EPR) effect.9,10
1.1 The enhanced permeability and retention (EPR) effect
Following administration to the body, small molecule drugs freely diffuse into tissue and away from the site of injection. In contrast, intravenously (i.v.) injected nanoparticles are restricted to the circulating blood flow, unable to cross the tightly packed endothelium due to their larger size. For optimal biodistribution, nanoparticles should be larger than 10 nm in diameter – below which they are filtered from the body via the kidneys11 – and smaller than 200 nm in diameter – above which they are rapidly recognized and phagocytosed by blood resident macrophages (principle cells of the mononuclear phagocyte system, MPS), within the liver and spleen, and are cleared from the body.12The EPR effect is a phenomena characterized by the ill-defined (‘leaky’) vasculature and poor lymphatic drainage of tumors that arises as a result of rapid angiogenesis (blood vessel growth) within tumor tissue.13 Circulating nanoparticles circulating through the tumor vasculature can therefore passively diffuse across gaps in the endothelium, accumulate within the tumor and remain there for extended periods of time. Once within the tumor, nanoparticle encapsulated drugs either passively diffuse from the nanoparticle or an endogenous or exogenous stimulus can be exploited to trigger release.
To maximize passive targeting of nanomedicines to solid tumors via the EPR effect, nanoparticles with long circulation lifetimes are sought. Put simply, the more times nanoparticles pass through the tumor vasculature, the more will accumulate there. Care must therefore be taken to minimize drug leakage from the nanoparticle en route to the tumor while ensuring therapeutically relevant concentrations of drugs are released once there. In the case of liposome-drug formulations, this involves careful choice of lipid reagents (e.g. cholesterol to rigidify fluid lipid membranes) to fine tune drug retention/release profiles while at the same time maximizing circulation lifetimes.14
1.2 Polyethylene glycol (PEG)
To achieve long circulation lifetimes, the principal biological barrier a nanoparticle must overcome is recognition and clearance by cells of the mononuclear phagocyte system (MPS).15 The principle organ of the MPS is the liver where hepatic macrophages – Kupffer cells – are highly proficient at recognizing and removing macromolecular, colloidal and pathogenic waste from circulation.16,17 Without any surface modification, up to 99% of systemically administered nanoparticles are cleared by the liver.18 In most cases, it is believed rapid adsorption of blood proteins to the surface of nanoparticles, (a process known as opsonisation), acts as the recognition beacon for MPS cells.19 For this reason, sterically shielding nanoparticle surfaces with biocompatible polymers, such as polyethylene glycol (PEG), has been effectively employed to minimize opsonisation and prolong blood circulation times of nanoparticles in vivo.20PEG is a synthetic polymer of repeating ethylene glycol units. Used as a reagent or additive in a wide range of biological, chemical and industrial settings,21,22 it is commercially available in a range of geometries (linear, branched, star, comb), molecular weights (from 300 Da – 6–7 repeating units – up to 10 MDa – >200![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 repeating units) and can be easily functionalized. PEGylation of nanoparticle surfaces has been shown to decrease serum protein adsorption, reduce nanoparticle uptake in the liver and prolong circulation lifetimes.23 Recently, reports have emerged to suggest PEG can elicit an immunogenic response in mammals.24 However, the extent of this response, caused by binding of anti-PEG antibodies, remains unclear.25 PEG remains an FDA approved polymer and is still the most widely used polymeric coating of nanomedicines, both in academic and industrial research. In terms of cancer nanomedicines, PEGylated liposomal-doxorubicin (Doxil®) has been used clinically for over 20 years in the treatment of select breast and ovarian cancers, multiple myeloma and AIDS-related Kaposi's sarcoma.22
000 repeating units) and can be easily functionalized. PEGylation of nanoparticle surfaces has been shown to decrease serum protein adsorption, reduce nanoparticle uptake in the liver and prolong circulation lifetimes.23 Recently, reports have emerged to suggest PEG can elicit an immunogenic response in mammals.24 However, the extent of this response, caused by binding of anti-PEG antibodies, remains unclear.25 PEG remains an FDA approved polymer and is still the most widely used polymeric coating of nanomedicines, both in academic and industrial research. In terms of cancer nanomedicines, PEGylated liposomal-doxorubicin (Doxil®) has been used clinically for over 20 years in the treatment of select breast and ovarian cancers, multiple myeloma and AIDS-related Kaposi's sarcoma.22
1.3 The PEG dilemma
While PEGylation prolongs circulation lifetimes, it also limits the cellular uptake of nanoparticles and therefore effective drug delivery to target cancer cells.26 This so-called ‘PEG dilemma’ has proved a major obstacle in the effective delivery of therapeutic cargos to cancer cells, particularly those that must be actively transported across the target cellular membrane (e.g. proteins and oligonucleotides).27 For instance, in the delivery of oligonucleotides (ODNs) or small interfering RNAs (siRNAs), significantly lower transfection/transduction efficiencies were observed for PEGylated vs. non-PEGylated DDS.28 To overcome this dilemma, strategies have been proposed to trigger the extracellular shedding of PEG (i.e. dePEGylation) from a nanoparticle surface upon reaching the target tumor. This leads to one of three scenarios (Fig. 1): (1) rupture of the nanoparticle and extracellular drug release; (2) cellular uptake (endocytosis) of the intact nanoparticle–drug complex or (3) in the case of liposomes, fusion with the target cellular membrane and drug release directly to the cell cytoplasm, crucially avoiding degradative endocytotic liposome uptake.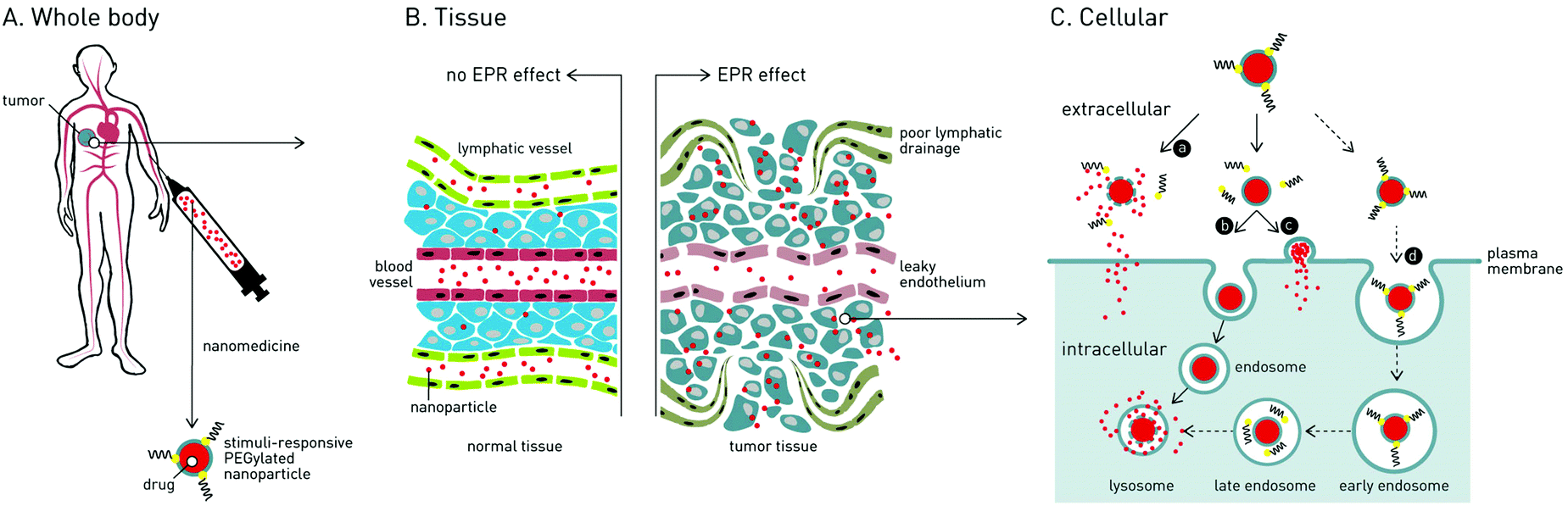 | ||
| Fig. 1 Following passive targeting of solid tumors via the enhanced permeability and retention (EPR) effect, stimuli-responsive dePEGylation of cancer nanomedicines can lead to various routes of enhanced drug delivery: route a – extracellular dePEGylation, nanocarrier rupture and extracellular drug delivery; route b – extracellular dePEGylation, endocytotic nanocarrier uptake and intracellular drug delivery; route c – extracellular dePEGylation, nanocarrier fusion with cancer cell membrane and direct cytosolic drug delivery (most relevant for liposomal nanomedicines); route d* – endocytotic nanocarrier uptake, intracellular dePEGylation and intracellular drug delivery. * this route does not overcome the “PEG dilemma” and the very limited uptake of PEGylated nanoparticles is a major drawback of these systems. | ||
In a significant number of reported dePEGylation strategies, it is required that PEGylated nanoparticles are first taken up by target cancer cells, whereupon the low pH, reductive and protease-rich environment of the late endosome/lysosome can be effectively exploited to trigger intracellular dePEGylation and drug release. However, these systems do not overcome the “PEG dilemma” and the very limited uptake of PEGylated nanoparticles remains a major drawback. As such, these systems will not be further discussed in this review but are included in the comprehensive summary of dePEGylation strategies presented in Table 1.
