DOI:
10.1039/C6RA02950G
(Paper)
RSC Adv., 2016,
6, 33036-33042
Cellulose/boron nitride core–shell microbeads providing high thermal conductivity for thermally conductive composite sheets†
Received
1st February 2016
, Accepted 24th March 2016
First published on 29th March 2016
Abstract
We fabricated sheets with high thermal conductivity in a way that required less filler. Our approach is as follows: (1) we used core–shell spherical microbeads as the thermally conductive filler; (2) we developed cellulose/h-boron nitride (BN) core–shell spherical microbeads using phase separation of a cellulose xanthate aqueous solution (viscose); (3) we hybridized the cellulose/h-BN core–shell microbeads with epoxy resin using compression molding. This process reduced the amount of h-BN required because the microbeads efficiently formed thermally conductive pathways among the shells in the insulating resin. The thermal conductivities of the resulting sheet in the thickness and in-plane directions were 10.6 and 15.6 W m−1 K−1, respectively, using only 48.5 vol% h-BN. In contrast, the thermal conductivities of the composite sheet with 75 vol% of naked h-BN particles were 6.31 and 22.9 W m−1 K−1 in the thickness and in-plane directions, respectively. This large difference resulted from the anisotropic structure of h-BN. The changes in thermal conductivity with h-BN content were inconsistent with percolation theory, when using the cellulose/h-BN core–shell microbeads as a filler. The thermally conductive sheets fabricated with microbeads exhibited thermal conductivities several times greater than that of sheets fabricated with naked h-BN. This indicated that thermally conductive pathways had formed in the insulating resin.
1. Introduction
As electronic devices demand higher performance, functionality, and density, they will consume more power and produce more heat. Various heat-dissipation techniques have been developed, but more effective methods are needed. One way to dissipate heat is to use a thermally conductive, electrically insulating sheet. Various thermally conductive inorganic materials have been explored,1–13 including diamond and boron nitride. Generally, thermal conduction in inorganic/resin composite sheets can be understood by percolation theory.14–16 This theory states that adding thermally conductive inorganic materials to an inorganic/resin composite sheet will not increase its thermal conductivity, unless the filler content is greater than the percolation threshold of 30 vol%.14 This phenomenon has been attributed to the contact resistance at the interface of the inorganic materials and resin.17,18 Unfortunately, using a high filler content is expensive, and it may worsen the final material by making it heavier and less flexible, with a rougher surface and higher mechanical friability. This is because the inorganic filler can agglomerate and become non-uniform.
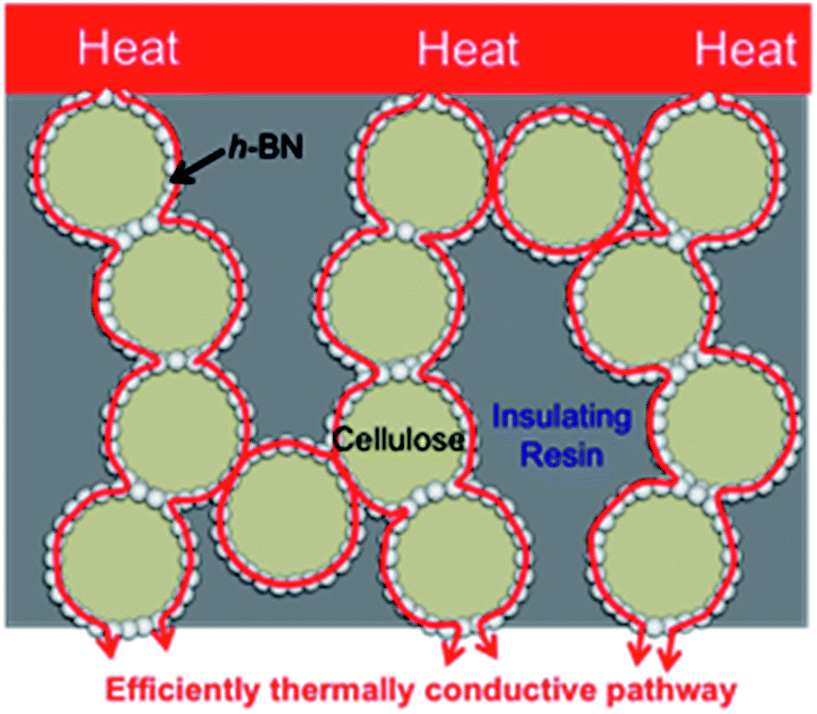
Boron nitride (BN) is a common thermally conductive material. It can adopt various structures such as hexagonal (h-BN), cubic (c-BN), rhombohedral (r-BN), and turbostratic (t-BN).19 For h-BN, while its thermal conductivity along its c-axis (thickness) is only 1–2 W m−1 K−1, along its a- and b-axises (in-plane) its thermal conductivity is more than 600 W m−1 K−1.20 In the current study, we investigated the use of h-BN as a thermally conductive material. We (1) used core–shell spherical microbeads as a thermally conductive filler; (2) developed these cellulose/h-BN core–shell spherical microbeads using phase separation of a cellulose xanthate aqueous solution (viscose). Cellulose is reportedly hybridized with inorganic materials such as TiO2,21,22 diamond, SiC, Al2O3, CeO2 (ref. 23) and various pigments;24 (3) hybridized the cellulose/h-BN core–shell microbeads with epoxy resin, using compression molding. This process reduced the required amount of h-BN. This is because the microbeads efficiently formed thermally conductive pathways among the shells in the insulating resin. This is shown in Fig. 1.
