 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Nanocomposite films and coatings using inorganic nanobuilding blocks (NBB): current applications and future opportunities in the food packaging sector
Ilke
Uysal Unalan
a,
Guido
Cerri
b,
Eva
Marcuzzo
c,
Carlo A.
Cozzolino
a and
Stefano
Farris
*a
aDeFENS, Department of Food, Environmental and Nutritional Sciences—Packaging Division, University of Milan, Via Celoria 2, 20133 Milan, Italy. E-mail: stefano.farris@unimi.it; Fax: +39 0250316672; Tel: +39 0250316654
bDepartment of Natural and Territorial Science, University of Sassari, via Piandanna 4, 07100 Sassari, Italy
cDepartment of Food Science, University of Udine, via Sondrio 2/A, 33100 Udine, Italy
First published on 17th June 2014
Abstract
The aim of this review is to provide an in-depth overview on the use of inorganic nano-sized entities for the generation of nanocomposite materials in the form of films and coatings for food packaging applications. According to recent trends toward “green” strategies, special focus has been dedicated to the development of nanocomposite coatings obtained using biopolymers as the main polymer matrix. After a first introductive part, the discussion has been addressed to the use of inorganic fillers, metals and metal-oxides, zeolites, and graphene. For each class of filler, a first ‘in-depth’ description of the most relevant physicochemical properties for the food packaging sector has been followed by case-by-case references to recent developments and envisaged implementations. The technical aspects that may be crucial in the design and end use of (bio)nanocomposite coatings have been covered in the last part of this work, which also includes an updated list of current applications on nano-sized inorganic fillers in the food packaging field.
 Ilke Uysal Unalan | Ilke Uysal Unalan received her M.S. in Food Engineering from Izmir Institute of Technology, Turkey, in 2008 discussing a thesis on active food packaging by edible films. In March–June, 2010 she was a Federation of European Microbiological Societies (FEMS) research fellow at the National Food Institute, Technical University of Denmark (DTU), in the Prof. P. Dalgaard's group, where she worked on shelf life studies based on modified atmosphere packaging strategies. She has been doing her PhD since January 2012 in the Food Packaging lab under the guidance of Dr S Farris. Her research topic deals with the design and development of graphene–biopolymer nanocomposite films and coatings for food packaging applications. From July 2013 till May 2014 she has been a visiting research fellow at the University of Warwick, UK, in the Nanocomposite Research Group of WMG led by Prof. T. McNally, under the supervision of Dr Chaoying Wan. |
 Carlo A. Cozzolino | Carlo Alessio Cozzolino received his M.S. in Agricultural Science & Technology in 2005 from the University of Sassari, Italy. In 2008 he joined the Food Packaging lab led by Prof. L. Piergiovanni at the University of Milan as a fellowship holder granted by the Government of Sardinia (September 2008–October 2009). In 2013 he earned his PhD in Food and Microbial Biotechnology at the University of Milan (granted by the University of Sassari), under the supervision of Dr S. Farris. His thesis defense focused on the development of an active packaging from biomacromolecules for the controlled release of natural antimicrobials. At present, he holds a research position at the Metalvuoto Spa Company, while keeping ongoing collaborations with the Food Packaging lab. |
 Stefano Farris | Stefano Farris received his M.S. in Agricultural Science & Technology from the University of Sassari, Italy, in 2004. In 2007 he earned his PhD in Food and Microbial Biotechnology. From 2007 to 2008 he was a postdoctoral fellow at Rutgers University (NJ) in the Food Packaging lab led by Prof. K. Yam, where he worked on the development of films and coatings from renewable resources. In 2011 he joined Prof. M. Hedenqvist's group at KTH, the Royal Institute of Technology in Stockholm, in the Department of Fibre and Polymer Technology. There, he worked on the development of hybrid materials by means of in situ polymerization through sol–gel approaches. At present he is Assistant Professor in the Department of Food, Environmental and Nutritional Sciences (DeFENS) at the University of Milan, Packaging Lab. His current research activity is mainly devoted to the development and deposition of high-performance coatings. |
1. Introduction
From the 60's to date, the concept of nanotechnology has developed into a multidisciplinary field of fundamental and applied science and technology. Over the last decade, in particular, nanotechnology has become one of the most appealing topics within the research programs of both public agencies (e.g., universities) and private companies. At least by researchers, nanotechnology is generally deemed as an exciting area in which to work, allowing creativity in obtaining new materials with deliberately induced nano-sized architectures. Eventually, this field will yield new and innovative features and functions that can be profitably exploited in everyday life. Some authors, however, have expressed some skepticism about nanotechnology, the main question being whether it is a real opportunity or just a hype to extract funds.1Although nanomaterials and nanoparticles have been consumed by humans for centuries, consumers' perceptions toward nanotechnology are not totally agreeable, mainly due to the still-existing uncertainties concerning the potential risks for humans and the environments associated with the use of nanomaterials for sensitive applications, such as many areas of the food industry, including packaging, nutrient delivery, and food quality.2 Nevertheless, in 2008 nanotechnology involved over $15 billion in worldwide research and development money (public and private) and employed over 400![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 researchers across the globe.3 Moreover, nanotechnologies will likely impact at least $3 trillion across the global economy by 2020, and worldwide nanotechnology industries may require at least 6 million workers to support them by the end of the decade.3 Despite the still open debate about nanotechnology, it is not rash to say that the world is in the middle of a nano-revolution.
000 researchers across the globe.3 Moreover, nanotechnologies will likely impact at least $3 trillion across the global economy by 2020, and worldwide nanotechnology industries may require at least 6 million workers to support them by the end of the decade.3 Despite the still open debate about nanotechnology, it is not rash to say that the world is in the middle of a nano-revolution.
The food industry seems to receive the largest benefits from nanotechnology, with potential uses already identified in virtually every segment of the sector (i.e., agriculture, food processing, food packaging, and nutrient supplements).4 Moreover, a number of the world's largest food companies are actively exploring the potential of nanomaterials for use in food applications.5 Within the food industry, however, packaging applications form the largest share of the current and short-term predicted market for nano-enabled products,6 with an estimated annual growth rate of ∼12% by 2014.7 One of the reasons for this trend is the consumers' willingness to embrace nanotechnology in “out-of-food” applications more than the direct addition of nanoparticles into the food.8,9 This is also why most nanotechnology applications for food and beverages are lagging behind applications for food packaging, which are already becoming a commercial reality.10
One of the most successful applications of nanotechnology in the field of packaging concerns the development of “nanocomposites”.11 In polymer science, while the term “composites” generally refers to mixtures of polymers with inorganic or organic additives having micron-length scale and certain geometries (fibers, flakes, spheres, and particulates), the use of nano-length-scale entities can be more specifically referred to as “nanocomposites.” These entities are well-defined nanobuilding blocks (NBB), which consist of preformed objects that keep their integrity in the final nanocomposite materials.12 These NBB can be clusters, nano-core shells, organically pre- or post-functionalized nanoparticles (e.g., metal oxides), and layered compounds (e.g., clays).13 The preferential route to produce nanocomposites lies in the “top-down” approach, which progressively reduces the size of nanocomposite bulk materials using grinding, milling, etching, lithography, or precision engineering.14 Nanocomposites represent a radical alternative to conventional polymer composites, because if properly manipulated, they can offer extra benefits such as low density, transparency, good flow, better surface properties, and recyclability, even at very low filler contents (generally lower than 5 wt%).15–18
A new subclass of nanocomposite materials has recently stemmed from the increasing endeavor to replace oil-based polymers with polymers partially or totally obtained from renewable resources. The term “bionanocomposites” refers to those materials in which the polymer matrix carrying the nano-sized fillers is a biopolymer (e.g., polysaccharides and proteins) or by a polymer of natural origin obtained by synthetic (e.g. poly-lactic acid, PLA) or biotechnological (pullulan, poly-hydroxyalkanoates–PHAs) routes. The use of NBB for the generation of bionanocomposites has enormous potential for overcoming the drawbacks exhibited by natural polymers, such as poor mechanical and thermal properties, sensitivity to moist environments, and inadequate barrier properties to gas and vapors; the development of bionanocomposites has thus drawn considerable attention in recent years. However, most examples concern the incorporation of the inorganic phase directly into the bulky biopolymer. Only very recently it has been proposed the use of fillers within coatings made of biopolymers to produce bionanocomposite coatings (i.e., thin layers of a biopolymer matrix loaded with a nanoparticle filler) to improve the properties of a plastic substrate without jeopardizing its original attributes and optimize cost efficiency.19–22
We conceived this review as guidance for tomorrow's research in the field of nanocomposite materials, especially in the form of coatings specifically intended for food packaging applications, with special emphasis on those solutions envisaging the use of biopolymer matrices. First, we provide a brief introduction, including key definitions and classifications. Second, we present an in-depth description of inorganic fillers with special focus to the following: inorganic clays (e.g. montmorillonite), which are the most widely used for food packaging purposes; metals and metal oxides, especially those obtained by sol–gel routes that have gained much attention for food packaging applications due to their high versatility; zeolites, which despite having many appealing features, are still under-exploited fillers; and graphene, which is one of the “fillers of the future”, owing to its promising and attractive properties for the next generation of materials—not only for packaging applications. Besides discussing the most relevant physicochemical properties, a detailed description of current developments for the food packaging sector is also reported for each class of filler. Among other aspects, in the final “technical” section, we first thoroughly discuss the processing of inorganic fillers within (bio)polymer matrices to obtain (bio)nanocomposite coatings. Successively, we describe the most important aspects that may impact the ultimate performance of (bio)nanocomposite films and coatings in light of their end use as food packaging materials. We also provide an outlook on future trends with the goal of prompting new research on (bio)nanocomposite coatings as potential alternatives to the currently used solutions. We have deliberately kept a discussion of the toxicological and regulatory aspects associated with the nano-dimension of the inorganic fillers out of the scope of this review, as these topics have been deeply addressed in other recent works.4,18,23–33
2. Classification of fillers
A first distinction within the huge field of fillers can be done according to their chemical composition. Accordingly, it is useful to distinguish between organic fillers, namely those containing carbon atoms and most often directly derived from biomass, and inorganic fillers, which may be also of natural origin but as a general rule do not consist of any carbon atoms. Graphene is an exception to this rule, as it is arbitrarily included within the inorganic filler class although it is only consists of carbon atoms. As both classes (organic and inorganic) encompass a wide assortment of examples, each including many different applications, it would be impossible to provide an exhaustive overview that takes into account all of them. Therefore, our focus will be only on the inorganic fillers.2.1 Inorganic fillers: physicochemical properties and main developments for food packaging applications
The use of inorganic nano-sized building blocks as fillers in the preparation of polymer/inorganic nanocomposites has attracted increasing interest in recent years, owing to their unique properties that find numerous applications in many industrial fields. As shown in Chart 1, a first arbitrary classification of inorganic fillers, primarily based on chemical composition and crystal structure, is between silicate and non-silicate minerals. Both families include several classes and many groups that, in turn, comprise different mineral species potentially suitable to produce nanocomposites. However, only few of these minerals have so far been exploited in practice. If, on one hand developers have thus far used layered silicates (e.g. clays) the most extensively as fillers to obtain nanocomposites,4,34–36 on the other hand, developers have introduced carbon-based nanofillers such as carbon black,37–39 expanded graphite,40–42 carbon nanotubes (CNTs),43–45 and carbon nanofibers46–48 in the preparation of polymer nanocomposites to achieve properties otherwise unattainable, such as electrical and thermal conductivity of the final nanocomposite material. Among these, CNTs have proven effective as conductive fillers.45,49–52 The only drawback of CNTs as a nanofiller is their relatively high production cost.43,51,53,54 Therefore, as Nicholos said,55 “When carbon fibers just won't do, but nanotubes are too expensive, where can a cost-conscious materials scientist go to find a practical conductive composite? The answer could lie with graphene sheets”.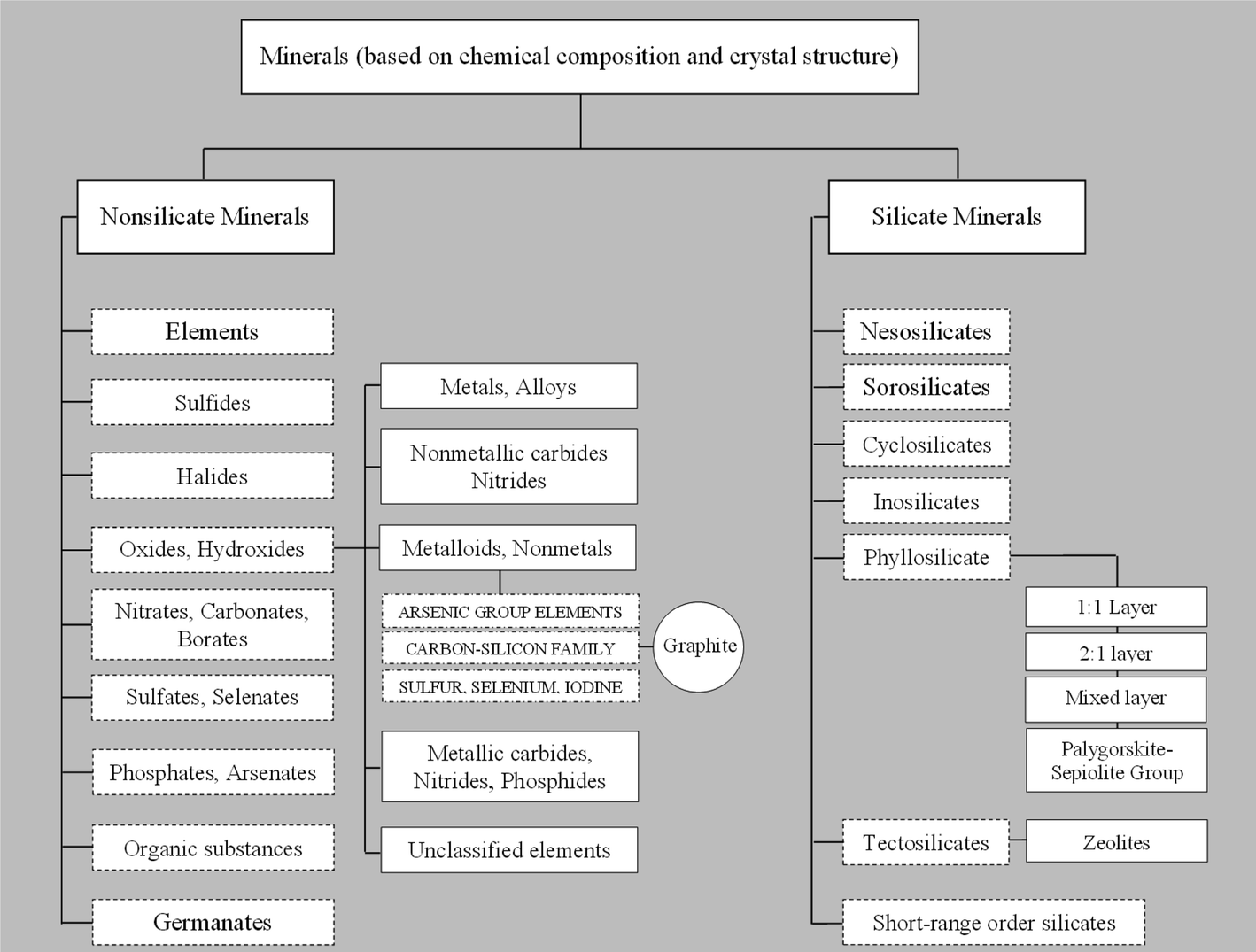 | ||
| Chart 1 Classification of inorganic fillers based on chemical composition and crystal structure. | ||
In this section, we first review the most widely used inorganic fillers, i.e. clays, with also a brief mention of other layered minerals of interest, i.e. micas and layered double hydroxides. Next, we discuss the current and potential use of zeolites, while the last part of this section will be dedicated to the rise of graphene as a promising candidate for the development of new advanced materials. We will place special emphasis on the structure–property relationships that eventually lead to the enhancement of material properties of interest for food packaging applications, such as barrier, mechanical and thermal properties.
:1 (T–O–T) and 1:1 (T–O) layered structures, where the sum of each sheet originates a layer (known as a platelet).57 In 2:1 layered structures, if isomorphic substitution of silicon and/or aluminum with a lower-valence atom takes place (either in the octahedral or tetrahedral sheets), the mineral is characterized by a negative surface charge, which, in some clay groups, will be naturally counterbalanced by positive inorganic ions (e.g. Li+, Na+, Ca2+, K+, Mg2+, etc.) in the interlayer. In these cases, the nature of the cations plays a crucial role in determining the swelling tendency of the clay upon water adsorption. As a general rule, the smaller is the cation and the lower is its charge, the more the clay swells. For example, in montmorillonite (a clay belonging to the smectite group), the swelling increases according with the following trend: Li+ > Na+ > Ca2+ > Fe2+ > K+.58 The K+ ion provides the lowest swelling capability because it stabilizes the structure due to the fact that, more than the other ions, its size well fits with the hosting size represented by the center of the hexagon defined by six Si-centered tetrahedrons.
The capability of layered silicates to exchange cations is quantified by the cation-exchange capacity (CEC), usually expressed as meq per 100 g. Ideally, the CEC of a clay mineral is equivalent to its (negative) layer charge. This parameter can greatly vary depending on the type and origin of the clays, as well as within the same clay, because the surface charge is locally randomly distributed (i.e. it may vary from layer to layer or within the same layer).59 The layers of mineral clays generally organize to form stacks with a regular van der Walls gap in between them called the “interlayer distance” or “gallery”, while the “d-spacing” (d001), also called the “basal spacing”, accounts for the sum of the interlayer and the platelet thickness (Fig. 1). When the hydrated cations are ion-exchanged with organic cations (e.g. more bulky alkyammoniums), which is normally done to increase the affinity towards more hydrophobic polymers such as polyolefins or PLA, the final result is a larger interlayer spacing, thus a larger “d” value. Three organization levels can be recognized within clays: (i) the layer (or platelet), with thickness of approximately 1 nm and length varying from a few hundreds of Å to several microns, depending on the type of clay; (ii) primary particle, composed of five to ten platelets stacked on top of one another; (iii) micro-aggregates (or tactoids), structures of approximately 0.1 to 10 μm formed through the association of randomly oriented several primary particles.60 One common feature of clays is the high aspect ratio (i.e., the ratio between length and thickness). Although this parameter can vary greatly depending on the type and origin of clays, it may be of the order of 1000.15
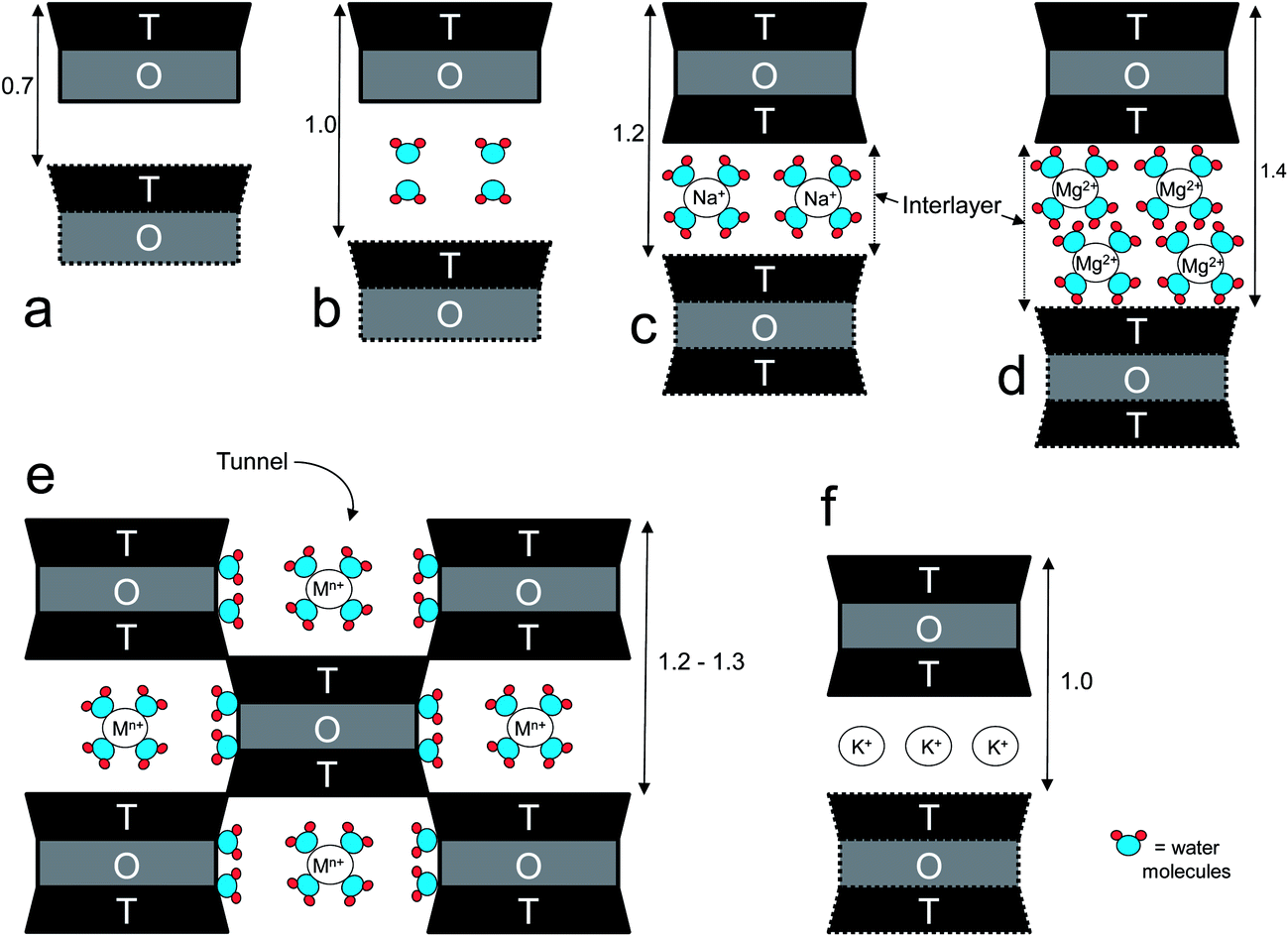 | ||
| Fig. 1 Schematic representation of some phyllosilicates viewed perpendicularly to the tetrahedral (T) and octahedral (O) sheets: (a) kaolinite; (b) halloysite; (c) Na+-montmorillonite (a smectite) partially hydrated; (d) Mg-vermiculite fully hydrated; (e) palygorskite–sepiolite structure; (f) muscovite (a flexible mica). Spacing in nm. Mn+ stands for a generic cation. | ||
Based on our bibliographic search, the four main groups of clay minerals of technological interest for the fabrication of nanocomposite materials are—in order of relevance for this field—smectite, kaolin, palygorskite–sepiolite, and vermiculite.
2.1.1.1 Smectite clays. The smectite group includes the most commonly used types of clays for the production of nanocomposites, due to their large availability, low cost and favorable technological attributes, such as ease of exfoliation, which results in optimal distribution in the polymer matrix.61–66 Smectites include 2
:1:1 (or T–O–T) hydrated clays primarily constituted of Si and Al and/or Mg. These clays consist of a central octahedral sheet containing Al3+, Fe3+, Mg2+ and Fe2+ sites coordinated to either oxygen anions and hydroxyl groups, sandwiched between two tetrahedral sheets constituted of Si4+, Al3+ and Fe3+ coordinated to oxygen anions arranged in a hexagonal fashion.67 The smectite group includes several types of clay minerals, among which montmorillonite, saponite, laponite, and hectorite have potential for the generation of nanocomposite materials. Although rectorite does not belong to the smectite group, it has arbitrarily been included in this section because it is partially constituted by montmorillonite layers.
2.1.1.1.1 Montmorillonite. Montmorillonite (MMT) (Fig. 2a) is the most widely exploited clay for the fabrication of polymer/clay nanocomposites.68 In MMT's structure, with a general formula (My+·nH2O)(Al2−yMgy)Si4O10(OH)2 (My+ refers to a generic monovalent interlayer cation),67 the negative surface charge coming from the partial substitution of the trivalent Al-cation in the octahedral gibbsite layer by the divalent Mg-cation is balanced by sodium and calcium ions, which exist hydrated in the interlayer.69 Because the layers are held together by relatively weak forces, water and other polar molecules can enter between the layers, causing the lattice to expand.70 A typical sodic MMT (Na+-MMT) is characterized by a d001 of 11.7 Å (at relative humidity values ∼50%), aspect ratio of 100–200, surface area of 700–800 m2 g−1, and CEC ranging between 70 and 120 meq per 100 g, depending on their extraction site.71,72
 | ||
| Fig. 2 TEM images of: (a) Na+-MMT; (b) REC (reproduced with permission from Elsevier Ltd191); (c) SAP (reproduced with permission from Elsevier Ltd197); (d) LAP (from http://www.laponite.com); (e) HEC (reproduced with permission from Elsevier Ltd220); (f) KAO (reproduced with permission from Elsevier Ltd224); (g) HNTs including tube cross-section in the inset (reproduced with permission from Elsevier Ltd230); (h) SEP (reproduced with permission from Elsevier Ltd263); (i) VMT (reproduced with permission from Elsevier Ltd272); (j) IMO (reproduced with permission from Elsevier Ltd280); (k) LDHs (reproduced with permission from Elsevier Ltd297); (l) synthetic mica (product name NTS-5, Topy Industries Limited, Tokyo, Japan). | ||
The development of oil-based polymer/MMT nanocomposites is well documented, and includes polymethylmethacrylate (PMMA),73 polyethylene terephthalate (PET),74–84 polypropylene (PP),85–99 ethylenvinylacetate (EVA),100–103 polyamide (PA),96,104–110 polyethylene (PE),90,100,111–121 polyethylenimine (PEI),122 polyvinylchloride (PVC),123,124 natural rubber latex,125 and polyurethane (PU),126–129 to provide some examples.
