
DFT study on the mechanism of phosphine-catalyzed ring-opening reaction of cyclopropyl ketones†
Xiaohan
Yu
a and
Yang
Wang
 *b
*b
aCollege of Chemical and Materials Engineering, Xuchang University, Xuchang, Henan Province, P. R. China
bDepartment of Material and Chemical Engineering, Zhengzhou University of Light Industry, 136 Science Avenue, 450001, Zhengzhou, Henan Province, P.R. China. E-mail: wangyang@zzuli.edu.cn; 2017017@email.zzuli.edu.cn
First published on 4th November 2024
Abstract
In the present study, the mechanism, origin of chemoselectivity, and substituent effects of the phosphine-catalyzed ring-opening reaction of cyclopropyl ketone have been investigated using the DFT method. Multiple pathways, including the formation of hydrofluorenone, the Cloke–Wilson product, and cyclopenta-fused product, were studied and compared. The computational results show that the pathway for the formation of hydrofluorenone is the most favorable one, which involves four processes: nucleophilic substitution to open the three-membered ring, an intramolecular Michael addition for the formation of an enolate intermediate, an intramolecular [1,5]-proton transfer to give ylide, and an intramolecular Wittig reaction to deliver the final product. For disclosing the origin of chemoselectivity, structural analysis and local reactivity index analysis were performed. Moreover, substituent effects were also considered using QTAIM analysis. The current study would provide useful insights for understanding phosphine-catalyzed chemoselective reactions.
Introduction
Cyclopropane frameworks are frequently found in natural products and pharmaceutical compounds. The strain release transformation of cyclopropane offers versatile synthons for constructing various complex molecules through cleavage of C–C single bonds.1 Compared to the extensively developed ring opening/transformation of activated cyclopropanes under transition metal catalysis,2 organocatalytic strain release transformation of activated cyclopropanes has attracted considerable attention.3 In the organocatalytic approach, Lewis acid catalysts are effective in breaking C–C single bonds of donor–acceptor cyclopropanes (DACs), which serve as C3-synthons (Scheme 1a) and can deliver various cyclic compounds through ring-opening/transformation reactions.4 Brønsted acids promote the ring-opening reaction of activated cyclopropanes by polarizing the C–C bond (Scheme 1b).5 Moreover, ring opening of electron-deficient cyclopropanes (such as formyl cyclopropane) happens under NHC organocatalysis through nucleophilic addition (Scheme 1c).6 To the best of our knowledge, these organocatalytic ring-opening reactions of cyclopropanes are focused on activated cyclopropanes; the ring opening/transformation of geminal cyclopropanes are rare. | ||
| Scheme 1 Organocatalytic ring-opening reaction of activated cyclopropane. | ||
Recently, Ramasastry and coworkers reported a first example of organophosphine-catalyzed ring opening/transformation of unactivated cyclopropyl ketone (Scheme 2).7 In this reaction, phosphine is utilized to promote the ring opening of unactivated cyclopropane, which fulfills the scope of organocatalytic C–C bond activation and also represents a new synthetic protocol for the formation of five-membered cyclic compounds through organocatalysis. As shown in Scheme 2, substrate R1 has multiple reactive sites that can undergo various types of reactions, leading to considerable complexity in the chemoselectivity of the reaction. Thus, elucidating the mechanism of the phosphine-catalyzed ring opening and recyclization of cyclopropyl ketone would be of interest and instructive for understanding phosphine-catalyzed chemoselective reactions. Moreover, using the phosphine catalyst (such as PPh2Me), the desired product P can be obtained in good yield (84%), whereas other possible products, such as the Cloke–Wilson product P1, cyclopenta-fused indanone P2, and the MBH product P3 were not observed. Therefore, the origin of chemoselectivity in this phosphine-catalyzed reaction should be disclosed theoretically. With mechanistic exploration of catalytic reactions at the core of the research activity in our group,8 we now perform a DFT study on the mechanism of phosphine-catalyzed ring-opening/transformation reaction of cyclopropyl ketone, which has been proved and widely utilized for the mechanistic investigation of organocatalytic9 and transition-metal-catalyzed reactions.10