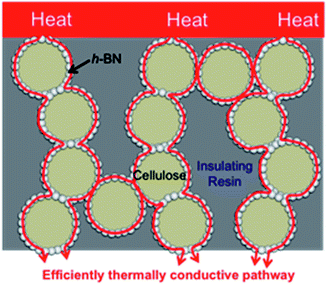 |
| | Fig. 1 Formation of thermal conductive network in resin sheet hybridized cellulose/BN core–shell microbeads. | |
2. Experimental
2.1. Materials
Cellulose/h-BN core–shell microbeads were prepared by the one-step phase separation of an aqueous solution of viscose (cellulose content 9.5 wt%; RENGO Co., Ltd., Osaka, Japan) and sodium polyacrylate (SPA; Aquaric DL-522, Mw = 170![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000, Nippon Shokubai Co., Ltd., Osaka, Japan). Two types of h-BN powder (BN-S and BN-L) were obtained from Denki Kagaku Kougyo Kabushiki Kaisha, Japan. Table 1 summarizes the characteristics of the h-BN powders and cellulose/h-BN core–shell microbeads. First, BN particles (BN-S and BN-L) were dispersed in an aqueous solution of sodium polyacrylate. Next, an aqueous solution of cellulose xanthate was added to the aqueous dispersion of BN particles and sodium polyacrylate. While stirring at 120 rpm, the suspension of BN and cellulose xanthate was heated to 80 °C over 1 h, then kept at 80 °C for 30 min. The obtained product was collected in filter cloth and washed with H2O. To remove sulfur from the microbeads, they were immersed in a stirred aqueous solution of 10 wt% HCl. The obtained microbeads were then washed with H2O and freeze dried.
000, Nippon Shokubai Co., Ltd., Osaka, Japan). Two types of h-BN powder (BN-S and BN-L) were obtained from Denki Kagaku Kougyo Kabushiki Kaisha, Japan. Table 1 summarizes the characteristics of the h-BN powders and cellulose/h-BN core–shell microbeads. First, BN particles (BN-S and BN-L) were dispersed in an aqueous solution of sodium polyacrylate. Next, an aqueous solution of cellulose xanthate was added to the aqueous dispersion of BN particles and sodium polyacrylate. While stirring at 120 rpm, the suspension of BN and cellulose xanthate was heated to 80 °C over 1 h, then kept at 80 °C for 30 min. The obtained product was collected in filter cloth and washed with H2O. To remove sulfur from the microbeads, they were immersed in a stirred aqueous solution of 10 wt% HCl. The obtained microbeads were then washed with H2O and freeze dried.
Table 1 Abbreviations of cellulose/BN microbeads and their loading ratios and contents of BN
| Cellulose/BN microbeads |
Loading ratio (wt%) |
Content (wt%) |
| BN-S: particle size 8 μm. BN-L: particle size 18 μm. |
| BN-Sa/Cell-1 |
30 |
27.5 |
| BN-S/Cell-2 |
50 |
44.9 |
| BN-Lb/Cell-3 |
70 |
67.7 |
2.2. Fabrication of composite sheet
The composite sheet was fabricated using a matrix of epoxy resin, namely 1,6-bis(2,3-epoxypropoxy)naphthalene (HP-4032; DIC Corp., Tokyo, Japan), and a filler of either the cellulose/h-BN core–shell microbeads or raw h-BN. We used a hardening agent of 1-cyanoethyl-2-ethyl-4-methylimidazole (2E4MZ-CN, Shikoku Chemicals Corp., Marugame, Japan). As summarized in Table 2, the epoxy resin, hardening agent, and fillers were mixed and deformed using a centrifugal stirrer under reducing pressure (Kakuhunter; Shashin Kagaku Co. Ltd., Tokyo, Japan). The mixture was then placed in a molding die with a volume of 1.0 cm3, and heated to 120 °C under a pressure of 2000 kg cm−2 using heating and compressing molding equipment (AH-10TD; As One, Tokyo, Japan). The contents of BN, cellulose and epoxy resin in the obtained sheets are summarized in Table S2.†
Table 2 Thermal conductivity of h-BN/epoxy resin composite sheet
| Sheet No. |
Filler |
Content of BN (wt%) |
Content of BN (vol%) |
Thermal conductivity (W m−1 K−1) |
In-plane direction/thickness direction |
| Filler type |
Thickness direction |
In-plane direction |
| BN content: 27.5 wt%. BN content: 44.9 wt%. BN content: 67.7 wt%. Measure temperature: 25 °C. |
| Sheet-1 |
BN-S |
39.9 |
27 |
1.20 |
2.0 |
1.66 |
| Sheet-2 |
63.3 |
49.0 |
2.74 |
4.43 |
1.62 |
| Sheet-3 |
80.2 |
69.3 |
4.30 |
10.1 |
2.35 |
| Sheet-4 |
BN-L |
19.6 |
12.0 |
0.70 |
0.60 |
0.85 |
| Sheet-5 |
57.1 |
42.6 |
2.40 |
4.50 |
1.88 |
| Sheet-6 |
84.4 |
75.0 |
6.31 |
22.9 |
3.62 |
| Sheet-7 |
BN-S/Cell-1a |
22.4 |
15.5 |
1.93 |
2.29 |
1.62 |
| Sheet-8 |
24.9 |
17.7 |
2.40 |
2.95 |
1.23 |
| Sheet-9 |
26.6 |
19.3 |
3.22 |
5.22 |
1.62 |
| Sheet-10 |
BN-S/Cell-2b |
31.6 |
22.1 |
1.78 |
2.27 |
1.28 |
| Sheet-11 |
39.3 |
29.3 |
4.17 |
5.30 |
1.27 |
| Sheet-12 |
42.5 |
32.5 |
4.65 |
7.77 |
1.67 |
| Sheet-13 |
BN-L/Cell-3c |
28.5 |
18.7 |
1.07 |
0.99 |
0.93 |
| Sheet-14 |
55.8 |
43.8 |
6.14 |
10.1 |
1.64 |
| Sheet-15 |
59.9 |
48.5 |
10.6 |
15.6 |
1.47 |
2.3. Characterization