In addition to “single polymer matrix” nanocomposites, researchers have also proposed blends of two or more polymeric materials (i.e. binary or ternary blends) for the development of MMT-based nanocomposites with enhanced physicochemical properties ensuing from synergistic effects. Blends of modified PA and nylon-6,130 PET and aromatic PA,131 PP and PA 6,6,132 nylon-6 and ethylenevinylalcohol (EVOH)133 were reinforced with MMT for the improvement of barrier, thermal and mechanical properties. Developers have also used MMT to build multifunctional thin films by alternating two or more layers of anionic and cationic polymer aqueous solutions or dispersions; this process could theoretically include an infinite number of layers. For instance, one study described the film growth and structure of assemblies made with cationic PEI and anionic MMT clay and polyacrylic acid (PAA), referring to one deposition sequence of PEI/PAA/PEI/MMT as a quadlayer. The study asserted the resulting polymer nanocomposite thin films have unprecedented barrier performance, especially in terms of oxygen permeability.134
Due to the increasing concerns about environmental issues linked to the waste disposal of plastics at the end of the service, research has focused on the possibility of extending the technical benefits of nanocomposites to polymers of natural origin, with the main goal of overcoming the traditional shortcomings of biopolymers, such as sensitivity to water or humid environments, as well as poor barrier and mechanical properties. More specifically, because the food industry is among the sectors that takes advantage of poorly biodegradable petroleum-based plastic materials, the development of eco-friendly bionanocomposite films and coatings is one of the greatest challenges that has become imperative to this industry. Within the food packaging sector, several studies have reported on the improvement of the mechanical, thermal, barrier, active, optical, and water-resistance properties of several bionanocomposites based on MMT, as reported in Table 1. Although MMT clays have attracted much attention due to their undeniable advantages over other type of clays, researchers have also explored the use of other clays belonging to the smectite group to fabricate bionanocomposites for food packaging applications.
| Inorganic filler | Properties | ||||||
|---|---|---|---|---|---|---|---|
| Mechanical | Thermal | Barrier | Active (antimicrobial, antioxidant, controlled release) | Optical | Interaction with water (solubility and absorption) | ||
| P O2 | P H2O | ||||||
| a Please refer to the main text for abbreviations. b Rectorite, an ordered interstratified clay mineral, is inserted in the smectite group because partially constituted by montmorillonite layers. | |||||||
| A Clay minerals | |||||||
| 1. Smectite Group | |||||||
| MMT | Starch5,63,135–142 | PLA148,150,160,161 | PLA149,150,164,165 | Chitosan144,145,168 | Agar156 | Starch136 | Starch136 |
| Chitosan143–146 | Chitosan144,145 | Wheat gluten169,170 | Chitosan176,177 | Agar156 | Agar156 | ||
| Alginate147 | PCL162 | CA166 | Starch135,138,142,171,172 | WPI178 | MC175 | Wheat gluten173 | |
| PLA148–150 | CKG158 | Fish gelatin154 | SPI152,153 | PLA179 | Chitosan174 | ||
| CA62,151 | SPI152 | SPI153 | Fish gelatin154 | Paper coated with wheat gluten181 | MC175 | ||
| SPI152,153 | Pectin155 | Pectin155,167 | Alginate157 | Wheat gluten + carvacrol180 | |||
| Fish gelatin154 | Alginate163 | CMC159 | Pectin155 | CMC + murta leaves extract159 | |||
| Pectin155 | Chitosan143 | PLA–PCL | Agar156 | Chitosan + rosemary essential oil146 | |||
| Agar156 | PLA–PCL | Blend150 | PLA165 | PLA or PLA–PCL blend + cyclodextrin on cheese150 | |||
| PCL157 | Blend150 | CMC159 | GCG + thymol on chicken breast182 | ||||
| CKG158 | Alginate + Ag on carrots183 | ||||||
| CMC159 | |||||||
| PLA–PCL blend150 | |||||||
| Rectoriteb | Alginate186 | Alginate186 | Starch190 | Chitosan + drug189 | Alginate186 | Chitosan + chitin whisker192 | |
| Starch190 | Starch190 | Chitosan189 | Chitosan + chitin whisker192 | Chitosan + chitin whisker192 | |||
| Chitosan + chitin whisker192 | Chitosan + chitin whisker192 | Chitosan189 | |||||
| Chitosan + PVOH blend191 | |||||||
| Saponite | PBS202 | PBS202 | |||||
| Laponite | BC207 | BC207 | Starch–PVOH blend218 | MFC217 | |||
| Starch–PVOH blend218 | MFC217 | ||||||
| MFC217 | |||||||
| Hectorite | Starch66 | PLA222 | |||||
| PLA222 | |||||||
| 2. Kaolin Group | |||||||
| Kaolinite | Corn starch225 | Cassava starch226,227 | PHB–PCL | PHB–PCL blend and also for D-limonene223 | Cassava | Corn starch225 | |
| Potato starch64 cassava starch226,227 | PHB–PCL blend223 | Blend223 | Starch226,227 | Cassava starch226,227 | |||
| PLA–PCL | Wheat starch228 | PLA–PCL | Wheat starch228 | ||||
| Blend229 | PLA–PCL blend229 | Blend229 | |||||
| Halloysite | Bovine gelatin244 | Low methoxyl pectin and hydroxypropyl | Bovine gelatin244 | Potato starch245 | Bovine gelatin244 | ||
| Potato starch245 | Cellulose232 | PLA248,249 | PLA248,249 | Cellulose250 | |||
| PLA246–249 | PLA246–249 | ||||||
| Cellulose250 | Cellulose250 | ||||||
| 3. Palygorskite–sepiolite group | |||||||
| Sepiolite | Starch265 | PLA263 | Arabinoxylan267 | Alginate269 | Gelatin-egg white + clove | ||
| Gelatin-egg white266 | PLA and MWNTs264 | Alginate269 | Essential oil266 | ||||
| Arabinoxylan267 | Arabinoxylan267 | ||||||
| Alginate269 | PLA and PCL268 | ||||||
| PLA and PCL268 | |||||||
| 4. Vermiculite Group | |||||||
| Vermiculite | Chitosan270 | ||||||
| PLLA–PMMO–PEO blend275,276 | |||||||
| PLLA277–279 | |||||||
| B Other layered silicate | |||||||
| Mica group | Carrageenan306 | PLLA309 | Carrageenan306 | Carrageenan306 | |||
| PLLA308,310 | Chitosan307 | ||||||
| MC307 | |||||||
| PLLA308 | |||||||
| C Other layered minerals | |||||||
| LDH | PLLA289,295,297 | PLLA295,297 | Pectin + benzoate | Pectin + benzoate derivatives302 | |||
| Pectin + benzoate derivatives302 | γ—PGA298 | Derivatives302 | PCL + benzoate derivatives299–301 | ||||
| Pectin + benzoate derivatives302 | |||||||
| PCL + benzoate derivatives299–301 | PCL + benzoate derivatives299–301 | PLLA295,297 | |||||
| PCL375 | PCL375 | ||||||
2.1.1.1.2 Rectorite. Rectorite (REC) (Fig. 2b) is a yellowish-white layered silicate consisting of a regular interstratification of 2
:1 dioctahedral mica layers (see paragraph 2.2.2.2) and 2:1 dioctahedral smectite (MMT-like) layers. The structure and properties of REC thus fall between completely expanded MMT and completely collapsed mica. The proportion of smectite layers in a mixed-layer clay determines the clay's expandability. In REC, since the distribution of smectite and mica layers in a clay crystallite is ordered (or regular), the final properties are virtually in between those of smectite and mica.184 The presence of mono- and divalent charge-compensating exchangeable cations in the smectite expandable sheet makes it possible to prepare polymer/REC nanocomposites by solution-mixing or melt-mixing processing techniques.185 Yang et al. reported for Na+-REC a CEC ∼45 meq per 100 g and d-spacing of 22.1 Å,186 while Jinhong et al. calculated a CEC of 32.2 meq per 100 g and d-spacing of 19.47 Å.187 The Brunnuer, Emmet and Teller (BET) surface area was ∼30 m2 g−1,188 whereas the length and thickness of REC layer were ∼100–200 nm and 2 nm, respectively, yielding an aspect ratio of 50–100, lower than MMT.189 Examples of bionanocomposite coatings based on REC are reported in Table 1.
2.1.1.1.3 Saponite. Saponite (SAP) (Fig. 2c) is another mineral clay belonging to the smectite group, with a chemical formula (Mx+·nH2O)Mg3(Si4−xAlx)O10(OH)2,67 surface area ∼200 m2 g−1,193 CEC of ∼80–120 meq per 100 g, aspect ratio of 50–60,17,194–197 and basal spacing between 12 Å (ref. 198) and 14.1 Å.197 Unlike MMT, which is a dioctahedral smectite, SAP has Mg2+ in the trioctahedral brucite-like sheet. Substitution of Mg2+ with Al3+ gives rise to a positive excess charge in the octahedral sheets,67,199 which is, however, over-compensated by the positive charge deficiency in the tetrahedral sheets due to substitutions of Si4+ with Al3+ (tetrahedral Al reported to be as high as 90% of the total Al in the sample).193 This eventually leads to an overall negative charge of SAP clays. The excess of positive charges in the octahedral sheet of SAP gives it a weaker aptitude to hydration compared with MMT, similar to vermiculite.194 However, the charge deficiency in the tetrahedral sheet of SAP has an important implication on the state of the interlayer water. In minerals in which the greatest part of the charge deficiency comes from tetrahedral substitutions (as in SAP), water molecules arrange in a three-dimensional organization mediated by the formation of hydrogen bonds between water and some negatively charged surface oxygen.200 Relatively rigid bridges are thus formed from one layer to another, the exchangeable cations acting as intermediaries.194 Therefore, with respect to water, SAP has a higher water affinity, but a lower hydration tendency than MMT. Another difference between SAP and MMT concerns the morphology. Whereas the structure of MMT is in the form of hexagonal lamellae, SAP shows a structure in the form of ribbons and laths.201
Developers have used SAP in combination with biodegradable polymers such as poly(butylene succinate) (PBS), in which the addition of the filler yielded better mechanical and oxygen permeability properties202 (Table 1). SAP used in combination with poly(vinyl alcohol) (PVOH) yielded PVOH/SAP nanocomposites with remarkable mechanical properties, thermal stability,197,203 water resistance,197,204 optical properties,203 and oxygen permeability.203
2.1.1.1.4 Laponite. Laponite (LAP) (Fig. 2d) is a fully synthetic smectite clay similar in structure and composition to natural hectorite (see next section). In this clay part of the Mg2+ in the trioctahedral sheet is replaced by Li+, resulting in a net negative charge of the layer, which is counterbalanced by Na+ cations located between adjacent layers in a stack.205 LAP is currently available in the market under two main types, with the following empirical formulae: [Si8(Mg5.5Li0.3)O20(OH)2.5F1.5]Na0.7, (type 1) and [Si8(Mg5.5Li0.3)O20(OH)4]Na0.7 (type 2).206 LAP has a plate-like shape with dimensions typically around 20–30 nm in length and 0.92 nm in width, basal spacing of ∼1.3 nm,207–209 estimated surface area of over 900 m2 g−1(206) and CEC typically 90 to 100 meq per 100 g for type 1 products and 50 to 60 meq per 100 g for type 2 products.210 One of the main technological advantages of LAP lies in the absence of silica and iron oxides in its composition, which allows producing clear and transparent colloidal suspensions of clay particles in water.211 Commercial application of LAP have thus far mainly involved household products, personal care items, horticulture, and the preparation of paper and polymer films.212 The materials science field has used LAP in combination with both hydrophobic and hydrophilic synthetic polymers, such as PE,213 PVC214 and PVOH,215,216 respectively. Developers have also used LAP in the production of bionanocomposites with claimed potential application in the food sector as biodegradable packaging films, as summarized in Table 1.
2.1.1.1.5 Hectorite. Hectorite (HEC) (Fig. 2e) is another 2
:1 trioctahedral smectite with a theoretical chemical formula (My+·nH2O)(Mg3−yLiy)Si4O10(OH)2;67 one study reported the CEC and surface area of HEC are 70–80 meq per 100 g and ∼200–300 m2 g−1 (measured by N2 adsorption at liquid nitrogen temperatures), respectively.219 Sànchez et al. measured a BET surface area of ∼220 m2 g−1 and basal spacing between 13.4 Å and 14.2 Å.220 Sinha Ray and Okamoto reported a CEC of ∼120 meq per 100 g.17 A more recent study reported a CEC of 43.9 meq per 100 g, a surface area of ∼65 m2 g−1 and basal spacing of 14.4 Å for Ca2+-HEC.66 Other research confirmed a surface area of 63 m2 g−1.221 HEC generally exhibits an aspect ratio of 200–300,196 which is higher than MMT and SAP. To the best of our knowledge, only two works have been published on HEC-based bionanocomposites with potential application for the food packaging sector (Table 1).
2.1.1.2 Kaolin clays. The term kaolin refers to a rock, white or near white in color, mainly constituted by kaolinite (i.e., KAO), which is the most important mineral within the group of kaolin clays. KAO is a 1
:1 (or T–O) layered silicate with a chemical formula Al2Si2O5(OH)4 consisting of one gibbsite-like dioctahedral sheet and one tetrahedral sheet layer (Fig. 2f).56 The ultimate structure is an asymmetric configuration with very little substitution in the structural lattice; thus it has a minimal layer charge and a low exchange capacity (∼3–15 meq per 100 g).148 This asymmetric structure allows the formation of hydrogen bonds between consecutive layers, providing a large cohesive energy.223 In turn, this makes intercalation of polymers more difficult, which is probably the main reason for the fewer occurrences of kaolin in the preparation of nanocomposite polymer materials, despite it being inexpensive, abundant, readily available, and environmental friendly. Another important technological property of KAO is the low absorption capacity, which relates to the minimal layer charge and the low surface area (5–20 g m−2) in comparison to smectite clays.148 Also, KAO exhibits a lower aspect ratio compared to smectite clays, usually around 10 but can vary widely from very low values ∼2 for laminated stacks (or “books”) to more than 11 up to 36 for very thin platelets in the 4–22 range.224 Although the largest use of kaolin is for coating paper,148 kaolinite has also been used to produce bionanocomposite materials especially in combination with corn starch derived from different sources (Table 1).
Within the kaolin group, halloysite (HAL) has recently gained researchers' attention. HAL is a 1:1 two-layered aluminum hydrated phyllosilicate with a predominantly hollow tubular structure in the submicron range.230 HAL may be regarded as a hydrated kaolinite Al2Si2O5(OH)4·2H2O (or even 4H2O) with curved layers and a basal spacing of 1 nm that decreases to about 0.7 nm upon dehydration.67 The neighboring alumina and silica layers and their waters of hydration, curve and form multilayer tubes with average diameters of 50 nm and inner lumen diameters of 15 nm, while the length may vary between 500 and 1000 nm.231 This peculiar tubular shape gives rise to the commonly used term “halloysite nanotubes” (HNTs) (Fig. 2g).232 HNTs have CEC of 5–10 meq per 100 g,233 whereas the typical surface area is 65 m2 g−1. The basal space of HNTs is 0.73 nm.234
HNTs are able to entrap a range of molecules with specific sizes and can serve as a viable nano-cage for active molecules, owing to the empty space inside the nanotubes (pores volumes of ∼1.25 mL g−1).235 For example, some studies asserted HAL as a suitable carrier for the controlled release of anticorrosion coating agents, herbicides, and fungicides.231,236–239 HNTs have also served as supports to immobilize catalyst molecules such as metallocomplexes.240 Like MMT nanosheets, HNTs can also be incorporated into polymers to modify their functional properties.241–243 However, we found very few works on this topic (Table 1). The results reported support the concept of bionanocomposite technology as a valuable tool to improve the water barrier, mechanical, and thermal properties of biopolymer films, with high potential for food packaging purposes.
2.1.1.3 Palygorskite and sepiolite clays. Palygorskite and sepiolite contain a continuous two-dimensional tetrahedral sheet (that is why they are considered phyllosilicates), whereas the octahedral sheet lacks of continuity. Their framework contains ribbons with a T–O–T structure, and the apices of tetrahedra belonging to adjacent ribbons point in opposite directions.67 This periodic inversion of the SiO4 tetrahedra determines channels or tunnels (extended along X-axis67) of rectangular section, which is the reason of the fibrous structure of these two non-swelling clays. The channels (wider in sepiolite) contain water and, due to the low CEC of both minerals, a small amount of exchangeable cations. Palygorskite (often called attapulgite in industrial and trade applications) is intermediate between di- and trioctahedral, whereas sepiolite is essentially trioctahedral. As far as their cell parameters, the major difference concerns b, respectively 1.78–1.80 nm and 2.68–2.72 nm.67
Sepiolite (SEP – Fig. 2h), with a theoretical half-unit-cell formula Mg8Si12O30(OH,F)4(OH2)4·8H2O,251 is a clay of relevance for the nanocomposites manufacturing. It shows a pore volume of ∼0.4 cm3 g−1,252–256 a surface area of ∼200–400 m2 g−1(257,258) and a CEC generally about 30 to 40 meq per 100 g.148 SEP is a “high silanols carrying filler”, with silanols spaced every 5 Å along the length of the SEP needle.259 This very high density of silanol groups (2.2 groups per 100 Å (260)) suggests that forces such as hydrogen bonding and van der Waals interactions are largely involved in coupling reactions with both polymers and organic surfactants as well as in adsorption phenomena at the interface.261 Both the excellent affinity with polymeric matrices and the strong anisotropy of this mineral account for the outstanding reinforcing effect on polymers, which results in a marked increase of the elastic modulus of the final compounds. However, the high concentration of hydroxyl groups on SEP surface is responsible for the strong catalytic effect of SEP on the thermal degradation of polyolefins.262 Compared to other inorganic fillers, SEP has found wide use in obtaining bionanocomposites with potential for food packaging applications (Table 1).
2.1.1.4 Other clay minerals.
2.1.1.4.1 Vermiculite. Vermiculite (VMT) (Fig. 2i) is a 2
:1 clay, generally trioctahedral. As in smectite, hydrated cations are located in the interlayer, although in VMT the negative layer charge, larger than that of smectite, arises mostly from substitution of Al3+ with Si4+ in the tetrahedral sheets.67 Occasionally isomorphic substitutions of Mg2+ with Al3+ determine an excess of positive charge in the brucite-like sheet (+0.62 electrostatic valence units), which is overcompensated by the excess of negative charge in the tetrahedral sheet (−1.81 electrostatic valence units).271 The location of the layer charge determines the strength of the Lewis base: VMT behaves as a hard base, whereas MMT, which presents relevant octahedral substitutions, behaves as a soft base.271 Furthermore, the intensity of negative charge and its position explain the overall tendency of VMT to swell less compared with MMT.194,271–274 Besides, VMT has a slight higher CEC compared with MMT, generally between 100 and 150 meq per 100 g.72 Brigatti et al. reported CEC = 84.4 meq per 100 g for MMT and 142 meq per 100 g for VMT,271 whereas Wang et al. reported CEC = 86.5 meq per 100 g for Na+-VMT.272 One study measured the basal spacing (d001) of vermiculite at 11.2 Å.272 Considering that the thickness of the Na+-VMT platelet is reported between 9.6 Å and 10 Å,273,274 it follows that the VMT interlayer distance (layer thickness subtracted from the basal spacing) ranges between 0.16 and 0.12 nm. Compared to MMT, VMT shows slightly lower surface area (500–700 m2 g−1), whereas Xu et al. reported VMT layers with thickness of ∼2–5 nm, and length of ∼200 nm.72 VMT is not as widely used as, for example, MMT for the generation of bionanocomposites. Few examples of VMT-based bionanocomposites are reported in Table 1.
2.1.1.4.2 Imogolite. Imogolite (IMO) is a clay-size hydrous alumino-silicate of short-range order with the chemical formula (OH)3Al2O3SiOH.67 IMO forms slender hollow tubules (Fig. 2j). The SiOH groups are located on the inner surface of the nanotube, while AlOH groups are on the outer surface. The hydroxyl groups on the walls and rims make the nanotube hydrophilic. Naturally occurring IMO is a few micrometers length with an internal diameter of about 1 nm, and an external diameter of about 2 nm.280–282 Synthetic IMO has a monodisperse nanotube length of 100 nm in solution, as determined by dynamic light scattering.283 These dimensions are considerably lower than HAL (halloysite); therefore IMO can provide higher surface areas than HAL for the same volume.
In IMO the tetrahedral sheets are located in the interior of the nanotubes, while the dioctahedral gibbsite-like sheets are on the exterior, with the hydroxyl groups of the Al2OH groups pointing outward, i.e. to the surrounding medium. As a consequence, the surface charge (and thus the exchange capacity) varies with the pH of the medium, which greatly affects the dissociation of hydroxyls. In this respect, the high pH at point-of-zero charge (pH > 11) on the outer parts of the tube and the anomalously high chloride adsorption of IMO may originate a weak positive charge on the outer tube walls, while a negative charge develops in the tubular pores.281 Therefore, IMO can have a positive charge under acidic conditions.284 However, chloride adsorption is largely equimolar with sodium adsorption when NaCl is added over a wide pH range,285,286 thus indicating the simultaneous presence of a weak positive charge and a weak negative charge along the outer and inner IMO nanotubes, respectively.
The surface area of IMO as measured by N2-BET adsorption is ∼300–400 m2 g−1.281 The repeating distance along the tubule axis is 0.84 nm.67 The formation of IMO occurs in a gibbsite sheet through the displacement of three hydroxyl groups surrounding a vacant octahedral site by an orthosilicate anion. This requires a considerable shortening of the O–O distances around the site (to <3 Å), which causes the gibbsite sheet to curl and form a tube. Doubly coordinated –Al2OH groups are found on the outer tube walls, whereas the inner tube walls carry SiOH groups.281 Although IMO does not swell under ordinary conditions, it retains 1.5 times more water than Na+-MMT due to micropores formed by intertwining fibrous particles.287
IMO is a “green” nanomaterial, although natural IMO is not sufficient to fulfill current demands. Recent advances thus concern IMO synthesis and new methods for large quantity production, which developers have performed without using toxic reagents or aggressive solvents, making this nanomaterial easily available.284
2.1.2.1 Layered double hydroxides. Layered double hydroxides (LDHs) (Fig. 2k), also called “non-silicate oxides and hydroxides”, have many physical and chemical properties that are surprisingly similar to those of clay minerals.288 LDHs have a general chemical formula represented by [M1−xMxIII(OH)2]y+[Ay/mm−·nH2O]; M is an univalent or a divalent metal ion such as Li+, Mg2+, Ni2+, Cu2+, Ca2+, or Zn2+; MIII is a trivalent metal ion such as Al3+, Fe3+, Cr3+, or Ga3+; y is the value of the layer charge for an octahedral unit; and Am− is an exchangeable anion with valence m like NO3−, Cl−, CO32−, or SO42−. Therefore y is equal to 2x − 1 or x for M is a univalent or a divalent metal ion.289
LDHs consist of stacked brucite [Mg(OH2)]-like trioctahedral sheets that are positively charged due to the replacement of Mg atoms by Al atoms, which is why LDHs are called “anionic clays” (indeed LDHs encase anionic exchangeable species intercalated between the layers). Hydrotalcite, Mg0.75Al0.25(OH)2(CO3)0.5·0.5H2O (formula often abbreviated as Mg–Al–CO3) is one of the most representative mineral of the group. LDHs possess a high anion exchange capacity (AEC) of 200–500 meq per 100 g,290 whereas the BET surface area for Mg–Al–CO3 LDH can be as high as 21.6 m2 g−1.291 The ion exchange or co-precipitation methods can introduce inorganic or organic anions between the octahedral hydroxide layers.292 Because many biomolecules (e.g. polysaccharides) are negatively charged, LDHs can find advantageous use in enhanced biopolymer/filler complexes.
Studies have reported the basal spacing of Mg–Al–CO3 (given by the sum of a brucite-like octahedral sheet and the interlayer spacing) as 7.8 Å (ref. 293) and 7.9 Å.289 Assuming the brucite-like layer of 4.8 Å,289,294 the interlayer spacing can be deducted as ∼3 Å thick, indicating the presence of strong interaction between the water molecules and the lattice OH groups of hydroxide layers.295 While modification of LDHs can be carried out to modify the surface properties, delamination of LDHs has been proven to be very difficult.296 One study derived a nominal aspect ratio of Mg–Al–NO3 LDH incorporated in PLA by the Nielsen model and calculated it as 50, while the same parameter increased to 85 for the same LDH after organo-modification with sodium γ-polyglutamate (γ-PGA).295 Several works have reported on the fabrication of LDHs-based bionanocomposites, indicating the potential application of these complexes for the food packaging field (Table 1).
2.1.2.2 Micas. The mica group (Fig. 2l) includes sheet silicates that are ubiquitous in nature, and often readily available at low cost, like muscovite. These minerals (natural or synthetic), are T–O–T phyllosilicates, and both di- and trioctahedral micas exist. In some naturally occurring micas, Si4+ nearly fills all of the tetrahedral sites, whereas in the most common species (muscovite and phlogopite) Al3+ substitutes for Si4+ in a ratio close to 1
:3.67 However, the Al3+ for Si4+ substitution can reach even a 1:1 ratio. This mineral group is subdivided in flexible (or true) micas, where the negative charge of the tetrahedral sheet is balanced by an interlayer monovalent cation (usually potassium), and brittle micas, where a bivalent cation (generally calcium) compensates a higher negative charge of the tetrahedral sheet. As a rule, the cations hosted in the mica interlayer are anhydrous. A typical example is muscovite (ideal formula KAl2[AlSi3O10](OH)2), which has layers well held together because the size of the K+ matches the hexagonal hole present in the tetrahedral mesh. Therefore, micas do not swell in water and, like pyrophyllite (a 2:1 dioctahedral phyllosilicate without any substitution of atoms), have no internal surface area.303,304 However, micas able to expand with very high aspect ratios (up to 1000) have been obtained by synthesis.305 Micas, in combination with different biopolymers, could serve as potential filler for the production of bionanocomposites for food packaging applications (Table 1).
