
The design, synthesis, and biological evaluation of 5,6,7,8-tetrahydropteridines as anti-inflammatory compounds†
Rachel M.
Chen
 *ab,
Stefan
Emming
b,
Roseanna
Cinnamon‡
a,
Jacob P.
Cameron
a,
Kate
Schroder
bc,
Bostjan
Kobe
abc and
Avril A. B.
Robertson
*ab
*ab,
Stefan
Emming
b,
Roseanna
Cinnamon‡
a,
Jacob P.
Cameron
a,
Kate
Schroder
bc,
Bostjan
Kobe
abc and
Avril A. B.
Robertson
*ab
aSchool of Chemistry and Molecular Biosciences, The University of Queensland, St Lucia, 4072, Australia. E-mail: uqrchen4@uq.edu.au; a.robertson3@uq.edu.au
bInstitute for Molecular Bioscience, The University of Queensland, St Lucia, 4072, Australia
cAustralian Infectious Diseases Research Centre, The University of Queensland, St Lucia, 4072, Australia
First published on 11th November 2024
Abstract
The NLRP3 inflammasome is implicated in the pathogenesis of a wide array of inflammatory diseases including cancer, type II diabetes, atherosclerosis, gout, and neurodegenerative disease. Research has shown that Bruton's tyrosine kinase (BTK) is a critical regulator of the NLRP3 inflammasome and that the pharmacological inhibition of BTK using the FDA-approved inhibitor ibrutinib diminishes NLRP3-dependent inflammatory response. Herein, we describe our pursuit towards novel anti-inflammatory compounds using a scaffold-hopping approach. In our drug discovery efforts, we identified 5,6,7,8-tetrahydropteridines as underutilized scaffolds in medicinal chemistry. We report the synthesis of 5,6,7,8-tetrahydropteridines with potential as anti-inflammatory compounds.
Introduction
Since their discovery in 2002,1 inflammasomes have emerged as principal regulators of innate and adaptive inflammatory responses. Inflammasomes sense pathogen- and damage-associated molecular patterns and initiate an inflammatory cascade through the activation of pro-inflammatory caspases. The NLRP3 inflammasome, one of the most well-characterized inflammasomes, is activated by a wide-range of stimuli including crystalline materials (e.g. monosodium urate, silica),2 peptide aggregates (e.g. amyloid β-sheets, fibrillar α-synuclein), bacterial toxins (e.g. nigericin), and mitochondrial dysfunction.3 The increasing number of stimuli that are able to trigger NLRP3 activation promote the hypothesis that the NLRP3 inflammasome acts as a sensor for cytosolic perturbance and cellular homeostasis rather than a receptor for specific molecular agonists.4 Since the NLRP3 inflammasome responds to a range of molecular patterns associated with ageing, metabolic disease, infection, obesity, and environmental factors, it is of particular importance as a target for pharmacological intervention.5 Key to this is an understanding of the mechanisms governing NLRP3 activation.Bruton's tyrosine kinase (BTK) is a member of the Tec family of cytoplasmic kinases that has drawn significant attention in the context of cancer and immunological disorders. BTK is implicated in toll-like receptor, Fc receptor, and B cell immunoreceptor signaling.6–8 Additionally, there is accumulating evidence pointing to BTK's involvement in driving innate immune responses: BTK directly interacts with the NLR family pyrin domain containing 3 (NLRP3) and apoptosis-associated speck-like protein containing a caspase recruitment domain (ASC) to promote the assembly of the NLRP3 inflammasome.9–11 BTK phosphorylates four tyrosine residues in the PYD-NACHT polybasic linker of NLRP3, inverting the charge of this region, causing dissociation from negatively-charged phosphatidylinositol phosphates on organelle membranes. These observations suggest that BTK plays a critical role in the dynamic localization of NLRP3 and the downstream assembly of the ASC speck in both murine and human cell lines.11 The ability for BTK to modulate NLRP3 activation positions this druggable kinase as an attractive target towards the treatment of NLRP3-driven inflammatory diseases.
The essential role of BTK in B cell malignancies has resulted in a surge in the BTK inhibitors (BTKis) being developed.12,13 Amongst BTKis, the FDA-approved ibrutinib (Imbruvica®) demonstrated micromolar inhibition of NLRP3-dependent production of IL-1β in murine and human cell lines.10,14 We were thus interested in exploring the pharmacophoric elements that drive the inhibitory activity of ibrutinib against the NLRP3 inflammasome.