The obtained microbeads were observed using field-emission scanning electron microscopy (SU-8000; Hitachi High Technologies, Co. Ltd., Tokyo, Japan). The zeta potential of the BN powder was measured using electrophoresis by the laser Doppler method (Zetasizer nano-ZS, Sysmex Corporation, Kobe, Japan). Thermal conductivity was measured using a thermal properties instrument by laser flashing (LFA-502, Kyoto Electronics Manufacturing Co. Ltd., Kyoto, Japan). The diameter and distribution of the cellulose/h-BN core–shell microbeads were measured using dynamic flow particle imaging (FPIA-3000, Malvern Instruments Ltd., Malvern, UK).
3. Results and discussion
3.1. Preparation of cellulose/h-BN core–shell microbeads
Cellulose/h-BN core–shell microbeads were prepared by viscose phase separation. The sphering mechanism of viscose phase separation was based on electrical repulsion between the dithio carbanion (CSS−) group of xanthate and the carbanion (COO−) of SPA. We previously reported that the distribution of inorganic particles in composite spherical microbeads prepared by viscose phase separation method depends on the zeta potential of the particles surface.21,22 The cellulose xanthate spherical droplet is produced by the electrostatic repulsion between the xanthate CSS−and SPA COO− groups. If the zeta potential of the inorganic particles in alkaline aqueous solution (viscose is a strong base) becomes negative and their charge distribution is narrow, the electrophoretic mobility of the inorganic particles becomes larger. This indicates that the inorganic particles become electrophoretic and will diffuse rapidly in alkaline aqueous solution. The inorganic particles diffuse toward the outside of the cellulose xanthate domain before the CSS moiety is eliminated by heat. The inorganic particles are also driven out as a result of the poly acrylic anion (COO−) negative charge around the cellulose xanthate domain. Therefore, the inorganic particles are located at the interface of the cellulose xanthate and poly acrylic anion domains, and are distributed on the surface of the obtained cellulose sphere. In contrast, if the zeta potential of the inorganic particles spans a wide positive–negative range, then the particles will tend to aggregate, which decreases their diffusion speed in the cellulose xanthate domain. Elimination of the CSS moiety and solidification of the cellulose occurs before inorganic particles move out of the cellulose xanthate domain. Thus, the inorganic particles become trapped within the cellulose sphere. The zeta potential of the current h-BN particles (BN-S) was −27.3 mV in the pH 13 aqueous solution. However, it was impossible to detect the distribution of electrophoretic mobility because the BN-S particles were too large to move in the aqueous solution. As such, we consider the electrophoretic mobility to have been very small. Phase separation between viscose and SPA occurred because intermolecular hydrogen bonding occurred among cellulose hydroxyl groups, as the CSS− moiety was eliminated from the cellulose xanthate upon heating. The distribution of inorganic particles in a cellulose sphere depends on their mobility in viscose and the CSS− elimination rate. To distribute the inorganic particles on the surface of the cellulose sphere, the inorganic particles must diffuse to the interface of the cellulose xanthate and the polyacrylic anion domains before the CSS moiety is eliminated. If the conversion from cellulose xanthate to cellulose occurs with the elimination of CSS− before the inorganic particles diffuse out during sphering, they remain within the cellulose sphere when the cellulose xanthate domain solidifies.21,22
Thus, we investigated the conditions for preparing microbeads with cellulose cores and h-BN particle shells. We examined two phase separation methods, one in which BN was dispersed in a cellulose xanthate aqueous solution (procedure-1), and another in which BN was dispersed in a SPA aqueous solution (procedure-2). These two procedures are described in the ESI, and are shown in Fig. S1.† In procedure-1, the cellulose xanthate aqueous solution, containing BN particles was added to the SPA aqueous solution, as reported previously.21–24 In procedure-2, the cellulose xanthate aqueous solution was added to the BN-S particle-SPA aqueous dispersion. Table S1† summarizes the loading ratios and contents of h-BN and cellulose. The detailed preparation conditions of the spherical microbeads are described in the ESI.† Fig. 2a and b shows SEM images of the cellulose/h-BN composite spherical microbeads obtained by procedure-1 and procedure-2, respectively. As expected for procedure-1, BN-S particles were involved in the cellulose sphere, similar to previous results concerning TiO2 and Al2O3.22,23 We attribute this result to the fact that solidification of the cellulose xanthate domain occurred with elimination of CSS−, unless the BN-S particles fully diffused to the interface between the cellulose and polyacrylic anion domains.
