
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
The virulence factor LecB varies in clinical isolates: consequences for ligand binding and drug discovery†
Roman
Sommer‡
ab,
Stefanie
Wagner‡
ab,
Annabelle
Varrot
c,
Corwin M.
Nycholat
d,
Ariane
Khaledi
e,
Susanne
Häussler
e,
James C.
Paulson
d,
Anne
Imberty
 c and
Alexander
Titz
*ab
c and
Alexander
Titz
*ab
aChemical Biology of Carbohydrates, Helmholtz Institute for Pharmaceutical Research Saarland (HIPS), D-66123 Saarbrücken, Germany. E-mail: alexander.titz@helmholtz-hzi.de; Web: http://www.helmholtz-hzi.de/cbch
bDeutsches Zentrum für Infektionsforschung (DZIF), Standort Hannover, Braunschweig, Germany
cCentre de Recherche sur les Macromolécules Végétales (CERMAV-UPR5301), CNRS and Université Grenoble Alpes, BP53, F-38041 Grenoble cedex 9, France
dDepartment of Cell and Molecular Biology and Department of Chemical Physiology, The Scripps Research Institute, 10550 North Torrey Pines Road, La Jolla, CA 92037, USA
eMolecular Bacteriology, Helmholtz Centre for Infection Research, D-38124 Braunschweig, Germany
First published on 11th May 2016
Abstract
P. aeruginosa causes a substantial number of nosocomial infections and is the leading cause of death of cystic fibrosis patients. This Gram-negative bacterium is highly resistant against antibiotics and further protects itself by forming a biofilm. Moreover, a high genomic variability among clinical isolates complicates therapy. Its lectin LecB is a virulence factor and necessary for adhesion and biofilm formation. We analyzed the sequence of LecB variants in a library of clinical isolates and demonstrate that it can serve as a marker for strain family classification. LecB from the highly virulent model strain PA14 presents 13% sequence divergence with LecB from the well characterized PAO1 strain. These differences might result in differing ligand binding specificities and ultimately in reduced efficacy of drugs directed towards LecB. Despite several amino acid variations at the carbohydrate binding site, glycan array analysis showed a comparable binding pattern for both variants. A common high affinity ligand could be identified and after its chemoenzymatic synthesis verified in a competitive binding assay: an N-glycan presenting two blood group O epitopes (H-type 2 antigen). Molecular modeling of the complex suggests a bivalent interaction of the ligand with the LecB tetramer by bridging two separate binding sites. This binding rationalizes the strong avidity (35 nM) of LecBPA14 to this human fucosylated N-glycan. Biochemical evaluation of a panel of glycan ligands revealed that LecBPA14 demonstrated higher glycan affinity compared to LecBPAO1 including the extraordinarily potent affinity of 70 nM towards the monovalent human antigen Lewisa. The structural basis of this unusual high affinity ligand binding for lectins was rationalized by solving the protein crystal structures of LecBPA14 with several glycans.
Introduction
Pseudomonas aeruginosa belongs to the ESKAPE pathogens, which cause a substantial number of nosocomial infections and successfully escape from antimicrobial effects of antibiotic treatments.1 The Gram-negative opportunistic pathogen P. aeruginosa is a major threat to immunocompromised patients and individuals suffering from cystic fibrosis. P. aeruginosa effectively applies a wide variety of defense mechanisms to escape from antimicrobials, e.g. genetic adaptation, efflux pumps, virulence factors and biofilm formation.2 The physical barrier of the biofilm matrix3 protects the bacteria embedded in these social colonies against host defense and antibiotic treatment impeding successful therapy.4 Biofilm embedded cells are 10- to 1000-fold more resistant towards a number of antimicrobial drugs than in planktonic culture.5 The dramatic increase of antibiotic resistances in the last decades is a central problem and new therapies are urgently needed.6 Antivirulence therapies are an alternative strategy that block bacterial virulence without killing or inhibiting bacterial growth and are therefore expected to avoid rapid development of resistance.7Two extracellular carbohydrate-binding proteins, LecA (PA-IL) and LecB (PA-IIL), are virulence factors and necessary for adhesion and biofilm formation.8–10 Disruption of the bacterial biofilm or inhibition of virulence factor function by blocking the lectins LecA or LecB (reviewed in ref. 11 and 12) is therefore an anti-virulence approach. LecA binds to D-galactose; LecB from the strain PAO1 shows high binding affinities for L-fucose (1) and reduced binding to D-mannose (4) and their glycoconjugates.13,14 Glycan-based LecB-directed inhibitors are able to disrupt established biofilms in vitro.15,16 To date, all reported LecB inhibitors focussed on the less virulent model strain P. aeruginosa PAO1 only (see for example ref. 15–22). The highly virulent P. aeruginosa patient isolate PA14 (ref. 23) has gained increasing attention recently. Despite a high genomic variability among the two strains and multiple other clinical isolates, the lecA and lecB genes are part of the commonly shared core genome.24,25 While the LecA sequence is conserved in these two model strains, the sequence of LecB shows a high degree of variation (Fig. 1A). Current anti-virulence drugs are directed towards LecB from PAO1, however, the efficacy of these therapies for other clinical isolates is a concern due to the observed variability of LecB, especially in the highly pathogenic strain PA14.
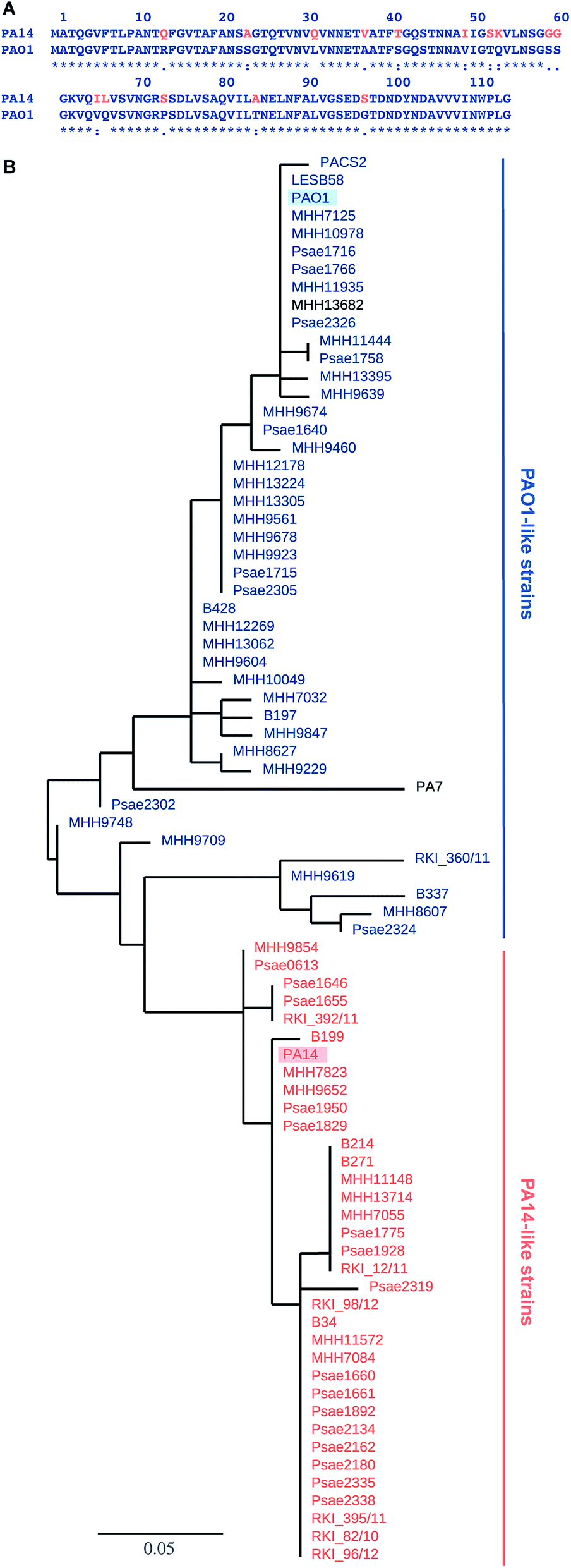 | ||
| Fig. 1 LecB sequence comparison: (A) alignment of LecB sequences from PA14 and PAO1. Amino acid numbering corresponds to the crystal structure of LecBPAO1, which lacks methionine; (B) phylogenetic tree based on LecB sequences from P. aeruginosa clinical isolates and reference strains PAO1, PA7, PA14, PACS2, LESB58. An independent phylogenetic analysis based on 200 core gene sequences allowed the classification of strains as PA14- or PAO1-like strains, illustrated in the color coding in orange (PA14) and blue (PAO1), PA7 and MHH13682 (shown in black) could not be assigned to either strain family. The branch length is proportional to the number of substitutions per site. | ||