However, the patent landscape covering BTKis has become crowded, making it challenging to discover novel pharmacophores that target the ATP-binding site of BTK. In fact, there are over 88 patents protecting the structure, pharmaceutical composition, synthesis, and therapeutic application of ibrutinib. Furthermore, the pyrazolopyrimidine core scaffold of ibrutinib has been intensely researched as a pharmacophore for a variety of biological targets.15
We used a scaffold-hopping approach to design novel analogues of ibrutinib. In particular, we sought to embed a fused pyrimidine scaffold as these scaffolds impart opportunities for chemical derivation and are renowned for their diverse biological activities in the context of cancer and inflammation.16 This led to the design of the 5,6,7,8-tetrahydropteridine scaffold (Fig. 1). Compared to ibrutinib, 1b showed an improvement in the calculated physicochemical properties that are important in drug discovery, such as lowered lipophilicity (CLogP), lowered topological polar surface area (TPSA), and greater fraction of sp3 carbons (Fsp3).17,18 We anticipated that the tetrahydropteridine scaffold could act as a bioisostere of adenine to mimic key hydrogen bond interactions with the ATP-binding hinge region of BTK (Fig. 2).19
 | ||
| Fig. 1 Scaffold hopping of ibrutinib to 1b comprising a 5,6,7,8-tetrahydropteridine scaffold. Physicochemical properties were calculated using SwissADME. | ||
 | ||
| Fig. 2 In silico analysis of 1b docked into BTK (PDB ID: 5P9J) using Autodock Vina and presented using PyMOL v2.5.3 (Schrödinger, LLC). Hydrogen bond distances (Å) to hinge residues are shown with dashed lines. | ||
There are limited studies that report on the synthesis of 5,6,7,8-tetrahydropteridine derivatives, supporting the notion that tetrahydropteridines are underutilized, novel scaffolds in medicinal chemistry. Currently, only 935 compound analogues of 5,6,7,8-tetrahydropteridines have been reported, compared to 224k compound analogues of pyrazolopyrimidines (SciFinder®; Chemical Abstracts Service).§ Among the 5,6,7,8-tetrahydropteridines, only 193 compound analogues of 5-N-aryl-5,6,7,8-tetrahydropteridines have been reported, none of which possess a 4-amino group. Hence, there are no synthetic routes toward 4-amino-5-N-aryl-tetrahydropteridines such as 1b in the peer reviewed literature. We were thus compelled to establish a synthetic route to these scaffolds to advance our own work and to broaden the applicability of the tetrahydropteridine scaffold.
Results and discussion
5,6,7,8-Tetrahydropteridines were first synthesized by Taylor and Sherman in 1959 through the reduction of aromatic pteridines.20 Tetrahydropteridines have also been synthesized from 4,5-diaminopyrimidines by condensation with 1,2-dicarbonyl compounds or by intramolecular cyclisation.21–23 We envisioned that 1b could be synthesized by cyclization of a 5-N-disubstituted pyrimidine, such as 2b.We utilized an approach toward the synthesis of 5-N-aryl-pyrimidine 2b, based on a patent disclosing the process-scale synthesis of tirabrutinib.¶![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 24 Early functionalization allowed for the installation of the 5-N-diphenyl ether and 3-(R)-N-piperidine or 3-(R)-N-pyrrolidine moieties. The 5,6-diaminopyrimidine intermediates were synthesized in 25% (2a) and 27% (2b) yields over four steps and were key intermediates to construct the 6:6 heterocyclic scaffold (Scheme 1).
24 Early functionalization allowed for the installation of the 5-N-diphenyl ether and 3-(R)-N-piperidine or 3-(R)-N-pyrrolidine moieties. The 5,6-diaminopyrimidine intermediates were synthesized in 25% (2a) and 27% (2b) yields over four steps and were key intermediates to construct the 6:6 heterocyclic scaffold (Scheme 1).