 |
| | Fig. 2 SEM images of cellulose/BN-S spherical microbeads prepared by procedure-1 (a) and -2 (b). (a) BN-S/Cell-0 (BN content: 41.6 wt%). (b) BN-S/Cell-1 (BN content: 44.9 wt%). | |
For procedure-2, the resulting microbeads were spherical and their surfaces were covered with h-BN particles, as shown in the SEM images of BN-S/Cell-1 in Fig. 2b. This indicates that BN-S was located on the surfaces of the cellulose spheres. They diffused to the area around the cellulose domain from the polyacrylic anion domain during sphering because of their electrostatic repulsion by polyacrylic anions. We prepared microbeads with cellulose cores and h-BN particle shells by phase separation of cellulose xanthate with BN dispersed in SPA aqueous media. This is shown in Fig. 3a–c for BN-S/Cell-1, BN-S/Cell-2, and BN-L/Cell-3, respectively. Table 1 summarizes the characteristics of the h-BN powder and cellulose/h-BN core–shell microbeads.
 |
| | Fig. 3 SEM images of three types of cellulose/h-BN core–shell microbeads. (a) BN-S/Cell-1 (BN content: 27.5 wt%), (b) BN-S/Cell-2 (BN content: 44.9 wt%), (c) BN-L/Cell-3 (BN content: 67.7 wt%). | |
3.2. Fabrication of composite sheets and their properties
The composite sheets were fabricated using heating and compression molding. The sheets were formed from a matrix of epoxy resin and a filler of either naked h-BN or the obtained cellulose/h-BN core–shell microbeads, as summarized in Table 2. The thermal conductivity of the fabricated sheets was lower in the absence of compression because of void formation. Compression helped ensure that voids did not form, and that shells were in close contact with each other. Fig. 4a shows cross-sectional SEM images of the composite sheets filled with naked h-BN. Fig. 4b show cross-sectional SEM image and EDX map of the composite sheets filled with cellulose/h-BN core–shell microbeads. Fig. 4a shows that the composite sheet fabricated using naked h-BN (sheet-3) had a layered structure. In contrast, the composite sheet fabricated using BN-S/Cell-1 (sheet-9) had a “stone-wall” structure. An increased contact area between shell layers of composite sheet was observed following compression. The h-BN particles on the boundaries of the core–shell microbeads were distributed because of the presence of nitrogen atoms, as shown in Fig. 4b. This distribution generated the stone-wall structure, in which the microbeads stacked three-dimensionally within the sheet.
 |
| | Fig. 4 Cross sectional images of h-BN/epoxy resin composite sheet. (a) Cross-sectional SEM image of sheet 3 (BN content: 69.3 vol%). (b) Cross-sectional SEM image and EDX map of sheet-9 (BN content: 19.3 vol%), green dots: N, red dots: C. | |
Fig. 5a and b shows the thermal conductivity as a function of h-BN content in h-BN/epoxy resin composite sheets filled with cellulose/h-BN (BN-S) core–shell microbeads and naked h-BN (BN-S), respectively. For the naked h-BN filler, the thermal conductivity of the composite sheet did not increase, unless the h-BN content was greater than the percolation threshold of 30 vol%. This was consistent with percolation theory.14–16 The composite sheet filled with cellulose/h-BN core–shell microbeads, including BN/Cell-1 and BN/Cell-2, showed much sharper increases in thermal conductivity in both the thickness and in-plane directions, with increasing h-BN content. The thermal conductivities of the composite sheets were estimated from the thermal conductivity curves in Fig. 5a and b. Those of the composite sheets filled with microbeads were much greater than that of the composite sheet filled with naked h-BN. Table 2 summarizes the thermal conductivities of the h-BN/epoxy resin composite sheets. The composite sheet containing 27.0 vol% naked h-BN sheet (sheet-1) exhibited a thermal conductivity in the thickness direction of only 1.2 W m−1 K−1. In contrast, the sheet filled containing 29.3 vol% cellulose/h-BN core–shell microbeads (sheet-11) exhibited a thermal conductivity in the thickness direction of 4.17 W m−1 K−1. This was approximately four times greater than that of the sheet filled with naked h-BN. The thermal conductivity of sheet-11 was comparable to that of the composite sheet filled with 69.3 vol% naked h-BN (sheet-3): 4.30 W m−1 K−1. The thermal conductivity of sheet-11 along the in-plane direction was 5.30 W m−1 K−1. We investigated the effect of the core–shell structure by comparing the thermal conductivities of sheet-9 (BN/Cell-1 filler) and sheet-10 (BN/Cell-2 filler). Although BN/Cell-1 had a much lower BN content than BN/Cell-2, sheet-9 had a much higher thermal conductivity than sheet-10. This indicates that the volume content of the BN/Cell-2 core–shell microbeads in sheet-10 (62.9 vol%) was sufficiently small that it could not form a complete, continuous thermally conductive pathway (95.8 vol% of the sheet volume). In contrast, the filler in BN/Cell-2 could form a complete, continuous thermal conductive pathway. This was because the occupied volume of the BN/Cell-1 core–shell microbeads in sheet-9 (95.8 vol%) was sufficient. Fig. 6 shows EDX maps of nitrogen and carbon atoms in the cross-sections of sheet-9 and sheet-10. Sheet-9 exhibited a more continuous thermal conductive pathway. BN-S/Cell-1 and BN-S/Cell-2 were incorporated into sheet-9 and sheet-10, respectively. As summarized in Table 2, the BN content of BN-S/Cell-2 was more than that of BN-S/Cell-1. The EDX cross-sections showed more N atoms in sheet-10 than in sheet-9. This was because the particle size of BN-S/Cell-2 was larger (mean particle diameter: 34 μm), and the shell of BN-S/Cell-2 was twice as thick as that of BN-S/Cell-1.