 | ||
| Scheme 2 Phosphine-catalyzed ring opening and recyclization of unactivated cyclopropane. | ||
Results and discussion
Reaction mechanism
As shown in Scheme 3, we have suggested a possible reaction mechanism of the title reaction based on experimental details. The ring opening of cyclopropyl ketone first occurs by nucleophilic addition of the phosphine catalyst PPh2Me to the C1 atom of R1. Then the zwitterionic intermediate M1 is generated through an SN2-type nucleophilic substitution. Intermediate M1 then undergoes intramolecular Michael-type addition to form the enolate intermediate M2. Subsequently, an intramolecular [1,5]-proton transfer occurs to give the ylide M3, which can transform to the final tetrahydrofluorenone product P through an intramolecular Wittig reaction. Alternatively, some active intermediates have the potential to transform to several possible products. For example: (1) the zwitterionic intermediate M1 can transform to the Cloke–Wilson product P1via an intramolecular SN2-type nucleophilic substitution, (2) the cyclopenta-fused indanone product P2 can be delivered through the transformation of enolate intermediate M2, and (3) the phosphine catalyst can also attack the C2 atom of the α,β-unsaturated ketone moiety to generate the Morita–Baylis–Hillman product P3. In the following section, we provide detailed analyses to disclose the mechanism and origin of chemoselectivity of the title reaction. | ||
| Scheme 3 Possible reaction mechanism of phosphine-catalyzed ring opening and recyclization of cyclopropyl ketone. | ||
Fig. 1 shows the relative Gibbs free energy profile of key transformation of cyclopropyl ketone R1 to the major product P. The nucleophilic attack of the C1 atom of R1 by the phosphine catalyst PPh2Me initiates the reaction. The zwitterionic intermediate M1 is produced through nucleophilic substitution via the transition state TS1. The energy barrier for this process is 28.9 kcal mol−1. Considering the reaction temperature and time, we think that the calculated energy barrier would be acceptable, which can be supported by previous theoretical studies.11 Then, the zwitterionic intermediate M1 undergoes the intramolecular Michael addition to generate enolate intermediate M2via the transition state TS2. The computational results show that this process suffers from an energy barrier of 15.8 kcal mol−1, which can easily occur under the experimental conditions.
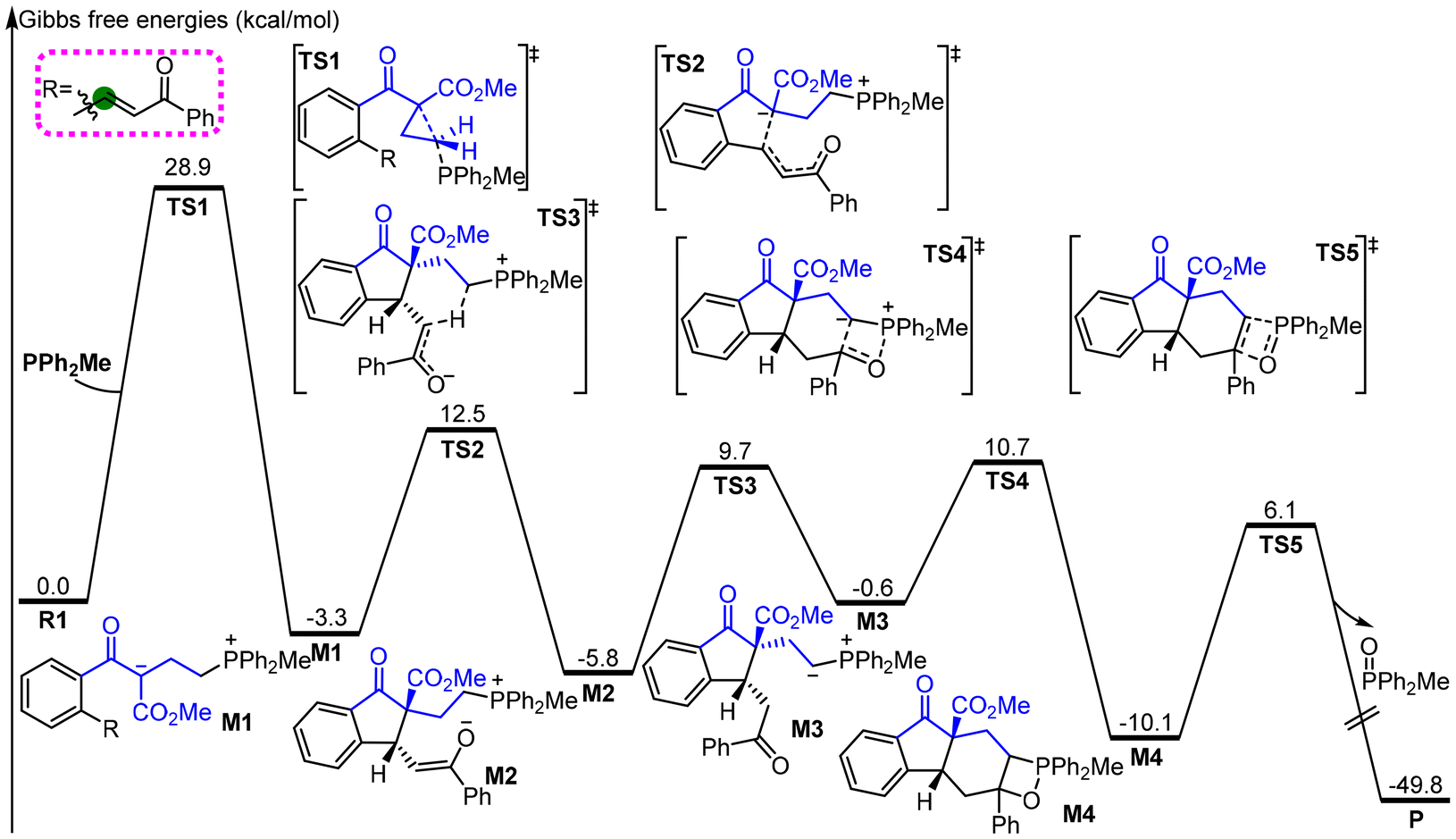 | ||
| Fig. 1 The relative Gibbs free energy profile for the title reaction. | ||
Subsequently, the phosphorous ylide M3 is formed through an intramolecular [1,5]-proton transfer. The energy barrier for this process is 15.5 kcal mol−1, associated with the transition state TS3. Next, the phosphorous ylide M3 undergoes an intramolecular Wittig reaction to deliver the final product P accompanied by the release of phosphine oxide MePh2P![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O. Based on the calculations, the Wittig reaction involves two processes: (1) formation of a four-membered oxaphosphetane intermediate M4via the transition state TS4 and (2) release of phosphine oxide MePh2PO to produce the final product via the transition state TS5. As shown in Fig. 1, the energy barriers for these two processes are 16.5 and 16.2 kcal mol−1, associated with transition states TS4 and TS5, respectively.