The subsequent N-alkylation of 2b required thorough optimisation. This challenge is likely due to the strong electron-withdrawing N-pyrimido and N-aryl substituents. Our initial attempts to alkylate 2b included the reaction with methyl bromoacetate using diisopropylethylamine (DIPEA) in DCM (Table 1, entry 1). However, this left the starting material intact. A similar result was observed under base-free conditions in refluxing toluene (entry 2).
|
|
|||
|---|---|---|---|
| Entry | Conditions | Time (h) | % product formationa |
| a Measured using peak integration by LCMS at UV254. Product identities determined from the m/z in the ES+ mass spectrum. NR denotes no reaction. | |||
| 1 | DIPEA, DCM, reflux | 17 | NR |
| 2 | Toluene, reflux | 17 | NR |
| 3 | NaH, THF, 0 °C to 35 °C | 2 | NR |
| 4 | BEMP, DCM, rt to 50 °C | 18 | 23 |
| 5 | Cs2CO3, TBAI, MeCN, 4 Å MS | 18 | 13 |
| 6 | DBU, TBAI, MeCN, reflux | 18 | NR |
| 7 | KOH, 18-crown-6 | 3 | 4 |
| 8 | NaOH, TBAHSO4, H2O/DCM (1:1) |
48 | NR |
| 9 | KOH (1.5 equiv.), TBAI, MeCN | 4 | 38 |
| 10 | KOH (2.5 equiv.), TBAI, MeCN | 2 | 97 |

Also tested was NaH in THF (entry 3) as well as other strong, non-nucleophilic bases such as 1,8-diazabicyclo[5.4.0]undec-7-ene (DBU), Cs2CO3, and 2-tert-butylimino-2-diethylamino-1,3-dimethylperhydro-1,3,2-diazaphosphorine (BEMP) (entries 4–6). These were also unsuccessful in producing 3b in a satisfactory yield.
Phase-transfer catalysts (PTCs) have been used to improve particularly challenging alkylations.25 Therefore, the PTCs tetrabutylammonium iodide (TBAI), 18-crown-6, and tetrabutylammonium hydrogen sulfate (TBAHSO4) were used (entries 5–10).25–27 Conditions specified in entries 5–8 were unsuccessful in yielding 3b in satisfactory amounts.
We found that the use of KOH and TBAI in MeCN was ideal for the alkylation of 2b and afforded the desired intermediate in excellent yields (86% isolated yield).26 This reaction required a finely powdered KOH base and vigorous stirring to maximize the surface area in the solid–liquid contacts.
Despite efforts to perform cyclisation of intermediate 3bin situ, this was not possible using the same conditions without leading to hydrolysis of the methyl ester. Therefore, the alkylated intermediate was isolated as a crude intermediate prior to cyclisation using Cs2CO3 in refluxing 1,4-dioxane. This gave the desired chloropteridinones 4a and 4b in 56% and 52% yield over two steps, respectively (Scheme 1).
With the 4-chloropteridinone 4b in hand, optimization studies on aminating the 4-chloro position were carried out. Commonly used amination conditions failed, including the reaction of 4b with ethanolic ammonia or sodium azide in ethanol followed by a Staudinger reduction.28,29 It appeared that substitution by ammonia or ethanol occurred at the more labile lactam of 4b. Changing the solvent to toluene and H2O (85:15) under phase transfer conditions was unsuccessful and led to an intractable mixture.30 An alternative approach was then pursued via the reaction of the 4-chloropteridinone 4b with the nucleophilic amine 2,4-dimethoxybenzylamine (2,4-DMB-NH2).31 The 4-(2,4-DMB)-pteridinones 6a and 6b were obtained in 98% and 68% yields, respectively, and could be deprotected at a later step under acidic conditions or by hydrogenolysis (Scheme 1).
The reduction of 6b to the tetrahydropteridine intermediate 8b was attempted using a range of hydride reagents (Scheme 2). Borane reducing agents such as BH3·THF and NaBH4 were unreactive towards 6b, and failed to reduce the amide in sufficient quantities (Scheme 2, reactions (A) and (B)).32,33 Furthermore, the reduction of 6b under the Wolff–Kishner conditions only returned the starting materials (reaction (C)).34 On the other hand, the reaction of 6b with LiAlH4 generated a partially-reduced hemiaminal intermediate, as evidenced by LCMS.35 This was unchanged by heating the reaction to 45 °C or 60 °C. It is possible that the electron-withdrawing effects of the pyrimidine ring hinder the imine transition state required for the reduction of amides to amines by LiAlH4. Consequently, the reduction of 6b was achieved in two steps (Scheme 2, steps 1 and 2). After reduction of 6b to the hemiaminal intermediate 9b, the hemiaminal was reduced using Et3SiH and TFA.36 Precise temperature control was required, as a rearrangement product 10b predominated at higher reaction temperatures (steps 2a vs. 2b). Key differences identified in the 1H NMR and the 2D NMR spectra facilitated the assignment of each isomer. Following reaction optimization, the target compounds 1a and 1b were then obtained by the reaction of 7a and 7b with acryloyl chloride (Scheme 1).