Results and discussion
Sequence analysis of clinical isolates: LecB as strain marker
The genes of both P. aeruginosa lectins lecA and lecB are part of the core genome25 and while LecA is highly conserved, LecB shows sequence variation in 15 amino acids in PA14 compared to PAO1 (Fig. 1A). To analyze the variability of LecB among patient isolates, we compared LecB sequences of our collection of clinical isolates.26 From these 151 isolates, a total of 79 strains showed a complete lecB transcriptomics sequence coverage. Five strains with mutations in the lecB gene resulting in frame shifts and thus in truncated amino acid sequences were discarded. The amino acid sequences of the remaining 74 strains were submitted to a multiple sequence alignment and the LecB sequences of P. aeruginosa strains PAO1, PA14, LESB58, PACS2 and PA7 obtained from Pseudomonas Genome Database27 were included in the analysis (Fig. S1†). The resulting phylogenetic tree (Fig. 1B), solely based on the LecB sequence, groups the strains into one branch containing the sequence of LecBPAO1 and a second branch containing LecBPA14, the sequence of the taxonomical outlier PA7 (ref. 28) is comparatively isolated. Surprisingly, the sequence distribution in this alignment of LecB sequences showed a striking correlation with an independent phylogenetic analysis26 of these clinical isolates based on 200 core genes, grouping the strains into two major families, PAO1-like and PA14-like (Fig. 1B, blue for PAO1 and orange for PA14). The sequence alignment shows that the sequence variations among the clinical isolates vary specifically at 15 positions where LecBPA14 and LecBPAO1 differ (Fig. S1,† see Fig. 1A for the individual mutations). In the taxonomical outlier PA7, sequence variations include two additional sites, not observed in any of the other strains. Besides those distinct differences observed between PA14 and PAO1, further variations are only present at two additional sites among all clinical isolates analyzed: A48G in 24 strains and A105S in 5 strains. Moreover, these additional substitutions are fully restricted to the PA14-like group. These findings show that the LecB sequence can serve as a genetic marker to classify clinical isolates into either PAO1- or PA14-like strains, a knowledge potentially useful for choosing a therapeutic action plan.Carbohydrate specificity
Three amino acid variations are either in direct proximity to (S23A, G97S) or may influence local secondary structure (P73S) at the carbohydrate binding site and could directly impact ligand binding specificity and affinity. LecBPA14 was cloned, recombinantly expressed and purified from E. coli. The carbohydrate specificity of LecBPA14 and potential differences in selectivity compared to LecBPAO1 was analyzed on the CFG mammalian glycan array, which provided a spectrum of 609 different carbohydrate epitopes.29 Both FITC-labeled lectins were analyzed and, surprisingly, the profile of the glycan binding specificities was very similar for both lectins with only subtle differences detected (Fig. 2 and S2†). Both lectins showed comparable binding to fucosylated oligosaccharides, such as fucosylated N-glycans and fucosylated N-acetyl lactosamine repeats, as well as to high-mannose-type structures. LecBPAO1 showed stronger binding to core-fucosylated structures, such as glycans 483 and 582. Interestingly, the highest apparent affinity of both LecB variants was detected for a bi-antennary H type II antigen on a di-LacNAc N-linked glycan structure (no. 543, Fig. 2 and S3†). This high affinity could result from a simultaneous bivalent binding to two binding sites on the tetramer, which is supported by its shortened but otherwise identical LacNAc analog (no. 360) showing a reduced apparent affinity in this assay.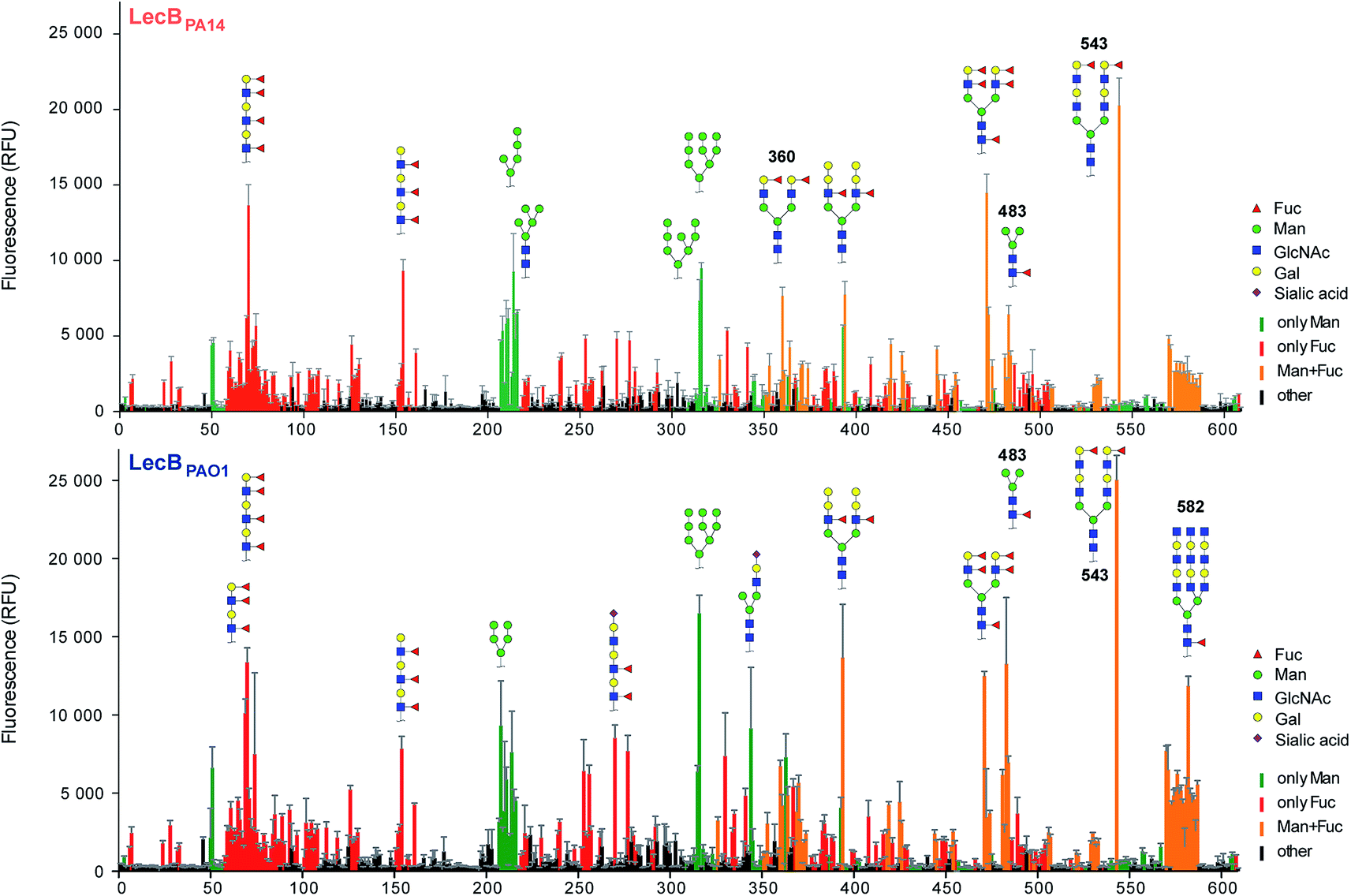 | ||
| Fig. 2 Profiling of glycan binding specificities of LecBPA14 (top) and LecBPAO1 (bottom) on the CFG mammalian glycan array. Glycans containing D-mannose are colored in green, those containing L-fucose in red and oligosaccharides with both moieties are colored in orange. Selected structures showing highest apparent affinities are illustrated in CFG notation. | ||
To quantify the binding affinities of the carbohydrates we adapted the competitive binding assay for LecBPA14 (Fig. S5†) previously described for LecBPAO1.18 Because the glycan array showed very similar carbohydrate specificities for both lectins, 21 selected ligands previously shown to bind to LecBPAO1 were analyzed for their potential of inhibiting LecBPA14 (Fig. 3 and S5†). Generally, three- to seven-fold lower IC50 values were obtained for the interaction between LecBPA14 and all tested mono- or disaccharides (1–11) than observed for LecBPAO1. Previously, the trisaccharide Lewisa (12), an antigen of the Lewis blood group system, was identified as a high affinity monovalent ligand for LecBPAO1 (Kd 212 nM).30 Furthermore, compound 543 (structure see Fig. S3†) identified from the glycan array screen bears a bi-antennary H type II blood group antigen, and therefore, the binding affinities of various blood group antigens were also examined (12–21, Fig. 3). The Lewis and H-type antigens (12–17) displayed nanomolar affinities. Lewisa (12) demonstrated very high affinity as a monovalent lectin ligand (IC50 78 nM) for LecBPA14 which was approximately two-fold higher than with LecBPAO1 (IC50 166 nM). The addition of galactose (blood group A, 18, 20) or N-acetyl galactosamine (blood group B, 19, 21) to the H-type antigen, and fucose to Lewisa or Lewisx (resulting in Lewisb16 and Lewisy17, respectively) led to reduced affinities.