| Site of dePEGylation within tumor | Stimulus | Example chemical structure | Nanocarrier | Drug release [ref.] | |
|---|---|---|---|---|---|
| Intracellular | Extracellular | ||||
| a Given the very limited uptake of PEGylated nanoparticles, systems reliant on intracellular triggers do not overcome the ‘PEG dilemma’ and are not further discussed in this review. | |||||
| Intracellulara | Low pH late endosome (pH < 6.5) lysosome (pH 5.5–6.5) |
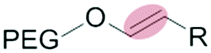
|
Liposomes | 46–49 | |
| Vinylether | Micelles | 50 | |||
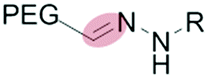
|
Liposomes | 52–54 | |||
| Hydrazone | Micelles | 55–61 | |||
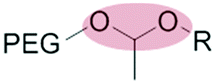
|
Liposomes | 62 | |||
| Acetal | Micelles | 63–69 | |||
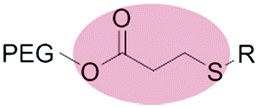
|
Micelles | 70 | |||
| β-Thiopropionate | |||||
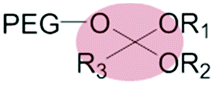
|
Liposomes | 71–73 | |||
| Ortho ester | |||||
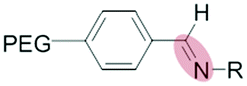
|
Micelles | 74–76 | |||
| Benzoic-imine | |||||
| Reduction glutathione (GSH) (2–10 mM) |

|
Liposomes | 93 | ||
| Polymersomes | 94 and 95 | ||||
| Micelles | 96–121 and 133 | ||||
| Graphene oxide | 122–124 | ||||
| Mesoporous silica nanoparticles (MSN) | 125–132 | ||||
| Magnetic nanoparticles | 134 | ||||
| Enzymatic cathepsin B |

|
Liposomes | 162 | ||
| (peptide consensus sequence) | |||||
For strategies involving extracellular dePEGylation within the target tumor, a key difference is whether dePEGylation causes destabilization of the nanocarrier and extracellular drug release (i.e. burst release), or intact nanocarrier internalization by target cancer cells and intracellular drug release. In the case of extracellular drug release, only drugs able to passively diffuse (or be actively transported) across target cancer cell membranes (e.g. membrane permeable doxorubicin) can be used. In the case of intracellular drug release, the delivery of membrane impermeable therapeutics (e.g. proteins, oligonucleotides) is possible. In either scenario, it is essential cancer cells are exposed to therapeutically relevant doses of cytotoxic drugs if improved therapeutic indices are to be achieved.
2. Physical dePEGylation
Two physical approaches to dePEGylate nanoparticle surfaces within target tissues have been investigated. The first, most relevant for liposomal nanomedicines, relies on the exchange of PEGylated lipids from a liposome membrane to a target membrane sink (e.g. target cancer cell membranes).29 Here, the rate at which exchange occurs is heavily dependent on the lipid anchor tethering PEG to the liposome membrane (i.e. how strongly it is held within the liposome membrane).30 The length and saturation of (phospho)lipid fatty acid (FA) chains determines both the thickness and rigidity of a lipid membrane. FA chain lengths within biological membranes typically vary between C12 and C30 – the number of carbon atoms.31 FA chains can be saturated (no double bonds) or unsaturated (1 or more double bond). Saturated FAs pack closely together to form rigid lipid membranes (gel state), whereas unsaturated FAs loosely pack to form fluid membranes liquid crystalline state.32 In addition, the shorter the FA chains, the more fluid the membrane. This is reflected in the liquid crystalline-to-gel transition temperatures (Tm) of individual (phospho)lipids.In a study of three different lipid–PEG conjugates, no lipid–PEG exchange was observed for long chain, saturated lipid anchors 1,2-distearoyl-sn-glycero-3-phosphoethanolamine (DSPE-PEG, C18:0 – 2 × 18 carbon FA chain; no double bonds) whereas exchange occurred in the time frame of hours for shorter saturated lipids 1,2-dimyristoyl-sn-glycero-3-phosphoethanolamine (DMPE-PEG; C14:0) or long chain, unsaturated lipids 1,2-dioleoyl-sn-glycero-3-phosphoethanolamine (DOPE-PEG; C18:1 – 2 × 18 carbon FA chain; each 1 double bond, ω9).33 This time frame enabled efficient accumulation of liposomes in tumor sites via the EPR effect (prior to dePEGylation) coupled with increased cellular uptake within the tumor (following dePEGylation). Conversely, a similar study found that only in the case of DSPE-PEG were circulation times prolonged enough to see efficient passive accumulation of nanoparticles within the tumor.34 These conflicting results highlight the fine balance required to achieve efficient passive accumulation within target tumors and subsequent dePEGylation via physical desorption of lipid–PEG reagents. The propensity for non-specific PEG exchange with biological membranes in vivo, prior to reaching the target tumor, has likely limited the widespread application of these approaches.
The second physical approach relies on non-covalent adsorption of PEG to a nanoparticle surface.35–39 For example, carboxylate-functionalized PEG adsorbed to a cationic nanoparticle surface.37 In this case, partial protonation of carboxylate groups within the acidic (pH 6.5–7) extracellular tumor microenvironment leads to dePEGylation and subsequent cellular nanoparticle uptake. While this approach is conceptually simple, the stability of the absorbed PEG corona in serum and the propensity of premature dePEGylation under physiological conditions (e.g. high salt) and/or through competition from other serum components has likely limited the widespread investigation of this approach.