Despite the wide range of nanofillers currently available on the market, selecting the most appropriate solution for a specific (bio)polymer system and for a given application may be not easy as it might seem. This choice requires accounting for a number of parameters with care, all of them generally relying on the maximization of the affinity between polymer matrix and filler, with the goal of making the latter an integrative part of the former. The chemical compatibility between the two phases (polymer and filler) is the first aspect to discriminate between natural and organo-modified fillers. The overall charge in its multifaceted aspects, namely type (negative or positive), amount (density of the charge), and location (tetrahedral or octahedral layers, bulk charge or edge charge), is another important parameter to consider especially if polyelectrolyte polymers (e.g. the negatively charged pectins, carrageenans, hyaluronic acid, or positively charged chitosan) have to be associated with the filler, or if active molecules (e.g. drugs, antimicrobials, antioxidants) must be hosted within the inorganic lattice. The water absorption and swelling capability of the filler may also be critical. While most often these properties coincide, they do not necessarily appear in the same filler; physical (e.g. porosity) and chemical (e.g. interlayer forces, type of counterions) characteristics may contribute in different ways. Thus, a “zero-point-charge” porous filler may absorb water but not swell and vice versa. Another important aspect is the shape of the filler and thus its aspect ratio, which becomes of utmost importance in light of the specific application. For example, selecting a needle-like shape or a platy shape can make a difference if the ultimate goal is to improve the mechanical properties or the barrier performance of the final nanocomposite material.
We report a summary of the information gathered during our literature search in an “all-inclusive” table (Table 2) that has never been proposed before. The aim is to provide a useful (although not exhaustive) tool for scientists working in the design and development of nanocomposite materials by offering the possibility to compare the most relevant properties or features of well-known silicate and non-silicate minerals (with the exclusion of metals and metal oxides) in one resource.
| Clay mineral | Type | Particle shape | Interlayer forces | Charge | IEC (meq per 100 g) | Swelling capability | Surface area (g m−2) | Basal spacing (Å) | Aspect ratio | Ref. |
|---|---|---|---|---|---|---|---|---|---|---|
| a This is due to the co-existence of both smectite-like and mica-like layers. b Once dehydrated. c Depending on the pH. d At pH = 7. | ||||||||||
| Na+-montmorillonite | 2:1 dioctahedral |
Lamellar | Very weak | − | 80–150 | Very high | 700–800 | ∼11.7 | 100–200 | 71 and 72 |
| Na+-rectorite | 2:1 dioctahedral |
Lamellar | Very weak/stronga | − | 32–45 | Medium | ∼30 | 20–22 | 50–100 | 186–189 |
| Saponite | 2:1 trioctahedral |
Lamellar | Weak | − | 80–120 | High | ∼200 | 12.0–14.1 | 50–60 | 193–198 |
| Laponite | 2:1 trioctahedral |
Rounded lamellae | Weak | − | 50–100 | High | ∼900 | ∼13 | 20–30 | 207–209 |
| Hectorite | 2:1 trioctahedral |
Lamellar | Weak | − | 45–120 | High | 65–300 | 12.6–14.4 | 200–300 | 17, 66 and 219–221 |
| Kaolinite | 1:1 dioctahedral |
Lamellar | Strong | − | 3–15 | Very low | 5–20 | ∼7.2 | 4–22 | 148 |
| Halloysite | 1:1 dioctahedral |
Tubular | Strong | − | 5–10 | Very low | ∼65 | 10 (∼7.2b) | 10–20 | 231–234 |
| Sepiolite (SEP) | 2:1 trioctahedral |
Rod | Strong | − | 3–20 | Low | 200–400 | ∼12 | 100–300 | 252–260 |
| Vermiculite | 2:1 trioctahedral |
Lamellar | Weak | − | 100–150 | High | 500–700 | 10–14 | 40–100 | 72, 194 and 271–274 |
| Imogolite | 1:1 dioctahedral |
Tubular | Strong | +/−c | 20–30d | Very low | 300–400 | ∼8.4 | ∼50 | 67 and 281 |
| Mica (natural) | 2:1 di-/tri-octahedral |
Lamellar | Strong | − | 10–40 | Low | 50–200 | ∼10 | 20–40 | 303 and 304 |
| Mg–Al–CO3 | 0:1 trioctahedral |
Lamellar | Strong | + | 200–500 | Medium | ∼22 | ∼8.0 | ∼50 | 289–295 |
Silver nanoparticles based on silver salts or metallic silver have been demonstrated to be readily incorporated into thermoplastic packaging polymeric materials such as PE, PP, PS, and nylon.311 Sánchez-Valdes et al. coated a five layer (PE/tie/PA-6/tie/PE) plastic film with an AgNPs/polyethylene nanocomposite layer and found antimicrobial activity against the fungus A. niger, a common food contaminant.312 From a practical point of view, the same authors interestingly observed that the activity was dependent on the coating method: methods that gave rise to a rougher surface (and hence more surface area for silver-ion release) had higher activity than those that resulted in a smoother surface. Colloidal silver particles have also been coated 90–150 nm thick onto paper using ultrasonic radiation, and this coated paper was shown to manifest excellent antimicrobial activity against E. coli and S. aureus, suggesting its potential application as a food packing material for longer shelf life.313 The incorporation of AgNPs also included biopolymer films such as alginate, chitosan, and starch, which exhibited strong antimicrobial activity against both Gram-positive and Gram-negative bacteria.314–317
These (bio)nanocomposite films and coatings with potent antimicrobial function have suggested the potential application in food packaging for the extension of shelf life and enhancement of the safety of packaged food.318–320 It has been pointed out that to make the Ag-based (bio)nanocomposite materials fully effective it is of utmost importance to consider various factors such as particle size, size distribution, degree of particle agglomeration, silver content, and the interaction of silver surface with the base polymer.321 This is because the antimicrobial efficacy of AgNPs, as any other nanocomposite antimicrobial system, strongly relies on the high surface-to-volume ratio and enhanced surface reactivity of the nano-sized antimicrobial particles, making them able to inactivate microorganisms more effectively than their micro- or macro-scale counterparts.322
Another metal with well-recognized antimicrobial properties is copper, although its potential biocidal activity is lower compared to Ag ions (about 10 mg Cu2+ kg−1 in water is necessary to kill 106 cells of Saccharomyces cerevisiae).323 The activity of copper ions has been exploited only in few polymer nanocomposites, such as Cu–PE nanocomposite films324 and Cu–chitosan bionanocomposite films intended for food packaging applications.325 The main reason for this restricted use is probably because copper is regarded as toxic in contact with food, and it would accelerate biochemical deterioration with foods due to its catalytic action of oxidation.326
Metal oxides also exhibit excellent antimicrobial properties against a wide spectrum of microorganisms. For this reason, (bio)polymer nanocomposites based especially on TiO2, ZnO, and MgO have been developed for food packaging applications. Among them, TiO2 nanocomposites are the most widely investigated. The antimicrobial properties of TiO2 lie in its photocatalytic activity, which is intimately linked to its crystal structure. There are three different forms of titanium dioxide (rutile or anatase, tetragonal, brookite, and orthorhombic) with different reactivity depending on their characteristic band gap. The irradiation of TiO2 at higher energies than the band gap generates electron-hole pairs, while giving rise to redox reactions. Negative electrons generate O2−, and positive electric holes generate hydroxyl radicals. Reactive oxygen species oxidize organic molecules, and kill bacteria and viruses.323 TiO2 coated polypropylene films illuminated with UV light sources, were effective in decreasing the counts of E. coli in in vitro experiments up to 3log10CFU g−1, but also during the storage of lettuce a reduction over 1log10CFU g−1 was observed.327 EVOH films loaded with TiO2 nanoparticles were produced by Cerrada et al., specifically for food packaging applications.328 The authors observed the effective photo-activated biocidal properties of the films against the nine microorganisms (bacteria and yeasts) cited to be involved in food poisoning and spoilage. From a practical point of view, the main limitation of TiO2 nanoparticles for the food packaging sector is due to its low photon utilization efficiency and necessity of the ultraviolet (UV) as an excitation source (i.e., TiO2-based antimicrobials are only active in the presence of UV light).329 Besides the antimicrobial activity, TiO2 nanoparticles may provide new features to the advantage of the overall performance of the final packaging material. In principle, food packaging films incorporating TiO2 nanoparticles may have the additional benefit of protecting food content from the oxidizing effects of UV irradiation while maintaining good optical clarity, as TiO2 nanoparticles are efficient short-wavelength light absorbers with high photostability.4 As for AgNPs and TiO2, their photoactivity can oxidize ethylene to H2O and CO2.323 TiO2 nanoparticles have also been used to produce oxygen scavenger films.330 In addition, incorporation of TiO2 into synthetic plastic matrix has shown to increase the biodegradability.331 Another interesting development concerns the ‘intelligent’ packaging nanocomposite films, i.e., those intended to monitor the condition of packaged food or the environment surrounding the food. Lee et al. developed an UV-activated colorimetric oxygen indicator, which uses nanoparticles of TiO2 to photosensitize the reduction of methylene blue (MB) by triethanolamine in a polymer encapsulation medium using UVA light. After UV irradiation, the sensor bleaches and remains colorless until it is exposed by oxygen, when its original blue color is restored. The rate of color recovery is proportional to the level of oxygen exposure.332
TiO2 thin films can be prepared on several substrates by various techniques such as chemical vapor deposition, evaporation, magnetron sputtering, ion beam technique, chemical spray pyrolysis, electro-deposition, and sol–gel method. The sol–gel process is suitable for producing composite materials of high purity without multiple steps.333
Zinc oxide (ZnO) particles are also known for their antimicrobial activity. Compared to AgNPs, ZnO have some advantages: first of all, the lower cost, which is related to the abundance of zinc as ubiquitous trace metal; and second, ZnO nanoparticles have also claimed to possess UV-blocking properties.334 Different ZnO-based nanocomposite films specifically intended for food packaging applications have been developed in recent years. For example, PVC films coated with ZnO nanoparticles were reported to have antimicrobial activities against E. coli and S. aureus.335 In a more recent work, Li et al. also verified the potential of the nano-packaging containing ZnO nanoparticles during the storage of Fuji apple cuts, observing a better preservation of quality indicators such as ascorbic acid and polyphenol content, and lower counts of typical altering microorganisms.336 Nanocomposite films of LDPE containing AgNPs and ZnO were claimed to exhibit antimicrobial activity, showing an impressive impact of the proposed nano-packaging on the shelf life of orange juice.337 It was also demonstrated that combinations of allylisothiocyanate, nisin, and zinc oxide nanoparticles coated on glass jars were able to effectively inactivate Salmonella in liquid egg albumen.338 It was observed that the antibacterial activity of ZnO nanoparticles increases with decreasing particle size.339 In addition, this activity does not require the presence of UV light (unlike TiO2), but it is stimulated by visible light.340
 | ||
| Fig. 3 (a) Crystals of phillipsite (a natural zeolite). Sample from Palombara quarry, Italy; (b) Schematic representation of phillipsite channel system plotted along X axis. Width of the channel is about 0.37 nm (http://izasc.ethz.ch/fmi/xsl/IZA-SC/ft.xsl). | ||
With regard to zeolitic membranes, despite many announcements in the last 15 years that have claimed the release of new commercial products for industrial applications,342 solvent dehydration remains to date the only industrial application able to exploit the multifaceted potential of zeolites profitably.343 This represents quite a surprising circumstance, in light of several excellent results in laboratory studies on zeolite membranes specifically intended for gas- and liquid-phase separations.343 Incorporation of zeolites into polymer gave rise to the fabrication of mixed-matrix membranes,344 which developers claimed were superior in selectivity compared to the traditional synthetic polymeric membranes used in the petroleum and petrochemical industries for separation applications345–347 such as pervaporation, e.g. for separation of volatile organic compounds (VOCs)–water mixtures,348–352 liquid separation by reverse osmosis,353 forward osmosis techniques,354 and ultrafiltration.355 Zeolites have also shown great potential for the catalysis of a variety of hydrocarbon reactions, including cracking, hydrocracking, alkylation, and isomerization, based on their combination of high thermal stability, high acidity and shape selectivity.356
Zeolite nanoparticles are of particular interest also for their adsorption capability. For example, zeolites successfully removed the pungent smell components identified in a coffee aroma-containing gas,357,358 reduced the odor and VOCs emissions of natural-flour-filled PP composites,359 and removed toxic and carcinogenic compounds (e.g. NO and NOx) from the main tobacco smoke stream by direct addition either in the cigarette filter or in the smoking tobacco leaves.360–365 Present and possible future applications of natural and synthetic zeolites also concern the biomedical field and the cosmetic sector.366
Silver ions (Ag+)-loaded zeolite micro- and nanocomposites are among the most widely used materials that include zeolites. In these composites, ionic exchange mechanisms replace the alkaline and alkaline earth metal ions naturally present within the zeolite with silver.323 Ag+ ions, due to their inherent antimicrobial activity, allow the production of potentially profitable new materials with antimicrobial features for use in many applications, including food contact and active food packaging scopes. For example, developers in Japan have used Ag-substituted zeolites as the most common antimicrobial agent associated with plastics to preserve packaged food products, in particular for those systems in which the use of volatile agents is not feasible, i.e. where the bioactivity is based on the migration of the antimicrobial molecule by direct contact with food.367 According with the Food and Drug Administration (FDA) Food Contact Substance Notification, in the USA, zeolite-based materials may be used in all types of polymers intended to come in contact with food,368 while the European Food Safety Agency expressed a positive opinion of in 2005 about the use of two different zeolites containing silver ions in food contact surfaces.369 However, the use of Ag+–zeolites for food packaging applications is limited to 0.05 mg Ag+ kg−1 of food, which represents the no observable adverse effect level for humans.370 The extensive research on the development of Ag+–zeolite antimicrobial food packaging systems has led to manifold applications over the last two decades, as reported in recent reviews.29,323 However, the use of Ag2+-zeolites nanoparticles for food packaging applications is still highly debated, due to some evidences pointing to potential risks associated with these new types of antimicrobial materials.323
Most recent uses of zeolites-based composites for food packaging applications concern membranes and films for fresh or minimally processed fruits and vegetables, which require selective permeation of carbon dioxide, oxygen, ethylene, and water vapor. Most research in this field involves ethylene, a plant growth regulator that plays a key role in physiological processes and during postharvest. Controlling the presence of ethylene in packages and storage environments could extend the shelf life of many fresh fruits and vegetables. Monprasit et al. incorporated a commercial hydrophobic zeolite (5 wt%) into low-density polyethylene (LDPE), achieving much higher ethylene permeability and selectivity, with concomitant appropriate tensile properties.371 In another study,372 the quality of broccoli packaged in LDPE-containing zeolite (as an ethylene absorber) resulted as the most similar to fresh broccoli. Boonruang et al. compared the effects of the use of various types of packaging films in maintaining the quality and extending the shelf life of mangoes fruit.373 The authors demonstrated that an ethylene-removing mechanism based on the use of zeolites incorporated into PE films extended the shelf life of mangoes up to 40 days, as compared with 20 days for the control (i.e. non-packaging treatment) and 5 days with common non-perforated PE film. Another study described the use of zeolite as CO2 absorber in combination with Na2CO3 to develop a suitable packaging system for kimchi, with controlled pressure and volume conditions.374 Metal-doped (e.g. iron, palladium, and platinum) zeolites are suitable oxygen scavengers for products sensitive to oxidation,323 as has also been demonstrated for zeolites with adsorbed terpenes or phenol derivatives loaded in packaging bags.375 Another study investigated the possibility of using zeolite modified with a series of organosilane compounds to control the gas permeability properties of membranes containing the inorganic filler.346 The addition of zeolite into PP films made PP a water- and water vapor-sensitive material.376 Developers have also produced PP/zeolite composite films coated with aluminum to obtain a packaging material impermeable to light, impermeable to moisture from the outer surface, and permeable to water and adsorbing moisture from the inner surface.377
The use of zeolites as inorganic filler may also improve the mechanical and thermal properties of plastics commonly used in food packaging. Biswas et al. reported that increasing zeolite 4A content up to 50 wt% increased the Young's modulus and yield stress of linear low density polyethylene (LLDPE) from 127 to 179 MPa and 9.4 to 11.1 MPa, respectively.378 Yuzay et al. reported that increasing zeolite 4A concentration up to 5 wt% resulted in increased Young's modulus of PLA from 1295 to 1465 MPa and tensile strength from 62.5 to 67.6 MPa, although elongation at break slightly decreased from 6.9 to 6.6%.379 Other studies attributed the poor mechanical properties of zeolite-loaded PP to the weak interfacial interactions between polymer and inorganic filler.376,380 To address this issue, developers have improved the filler compatibility and mechanical properties of the composites by surface treatments of natural zeolite.381 Similarly, studies have reported that an activated natural zeolite has a compatibilizing effect on a recycled tetra-component polymer blend consisting of PP (40 wt%), LDPE, high density polyethylene (HDPE) (15 and 40 wt%, respectively), and polystyrene (PS) (5 wt%).382
Zeolites have also improved the thermal properties of oil-based polymers such as PP,359,376,383 polymers of natural origin such as PLA,384 sodium alginate,385 and blends of oil-based and natural-origins polymers, such as PE/thermoplastic starch (PE/TPS).386
000 cm2 V−1 s−1),392 specific surface area (calculated value, ∼2630 m2 g−1),393 fascinating transport phenomena (e.g., the quantum Hall effect394), and gas impermeability.395 These properties make graphene one of today's most exciting materials, as it represents the best candidate for the enhancement of electrical, mechanical, thermal and gas barrier properties for advantageous exploitation in many areas, such as photovoltaics, biosensors, supercapacitors, super adsorbents and fuel cells,396 just to provide some examples. Despite the broad spectrum of potential applications, there are still many challenges for graphene to reach its full potential. Among others, production costs represent the main limitation to large scale utilization, mainly due to the highly expensive and low-yielding methods and procedures to obtain graphite monolayers.
 | ||
| Fig. 4 (a) TEM image of graphene sheets (dark areas denote overlapping zones); (b) HRTEM image of 3 overlapping zigzag-armchair edges obtained by applying Joule heating to a graphitic nanoribbon inside a HRTEM; hexagonal models are depicted for clarity (reproduced with permission from Elsevier Ltd387). | ||
The first attempt to produce graphene was based on the chemical conversion of graphite into graphite oxide by chemical oxidation, with subsequent dispersion and exfoliation in water or suitable organic solvents. Several researchers, such as Staudenmaier,397 Hummers and Offeman,398 developed oxidation methods to produce graphene oxide (GO) from graphite flakes. The main drawback involved in the chemical oxidation process is the use of hazardous strong oxidizing reagents (e.g., HNO3, KMnO4, and/or H2SO4) necessary to overcome the enormous van der Waals-like forces between graphene layers to yield a complete exfoliation of graphite flakes and to disperse the resulting graphene sheets stably in a liquid media.398–400
The new functional groups arising from chemical oxidation are mostly in the form of hydroxyl and epoxy groups on the basal plane, with smaller amounts of carboxyl, carbonyl, phenol, lactone, and quinone at the sheet edges.401,402 These highly reactive oxygen functionalities, besides making possible the grafting of both small molecules and polymers onto GO sheets, are responsible for the strongly hydrophilic character of GO, such that intercalation of water molecules between the layers readily occurs, leading to an interlayer spacing between 6 and 10 Å, depending on the water content.403
Most recent methods to produce graphene fall within either the bottom-up or the top-down strategies. The bottom-up approach, based on the organic synthesis starting from small precursors (e.g. atoms, molecules, and so on) includes chemical vapor deposition (CVD),404,405 epitaxial growth on silicon carbide406–408 and metals,409,410 reduction of CO,411 and conversion of CO2.412 In particular, CVD and epitaxial growth produce relatively good-quality large graphene sheets with fewer defects, but are not suitable for the fabrication of polymer nanocomposites, because these require large quantities of graphene sheets.
Top-down methods are based on the exfoliation of graphite into few or single-layered graphene sheets by breaking the van der Waals-like forces between graphite layers. Top-down methods account for two main routes. The first route, which includes a micromechanical cleavage based on “Scotch tape”389 method and mechano-chemical methods such as ball milling413 and roll milling,414 consists of the separation of strongly layered graphite into individual graphene sheets without basal plane distortion. The second route involves the basal plane distortion of graphene sheets and includes sono-chemical methods such as sonication415 and ultrasonication.416 Micromechanical cleavage of graphite, referred to as “Scotch tape” method, gave birth to renewed interest in graphene because it disclosed the huge potential of the material.389 Although the Scotch tape method provides large and high-quality sheets, it enables only limited production, which makes it suitable exclusively for fundamental studies.389 By contrast, sonication and milling techniques allow industrial-scale outputs that make them effective and low-cost strategies for mass production of high-quality graphene sheets.
The use of graphene even at low loading, makes it possible to obtain nanocomposite polymer systems with highly improved properties, such as tensile strength and elastic modulus, electrical and thermal conductivity, thermal stability, gas barrier, and flame retardance. Due to this broad multifunctionality, graphene/polymer nanocomposites can find application in various fields. These include the following: advanced biochemical and electrochemical applications with flexible and electrically conductive properties;417,418 sensors, including electrochemical biosensors419,420 and prototype gas sensors;421 thermal interface applications with high thermal conductivity, low electrical conductivity, and highly compliant nature (e.g. thermal pads and thermal adhesives);422,423 electric and electronic engineering materials, such as touch panels or flexible displays, where high optical transparency, good barrier properties, thermal stability, and mechanical strength are required;404,424–427 automotive engineering, e.g. parts where thermal, mechanical, and flame-retardant properties are necessary;428 highly efficient visible-light plasmonic photocatalysts;429,430 biomedical applications, where controlled release behavior, long term biocompatibility, better mechanical properties, faster tissue regeneration, and less thrombogenicity are sought-after properties;431,432 transportation and space technologies with high thermal conductivity;433 piezoelectric applications;434 aerospace and radar evasion applications with electromagnetic wave shielding effects at microwave frequencies;435,436 electronic, energy storage, and photovoltaic devices with enhanced electrical conductivity;405,437,438 package-free flexible photovoltaic devices;432,439 and anticorrosion coatings with outstanding barrier properties against both O2 and H2O.440
Potential use of graphene-based polymer nanocomposites also includes food packaging applications due to the expected enhancement of mechanical,441–450 thermal,442,444,445,447–449,451,452,454 and thermo-mechanical properties454,455 of the final materials. Another benefit arising from the use of graphene that can boost its application in the food packaging field relies on its gas-impermeable atomic membrane, which would allow producing barrier materials against permeation of O2,445,455,456 N2,444,455 and He.444 Graphene exhibits a unique tunable water-permeable atomic membrane, which is of paramount importance for food packaging applications. For example, Geim et al. recently reported the tunable permeation rate of water across a GO membrane that is completely impermeable to other liquids, vapors, and gases.457
Yoo et al. have recently published a comprehensive review on the potential applications of graphene and GO to produce barrier polymers.458 In addition, a recent study tested the transparency and water resistance of graphene-based polymers,450,456 making this the preferential filler for exploring new packaging applications. Although only few works have clearly addressed the potential impact of graphene/polymer nanocomposites in the food packaging area (Table 3), this number is increasing considerably, demonstrating the interest in graphene-based nanocomposites also for this sector.