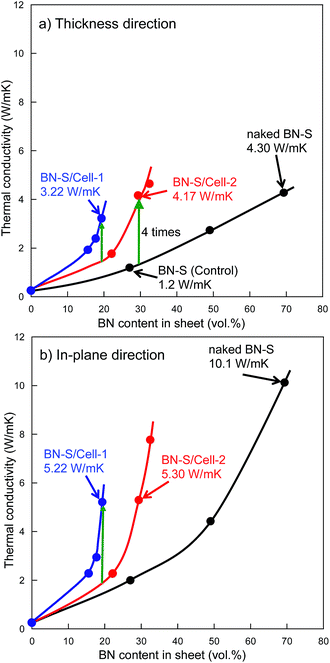 |
| | Fig. 5 Thermal conductivity against BN content in sheet. (a) Thickness direction, (b) in-plane direction. ( ): Sheet fabricated using BN-S/Cell-1. ( ): Sheet fabricated using BN-S/Cell-1. ( ): Sheet fabricated using BN-S/Cell-2. ( ): Sheet fabricated using BN-S/Cell-2. ( ): Sheet fabricated using naked BN-S. ): Sheet fabricated using naked BN-S. | |
 |
| | Fig. 6 Cross sectional EDX maps of composite sheet. Green dots: N, red dots: C. (a) Sheet-9 prepared using BN-S/Cell-1 (BN content: 19.3 vol%). (b) Sheet-10 prepared using BN-S/Cell-2 (BN content: 22.1 vol%). | |
Increasing the particle size of BN in a composite sheet also increases the sheet's thermal conductivity.25 We attempted using fillers containing larger BN particles to fabricate sheets with higher thermal conductivity, sheet-13, sheet-14, and sheet-15 contained the filler of BN-L/Cell-3 (BN size: 18 μm). Core–shell microbeads (BL-L/Cell-3) using BN-L were prepared, as shown in Fig. 3. The surface of BL-L/Cell-3 was covered with h-BN particles, similar to the surface of BN-S/Cell-2. As shown in Fig. 7, the thermal conductivities of the composite sheet with naked BN-L particles (sheet-6) in the thickness and in-plane directions were 6.31 and 22.9 W m−1 K−1, respectively. In contrast, the thermal conductivities of the composite sheet with BL-L/Cell-3 in the thickness and in-plane directions were 10.6 and 15.6 W m−1 K−1, respectively. Sheet-15 had a higher thermal conductivity in the thickness direction (10.6 W m−1 K−1) than sheet-6 (6.31 W m−1 K−1), despite the h-BN content of sheet-15 being only 48.5 vol%. Sheet-15 had the highest thermal conductivity in the thickness direction among all the sheets. The large difference in thermal conductivity between the thickness and in-plane directions arises from the anisotropic structure of h-BN. As shown by the results of naked BN-S and BN-L in Table 2, the difference between the conductivity in the thickness and in-plane directions increased with increasing h-BN content and thermal conductivity. The conductivities along the in-plane direction of sheets filled with core–shell microbeads became smaller than those of sheets filled with naked h-BN, as evidenced by comparing sheets with comparable thermal conductivities in the thickness direction: sheet-8 versus sheet-2, sheet-12 versus sheet-3, sheet-14 versus sheet-6. This behavior indicates that the anisotropy of crystalline h-BN in the insulating resin was suppressed in the composite sheet. This was because the h-BN particles were distributed around the cellulose spheres. Furthermore, the changes in thermal conductivity with h-BN content for the microbead-filled sheets were inconsistent percolation theory. The sheets filled with core–shell microbeads were several times more thermally conductive than the sheets filled with naked h-BN.