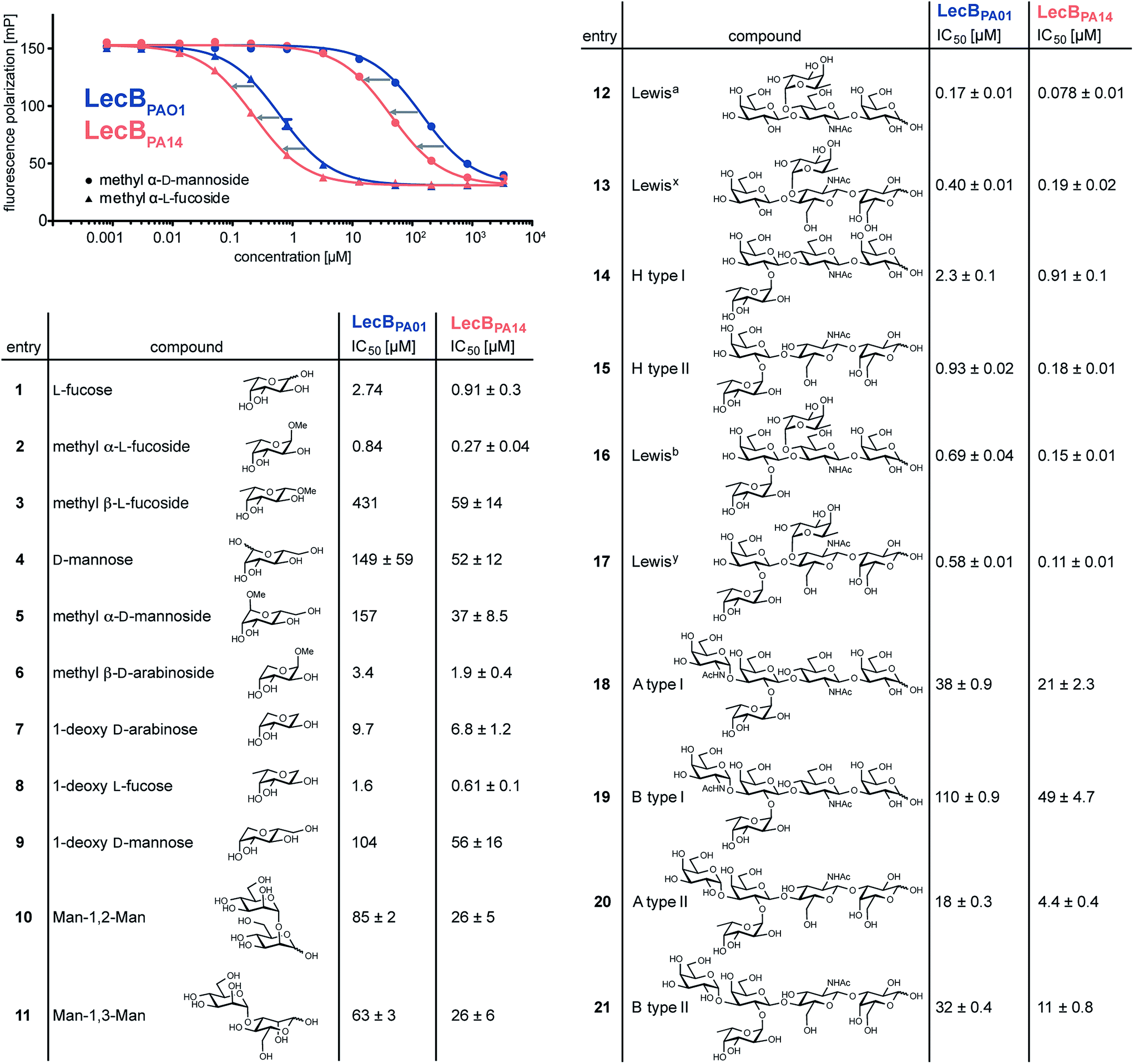 | ||
| Fig. 3 Biophysical evaluation of LecBPA14 and LecBPAO1 by a competitive binding assay. Data for LecBPAO1 with 1–3, 5–9 are from the literature.18,20 | ||
The interactions of methyl α-L-fucoside (2), methyl α-D-mannoside (5), the high affinity ligand Lewisa (12) and the H-type II antigen (15) with LecBPA14 were further studied by isothermal microcalorimetry (Fig. 4, S6 and S7†). The interaction of 15 with LecBPAO1 was also analyzed by ITC (Fig. 4 and S7†). Generally, increased affinities of the ligands to LecBPA14 were observed by two- to three-fold lower Kd values for LecBPA14 compared to LecBPAO1. The high affinity of the Lewisa antigen (12, IC50 78 nM) observed in the competitive binding assay was confirmed (Kd 70 nM for LecBPA14) and resulted from mainly enthalpy driven binding. To the best of our knowledge, this is the highest affinity observed for native monovalent glycan ligands and lectins in general. Stronger lectin binding affinities of monovalent ligands have been reported only for lipophilic glycoconjugates with FimH31,32 or cholera toxin,33 or glycomimetics.34 In contrast to LecBPAO1, the binding of methyl α-L-fucoside (2, Kd 202 nM for LecBPA14), methyl α-D-mannoside (5, Kd 16 μM for LecBPA14) and the H-type II antigen (15, Kd 199 nM for LecBPA14) revealed reduced enthalpic contributions to binding compensated with favorable entropic contribution for LecBPA14.
 | ||
| Fig. 4 Isothermal titration calorimetry (ITC) of LecBPA14 with 2, 5, 12 and 15 and comparison with the titration of 15 against LecBPAO1 or literature data for 2, 5 and 12 with LecBPAO1.14,30 | ||
Synthesis and evaluation of a bivalent N-glycan ligand for LecB
LecB from both PAO1 and PA14 showed strong binding on the glycan microarray to compound 543 which is a biantennary di-LacNAc N-linked glycan bearing the H-type II antigen on the terminus of both branches (see 23, Scheme 1). Glycosylated asparagine 22 (ref. 35) bearing the biantennary di-LacNAc N-linked glycan in presence of GDP-fucose was exposed to the insect cell culture supernatant of a baculovirus-infected expression culture of FUT-II36 (Scheme 1). The reaction required extended reaction times (72 h) for completion, leading to a removal of the asparagine moiety through other components in the complex culture media. However, sufficient quantities of the difucosylated free oligosaccharide 23 were obtained as pure compound in 20% yield after two chromatographic purifications. 23 was then tested in the competitive binding assays. Compound 23 was found to be a potent ligand for both LecBPAO1 (IC50 = 114 nM) and LecBPA14 (IC50 = 35 nM) (Scheme 1), with a strong avidity for LecB compared to its monovalent derivative 15, displaying IC50s of 930 and 180 nM, respectively.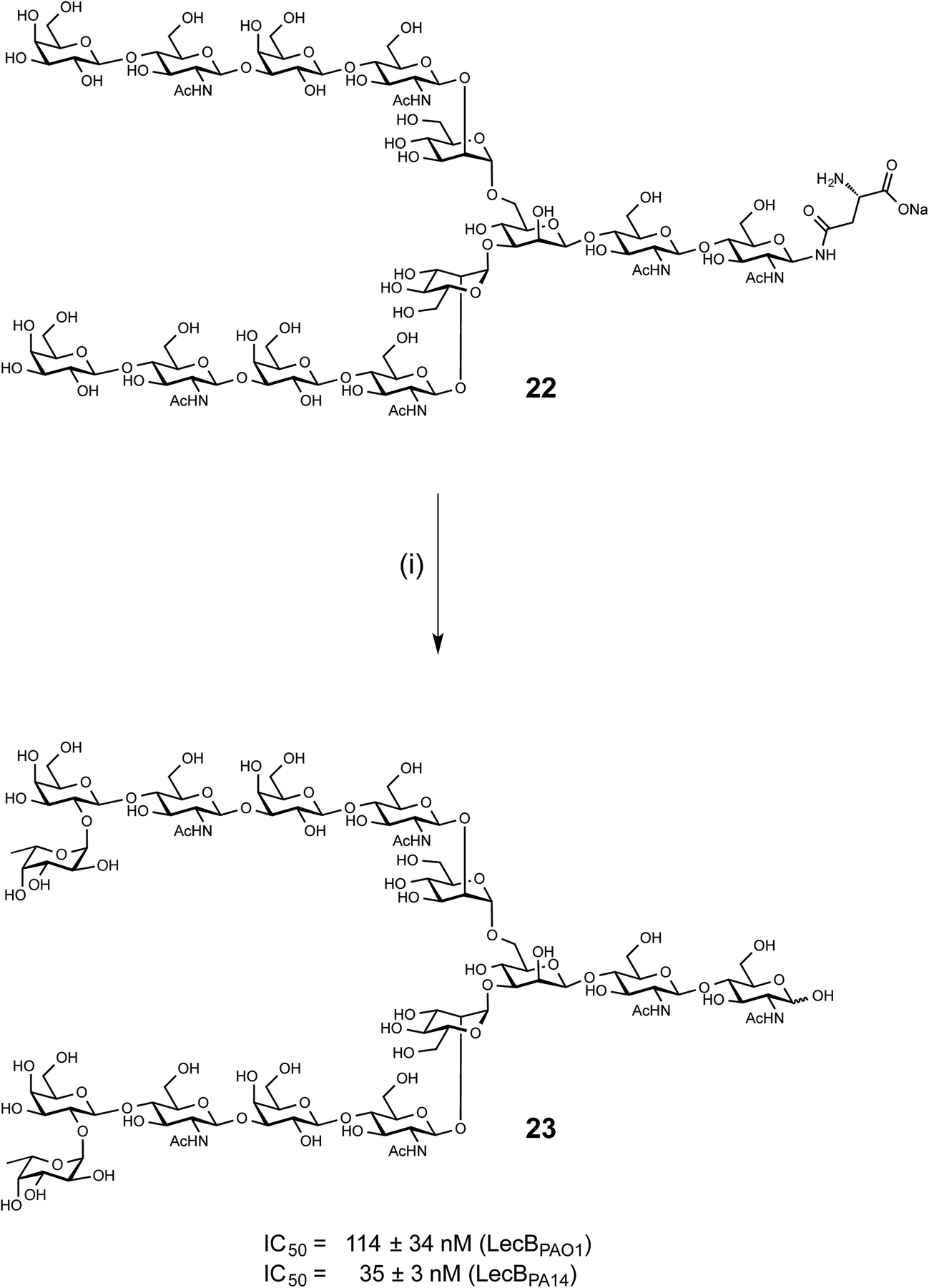 | ||
| Scheme 1 Chemoenzymatic synthesis of bivalent ligand 23 and inhibition of LecBPAO1 and LecBPA14. (i) GDP-Fuc, FUT-II (culture medium), Tris buffer, 37 °C, 72 h. | ||
The binding mode of compound 543 of the mammalian glycan array was analyzed by molecular docking experiments with LecBPAO1. Docking of the bi-antennary H type II ligand 23 to LecBPAO1 revealed that a bivalent binding mode is possible (Fig. 5). Here, both fucosides in one ligand molecule can simultaneously bind to two binding sites on the vertices of one LecB tetramer. The arrangement of the two extended di-LacNAc antenna of this N-glycan allows optimal positioning of the two fucose residues into two very distant binding sites of the tetramer. In contrast to reports for other multimeric lectins, e.g. wheat germ agglutinin,37 shiga toxin,38 LecA39,40 and others, this is the first proposal of a chelating binding mode for LecB.