| Polymer/graphene type | Preparation method | Concentration | Properties | Ref. |
|---|---|---|---|---|
| a Please refer to the main text for abbreviations. b Ti: initial degradation temperature, TRG: thermally reduced graphene, EFG: exfoliated graphite, HD-GNR: hexadecyl-functionalized low-defect graphene nanoribbons; DA-GO: dodecyl amine (DA) functionalized graphene oxide and DA-RGO: dodecyl amine functionalized reduced graphene oxide; E′: storage modulus. | ||||
| PDN/AFG | Solvent casting | 0.2–5.0 wt% | Transmittance at 0.5% concentration is over 82% | 453 |
| Transmittance sharply decreases at 1.0% concentration | ||||
| Oxygen permeability (23 °C, 0% RH) at 5% concentration decreases by 20.7% of neat PDN | ||||
| Young's modulus and tensile strength at 5% concentration increase by 49.1% and 63%, respectively; elongation at break decreases by 58% of PDN film | ||||
| PS/PIFG | Hot pressing | 0.02–2.27 vol% | Transmittance ∼40% at 0.02% concentration; full opacity at 0.24% concentration | 459 |
| Oxygen permeability (23 °C, 0% RH) at 0.02% concentration ∼80% of neat PS | ||||
| PLA/GO or PLA/GNP | Doctor blade | 0.2–0.8 wt% GO | Transmittance ∼72% and ∼23% at 0.6% concentration GO and GNP, respectively | 460 |
| 0.2–1.0 wt% GNP | The largest decreases in oxygen and nitrogen permeabilities at 0.4% GO and GNP concentration are 70% and 60% of neat PLA, respectively | |||
| Young's modulus and yield strength at 0.3% GO concentration increases by 115% and 95% compared to the neat PLA film | ||||
| Young's modulus and yield strength at 0.4% GNP concentration increases by 156% and 129% compared to the neat PLA film | ||||
| PPC/GO | Compression molding | 0.1–1.0 wt% | Young's modulus and tensile strength at 1% concentration increase by 1800% and 1000%, respectively; elongation at break decreases by ∼50% compared to neat PPC film, though still remaining as high as 700% | 461 |
| Storage modulus at 1% concentration at 30 °C increases up to 1600 MPa (storage modulus of neat PPC ∼30 MPa) | ||||
| T g of PPC increases from 29 °C to 39.5 °C after loading 1% GO | ||||
| Chitosan/GNP or starch/GNP | Solvent casting | 0.2–3.0 wt% | Young's modulus and tensile strength at 1.5% concentration increase, further concentration increase results in decrease in both chitosan and starch films; elongation at break decreases as concentration increases compared to the neat chitosan and starch films | 462 |
| The Tg values do not exhibit a significant effect attributable to GNP (Tg ∼ 72 °C) WVTR of starch and chitosan (25 °C, 50% RH) at 3% concentration decreases by 24% and 17%, respectively, of neat films | ||||
| PPC/EFG | Ultrasonication/solvent casting | 0.5–5.0 wt% | The maximum increase in tensile strength is at 2% EFG by 50%; elongation at break decreases by 89% as concentration increases compared to the neat film | 463 |
| T g of PPC increases from 48.3 °C to 52.1 °C after loading 5% EFG | ||||
| OTR (23 °C, 0% RH) and WVTR (37.5 °C, 90% RH) at 5% concentration decreases by 45% and 18%, respectively, of neat PPC | ||||
| Oxygen and CO2 permeability (room temperature, 50% RH) at 1.37% concentration decreases by 45% and 68%, respectively | ||||
| PLA/GO | Ultrasonication/compression molding | 0.17–1.37 vol% | T g is increased by 15.6 °C at 1.37% retarding the degradation of the PLA | 464 |
| Transmittance of neat PLA film is about 60%, loading is above 0.35%, and nearly no UV light is penetrated | ||||
| EVOH/TRG | Ultrasonication/solvent casting | 0.5–4.0 wt% | Young's modulus and tensile strength at 4% concentration increase by 400% and 280%, respectively, elongation at break decreases by ∼87% compared to neat EVOH film | 465 |
| Only 0.5 wt% TRG loading is significantly decreased oxygen permeability (room temperature, 37% RH to 8.517 × 10−15 cm3 cm cm−2 s−1 Pa−1), which is nearly 1670 times lower than that of neat EVOH films (3.911 × 10−11 cm3 cm cm−2 s−1 Pa−1) | ||||
| T g of EVOH films increases from 65.31 °C to 73.81 °C after loading 1.5% TRG and then gradually shifts to high temperatures with increasing TRG loading | ||||
| PU/HD-GNR | Sonication/solvent casting | 0.1–0.5 wt% | Nitrogen gas effective diffusivity of TPU was decreased by 3 orders magnitude with only 0.5 wt% HD-GNRs | 466 |
| The modulus increased and peaked at 0.1 wt% HD-GNRs, and then gradually decreased upon further filler additions | ||||
| PET/GNP | Melt compounding | 0.1–1.5 wt% | OTR (23 °C, 0% RH) at 1.5% concentration decreases by more than 99% compared to neat PET | 467 |
| T g does not exhibit a significant GNP effect. GNP stabilizes the PET—manifested in increasing its char residue by 5 wt% at 1.5 wt% GNP content at 600 °C | ||||
| PBS/GO | Ultrasonication/compression molding | 0.3–2.0 wt% | Young's modulus, tensile strength, elongation at break, and fracture energy of PBS at 2% concentration increase by 70, 53, 12, and 100%, respectively | 468 |
| Tm does not exhibit a significant GO effect while Tg increased by 5 °C with addition of 2.0% GO | ||||
| (PEI/GO)n multilayer PLA | Sonication/dipping | 0.02–0.06 wt% | Compared with the pure PLA film (the transmittance of 92.3%), the multilayer film coated with (PEI/GO)6 still remained a high visible light transmittance (84.4%) | 469 |
| OTR (23 °C, 0% RH) at 0.06% GO of PLA film coated with 4 layer decreases by 98.7% compared to neat PLA | ||||
| EVOH/EFG | Ultrasonication/solvent casting | 0.1–2.0 wt% | T g and Tm do not exhibit a significant EFG effect. Ti is increased by 14.4 °C at 0.5% retarding the degradation of the EVOH and further loading results in decreasing | 470 |
| OTR at 23 °C, 0% RH and at 23 °C, 80% RH at 2% concentration decreases by 97% and 77%, respectively, compared to neat EVOH | ||||
| HDPE/DA-RGO or HDPE/DA-GO | Ultrasonication/hot press | 0.1–1.0 wt% | At 25 °C, the E′ of the DA-GO/HDPE and DA-RGO/HDPE nanocomposites with only 0.1 wt% filler content increase by 7.1% and 7.4% from, respectively, whereas the E′ of the nanocomposites with 1.0 wt% filler increases by 17.3% | 471 |
| With only 0.1 wt% DA-GO loading the PO2 of neat HDPE decrease by 62.3%. Only 66.7% decrease of PO2 can be obtained by addition of 1.0 wt% DA-GO. With 0.1 wt% and 1.0 wt% DA-RGO contents, the PO2 of DA-RGO/HDPE nanocomposites decreased by 63.6% and 67.0%, respectively | ||||
| The temperature at 50% weight loss of HDPE only increase by 7.2 and 6.4 °C at 1% DA-GO and DA-RGO nanocomposite, respectively | ||||
| HPMC/GO | Ultrasonication/solvent casting | 0.02–1.3 wt% | Young's modulus, tensile strength, elongation at break, and fracture energy of PBS at 1.3% concentration increase by 201, 122, 120, respectively | 472 |
| The temperature at 50% weight loss of HPMC increase by 8 °C at 1.3% GO | ||||
| WVTR (75% RH) at 1.3% concentration decreases by 44% compared to neat HPMC | ||||
The impact at biological level of the newly developed graphene-based nanocomposites is not yet a major focus of research. Because a thorough understanding of the biological behavior of nanomaterials would guarantee the sustainability of nanotechnology,473–478 future research toward this topic must fill in the current lack of this important information.
In light of the recently obtained exciting results, which may make the production costs of graphene accessible, the use of graphene, even though it is in its early stages, may be the most promising approach to guide future advancement in the development of new, revolutionary packaging materials.
3. Fabrication of (bio)nanocomposites
To take full advantage of the great potential of (bio)nanocomposites for real applications, the integration of the selected nano building blocks in the polymer matrices is of primary importance. Many factors, including the exfoliation degree of the fillers, their spatial arrangement (wrinkling or stretched), the morphology of the composite, and the dispersion state in the polymer matrix (stacked or agglomerated)—hence the extent of the interfacial interactions between filler and polymer—may dictate the final performance of the nanocomposite packaging material.479 Accordingly, for example, the ultimate barrier properties of nanocomposite polymers will be affected by nanosized fillers in two specific ways: (i) a more tortuous path for diffusion of the permeant and (ii) local changes in the polymer matrix properties (e.g., molecular mobility) at the interfacial (polymer nanoparticle) regions.4Historically, the nanofiller incorporation into the polymer matrix takes place through three main methods, i.e. (i) the in situ polymerization, (ii) the solution casting or (iii) the melt processing.15,60,480,481 Most recently, sonication and high-shear mixing have been proposed as alternative techniques to prepare (bio)nanocomposite materials.
3.1 In situ polymerization
This process involves the polymerization of monomer species in the presence of the layered materials. In this process, the nanoclays are first swollen within the liquid monomer or monomer solution, which is followed by its polymerization in between the intercalated sheets (Fig. 5a). Polymerization can be initiated either by heat or radiation, by the diffusion of a suitable initiator or by an organic initiator or catalyst fixed through cationic exchange inside the interlayer and before the engorgement step when required.17 The macromolecules molecular weight increases, leading to a d001 increase and sometimes to an almost fully exfoliated morphology for some studied systems.17 One of the main drawbacks of this method lies in the tendency of inorganic particles to phase separate and sediment quickly from the organic polymer. To enhance the interaction at the solvent/filler interface, specific groups have to be linked onto their surface to stabilize the nanoparticle dispersions.482 Another relevant aspect concerns the unsuitableness of this method for biomacromolecules such as proteins and polysaccharides (that are already extracted as ‘polymerized’ entities), which indeed represent target polymers for the generation of bionanocomposites.60 | ||
| Fig. 5 Schematic representation of (a) in situ polymerization, (b) melt processing, and (c) solution casting. | ||
3.2 Melt processing
In the melt processing technique, the nanoparticles are mixed with the polymer in the molten state (Fig. 5b). More specifically, the process involves mixing the particles with the polymer and heating the mixture above the softening point of the polymer, statically or under shear interlayers.483,484 The main advantages of the melt processing method are the absence of any solvent throughout the process and its compatibility with current industrial processes, such as extrusion and injection molding. Several factors may affect the extent of exfoliation/intercalation by melt processing, such as the thermodynamic interaction between the polymer and the nanoparticle and the transport/diffusion of polymer chains from the bulk melt into the silicate interlayers.483,484 To increase the compatibility between polymer and nanoparticles to ensure proper dispersion, two main factors have to be taken into consideration, namely the favorable enthalpic interaction between the polymer and the nanoparticle (which can be manipulated by chemical modification of the filler and/or the polymer) and proper processing conditions.481 In this regard, shearing is necessary to induce the platelets delamination from tactoids, while an extended residence time is needed to allow the polymer chains to diffuse into the inter-layer gallery and then obtain an exfoliated morphology.485However, in the case of most bionanocomposites (e.g., PLA-, chitosan-, and pullulan-based nanocomposites), the melt processing technique is not adequate in principle because these polymers tend to degrade due to either the mechanical shearing force or the temperature applied during processing. For example, PLA experiences processing instability (i.e., thermal, oxidative, and hydrolytic degradations) may occur during processing, which leads to the cleavage of polymer chains and consequently to a decrease in molecular weight. This degradation could even be accelerated in nanocomposites that have organically modified clays, which could initiate the degradation.481 Therefore, careful optimization of the processing parameters is required for the majority of thermally sensitive biopolymers.60
3.3 Solution casting
The solution casting method is based on a solvent system in which the polymer (or pre-polymer, in case of insoluble polymers) and any other component of the mixture (e.g., surfactants) is soluble. The polymer is usually dissolved in a suitable solvent while the nano-fillers are dispersed in the same or a different solvent before the two are mixed together to generate a homogeneous dispersion (Fig. 5c). The main advantage of this method is the relatively rapid exfoliation of the stacked layers by the use of an appropriate solvent.17,484 The successive addition of polymer solution to the dispersion of the complete delaminated nanoparticles (e.g., platelets) leads to the strong interaction between macromolecules and individual layers. The driving force for the intercalation of the biopolymer into the clay galleries from solution is the entropy gained from the desorption of the solvent molecules, which compensates for the decreased entropy of the confined, intercalated chains. When the solvent is evaporated, the intercalated structure remains, which results in the final nanocomposites.481 Due to the large amount of the solvent required, this method is perceived as unsafe and non-environmentally benign when organic solvents are required (e.g., for non-polar or highly hydrophobic polymers).486 Conversely, this method has gained increasing attention for water-soluble polymers such as PVOH, especially in the form of thin coatings, which reduces the amount of water used throughout the process. More recently, the solution casting method has been adopted for the generation of bionanocomposites, for which both in situ polymerization and melt intercalation are often unsuitable due to the inherent characteristics of most biopolymers, as discussed before. From a practical point of view, the fabrication of (bio)nanocomposite films and coatings through the solution casting method requires special attention during the removal of the solvent (evaporation) step. Indeed, if a small amount of solvent remains entrapped in the final product, a lower interfacial interaction between the polymer and the filler can arise.487 For this reason, coupling infra-red lamps with high performance air ovens is the best strategy to prevent this potential drawback.4883.4 Sonication
Sonication is increasingly used in the top-down generation of nanoparticles. This is achieved through deagglomeration and reduction of microsized particles (e.g., tactoids) by means of sound waves (more frequently, ultrasound waves) as a result of the mechanical effects of the phenomenon called cavitation, which refers to the formation, growth, and implosive collapse of bubbles in a liquid (Fig. 6).489 The speed of sound in a typical liquid is 1000 to 1500 m s−1, and ultrasonic wavelengths will vary from roughly 10 cm down to 100 mm over a frequency range of 20 kHz to 15 MHz, much larger than the molecular size scale. The chemical and physical effects of ultrasound therefore arise not from a direct interaction between chemical species and sound waves, but rather from the physical phenomenon of acoustic cavitation.490–492 | ||
| Fig. 6 Schematic illustration of the ultrasonication method based on the acoustic cavitation. The sonotrode of an ultrasonicator generates high intensity ultrasound waves of alternating expansive and compressive cycles. This causes the formation, growth, and collapse of bubbles, which in turn yields intense shockwaves that promote interparticle collisions and, eventually, the exfoliation of the layered filler. | ||
After bubbles collapse, a number of major local events prompt the deagglomeration of microsized particles dispersed in the medium: heating (∼5000 K), high pressure (∼1000 atm), huge heating–cooling rates (>109 K·s−1), and abrupt liquid jet streams (∼400 km·h−1).493–495 The extreme, transient conditions produced during acoustic cavitation allow the formation of unique materials and also permit syntheses on the benchtop in a room-temperature liquid that would otherwise require high temperatures, high pressures, or long reaction times. The most important physical phenomena for the preparation or modification of nanomaterials are microjets and shock waves. Microjets occur when bubbles collapse near an extended surface (i.e., surfaces several times larger than the bubble radius).496 In addition, shock waves can accelerate solid particles suspended in the liquid. Interparticle collisions can reach velocities of hundreds of meters per second, which is the main cause, among other phenomena, of the exfoliation of layered materials into 2-D layers.497–499
Ultrasonication, first adopted to develop nanocomposites based on oil-derived polymer/inorganic clay systems, has gradually been extended to polymers of natural origin (e.g., polysaccharides, proteins, and lipids) for the development of bionanocomposites.19,22,167,500–503 More recently, ultrasonication has been suggested as a valuable tool for the preparation of graphene-based nanocomposites starting from graphite flakes or particles dispersed in a liquid medium, both non-aqueous and aqueous systems. Due to the significant advantages of aqueous systems over non-aqueous systems (e.g., lower costs and fewer potential health risks and environmental issues), the former approach (i.e., using water as a solvent) has been attracting much attention over the recent years. Lotya et al.504 firstly reported the ultrasonication-assisted exfoliation of graphite in an aqueous system using a cationic surfactant, i.e., sodium dodecyl benzene sulfonate (SDBS). A later development accounted for a ‘one-pot’ procedure to obtain graphene-based bionanocomposite materials, which involves the use of biopolymers directly in the ultrasonication reactor, without the necessity to recover the nanoparticles in a second step. According to this method, it is believed that biopolymer works similarly to surfactants (i.e., as a spacer), but without the formation of micelles. More specifically, the biopolymer adsorbs onto the exposed surfaces of the graphene layers, thus preventing any possible aggregation or restacking phenomenon, which would represent the simplest, most efficient, and environmentally friendly strategy for the preparation of bionanocomposite materials.505 Examples of direct exfoliation of graphite into few layers graphene sheets by (bio)polymer-assisted ultrasonication concerned poly(vinyl pyrrolidone) (PVP) and PVOH,506 a pyrene-functionalized amphiphilic block copolymer,507 pyrene polymers,508 gum arabic,509–511 and acrylate polymers.512
3.5 High-shear mixing
High-shear mixing methods for the exfoliation of layered materials have been developed more recently compared to other routes in an attempt to provide an alternative approach that is able to boost the progress from the laboratory to commercial applications. High-shear mixing methods, although still at an early stage, have shown the great potential for the exfoliation of graphene and other two-dimensional materials, providing a viable route for the industrial scaling of applications based on these layered crystals in large amounts.513–515 A recent study has demonstrated that graphite can be exfoliated to give graphene by generating shear in a very thin liquid layer in a rapidly rotating tube.516 However, this method gives very small quantities of graphene and is inherently un-scalable. Recently, Paton et al. demonstrated a method to exfoliate graphite into graphene nanosheets by high-shear mixing using a closely spaced rotor/stator combination.517 This method is claimed to be many times more efficient than sonication in terms of yield, enabling large quantities of defect-free, unoxidized graphene to be obtained. In addition, the same method can be scaled-up to an industrial level. The same authors developed a simple model showing that exfoliation occurs once the local shear rate exceeds 104 s−1 in liquid volumes from hundreds of milliliters, up to hundreds of liters and beyond.Ball milling is another commonly used high-shear mixing technique, which involves a solid-state mixing at room temperature.518 It is a simple and efficient technique based on the pure shear among balls of various diameters, which is able to break the van der Waals interactions between layers.519 Clay dispersion is thus promoted by the energy transfer between milling tools (generally balls) and polymer/clay mixture, which in turns results grinded and intimately mixed.513 At the same time, the newly obtained layer would be wrapped up by polymer matrix, not only preventing the layers from sticking together, but also keeping the inherent structure of the layers.519 Among the available techniques used for nanocomposites preparation, ball milling has the considerable advantage of not requiring the use of high temperature or solvent treatments, which makes the composites preparation more green, convenient and effective.520 The ball milling method was introduced to fabricate bionanocomposites such as MMT/pectin513 and LDH/PCL.514
4. Technological aspects linked to the manufacture of (bio)nanocomposite coatings
The success of (bio)nanocomposite materials strongly depends on some important factors during the design and development steps. Besides significant costs, technological aspects may act as the “go-no go” gate before market applications. These aspects concern the coating system both before (e.g., the coating suspension) and after (i.e., the dry (bio)nanocomposite coating) the deposition on the selected substrate.4.1 Compatibility between polymer and filler
The affinity between organic and inorganic components at the hybrid interface plays a key role in determining the final performance of (bio)nanocomposite polymer systems. Maximizing the advantages of the interface effect between polymer and filler therefore requires selecting the best possible starting materials. In this respect, a significant difference applies between petrol-based polymers and biopolymers. Most plastic polymers derived from oil are inherently hydrophobic (e.g., polyolefins), while inorganic fillers (e.g., clays) are naturally hydrophilic, thereby resulting in a marked incompatibility between phases.To enhance the compatibility with oil-based polymers, production methods generally replace the alkali or alkaline-earth interlayer cations by means of ion exchange reactions with organic ammonium and phosphonium cations to reduce the surface energy of the inorganic phase.13 In other circumstances, an alternative approach consists in the tailored functionalization of the filler by grafting new moieties with high affinity for the polymer. For example, researchers have achieved the interfacial adhesion between graphene and the polymer matrix by chemical functionalization of graphene surface via “grafting from”,521–523 “grafting on”,399,524,525 and “grafting to”526 techniques. Unlike common plastics, most biopolymers are hydrophilic, which makes their miscibility easier with natural fillers such as cellulose-derivatives, protein-derived whiskers, and especially clays.
In practice, enhancing the compatibility between organic and inorganic components allows the minimization of phase separation during processing as well as successful intercalation, swelling, and exfoliation of nanoclays. In turn, this will yield an even dispersion and distribution of the filler within the polymer matrix, avoiding the formation of traditional filled polymers with fewer applications due to the depression of the “interface effect”. Eventually, efficient dispersion of the filler in the hosting matrices should enable low-cost solutions527–530 for sustainable and lightweight nanocomposite materials.
4.2 Rheological properties
Manufacturers of nanocomposite polymer systems widely use rheological measurements, as they represent an indirect method to assess the state of dispersion of the nano-objects (i.e., fillers), such as the degree of intercalation, exfoliation, and dispersion in the polymer matrix.531 Because most synthetic routes adopted to obtain the final nanocomposite polymer system (i.e., solution processing, melt processing, in situ polymerization, and mesophase mediated processing) involve flow, rheological properties of polymer nanocomposites may help to achieve optimum process conditions17,532,533 while an understanding of the rheological behavior can provide insightful information on the structure–property relationship in the final materials.17Although melt intercalation is the most widely adopted approach for the preparation of nanocomposite materials, the use of solvent-based systems is predominant within the coating technology, whereby water is the main (sometimes the unique) solvent when biopolymers are used as the organic matrix. The study of the rheology of nanoclay suspensions has a relatively long history because of the application of nanoclays as thickening and thixotropic agents in different fields than food packaging, such as cosmetics, inks, and paints. Even filler concentrations as low as 4–5 wt% can drastically affect the rheological properties of these suspensions, particularly the viscoelastic behavior. The association of filler particles (which generally takes place for concentrations above 3 wt% for clay platelets) induces the formation of a continuous structure, leading to gel-like behavior.534–536 Van Olphen (1964) first described different nanoplatelet associations: (i) face-to-face, (ii) edge-to-face, and (iii) edge-to-edge aggregations (Fig. 7).537 Face-to-face association leads to thicker and larger tactoids, whereas edge-to-face and edge-to-edge lead to three-dimensional “house of cards” structures. Aggregation is described by the face-to-face association, whereas the continuous gel-like structure is a consequence of edge-to-face and edge-to-edge association. Moreover, the nanoclay dispersion could be destabilized by salts (coagulation) or by polymers (flocculation). One of these systems has to be realized as a function of the final application. To achieve a true nanocomposite polymer system with improved properties compared to conventional microcomposites, the filler must be uniformly dispersed and not aggregated. This will first allow developing the highest proportion of an interfacial zone, thus maximizing the adsorption of the polymer chains onto the sheets.535 Secondly, efficient dispersion of the filler is the key to obtaining suspensions that are stable over time. Generally, stable suspensions show liquid-like (viscous) non-Newtonian flow behavior with a shear thinning or thickening character; the presence of colloidal aggregates imparts viscoelastic behavior to colloidal suspensions, which frequently show a time-dependent flow behavior known as thixotropy.538–540 In fact, during rest periods, all solid particles connect and form a gel characterized by a perceptible limiting shear stress, or yield stress, at which the material begins to flow: if the system is subjected to a constant shear rate, viscosity decreases with time as the gel structure is broken down, until equilibrium viscosity is reached. Most gelling colloidal systems follow Herschel Bulkley's rheological model; in contrast, most non-gelling colloidal dispersions follow Ostwald's rheological model (or power law).536 While gelation is a sought-after property in certain systems to stabilize the structure (e.g., food systems) it must be carefully controlled in all those applications requiring ease of flow, e.g. in coating technology.534–541
 | ||
| Fig. 7 Modes of particle association in clay suspensions: (a) dispersed; (b) face-to-face; (c) edge-to-face; (d) edge-to-edge (adapted from ref. 537). | ||
4.3 Optical properties
Optical properties of materials are particularly important in certain sectors, where they can impact either the performance of the final material or the consumer's choice. In the food packaging field, both aspects are relevant and worth high consideration when designing a new (bio)nanocomposite coating. On one hand, UV radiation (wavelengths below ∼340 nm) should be prevented, because it may cause photo-oxidation of photosensitive foods such as meat, beer, and milk, resulting in changes in color, flavor, and taste. On the other hand, high transmittance of visible light (wavelengths between ∼340 nm and ∼800 nm) should be guaranteed at the same time, as it allows consumers to see through the package (visual inspection of the packaged food).Researchers have developed (bio)nanocomposite systems (e.g., coatings) with UV-shielding/absorbing properties based on the use of metal oxides such as TiO2 (ref. 542 and 543) and ZnO544 or clays.167 However, after coating deposition and solvent removal by evaporation, the optical properties (e.g., transparency) of the substrate beneath can dramatically be impaired due to the presence of the filler. This can be due to three main reasons: (i) unsuccessful dispersion and distribution of the filler within the polymer matrix, generally caused by either erroneous selection of the (bio)polymer/filler system (incompatibility) or inefficient deagglomeration procedure/set up; (ii) high concentration of the filler—increasing the filler volume fraction leads to a proportional increase in haze due to an increase in scattering centers,19 and reaggregation phenomena—for the same filler volume fraction, the reaggregation of previously-exfoliated platelets may occur as a consequence of high energy input to the system (e.g., prolonged ultrasonication time), with subsequent restoration of the original macro-sized tactoids. In this case, also the size of the scattering centers play a role in decreasing the transparency of the (bio)nanocomposite coating.19
Therefore, the effect of the (bio)nanocomposite coatings on the optical properties of the substrate (e.g., plastic films) can be controlled by two main routes: (i) selecting suitable methods and procedures throughout the overall manufacturing process, and (ii) selecting the most appropriate filler type. Regarding procedure selection for example, in the exfoliation step of the filler, physical, chemical, or mechanical methods can be more or less efficient depending on the filler. Moreover, regardless of the specific methods, pinpointing the best process conditions (e.g., time, temperature, energy input, amount of additives, solvents and co-solvents, and so on) may dictate the final size of the filler (macro-sized aggregates or nano-sized entities). In selecting the most appropriate filler type, depending on the final application, the final choice will affect the optical properties of the final materials differently. For example, if the transparency of the final material is mandatory, the synthetic clay LAP offers higher performance over sheet-like and needle-like clays such as MMT and SEP, respectively, due to both its well-defined chemical composition and dimensions and lack of common inorganic components (e.g. silica and iron oxides) generally carried in natural clays, which allows the production of clear and transparent colloidal water suspensions.
4.4 Surface properties and the scalping effect
The addition of fillers to the formulation of coatings may also have an impact on the surface properties of the final material. In food packaging, the surface properties play a major role in several converting operations, such as printing, lamination, and co-extrusion. As a general rule, the addition of an inorganic filler, especially for a high filler volume fraction, is first reflected by changes in the surface topography, i.e. an increase in the surface roughness, which is linked to the spatial distribution of the fillers within the coating thickness (Fig. 8).19,167,545 For example, platy clays with large surface areas often display a “house-of-cards” or “cell-like” configuration after solvent removal.19 An increase in roughness, besides leading to an increase in the haze of the final material,546 may have great impact on the wettability attribute of the surface, as described by both Wenzel's and Cassie-Baxter’s theories.547,548 In addition, highly porous fillers (e.g., zeolites) as well as fillers with high affinity towards water molecules (e.g., natural MMT) might boost absorption phenomena at the solid/liquid interface.167 | ||
| Fig. 8 Influence of the clay loading on the surface roughness of biopolymer coatings. AFM height (upper) and 3D (lower) images (10 × 10 μm2) of pure pullulan coating (a) and pullulan/Na+-MMT nanocomposite coating (b) (filler volume fraction φ = 0.12) (reproduced with permission from American Chemical Society19). | ||
Not only the surface properties, but also mechanical properties can be influenced by an increase in roughness. Depending on both filler loading and the nature of the (bio)polymer carrying the filler, an increase in roughness may be associated with either an increase or a decrease in the slipping properties of the surface, which is quantitatively described by a decrease and an increase in the coefficient of friction, respectively.549–551 Whether this change is positive or negative strongly depends on the final application. For example, low coefficient of friction values aid the unwinding operations of plastic films on industrial lines, to avoid the “blocking effect” on the reels and decreasing overall throughput. Conversely, lower coefficient of friction values are problematic if the final packages (e.g., plastic bags) have to be stacked on top one of another; high coefficients of friction can keep the stack from collapsing.