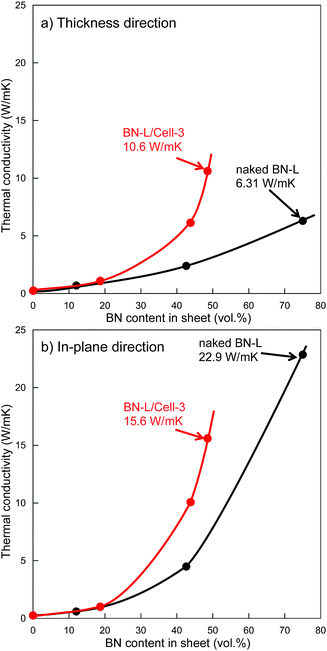 |
| | Fig. 7 Thermal conductivity against BN content in sheet. (a) Thickness direction, (b) in-plane direction. ( ): Sheet fabricated using BN-L/Cell-3. ( ): Sheet fabricated using BN-L/Cell-3. ( ): Sheet fabricated using naked BN-L. ): Sheet fabricated using naked BN-L. | |
Table S2† summarizes the physical properties of all the composite sheets, including their gravities. The gravity of the composite sheet increased with increasing BN content (gravity: 2.26 g cm−3), thus the composite sheets became heavier. For example, the thermal conductivities of sheet-6 and sheet-14 in the thickness direction were comparable, but their gravities were 1.97 and 1.74, respectively. A thermal conductive sheet of lower gravity is one advantage of using core–shell microbeads as the filler.
4. Conclusions
We prepared cellulose microbeads covered with h-BN particles by viscose phase separation. We fabricated composite sheets filled with naked h-BN or core–shell composite microbeads by compression. Incorporating 48.5 vol% of 18 μm-diameter h-BN into the resin sheet increased its thermal conductivity in the thickness and in-plane directions increased to 10.6 and 15.6 W m−1 K−1, respectively. This sheet had a comparatively low h-BN content of 48.5 vol%, yet its thermal conductivity in the thickness direction was the highest of all prepared sheets. This indicates that thermally conductive three-dimensional networks were formed. The composite sheet fabricated with 75.0 vol% of naked h-BN exhibited a thermal conductivity in the thickness and in-plane directions of 6.31 and 22.87 W m−1 K−1, respectively. We attributed the large difference in the conductivities in different directions to the anisotropic structure of h-BN. However, this difference was smaller when core–shell microbeads were used as the filler. This indicates that the anisotropy of crystalline h-BN was suppressed because h-BN particles were located around the cellulose sphere. Furthermore, changes in the thermal conductivity with changing h-BN content were inconsistent with percolation theory. The fabricated thermally conductive sheets had a much higher thermal conductivity than those fabricated with naked h-BN. This indicates that favorable thermal conductive pathways were formed in the insulating resin. Given the high filler cost, these findings represent a more cost-effective production of high thermal conductivity sheets.
Acknowledgements
This study was supported by JSPS KAKENHI (grant no. 24350102). We thank Mr Nobuo Sonoda, Mr Takayuki Otsuka and Mr Hideaki Matsuda of Ogic Technologies Co., Ltd., for detailed discussions. We also thank Mrs Erika Tsuda of Kumamoto University for preliminary experiment of cellulose/Dia core–shell microbeads.
Notes and references
- Y. Xu, D. D. L. Chung and C. Mroz, Comp. Biochem. Physiol., Part A: Mol. Integr. Physiol., 2001, 32, 1749–1757 Search PubMed
 .
. - Y. Hwang, M. Kim and J. Kim, RSC Adv., 2014, 4, 17015–17021 RSC .
- R. Qian, J. Yu, C. Wu, X. Zhai and P. Jiang, RSC Adv., 2013, 3, 17373–17379 RSC .
- J. Jaramillo-Fernandez, J. Ordonez-Miranda, E. Ollier and S. Volz, Phys. Chem. Chem. Phys., 2015, 17, 8125–8137 RSC .
- K. Sato, H. Horibe, T. Shirai, Y. Hotta, H. Nakano, H. Nagai, K. Mitsuishi and K. Watari, J. Mater. Chem., 2010, 20, 2749–2752 RSC .
- L. Fang, C. Wu, R. Qian, L. Xie, K. Yanga and P. Jiang, RSC Adv., 2014, 4, 21010–21017 RSC .
- G. Droval, J. F. Feller, P. Salagnac and P. Glouannec, Polym. Adv. Technol., 2006, 17, 732–745 CrossRef CAS .
- B. H. Xie, X. Huang and G. J. Zhang, Compos. Sci. Technol., 2013, 85, 98–103 CrossRef CAS .
- J. Hou, G. Li, N. Yang, L. Qin, M. E. Grami, Q. Zhang, N. Wang and X. Qu, RSC Adv., 2014, 4, 44282–44290 RSC .
- H. Liem and H. S. Choy, Solid State Commun., 2013, 163, 41–45 CrossRef CAS .
- H. L. Lee, O. H. Kwon, S. M. Ha, B. G. Kim, Y. S. Kim, J. C. Won, J. Kim, J. Han Choi and Y. Yoo, Phys. Chem. Chem. Phys., 2014, 16, 20041–20046 RSC .
- R. F. Hill and P. H. Supancic, J. Am. Ceram. Soc., 2002, 85, 851–857 CrossRef CAS .
- Y. X. Fu, Z. X. He, D. C. Mo and S. S. Lu, Appl. Therm. Eng., 2014, 66, 493–496 CrossRef CAS .
- D. Yorifuji and S. Ando, J. Mater. Chem., 2011, 21, 4402–4407 RSC .
- R. A. Hauser, J. M. Keith, J. A. King and J. L. Holdren, J. Appl. Polym. Sci., 2008, 110, 2914–2923 CrossRef CAS .