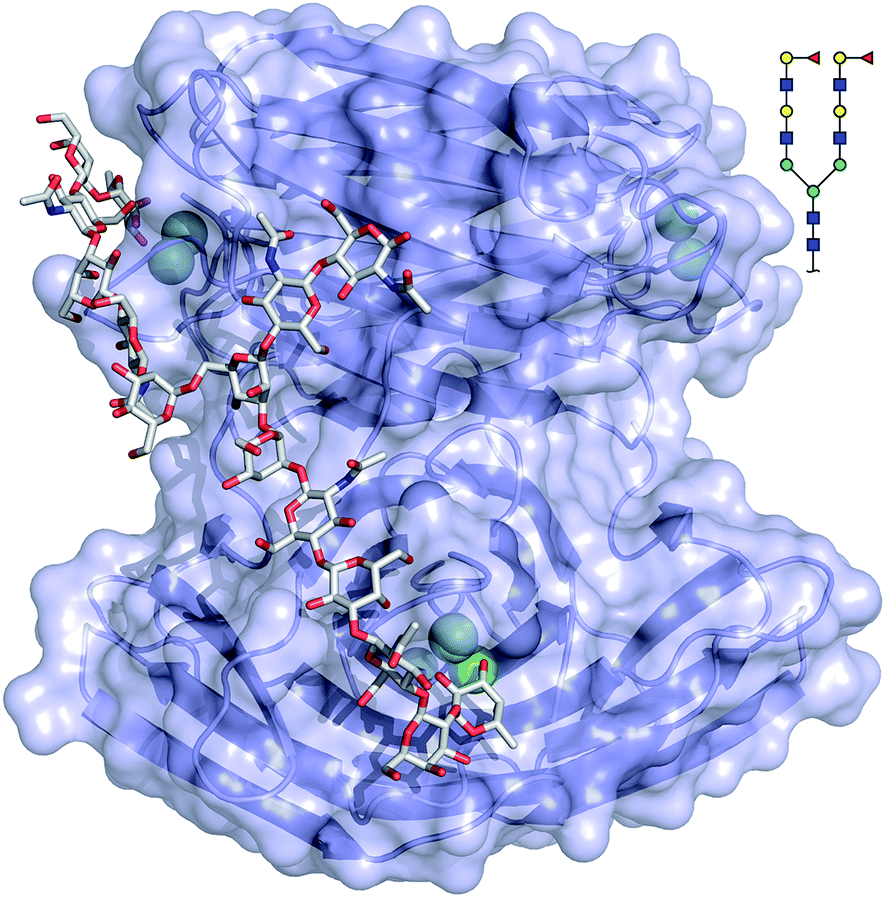 | ||
| Fig. 5 Docking of the bi-antennary H type II ligand (CFG compound 543) to LecBPAO1. This bivalent ligand can simultaneously bind to two binding sites of LecBPAO1, which are presented on the vertices of a tetrahedron in the tetramer. Carbohydrate ligands and amino acid side chains are shown as sticks colored by elements (C: grey, N: blue, O: red). Ca2+-ions are shown as green spheres. | ||
The crystal structure of LecB from P. aeruginosa PA14
LecBPA14 was subsequently crystallized and its structure was solved by X-ray crystallography (Fig. 6 and Table S1†). In analogy to LecBPAO1,41 the 1.70 Å resolution native structure of LecBPA14 shows the lectin as homotetramer with the four carbohydrate recognition domains (CRD) containing two Ca2+-ions each located on the vertices of a pseudotetrahedron (Fig. 6). A direct binding of G114 to one Ca2+-ion from the neighboring monomer stabilizes the tetrameric structure of the lectin, which was confirmed in solution by dynamic light scattering experiments (Fig. S4†). Notably, the structural alignment of LecBPA14 and LecBPAO1 revealed that all amino acid variations in LecBPA14 are located exclusively on the outer surface of the tetramer (Fig. 6). Mutations at the interfaces of the tetramer subunits are not present, and thus the oligomerization of LecB is conserved. Oligomerization of lectins is a prerequisite for their function as cross-linking agents, a feature that is probably important for the biological function of LecB.17,42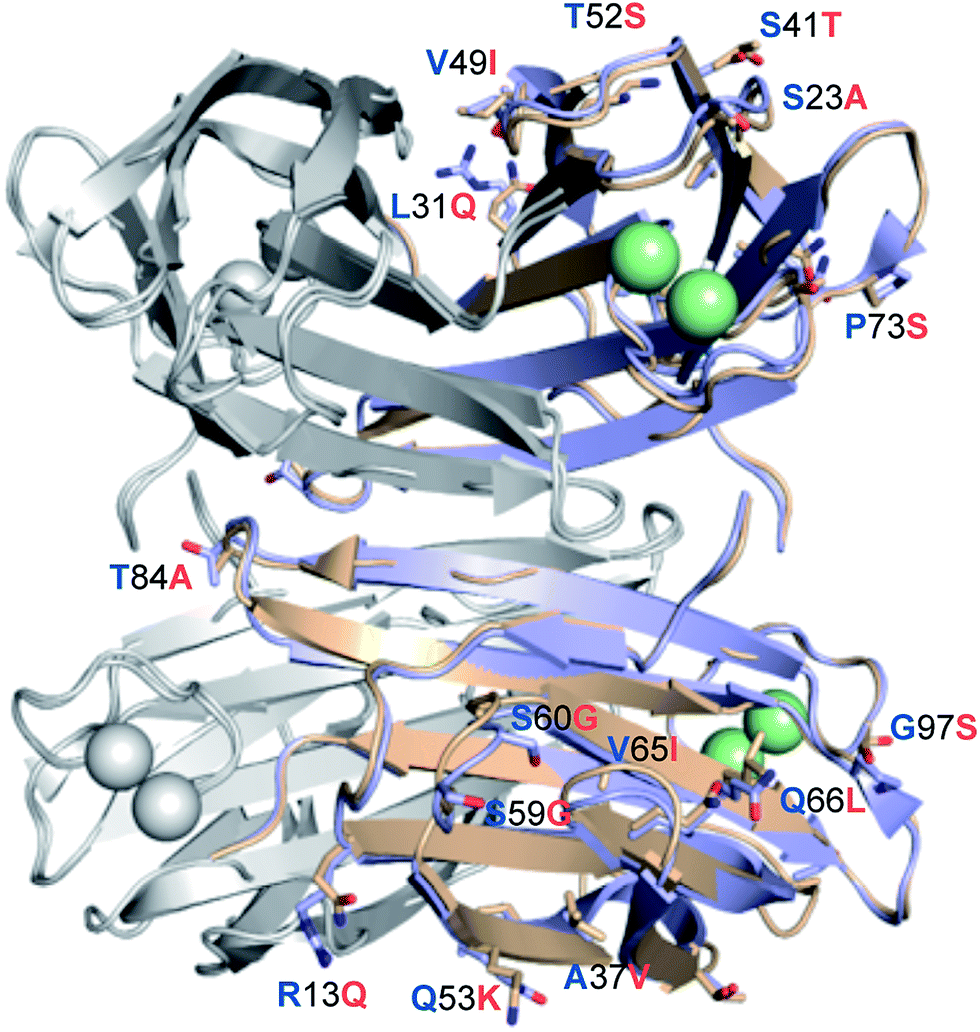 | ||
| Fig. 6 Crystal structure of LecBPA14. Superposition of the crystal structures LecBPA14 (orange, resolution 1.70 Å) and LecBPAO1 (blue; PDB code: 1OXC44). For clarity of illustration, differing amino acids are not shown in each monomer but are depicted only once per tetramer as labeled sticks (amino acid one letter code: blue for PAO1, orange for PA14) and colored by elements (N: blue, O: red). Ca2+-ions are shown as green spheres. | ||
Structure of LecBPA14 in complex with carbohydrate ligands
Co-crystals of LecBPA14 were obtained and the structures of LecBPA14 in complex with methyl α-L-fucoside (2, Fig. 7A), α-1,3-D-mannosyl D-mannose (11, Fig. 7B), and the high affinity ligand Lewisa (12, Fig. 7C) were solved by X-ray crystallography at resolutions between 1.40 and 1.55 Å (Table S1†). Both Ca2+-ions in LecBPA14 mediate the binding with the carbohydrates by complexation of three hydroxy groups in the saccharide ligand. Additionally, a direct contact of these hydroxy groups with D96, D99 and the C-terminal G114 of the neighboring monomer is observed. In the fucose-containing ligands (2, 12) the C-6 methyl group is embedded in a lipophilic pocket formed by T45 and A23. This latter variation in the CRD, S23A, allows additional van der Waals interactions between A23 and aglycones of the Ca2+-coordinated carbohydrate moiety. In contrast to LecBPAO1, the G97S variation in LecBPA14 offers an additional hydroxy group with hydrogen bonding potential in the CRD. In each of the three LecBPA14 complexes, the carbohydrate ligands establish one water-mediated hydrogen bond with S97. In addition to the interactions described, the high affinity ligand Lewisa (12) forms numerous hydrogen bonds with LecBPA14, involving two water molecules that simultaneously establish several hydrogen bonds with protein and ligand. While the reducing end galactose of the tetrasaccharide Lewisa does not interact with the protein, all other three monosaccharides in Lewisa contribute to the binding to LecBPA14. One water-mediated interaction of GlcNAc O-6 with D96, as well as another water-mediated hydrogen bond between O-6 of the terminal D-galactose moiety in Lewisa (12) with S97 are observed in the crystal structure. This latter water molecule additionally donates one hydrogen to establish a hydrogen bond with the anomeric oxygen of fucose. The extensive set of hydrophobic and hydrophilic interactions of Lewisa with LecBPA14 forms the molecular basis for its high affinity binding.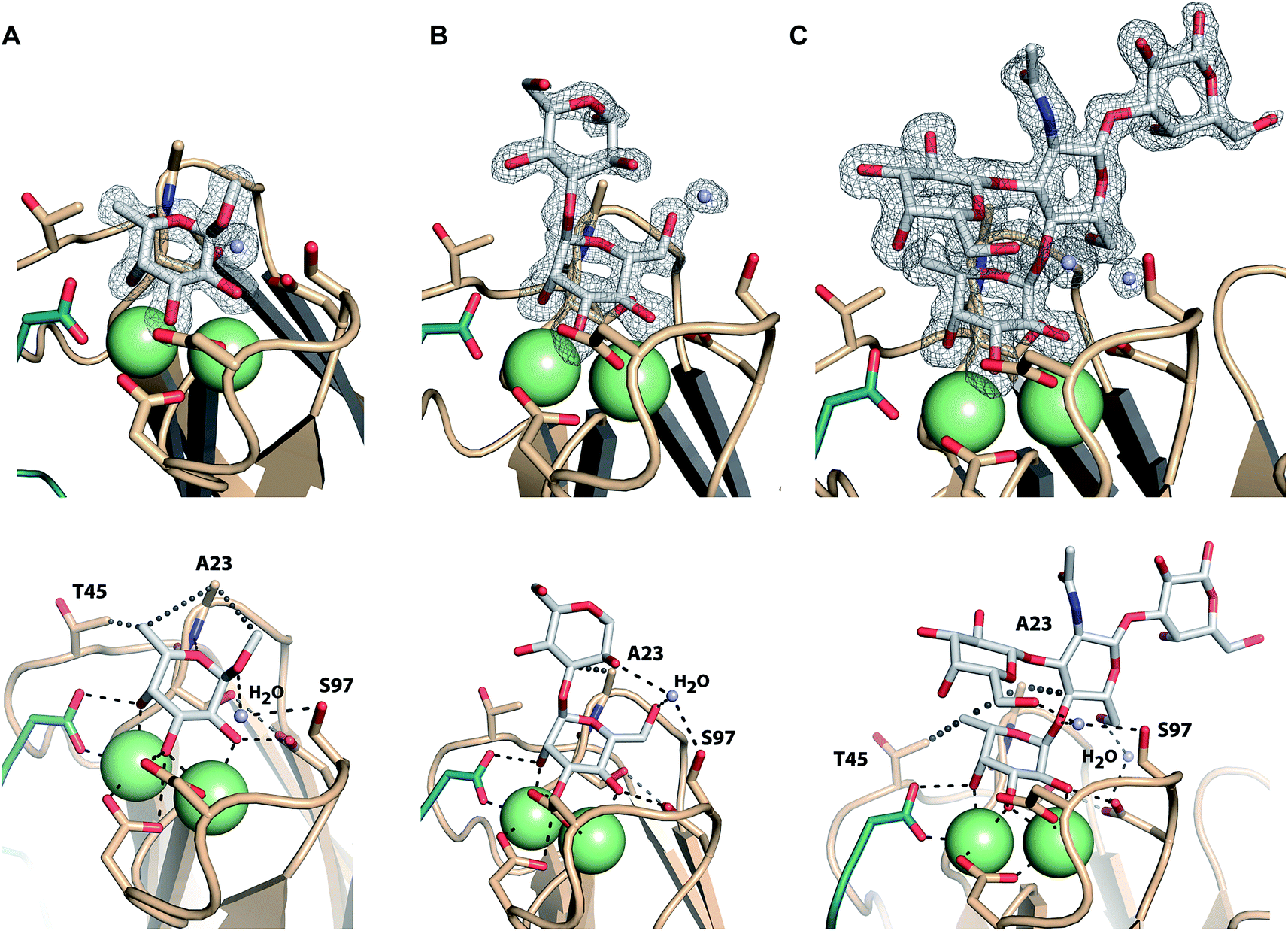 | ||