Finally, but not less important, is the adverse effect that can potentially arise from the use of (bio)nanocomposite coatings on the overall quality of packaged food. “Scalping” is the sorption of food constituents, especially aroma compounds, by the packaging materials. Both porosity and chemical affinity of the filler for aromas and volatile molecules must be taken into account in the design of a (bio)nanocomposite coating, especially if it will face the inner side of the package and be close to the food. Scalping phenomena have received considerable attention within the food packaging industry as they may negatively influence consumer acceptance of the food product due to loss of aroma intensity or the development of an unbalanced flavor profile.552
5. Advantages of bionanocomposite coatings in food packaging
From an industrial perspective, the development of (bio)nanocomposite coatings has aimed at improving three main properties of the base resin: mechanical (stiffness, impact, and wear resistance), flame-retardance (anti-flammable materials), and barrier properties (against gases, vapors, and radiation). In the food and beverage packaging sector, however, considerable industrial and research developments of nanocomposite coatings have focused on improving the barrier properties, which has led to several applications in recent years. The excellent barrier properties of (bio)nanocomposite coatings may be explained by the “tortuous path” theory,25i.e. the formation of a labyrinth ensuing from the spatial dispersion of nano-clay platelets within the polymeric structure, which physically decreases the diffusion of the permeant (e.g., gases and vapors) across the thickness of the material.Studies have reported examples of applications of modified atmosphere packaging for food (meats, cheese, confectionery, cereals and “boil-in-the-bag” foods), extrusion-coating applications in association with paperboard for fruit juice and dairy products, and in combination with co-extrusion processes for the manufacture of beer and carbonated drink bottles.553 Several nanocomposite materials intended for food packaging applications are available in the market. Nanocor and Mitsubishi Gas Chemical Company developed the MXD6 high barrier semi-aromatic nylons in nanocomposite form.554 The latest development of the resin MDX6 led to the Imperm® products, a family of superior gas barrier resins particularly useful in extending package shelf life.555 Honeywell has developed a polyamide-based nanocomposite with passive barrier properties against oxygen under the Aegis® trade name. Passive barriers use clay particles, which render the oxygen transmission inside the composite more difficult.556 The U.S. military and NASA, in conjunction with Triton Systems, Inc., worked together on nanoclays as barrier enhancers for ethylene vinyl alcohol (EVOH) in long-shelf life packaging (i.e., non-refrigerated food). This collaboration led to an experimental thermoformed food tray from EVOH loaded with 3% of Southern Clay's Cloisite® in a layer sandwiched between two PP layers. It reportedly imparts a three- to five-years shelf life without refrigeration, plus good clarity, processability, and recyclability.557 InMat Inc. developed Nanolok™, a high barrier, water-based, environmentally friendly nanocomposite barrier coating for transparent packaging applications. They claim the nanocomposite coating provides an excellent oxygen barrier up to 80% relative humidity and is more cost effective than EVOH.558 Finally, NanoPack Inc. has developed a water-based coating made of PVOH and VMT. This coating, sold under the NanoSeal™ trade name, is specifically intended for food packaging applications where protection of food against oxygen and aromas is required.559
Besides advantages directly linked to their functional properties, the use of (bio)nanocomposite coatings may indirectly provide additional benefits, often underestimated or neglected. For example, the weight advantage could present a significant impact on environmental concerns. According to the “packaging optimization” principle,560 the use of high-performance thin layers may be a valuable approach to down-gauging current packaging structures, e.g. laminates and/or co-extruded materials (Fig. 9). (Bio)nanocomposite coatings thus have the great potential to allow reducing the thickness of the package without jeopardizing (rather improving) the overall performance, at reasonable cost. In turn, this would mean an upstream reduction of plastics, hence a less amount of plastics dumped into the environment, which would help in facing the waste disposal issue. From a strict economical point of view, the use of (bio)nanocomposite coatings to obtain lighter packages has the additional advantage to reduce the energy inputs for production, transport, and storage. Of course, the higher the degree of multi-functionality integrated in the (bio)nanocomposite coatings, the wider the environmental and economic advantages.
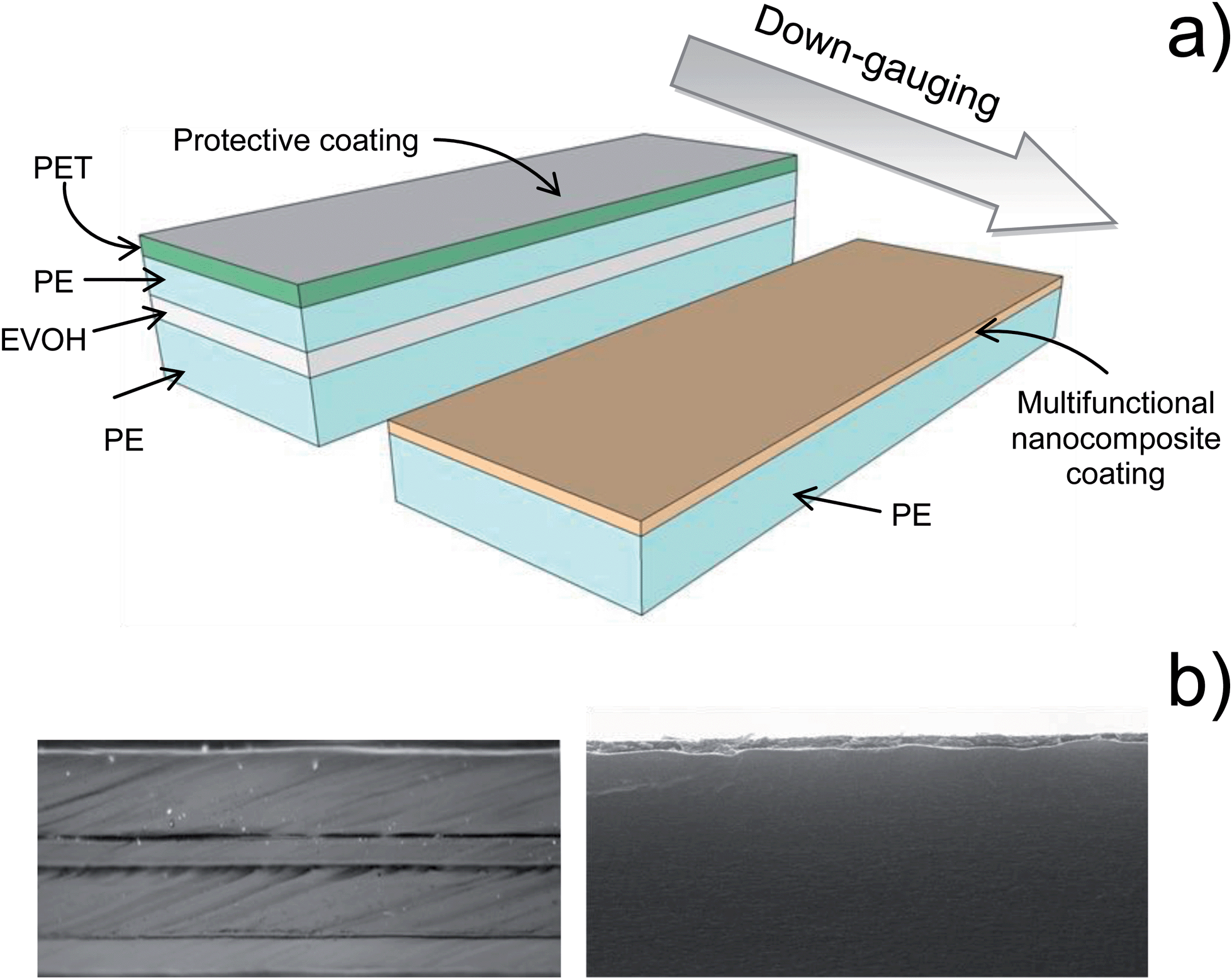 | ||
| Fig. 9 (a) Schematic illustration of the ‘packaging lightweighting’ concept: the use of multifunctional nanocomposite coatings makes possible avoiding multiple layers packages; (b) optical microscope cross-section of a multi-layer (total thickness 90 μm) package (left) and SEM of a nanocomposite coating (∼0.7 μm) on a 12 μm PET substrate (right). | ||
6. Concluding remarks
The development of new packaging materials offering new functionalities, less environmental impact, and economical benefits is now an urgent necessity. The driving forces behind this pressing task are manifold. On one hand, the shelf life extension of packaged products can be an answer to the exponential increase of worldwide demand for food. On the other hand, the increase in the price of crude oil, together with the uncertainty related to its durability, has imposed the necessity to manufacture new structures to replace oil-derived polymers partially or totally in the future. Finally, consumers' awareness towards environmental issues increasingly pushes industries to look with renewed interest to “green” solutions.Coating technology is one of the most efficient and convenient approaches to exploit toward this goal. This versatile technique enables manipulating matrices of completely different origins and compositions (e.g., water-based/organic solvent-based, biopolymer/oil-derived polymers, solutions/dispersions) to yield thin layers that ennoble the substrate beneath (e.g., plastic webs) by conferring new (sometimes unexpected) functional properties. Within a shelf life extension perspective, the generation of biopolymer nanocomposite coatings is one of the most striking and promising outcomes of coating technology for tomorrow's challenges.
Abbreviations
| AFG | Amine-functionalized graphene |
| BET | Brunnuer, Emmet and Teller |
| BC | Bacterial cellulose |
| CEC | Cation-exchange capacity |
| CMC | Carboxymethyl cellulose |
| CMKG | Carboxymethyl konjac glucomannan |
| CNCs | Cellulose nanocrystals |
| CNTs | Carbon nanotubes |
| CVD | Chemical vapor deposition |
| EVA | Ethylene vinyl acetate |
| EVOH | Ethylene vinyl alcohol |
| GCG | Gelidium corneum–gelatin |
| GNP | Graphene nanoplatelet |
| GO | Graphene oxide |
| HAL | Halloysite |
| HDPE | High density polyethylene |
| HEC | Hectorite |
| HNTs | Halloysite nanotubes |
| HRTEM | High-resolution transmission electron microscopy |
| IMO | Imogolite |
| KAO | Kaolinite |
| LAP | Laponite |
| LDHs | Layered double hydroxides |
| LDPE | Low density polyethylene |
| LLDPE | Linear low density polyethylene |
| MFC | Microfibrillated cellulose |
| MMT | Montmorillonite |
| MWNTs | Multi walled carbon nanotubes |
| PA | Polyamide |
| PBS | Poly(butylene succinate) |
| PCL | Polycaprolactone |
| PE | Polyethylene |
| PEI | Polyethylenimine |
| PEO | Poly(ethylene oxide) |
| PET | Poly(ethylene terephthalate) |
| γ-PGA | γ-Polyglutamate |
| PHAs | Poly-hydroxyalkanoates |
| PHB | Poly(3-hydroxybutyrate) |
| PHBV | Poly(3-hydroxybutyrate-co-3-hydroxyvalerate) |
| PIFG | Phenyl isocyanate-functionalized graphene |
| PLA | Poly(lactic acid) |
| PLLA | Poly(L-lactic acid) |
| PMMA | Poly(methyl methacrylate) |
| PND | Polynorbornene dicarboximide |
| Poly(S-co-BuA) | Poly(styrene-co-butyl acrylate) |
| PP | Polypropylene |
| PPC | Poly(propylene carbonate) |
| PS | Polystyrene |
| PU | Polyurethane |
| PVC | Poly(vinyl chloride) |
| PVOH | Poly(vinyl alcohol) |
| REC | Rectorite |
| SAP | Saponite |
| SEP | Sepiolite |
| SNCs | Starch nanocrystals |
| SPI | Soy protein isolate |
| VMT | Vermiculite |
| VOCs | Volatile organic compounds |
| WPI | Whey protein isolate |
References
- D. R. Baer, P. E. Burrows and A. A. El-Azab, Prog. Org. Coat., 2003, 47, 342 CrossRef CAS.
- A. L. Chun, Nat. Nanotechnol., 2009, 4, 790 CrossRef CAS PubMed.
- M. C. Roco, C. A. Mirkin and M. C. Hersam, J. Nanopart. Res., 2011, 13, 897 CrossRef.
- T. V. Duncan, J. Colloid Interface Sci., 2011, 363, 1 CrossRef CAS PubMed.
- Q. Chaudhry, M. Scotter, J. Blackburn, B. Ross, A. Boxall, L. Castle, R. Aitken and R. Watkin, Food Addit. Contam., Part A, 2008, 25, 241 CrossRef CAS PubMed.
- Cientifica, Nanotechnologies in the food industry, 2006, http://www.cientifica.com/www/details.php?id=47, accessed December 2012 Search PubMed.
- IRAP, Nano-enabled Packaging for Food and Beverage Industry, 2009, http://www.innoresearch.net/report_summary.aspx?id=68%26pg=107%26rcd=FT102%26pd=7/1/2009, accessed December 2012 Search PubMed.
- M. Siegrist, M. E. Cousin, H. Kastenholz and A. Wiek, Appetite, 2007, 49, 459 CrossRef PubMed.
- M. Siegrist, N. Stampfli, H. Kastenholz and C. Keller, Appetite, 2008, 51, 283 CrossRef PubMed.
- Q. Chaudhry, R. Watkins and L. Castle, in Nanotechnologies in Food, ed. Q. Chaudhry, R. Watkins and L. Castle, RSC Publishing, Cambridge, 2010, p. 1 Search PubMed.
- J. M. Lagaron, L. Cabedo, D. Cava, J. L. Feijoo, R. Gavara and E. Gimenez, Food Addit. Contam., 2005, 22, 994 CrossRef CAS PubMed.
- C. Sanchez, G. J. de A. A. Soler-Illia, F. Ribot, T. Lalot, C. R. Mayer and V. Cabuil, Chem. Mater., 2001, 13, 3061 CrossRef CAS.
- C. Sanchez, B. Julian, P. Belleville and M. Popall, J. Mater. Chem., 2005, 15, 3559 RSC.
- P. Sanguansri and M. A. Augustin, Trends Food Sci. Technol., 2006, 17, 547 CrossRef CAS PubMed.
- M. Alexandre and P. Dubois, Mater. Sci. Eng., 2000, 28, 1 CrossRef.
- E. P. Giannelis, Adv. Mater., 1996, 8, 29 CrossRef CAS.
- S. Sinha Ray and M. Okamoto, Prog. Polym. Sci., 2003, 28, 1539 CrossRef PubMed.
- A. Sorrentino, G. Gorrasi and V. Vittoria, Trends Food Sci. Technol., 2007, 18, 84 CrossRef CAS PubMed.
- L. Introzzi, T. O. Blomfeldt, S. Trabattoni, S. Tavazzi, N. Santo, A. Schiraldi, L. Piergiovanni and S. Farris, Langmuir, 2012, 28, 11206 CrossRef CAS PubMed.
- L. Introzzi, J. M. Fuentes-Alventosa, C. A. Cozzolino, S. Trabattoni, S. Tavazzi, C. L. Bianchi, A. Schiraldi, L. Piergiovanni and S. Farris, ACS Appl. Mater. Interfaces, 2012, 4, 3692 CAS.
- S. Farris, L. Introzzi, J. M. Fuentes-Alventosa, N. Santo, R. Rocca and L. Piergiovanni, J. Agric. Food Chem., 2012, 60, 782 CrossRef CAS PubMed.
- J. M. Fuentes-Alventosa, L. Introzzi, N. Santo, G. Cerri, A. Brundu and S. Farris, RSC Adv., 2013, 25086 RSC.
- M. Imran, A.-M. Revol-Junelles, A. Martyn, E. A. Tehrany, M. Jacquot, M. Linder and S. Desobry, Crit. Rev. Food Sci. Nutr., 2010, 50, 799 CrossRef CAS PubMed.
- E. L. Bradley, L. Castle and Q. Chaudhry, Trends Food Sci. Technol., 2011, 22, 604 CrossRef CAS PubMed.
- C. Silvestre, D. Duraccio and S. Cimmino, Prog. Polym. Sci., 2011, 36, 1766 CrossRef CAS PubMed.
- J. M. Lagaron and A. Lopez-Rubio, Trends Food Sci. Technol., 2011, 22, 611 CrossRef CAS PubMed.
- N. B. Hatzigrigoriou and C. D. Papaspyrides, J. Appl. Polym. Sci., 2011, 122, 3720 CrossRef CAS.
- B. A. Magnuson, T. S. Jonaitis and J. W. Card, J. Food Sci., 2011, 76, 126 CrossRef PubMed.
- M. Cushen, J. Kerry, M. Morris, M. Cruz-Romero and E. Cummins, Trends Food Sci. Technol., 2012, 24, 30 CrossRef CAS PubMed.
- J.-W. Rhim, H.-M. Park and C.-S. Ha, Prog. Polym. Sci., 2013, 38, 1629 CrossRef CAS PubMed.
- J. Ging, R. Tejerina-Anton, G. Ramakrishnan, M. Nielsen, K. Murphy, J. M. Gorham, T. Nguyen and A. Orlov, Sci. Total Environ., 2014, 473–474, 9 CrossRef CAS PubMed.
- S. Maisanaba, S. Pichardo, M. Jordá-Beneyto, S. Aucejo, A. M. Cameán and Á. Jos, Food Chem. Toxicol., 2014, 66, 366 CrossRef CAS PubMed.
- X. Guo and N. Mei, J. Food Drug Anal., 2014, 22, 105 CrossRef CAS PubMed.
- X. Liu and Q. Wu, Polymer, 2001, 42, 10013 CrossRef CAS.
- K. Sterky, H. Jacobsen, I. Jakubowicz, N. Yarahmadi and T. Hjertberg, Eur. Polym. J., 2010, 46, 1203 CrossRef CAS PubMed.
- S. Pavlidou and C. D. Papaspyrides, Prog. Polym. Sci., 2008, 33, 1119 CrossRef CAS PubMed.
- M. Maiti, S. Sadhu and A. K. Bhowmick, J. Appl. Polym. Sci., 2005, 96, 443 CrossRef CAS.
- T. V. Kosmidou, eXPRESS Polym. Lett., 2008, 2, 364 CrossRef CAS.
- X. Ye, P. J. Yao, M. M. Xi and L. Q. Zhang, Plast., Rubber Compos., 2013, 42, 50 CrossRef CAS PubMed.
- A. Al-Jabareen, H. Al-Bustami, H. Harel and G. Marom, J. Appl. Polym. Sci., 2013, 128, 1534 CAS.
- K. Kalaitzidou, H. Fukushima and L. T. Drzal, Composites, Part A, 2007, 38, 1675 CrossRef PubMed.
- K. Fukushima, M. Murariu, G. Camino and P. Dubois, Polym. Degrad. Stab., 2010, 95, 1063 CrossRef CAS PubMed.
- J. N. Coleman, U. Khan and Y. K. Gun'ko, Adv. Mater., 2006, 18, 689 CrossRef CAS.
- M. T. Byrne and Y. K. Gunko, Adv. Mater., 2010, 22, 1672 CrossRef CAS PubMed.
- Y. X. Zhou, eXPRESS Polym. Lett., 2008, 2, 40 CrossRef CAS.
- S. P. Bao and S. C. Tjong, Polym. Compos., 2009, 30, 1749 CrossRef CAS.
- X. Chen, S. Wei, A. Yadav, R. Patil, J. Zhu, R. Ximenes, L. Sun and Z. Guo, Macromol. Mater. Eng., 2011, 296, 434 CrossRef CAS.
- L. Sun, J. Y. O'Reilly, C. W. Tien and H. J. Sue, J. Chem. Educ., 2008, 85, 1105 CrossRef CAS.
- E. T. Thostenson, Z. Ren and T. W. Chou, Compos. Sci. Technol., 2001, 61, 1899 CrossRef CAS.
- B. E. Kilbride, J. N. Coleman, J. Fraysse, P. Fournet, M. Cadek, A. Drury, S. Hutzler, S. Roth and W. J. Blau, J. Appl. Phys., 2002, 92, 4024 CrossRef CAS PubMed.
- M. J. Biercuk, M. C. Llaguno, M. Radosavljevic, J. K. Hyun, A. T. Johnson and J. E. Fischer, Appl. Phys. Lett., 2002, 80, 2767 CrossRef CAS PubMed.
- O. Regev, P. N. B. ElKati, J. Loos and C. E. Koning, Adv. Mater., 2004, 16, 248 CrossRef CAS.
- B. K. Price, J. L. Hudson and J. M. Tour, J. Am. Chem. Soc., 2005, 127, 14867 CrossRef CAS PubMed.
- K. S. Coleman, Annu. Rep. Prog. Chem., Sect. A: Inorg. Chem., 2012, 108, 478 RSC.
- N. A. Kotov, Nature, 2006, 442, 254 CrossRef CAS PubMed.
- H. H. Murray, Appl. Clay Sci., 2000, 17, 207 CrossRef CAS.
- J. C. Miranda-Trevino and C. A. Coles, Appl. Clay Sci., 2003, 23, 133 CrossRef CAS.
- D. H. Powell, H. E. Fischer and N. T. Skipper, J. Phys. Chem. B, 1998, 102, 10899 CrossRef CAS.
- X. Kornmann, H. Lindberg and L. A. Berglund, Polymer, 2001, 42, 1303 CrossRef CAS.
- F. Chivrac, E. Pollet and L. Avérous, Mater. Sci. Eng., R, 2009, 67, 1 CrossRef PubMed.
- H. M. Park, W. K. Lee, C. Y. Park, W. J. Cho and C. Ha, J. Mater. Sci., 2003, 38, 909 CrossRef CAS.
- H. M. Park, M. Misra, L. T. Drzal and A. K. Mohanty, Biomacromolecules, 2004, 5, 2281 CrossRef CAS PubMed.
- M. Avella, J. J. De Vlieger, M. E. Errico, S. Fischer, P. Vacca and M. G. Volpe, Food Chem., 2005, 93, 467 CrossRef CAS PubMed.
- B. Chen and J. R. G. Evans, Carbohydr. Polym., 2005, 61, 455 CrossRef CAS PubMed.
- B. Chen and J. R. G. Evans, Polym. Int., 2005, 54, 807 CrossRef CAS.
- H. M. Wilhelm, M. R. Sierakowski, G. P. Souza and F. Wypych, Carbohydr. Polym., 2003, 52, 101 CrossRef CAS.
- M. F. Brigatti, E. Galan and B. K. G. Theng, in Developments in Clay Science, ed. F. Bergaya, B. K. G. Then and G. Lagaly, Elsevier, 2006, vol. 1, pp. 19–86 Search PubMed.
- J. Elzea and H. Murray, in Industrial Minerals and Rocks, ed. D. Carr, SME, Littleton, CO, 1994, pp. 233–246 Search PubMed.
- E. Manias, A. Touny, L. Wu, K. Strawhecker, B. Lu and T. Chung, Chem. Mater., 2001, 13, 3516 CrossRef CAS.
- I. J. Chin, T. Thurn-Albrecht, H. C. Kim, T. P. Russell and J. Wang, Polymer, 2001, 42, 5947 CrossRef CAS.
- F. Thomas, L. J. Michot, D. Vantelon, E. Montarges, B. Prélot, M. Cruchaudet and J. F. Delon, Colloids Surf., A, 1999, 159, 351 CrossRef CAS.
- J. Xu, R. K. Y. Li, Y. Xu, L. Li and Y. Z. Meng, Eur. Polym. J., 2005, 41, 881 CrossRef CAS PubMed.
- T. Nogueira, R. Botan, F. Wypych and L. Lona, J. Appl. Polym. Sci., 2012, 124, 1764 CrossRef CAS.
- C. H. Davis, L. J. Mathias, J. W. Gilman, D. A. Schiraldi, J. R. Shields, P. Trulove, T. E. Sutto and H. C. Delong, J. Polym. Sci., Part B: Polym. Phys., 2002, 40, 2661 CrossRef CAS.
- W. Joon Choi, H. J. Kim, K. Han Yoon, O. Hyeong Kwon and C. Ik Hwang, J. Appl. Polym. Sci., 2006, 100, 4875 CrossRef.
- L. V. Todorov and J. C. Viana, J. Appl. Polym. Sci., 2007, 106, 1659 CrossRef CAS.
- S. Martínez-Gallegos, M. Herrero, C. Barriga, F. M. Labajos and V. Rives, Appl. Clay Sci., 2009, 45, 44 CrossRef PubMed.
- R. S. Rajeev, E. Harkin-Jones, K. Soon, T. McNally, G. Menary, C. G. Armstrong and P. J. Martin, Eur. Polym. J., 2009, 45, 332 CrossRef CAS PubMed.
- K. Soon, E. Harkin-Jones, R. S. Rajeev, G. Menary, P. J. Martin and C. G. Armstrong, Polym. Eng. Sci., 2012, 52, 532 CAS.
- C. E. Corcione, A. Cavallo, E. Pesce, A. Greco and A. Maffezzoli, Polym. Eng. Sci., 2011, 51, 1280 CAS.
- M. Herrero, S. Martínez-Gallegos, F. M. Labajos and V. Rives, J. Solid State Chem., 2011, 184, 2862 CrossRef CAS PubMed.
- R. Scaffaro, L. Botta, M. Ceraulo and F. P. La Mantia, J. Appl. Polym. Sci., 2011, 122, 384 CrossRef CAS.
- S. Hayrapetyan, A. Kelarakis, L. Estevez, Q. Lin, K. Dana, Y. L. Chung and E. P. Giannelis, Polymer, 2012, 53, 422 CrossRef CAS PubMed.
- R. Labde, E. A. Lofgren and S. A. Jabarin, J. Appl. Polym. Sci., 2012, 125, E369 CrossRef CAS.
- M. Modesti, A. Lorenzetti, D. Bon and S. Besco, Polymer, 2005, 46, 10237 CrossRef CAS PubMed.
- M. J. Dumont, A. Reyna-Valencia, J. P. Emond and M. Bousmina, J. Appl. Polym. Sci., 2007, 103, 618 CrossRef CAS.
- A. Mirzadeh and M. Kokabi, Eur. Polym. J., 2007, 43, 3757 CrossRef CAS PubMed.
- M. A. Osman, V. Mittal and U. W. Suter, Macromol. Chem. Phys., 2007, 208, 68 CrossRef CAS.
- D. A. Pereira de Abreu, P. Paseiro Losada, I. Angulo and J. M. Cruz, Eur. Polym. J., 2007, 43, 2229 CrossRef CAS PubMed.
- N. Touati, M. Kaci, H. Ahouari, S. Bruzaud and Y. Grohens, Macromol. Mater. Eng., 2007, 292, 1271 CrossRef CAS.
- M. A. Treece and J. P. Oberhauser, J. Appl. Polym. Sci., 2007, 103, 884 CrossRef CAS.
- S. Cimmino, D. Duraccio, C. Silvestre and M. Pezzuto, Appl. Surf. Sci., 2009, 256, S40 CrossRef CAS PubMed.
- M. A. Perez, B. L. Rivas, S. M. Rodrigues, Á. Maldonado and C. Venegas, J. Chil. Chem. Soc., 2010, 55, 440 CrossRef CAS.
- R. N. Choi, C. I. Cheigh, S. Y. Lee and M. S. Chung, J. Food Sci., 2011, 76, N62 CrossRef CAS PubMed.
- S. Cimmino, C. Silvestre, D. Duraccio and M. Pezzuto, J. Appl. Polym. Sci., 2011, 119, 1135 CrossRef CAS.