- H. He, R. Fu, Y. Hun, Y. Shen and X. Song, J. Mater. Sci., 2007, 42, 6749–6754 CrossRef CAS .
- T. Zhou, X. Wang, G. U. Mingyuan and X. Liu, Polymer, 2008, 49, 4666–4672 CrossRef CAS .
- F. H. Gojny, M. H. G. Wichmann, B. Fiedler, I. A. Kinloch, W. Bauhofer, A. H. Windle and K. Schulte, Polymer, 2006, 47, 2036–2045 CrossRef CAS .
- T. Oku, K. Hiraga, T. Matsuda, T. Hirai and M. Hirabayashi, Diamond Relat. Mater., 2003, 12, 1138–1145 CrossRef CAS .
- H. B. Cho, T. Nakayama, Y. Tokoi, S. Endo, S. Tanaka, T. Suzuki, W. Jiang, H. Suematsu and K. Niihara, Compos. Sci. Technol., 2010, 70, 1681–1686 CrossRef CAS .
- S. Nagaoka, Y. Hamasaki, S. Ishihara, M. Nagata, K. Iio, C. Nagasawa and H. Ihara, J. Mol. Catal. A: Chem. A., 2002, 177, 255–263 CrossRef CAS .
- S. Nagaoka, K. Arinaga, H. Kubo, S. Hamaoka, T. Sakurai, M. Takafuji and H. Ihara, Polym. J., 2005, 37, 186–191 CrossRef CAS .
- S. Nagaoka, K. Hirakawa, S. Kobayashi, K. Satoh, M. Nagata, M. Takafuji and H. Ihara, Kobunshi Ronbunshu, 2008, 65, 80–89 CrossRef CAS .
- S. Nagaoka, M. Nagata, K. Arinaga, K. Shigemori, M. Takafuji and H. Ihara, Color. Technol., 2007, 123, 344–350 CAS .
- M. Tanimoto, T. Yamagata, K. Miyata and S. Ando, ACS Appl. Mater. Interfaces, 2013, 5, 4374–4382 CAS .
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c6ra02950g |
|
| This journal is © The Royal Society of Chemistry 2016 |




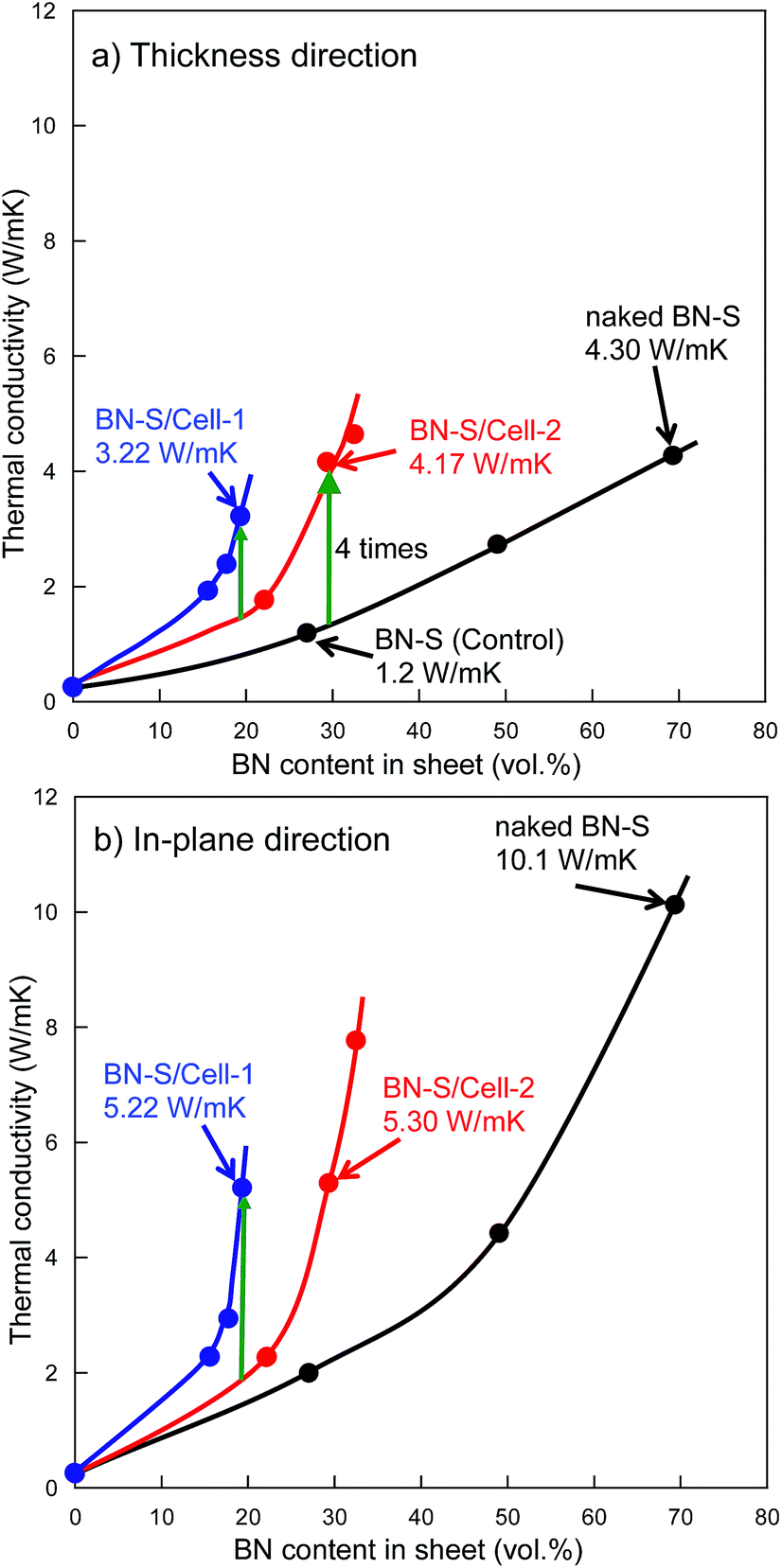
 ): Sheet fabricated using BN-S/Cell-1. (
): Sheet fabricated using BN-S/Cell-1. ( ): Sheet fabricated using BN-S/Cell-2. (
): Sheet fabricated using BN-S/Cell-2. ( ): Sheet fabricated using naked BN-S.