3. Chemical dePEGylation strategies
By far the most common method to achieve extracellular dePEGylation of nanoparticle surfaces, within the tumor microenvironment, is through chemical approaches. In these cases, PEG is grafted to the nanoparticle via a stimuli-responsive covalent chemical bond (Tables 1 and 2).40 Stimuli can be both endogenous and exogenous. In the case of endogenous stimuli, intrinsic differences in the pathophysiology of tumor and healthy tissues are exploited, namely the low pH,41 reducing42 and matrix metalloprotease (MMP)-rich environment43 of certain solid tumors. Exogenous stimuli, including light and heat, have the benefit of being under complete user control in both time and space.44 In a clinical setting however, these approaches rely on the ability to efficiently deliver stimuli (e.g. light) to tissues often deep within the body. The various stimuli-responsive chemistries commonly used in both the intra- and extracellular dePEGylation of nanoparticles are summarized in Tables 1 and 2.| Site of dePEGylation within tumor | Stimulus | Example chemical structure | Nanocarrier | Drug release [ref.] | |
|---|---|---|---|---|---|
| Intracellular | Extracellular | ||||
| Extracellular | Low pH (pH 6.5–7) |

|
Polymeric nanoparticles | 77 | |
| Benzoic-imine | Magnetic nanoparticles | 78–80 | |||
| Micelles | 81 | 82 | |||
| Reduction glutathione (GSH) (4–80 μM) |

|
Liposomes | 88–92 | ||
| Disulfide | |||||
| Enzymatic (non-specific) esterase |
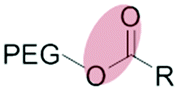
|
Liposomes | 137 and 138 | ||
| Enzymatic (specific) matrix metallo-proteinases (MMPs) |

|
Liposomes | 139–145 | ||
| Quantum dots (QDs) | 157 | ||||
| Polymersomes | 158 | ||||
| (peptide consensus sequence) | Magnetic nanoparticles | 159 | |||
| Micelles | 146–156 | 160 and 161 | |||
| External light |
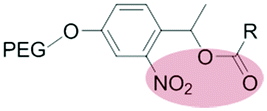
|
Liposomes | 163 | ||
| MSN | 164 | ||||
| Micelles | 168 | 63, 165–167 and 169 | |||
3.1 pH-Sensitive dePEGylation
The mildly acidic (pH 6.5–7.2) extracellular environment of hypoxic tumors – a result of increased glucose catabolism and efflux of H+ by cancer cells – has been exploited to trigger extracellular dePEGylation of nanoparticle surfaces.45 For this, chemical functionalities stable at physiological pH (pH 7.4) but labile at lower pH are required. The most commonly used acid labile chemical groups are vinyl ethers,46–50 hydrazones,51–61 acetals,62–69 β-thiopropionates,70ortho esters71–73 and benzoic imines.74–82 Here however, it is important to differentiate between the mildly acidic extracellular pH within the tumor microenvironment (pH 6.5–7) and the strongly acidic intracellular pH within late endosomes/lysosomes (pH 4.5–5.5) and to stress that optimal sensitivity (and subsequent dePEGylation efficiency) of these acid-labile functionalities is typically at pH 5–5.5. Therefore all these pH-sensitive systems demonstrate inefficient/sluggish acidolytic dePEGylation within the extracellular tumor microenvironment. This can be exploited to achieve prolonged and sustained drug release within the tumor and/or partial dePEGylation may still generate the desired outcome. For example, Gu et al. reported pH dependent dePEGylation of polycationic micelles through grafting of PEG, via benzoic imine linkages, to poly-L-lysine (PLL)/cholic acid co-polymers.82 By measuring changes in surface charge (zeta potential), the authors were able to show colloidal stability at physiological pH as well as increasing rates of dePEGylation with decreasing pH (complete acidolysis at pH 5.5 within 10 min). At pH 6.5–7 (i.e. pH of the extracellular tumor microenvironment) only partial dePEGylation was observed, however this was accompanied by a significant increase in hemolytic activity suggesting partial dePEGylation was sufficient to endow these particles with the desired function. As this system was not tested in cancer models in vivo, it remains to be seen whether this slow rate of acidolysis will adversely affect function and efficacy. Indeed, the individual successes of pH responsive dePEGylation systems ultimately depends on the ability to deliver therapeutically relevant drug doses to cancer cells above and beyond those of the administered free drug alone. It is worth noting, however, these technologies – as with any system exploiting endogenous stimuli – will likely demonstrate significant variations in efficacy due to patient-to-patient heterogeneity of tumor pathologies.83,843.2 Redox-sensitive dePEGylation
Glutathione (GSH), is an abundant reducing agent (2–10 mM) in most mammalian cells, including cancer cells.85 Extracellular GSH concentrations in healthy tissue are approximately 1000× lower (2–20 μM),86 however this value can increase up to 4-fold (4–80 μM) within the tumor microenvironment.87 There are conflicting reports as to whether this small differential in extracellular GSH concentrations can indeed be exploited to trigger extracellular dePEGylation. While a small number of studies report (partial) extracellular cleavage of disulfide linked PEG constructs within the tumor microenvironment,88–92 most exploit GSH as an intracellular trigger only.93–134 In these cases, the very large differential between extra- and intracellular GSH concentrations is a readily exploitable endogenous trigger. Indeed, for systems designed to exploit intracellular GSH levels, extracellular stability (i.e. very limited reduction) of disulfide–PEG constructs is often reported as a key feature in maintaining colloidal stability of nanoparticles in circulation and en route to the target tumor. In our critical opinion, exploiting the marginally elevated extracellular GSH levels of the tumor microenvironment is an ineffective strategy to overcome the ‘PEG dilemma’.3.3 Protease-sensitive dePEGylation
Within the tumor microenvironment, there are high levels of extracellular matrix metalloproteinases (MMPs). These lytic enzymes are secreted at high levels by tumor cells to degrade the extracellular matrix (ECM) and aid cancer cell migration.135,136 Short peptides containing enzyme-consensus sequences, linking PEG to a nanoparticle surface, have been effectively used to dePEGylate nanoparticles within the tumor microenvironment.137–161 Torchilin et al. have reported two elegant examples of MMP-triggered dePEGylation. The first employed a multifunctional liposomal formulation comprising longer, MMP-cleavable lipid–PEG3400 constructs and shorter, non-cleavable TAT-functionalised lipid–PEG2000 constructs.141 In the absence of MMPs, longer PEG3400 chains effectively shielded the cell penetrating function of the underlying TAT peptide and liposomes were sparingly taken up by cells. Upon MMP-mediated dePEGylation however, the newly revealed TAT-functionalised liposomes were avidly taken up by 4T1 breast cancer cells. Going one step further, the same group reported a similar strategy of exploiting MMP-mediated dePEGylation to reveal newly functional drug polymer micelles.146 Crucially in this approach, dePEGylation did not destroy the integrity of the underlying drug-filled micelle leading to efficient stimuli responsive, intracellular drug delivery to cancer cells, as demonstrated in mice models.It is worth noting here that both cathepsin B (protease)162 and esterases138 have also been exploited to trigger dePEGylation of nanomedicines. However, cathepsin B is only found at high levels within (intracellular) cancer cells, while esterases are widely distributed in plasma and healthy tissues and not therefore specific to the tumor microenvironment. In our opinion, MMP-mediated dePEGylation of nanoparticles within the tumor microenvironment represents the most selective and efficient strategy to enhance the efficacy of cancer nanomedicines exploiting endogenous stimulus.
3.4 Light-sensitive dePEGylation
Photolabile chemical bonds have been extensively used, in both chemistry and biological contexts, to precisely control where and when new functionality is revealed. Unlike endogenous stimuli such as pH, redox and enzymatic cleavage, the application of light can be precisely controlled in both time, space and intensity (i.e. is user defined) and requires no other reactive species (other than, in some cases, water). In addition, photolysis is generally rapid (few seconds, pulsed laser), quantitative and clean.For light triggered dePEGylation of potential cancer nanomedicines, o-nitrobenzyl (o-Nb),63,163–166 platinum–azide complexes167 and azobenzene168 functionalities have all been explored.169 In the case of o-Nb functionalities, non-hydrolytic photolysis proceeds through a cyclic intermediate followed by the release of the desired alcohol and a nitroso by-product.170 To increase biological compatibility, methoxy substitution of the aryl ring results in reduced toxicity of nitroso byproducts.171 We have recently reported two separate strategies in which light triggered dePEGylation was successfully used to initiate efficient drug delivery to target cancer cells. In the first example, we created 100 nm, loose core shell micelles composed exclusively of photolabile doxorubicin–PEG2000 reagents.165 In the absence of light, micelles were stable, non-toxic (i.e. not taken up by cells in vitro) and no doxorubicin release was observed over time. Upon light (365 nm) activation triggered dePEGylation, micelle destabilisation and subsequent burst drug release resulted in in vitro cytotoxicity comparable to free doxorubicin. In addition, we were able to demonstrate precise spatiotemporal control of doxorubicin delivery to cells in vitro through light templated activation. We are currently assessing this system in vivo to determine circulation lifetimes and tumor accumulation of PEGylated doxorubicin prodrug micelles prior to light triggered dePEGylation.