- D. Fischer, J. Müller, S. Kummer and B. Kretzschmar, Macromol. Symp., 2011, 305, 10 CrossRef CAS.
- M. R. Manikantan and N. Varadharaju, Packag. Technol. Sci., 2011, 24, 191–209 CrossRef CAS.
- S. P. Lonkar, S. Therias, F. Leroux, J. L. Gardette and R. P. Singh, Polym. Eng. Sci., 2012, 52, 2006 CAS.
- K. Srithammaraj, R. Magaraphan and H. Manuspiya, Packag. Technol. Sci., 2012, 25, 63 CrossRef CAS.
- Y. Zhong and D. De Kee, Polym. Eng. Sci., 2005, 45, 469 CAS.
- M. Shafiee and A. Ramazani S.A., Macromol. Symp., 2008, 274, 1 CrossRef CAS.
- S. Joseph and W. W. Focke, Polym. Compos., 2011, 32, 252 CrossRef CAS.
- R. Wilson, T. S. Plivelic, A. S. Aprem, C. Ranganathaiagh, S. A. Kumar and S. Thomas, J. Appl. Polym. Sci., 2012, 123, 3806 CrossRef CAS.
- P. M. Gyoo, S. Venkataramani and S. C. Kim, J. Appl. Polym. Sci., 2006, 101, 1711 CrossRef CAS.
- A. Ammala, A. J. Hill, K. A. Lawrence and T. Tran, J. Appl. Polym. Sci., 2007, 104, 1377 CrossRef CAS.
- A. Ammala, S. J. Pas, K. A. Lawrence, R. Stark, R. I. Webb and A. J. Hill, J. Mater. Chem., 2008, 18, 911 RSC.
- K. Magniez, B. L. Fox and M. G. Looney, J. Polym. Sci., Part B: Polym. Phys., 2009, 47, 1300 CrossRef CAS.
- D. Pereira, P. P. Losada, I. Angulo, W. Greaves and J. M. Cruz, Polym. Compos., 2009, 30, 436 CrossRef CAS.
- F. Touchaleaume, J. Soulestin, M. Sclavons, J. Devaux, M. F. Lacrampe and P. Krawczak, Polym. Degrad. Stab., 2011, 96, 1890 CrossRef CAS PubMed.
- I. Isik-Gulsac, U. Yilmazer and G. Bayram, J. Appl. Polym. Sci., 2012, 125, 4060 CrossRef CAS.
- S. Sánchez-Valdes, M. L. López-Quintanilla, E. Ramírez-Vargas, F. J. Medellín-Rodríguez and J. M. Gutierrez-Rodriguez, Macromol. Mater. Eng., 2006, 291, 128 CrossRef.
- S. K. Swain and A. I. Isayev, Polymer, 2007, 48, 281 CrossRef CAS PubMed.
- C. Lotti, C. S. Isaac, M. C. Branciforti, R. M. V. Alves, S. Liberman and R. E. S. Bretas, Eur. Polym. J., 2008, 44, 1346 CrossRef CAS PubMed.
- S. Coiai, M. Scatto, L. Conzatti, F. Azzurri, L. Andreotti, E. Salmini, P. Stagnaro, A. Zanolin, F. Cicogna and E. Passaglia, Polym. Adv. Technol., 2001, 22, 2285 CrossRef.
- S. M. Ali Dadfar, I. Alemzadeh, S. M. Reza Dadfar and M. Vosoughi, Mater. Des., 2011, 32, 1806 CrossRef PubMed.
- Q. Hu, Y. Fang, Y. Yang, N. Ma and L. Zhao, Food Res. Int., 2011, 44, 1589 CrossRef CAS PubMed.
- M. H. Abdolrasouli, E. Behzadfar, H. Nazockdast and F. Sharif, J. Appl. Polym. Sci., 2012, 125, E435 CrossRef CAS.
- M. A. Busolo and J. M. Lagaron, Innovative Food Sci. Emerging Technol., 2012, 16, 211 CrossRef CAS PubMed.
- S. I. Hong and J. W. Rhim, LWT--Food Sci. Technol., 2012, 48, 43 CrossRef CAS PubMed.
- P. Santamaría and J. I. Eguiazabal, Polym. Test., 2012, 31, 367 CrossRef PubMed.
- L. Zhao, F. Li, G. Chen, Y. Fang, X. An, Y. Zheng, Z. Xin, M. Zhang, Y. Yang and Q. Hu, Int. J. Food Sci. Technol., 2012, 47, 572 CrossRef CAS PubMed.
- M. A. Priolo, K. M. Holder, D. Gamboa and J. C. Grunlan, Langmuir, 2011, 27, 12106 CrossRef CAS PubMed.
- C. H. Chen, C. C. Teng and C. H. Yang, J. Polym. Sci., Part B: Polym. Phys., 2005, 43, 1465 CrossRef CAS.
- M. Ge and D. Jia, J. Thermoplast. Compos. Mater., 2010, 23, 855 CrossRef CAS PubMed.
- A. Bendahou, H. Kaddami, E. Espuche, F. Gouanvé and A. Dufresne, Macromol. Mater. Eng., 2011, 296, 760 CrossRef CAS.
- C. Esposito Corcione, P. Prinari, D. Cannoletta, G. Mensitieri and A. Maffezzoli, Int. J. Adhes. Adhes., 2008, 28, 91 CrossRef CAS PubMed.
- C. Esposito Corcione, G. Mensitieri and A. Maffezzoli, Polym. Eng. Sci., 2009, 49, 1708 CAS.
- A. K. Barick and D. K. Tripathy, J. Appl. Polym. Sci., 2010, 117, 639 CrossRef CAS.
- Y. Qian, C. I. Lindsay, C. Macosko and A. Stein, ACS Appl. Mater. Interfaces, 2011, 3, 3709 CAS.
- J. T. Yeh, C. J. Chang, F. C. Tsai, K. N. Chen and K. S. Huang, Appl. Clay Sci., 2009, 45, 1 CrossRef CAS PubMed.
- S. Donadi, M. Modesti, A. Lorenzetti and S. Besco, J. Appl. Polym. Sci., 2011, 122, 3290 CrossRef CAS.
- B. Kouini and A. Serier, Mater. Des., 2012, 34, 313 CrossRef CAS PubMed.
- N. Artzi, A. Tzur, M. Narkis and A. Siegmann, Polym. Compos., 2005, 26, 343 CrossRef CAS.
- M. A. Priolo, D. Gamboa, K. M. Holder and J. C. Grunlan, Nano Lett., 2010, 10, 4970 CrossRef CAS PubMed.
- H. M. Park, X. Li, C. Z. Jin, C. Y. Park, W. J. Cho and C. S. Ha, Macromol. Mater. Eng., 2002, 287, 553 CrossRef CAS.
- P. Kampeerapappun, D. Aht-ong, D. Pentrakoon and K. Srikulkit, Carbohydr. Polym., 2007, 67, 155 CrossRef CAS PubMed.
- V. P. Cyras, L. B. Manfredi, M. T. Ton-That and A. Vázquez, Carbohydr. Polym., 2008, 73, 55 CrossRef CAS PubMed.
- N. Wang, X. Zhang, N. Han and S. Bai, Carbohydr. Polym., 2009, 76, 68 CrossRef CAS PubMed.
- M. Hassan Nejad, J. Ganster and B. Volkert, J. Appl. Polym. Sci., 2010, 118, 503 CrossRef CAS.
- M. Hassan Nejad, J. Ganster, A. Bohn, B. Volkert and A. Lehmann, Carbohydr. Polym., 2011, 84, 90 CrossRef CAS PubMed.
- K. Majdzadeh-Ardakani, A. H. Navarchian and F. Sadeghi, Carbohydr. Polym., 2010, 79, 547 CrossRef CAS PubMed.
- W. Gao, H. Dong, H. Hou and H. Zhang, Carbohydr. Polym., 2012, 88, 321 CrossRef CAS PubMed.
- S. F. Wang, L. Shen, Y. J. Tong, L. Chen, I. Y. Phang, P. Q. Lim and T. X. Lu, Polym. Degrad. Stab., 2005, 90, 123 CrossRef CAS PubMed.
- S. I. Hong, J. H. Lee, H. J. Bae, S. Y. Koo, H. S. Lee, J. H. Choi, D. H. Kim, S. H. Park and H. J. Park, J. Appl. Polym. Sci., 2011, 119, 2742 CrossRef CAS.
- H. Oguzlu and F. Tihminlioglu, Macromol. Symp., 2010, 298, 91 CrossRef CAS.
- M. Abdollahi, M. Rezaei and G. Farzi, J. Food Eng., 2012, 111, 343 CrossRef CAS PubMed.
- M. Alboofetileh, M. Rezaei, H. Hosseini and M. Abdollahi, J. Food Eng., 2013, 117, 26 CrossRef CAS PubMed.
- P. Krishnamachari, J. Zhang, J. Lou, J. Yan and L. Uitenham, Int. J. Polym. Anal. Charact., 2009, 14, 336 CrossRef CAS.
- V. P. Martino, A. Jiménez, R. A. Ruseckaite and L. Avérous, Polym. Adv. Technol., 2011, 22, 2206 CrossRef CAS.
- D. V. Plackett, V. K. Holm, P. Johansen, S. Ndoni, P. V. Nielsen, T. Sipilainen-Malm, A. Södergård and S. Verstichel, Packag. Technol. Sci., 2006, 19, 1 CrossRef CAS.
- A. C. Wibowo, M. Misra, H. M. Park, L. T. Drzal, R. Schalek and A. K. Mohanty, Composites, Part A, 2006, 37, 1428 CrossRef PubMed.
- P. Kumar, K. P. Sandeep, S. Alavi, V. D. Truong and R. E. Gorga, J. Food Sci., 2010, 75, N46 CrossRef CAS PubMed.
- J. E. Lee and K. M. Kim, J. Appl. Polym. Sci., 2010, 118, 2257 CAS.
- H. J. Bae, H. J. Park, D. O. Darby, R. M. Kimmel and W. S. Whiteside, Packag. Technol. Sci., 2009, 22, 371 CrossRef CAS.
- P. Mangiacapra, G. Gorrasi, A. Sorrentino and V. Vittoria, Carbohydr. Polym., 2006, 64, 516 CrossRef CAS PubMed.
- J. W. Rhim, S. B. Lee and S. I. Hong, J. Food Sci., 2011, 76, N40 CrossRef CAS PubMed.
- B. Chen and J. R. Evans, Macromolecules, 2006, 39, 747 CrossRef CAS.
- J. Lu and C. Xiao, J. Appl. Polym. Sci., 2007, 103, 2954 CrossRef CAS.
- M. Quilaqueo Gutiérrez, I. Echeverría, M. Ihl, V. Bifani and A. N. Mauri, Carbohydr. Polym., 2012, 87, 1495 CrossRef PubMed.
- M. A. Paul, M. Alexandre, P. Degée, C. Henrist, A. Rulmont and P. Dubois, Polymer, 2003, 44, 443 CrossRef CAS.
- K. Sermsantiwanit and S. Phattanarudee, Prog. Org. Coat., 2912, 74, 660 CrossRef PubMed.
- J. Ahmed, R. Auras, T. Kijchavengkul and S. K. Varshney, J. Food Eng., 2012, 111, 580 CrossRef CAS PubMed.
- F. Tezcan, E. Gunister, G. Ozen and F. B. Erim, Int. J. Biol. Macromol., 2012, 50, 1165 CrossRef CAS PubMed.
- I. Siró, D. Plackett and P. Sommer-Larsen, Packag. Technol. Sci., 2010, 23, 301 CrossRef.
- M. Żenkiewicz, J. Richert and A. Różański, Polym. Test., 2010, 29, 251 CrossRef PubMed.
- F. J. Rodríguez, M. J. Galotto, A. Guarda and J. E. Bruna, J. Food Eng., 2012, 110, 262 CrossRef PubMed.
- J. Vartiainen, M. Tuominen and K. Nättinen, J. Appl. Polym. Sci., 2010, 116, 3638 CAS.
- M. Lavorgna, F. Piscitelli, P. Mangiacapra and G. G. Buonocore, Carbohydr. Polym., 2010, 82, 291 CrossRef CAS PubMed.
- I. Olabarrieta, M. Gällstedt, I. Ispizua, J. R. Sarasua and M. S. Hedenqvist, J. Agric. Food Chem., 2006, 54, 1283 CrossRef CAS PubMed.
- M. R. Guilherme, L. H. C. Mattoso, N. Gontard, S. Guilbert and E. Gastaldi, Composites, Part A, 2010, 41, 375 CrossRef PubMed.
- H. Dai, P. R. Chang, F. Geng, J. Yu and X. Ma, J. Polym. Environ., 2009, 17, 225 CrossRef CAS.
- A. C. Souza, R. Benze, E. S. Ferrão, C. Ditchfield, A. C. V. Coelho and C. C. Tadini, LWT--Food Sci. Technol., 2012, 46, 110 CrossRef CAS PubMed.
- S. Tunc, H. Angellier, Y. Cahyana, P. Chalier, N. Gontard and E. Gastaldi, J. Membr. Sci., 2007, 289, 159 CrossRef CAS PubMed.
- A. Casariego, B. W. S. Souza, M. A. Cerqueira, J. A. Teixeira, L. Cruz, R. Díaz and A. A. Vicente, Food Hydrocolloids, 2009, 23, 1895 CrossRef CAS PubMed.
- S. Tunç and O. Duman, Appl. Clay Sci., 2010, 48, 414 CrossRef PubMed.
- J. W. Rhim, S. I. Hong, H. M. Park and P. K. Ng, J. Agric. Food Chem., 2006, 54, 5814 CrossRef CAS PubMed.
- S. I. Hong and J. W. Rhim, J. Nanosci. Nanotechnol., 2008, 8, 5818 CrossRef CAS PubMed.
- R. Sothornvit, S. I. Hong, D. J. An and J. W. Rhim, LWT--Food Sci. Technol., 2010, 43, 279 CrossRef CAS PubMed.
- J. W. Rhim, S. I. Hong and C. S. Ha, LWT--Food Sci. Technol., 2009, 42, 612 CrossRef CAS PubMed.
- E. Mascheroni, P. Chalier, N. Gontard and E. Gastaldi, Food Hydrocolloids, 2010, 24, 406 CrossRef CAS PubMed.
- E. Mascheroni, V. Guillard, E. Gastaldi, N. Gontard and P. Chalier, Food Control, 2011, 22, 1582 CrossRef CAS PubMed.
- S. A. Jang, G. O. Lim and K. B. Song, Int. J. Food Sci. Technol., 2010, 45, 1883 CrossRef CAS PubMed.
- C. Costa, A. Conte, G. G. Buonocore, M. Lavorgna and M. A. Del Nobile, Food Res. Int., 2012, 48, 164 CrossRef CAS PubMed.
- D. Eberl, Clays Clay Miner., 1978, 26, 327 CAS.
- X. Ma, H. Lu, G. Liang and H. Yan, J. Appl. Polym. Sci., 2004, 93, 608 CrossRef CAS.
- L. Yang, G. Liang, Z. Zhang, S. He and J. Wang, J. Appl. Polym. Sci., 2009, 114, 1235 CrossRef CAS.
- Z. Jinhong, L. Xiancai, Z. Jianxi, L. Xiandong, W. Jingming, Z. Qing, Y. Peng and H. Hongping, J. Phys. Chem. C, 2012, 116, 13071 Search PubMed.
- J. He, S. Hong, L. Zhang, F. Gan and Y. S. Ho, Fresenius Environ. Bull., 2010, 19, 2651 CAS.
- X. Wang, Y. Du, J. Luo, B. Lin and J. F. Kennedy, Carbohydr. Polym., 2007, 69, 41 CrossRef CAS PubMed.
- P. R. Chang, D. Wu, D. P. Anderson and X. Ma, Carbohydr. Polym., 2012, 89, 687 CrossRef CAS PubMed.
- H. Deng, P. Lin, S. Xin, R. Huang, W. Li, Y. Du, X. Zhou and J. Yang, Carbohydr. Polym., 2012, 89, 307 CrossRef CAS PubMed.
- X. Li, X. Li, B. Ke, X. Shi and Y. Du, Carbohydr. Polym., 2011, 85, 747 CrossRef CAS PubMed.
- B. Casal, J. Merino, E. Ruiz-Hitzky, E. Gutierrez and A. Alvarez, Clay Miner., 1997, 32, 41 CAS.
- H. Suquet, C. de la Calle and H. Pezerat, Clays Clay Miner., 1975, 23, 1 CAS.
- D. W. Kim, B. C. Ku, D. Steeves, R. Nagarajan, A. Blumstein, J. Kumar, P. W. Gibson, J. A.Ratto and L. A. Samuelson, J. Membr. Sci., 2006, 275, 12 CrossRef CAS PubMed.
- L. A. Utracki, M. Sepehr and E. Boccaleri, Polym. Adv. Technol., 2007, 18, 1 CrossRef CAS.
- W. Zhen, C. Lu, C. Li and M. Liang, Appl. Clay Sci., 2012, 57, 64 CrossRef CAS PubMed.
- W. L. Hemme, W. Fujita, K. Awaga and H. Eckert, Dalton Trans., 2009, 7995 RSC.
- F. Bergaya and G. Lagaly, in Developments in Clay Science, ed. B. K. G. T. Faïza Bergaya and L. Gerhard, Elsevier, 2006, pp. 1–18 Search PubMed.
- V. C. Farmer and J. D. Russell, Trans. Faraday Soc., 1971, 67, 2737 RSC.
- A. Okada and A. Usuki, Mater. Sci. Eng., C, 1995, 3, 109 CrossRef.
- S. Sinha Ray, K. Okamoto and M. Okamoto, Macromolecules, 2003, 36, 2355 CrossRef.
- J. H. Yeun, G. S. Bang, B. J. Park, S. K. Ham and J. H. Chang, J. Appl. Polym. Sci., 2006, 101, 591 CrossRef CAS.
- W. Zhen and C. Lu, Appl. Surf. Sci., 2012, 258, 6969 CrossRef CAS PubMed.
- N. Willenbacher, J. Colloid Interface Sci., 1996, 182, 501 CrossRef CAS.
- Technical Brochure “Laponite as a film forming agent”, Laponite: performance additives, Rockwood Additives Limited, Cheshire, United Kingdom. pp. 1–22.
- P. D. Kaviratna, T. J. Pinnavaia and P. A. Schroeder, J. Phys. Chem. Solids, 1996, 57, 1897 CrossRef CAS.
- G. F. Perotti, H. S. Barud, Y. Messaddeq, S. J. L. Ribeiro and V. R. L. Constantino, Polymer, 2011, 52, 157 CrossRef CAS PubMed.
- A. Mourchid, E. Lécolier, H. Van Damme and P. Levitz, Langmuir, 1998, 14, 4718 CrossRef CAS.
- Laponite Additives, Synthetic Silicate Additives, http://www.laponite.com/faqs.asp#_What_is_the_5, accessed May 2013 Search PubMed.
- Technical Bulletin Laponite L104/90/A, Laponite: Structure, chemistry and relationship to natural clays, Laporte Absorbents, Cherise, United Kingdom, 1990, pp. 1–15 Search PubMed.
- Laponite® Technical Brochure, Laponite: performance additives, Rockwood Additives Limited: A Rockwood Holdings, Inc. Company, Cheshire, United Kingdom, pp. 1–22, http://www.byk.com/en/additives/product-groups/rheology/former-rockwood-additives/technical-brochures.html, accessed February 2013.
- K. Wang, M. Xu, Y. Choi and I. Chung, Polym. Bull., 2001, 46, 499 CrossRef CAS.
- H. A. Essawy, N. A. Abd El-Wahab and M. A. Abd El-Ghaffar, Polym. Degrad. Stab., 2008, 93, 1472 CrossRef CAS PubMed.
- S. H. Nair, K. C. Pawar, J. P. Jog and M. V. Badiger, J. Appl. Polym. Sci., 2007, 103, 2896 CrossRef CAS.
- T. U. Patro and H. D. Wagner, Nanotechnology, 2011, 22, 455706 CrossRef PubMed.
- M. Gabr, N. Phong, M. Abdelkareem, K. Okubo, K. Uzawa, I. Kimpara and T. Fujii, Cellulose, 2013, 20, 819 CrossRef CAS.
- X. Tang and S. Alavi, J. Agric. Food Chem., 2012, 60, 1954 CrossRef CAS PubMed.
- M. F. Traynor, M. M. Mortland and T. J. Pinnavaia, Clays Clay Miner., 1978, 26, 318 Search PubMed.
- T. Sánchez, F. B. Gebretsadik, P. Salagre, Y. Cesteros, N. Guillén-Hurtado, A. García-García and A. Bueno-López, Appl. Clay Sci., 2013, 77–78, 40 CrossRef PubMed.
- H. Van Olphen and J. J. Fripiar, Soil Sci., 1981, 131 Search PubMed.
- K. Fukushima, D. Tabuani and G. Camino, Mater. Sci. Eng., C, 2012, 32, 1790 CrossRef CAS PubMed.
- M. D. Sanchez-Garcia, E. Gimenez and J. M. Lagaron, J. Appl. Polym. Sci., 2008, 108, 2787 CrossRef CAS.
- M. S. Żbik, N. A. Raftery, R. St. C. Smart and R. L. Frost, Appl. Clay Sci., 2010, 50, 299 CrossRef PubMed.
- A. J. F. de Carvalho, A. A. S. Curvelo and J. A. M. Agnelli, Carbohydr. Polym., 2001, 45, 189 CrossRef CAS.
- K. Kaewtatip and V. Tanrattanakul, Mater. Des., 2012, 37, 423 CrossRef CAS PubMed.
- J. A. Mbey, S. Hoppe and F. Thomas, Carbohydr. Polym., 2012, 88, 213 CrossRef CAS PubMed.
- P. Lu, M. Zhang, P. Qian and Q. Zhu, Polym. Compos., 2012, 33, 889 CrossRef CAS.
- L. Cabedo, J. Luis Feijoo, M. Pilar Villanueva, J. M. Lagarón and E. Giménez, Macromol. Symp., 2006, 233, 191 CrossRef CAS.
- Y. Lvov and E. Abdullayev, Prog. Polym. Sci., 2013, 38, 1690 CrossRef CAS PubMed.
- Y. M. Lvov, D. G. Shchukin, H. Mohwald and R. R. Price, ACS Nano, 2008, 2, 814 CrossRef CAS PubMed.
- G. Cavallaro, D. I. Donato, G. Lazzara and S. Milioto, J. Phys. Chem. C, 2011, 115, 20491 CAS.
- D. Carroli, Geol. Soc. Am. Bull., 1959, 70, 749 CrossRef.
- Y. Lvov, R. Price, B. Gaber and I. Ichinose, Colloids Surf., A, 2002, 198–200, 375 CrossRef CAS.
- Y. Lvov and R. Price, Halloysite nanotubes in nanomaterials research, Technical Note, http://www.sigmaaldrich.com/materials-science/nanomaterials/nanoclay-building/halloysite-nanotubes.html, accessed May 2013 Search PubMed.
- R. R. Price, B. P. Gaber and Y. Lvov, J. Microencapsulation, 2001, 18, 713 CrossRef CAS PubMed.
- D. G. Shchukin and H. Möhwald, Adv. Funct. Mater., 2007, 17, 1451 CrossRef CAS.
- D. G. Shchukin, S. V. Lamaka, K. A. Yasakau, M. L. Zheludkevich, M. G. S. Ferreira and H. Mohwald, J. Phys. Chem. C, 2008, 112, 958 CAS.
- E. Abdullayev, R. Price, D. Shchukin and Y. Lvov, ACS Appl. Mater. Interfaces, 2009, 1, 1437 CAS.
- G. S. Machado, K. A. D. de Freitas Castro, F. Wypych and S. Nakagaki, J. Mol. Catal. B: Enzym., 2008, 283, 99 CrossRef CAS PubMed.
- H. Ismail, P. Pasbakhsh, M. N. A. Fauzi and A. Abu Bakar, Polym. Test., 2008, 27, 841 CrossRef CAS PubMed.
- P. Pasbakhsh, H. Ismail, M. N. A. Fauzi and A. A. Bakar, Polym. Test., 2009, 28, 548 CrossRef CAS PubMed.
- P. Pasbakhsh, H. Ismail, M. N. A. Fauzi and A. A. Bakar, Appl. Clay Sci., 2010, 48, 405 CrossRef CAS PubMed.
- H. C. Voon, R. Bhat, A. M. Easa, M. T. Liong and A. A. Karim, Food Bioprocess Technol., 2010, 5, 1766 CrossRef.
- Y. He, W. Kong, W. Wang, T. Liu, Y. Liu, Q. Gong and J. Gao, Carbohydr. Polym., 2012, 87, 2706 CrossRef CAS PubMed.
- W. Wu, X. Cao, Y. Zhang and G. He, J. Appl. Polym. Sci., 2013, 130, 443 CrossRef CAS.
- K. Prashantha, B. Lecouvet, M. Sclavons, M. F. Lacrampe and P. Krawczak, J. Appl. Polym. Sci., 2013, 128, 1895 CAS.
- G. Gorrasi, R. Pantani, M. Murariu and P. Dubois, Macromol. Mater. Eng., 2014, 299, 104 CrossRef CAS.
- P. Russo, S. Cammarano, E. Bilotti, T. Peijs, P. Cerruti and D. Acierno, J. Appl. Polym. Sci., 2014, 131, 39798 CrossRef.
- M. Soheilmoghaddam and M. U. Wahit, Int. J. Biol. Macromol., 2013, 58, 133 CrossRef CAS PubMed.
- P. Aranda, R. Kun, M. A. Martín-Luengo, S. Letaïef, I. Dékány and E. Ruiz-Hitzky, Chem. Mater., 2008, 20, 84 CrossRef CAS.
- L. González, A. Rodríguez, A. Marcos-Fernández and A. Del Campo, J. Appl. Polym. Sci., 2001, 79, 714 CrossRef.
- R. L. Frost and Z. Ding, Thermochim. Acta, 2003, 397, 119 CrossRef CAS.
- L. Bokobza, A. Burr, G. Garnaud, M. Y. Perrin and S. Pagnotta, Polym. Int., 2004, 53, 1060 CrossRef CAS.