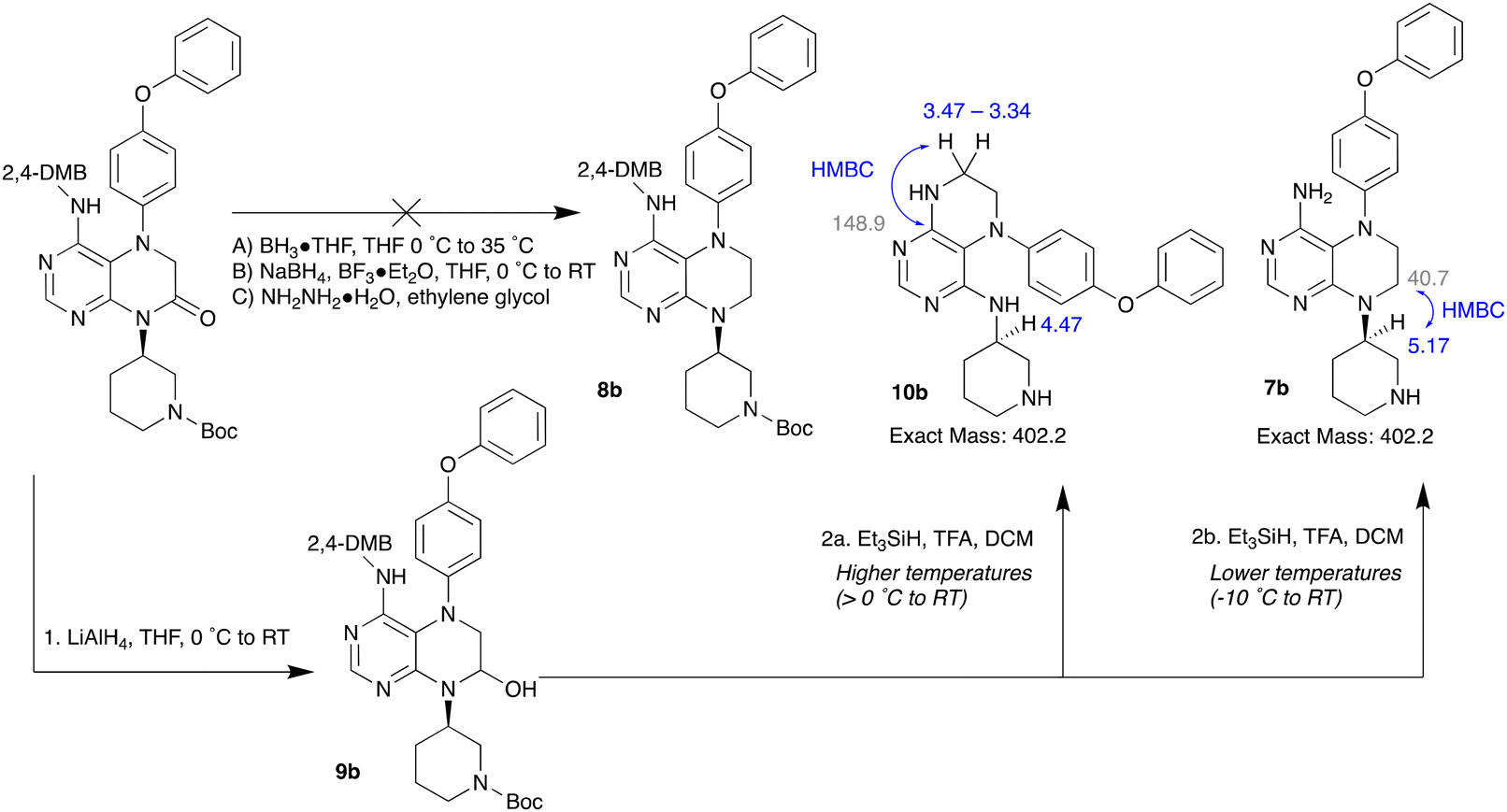 | ||
| Scheme 2 Conditions trialled for the reduction of 6b to 5,6,7,8-tetrahydropteridine intermediates. 13C NMR chemical shifts (ppm) shown in grey, 1H NMR chemical shifts (ppm) shown in blue. | ||
In addition to the tetrahydropteridines 1a and 1b, we identified that the 4-chloropteridinones, 13a and 13b, and 4-aminopteridinones, 14a and 14b, would contribute to the overall structure–activity relationship of this scaffold (Scheme 3). Boc-protected chloropteridinones 12a and 12b were deprotected under acidic conditions, followed by acylation with acryloyl chloride to give 14a and 14b in 8% and 23% yields over two steps, respectively. The aminopteridinones 11a and 11b required stronger acidic conditions to liberate both 2,4-DMB and the Boc protecting groups. The corresponding acrylamide-containing aminopteridinones 13a and 13b were synthesized in 23% and 34% yields, respectively. Notably, the aminopteridinones were not stable to RP-HPLC using 0.1% TFA in MeCN/H2O. However, the stability of these analogues was improved after purification by RP-HPLC using 20 mM ammonium acetate (pH 5.2) in H2O/MeCN, such that no degradation was seen in five days of storage.
 | ||
| Scheme 3 Deprotection and acylation of chloropteridinones and aminopteridinones. | ||
A selection of acrylamide-containing 5,6,7,8-tetrahydropteridines and 5,6-dihydropteridinones were tested for their inhibitory activity against BTK (Table 2). Compared to ibrutinib, the acrylamide-containing compounds synthesized in this study showed reduced inhibitory activity against BTK, indicating a strong preference for the pyrazolopyrimidine scaffold. Of the six acrylamide compounds synthesized, 13b showed the highest inhibitory activity against BTK with an IC50 value of 26 μM. Consistent with a previous