In the second example, light triggered dePEGylation was used to precisely control, in time and space, the function of a simplified membrane fusion system. This system comprises two complementary peptides – peptide E and K – displayed from opposing lipid membranes (either liposome–liposome or liposome–cell).163 In this case, PEGylation (via a photolabile cholesterol–PEG construct) of one lipid membrane effectively shielded the interaction between complementary peptides. However, upon light triggered dePEGylation regain of fusion function was instantaneous. We have subsequently shown our simplified membrane fusion system can be used to selectively deliver liposome-encapsulated cargos, via membrane fusion, to target (xenografted) cancer cells in vivo (zebrafish larvae).172 Extending this approach to include light triggered dePEGylation, to enable precise user control of drug delivery, is the subject of current investigations in the group.
The use of light does, of course, raise valid concerns going forward into the clinic. In all reported examples of light triggered dePEGylation, systems are most sensitive to high energy UV-A light (<400 nm). Short wavelength UV light suffers from poor tissue penetration (100–200 μm) and, following prolonged exposure, can elicit significant photocytotoxicity.173 Only for polymeric systems containing platinum–azide complexes167 was photolytic dePEGylation investigated using visible light irradiation. Here, decreasing photolytic efficiency correlated with longer light wavelengths. Here however, it is important to note that photodynamic therapies,174 combining chemical photosensitizers and light activation, are already widely used in the clinic to treat a range of medical conditions, including acne, atherosclerosis and cancer.175 Furthermore, advances in fibre optic technologies (to deliver UV light deep within tissue),176 the development of photolabile chemical bonds sensitive to longer wavelength light177 and the optimization of photosensitive chemical functionality to minimize light exposure, will only further the clinical applicability of light. One promising development has been photolabile chemical groups sensitive to two photon light,178 to not only increasing tissue penetration (>1 cm) of light and minimising induced photocytotoxicity but, by restricting light activation to the focal point of two photon beams, enabling activation volumes in patients of <1 femtolitre. In this vein, we and others have also shown it is possible to cleave o-Nb groups using 2-photon light.179 There are currently no examples of responsive dePEGylation of nanoparticles using alternative external stimuli (e.g. heat or ultrasound).
4. Conclusion
Stimuli-responsive dePEGylation is a proven strategy to increase the efficacy of cancer nanomedicines passively targeting solid tumors via the EPR effect. This approach has the dual advantage of both extended circulation lifetimes of PEGylated nanoparticles (to enhance passive targeting efficiency to tumors) as well as enhanced drug delivery profiles of non-PEGylated (or ruptured) nanoparticles within the tumor microenvironment. To achieve maximal effect, nanomedicines must remain PEGylated en route to the tumor (i.e. are serum stable) and be efficiently dePEGylated within the extracellular tumor microenvironment. Given the very low cellular uptake of PEGylated nanoparticles, strategies that report stimuli-responsive intracellular dePEGylation should not be considered effective. In our view, the most promising stimuli-responsive nanomedicines to date have exploited the MMP-rich microenvironment of solid tumors to trigger targeted and extracellular dePEGylation. However, by exploiting endogenous pathophysiological differences between healthy and diseased tissue, such as differences in MMP concentrations, the efficacy of these stimuli-responsive systems in patients will likely vary due to patient-to-tumor tumor heterogeneity.83 In contrast, dePEGylation triggered by external stimuli, such as light, is exclusively determined by the user. While these approaches negate potential differences in efficacy driven by tumor heterogeneity, the current technological limitations of delivering external stimuli to site specific locations in patients remains a major drawback. However, the continued advance and optimisation of fibre-optic technologies as well more advanced photolabile chemical groups will only promote further the future application of light triggered cancer nanomedicines.Conflicts of interest
There are no conflicts to declare.References
- R. M. McQuade, V. Stojanovska, R. Abalo, J. C. Bornstein and K. Nurgali, Front. Pharmacol., 2016, 7, 414–427 CrossRef CAS PubMed.
- A. M. Rahman, S. W. Yusuf and M. S. Ewer, Int. J. Nanomed., 2007, 2, 567–583 CAS.
- Y. H. Bae and K. Park, J. Controlled Release, 2011, 153, 198–205 CrossRef CAS PubMed.
- A. Beck, L. Goetsch, C. Dumontet and N. Corvaia, Nat. Rev. Drug Discovery, 2017, 16, 315–337 CrossRef CAS PubMed.
- H. I. Chang and M. K. Yeh, Int. J. Nanomed., 2012, 7, 49–60 CAS.
- S. C. Alley, N. M. Okeley and P. D. Senter, Curr. Opin. Chem. Biol., 2010, 14, 529–537 CrossRef CAS PubMed.
- H. L. Perez, P. M. Cardarelli, S. Deshpande, S. Gangwar, G. M. Schroeder, G. D. Vite and R. M. Borzilleri, Drug Discovery Today, 2014, 19, 869–881 CrossRef CAS PubMed.
- J. Shi, P. W. Kantoff, R. Wooster and O. C. Farokhzad, Nat. Rev. Cancer, 2017, 17, 20–37 CrossRef CAS PubMed.
- H. Maeda, J. Wu, T. Sawa, Y. Matsumura and K. Hori, J. Controlled Release, 2000, 65, 271–284 CrossRef CAS PubMed.
- K. Greish, Methods Mol. Biol., 2010, 624, 25–37 CrossRef CAS PubMed.
- M. Longmire, P. L. Choyke and H. Kobayashi, Nanomedicine, 2008, 3, 703–717 CrossRef CAS PubMed.
- E. Blanco, H. Shen and M. Ferrari, Nat. Biotechnol., 2015, 33, 941–951 CrossRef CAS PubMed.
- H. Maeda, H. Nakamura and J. Fang, Adv. Drug Delivery Rev., 2013, 65, 71–79 CrossRef CAS PubMed.
- A. Akbarzadeh, R. Rezaei-Sadabady, S. Davaran, S. W. Joo, N. Zarghami, Y. Hanifehpour, M. Samiei, M. Kouhi and K. Nejati-Koshki, Nanoscale Res. Lett., 2013, 8, 102–110 CrossRef PubMed.
- D. A. Hume, Curr. Opin. Immunol., 2006, 18, 49–53 CrossRef CAS PubMed.
- A. J. Tavares, W. Poon, Y. N. Zhang, Q. Dai, R. Besla, D. Ding, B. Ouyang, A. Li, J. Chen, G. Zheng, C. Robbins and W. C. W. Chan, Proc. Natl. Acad. Sci. U. S. A., 2017, 114, 10871–10880 CrossRef PubMed.
- K. M. Tsoi, S. A. MacParland, X. Z. Ma, V. N. Spetzler, J. Echeverri, B. Ouyang, S. M. Fadel, E. A. Sykes, N. Goldaracena, J. M. Kaths, J. B. Conneely, B. A. Alman, M. Selzner, M. A. Ostrowski, O. A. Adeyi, A. Zilman, I. D. McGilvray and W. C. Chan, Nat. Mater., 2016, 15, 1212–1221 CrossRef CAS PubMed.
- Y. N. Zhang, W. Poon, A. J. Tavares, I. D. McGilvray and W. C. W. Chan, J. Controlled Release, 2016, 240, 332–348 CrossRef CAS PubMed.
- P. Aggarwal, J. B. Hall, C. B. McLeland, M. A. Dobrovolskaia and S. E. McNeil, Adv. Drug Delivery Rev., 2009, 61, 428–437 CrossRef CAS PubMed.
- J. S. Suk, Q. G. Xu, N. Kim, J. Hanes and L. M. Ensign, Adv. Drug Delivery Rev., 2016, 99, 28–51 CrossRef CAS PubMed.