- G. Tartaglione, D. Tabuani and G. Camino, Microporous Mesoporous Mater., 2008, 107, 161 CrossRef CAS PubMed.
- N. Volle, L. Challier, A. Burr, F. Giulieri, S. Pagnotta and A. M. Chaze, Compos. Sci. Technol., 2011, 71, 1685 CrossRef CAS PubMed.
- H. Shariatmadari, A. Mermut and M. Benke, Clays Clay Miner., 1999, 47, 44 CAS.
- J. Jog, Crystallization in Polymer Nanocomposites, Optimization of Polymer Nanocomposite Properties, Wiley-VCH Verlag GmbH & Co. KGaA, 2010, pp. 279–300 Search PubMed.
- J. L. Ahlrichs, C. Serna and J. M. Serratosa, Clays Clay Miner., 1975, 23, 119 CAS.
- M. C. Hermosín and J. Cornejo, Clays Clay Miner., 1986, 34, 591 Search PubMed.
- G. Tartaglione, D. Tabuani, G. Camino and M. Moisio, Compos. Sci. Technol., 2008, 68, 451 CrossRef CAS PubMed.
- A. Marcilla, A. Gómez, S. Menargues and R. Ruiz, Polym. Degrad. Stab., 2005, 88, 456 CrossRef CAS PubMed.
- M. Liu, M. Pu and H. Ma, Compos. Sci. Technol., 2012, 72, 1508 CrossRef CAS PubMed.
- T. D. Hapuarachchi and T. Peijs, Composites, Part A, 2010, 41, 954 CrossRef PubMed.
- F. Chivrac, E. Pollet, M. Schmutz and L. Avérous, Carbohydr. Polym., 2010, 80, 145 CrossRef CAS PubMed.
- B. Giménez, M. C. Gómez-Guillén, M. E. López-Caballero, J. Gómez-Estaca and P. Montero, Food Hydrocolloids, 2012, 27, 475 CrossRef PubMed.
- Z. Sárossy, T. O. J. Blomfeldt, M. S. Hedenqvist, C. B. Koch, S. S. Ray and D. Plackett, ACS Appl. Mater. Interfaces, 2012, 4, 3378 Search PubMed.
- K. Fukushima, D. Tabuani and G. Camino, Mater. Sci. Eng., C, 2009, 29, 1433 CrossRef CAS PubMed.
- A. C. S. Alcântara, M. Darder, P. Aranda and E. Ruiz-Hitzky, Eur. J. Inorg. Chem., 2012, 5216 CrossRef.
- K. Zhang, J. Xu, K. Y. Wang, L. Cheng, J. Wang and B. Liu, Polym. Degrad. Stab., 2009, 94, 2121 CrossRef CAS PubMed.
- M. F. Brigatti, S. Colonna, D. Malferrari, L. Medici and L. Poppi, Appl. Clay Sci., 2005, 28, 1 CrossRef CAS PubMed.
- L. Wang, X. Wang, Z. Chen and P. Ma, Appl. Clay Sci., 2013, 75–76, 74 CrossRef CAS PubMed.
- S. Williams-Daryn and R. K. Thomas, J. Colloid Interface Sci., 2002, 255, 303 CrossRef CAS.
- C. Marcos, Y. Arango and I. Rodriguez, Appl. Clay Sci., 2009, 42, 368 CrossRef CAS PubMed.
- A. Auliawan and E. M. Woo, Polym. Compos., 2011, 32, 1916 CrossRef CAS.
- A. Auliawan and E. M. Woo, J. Appl. Polym. Sci., 2012, 125, E444 CrossRef CAS.
- M. J. Fernández, M. D. Fernández and I. Aranburu, Appl. Clay Sci., 2013, 80–81, 372 CrossRef PubMed.
- M. J. Fernández, M. D. Fernández and I. Aranburu, Eur. Polym. J., 2013, 49, 1257 CrossRef PubMed.
- J. Drelich, B. Li, P. Bowen, J. Y. Hwang, O. Mills and D. Hoffman, Appl. Surf. Sci., 2011, 257, 9435 CrossRef CAS PubMed.
- Y. Tsujimoto, A. Yoshida, M. Kobayashi and Y. Adachi, Colloids Surf., A, 2013, 435, 109 CrossRef CAS PubMed.
- J. P. Gustafsson, Clays Clay Miner., 2001, 49, 73 CAS.
- F. V. C. Cradwick, J. D. Russell, C. R. Masson, K. Wada and N. Yoshinaga, Nat. Phys. Sci., 1972, 240, 187 CrossRef.
- S. Mukherjee, V. M. Bartlow and S. Nair, Chem. Mater., 2005, 17, 4900 CrossRef CAS.
- W. Ma, W. O. Yah, H. Otsuka and A. Takahara, J. Mater. Chem., 2012, 22, 11887 RSC.
- C. Clark and M. B. McBride, Clays Clay Miner., 1984, 32, 291 CAS.
- C. Su and J. B. Harsh, Clays Clay Miner., 1993, 41, 461 CAS.
- J. Karube and Y. Abe, Clays Clay Miner., 1998, 46, 322 CAS.
- C. Forano, T. Hibino, F. Leroux and C. Taviot-Guého, in Developments in Clay Science, ed. F. Bergaya, B. K. G. Theng and G. Lagaly, Elsevier, 2006, vol. 1, pp. 1021–1095 Search PubMed.
- M. F. Chiang and T. M. Wu, Compos. Sci. Technol., 2010, 70, 110 CrossRef CAS PubMed.
- F. Li and X. Duan, in Layered Double Hydroxides, ed. X. Duan and D. Evans, Springer, Berlin, 2006, pp. 193–223 Search PubMed.
- L. Moyo, N. Nhlapo and W. Focke, J. Mater. Sci., 2008, 43, 6144 CrossRef CAS PubMed.
- P. Ding, M. Zhang, J. Gai and B. Qu, J. Mater. Chem., 2007, 17, 1117 RSC.
- T. Kameda, T. Yamazaki and T. Yoshioka, Microporous Mesoporous Mater., 2008, 114, 410 CrossRef CAS PubMed.
- T. Sato, H. Fujita, T. Endo, M. Shimada and A. Tsunashima, React. Solids, 1988, 5, 219 CrossRef CAS.
- M. F. Chiang, E. C. Chen and T. M. Wu, Polym. Degrad. Stab., 2012, 97, 995 CrossRef CAS PubMed.
- S. O'Leary, D. O'Hare and G. Seeley, Chem. Commun., 2002, 1506 RSC.
- M. F. Chiang and T. M. Wu, Composites, Part B, 2012, 43, 2789 CrossRef CAS PubMed.
- M. F. Chiang and T. M. Wu, Appl. Clay Sci., 2011, 51, 330 CrossRef CAS PubMed.
- V. Bugatti, U. Costantino, G. Gorrasi, M. Nocchetti, L. Tammaro and V. Vittoria, Eur. Polym. J., 2010, 46, 418 CrossRef CAS PubMed.
- R. Pucciariello, L. Tammaro, V. Villani and V. Vittoria, J. Polym. Sci., Part B: Polym. Phys., 2007, 45, 945 CrossRef CAS.
- V. Bugatti, G. Gorrasi, F. Montanari, M. Nocchetti, L. Tammaro and V. Vittoria, Appl. Clay Sci., 2011, 52, 34 CrossRef CAS PubMed.
- G. Gorrasi, V. Bugatti and V. Vittoria, Carbohydr. Polym., 2012, 89, 132 CrossRef CAS PubMed.
- J. B. Dixon, Appl. Clay Sci., 1991, 5, 489 CrossRef CAS.
- H. Van Olphen, An introduction to clay colloid chemistry: For clay technologists, geologists, and soil scientists, Interscience (Wiley), New York, 1977 Search PubMed.
- http://www.topy.co.jp/english/dept/mica/industrial.html, last access December 2013.
- P. Dallas, V. K. Sharma and R. Zboril, Adv. Colloid Interface Sci., 2011, 166, 119 CAS.
- A. D. Russell and W. B. Hugo, Prog. Med. Chem., 1994, 31, 351 CrossRef CAS.
- Q. L. Feng, J. Wu, G. Q. Chen, F. G. Cui, T. N. Kim and J. O. Kim, J. Biomed. Mater. Res., 2000, 52, 662 CrossRef CAS.
- I. Sondi and B. Salopek-Sondi, J. Colloid Interface Sci., 2004, 275, 177 CrossRef CAS PubMed.
- S. Egger, R. P. Lehmann, M. J. Height, M. J. Loessner and M. Schuppler, Appl. Environ. Microbiol., 2009, 75, 2973 CrossRef CAS PubMed.
- P. Appendini and J. H. Hotchkiss, Innovative Food Sci. Emerging Technol., 2002, 3, 113 CrossRef CAS.
- S. Sánchez-Valdes, H. Ortega-Ortiz, L. F. Ramos-de Valle, F. J. Medellín-Rodríguez and R. Guedea-Miranda, J. Appl. Polym. Sci., 2009, 111, 953 Search PubMed.
- R. Gottesman, S. Shukla, N. Perkas, L. A. Solovyov, Y. Nitzan and A. Gedanken, Langmuir, 2011, 27, 720 CrossRef CAS PubMed.
- A. M. Fayaz, K. Balaji, M. Girilal, P. T. Kalaichelvan and R. Venkatesan, J. Agric. Food Chem., 2009, 57, 6246 CrossRef PubMed.
- R. Yoksan and S. Chirachanchai, Mater. Sci. Eng., C, 2010, 30, 891 CrossRef CAS PubMed.
- K. Vimala, Y. M. Mohan, K. S. Sivudu, K. Varaprasad, S. Ravindra, N. N. Reddy, Y. Padma, B. Sreedhar and K. MohanaRaju, Colloids Surf., B, 2010, 76, 248 CrossRef CAS PubMed.
- S. Tripathi, G. K. Mehrotra and P. K. Dutta, Bull. Mater. Sci., 2011, 34, 29 CrossRef CAS PubMed.
- R. Tankhiwale and S. K. Bajpai, Colloids Surf., B, 2009, 69, 164 CrossRef CAS PubMed.
- C. Zhu, J. Xue and J. He, J. Nanosci. Nanotechnol., 2009, 9, 3067 CrossRef CAS PubMed.
- S. T. Dubas, P. Kumlangdudsana and P. Potiyaraj, Colloids Surf., A, 2006, 289, 105 CrossRef CAS PubMed.
- J. S. Kim, K. E. Kuk, K. N. Yu, J. H. Kim, S. J. Park, H. J. Lee, S. H. Kim, Y. K. Park, Y. H. Park, C. Y. Hwang, Y. K. Kim, Y. S. Lee, D. H. Jeong and M. H. Cho, J. Nanomed. Nanotechnol., 2007, 3, 95 CrossRef CAS PubMed.
- C. Damm, H. Münsted and A. Rösch, Mater. Chem. Phys., 2008, 108, 61 CrossRef CAS PubMed.
- A. Llorens, E. Lloret, P. A. Picouet, R. Trbojevich and A. Fernandez, Trends Food Sci. Technol., 2012, 24, 19 CrossRef CAS PubMed.
- N. M. Ushakov, A. N. Ulzutuev and I. D. Kosobudskii, Tech. Phys., 2008, 53, 1597 CrossRef CAS.
- G. Cardenas, J. Diaz, M. F. Melendrez, C. Cruzat and A. Garcia Cancino, Polym. Bull., 2009, 62, 511 CrossRef CAS PubMed.
- A. Fernández, E. Soriano, P. Hernández-Munoz and R. Gavara, J. Food Sci., 2010, 75, 186 CrossRef PubMed.
- C. Chawengkijwanich and Y. Hayata, Int. J. Food Microbiol., 2008, 123, 288 CrossRef CAS PubMed.
- M. L. Cerrada, C. Serrano, M. Sanchez-Chaves, M. Fernandez-Garcia, F. Fernandez-Martin, A. De Andres, R. J. J. Riobóo, A. Kubacka, M. Ferrer and M. Fernández-García, Adv. Funct. Mater., 2008, 18, 1949 CrossRef CAS.
- W. Zhang, Y. Chen, S. Yu, S. Chen and Y. Yin, Thin Solid Films, 2008, 516, 4690 CrossRef CAS PubMed.
- L. Xiao-e, A. N. M. Green, S. A. Haque, A. Mills and J. R. Durrant, J. Photochem. Photobiol., A, 2004, 162, 253 CrossRef PubMed.
- A. Kubacka, C. Serrano, M. Ferrer, H. Lunsdorf, P. Bielecki, M. L. Cerrada, M. Fernández-Garcia and M. Fernández-Garcia, Nano Lett., 2007, 7, 2529 CrossRef CAS PubMed.
- S. W. Lee, C. Mao, C. E. Flynn and A. M. Belcher, Science, 2002, 296, 892 CrossRef CAS PubMed.
- E. Celik, Z. Gokcen, N. F. Ak Azem, M. Tanoglu and O. F. Emrullahoglu, Mater. Sci. Eng., B, 2006, 132, 258 CrossRef CAS PubMed.
- R. Dastjerdi and M. Montazer, Colloids Surf., B, 2010, 79, 5 CrossRef CAS PubMed.
- X. Li, Y. Xing, Y. Jiang, Y. Ding and W. Li, Int. J. Food Sci. Technol., 2009, 44, 2161 CrossRef CAS PubMed.
- X. Li, W. Li, Y. Xing, Y. Jiang, Y. Ding and P. Zhang, Adv. Mater. Res., 2011, 152–153, 450 CAS.
- A. Emamifar, M. Kadivar, M. Shahedi and S. Soleimanian-Zad, Innovative Food Sci. Emerging Technol., 2010, 11, 742 CrossRef CAS PubMed.
- T. Jin and J. B. Gurtler, J. Appl. Microbiol., 2010, 110, 704 CrossRef PubMed.
- O. Yamamoto, Int. J. Inorg. Mater., 2001, 3, 643 CrossRef CAS.
- N. Jones, B. Ray, K. K. T. Ranjit and A. C. Manna, FEMS Microbiol. Lett., 2008, 279, 71 CrossRef CAS PubMed.
- L. H. Chen, X. Y. Li, J. C. Rooke, Y. H. Zhang, X. Y. Yang, Y. Tang, F. S. Xiao and B. L. Su, J. Mater. Chem., 2012, 22, 17381 RSC.
- J. Gascon, F. Kapteijn, B. Zornoza, V. Sebastián, C. Casado and J. Coronas, Chem. Mater., 2012, 24, 2829 CrossRef CAS.
- M. P. Pina, R. Mallada, M. Arruebo, M. Urbiztondo, N. Navascués, O. de la Iglesia and J. Santamaria, Microporous Mesoporous Mater., 2011, 144, 19 CrossRef CAS PubMed.
- C. M. Zimmerman, A. Singh and W. J. Koros, J. Membr. Sci., 1997, 137, 145 CrossRef CAS.
- Z. Huang, J. F. Su, X. Q. Su, Y. H. Guo, L. J. Teng and C. Min Yang, J. Appl. Polym. Sci., 2009, 112, 9 CrossRef CAS.
- D. Sirikittikul, A. Fuongfuchat and W. Booncharoen, Polym. Adv. Technol., 2009, 20, 802 CrossRef CAS.
- Y. Shen and A. C. Lua, Polym. Eng. Sci., 2012, 52, 2106 CAS.
- A. L. Ahmad, M. G. Mohd Nawawi and L. K. So, J. Appl. Polym. Sci., 2006, 99, 1740 CrossRef CAS.
- O. G. Nik, A. Moheb and T. Mohammadi, Chem. Eng. Technol., 2006, 29, 1340 CrossRef CAS.
- J. G. Varghese, A. A. Kittur and M. Y. Kariduraganavar, J. Appl. Polym. Sci., 2009, 111, 2408 CrossRef CAS.
- C. V. Prasad, B. Y. Swamy, H. Sudhakar, T. Sobharani, K. Sudhakar, M. C. S. Subha and K. C. Rao, J. Appl. Polym. Sci., 2011, 121, 1521 CrossRef CAS.
- H. Sudhakar, C. Venkata Prasad, K. Sunitha, K. Chowdoji Rao, M. C. S. Subha and S. Sridhar, J. Appl. Polym. Sci., 2011, 121, 2717 CrossRef CAS.
- M. L. Lind, A. K. Ghosh, A. Jawor, X. Huang, W. Hou, Y. Yang and E. M. V. Hoek, Langmuir, 2009, 25, 10139 CrossRef CAS PubMed.
- N. Ma, J. Wei, R. Liao and C. Y. Tang, J. Membr. Sci., 2012, 405–406, 149 CrossRef CAS PubMed.
- G. J. Dahe, R. S. Teotia and J. R. Bellare, Chem. Eng. J., 2012, 197, 398 CrossRef CAS PubMed.
- S. Polarz and B. Smarsly, J. Nanosci. Nanotechnol., 2002, 2, 581 CrossRef CAS PubMed.
- T. Sakano, H. Tamon, M. Miyahara and M. Okazaki, Food Sci. Technol. Int., 1996, 2, 174 CrossRef CAS.
- T. Sakano, K. Yamamura, H. Tamon, M. Miyahara and M. Okazaki, J. Food Sci., 1996, 61, 473 CrossRef CAS PubMed.
- H. S. Kim and H. J. Kim, J. Appl. Polym. Sci., 2008, 110, 3247 CrossRef CAS.
- N. Cvetkovic, B. Adnadjevic and M. Nikolic, Beiträge zur Tabakforschung International, 2002, 20, 43 CAS.
- Y. Xu, J. H. Zhu, L. L. Ma, A. Ji, Y. L. Wei and X. Y. Shang, Microporous Mesoporous Mater., 2003, 60, 125 CrossRef CAS.
- L. Gao, Y. Wang, Y. Xu, S. L. Zhou, T. T. Zhuang, Z. Y. Wu and J. H. Zhu, Clean: Soil, Air, Water, 2008, 36, 270 CrossRef CAS.
- L. Gao, Y. Cao, S. L. Zhou, T. T. Zhuang, Y. Wang and J. H. Zhu, J. Hazard. Mater., 2009, 169, 1034 CrossRef CAS PubMed.
- A. Marcilla, A. Gómez-Siurana, D. Berenguer, I. Martínez-Castellanos and M. I. Beltrán, Microporous Mesoporous Mater., 2012, 161, 14 CrossRef CAS PubMed.
- W. M. Meier and K. Siegmann, Microporous Mesoporous Mater., 1999, 33, 307 CrossRef CAS.
- G. Cerri, M. Degennaro, M. Bonferoni and C. Caramella, Appl. Clay Sci., 2004, 27, 141 CrossRef CAS PubMed.
- S. Quintavalla and L. Vicini, Meat Sci., 2002, 62, 373 CrossRef CAS.
- FDA, Inventory of effective food contact substance (FCS) notifications, http://www.accessdata.fda.gov/scripts/fcn/fcnDetailNavigation.cfm?rpt=fcsListing%26id=697, accessed May 2013 Search PubMed.
- EFSA, Opinion of the scientific panel on food additives, flavourings, processing aids and materials in contact with food (AFC) on a request from the Commission related to a 7th list of substances for food contact materials, EFSA J., 2005, 201, 1 Search PubMed.
- EFSA, Scientific Opinion on the safety evaluation of the substance, silver zeolite A (silver zinc sodium ammonium alumino silicate), silver content 2–5%, for use in food contact materials, EFSA J., 2011, 9, 1999 Search PubMed.
- P. Monprasit, C. Ritvirulh, T. Sooknoi, S. Rukchonlatee, A. Fuongfuchat and D. Sirikittikul, Polym. Eng. Sci., 2011, 51, 1264 CAS.
- A. Jacobsson, T. Nielsen, I. Sjöholm and K. Wendin, Food Qual. Prefer., 2004, 15, 301 CrossRef.
- K. Boonruang, V. Chonhenchob, S. P. Singh, W. Chinsirikul and A. Fuongfuchat, Packag. Technol. Sci., 2012, 25, 107 CrossRef CAS.
- D. H. Shin, H. S. Cheigh and S. L. Dong, J. Food Eng., 2002, 53, 229 CrossRef.
- E. Frydrych, Z. Foltynowicz, S. Kowalak and E. Janiszewska, in Studies in Surface Science and Catalysis, ed. R. Xu, Z. Gao, J. Chen and W. Yan, Elsevier, 2007, vol. 170, pp. 1597–1604 Search PubMed.
- F. Ozmihci, D. Balkose and S. Ulku, J. Appl. Polym. Sci., 2001, 82, 2913–2921 CrossRef CAS.
- D. Balköse, K. Oguz, L. Ozyuzer, S. Tari, E. Arkis and F. O. Omurlu, J. Appl. Polym. Sci., 2011, 120, 1671 CrossRef.
- J. Biswas, H. Kim, S. Shim and G. Kim, J. Ind. Eng. Chem., 2004, 10, 582 CAS.
- I. E. Yuzay, R. Auras and S. Selke, J. Appl. Polym. Sci., 2010, 115, 2262 CrossRef CAS.
- H. Pehlivan, D. Balköse, S. Ülkü and F. Tihminlioğlu, Compos. Sci. Technol., 2005, 65, 2049 CrossRef CAS PubMed.
- D. Metın, F. Tihminlioğlu, D. Balköse and S. Ülkü, Composites, Part A, 2004, 35, 23 CrossRef PubMed.
- S. Djoumaliisky and P. Zipper, Macromol. Symp., 2004, 217, 391 CrossRef CAS.
- H. Pehlivan, D. Balköse, S. Ülkü and F. Tıhmınlıoğlu, J. Appl. Polym. Sci., 2006, 101, 143 CrossRef CAS.
- I. E. Yuzay, R. Auras, H. Soto-Valdez and S. Selke, Polym. Degrad. Stab., 2010, 95, 1769 CrossRef CAS PubMed.
- H. Zhen, Y. Xiaoxu, P. Xi, Z. Yulong, F. Meifen and Z. Qiang, Thermal degradation study of sodium alginate-zeolite 4A composites, Proc. the 17th IAPRI World Conference on Packag., Tianjin, China, Scientific Research Publishing, USA, 2010, pp. 406–409 Search PubMed.
- R. Thipmanee and A. Sane, J. Appl. Polym. Sci., 2012, 126, 252 CrossRef.
- M. Terrones, A. R. Botello-Méndez, J. Campos-Delgado, F. López-Urías, Y. I. Vega-Cantú, F. J. Rodríguez-Macías, A. L. Elías, E. Muñoz-Sandoval, A. G. Cano-Márquez, J.-C. Charlier and H. Terrones, Nano Today, 2010, 5, 351 CrossRef PubMed.
- M. Eizenberg and J. M. Blakely, Surf. Sci., 1979, 82, 228 CrossRef CAS.
- K. S. Novoselov, A. K. Geim, S. V. Morozov, D. Jiang, Y. Zhang, S. V. Dubonos, I. V. Grigorieva and A. A. Firsov, Science, 2004, 306, 666 CrossRef CAS PubMed.
- C. Lee, X. Wei, J. W. Kysar and J. Hone, Science, 2008, 321, 385 CrossRef CAS PubMed.
- A. A. Balandin, S. Ghosh, W. Bao, I. Calizo, D. Teweldebrhan, F. Miao and C. N. Lau, Nano Lett., 2008, 8, 902 CrossRef CAS PubMed.
- K. I. Bolotin, K. J. Sikes, Z. Jiang, M. Klima, G. Fudenberg, J. Hone, P. Kim and H. L. Stormer, Solid State Commun., 2008, 146, 351 CrossRef CAS PubMed.
- M. D. Stoller, S. Park, Y. Zhu, J. An and R. S. Ruoff, Nano Lett., 2008, 8, 3498 CrossRef CAS PubMed.
- Y. Zhang, Y. W. Tan, H. L. Stormer and P. Kim, Nature, 2005, 438, 201 CrossRef CAS PubMed.
- J. S. Bunch, S. S. Verbridge, J. S. Alden, A. M. van der Zande, J. M. Parpia, H. G. Craighead and P. L. McEuen, Nano Lett., 2008, 8, 2458 CrossRef CAS PubMed.
- L. Imperiali, C. Clasen, J. Fransaer, C. W. Macosko and J. Vermant, Mater. Horiz., 2014, 1, 139 RSC.
- L. Staudenmaier, Ber. Dtsch. Chem. Ges., 1898, 31, 1481 CrossRef CAS.
- W. S. Hummers and R. E. Offeman, J. Am. Chem. Soc., 1958, 80, 1339 CrossRef CAS.
- S. Niyogi, E. Bekyarova, M. E. Itkis, J. L. McWilliams, M. A. Hamon and R. C. Haddon, J. Am. Chem. Soc., 2006, 128, 7720 CrossRef CAS PubMed.
- G. Eda, G. Fanchini and M. Chhowalla, Nat. Nanotechnol., 2008, 3, 270 CrossRef CAS PubMed.
- X. Li, G. Zhang, X. Bai, X. Sun, X. Wang, E. Wang and H. Dai, Nat. Nanotechnol., 2008, 3, 538 CrossRef CAS PubMed.
- G. Eda and M. Chhowalla, Adv. Mater., 2010, 22, 2392 CrossRef CAS PubMed.
- A. Buchsteiner, A. Lerf and J. Pieper, J. Phys. Chem. B, 2006, 110, 22328 CrossRef CAS PubMed.
- S. Bae, H. Kim, Y. Lee, X. Xu, J. S. Park, Y. Zheng, J. Balakrishnan, T. Lei, H. R. Kim, Y. I. Song, Y. J. Kim, K. S. Kim, B. Özyilmaz, J. H. Ahn, B. H. Hong and S. Iijima, Nat. Nanotechnol., 2010, 5, 574 CrossRef CAS PubMed.
- L. Gomez De Arco, Y. Zhang, C. W. Schlenker, K. Ryu, M. E. Thompson and C. Zhou, ACS Nano, 2010, 4, 2865 CrossRef CAS PubMed.
- C. Berger, Z. Song, T. Li, X. Li, A. Y. Ogbazghi, R. Feng, Z. Dai, A. N. Marchenkov, E. H. Conrad, P. N. First and W. A. de Heer, J. Phys. Chem. B, 2004, 108, 19912 CrossRef CAS.
- M. Sprinkle, M. Ruan, Y. Hu, J. Hankinson, M. Rubio-Roy, B. Zhang, X. Bu, C. Berger and W. A. de Heer, Nat. Nanotechnol., 2010, 5, 727 CrossRef CAS PubMed.