report,37 the analogous compounds containing a 5-membered pyrrolidine-linked acrylamide were less potent, with 14a showing minimal activity in the concentrations tested, while 13a showed an IC50 greater than 40 μM. Compounds possessing the tetrahydropteridine scaffold, 1a and 1b, showed minimal inhibition of BTK. However, the aminopteridinone and chloropteridinone core scaffolds conferred higher, albeit only modest, activity against BTK.
| Compound | R1 | X | A | BTK IC50a (μM) |
NLRP3 IC50b (μM) |
Overall structure |
|---|---|---|---|---|---|---|
| a Inhibitory values were obtained using the ADP-Glo kinase assay (Promega). b Inhibitory values measured by ELISA of IL-1β production from PMA-THP-1 cells stimulated with lipopolysaccharide (LPS) and nigericin. ND denotes IC50 not determined in the concentrations tested (see the ESI† for details of biochemical assays). | ||||||
| 1b |

|
NH2 | CH2 | ND | >10 |

|
| 13b | NH2 | C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O O |
26 | >10 | ||
| 14b | Cl | CO |
>40 | >3 | ||
| 1a |

|
NH2 | CH2 | >80 | >10 | |
| 13a | NH2 | CO |
>40 | >30 | ||
| 14a | Cl | CO |
ND | >10 | ||
| Ibrutinib |

|
— | — | 0.012 | >30 |

|
Michael acceptor moieties, such as the acrylamide group, can directly target NLRP3 by alkylation of important cysteine residues located in the ATPase site or in other domains.38 We were thus motivated to explore the ability of the acrylamide-containing 5,6,7,8-tetrahydropteridines and 5,6-dihydropteridinones to inhibit the NLRP3 inflammasome. This was assessed using phorbol 12-myristate 13-acetate (PMA)-differentiated THP-1 cells, a model cell line for human monocytes (Table 2). The cells were first primed with lipopolysaccharide (LPS) and then treated with the inhibitors one hour before stimulation with the NLRP3 activator nigericin.