- G. Pasut and F. M. Veronese, J. Controlled Release, 2012, 161, 461–472 CrossRef CAS PubMed.
- P. L. Turecek, M. J. Bossard, F. Schoetens and I. A. Ivens, J. Pharm. Sci., 2016, 105, 460–475 CrossRef CAS PubMed.
- Y. Maitani, J. Drug Delivery Sci. Technol., 2011, 21, 27–34 CrossRef CAS.
- R. P. Garay, R. El-Gewely, J. K. Armstrong, G. Garratty and P. Richette, Expert Opin. Drug Delivery, 2012, 9, 1319–1323 CrossRef CAS PubMed.
- H. Schellekens, W. E. Hennink and V. Brinks, Pharm. Res., 2013, 30, 1729–1734 CrossRef CAS PubMed.
- H. Hatakeyama, H. Akita and H. Harashima, Biol. Pharm. Bull., 2013, 36, 892–899 CrossRef CAS.
- H. Hatakeyama, H. Akita and H. Harashima, Adv. Drug Delivery Rev., 2011, 63, 152–160 CrossRef CAS PubMed.
- H. Y. Xue, P. Guo, W. C. Wen and H. L. Wong, Curr. Pharm. Des., 2015, 21, 3140–3147 CrossRef CAS PubMed.
- J. W. Holland, C. Hui, P. R. Cullis and T. D. Madden, Biochemistry, 1996, 35, 2618–2624 CrossRef CAS PubMed.
- J. R. S. a. M. J. Zuckermann, Biochemistry, 1993, 32, 3153–3161 CrossRef.
- A. S. Janoff, Lab. Invest., 1992, 66, 655–658 CAS.
- S. Leekumjorn, H. J. Cho, Y. F. Wu, N. T. Wright, A. K. Sum and C. Chan, Biochim. Biophys. Acta, Biomembr., 2009, 1788, 1508–1516 CrossRef CAS PubMed.
- W. M. Li, L. Xue, L. D. Mayer and M. B. Bally, Biochim. Biophys. Acta, Biomembr., 2001, 1513, 193–206 CrossRef CAS.
- G. Adlakha-Hutcheon, M. B. Bally, C. R. Shew and T. D. Madden, Nat. Biotechnol., 1999, 17, 775–779 CrossRef CAS PubMed.
- F. Fan, Y. Yu, F. Zhong, M. Gao, T. Sun, J. Liu, H. Zhang, H. Qian, W. Tao and X. Yang, Theranostics, 2017, 7, 1290–1302 CrossRef CAS PubMed.
- M. Barattin, A. Mattarei, A. Balasso, C. Paradisi, L. Cantu, E. Del Favero, T. Viitala, F. Mastrotto, P. Caliceti and S. Salmaso, ACS Appl. Mater. Interfaces, 2018, 10, 17646–17661 CrossRef CAS PubMed.
- C. Zhao, L. Shao, J. Lu, X. Deng and Y. Wu, ACS Appl. Mater. Interfaces, 2016, 8, 6400–6410 CrossRef CAS PubMed.
- M. Fan, Y. Zeng, H. Ruan, Z. Zhang, T. Gong and X. Sun, Mol. Pharmaceutics, 2017, 14, 3152–3163 CrossRef CAS PubMed.
- A. Pourjavadi, Z. M. Tehrani and C. Bennett, Int. J. Polym. Mater. Polym. Biomater., 2015, 64, 570–577 CrossRef CAS.
- B. Romberg, W. E. Hennink and G. Storm, Pharm. Res., 2008, 25, 55–71 CrossRef CAS PubMed.
- B. A. Webb, M. Chimenti, M. P. Jacobson and D. L. Barber, Nat. Rev. Cancer, 2011, 11, 671–677 CrossRef CAS PubMed.
- G. Ilangovan, H. Q. Li, J. L. Zweier and P. Kuppusamy, Mol. Cell. Biochem., 2002, 234, 393–398 CrossRef PubMed.
- C. Mehner, A. Hockla, E. Miller, S. Ran, D. C. Radisky and E. S. Radisky, Oncotarget, 2014, 5, 2736–2749 CrossRef PubMed.
- S. Mura, J. Nicolas and P. Couvreur, Nat. Mater., 2013, 12, 991–1003 CrossRef CAS PubMed.
- M. Meyer and E. Wagner, Expert Opin. Drug Delivery, 2006, 3, 563–571 CrossRef CAS PubMed.
- H. K. Kim, J. Van den Bossche, S. H. Hyun and D. H. Thompson, Bioconjugate Chem., 2012, 23, 2071–2077 CrossRef CAS PubMed.
- J. Shin, J. Controlled Release, 2003, 91, 187–200 CrossRef CAS.
- N. Bergstrand, M. C. Arfvidsson, J. M. Kim, D. H. Thompson and K. Edwards, Biophys. Chem., 2003, 104, 361–379 CrossRef CAS.
- J. A. Boomer, M. M. Qualls, H. D. Inerowicz, R. H. Haynes, V. S. Patri, J. M. Kim and D. H. Thompson, Bioconjugate Chem., 2009, 20, 47–59 CrossRef CAS PubMed.
- Z. Xu, W. Gu, L. Chen, Y. Gao, Z. Zhang and Y. Li, Biomacromolecules, 2008, 9, 3119–3126 CrossRef CAS PubMed.
- M. Kanamala, B. D. Palmer, W. R. Wilson and Z. Wu, Int. J. Pharm., 2018, 548, 288–296 CrossRef CAS PubMed.
- C. L. Chan, R. N. Majzoub, R. S. Shirazi, K. K. Ewert, Y. J. Chen, K. S. Liang and C. R. Safinya, Biomaterials, 2012, 33, 4928–4935 CrossRef CAS PubMed.
- A. Apte, E. Koren, A. Koshkaryev and V. P. Torchilin, Cancer Biol. Ther., 2014, 15, 69–80 CrossRef CAS PubMed.
- L. Zhang, Y. Wang, Y. Yang, Y. Liu, S. Ruan, Q. Zhang, X. Tai, J. Chen, T. Xia, Y. Qiu, H. Gao and Q. He, ACS Appl. Mater. Interfaces, 2015, 7, 9691–9701 CrossRef CAS PubMed.
- G. F. Walker, C. Fella, J. Pelisek, J. Fahrmeir, S. Boeckle, M. Ogris and E. Wagner, Mol. Ther., 2005, 11, 418–425 CrossRef CAS PubMed.
- F. Li, J. He, M. Zhang, K. C. Tam and P. Ni, RSC Adv., 2015, 5, 54658–54666 RSC.
- F. Li, J. He, M. Zhang and P. Ni, Polym. Chem., 2015, 6, 5009–5014 RSC.
- D. Chen, Q. Tang, J. Zou, X. Yang, W. Huang, Q. Zhang, J. Shao and X. Dong, Adv. Healthcare Mater., 2018, 7, 1701272 CrossRef PubMed.
- M. Yang, L. Yu, R. Guo, A. Dong, C. Lin and J. Zhang, Nanomaterials, 2018, 8, 167–184 CrossRef PubMed.
- N. Sun, C. Zhao, R. Cheng, Z. Liu, X. Li, A. Lu, Z. Tian and Z. Yang, Mol. Pharmaceutics, 2018, 15, 3343–3355 CrossRef CAS PubMed.
- B. Balcı and A. Top, J. Polym. Res., 2018, 25, 104–115 CrossRef.
- J. A. Boomer, H. D. Inerowicz, Z. Y. Zhang, N. Bergstrand, K. Edwards, J. M. Kim and D. H. Thompson, Langmuir, 2003, 19, 6408–6415 CrossRef CAS.
- N. Kalva, N. Parekh and A. V. Ambade, Polym. Chem., 2015, 6, 6826–6835 RSC.