- W. A. de Heer, C. Berger, X. Wu, M. Sprinkle, Y. Hu, M. Ruan, J. A. Stroscio, P. N. First, R. Haddon, B. Piot, C. Faugeras, M. Potemski and J. S. Moon, J. Phys. D: Appl. Phys., 2010, 43, 374007 CrossRef.
- J. Wintterlin and M. L. Bocquet, Surf. Sci., 2009, 603, 1841 CrossRef CAS PubMed.
- A. Zangwill and D. D. Vvedensky, Nano Lett., 2011, 11, 2092 CrossRef CAS PubMed.
- C. D. Kim, B. K. Min and W. S. Jung, Carbon, 2009, 47, 1610 CrossRef CAS PubMed.
- A. Chakrabarti, J. Lu, J. C. Skrabutenas, T. Xu, Z. Xiao, J. A. Maguire and N. S. Hosmane, J. Mater. Chem., 2011, 21, 9491 RSC.
- W. Zhao, M. Fang, F. Wu, H. Wu, L. Wang and G. Chen, J. Mater. Chem., 2010, 20, 5817 RSC.
- J. Chen, M. Duan and G. Chen, J. Mater. Chem., 2012, 22, 19625 RSC.
- Y. Hernandez, V. Nicolosi, M. Lotya, F. M. Blighe, Z. Sun, S. De, I. T. McGovern, B. Holland, M. Byrne, Y. K. Gun'Ko, J. J. Boland, P. Niraj, G. Duesberg, S. Krishnamurthy, R. Goodhue, J. Hutchison, V. Scardaci, A. C. Ferrari and J. N. Coleman, Nat. Nanotechnol., 2008, 3, 563 CrossRef CAS PubMed.
- A. Ciesielski and P. Samorì, Chem. Soc. Rev., 2014, 43, 381 RSC.
- W. Yang, K. R. Ratinac, S. P. Ringer, P. Thordarson, J. J. Gooding and F. Braet, Angew. Chem., Int. Ed., 2010, 49, 2114 CrossRef CAS PubMed.
- Y. Feng, X. Zhang, Y. Shen, K. Yoshino and W. Feng, Carbohydr. Polym., 2012, 87, 644 CrossRef CAS PubMed.
- X. Huang, Z. Yin, S. Wu, X. Qi, Q. He, Q. Zhang, Q. Yan, F. Boey and H. Zhang, Small, 2011, 7, 1876 CrossRef CAS PubMed.
- F. Liu, Y. Piao, K. S. Choi and T. S. Seo, Carbon, 2012, 50, 123 CrossRef CAS PubMed.
- Y. Yao, Z. Lin, Z. Li, X. Song, K. S. Moon and C. P. Wong, J. Mater. Chem., 2012, 22, 13494 RSC.
- M. A. Raza, A. Westwood, A. Brown, N. Hondow and C. Stirling, Carbon, 2011, 49, 4269 CrossRef CAS PubMed.
- M. A. Raza, A. V. K. Westwood, A. P. Brown and C. Stirling, Compos. Sci. Technol., 2012, 72, 467 CrossRef CAS PubMed.
- Y. H. Yang, L. Bolling, M. A. Priolo and J. C. Grunlan, Adv. Mater., 2013, 25, 503 CrossRef CAS PubMed.
- M. H. Tsai, I. H. Tseng, Y. F. Liao and J. C. Chiang, Polym. Int., 2013, 62, 1302 CrossRef CAS.
- M. H. Tsai, C. J. Chang, H. H. Lu, Y. F. Liao and I. H. Tseng, Thin Solid Films, 2013, 544, 324 CrossRef CAS PubMed.
- I. H. Tseng, Y. F. Liao, J. C. Chiang and M. H. Tsai, Mater. Chem. Phys., 2012, 136, 247 CrossRef CAS PubMed.
- K. H. Liao, Y. Qian and C. W. Macosko, Polymer, 2012, 53, 3756 CrossRef CAS PubMed.
- V. Štengl, D. Popelková and P. Vláčil, J. Phys. Chem. C, 2011, 115, 25209 Search PubMed.
- Y. Zhu, S. Murali, M. D. Stoller, K. J. Ganesh, W. Cai, P. J. Ferreira, A. Pirkle, R. M. Wallace, K. A. Cychosz, M. Thommes, D. Su, E. A. Stach and R. S. Ruoff, Science, 2011, 332, 1537 CrossRef CAS PubMed.
- V. K. Rana, M. C. Choi, J. Y. Kong, G. Y. Kim, M. J. Kim, S. H. Kim, S. Mishra, R. P. Singh and C. S. Ha, Macromol. Mater. Eng., 2011, 296, 131 CrossRef CAS.
- A. M. Pinto, S. Moreira, I. C. Gonçalves, F. M. Gama, A. M. Mendes and F. D. Magalhães, Colloids Surf., B, 2013, 104, 229 CrossRef CAS PubMed.
- W. L. Song, L. M. Veca, C. Y. Kong, S. Ghose, J. W. Connell, P. Wang, L. Cao, Y. Lin, M. J. Meziani, H. Qian, G. E. LeCroy and Y. P. Sun, Polymer, 2012, 53, 3910–3916 CrossRef CAS PubMed.
- M. El Achaby, F. Z. Arrakhiz, S. Vaudreuil, E. M. Essassi and A. Qaiss, Appl. Surf. Sci., 2012, 258, 7668 CrossRef CAS PubMed.
- A. A. Al-Ghamdi, F. El-Tantawy, N. Abdel Aal, E. H. El-Mossalamy and W. E. Mahmoud, Polym. Degrad. Stab., 2009, 94, 980 CrossRef CAS PubMed.
- A. A. Al-Ghamdi and F. El-Tantawy, Composites, Part A, 2010, 41, 1693 CrossRef PubMed.
- S. Mao, H. Pu and J. Chen, RSC Adv., 2012, 2, 2643 RSC.
- Z. S. Wu, G. Zhou, L. C. Yin, W. Ren, F. Li and H. M. Cheng, Nano Energy, 2012, 1, 107 CrossRef CAS PubMed.
- Z. Liu, J. Li and F. Yan, Adv. Mater., 2013, 25, 4296 CrossRef CAS PubMed.
- C. H. Chang, T. C. Huang, C. W. Peng, T. C. Yeh, H. I. Lu, W. I. Hung, C. J. Weng, T. I.Yang and J. M. Yeng, Carbon, 2012, 50, 5044 CrossRef CAS PubMed.
- N. H. Kim, T. Kuila and J. H. Lee, J. Mater. Chem. A, 2013, 1, 1349 CAS.
- B. Mortazavi, F. Hassouna, A. Laachachi, A. Rajabpour, S. Ahzi, D. Chapron, V. Toniazzo and D. Ruch, Thermochim. Acta, 2013, 552, 106 CrossRef CAS PubMed.
- W. Zheng, X. Lu and S. C. Wong, J. Appl. Polym. Sci., 2004, 91, 2781 CrossRef CAS.
- H. Kim and C. W. Macosko, Polymer, 2009, 50, 3797 CrossRef CAS PubMed.
- P. J. Hubert, K. Kathiresan and K. Wakabayashi, Polym. Eng. Sci., 2011, 51, 2273 CAS.
- S. Bandla and J. C. Hanan, J. Mater. Sci., 2011, 47, 876 CrossRef.
- D. Cai, J. Jin, K. Yusoh, R. Rafiq and M. Song, Compos. Sci. Technol., 2012, 72, 702 CrossRef CAS PubMed.
- M. El Achaby, F. E. Arrakhiz, S. Vaudreuil, A. el Kacem Qaiss, M. Bousmina and O. Fassi-Fehri, Polym. Compos., 2012, 33, 733 CrossRef CAS.
- X. Yang, Y. Zhan, R. Zhao and X. Liu, J. Appl. Polym. Sci., 2012, 124, 1723 CrossRef CAS.
- J. Wang, X. Wang, C. Xu, M. Zhang and X. Shang, Polym. Int., 2011, 60, 816 CrossRef CAS.
- H. J. Salavagione, G. Martínez and M. A. Gómez, J. Mater. Chem., 2009, 19, 5027 RSC.
- H. J. Salavagione and G. Martínez, Macromolecules, 2011, 44, 2685 CrossRef CAS.
- D. Lee, M. C. Choi and C. S. Ha, J. Polym. Sci., Part A: Polym. Chem., 2012, 50, 1611 CrossRef CAS.
- T. Kuila, P. Khanra, A. K. Mishra, N. H. Kim and J. H. Lee, Polym. Test., 2012, 31, 282 CrossRef CAS PubMed.
- T. Kuila, S. Bose, A. K. Mishra, P. Khanra, N. H. Kim and J. H. Lee, Polym. Test., 2012, 31, 31 CrossRef CAS PubMed.
- H. Kim, S. Kobayashi, M. A. AbdurRahim, M. J. Zhang, A. Khusainova, M. A. Hillmyer, A. A. Abdala and C. W. Macasko, Polymer, 2011, 52, 1837 CrossRef CAS PubMed.
- R. R. Nair, H. A. Wu, P. N. Jayaram, I. V. Grigorieva and A. K. Geim, Science, 2012, 335, 442 CrossRef CAS PubMed.
- B. Min Yoo, H. Jin Shin, H. Wook Yoon and H. Bum Park, J. Appl. Polym. Sci., 2014, 131, 39628 Search PubMed.
- O. C. Compton, S. Kim, C. Pierre, J. M. Torkelson and S. T. Nguyen, Adv. Mater., 2010, 22, 4759 CrossRef CAS PubMed.
- A. M. Pinto, J. Cabral, D. A. P. Tanaka, A. M. Mendesa and F. D. Magalhães, Polym. Int., 2013, 62, 33 CrossRef CAS.
- J. Gao, F. Chen, K. Wang, H. Deng, Q. Zhang, H. Bai and Q. Fu, J. Mater. Chem., 2011, 21, 17627 RSC.
- A. Ashori, Polym. Eng. Sci., 2013 DOI:10.1002/pen.23774 , in press.
- Y. Lee, D. Kim, J. Seo, H. Han and S. B. Khan, Polym. Int., 2013, 62, 1386 CrossRef CAS.
- H.-D. Huang, P.-G. Ren, J.-Z. Xu, L. Xu, G.-J. Zhong, B. S. Hsiao and Z.-M. Li, J. Membr. Sci., 2014, 464, 110 CrossRef CAS PubMed.
- J. Yang, L. Bai, G. Feng, X. Yang, M. Lv, C. Zhang, H. Hu and X. Wang, Ind. Eng. Chem. Res., 2013, 52, 16745 CrossRef CAS.
- C. Xiang, P. J. Cox, A. Kukovecz, B. Genorio, D. P. Hashim, Z. Yan, Z. Peng, C.-C. Hwang, G. Ruan, E. L. G. Samuel, P. M. Sudeep, Z. Konya, X. R. Vajtai, P. M. Ajayan and J. M. Tour, ACS Nano, 2013, 7, 10380 CrossRef CAS PubMed.
- A. Al-Jabareen, H. Al-Bustami, H. Harel and G. Marom, J. Appl. Polym. Sci., 2013, 128, 1534 CAS.
- C. Wan and B. Chen, J. Appl. Polym. Sci., 2013, 127, 5094 CrossRef CAS.
- L.-L. Wu, J. Wang, X. He, T. Zhang and H. Sun, Packag. Technol. Sci., 2014 DOI:10.1002/pts.2058 , in press.
- D. Kim, H. Kwon and J. Seo, Polym. Compos., 2014, 35, 644 CrossRef CAS.
- P.-G. Ren, H. Wang, H.-D. Huang, D.-X. Yan and Z.-M. Li, J. Appl. Polym. Sci., 2014, 131, 39803 Search PubMed.
- T. K. Ghosh, S. Gope, D. Mondal, B. Bhowmik,Md, M. R. Mollick, D. Maity, I. Roy, G. Sarkar, S. Sadhukhan, D. Rana, M. Chakraborty and D. Chattopadhyay, Int. J. Biol. Macromol., 2014, 66, 338 CrossRef CAS PubMed.
- G. Oberdorster, A. Maynard, K. Donaldson, V. Castranova, J. Fitzpatrick, K. Ausman, J. Carter, B. Karn, W. Kreyling, D. Lai, S. Olin, N. Monteiro-Riviere, D. Warheit and H. Yang, Part. Fibre Toxicol., 2005, 2, 8 CrossRef PubMed.
- G. Oberdörster, E. Oberdörster and J. Oberdörster, Environ. Health Perspect., 2005, 113, 823 CrossRef.
- A. Nel, T. Xia, L. Madler and N. Li, Science, 2006, 311, 622 CrossRef CAS PubMed.
- K. L. Aillon, Y. Xie, N. El-Gendy, C. J. Berkland and M. L. Forrest, Adv. Drug Delivery Rev., 2009, 61, 457 CrossRef CAS PubMed.
- S. M. Hussain, L. K. Braydich-Stolle, A. M. Schrand, R. C. Murdock, K. O. Yu, D. M. Mattie, J. J. Schlager and M. Terrones, Adv. Mater., 2009, 21, 1549 CrossRef CAS.
- T. Xia, N. Li and A. E. Nel, Annu. Rev. Public Health, 2009, 30, 137 CrossRef PubMed.
- S. G. Prolongo, A. Jiménez-Suárez, R. Moriche and A. Ureñaet, Eur. Polym. J., 2014, 53, 292 CrossRef CAS PubMed.
- S. Li, M. M. Lin, M. S. Toprak, D. K. Kim and M. Muhammed, Nano Rev., 2010, 1, 5214 Search PubMed.
- V. Ojijo and S. Sinha Raya, Prog. Polym. Sci., 2013, 38, 1543 CrossRef CAS PubMed.
- H. Althues, J. Henle and S. Kaskel, Chem. Soc. Rev., 2007, 36, 1454 RSC.
- R. A. Vaia, K. D. Jandt, E. J. Kramer and E. P. Giannelis, Chem. Mater., 1996, 8, 2628 CrossRef CAS.
- R. A. Vaia and E. P. Giannelis, Macromolecules, 1997, 30, 7990 CrossRef CAS.
- H. R. Dennis, D. L. Hunter, D. Chang, S. Kim, J. L. White, J. W. Cho and D. R. Paul, Polymer, 2001, 42, 9513 CrossRef CAS.
- M. M. Reddy, S. Vivekanandhan, M. Misra, S. K. Bhatia and A. K. Mohanty, Prog. Polym. Sci., 2013, 38, 1653 CrossRef CAS PubMed.
- Y. H. Jin, H. J. Park, S. S. Im, S. Y. Kwak and S. Kwak, Macromol. Rapid Commun., 2002, 23, 135 CrossRef CAS.
- S. Farris, in The Wiley Encyclopedia of Packaging Technology, ed. K. L. Yam, Wiley & Sons, 3rd edn, 2009, pp. 285–294 Search PubMed.
- T. Hielscher, Ultrasonic Production of Nano-size Dispersions and Emulsions, Proceedings of European Nanosystems Conference ENS '05, Paris, 2005 Search PubMed.
- K. S. Suslick, Sci. Am., 1989, 260, 80 CrossRef CAS PubMed.
- C. R. Brenner, Cavitation and Bubble Dynamics, Oxford University Press, 1995 Search PubMed.
- T. G. Leighton, The Acoustic Bubble, Academic Press, 1994 Search PubMed.
- K. S. Suslick, Science, 1990, 247, 1439 CAS.
- K. S. Suslick, Kirk-Othmer Encyclopedia of Chemical Technology, Wiley & Sons, 4th edn, 1998 Search PubMed.
- K. S. Suslick and D. J. Flannigan, Annu. Rev. Phys. Chem., 2008, 59, 659 CrossRef CAS PubMed.
- J. R. Blake and D. C. Gibson, Annu. Rev. Fluid Mech., 1987, 19, 99 CrossRef.
- K. S. Suslick and G. J. Price, Annu. Rev. Mater. Sci., 1999, 29, 295 CrossRef CAS.
- J. H. Bang and K. S. Suslick, Adv. Mater., 2010, 22, 1039 CrossRef CAS PubMed.
- D. G. Shchukin, D. Radziuk and H. Mohwald, Annu. Rev. Mater. Res., 2010, 40, 345 CrossRef CAS.
- E. Ahmadi, S. Sareminezhad and M. H. Azizi, Food Hydrocolloids, 2011, 25, 1399 CrossRef CAS PubMed.
- F. A. Aouada, L. H. C. Mattoso and E. Longo, Ind. Crops Prod., 2013, 50, 449 CrossRef CAS PubMed.
- X. Feng, X. Wang, W. Xing, K. Zhou, L. Song and Y. Hu, Compos. Sci. Technol., 2014, 93, 76 CrossRef CAS PubMed.
- M. Soheilmoghaddam, M. Uzir Wahit, W. T. Whye, N. I. Akos, R. H. Pour and A. A. Yussuf, Carbohydr. Polym., 2014, 106, 326 CrossRef CAS PubMed.
- M. Lotya, Y. Hernandez, P. J. King, R. J. Smith, V. Nicolosi, L. S. Karlsson, F. M. Blighe, S. De, Z. M. Wang, I. T. McGovern, G. S. Duesberg and J. N. Coleman, J. Am. Chem. Soc., 2009, 131, 3611 CrossRef CAS PubMed.
- D. Nuvoli, M. Rassu, V. Alzari, R. Sanna, G. Malucelli and A. Mariani, Compos. Sci. Technol., 2014, 96, 97 CrossRef CAS PubMed.
- F. Delbecq, F. Kono and T. Kawai, Eur. Polym. J., 2013, 49, 2654 CrossRef CAS PubMed.
- Z. Liu, J. Liu, L. Cui, R. Wang, X. Luo, C. J. Barrow and W. Yang, Carbon, 2013, 51, 148 CrossRef CAS PubMed.
- X. Zheng, Q. Xu, J. Li, L. Li and J. Wei, RSC Adv., 2012, 2, 10632 RSC.
- V. Chabot, B. Kim, B. Sloper, C. Tzoganakis and A. Yu, Sci. Rep., 2013, 3, 1378 CrossRef PubMed.
- J. Fan, Z. Shi, J. Wang and J. Yin, Polymer, 2013, 54, 3921 CrossRef CAS PubMed.
- J. Fan, Z. Shi, Y. Ge, J. Wang, Y. Wang and J. Yin, J. Mater. Chem., 2012, 22, 13764 RSC.
- Z. Sun, S. Pöller, X. Huang, D. Guschin, C. Taetz, P. Ebbinghaus, J. Masa, A. Erbe, A. Kilzer, W. Schuhmann and M. Muhler, Carbon, 2013, 64, 288 CrossRef CAS PubMed.
- P. Mangiacapra, G. Gorrasi, A. Sorrentino and V. Vittoria, Carbohydr. Polym., 2006, 64, 516 CrossRef CAS PubMed.
- U. Costantino, V. Bugatti, G. Gorrasi, F. Montanari, M. Nocchetti, L. Tammaro and V. Vittoria, ACS Appl. Mater. Interfaces, 2009, 1, 668 CAS.
- J. M. Tour, Nat. Mater., 2014, 13, 545 CrossRef CAS PubMed.
- X. J. Chen, J. F. Dobson and C. L. Raston, Chem. Commun., 2012, 48, 3703 RSC.
- K. R. Paton, E. Varrla, C. Backes, R. J. Smith, U. Khan, A. O’Neill, C. Boland, M. Lotya, O. M. Istrate, P. King, T. Higgins, S. Barwich, P. May, P. Puczkarski, I. Ahmed, M. Moebius, H. Pettersson, E. Long, J. Coelho, S. E. O’Brien, E. K. McGuire, B. Mendoza Sanchez, G. S. Duesberg, N. McEvoy, T. J. Pennycook, C. Downing, A. Crossley, V. Nicolosi and J. N. Coleman, Nat. Mater., 2014, 13, 624 CrossRef CAS PubMed.
- A. Sorrentino, G. Gorrasi, M. Tortora, V. Vittoria, U. Costantino, F. Marmottini and F. Padella, Polymer, 2005, 46, 1601 CrossRef CAS PubMed.
- W. Guo and G. Chen, J. Appl. Polym. Sci., 2014, 131, 40565 Search PubMed.
- L. Tammaro, V. Vittoria and V. Bugatti, Eur. Polym. J., 2014, 52, 172 CrossRef CAS PubMed.
- M. Fang, K. Wang, H. Lu, Y. Yang and S. Nutt, J. Mater. Chem., 2009, 19, 7098 RSC.
- G. Gonçalves, P. A. A. P. Marques, A. Barros-Timmons, I. Bdkin, M. K. Singh, N. Emami and J. Grácio, J. Mater. Chem., 2010, 20, 9927 RSC.
- B. Shen, W. Zhai, D. Lu, J. Wang and W. Zheng, RSC Adv., 2012, 2, 4713 RSC.
- S. Stankovich, R. D. Piner, X. Chen, N. Wu, S. T. Nguyen and R. S. Ruoff, J. Mater. Chem., 2006, 16, 155 RSC.
- M. Quintana, A. Montellano, A. E. del Rio Castillo, G. Van Tendeloo, C. Bittencourt and M. Prato, Chem. Commun., 2011, 47, 9330 RSC.
- B. Shen, D. Lu, W. Zhai and W. Zheng, J. Mater. Chem. C, 2013, 1, 50 RSC.
- H. Kim, A. A. Abdala and C. W. Macosko, Macromolecules, 2010, 43, 6515 CrossRef CAS.
- J. R. Potts, D. R. Dreyer, C. W. Bielawski and R. S. Ruoff, Polymer, 2011, 52, 5 CrossRef CAS PubMed.
- D. Cai and M. Song, J. Mater. Chem., 2010, 20, 7906 RSC.
- Y. Cao, Z. Lai, J. Feng and P. Wu, J. Mater. Chem., 2011, 21, 9271 RSC.
- A. M. Rezadoust, M. Esfandeh, M. H. Beheshty and G. Heinrich, Polym. Eng. Sci., 2013, 53, 809 CAS.
- C. Dazhu, Y. Haiyang, H. Pingsheng and Z. Weian, Compos. Sci. Technol., 2005, 65, 1593 CrossRef PubMed.
- S. Zhu, J. Chen, H. Li and Y. Cao, J. Appl. Polym. Sci., 2013, 128, 3876 CrossRef CAS.
- S. Abend and G. Lagaly, Appl. Clay Sci., 2000, 16, 201 CrossRef CAS.
- D. Burgentzlé, J. Duchet, J. F. Gérard, A. Jupin and B. Fillon, J. Colloid Interface Sci., 2004, 278, 26 CrossRef PubMed.
- J. Labanda and J. Llorens, Rheol. Acta, 2006, 45, 305 CrossRef CAS PubMed.
- H. Van Olphen, J. Colloid Sci., 1964, 19, 313 CrossRef CAS.
- H. Van Olphen, J. Colloid Sci., 1962, 17, 660 CrossRef CAS.
- P. F. Luckham and S. Rossi, Adv. Colloid Interface Sci., 1999, 82, 43 CrossRef CAS.
- E. Tombácz and M. Szekeres, Appl. Clay Sci., 2004, 27, 75 CrossRef PubMed.
- B. Dobias, Coagulation and Flocculation: Theory and Applications, CRC Press, 1993 Search PubMed.
- B. Seentrakoon, B. Junhasavasdikul and W. Chavasiri, Polym. Degrad. Stab., 2013, 98, 566 CrossRef CAS PubMed.
- S. H. Hsieh, F. R. Zhang and H. S. Li, J. Appl. Polym. Sci., 2006, 100, 4311 CrossRef CAS.
- Q. G. Du, G. Alagappan, H. Dai, H. V. Demir, H. Y. Yu, X. W. Sun and C. H. Kam, Opt. Commun., 2012, 285, 3238 CrossRef CAS PubMed.
- J. W. Rhim, Carbohydr. Polym., 2011, 86, 691 CrossRef CAS PubMed.
- A. J. Peacock and A. R. Calhoun, Polymer Chemistry: Properties and Applications, Hanser Publishers, 2006 Search PubMed.
- R. N. Wenzel, Ind. Eng. Chem., 1936, 28, 988 CrossRef CAS.
- A. B. D. Cassie and S. Baxter, Trans. Faraday Soc., 1944, 40, 546 RSC.
- K. Chen, T. Y. Zhang and W. Wei, Adv. Mater. Res., 2011, 311, 92 CrossRef PubMed.
- S. Bhuyan, S. Sundararajan, Y. Lu and R. Larock, Wear, 2010, 268, 797 CrossRef CAS PubMed.
- G. Malucelli and F. Marino, in Abrasion Resistance of Materials, ed. M. Adamiak, InTech, 2012, ISBN: 978-953-51-0300-4, http://www.intechopen.com/books/abrasion-resistance-of-materials/abrasion-resistance-of-polymernanocomposites Search PubMed.
- T. Nielsen and M. Jägerstad, Trends Food Sci. Technol., 1994, 5, 353 CrossRef CAS.
- C. Sanchez, P. Belleville, M. Popall and L. Nicole, Chem. Soc. Rev., 2011, 40, 696 RSC.
- Nanocor, Multilayer containers featuring nano-nylon MDX6 barrier layers with superior performance and clarity, 2003, http://www.nanocor.com/tech_papers/NOVAPACK03.pdf, last access May 2013 Search PubMed.
- Nanocor, Technical Bulletin, Film and Applications, http://www.nanocor.com/tech_sheets/I105.pdf, last access October 2013 Search PubMed.
- Plastics Technology, Chasing Nanocomposites, 2004, http://www.ptonline.com/articles/chasing-nanocomposites, accessed August 2013 Search PubMed.
- Triton Systems, http://www.tritonsys.com/, last access October 2013.
- InMat, Ultrahigh barrier coating breakthrough from InMat® reduces food packaging costs and environmental impact, 2009, http://www.inmat.com/upload/files/inmat_press_release_05072009.pdf, last access September 2013 Search PubMed.
- NanoPack, NanoSeal™ – Barrier Coating, OPP Barrier Coated Film, Polypropylene Film, Liquid Coating, http://www.nanopackinc.com/products.asp, last access October 2013.
- S. Farris, C. A. Cozzolino, L. Introzzi and L. Piergiovanni, Packag. Technol. Sci., 2009, 22, 359 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2014 |