The acrylamide-containing 5,6,7,8-tetrahydropteridines and 5,6-dihydropteridinones showed good inhibition against the NLRP3 inflammasome (Table 2). 1a, 1b, and 13b showed the highest reduction of IL-1β at 30 μM by 69%, 68%, and 68%, respectively. 14a, 14b, and 13a led to a 65%, 59% and 50% reduction of IL-1β, respectively (Fig. 3A). This inhibition was greater than that of ibrutinib, which showed 43% inhibition at 30 μM (Fig. 3B). Intriguingly, the ability of 1a and 1b to inhibit the NLRP3 inflammasome without showing a strong ability to inhibit BTK indicates that the activity of these compounds is likely decoupled from BTK. Instead, it is possible that these compounds directly target NLRP3 or other proteins involved in the activation of IL-1β. The identification of these proteins is cause for future work.
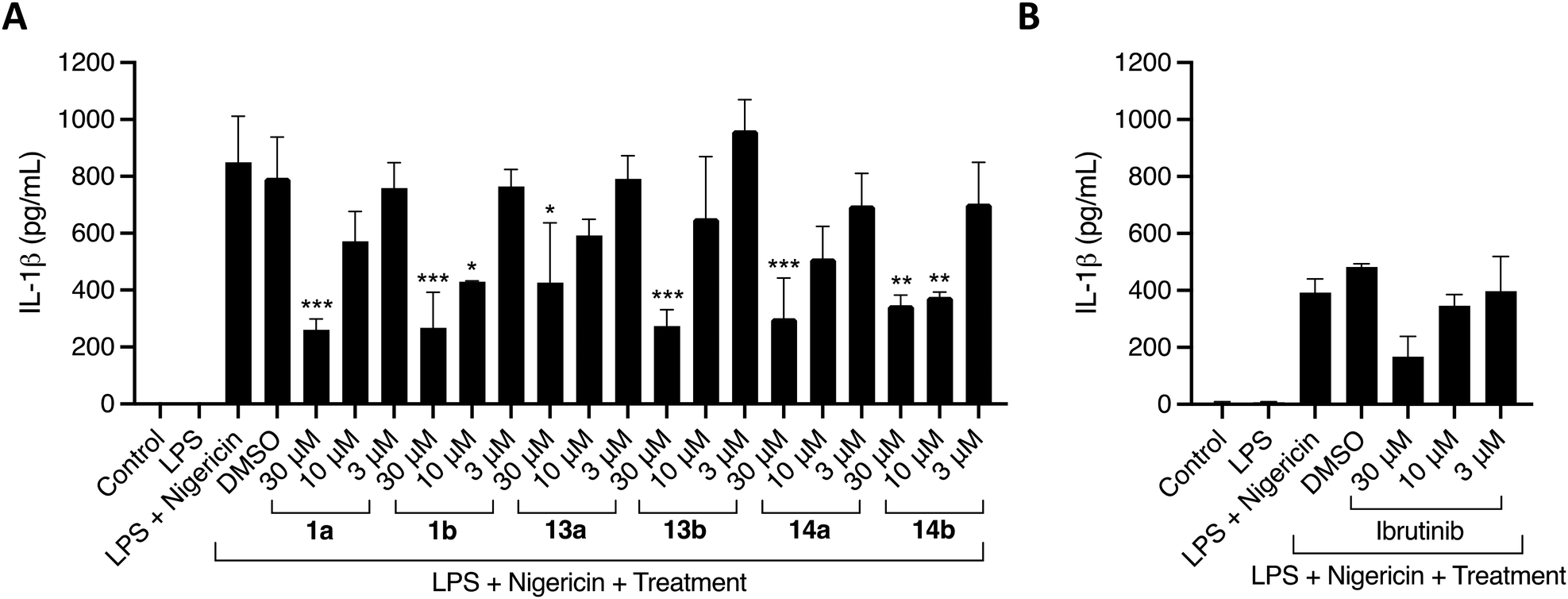 | ||
| Fig. 3 Acrylamide-containing compounds inhibit the NLRP3 inflammasome. PMA-THP-1 cells were stimulated with LPS (100 ng mL−1) for three hours and pretreated with DMSO, compounds (A) or ibrutinib (B) for 1 hour before stimulation with nigericin (10 mM) for 2 hours to activate the NLRP3 inflammasome. Data are expressed as the mean ± standard deviation of two independent experiments carried out in duplicate. Data were analysed by a one-way ANOVA followed by a Dunnett multiple comparisons test. *p < 0.05, **p < 0.01, ***p < 0.001. | ||
Conclusions
In summary, BTKis are intensely researched owing to their efficacy against B-cell hematologic disorders and inflammatory disease. As a result, the patent landscape is crowded, necessitating the design for novel and distinct molecular scaffolds. Fused pyrimidines have proved to be a valuable molecular scaffold in the context of cancer, inflammation, and BTKi development. In this article, we use a scaffold hopping approach to design a novel NLRP3 inflammasome inhibitor comprising a 5,6,7,8-tetrahydropteridine scaffold. We demonstrate that analogues containing the 5,6,7,8-tetrahydropteridine and 5,6-dihydropteridinone scaffolds exhibit marked anti-inflammatory activity in a human cell line.In our