): Sheet fabricated using naked BN-S.
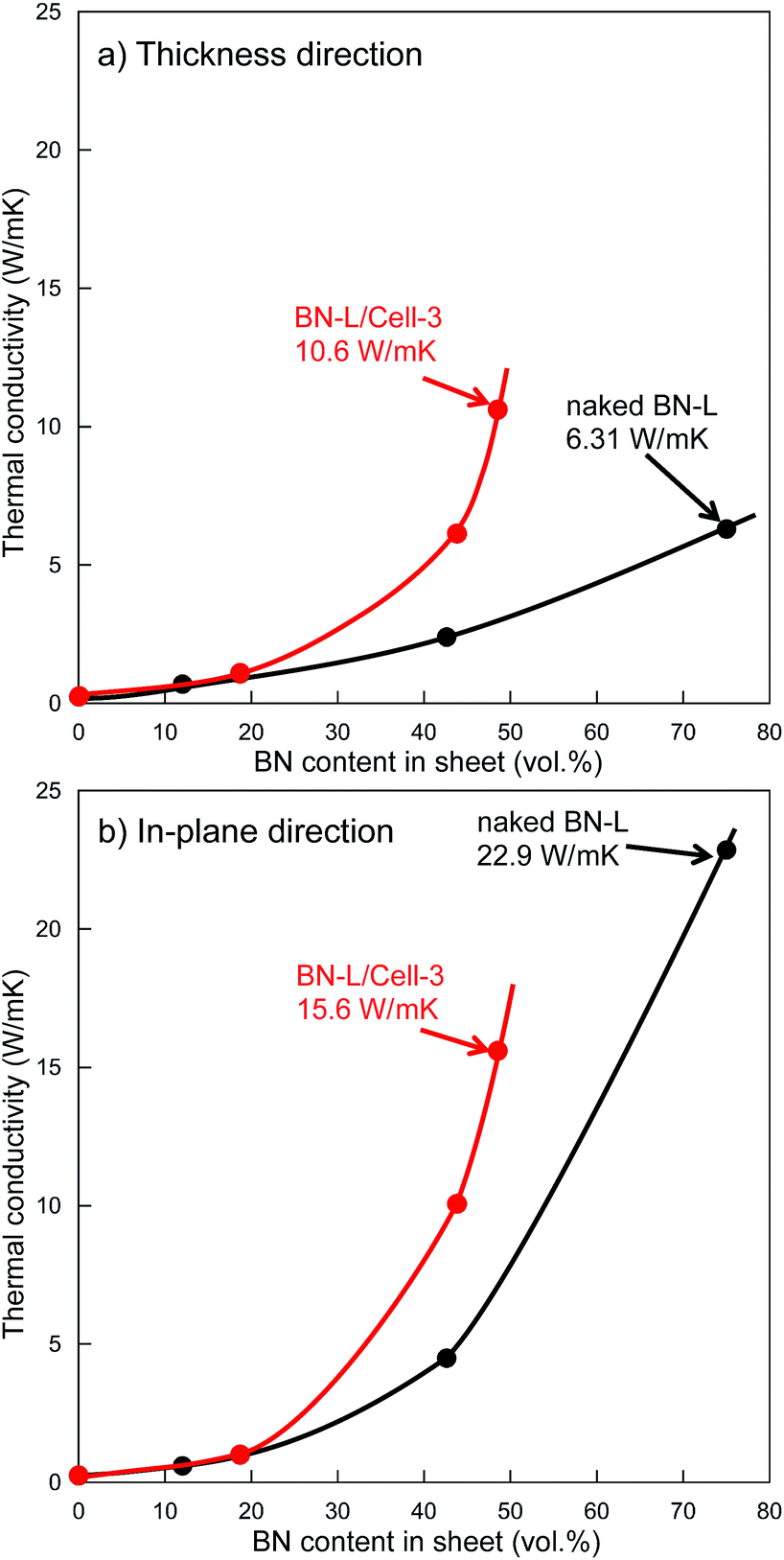
 ): Sheet fabricated using BN-L/Cell-3. (
): Sheet fabricated using BN-L/Cell-3. ( ): Sheet fabricated using naked BN-L.
): Sheet fabricated using naked BN-L.