O. Based on the calculations, the Wittig reaction involves two processes: (1) formation of a four-membered oxaphosphetane intermediate M4via the transition state TS4 and (2) release of phosphine oxide MePh2PO to produce the final product via the transition state TS5. As shown in Fig. 1, the energy barriers for these two processes are 16.5 and 16.2 kcal mol−1, associated with transition states TS4 and TS5, respectively.
As shown in Scheme 3, the substrate R1 and active intermediates M1 and M2 have multiple active sites, which would undergo various transformations. As shown in Scheme 4, intermediates M1 and M2 can undergo nucleophilic substitution to deliver the Cloke–Wilson product P1 and the cyclopenta-fused indanone product P2 through transition states TS2-1 and TS3-2, respectively. The computational results indicate that these two possible transformations need to overcome the energy barriers of 37.3 and 34.3 kcal mol−1 respectively, which are 21.5 and 18.8 kcal mol−1 higher than those of the intramolecular Michael addition process (ΔG‡ = 15.8 kcal mol−1, Fig. 1) and the [1,5]-proton transfer process (ΔG‡ = 15.5 kcal mol−1, Fig. 1), respectively. It should be noted that the formation of the Cloke–Wilson product and the intramolecular Michael addition process share the same ring-opening step, while the formation of the cyclopenta-fused indanone product and the [1,5]-proton transfer process share the same ring-opening and Michael addition steps. Thus, we think that the competitive formations of P1 and P2 are energetically unfavorable and impossible to occur under the experimental conditions. Alternatively, nucleophilic phosphine can also attack the β-carbon of the ketone moiety to deliver a zwitterionic intermediate M1-3 through nucleophilic addition via the transition state TS1-3. The energy barrier for this step is 17.7 kcal mol−1, which shows that the nucleophilic addition reaction can occur easily under the experimental conditions. Subsequently, M1-3 undergoes the Michael addition to afford a fused five-membered ring intermediate M2-3 through the transition state TS2-3, which needs to overcome an energy barrier of 17.5 kcal mol−1. The sequential proton transfer process occurs to give the enol intermediate M3-3. The computational results show that the direct [1,3]-proton transfer suffers from an extremely high energy barrier of 36.9 kcal mol−1 (Fig. S1 of the ESI†), which is impossible to occur under the experimental conditions. Inspired by the elegant work of Yu,12 we then considered that a trace amount of water would participate in the reaction. As shown in Scheme 4, the energy barrier for water-assisted proton transfer is 27.2 kcal mol−1, associated with the transition state TS3-3. Finally, dissociation of the phosphine catalyst would release product P3. The energy barrier for this step via the transition state TS4-3 is 19.5 kcal mol−1. Overall, the entire energy barrier for the formation of the MBH product P3 is 27.2 kcal mol−1. Although the energy barrier for the formation of the MBH product is 3.5 kcal mol−1 lower than that for the formation of the hydrofluorenone product, the relative Gibbs free energy of product P3 is only −2.0 kcal mol−1, which is 47.8 kcal mol−1 higher than that of P. Considering the experimental conditions, the reaction for forming the MBH product would be reversible and the formation of the hydrofluorenone product is irreversible. Thus, we think that the pathway leading to the formation of MBH product would be controlled by kinetics and such a product would be unstable under the experimental conditions. The above discussions show that the possible transformation processes for the formation of P1, P2, and P3 are less favorable, which align well with the experimental observations.