| Fig. 7 Crystal structures of LecBPA14 in complex with 2 (A), 11 (B) and 12 (C). (Upper panel) Display of electron density for the ligands at 1σ. Ca2+-ions are shown as green spheres. ((A) lower panel) Methyl α-L-fucoside (2) establishes a lipophilic interaction with T45 and A23. The crystal structure reveals a water-mediated hydrogen bond between the anomeric oxygen and S97 and an additional hydrophobic interaction of the aglycon with A23. ((B) lower panel) The crystal structure with α-1,3-D-mannosyl D-mannose (11) indicates one water mediated hydrogen bond between the 6-OH group and S97, as well as an interaction of the same water with the 2-OH group of the second mannose. An additional lipophilic contact between the aglycon and A23 is observed. ((C) lower panel) The tetrasaccharide Lewisa (12) establishes water-mediated hydrogen bonds via two water molecules and lipophilic contacts with T45 and A23. The C-terminus of each chain involved in the CRD of the neighboring chain is shown as cartoon in green. Ca2+-ions are shown as green spheres. | ||
Both sequence variations that are located directly in the carbohydrate binding site, S23A and G97S, are involved in extensive interactions with all carbohydrate ligands in LecBPA14 which is not observed in LecBPAO1 to this extent. Therefore, both variations are likely to contribute to the increased affinity of LecBPA14 for its ligands compared to LecBPAO1.
Conclusion
The adhesins LecA and LecB are virulence factors of P. aeruginosa and play an important role in biofilm formation and thus chronic infections. To date, only LecB from PAO1 has been studied.13,41,43,44 The sequence of LecB, however, differs significantly between the evolutionarily diverged PAO1- or PA14-like strains. Here, we showed that its sequence is a direct marker and correlates with the classification of P. aeruginosa into either PA14- or PAO1-like family of strains. This property could serve clinically for rapid choice of the appropriate therapies in accordance to the strain family which have different virulence properties. Despite high sequence variations, biophysical characterization uncovered that both variants possess very similar glycan binding preferences, although very surprisingly LecBPA14 generally displays 2- to 7-fold higher affinities. LecBPA14 showed a very high affinity of 70 nM for Lewisa, an unprecedented high affinity for a monovalent natural glycan–lectin interaction in general. Chemoenzymatic synthesis of a common high affinity ligand identified in the glycan array screen allowed evaluation of 23 in the competitive binding assays and potencies up to 35 nM for LecBPA14 were observed. Due to the high avidity of 23 and a docking model with LecB, this is first bivalent ligand likely to bridge simultaneously two binding sites in the tetramer of LecB. The crystal structure of LecBPA14 and comparison to its PAO1 homolog revealed the localization of all mutated amino acids on the outer surface of the LecB tetramer some of which are located in or near the carbohydrate binding site. The molecular basis for the superior affinity of LecBPA14 for its carbohydrate ligands was established on the basis of three crystal structures of LecBPA14 in complex with its ligands, revealing an extensive set of attractive interactions that are possible due to the two amino acid variations in the carbohydrate binding site.Importantly, based on the knowledge obtained in this study, carbohydrate-derived inhibitors targeting LecB of both P. aeruginosa families, PAO1 and PA14, that equally constitute the collection of strains analyzed, can now be developed into future anti-adhesion therapeutics against a broad range of clinical isolates.