- J. A. Boomer, H. D. Inerowicz, Z.-Y. Zhang, N. Bergstrand, K. Edwards, J.-M. Kim and D. H. Thompson, Langmuir, 2003, 19, 6408–6415 CrossRef CAS.
- H. Wang, J. He, M. Zhang, Y. Tao, F. Li, K. C. Tam and P. Ni, J. Mater. Chem. B, 2013, 1, 6596–6607 RSC.
- J. Hu, J. He, M. Zhang and P. Ni, Polym. Chem., 2015, 6, 1553–1566 RSC.
- S. Zhang, J. Xu, H. Chen, Z. Song, Y. Wu, X. Dai and J. Kong, Macromol. Biosci., 2017, 17, 1600258 CrossRef PubMed.
- L. Xiao, L. Huang, F. Moingeon, M. Gauthier and G. Yang, Biomacromolecules, 2017, 18, 2711–2722 CrossRef CAS PubMed.
- A. M. Jazani and J. K. Oh, Macromolecules, 2017, 50, 9427–9436 CrossRef CAS.
- M. Oishi, F. Nagatsugi, S. Sasaki, Y. Nagasaki and K. Kataoka, ChemBioChem, 2005, 6, 718–725 CrossRef CAS PubMed.
- X. Guo, J. A. MacKay and F. C. Szoka, Jr., Biophys. J., 2003, 84, 1784–1795 CrossRef CAS PubMed.
- W. Li, Z. Huang, J. A. MacKay, S. Grube and F. C. Szoka, Jr., J. Gene Med., 2005, 7, 67–79 CrossRef CAS PubMed.
- J. S. Choi, J. A. MacKay and F. C. Szoka, Jr., Bioconjugate Chem., 2003, 14, 420–429 CrossRef CAS PubMed.
- H. Rongbin, X. Lei, L. Ying, D. Xiangping, C. Xuan, L. Lanfang, Y. Cuiyun, C. Yanming and T. Guotao, J. Pharm. Pharmacol., 2016, 68, 751–761 CrossRef PubMed.
- S. Wu, L. Zheng, C. Li, Y. Xiao, S. Huo and B. Zhang, J. Polym. Sci., Part A: Polym. Chem., 2017, 55, 2036–2046 CrossRef CAS.
- S. Yang, F. Zhu, Q. Wang, F. Liang, X. Qu, Z. Gan and Z. Yang, J. Mater. Chem. B, 2015, 3, 4043–4051 RSC.
- Y. Guan, H. Lu, W. Li, Y. Zheng, Z. Jiang, J. Zou and H. Gao, ACS Appl. Mater. Interfaces, 2017, 9, 26731–26739 CrossRef CAS PubMed.
- J. Wang, C. Gong, Y. Wang and G. Wu, Colloids Surf., B, 2014, 118, 218–225 CrossRef CAS PubMed.
- J. Wang, C. Gong, Y. Wang and G. Wu, RSC Adv., 2014, 4, 15856–15863 RSC.
- M. Zhang, J. Liu, Y. Kuang, Q. Li, H. Chen, H. Ye, L. Guo, Y. Xu, X. Chen, C. Li and B. Jiang, J. Mater. Chem. B, 2016, 4, 3387–3397 RSC.
- X. Guan, Z. Guo, L. Lin, J. Chen, H. Tian and X. Chen, Nano Lett., 2016, 16, 6823–6831 CrossRef CAS PubMed.
- W.-P. C. Jingxia Gu, X. z. Qu, J. Liu, S.-Y. Lo and Z. Yang, Biomacromolecules, 2008, 9, 255–262 CrossRef PubMed.
- M. W. Dewhirst and T. W. Secomb, Nat. Rev. Cancer, 2017, 17, 738–750 CrossRef CAS PubMed.
- J. I. Hare, T. Lammers, M. B. Ashford, S. Puri, G. Storm and S. T. Barry, Adv. Drug Delivery Rev., 2017, 108, 25–38 CrossRef CAS PubMed.
- T. Sun, A. Morger, B. Castagner and J. C. Leroux, Chem. Commun., 2015, 51, 5721–5724 RSC.
- Q. L. Li, S. H. Xu, H. Zhou, X. Wang, B. A. Dong, H. Gao, J. Tang and Y. W. Yang, ACS Appl. Mater. Interfaces, 2015, 7, 28656–28664 CrossRef CAS PubMed.
- P. Kuppusamy, H. Q. Li, G. Ilangovan, A. J. Cardounel, J. L. Zweier, K. Yamada, M. C. Krishna and J. B. Mitchell, Cancer Res., 2002, 62, 307–312 CAS.
- P. S. Kulkarni, M. K. Haldar, R. R. Nahire, P. Katti, A. H. Ambre, W. W. Muhonen, J. B. Shabb, S. K. Padi, R. K. Singh, P. P. Borowicz, D. K. Shrivastava, K. S. Katti, K. Reindl, B. Guo and S. Mallik, Mol. Pharmaceutics, 2014, 11, 2390–2399 CrossRef CAS PubMed.
- K. M. McNeeley, E. Karathanasis, A. V. Annapragada and R. V. Bellamkonda, Biomaterials, 2009, 30, 3986–3995 CrossRef CAS PubMed.
- W. Y. Rui Kuai, Y. Qin, H. Chen, J. Tang, M. q. Yuan, Z. Zhang and Q. He, Mol. Pharmaceutics, 2010, 7, 1816–1826 CrossRef PubMed.
- L. Mei, L. Fu, K. Shi, Q. Zhang, Y. Liu, J. Tang, H. Gao, Z. Zhang and Q. He, Int. J. Pharm., 2014, 468, 26–38 CrossRef CAS PubMed.
- J. Tang, H. Fu, Q. Kuang, L. Zhang, Q. Zhang, Y. Liu, R. Ran, H. Gao, Z. Zhang and Q. He, J. Drug Targeting, 2014, 22, 313–326 CrossRef CAS PubMed.
- J. Tang, L. Zhang, H. Gao, Y. Liu, Q. Zhang, R. Ran, Z. Zhang and Q. He, Drug Delivery, 2016, 23, 1130–1143 CAS.
- L. Jia, D. Cui, J. Bignon, A. Di Cicco, J. Wdzieczak-Bakala, J. Liu and M. H. Li, Biomacromolecules, 2014, 15, 2206–2217 CrossRef CAS PubMed.
- T. Ren, W. Wu, M. Jia, H. Dong, Y. Li and Z. Ou, ACS Appl. Mater. Interfaces, 2013, 5, 10721–10730 CrossRef CAS PubMed.
- W. Hou, F. Xia, C. S. Alves, X. Qian, Y. Yang and D. Cui, ACS Appl. Mater. Interfaces, 2016, 8, 1447–1457 CrossRef CAS PubMed.
- H. Sun, B. Guo, R. Cheng, F. Meng, H. Liu and Z. Zhong, Biomaterials, 2009, 30, 6358–6366 CrossRef CAS PubMed.
- X. Q. Li, H. Y. Wen, H. Q. Dong, W. M. Xue, G. M. Pauletti, X. J. Cai, W. J. Xia, D. Shi and Y. Y. Li, Chem. Commun., 2011, 47, 8647–8649 RSC.
- X.-J. Cai, H.-Q. Dong, W.-J. Xia, H.-Y. Wen, X.-Q. Li, J.-H. Yu, Y.-Y. Li and D.-L. Shi, J. Mater. Chem., 2011, 21, 14639–14645 RSC.
- T.-B. Ren, W.-J. Xia, H.-Q. Dong and Y.-Y. Li, Polymer, 2011, 52, 3580–3586 CrossRef CAS.
- H. Y. Wen, H. Q. Dong, W. J. Xie, Y. Y. Li, K. Wang, G. M. Pauletti and D. L. Shi, Chem. Commun., 2011, 47, 3550–3552 RSC.
- Q. Guo, P. Luo, Y. Luo, F. Du, W. Lu, S. Liu, J. Huang and J. Yu, Colloids Surf., B, 2012, 100, 138–145 CrossRef CAS PubMed.