 | ||
| Scheme 4 Other possible transformations for the formation of P1, P2, and P3 (values presented in parentheses are the relative Gibbs free energies and given in kcal mol−1). | ||
Origin of chemoselectivity
The above discussions show that the energy barriers for the formation of the Cloke–Wilson product P1 and the cyclopenta-fused indanone product P2 are quite higher than the corresponding Michael addition and [1,5]-proton transfer processes. For identifying the chemoselectivity, we have performed local reactivity analysis.13 As shown in Fig. 2A, the nucleophilic reactivities of the C3 and O1 atoms are 0.78 and 0.30, respectively, while the electrophilic reactivities of the C1 and C4 atoms are 0.00 and 0.27, respectively. The local reactivity analysis results indicate that the nucleophilicity of C3 is larger than that of O1 and the electrophilicity of C4 is larger than that of C1. Thus, the Michael addition reaction that occurred for the formation of the C3–C4 bond is more preferential than that for the formation of the O1–C1 bond through nucleophilic substitution. In intermediate M2, the nucleophilicity of the C5 atom is 0.80, while the electrophilicities of the C1 and H1 atoms are the same, indicating that proton transfer and nucleophilic substitution are competitive. However, as shown in Fig. 2B, intramolecular [1,5]-proton transfer occurs through the six-membered ring transition state TS3 and the intramolecular nucleophilic substitution happens via the five-membered ring transition state TS3-2. This phenomenon shows that the six-membered ring transition structure has a lower ring strain and thus makes TS3 more stable. Furthermore, the energy barrier for the formation of P3 is much higher than that for the formation of P, which shows that it is impossible for[1,3]-proton transfer to occur under non-protic conditions. In addition, the relative Gibbs free energy of P3 is −2.0 kcal mol−1, which is higher than that of P (−49.8 kcal mol−1, Fig. 1). These results show that the formation of the Morita–Baylis–Hillman product P3 is not favored under the experimental conditions. | ||
| Fig. 2 (A) Local reactivity indexes of intermediates M1 and M2 and (B) structures of chemoselective transition states TS3 and TS3-2. | ||
Substituent effects
Based on the experimental observations, the bi-activated cyclopropane is effective for the reaction and it is difficult for the mono-activated cyclopropane to undergo the ring-opening/recyclization reaction under the experimental conditions. As shown in Fig. 3A, the energy barriers for the ring-opening process of R1a and R1b are 40.8 and 37.0 kcal mol−1via transition states TS1a and TS1b, respectively. The computational results show that the substituent would have significant influence on the energy barrier for the ring-opening process. For further identifying the substituent effects, we have performed QTAIM (quantum theory of atoms-in-molecule) analysis for substrates R1, R1a, and R1b, which has been successfully utilized to disclose the role of the catalyst and the substituent effect.14 As shown in Fig. 3B, the contour line maps of the Laplacian of electron density with the positions of QTAIM bcps of the three substrates are represented using QTAIM analysis. The corresponding values of the Laplacian of electron density (ρ(bcp)) were calculated at the bond critical points (BCPs). The values of the Laplacian of electron density for the three substrates are −0.33, −0.37, and −0.36 respectively, showing an internuclear region. Moreover, the C1–C3 bond in R1 has the lowest Laplacian of electron density, which indicates that the strength of the C1–C3 bond in R1 is weaker than those in the other two substrates; the corresponding energy barriers for ring opening are 28.9, 40.8, and 37.0 kcal mol−1, respectively. These results show that the energy barriers for the ring-opening process involved in different substrates are correlated with the Laplacian of electron density. The disubstituted cyclopropane has a larger Laplacian of electron density associated with a lower energy barrier. Moreover, the substrate with the electron-withdrawing group facilitates the cleavage of the C1–C3 bond by reducing the electron density of the C1–C3 bond. | ||
| Fig. 3 (A) Energy barriers for TS1, TS1a, and TS1b; (B) QTAIM analyses of R1, R1a, and R1b (d(C1–C3) and ρ(bcp) represent the distance of the C1–C3 bond and the Laplacian of electron density between the C1 and C3 atoms, respectively; units of distance and electron density are Å and e Å−3). | ||
In addition, the experimental results also indicate that the substituents on the ketone moiety also affect the reaction. Based on the experimental observations, no desired products can be obtained when R1-1t, R1-1u, and R1-1v are employed as reactants. For an in-depth understanding of the substituent effects, we then calculated and compared the energy barriers involved in the ring-opening and Michael addition processes for other substrates (i.e., R1-1t, R1-1u, and R1-1v). As summarized in Table S2,† the energy barriers associated with TS1-1t, TS1-1u, and TS1-1v are 33.8, 32.2, and 34.3 kcal mol−1 respectively, which are higher than that involved in TS1 (ΔG‡ = 28.9 kcal mol−1, Fig. 1), while the energy barriers for the subsequent Michael addition viaTS2-1t, TS2-1u, and TS2-1v are 16.9, 18.4, and 19.3 kcal mol−1, which are also unfavorable compared to that of TS2 (ΔG‡ = 15.8 kcal mol−1, Fig. 1). These results show that the substituent on the ketone moiety would also affect the reaction through the Michael addition process.