The evolutionary reasons for the diverged protein sequence families (LecB from PAO1 vs. PA14) remain unclear. One hypothesis is that the LecBPA14 compensates for the absence of the exopolysaccharide psl in PA14. However, analysis of the clinical isolates showed that only PA14 lacks pslA-D from the psl operon, whereas other strains in this family generally possessed the full psl operon.26 Whether the highly conserved sequence variations between the PAO1- and PA14-like LecB sequence families and the resulting higher affinities for carbohydrate ligands are directly responsible for the enhanced virulence of P. aeruginosa PA14 compared to PAO1, as observed for the pathoadaptive adhesin FimH in E. coli,45 is subject to future investigations.
Materials and methods
Chemicals
D-Mannose (4) and methyl α-D-mannoside (5) were purchased from Sigma Aldrich (Germany), L-fucose (1), α-1,2-D-mannosyl D-mannose (10), α-1,3-D-mannosyl D-mannose (11) from Dextra Laboratories (Reading, UK), methyl β-D-arabinoside (6) from TCI Europe, methyl α-L-fucoside (2) and methyl β-L-fucoside (3) from Carbosynth Ltd. (UK). Blood group antigens 12–21 were purchased from Elicityl OligoTech (France).Chemoenzymatic synthesis of bivalent ligand α-L-fucopyranosyl-(1→2)-β-D-galactopyranosyl-(1→4)-2-acetamido-2-deoxy-β-D-glucopyranosyl-(1→3)-β-D-galactopyranosyl-(1→4)-2-acetamido-2-deoxy-β-D-glucopyranosyl-(1→2)-α-D-mannopyranosyl-(1→3)-(α-L-fucopyranosyl-(1→2)-β-D-galactopyranosyl-(1→4)-2-acetamido-2-deoxy-β-D-glucopyranosyl-(1→3)-β-D-galactopyranosyl-(1→4)-2-acetamido-2-deoxy-β-D-glucopyranosyl-(1→2)-α-D-mannopyranosyl-(1→6))-β-D-mannopyranosyl-(1→4)-2-acetamido-2-deoxy-β-D-glucopyranosyl-(1→4)-2-acetamido-2-deoxy-D-glucopyranose (23)
Compound 22 (ref. 35) (11.9 mg, 4.76 μmol) and GDP-Fuc (11.2 mg, 19.1 μmol, 4 eq.) were dissolved in 500 μL Tris–HCl buffer (100 mM, pH 7.5, 20 mM MgCl2). FUT-II36 culture media (∼10 U mmol−1 substrate) was added then the reaction was incubated for 48 hours at 37 °C. Additional GDP-Fuc (2 eq.) and FUT-II were added and the reaction continued for 24 hours. The reaction was centrifuged and the supernatant subjected to size exclusion chromatography using a Sephadex™ G-25 column (1 × 140 cm), equilibrated and eluted with H2O. Fractions containing product were combined and lyophilized. The solid was dissolved in H2O then subjected to further purification using an All-Tech™ Carbograph column (150 mg) eluted with H2O with increasing concentration of methanol (0 to 50%) to give pure α/β fucosylated product 23 (2.5 mg, 20%) as a white amorphous solid. The asparagine aglycone of 22 was hydrolyzed during the reaction; Rf 0.05 (6![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :3:2, iPrOH–H2O–NH4OH); 1H NMR (D2O, 600 MHz) δ 5.22 (2H, m), 5.11 (1H, s, H-1 GlcNAc-α-OH), 5.04 (1H, s), 4.85 (1H, s), 4.63–4.61 (2H, m), 4.54–4.46 (5H, m), 4.38 (2H, m), 4.17–4.03 (6H, m), 3.91–3.39 (81H, m), 2.00 (3H, s), 1.96 (15H, m), 1.15 (6H, m); MS (MALDI-TOF) m/z [M + Na]+ calcd for C102H170N6NaO74 2686, obs 2688.
:3:2, iPrOH–H2O–NH4OH); 1H NMR (D2O, 600 MHz) δ 5.22 (2H, m), 5.11 (1H, s, H-1 GlcNAc-α-OH), 5.04 (1H, s), 4.85 (1H, s), 4.63–4.61 (2H, m), 4.54–4.46 (5H, m), 4.38 (2H, m), 4.17–4.03 (6H, m), 3.91–3.39 (81H, m), 2.00 (3H, s), 1.96 (15H, m), 1.15 (6H, m); MS (MALDI-TOF) m/z [M + Na]+ calcd for C102H170N6NaO74 2686, obs 2688.
Sequence analysis
The amino acid sequences of the P. aeruginosa strains (clinical isolates26 and reference strains27) were submitted to a multiple sequence alignment resulting in a phylogenetic tree using the http://phylogeny.lirmm.fr platform.46 The alignment was performed using MUSCLE,47 Gblocks for alignment curation48 and PhyML for phylogeny.49 See Fig. S1, ESI† for the alignment.Bacterial strains and growth conditions
E. coli XL1 blue was used for amplification and E. coli BL21 (DE3) for expression of plasmid pRS01.4 carrying the sequence of LecBPA14. Bacteria were grown in lysogeny broth (LB) supplemented with ampicillin (100 mg L−1).Molecular cloning and expression of LecBPA14
Genomic DNA from P. aeruginosa UCBPP-PA14 was isolated using Gen Elute Bacterial Genomic DNA Kit (Sigma Aldrich). PCR amplification was performed with Phusion polymerase (New England Biolabs) and primers introducing NdeI (5′-GGAATTCCATATGGCAACAAGGAGTG-3′) and HindIII (5′-CCCAAGCTTCTAGCCGAGCGGCCAG-3′) restrictions sites. After digestion with NdeI and HindIII restriction enzymes (New England Biolabs) the DNA fragment was ligated into the multiple cloning site of digested pET22b(+) (Novagen) with T4 DNA ligase (New England Biolabs) and resulted in plasmid pRS01.4. The sequence was confirmed by sequencing (GATC Biotech) with primers T7 promotor (5′-TAATACGACTCACTATATAGG-3′) and T7 terminator (5′-GCTAGTTATTGCTCAGCGG-3′). Expression and purification of the protein was performed in analogy to LecBPAO1 (ref. 18) and the protein was dialyzed against TBS/Ca (20 mM Tris, 137 mM NaCl, 2.6 mM KCl at pH 7.4 supplemented with 1 mM CaCl2) and stored at −20 °C.Fluorescence labeling of LecB and CFG mammalian glycan array
Each LecB variant (700 μL, 58 μM in Na2CO3 buffer, pH 9.3) was incubated at r.t. under shaking (500 rpm) with FITC (33 μL, 3 mg mL−1, in Na2CO3 buffer, pH 9.3) for 1 h. Purification was performed as described for unlabeled protein above, the protein concentration was determined as described in Fig. S2.† FITC-labeled LecBPAO1 and LecBPA14 were tested on the Consortium for Functional Glycomics (CFG) mammalian glycan array (Core H) version 5.2. Standard procedures of Core H were run at 200 μg mL−1, 20 μg mL−1, and 2 μg mL−1 protein concentration.Dynamic light scattering (DLS) measurements
DLS measurements were performed on a Zetasizer Nano-ZS (Malvern Instruments, UK). Stock solutions were filtered with a syringe filter before measurements. 50 μL of LecB (100 μM) in TBS/Ca (20 mM Tris, 137 mM NaCl, 2.6 mM KCl at pH 7.4 supplemented with 1 mM CaCl2) was measured at 25 °C.Competitive binding assay
The competitive binding assay based on fluorescence polarization was performed as described previously for PAO1.18 Briefly, 20 μL of a stock solution of LecBPA14 (150 nM) and fluorescent reporter ligand N-(fluorescein-5-yl)-N′-(α-L-fucopyranosyl ethylen)-thiocarbamide (15 nM) in TBS/Ca (20 mM Tris, 137 mM NaCl, 2.6 mM KCl at pH 7.4 supplemented with 1 mM CaCl2) were mixed with 10 μL serial dilutions (1 mM to 12.8 nM) of testing compounds in TBS/Ca in triplicates in black 384-well microtiter plates (Greiner Bio-One, Germany, cat no 781900). After addition of the reagents, the plate was incubated for 8–22 h at r.t. in a humidity chamber. Fluorescence emission parallel and perpendicular to the excitation plane was measured on a PheraStar FS (BMG Labtech, Germany) plate reader with excitation filters at 485 nm and emission filters at 535 nm. The measured intensities were reduced by buffer values and fluorescence polarization was calculated. The data were analyzed using BMG Labtech MARS software and/or with Graphpad Prism and fitted according to the four parameter variable slope model. Bottom and top plateaus were defined by the standard compounds L-fucose (1) and methyl α-D-mannoside (5) respectively and the data was reanalyzed with these values fixed. A minimum of three independent measurements of triplicates each was performed for every ligand.Isothermal titration calorimetry (ITC)
The concentration of the monomer of LecBPA14 (dissolved in TBS/Ca (20 mM Tris, 137 mM NaCl, 2.6 mM KCl at pH 7.3 supplemented with 1 mM CaCl2)) was determined by UV spectroscopy at 280 nm using a molar extinction coefficient of 6990 M−1 cm−1 (obtained from ProtParam50). The temperature of the sample cell was 25 °C. The titration was performed with a solution of ligands in the same buffer, concentrations of protein and ligands is given in Fig. S6 and S7.† ITC was performed on a Microcal ITC200 (GE) and the data was analyzed according to the one site binding model using the Microcal Origin software. A minimum of three independent titrations was performed for each ligand.Docking
Bi-antennary oligosaccharide 543 was built in the Sybyl graphic editor (Certara) using the monosaccharides structures available on the glyco3D portal (http://glyco3D.cermav.cnrs.fr). One fucose was located in the binding site as observed in crystal structure of LecBPAO1 (ref. 41) and each monosaccharide was stepwise included with conformation in agreement with published energy maps51 and general direction towards one of the two neighboring binding site. Only one of the two sites was accessible after building the whole oligosaccharide in extended conformation. Final optimization was performed with distance constraints to maintain both fucoses in the crystallographic position and energy minimization with the Tripos force-field.Crystallization and structure determination
For crystallization of LecBPA14 with ligands 2, 11 or 12, a protein solution in water (10 mg mL−1) was incubated in a 9:1 ratio with compound (10 mM compound in 20 mM Tris, 137 mM NaCl, 2.6 mM KCl at pH 7.3 supplemented with 1 mM CaCl2) for minimum 1 h prior crystallization. Crystallization was performed by the hanging drop vapor diffusion method using 1 μL of protein plus ligand + 1 μL of reservoir solution at 19 °C in a 24 well plate. Crystals were formed after one or two days and were frozen in liquid nitrogen after the addition of appropriate cryoprotectant (Table S1†). Data were collected on beamline BM30A at ESRF (Grenoble) using a ADSC Q315r CCD detector apart from the structure of 11, which was collected on ID23-2 using a pilatus detector. Data were processed using XDS and all further computing was performed using the CCP4 suite.52,53 All structures were solved by molecular replacement using PHASER.54 The tetramer coordinates of PDB 1UZV were used as model for the native structure of LecBPA14 which was then used as model for all the complex structures. Manual corrections of the model were performed using Coot55 and interspersed with cycles of maximum likelihood refinement with REFMAC5.8.56 Coordinates and structure factors have been deposited in the Protein Data Bank under accession codes 5A6Q, 5A6X, 5A6Y, 5A6Z.