- Y. Zhong, W. Yang, H. Sun, R. Cheng, F. Meng, C. Deng and Z. Zhong, Biomacromolecules, 2013, 14, 3723–3730 CrossRef CAS PubMed.
- X. Wang, H. Sun, F. Meng, R. Cheng, C. Deng and Z. Zhong, Biomacromolecules, 2013, 14, 2873–2882 CrossRef CAS PubMed.
- C. Cui, Y. N. Xue, M. Wu, Y. Zhang, P. Yu, L. Liu, R. X. Zhuo and S. W. Huang, Biomaterials, 2013, 34, 3858–3869 CrossRef CAS PubMed.
- Y. Ping, Q. Hu, G. Tang and J. Li, Biomaterials, 2013, 34, 6482–6494 CrossRef CAS PubMed.
- J. Ding, J. Chen, D. Li, C. Xiao, J. Zhang, C. He, X. Zhuang and X. Chen, J. Mater. Chem. B, 2013, 1, 69–81 RSC.
- T. Thambi, G. Saravanakumar, J.-U. Chu, R. Heo, H. Ko, V. G. Deepagan, J.-H. Kim and J. H. Park, Macromol. Res., 2012, 21, 100–107 CrossRef.
- L. Jia, Z. Li, D. Zhang, Q. Zhang, J. Shen, H. Guo, X. Tian, G. Liu, D. Zheng and L. Qi, Polym. Chem., 2013, 4, 156–165 RSC.
- K. Wang, Y. Liu, W.-J. Yi, C. Li, Y.-Y. Li, R.-X. Zhuo and X.-Z. Zhang, Soft Matter, 2013, 9, 692–699 RSC.
- H. Zhu, C. Dong, H. Dong, T. Ren, X. Wen, J. Su and Y. Li, ACS Appl. Mater. Interfaces, 2014, 6, 10393–10407 CrossRef CAS PubMed.
- X. Ai, J. Sun, L. Zhong, C. Wu, H. Niu, T. Xu, H. Lian, X. Han, G. Ren, W. Ding, J. Wang, X. Pu and Z. He, Macromol. Biosci., 2014, 14, 1415–1428 CrossRef CAS PubMed.
- H. Dong, C. Dong, W. Xia, Y. Li and T. Ren, Med. Chem. Commun., 2014, 5, 147–152 RSC.
- C. Cui, P. Yu, M. Wu, Y. Zhang, L. Liu, B. Wu, C. X. Wang, R. X. Zhuo and S. W. Huang, Colloids Surf., B, 2015, 129, 137–145 CrossRef CAS PubMed.
- H. Wen, H. Dong, J. Liu, A. Shen, Y. Li and D. Shi, J. Mater. Chem. B, 2016, 4, 7859–7869 RSC.
- Y. Zhu, X. Wang, J. Zhang, F. Meng, C. Deng, R. Cheng, J. Feijen and Z. Zhong, J. Controlled Release, 2017, 250, 9–19 CrossRef CAS PubMed.
- H. Fan, Y. Li, J. Yang and X. Ye, J. Phys. Chem. B, 2017, 121, 9708–9717 CrossRef CAS PubMed.
- J. Li, Y. J. Ma, Y. Wang, B. Z. Chen, X. D. Guo and C. Y. Zhang, Chem. Eng. J., 2018, 341, 450–461 CrossRef CAS.
- H. Wang, M. Sun, D. Li, X. Yang, C. Han and W. Pan, Artif. Cells, Nanomed., Biotechnol., 2018, 46, 313–322 CrossRef CAS PubMed.
- W. Chen, P. Zhong, F. Meng, R. Cheng, C. Deng, J. Feijen and Z. Zhong, J. Controlled Release, 2013, 169, 171–179 CrossRef CAS PubMed.
- Y. Cao, J. Zhao, Y. Zhang, J. Liu, J. Liu, A. Dong and L. Deng, RSC Adv., 2015, 5, 28060–28069 RSC.
- Y. Li, Z. Wu, D. Du, H. Dong, D. Shi and Y. Li, RSC Adv., 2016, 6, 6516–6522 RSC.
- H. Xiong, Z. Guo, W. Zhang, H. Zhong, S. Liu and Y. Ji, J. Photochem. Photobiol., B, 2014, 138, 191–201 CrossRef CAS PubMed.
- H. Wen, C. Dong, H. Dong, A. Shen, W. Xia, X. Cai, Y. Song, X. Li, Y. Li and D. Shi, Small, 2012, 8, 760–769 CrossRef CAS PubMed.
- J. Jiao, X. Li, S. Zhang, J. Liu, D. Di, Y. Zhang, Q. Zhao and S. Wang, Mater. Sci. Eng., C, 2016, 67, 26–33 CrossRef CAS PubMed.
- Y. Wang, N. Han, Q. Zhao, L. Bai, J. Li, T. Jiang and S. Wang, Eur. J. Pharm. Sci., 2015, 72, 12–20 CrossRef CAS PubMed.
- H. M. Gong, Z. F. Xie, M. X. Liu, H. H. Sun, H. D. Zhu and H. L. Guo, Colloid Polym. Sci., 2015, 293, 2121–2128 CrossRef CAS.
- L. Chen, Z. Zheng, J. Wang and X. Wang, Microporous Mesoporous Mater., 2014, 185, 7–15 CrossRef CAS.
- H. He, H. Kuang, L. Yan, F. Meng, Z. Xie, X. Jing and Y. Huang, Phys. Chem. Chem. Phys., 2013, 15, 14210–14218 RSC.
- Y. Cui, H. Dong, X. Cai, D. Wang and Y. Li, ACS Appl. Mater. Interfaces, 2012, 4, 3177–3183 CrossRef CAS PubMed.
- H. Kim, S. Kim, C. Park, H. Lee, H. J. Park and C. Kim, Adv. Mater., 2010, 22, 4280–4283 CrossRef CAS PubMed.
- H. Gong, Z. Xie, M. Liu, H. Zhu and H. Sun, RSC Adv., 2015, 5, 59576–59582 RSC.
- Y. Dong, X. Ma, H. Huo, Q. Zhang, F. Qu and F. Chen, J. Appl. Polym. Sci., 2018, 135, 46675–46685 CrossRef.
- J. Yu, X. Li, Y. Luo, W. Lu, J. Huang and S. Liu, Colloids Surf., B, 2013, 107, 213–219 CrossRef CAS PubMed.
- R. K. E. Charles, C. Pak, P. L. Ahl, A. S. Jano and P. Meers, Biochim. Biophys. Acta, 1999, 1419, 111–126 Search PubMed.
- L. L. H. Benjamin, E. Turk, E. T. Piro and L. C. Cantley, Nat. Biotechnol., 2001, 19, 661–667 CrossRef PubMed.
- D. Chen, W. Liu, Y. Shen, H. Mu, Y. Zhang, R. Liang, A. Wang, K. Sun and F. Fu, Int. J. Nanomed., 2011, 6, 2053–2061 CrossRef CAS PubMed.
- H. Xu, Y. Deng, D. Chen, W. Hong, Y. Lu and X. Dong, J. Controlled Release, 2008, 130, 238–245 CrossRef CAS PubMed.
- T. Terada, M. Iwai, S. Kawakami, F. Yamashita and M. Hashida, J. Controlled Release, 2006, 111, 333–342 CrossRef CAS PubMed.
- F. Zhou, B. Feng, T. Wang, D. Wang, Q. Meng, J. Zeng, Z. Zhang, S. Wang, H. Yu and Y. Li, Adv. Funct. Mater., 2017, 27, 1606530 CrossRef.
- P. K. Lin Zhu and V. P. Torchilin, ACS Nano, 2012, 6, 3491–3498 CrossRef PubMed.
- M. R. Gordon, B. Zhao, F. Anson, A. Fernandez, K. Singh, C. Homyak, M. Canakci, R. W. Vachet and S. Thayumanavan, Biomacromolecules, 2018, 19, 860–871 CrossRef CAS PubMed.