Conclusions
In summary, the mechanism, origin of chemoselectivity, and substituent effects of the phosphine-catalyzed ring-opening reaction of cyclopropyl ketone have been extensively explored using the DFT method. The computational results show that the most energetically favorable pathway involves four processes: (1) nucleophilic substitution to open the three-membered ring, (2) an intramolecular Michael addition for the formation of an enolate intermediate, (3) an intramolecular [1,5]-proton transfer to give the ylide, and (4) an intramolecular Wittig reaction to deliver the final product. The nucleophilic substitution is the rate-determining step, and the local reactivity index analysis shows that the reactivity of potential active sites determines the chemoselectivity. The substituent effects show that the electron-withdrawing group can reduce the electron density and thus facilitates the ring opening of cyclopropanes through ELF analysis.Experimental
Computational method
The Gaussian 09 program was utilized to perform the calculations.15 The M06-2X functional16 combined with the 6-31G(d,p) basis set17 was selected to optimize all the stationary points involved in the reaction. Frequency calculations were also conducted to obtain Gibbs free energy corrections and confirm that the obtained reactants, intermediates, and products have no imaginary frequency; correspondingly, the transition states have one and only one imaginary frequency. During the optimizations, the solvation effects are also considered using the IEF-PCM solvation model18 with acetonitrile (ε = 35.688) as a solvent. Moreover, the energies obtained at the M06-2X/6-31G(d,p)//IEF-PCMMeCN level were further refined at the M06-2X/6-311++G(2df,2pd)//IEF-PCMMeCN level through single-point energy calculations. In addition, a correction of 1.89 kcal mol−1 was added to the energies of all the stationary points involved in the discussions to correct the Gibbs free energies in the gas phase (1 atm) to the standard state in solution (1 mol L−1). The QTAIM analysis was performed using Multiwfn software.19In addition, we have also performed benchmark studies involved in the rate-determining step using different functionals, such as B3LYP,20 Cam-B3LYP,21 B3PW91,22 and ωB97X-D.23 The computational results are summarized in Table S1 of the ESI.† By comparison, the energy barriers for the nucleophilic substitution step are similar with the results obtained by M06-2X functional and indicate that the results obtained from the selected functional discussed in the text are reliable.
Author contributions
Xiaohan Yu: data curation, formal analysis, funding acquisition, and investigation; Yang Wang: conceptualization, funding acquisition, writing – original draft, and writing – review and editing.Data availability
The data supporting this article have been included as part of the ESI.†Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors gratefully thank the financial support from the National Natural Science Foundation of China (No. 21703195), the Key R&D and Promotion Projects (Science and Technology Key Projects) of Henan Province (No. 242102310470), and the Key Projects of Colleges and Universities in Henan Province (No. 24B150045).References
- For recent reviews: (a) V. Pirenne, B. Muriel and J. Waser, Chem. Rev., 2021, 121, 227–263 CrossRef CAS; (b) J. Caille and R. Robiette, Org. Biomol. Chem., 2021, 19, 5702–5724 RSC; (c) B. Biletskyi, P. Colonna, K. Masson, J.-L. Parriain, L. Commeiras and G. Chouraqui, Chem. Soc. Rev., 2021, 50, 7513–7538 RSC.
- (a) Y. X. Zeng, Z.-T. Jiang and Y. Xia, Chem. Commun., 2024, 60, 3764–3773 RSC; (b) H. Yang, Y. X. Zeng, X. Y. Song, L. Che, Z.-T. Jiang, G. Lu and Y. Xia, Angew. Chem., Int. Ed., 2024, 63, e202403602 CrossRef CAS PubMed; (c) Z.-X. Yang, Y.-Q. Wei, C. Yang, Y.-F. Li, C.-H. Ding and B. Xu, Asian J. Org. Chem., 2022, 11, e202100683 CrossRef CAS; (d) W.-D. Chu, Y.-T. Wang, T.-T. Liang, T. Long, J.-Y. Zuo, Z. H. Shao, B. Chen, C.-Y. He and Q.-Z. Liu, Org. Lett., 2022, 24, 3965–3969 CrossRef CAS PubMed; (e) S. Nicolai and J. Waser, Angew. Chem., Int. Ed., 2022, 61, e202209006 CrossRef CAS; (f) C. Zhang and C. Mazet, Org. Lett., 2024, 26, 5386–5390 CrossRef PubMed; (g) A. S. Adhikari, S. Pandit and N. Majumdar, ACS Catal., 2023, 13, 6261–6267 CrossRef CAS; (h) J. Wu, Y. H. Tang, W. Wei, Y. Wu, Y. Li, J. J. Zhang, Y. S. Zheng and S. L. Xu, Angew. Chem., Int. Ed., 2018, 57, 6284–6288 CrossRef CAS PubMed.