Author contributions
R. S. and S. W. cloned and generated recombinant protein; R. S. and A. V. performed the structural-biology experiments; R. S. performed binding assays; A. I. performed modeling experiments; S. W. analyzed glycan array data and sequencing data; C. N. and J. P. synthesized divalent H-type II antigen, A. K. and S. H. provided sequencing data; A. I. and S. H. gave conceptual advice; R. S., S. W. and A. T. conceived the experiments and wrote the paper.Conflict of interest
The authors declare no competing financial interest.Acknowledgements
The authors are grateful to Dr Martin Frank for initial help with the 3D-structure of cmp 543 and to the Consortium for Functional Glycomics (Core H, Protein-glycan Interaction Resource of the CFG and the supporting grant R24 GM098791). Crystal data collection was performed at the European Synchrotron Radiation Facility, Grenoble, France and we are grateful for access and technical support to beamline BM30A (proposal 20130700/30-01/925) and ID23-2. We thank the Helmholtz Association (to AT, grant no VH-NG-934), the Konstanz Research School Chemical Biology (to RS, AT), COST action BM1003 (to RS), and the Deutsche Forschungsgemeinschaft (to AT, grant no Ti756/2-1) for financial support. AI and AV acknowledge support from Labex ARCANE (ANR-11-LABX-0003-01) and GDR Pseudomonas. JCP acknowledges financial support from The National Heart, Lung and Blood Institute (HL107151).References
- L. B. Rice, J. Infect. Dis., 2008, 197(8), 1079–1081 CrossRef PubMed.
- K. Poole, Front. Microbiol., 2011, 2, 65 CAS.
- E. E. Mann and D. J. Wozniak, FEMS Microbiol. Rev., 2012, 36(4), 893–916 CrossRef CAS PubMed.
- T. Bjarnsholt, O. Ciofu, S. Molin, M. Givskov and N. Høiby, Nat. Rev. Drug Discovery, 2013, 12(10), 791–808 CrossRef CAS PubMed.
- D. Davies, Nat. Rev. Drug Discovery, 2003, 2(2), 114–122 CrossRef CAS PubMed.
- M. A. Cooper and D. Shlaes, Nature, 2011, 472(7341), 32 CrossRef CAS PubMed.
- A. E. Clatworthy, E. Pierson and D. T. Hung, Nat. Chem. Biol., 2007, 3(9), 541–548 CrossRef CAS PubMed.
- S. P. Diggle, R. E. Stacey, C. Dodd, M. Cámara, P. Williams and K. Winzer, Environ. Microbiol., 2006, 8(6), 1095–1104 CrossRef CAS PubMed.
- D. Tielker, S. Hacker, R. Loris, M. Strathmann, J. Wingender, S. Wilhelm, F. Rosenau and K.-E. Jaeger, Microbiology, 2005, 151(5), 1313–1323 CrossRef CAS PubMed.
- C. Chemani, A. Imberty, S. de Bentzmann, M. Pierre, M. Wimmerová, B. P. Guery and K. Faure, Infect. Immun., 2009, 77(5), 2065–2075 CrossRef CAS PubMed.
- R. Sommer, I. Joachim, S. Wagner and A. Titz, Chimia, 2013, 67(4), 286–290 CrossRef CAS PubMed.
- S. Wagner, R. Sommer, S. Hinsberger, C. Lu, R. W. Hartmann, M. Empting and A. Titz, J. Med. Chem., 2016 DOI:10.1021/acs.jmedchem.5b01698.
- N. Gilboa-Garber, Methods Enzymol., 1982, 83, 378–385 CAS.
- C. Sabin, E. P. Mitchell, M. Pokorná, C. Gautier, J.-P. Utille, M. Wimmerová and A. Imberty, FEBS Lett., 2006, 580(3), 982–987 CrossRef CAS PubMed.
- A. M. Boukerb, A. Rousset, N. Galanos, J.-B. Méar, M. Thepaut, T. Grandjean, E. Gillon, S. Cecioni, C. Abderrahmen, K. Faure, D. Redelberger, E. Kipnis, R. Dessein, S. Havet, B. Darblade, S. E. Matthews, S. de Bentzmann, B. Guéry, B. Cournoyer, A. Imberty and S. Vidal, J. Med. Chem., 2014, 57(24), 10275–10289 CrossRef CAS PubMed.
- E. M. V. Johansson, S. A. Crusz, E. Kolomiets, L. Buts, R. U. Kadam, M. Cacciarini, K.-M. Bartels, S. P. Diggle, M. Cámara, P. Williams, R. Loris, C. Nativi, F. Rosenau, K.-E. Jaeger, T. Darbre and J.-L. Reymond, Chem. Biol., 2008, 15(12), 1249–1257 CrossRef CAS PubMed.
- S. Cecioni, A. Imberty and S. Vidal, Chem. Rev., 2015, 115(1), 525–561 CrossRef CAS PubMed.
- D. Hauck, I. Joachim, B. Frommeyer, A. Varrot, B. Philipp, H. M. Möller, A. Imberty, T. E. Exner and A. Titz, ACS Chem. Biol., 2013, 8(8), 1775–1784 CrossRef CAS PubMed.
- A. Hofmann, R. Sommer, D. Hauck, J. Stifel, I. Göttker-Schnetmann and A. Titz, Carbohydr. Res., 2015, 412, 34–42 CrossRef CAS PubMed.
- R. Sommer, T. E. Exner and A. Titz, PLoS One, 2014, 9(11), e112822 Search PubMed.
- R. Sommer, D. Hauck, A. Varrot, S. Wagner, A. Audfray, A. Prestel, H. M. Möller, A. Imberty and A. Titz, ChemistryOpen, 2015, 4(6), 756–767 CrossRef CAS.
- A. Titz, Carbohydrate-Based Anti-Virulence Compounds Against Chronic Pseudomonas aeruginosa Infections with a Focus on Small Molecules, in Topics in Medicinal Chemistry, ed. Seeberger H. Peter and C. Rademacher, Springer, Berlin Heidelberg, 2014, vol. 12, Carbohydrates as Drugs, pp. 169–186 Search PubMed.
- L. G. Rahme, E. J. Stevens, S. F. Wolfort, J. Shao, R. G. Tompkins and F. M. Ausubel, Science, 1995, 268(5219), 1899–1902 CAS.
- C. K. Stover, X. Q. Pham, A. L. Erwin, S. D. Mizoguchi, P. Warrener, M. J. Hickey, F. S. Brinkman, W. O. Hufnagle, D. J. Kowalik, M. Lagrou, R. L. Garber, L. Goltry, E. Tolentino, S. Westbrock-Wadman, Y. Yuan, L. L. Brody, S. N. Coulter, K. R. Folger, A. Kas, K. Larbig, R. Lim, K. Smith, D. Spencer, G. K. Wong, Z. Wu, I. T. Paulsen, J. Reizer, M. H. Saier, R. E. Hancock, S. Lory and M. V. Olson, Nature, 2000, 406(6799), 959–964 CrossRef CAS PubMed.