- F. Guo, J. Wu, W. Wu, D. Huang, Q. Yan, Q. Yang, Y. Gao and G. Yang, J. Nanobiotechnol., 2018, 16, 57–69 CrossRef PubMed.
- P. Yingyuad, M. Mevel, C. Prata, S. Furegati, C. Kontogiorgis, M. Thanou and A. D. Miller, Bioconjugate Chem., 2013, 24, 343–362 CrossRef CAS PubMed.
- H. Hatakeyama, H. Akita, K. Kogure, M. Oishi, Y. Nagasaki, Y. Kihira, M. Ueno, H. Kobayashi, H. Kikuchi and H. Harashima, Gene Ther., 2007, 14, 68–77 CrossRef CAS PubMed.
- L. Zhu, T. Wang, F. Perche, A. Taigind and V. P. Torchilin, Proc. Natl. Acad. Sci. U. S. A., 2013, 110, 17047–17052 CrossRef CAS PubMed.
- L. Zhu, F. Perche, T. Wang and V. P. Torchilin, Biomaterials, 2014, 35, 4213–4222 CrossRef CAS PubMed.
- K. L. Veiman, K. Kunnapuu, T. Lehto, K. Kiisholts, K. Parn, U. Langel and K. Kurrikoff, J. Controlled Release, 2015, 209, 238–247 CrossRef CAS PubMed.
- H. Zhou, H. Sun, S. Lv, D. Zhang, X. Zhang, Z. Tang and X. Chen, Acta Biomater., 2017, 54, 227–238 CrossRef CAS PubMed.
- Y. Tu and L. Zhu, J. Controlled Release, 2015, 212, 94–102 CrossRef CAS PubMed.
- W. Ke, J. Li, K. Zhao, Z. Zha, Y. Han, Y. Wang, W. Yin, P. Zhang and Z. Ge, Biomacromolecules, 2016, 17, 3268–3276 CrossRef CAS PubMed.
- G. Salzano, D. F. Costa, C. Sarisozen, E. Luther, G. Mattheolabakis, P. P. Dhargalkar and V. P. Torchilin, Small, 2016, 12, 4837–4848 CrossRef CAS PubMed.
- J. Yoo, N. Sanoj Rejinold, D. Lee, S. Jon and Y. C. Kim, J. Controlled Release, 2017, 264, 89–101 CrossRef CAS PubMed.
- Z. Dai, Y. Tu and L. Zhu, J. Biomed. Nanotechnol., 2016, 12, 1199–1210 CrossRef CAS PubMed.
- Y. Zeng, Z. Zhou, M. Fan, T. Gong, Z. Zhang and X. Sun, Mol. Pharmaceutics, 2017, 14, 81–92 CrossRef CAS PubMed.
- J. M. Shin, S. J. Oh, S. Kwon, V. G. Deepagan, M. Lee, S. H. Song, H. J. Lee, S. Kim, K. H. Song, T. W. Kim and J. H. Park, J. Controlled Release, 2017, 267, 181–190 CrossRef CAS PubMed.
- H. Han, D. Valdeperez, Q. Jin, B. Yang, Z. Li, Y. Wu, B. Pelaz, W. J. Parak and J. Ji, ACS Nano, 2017, 11, 1281–1291 CrossRef CAS PubMed.
- J. Li, S. Xiao, Y. Xu, S. Zuo, Z. Zha, W. Ke, C. He and Z. Ge, ACS Appl. Mater. Interfaces, 2017, 9, 17727–17735 CrossRef CAS PubMed.
- C. Nazli, G. S. Demirer, Y. Yar, H. Y. Acar and S. Kizilel, Colloids Surf., B, 2014, 122, 674–683 CrossRef CAS PubMed.
- Z. Dai, Q. Yao and L. Zhu, ACS Appl. Mater. Interfaces, 2016, 8, 12661–12673 CrossRef CAS PubMed.
- D. Guarnieri, M. Biondi, H. Yu, V. Belli, A. P. Falanga, M. Cantisani, S. Galdiero and P. A. Netti, Biotechnol. Bioeng., 2015, 112, 601–611 CrossRef CAS PubMed.
- J. X. Zhang, S. Zalipsky, N. Mullah, M. Pechar and T. M. Allen, Pharmacol. Res., 2004, 49, 185–198 CrossRef CAS PubMed.
- L. Kong, S. H. C. Askes, S. Bonnet, A. Kros and F. Campbell, Angew. Chem., Int. Ed., 2016, 55, 1396–1400 CrossRef CAS PubMed.
- F. Zhang, L. Kong, D. Liu, W. Li, E. Mäkilä, A. Correia, R. Lindgren, J. Salonen, J. J. Hirvonen, H. Zhang, A. Kros and H. A. Santos, Adv. Ther., 2018, 1, 1800013 CrossRef.
- L. Kong, D. Poulcharidis, G. F. Schneider, F. Campbell and A. Kros, Int. J. Mol. Sci., 2017, 18, 2033–2040 CrossRef PubMed.
- Q. Jin, T. Cai, H. Han, H. Wang, Y. Wang and J. Ji, Macromol. Rapid Commun., 2014, 35, 1372–1378 CrossRef CAS PubMed.
- D. Zhou, J. Guo, G. B. Kim, J. Li, X. Chen, J. Yang and Y. Huang, Adv. Healthcare Mater., 2016, 5, 2493–2499 CrossRef CAS PubMed.
- J. Wang, Y. Ouyang, S. Li, X. Wang and Y. He, RSC Adv., 2016, 6, 57227–57231 RSC.
- G. Saravanakumar, H. Park, J. Kim, D. Park, S. Pramanick, D. H. Kim and W. J. Kim, Biomacromolecules, 2018, 19, 2202–2213 CrossRef CAS PubMed.
- Y. V. Il'ichev, M. A. Schworer and J. Wirz, J. Am. Chem. Soc., 2004, 126, 4581–4595 CrossRef PubMed.
- M. P. Hay, B. M. Sykes, W. A. Dennya and C. J. O'Connor, J. Chem. Soc., Perkin Trans. 1, 1999, 2759–2770 RSC.
- J. Yang, Y. Shimada, R. C. L. Olsthoorn, B. E. Snaar-Jagalska, H. P. Spaink and A. Kros, ACS Nano, 2016, 10, 7428–7435 CrossRef CAS PubMed.
- T. J. McMillan, E. Leatherman, A. Ridley, J. Shorrocks, S. E. Tobi and J. R. Whiteside, J. Pharm. Pharmacol., 2008, 60, 969–976 CrossRef CAS PubMed.
- A. P. Castano, P. Mroz and M. R. Hamblin, Nat. Rev. Cancer, 2006, 6, 535–545 CrossRef CAS PubMed.
- C. M. Allen, W. M. Sharman and J. E. Van Lier, J. Porphyrins Phthalocyanines, 2001, 5, 161–169 CrossRef CAS.
- S. H. Yun and S. J. J. Kwok, Nat. Biomed. Eng., 2017, 1, 8–15 CrossRef PubMed.
- L. Fournier, C. Gauron, L. Xu, I. Aujard, T. Le Saux, N. Gagey-Eilstein, S. Maurin, S. Dubruille, J. B. Baudin, D. Bensimon, M. Volovitch, S. Vriz and L. Jullien, ACS Chem. Biol., 2013, 8, 1528–1536 CrossRef CAS PubMed.
- X. M. M. Weyel, M. A. H. Fichte and A. Heckel, ACS Chem. Biol., 2017, 12, 2183–2190 CrossRef CAS PubMed.
- K. Peng, I. Tomatsu, B. van den Broek, C. Cui, A. V. Korobko, J. van Noort, A. H. Meijer, H. P. Spaink and A. Kros, Soft Matter, 2011, 7, 4881–4887 RSC.
| This journal is © The Royal Society of Chemistry 2019 |