- (a) E. Reyes, U. Uria, L. Prieto, L. Carrillo and J. L. Vicario, Chem. Commun., 2024, 60, 7288–7298 RSC; (b) N. Yadav, A. Hazra, P. Singh and P. Banerjee, Adv. Synth. Catal., 2024, 366, 1113–1119 CrossRef CAS; (c) A. Hazra, A. Ghosh, N. Yadav and P. Banerjee, Chem. Commun., 2023, 59, 11133–11136 RSC; (d) Y. F. Meng, J. Y. Gu, M. X. Xin, Y. Jiang, Z. B. Du, G. Q. Lu, J. Y. Jiang, A. S. C. Chan, Z. F. Ke and Y. Zou, J. Org. Chem., 2024, 89, 2726–2740 CrossRef CAS PubMed; (e) K. Biswas, A. Khamrai, S. Malik and V. Ganesh, Org. Lett., 2023, 25, 1805–1810 CrossRef CAS PubMed.
- (a) Y. Xia, X. H. Liu and X. M. Feng, Angew. Chem., Int. Ed., 2021, 60, 9192–9204 CrossRef CAS PubMed; (b) D. X. Zhang, Q. H. Cheng, L. J. Chen, H. Q. Deng, H. Cai and Q.-F. Zhang, Org. Biomol. Chem., 2021, 19, 9645–9648 RSC; (c) Y. Jiang, H.-J. Ma, C.-Y. Zhai and X.-L. Wang, Org. Lett., 2024, 26, 1672–1676 CrossRef CAS; (d) A. E. Vartanova, I. I. Levina, N. K. Ratmanova, I. A. Andreev, O. A. Ivanova and I. V. Trushkov, Org. Biomol. Chem., 2022, 20, 7795–7802 RSC; (e) P.-C. Tu, L. Zhou, A. M. Kirillov, R. Fang and L. Z. Yang, Org. Chem. Front., 2018, 5, 1702–1712 RSC; (f) S. Thangamalar, M. Thangamani and K. Srinivasan, Org. Biomol. Chem., 2022, 20, 3145–3153 RSC; (g) P. R. Singh, B. Gopal, M. Kumar and A. Goswami, Org. Biomol. Chem., 2022, 20, 4933–4941 RSC.
- (a) Z.-J. Yu, S. Yan, X.-L. Zhao, J. Zhang and M.-X. Zhao, J. Org. Chem., 2024, 89, 8691–8705 CrossRef CAS PubMed; (b) Y. Chen, Y. Zhang and Y. Xue, Org. Biomol. Chem., 2022, 20, 4006–4015 RSC; (c) Y. Z. Zhou, R.-C. Xue, Y. Q. Feng and L. Zhang, Asian J. Org. Chem., 2020, 9, 311–316 CrossRef CAS; (d) L. C. Irwin, C. R. Renwick and M. A. Kerr, J. Org. Chem., 2018, 83, 6235–6242 CrossRef CAS PubMed; (e) E. Richmond, V. D. Vukovic and J. Moran, Org. Lett., 2018, 20, 574–577 CrossRef CAS PubMed.
- (a) L. Prieto, E. Sanchez-Diez, U. Uria, E. Reyes, L. Carrillo and J. L. Vicario, Adv. Synth. Catal., 2018, 359, 1678–1683 CrossRef; (b) L. X. Li, D. Du, J. Ren and Z. W. Wang, Eur. J. Org. Chem., 2011, 614–618 CrossRef CAS; (c) Y. Wang, Y. Qiao, Y. Lan and D. H. Wei, Catal. Sci. Technol., 2021, 11, 332–337 RSC; (d) H. Lv, J. M. Mo, X. Q. Fang and Y. R. Chi, Org. Lett., 2011, 13, 5366–5369 CrossRef CAS.
- J. P. Maurya and S. S. V. Ramasastry, Org. Lett., 2024, 26, 2282–2286 CrossRef CAS PubMed.