- D. G. Lee, J. M. Urbach, G. Wu, N. T. Liberati, R. L. Feinbaum, S. Miyata, L. T. Diggins, J. He, M. Saucier, E. Déziel, L. Friedman, L. Li, G. Grills, K. Montgomery, R. Kucherlapati, L. G. Rahme and F. M. Ausubel, GenomeBiology, 2006, 7(10), R90 CrossRef PubMed.
- A. Dötsch, M. Schniederjans, A. Khaledi, K. Hornischer, S. Schulz, A. Bielecka, D. Eckweiler, S. Pohl and S. Häussler, mBio, 2015, 6(4), e00749-15 CrossRef PubMed.
- G. L. Winsor, D. K. W. Lam, L. Fleming, R. Lo, M. D. Whiteside, N. Y. Yu, R. E. W. Hancock and F. S. L. Brinkman, Nucleic Acids Res., 2011, 39, D596–D600 CrossRef CAS PubMed.
- P. H. Roy, S. G. Tetu, A. Larouche, L. Elbourne, S. Tremblay, Q. Ren, R. Dodson, D. Harkins, R. Shay, K. Watkins, Y. Mahamoud and I. T. Paulsen, PLoS One, 2010, 5(1), e8842 Search PubMed.
- O. Blixt, S. Head, T. Mondala, C. Scanlan, M. E. Huflejt, R. Alvarez, M. C. Bryan, F. Fazio, D. Calarese, J. Stevens, N. Razi, D. J. Stevens, J. J. Skehel, I. van Die, D. R. Burton, I. A. Wilson, R. Cummings, N. Bovin, C.-H. Wong and J. C. Paulson, Proc. Natl. Acad. Sci. U. S. A., 2004, 101(49), 17033–17038 CrossRef CAS PubMed.
- S. Perret, C. Sabin, C. Dumon, M. Pokorná, C. Gautier, O. Galanina, S. Ilia, N. Bovin, M. Nicaise, M. Desmadril, N. Gilboa-Garber, M. Wimmerová, E. P. Mitchell and A. Imberty, Biochem. J., 2005, 389(2), 325–332 CrossRef CAS PubMed.
- T. Klein, D. Abgottspon, M. Wittwer, S. Rabbani, J. Herold, X. Jiang, S. Kleeb, C. Lüthi, M. Scharenberg, J. Bezençon, E. Gubler, L. Pang, M. Smiesko, B. Cutting, O. Schwardt and B. Ernst, J. Med. Chem., 2010, 53(24), 8627–8641 CrossRef CAS PubMed.
- C. K. Cusumano, J. S. Pinkner, Z. Han, S. E. Greene, B. A. Ford, J. R. Crowley, J. P. Henderson, J. W. Janetka and S. J. Hultgren, Sci. Transl. Med., 2011, 3(109), 109ra115 Search PubMed.
- G. M. Kuziemko, M. Stroh and R. C. Stevens, Biochemistry, 1996, 35(20), 6375–6384 CrossRef CAS PubMed.
- J. Egger, C. Weckerle, B. Cutting, O. Schwardt, S. Rabbani, K. Lemme and B. Ernst, J. Am. Chem. Soc., 2013, 135(26), 9820–9828 CrossRef CAS PubMed.
- C. M. Nycholat, R. McBride, D. C. Ekiert, R. Xu, J. Rangarajan, W. Peng, N. Razi, M. Gilbert, W. Wakarchuk, I. A. Wilson and J. C. Paulson, Angew. Chem., Int. Ed., 2012, 51(20), 4860–4863 CrossRef CAS PubMed.
- D. Vasiliu, N. Razi, Y. Zhang, N. Jacobsen, K. Allin, X. Liu, J. Hoffmann, O. Bohorov and O. Blixt, Carbohydr. Res., 2006, 341(10), 1447–1457 CrossRef CAS PubMed.
- D. Schwefel, C. Maierhofer, J. G. Beck, S. Seeberger, K. Diederichs, H. M. Möller, W. Welte and V. Wittmann, J. Am. Chem. Soc., 2010, 132(25), 8704–8719 CrossRef CAS PubMed.
- P. I. Kitov, J. M. Sadowska, G. Mulvey, G. D. Armstrong, H. Ling, N. S. Pannu, R. J. Read and D. R. Bundle, Nature, 2000, 403(6770), 669–672 CrossRef CAS PubMed.
- F. Pertici and R. J. Pieters, Chem. Commun., 2012, 48(33), 4008–4010 RSC.
- A. Novoa, T. Eierhoff, J. Topin, A. Varrot, S. Barluenga, A. Imberty, W. Römer and N. Winssinger, Angew. Chem., Int. Ed., 2014, 53(34), 8885–8889 CrossRef CAS PubMed.
- E. Mitchell, C. Houles, D. Sudakevitz, M. Wimmerova, C. Gautier, S. Pérez, A. M. Wu, N. Gilboa-Garber and A. Imberty, Nat. Struct. Biol., 2002, 9(12), 918–921 CrossRef CAS PubMed.
- T. K. Dam and C. F. Brewer, Glycobiology, 2010, 20(3), 270–279 CrossRef CAS PubMed.
- E. P. Mitchell, C. Sabin, L. Snajdrová, M. Pokorná, S. Perret, C. Gautier, C. Hofr, N. Gilboa-Garber, J. Koca, M. Wimmerová and A. Imberty, Proteins: Struct., Funct., Bioinf., 2004, 58(3), 735–746 CrossRef PubMed.
- R. Loris, D. Tielker, K.-E. Jaeger and L. Wyns, J. Mol. Biol., 2003, 331(4), 861–870 CrossRef CAS PubMed.
- D. J. Schwartz, V. Kalas, J. S. Pinkner, S. L. Chen, C. N. Spaulding, K. W. Dodson and S. J. Hultgren, Proc. Natl. Acad. Sci. U. S. A., 2013, 110(39), 15530–15537 CrossRef CAS PubMed.
- A. Dereeper, V. Guignon, G. Blanc, S. Audic, S. Buffet, F. Chevenet, J.-F. Dufayard, S. Guindon, V. Lefort, M. Lescot, J.-M. Claverie and O. Gascuel, Nucleic Acids Res., 2008, 36, W465–W469 CrossRef CAS PubMed.
- R. C. Edgar, Nucleic Acids Res., 2004, 32(5), 1792–1797 CrossRef CAS PubMed.
- J. Castresana, Mol. Biol. Evol., 2000, 17(4), 540–552 CrossRef CAS PubMed.
- S. Guindon, J.-F. Dufayard, V. Lefort, M. Anisimova, W. Hordijk and O. Gascuel, Syst. Biol., 2010, 59(3), 307–321 CrossRef CAS PubMed.
- M. R. Wilkins, E. Gasteiger, A. Bairoch, J. C. Sanchez, K. L. Williams, R. D. Appel and D. F. Hochstrasser, Methods Mol. Biol., 1999, 112, 531–552 CAS.
- A. Imberty, S. Gerber, V. Tran and S. Perez, Glycoconjugate J., 1990, 7(1), 27–54 CrossRef CAS.
- M. D. Winn, C. C. Ballard, K. D. Cowtan, E. J. Dodson, P. Emsley, P. R. Evans, R. M. Keegan, E. B. Krissinel, A. G. W. Leslie, A. McCoy, S. J. McNicholas, G. N. Murshudov, N. S. Pannu, E. A. Potterton, H. R. Powell, R. J. Read, A. Vagin and K. S. Wilson, Acta Crystallogr., Sect. D: Biol. Crystallogr., 2011, 67(4), 235–242 CrossRef CAS PubMed.
- W. Kabsch, Acta Crystallogr., Sect. D: Biol. Crystallogr., 2010, 66(2), 125–132 CrossRef CAS PubMed.
- A. J. McCoy, R. W. Grosse-Kunstleve, P. D. Adams, M. D. Winn, L. C. Storoni and R. J. Read, J. Appl. Crystallogr., 2007, 40(4), 658–674 CrossRef CAS PubMed.
- P. Emsley, B. Lohkamp, W. G. Scott and K. Cowtan, Acta Crystallogr., Sect. D: Biol. Crystallogr., 2010, 66(4), 486–501 CrossRef CAS PubMed.
- G. N. Murshudov, P. Skubák, A. A. Lebedev, N. S. Pannu, R. A. Steiner, R. A. Nicholls, M. D. Winn, F. Long and A. A. Vagin, Acta Crystallogr., Sect. D: Biol. Crystallogr., 2011, 67(4), 355–367 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available: Multiple sequence alignment of LecB from clinical isolates. Concentration determination of FITC-labeled lectins LecBPAO1 and LecBPA14. Chemical structure drawing of compound 543. Dynamic light scattering of LecBPA14. Titration curves for fluorescence polarization-based direct and competitive binding assay. Titration figures for ITC experiments, as well as for compound 23 MALDI-MS and 1H-NMR. Data collection and refinement statistics for LecBPA14 structures. See DOI: 10.1039/c6sc00696e |
| ‡ Both authors contributed equally. |
| This journal is © The Royal Society of Chemistry 2016 |