- (a) J. Ye and Y. Wang, Eur. J. Org. Chem., 2023, e202300520 CrossRef CAS; (b) Y. Wang, Y. L. Luo, M. Zhao and Y. Qiao, Mol. Catal., 2023, 551, 113621 CrossRef CAS; (c) L. J. Zhang and Y. Wang, Comput. Theor. Chem., 2023, 1220, 114007 CrossRef CAS; (d) Y. Wang and Y. Lan, Chin. Chem. Lett., 2020, 31, 736–738 CrossRef CAS; (e) S.-L. Liu, X. Q. Liu, Y. Wang and D. H. Wei, Mol. Catal., 2022, 519, 112122 CrossRef CAS; (f) S.-L. Liu, Y. Qiao and Y. Wang, Org. Chem. Front., 2023, 10, 2670–2679 RSC; (g) Y.-N. Wang and Y. Wang, New J. Chem., 2024, 48, 11360–11365 RSC; (h) Y. Wang and D. H. Wei, Mol. Catal., 2020, 489, 110944 CrossRef CAS; (i) Y. Wang, K. L. Gong, H. Zhang, Y. Liu and D. H. Wei, Catal. Sci. Technol., 2022, 12, 4380–4387 RSC; (j) Y. Wang, H. Zhang, Y. Liu, K. L. Gong and D. H. Wei, Mol. Catal., 2022, 527, 112410 CrossRef CAS; (k) Y. L. Luo, M. Zhao and Y. Wang, J. Phys. Chem. A, 2024, 128, 6190–6198 CrossRef CAS PubMed; (l) P. X. Liang, D. Y. Shi and Y. Wang, Catal. Sci. Technol., 2024, 14, 4302–4310 RSC; (m) M.-Q. Yang and Y. Wang, Mol. Catal., 2024, 567, 114445 CrossRef CAS.
- (a) P. X. Liang, H. R. Yang and Y. Wang, Phys. Chem. Chem. Phys., 2024, 26, 4320–4328 RSC; (b) D. C. Li and Y. Wang, Org. Biomol. Chem., 2024, 22, 2662–2669 RSC; (c) Q. Q. Shi, Z. P. Pei, J. S. Song, S.-J. Li, D. H. Wei, M. L. Coote and Y. Lan, J. Am. Chem. Soc., 2022, 144, 3137–3145 CrossRef CAS PubMed; (d) C. H. Liu, P. L. Han, X. X. Hou, S. X. Ge and D. H. Wei, Org. Chem. Front., 2024, 11, 3952–3961 RSC; (e) C. H. Liu, P. L. Han, X. S. Zhang, Y. Qiao, Z. H. Xu, Y. G. Zhang, D. P. Li, D. H. Wei and Y. Lan, J. Org. Chem., 2022, 87, 7989–7994 CrossRef CAS PubMed; (f) X.-X. Hou and D. H. Wei, J. Org. Chem., 2024, 89, 3133–3142 CrossRef CAS PubMed; (g) J. M. Zhang, Q. Y. Qiao, Z. J. Wu, Z. Pang, Q. Q. Shi, Y. Y. Wang, Y. Qiao and D. H. Wei, Org. Biomol. Chem., 2022, 20, 1662–1670 RSC.
- (a) X. H. Wang, P. Jin, S. Q. Li, Y. Q. Wen, F. K. Wang, H. J. Wei and D. H. Wei, Org. Biomol. Chem., 2023, 21, 7410–7418 RSC; (b) Y. Wang, C. Du, Y. Y. Wang, X. K. Guo, L. Fang, M.-P. Song, J.-L. Niu and D. H. Wei, Adv. Synth. Catal., 2018, 360, 2668–2677 CrossRef CAS; (c) S.-S. Dai, X.-J. Yang, R. Fang, A. M. Kirillov and L. Z. Yang, Org. Chem. Front., 2023, 10, 1948–1958 RSC; (d) K. Lv and X. G. Bao, Org. Chem. Front., 2022, 9, 5237–5245 RSC; (e) H. B. Liu, H. P. Shi, P. D. Han, Z. T. Meng, T. Liu and L. L. Han, Org. Biomol. Chem., 2022, 20, 7294–7301 RSC.
- (a) C. Patel, V. Abraham and R. B. Sunoj, Organometallics, 2017, 36, 151–158 CrossRef CAS; (b) S. Singh, K. Surya and R. B. Sunoj, J. Org. Chem., 2017, 82, 9619–9626 CrossRef CAS PubMed; (c) Z. Y. Yu and Y. Lan, J. Org. Chem., 2013, 78, 11501–11507 CrossRef CAS PubMed; (d) I. Nohira, S. Liu, R. B. Bai, Y. Lan and N. Chatani, J. Am. Chem. Soc., 2020, 142, 17306–17311 CrossRef CAS PubMed; (e) G. Y. Tan, L. Zhu, X. R. Liao, Y. Lan and J. S. You, J. Am. Chem. Soc., 2017, 139, 15724–15737 CrossRef CAS PubMed; (f) Y. Wang, D. H. Wei, W. J. Zhang, Y. Y. Wang, Y. Y. Zhu, Y. Jia and M. S. Tang, Org. Biomol. Chem., 2014, 12, 7503–7514 RSC.
- Y. Z. Xia, Y. Liang, Y. Y. Chen, M. Wang, L. Jiao, F. Huang, S. Liu, Y. H. Li and Z.-X. Yu, J. Am. Chem. Soc., 2007, 129, 3470–3471 CrossRef CAS PubMed.
- (a) L. R. Domingo, M. Ríos-Gutierrez and P. Perez, Molecules, 2016, 21, 748 CrossRef PubMed; (b) L. R. Domingo, E. Chamorro and P. Perez, J. Org. Chem., 2008, 73, 4615–4624 CrossRef CAS PubMed.
- (a) Q.-Y. Zhang, Y. Wang, S.-J. Li, Y. Y. Wang and D. H. Wei, Catal. Sci. Technol., 2022, 12, 947–953 RSC; (b) J. Ye, Y. L. Luo, G. L. Huang and Y. Wang, New J. Chem., 2023, 47, 16636–16642 RSC; (c) Y. R. Zhang and Y. Wang, Org. Chem. Front., 2024, 11, 4391–4401 RSC; (d) J. J. Wang, D. H. Wei, Z. Duan and F. Mathey, J. Am. Chem. Soc., 2020, 142, 20973–20978 CrossRef CAS PubMed; (e) Y. Wang, Y. Liu, K. L. Gong, H. Zhang, Y. Lan and D. H. Wei, Org. Chem. Front., 2021, 8, 5352–5360 RSC.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, B. Mennucci, G. A. Petersson, H. Nakatsuji, M. Caricato, X. Li, H. P. Hratchian, A. F. Izmaylov, J. Bloino, G. Zheng, J. L. Sonnenberg, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, J. A. Montgomery Jr., J. E. Peralta, F. Ogliaro, M. Bearpark, J. J. Heyd, E. Brothers, K. N. Kudin, V. N. Staroverov, T. Keith, R. Kobayashi, J. Normand, K. Raghavachari, A. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, N. Rega, J. M. Millam, M. Klene, J. E. Knox, J. B. Cross, V. Bakken, C. Adamo, J. Jaramillo, R. Gomperts, R. E. Stratmann, O. Yazyev, A. J. Austin, R. Cammi, C. Pomelli, J. W. Ochterski, R. L. Martin, K. Morokuma, V. G. Zakrzewski, G. A. Voth, P. Salvador, J. J. Dannenberg, S. Dapprich, A. D. Daniels, O. Farkas, J. B. Foresman, J. V. Ortiz, J. Cioslowski and D. J. Fox, Gaussian 09, Revision D.01, Gaussian, Inc., Wallingford CT, 2013 Search PubMed.
- (a) Y. Zhao and D. G. Truhlar, Theor. Chem. Acc., 2008, 120, 215–241 Search PubMed; (b) Y. Zhao and D. G. Truhlar, Acc. Chem. Res., 2008, 41, 157–167 CrossRef CAS PubMed.
- (a) W. J. Hehre, R. Ditchfield and J. A. Pople, J. Chem. Phys., 1972, 56, 2257–2261 CrossRef CAS; (b) R. Ditchfield, W. J. Hehre and J. A. Pople, J. Chem. Phys., 1971, 54, 724–728 CrossRef CAS.
- (a) S. Miertuš, E. Scrocco and J. Tomasi, Chem. Phys., 1981, 55, 117–129 CrossRef; (b) S. Miertuš and J. Tomasi, Chem. Phys., 1982, 65, 239–245 CrossRef; (c) J. L. Pascual-Ahuir, E. Silla and I. Tuñón, J. Comput. Chem., 1994, 15, 1127–1138 Search PubMed.
- T. Lu and F. W. Chen, J. Comput. Chem., 2012, 33, 580–592 CrossRef CAS PubMed.
- (a) A. D. Backe, J. Chem. Phys., 1993, 98, 5648–5652 CrossRef; (b) B. Miehlich, A. Savin, H. Stoll and H. Preuss, Chem. Phys. Lett., 1989, 157, 200–206 CrossRef CAS; (c) C. T. Lee, W. T. Yang and R. G. Parr, Phys. Rev. B: Condens. Matter Mater. Phys., 1988, 37, 785–789 CrossRef CAS PubMed.
- T. Yanai, D. Tew and N. Handy, Chem. Phys. Lett., 2004, 393, 51–57 CrossRef CAS.
- (a) J. P. Perdew, J. A. Chevary, S. H. Vosko, K. A. Jackson, M. R. Pederson, D. J. Singh and C. Fiolhais, Phys. Rev. B: Condens. Matter Mater. Phys., 1992, 46, 6671–6687 CrossRef CAS PubMed; (b) J. P. Perdew and Y. Wang, Phys. Rev. B: Condens. Matter Mater. Phys., 1996, 54, 16533–16539 CrossRef CAS PubMed.
- (a) J.-D. Chai and M. Head-Gordon, J. Chem. Phys., 2008, 128, 0804106 CrossRef PubMed; (b) J.-D. Chai and M. Head-Gordon, Phys. Chem. Chem. Phys., 2008, 10, 6615–6620 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4ob01459f |
| This journal is © The Royal Society of Chemistry 2025 |