pursuit of a novel fused pyrimidine scaffold, we have established a reliable synthetic route to access 5′ and 8′ N-substituted 5,6,7,8-tetrahydropteridines at the 1 mmol scale. To the best of our knowledge, the present study discloses the first synthetic route toward 4-amino-5-N-aryl-tetrahydropteridines in the peer reviewed literature. It is appreciated that while these fused pyrimidine scaffolds were designed to target BTK and NLRP3, they may provide more opportunities for drug discovery against other therapeutically-important protein targets.
Experimental
Synthesis of tetrahydropteridines and related analogues
O). 1H NMR (600 MHz, chloroform-d) δ 8.52 (s, 1H), 7.38–7.29 (m, 2H), 7.14–7.05 (m, 1H), 7.04–6.97 (m, 2H), 6.97–6.88 (m, 2H), 6.83–6.77 (m, 2H), 4.95 (tt, J = 11.8, J = 4.1 Hz, 1H), 4.37 (d, Jgem = 16.6 Hz, 1H), 4.33 (d, Jgem = 16.6 Hz, 1H), 4.17–4.07 (m, 1H), 4.07–3.98 (m, 1H), 3.80 (dd, J = 11.8, Jgem = 11.8 Hz, 1H), 2.75 (dd, Jgem = 12.8, J = 12.8 Hz, 1H), 2.60 (qd, Jgem = 12.8, J = 12.8, J = 12.5, J = 4.2 Hz, 1H), 1.86–1.74 (m, 2H), 1.64 (tdt, J = 12.8, Jgem = 8.8, J = 4.3, 2H), 1.46 (s, 9H). 13C NMR (151 MHz, CDCl3) δ 167.5, 157.4, 154.9, 154.0, 153.7, 152.6, 151.8, 140.8, 129.9, 124.0, 123.4, 121.9, 120.1, 118.8, 80.0, 55.8, 52.4, 44.9, 44.3, 28.6, 27.0, 25.7. HRMS (ESI) m/z [M + H]+ calcd for [C28H31ClN5O4]+ 536.2060; found 536.2030.
4a was prepared following the same procedure as for 4b, using 2a (2.28 g, 4.73 mmol).¶ The product was purified by column chromatography on silica gel with EtOAc/hexane (0–40% gradient) as eluent to afford a white foam of 4a (1.38 g, 56% over two steps). FTIR (ATR, neat) λmax (cm−1) 2972 (aromC–H), 1689 (CONR2). 1H NMR (600 MHz, MeOD4) δ 8.54 (s, 1H), 7.39–7.31 (m, 2H), 7.13–7.07 (m, 1H), 7.00–6.93 (m, 6H), 5.77 (p, J = 8.4, 1H), 4.45 (s, 2H), 3.88–3.77 (m, 1H), 3.77–3.71 (m, 1H), 3.71–3.62 (m, 1H), 3.49–3.38 (m, 1H), 2.75–2.59 (m, 1H), 2.30–2.14 (m, 1H), 1.50 (s, 4H*), 1.47 (s, 5H). 13C NMR (151 MHz, MeOD) δ 169.5, 158.9, 156.4, 155.6, 154.8, 153.1, 152.7, 142.6, 130.9, 125.7, 124.3, 123.1, 120.9, 119.6, 81.0, 56.4, 53.4, 52.8*, 48.0, 47.8*, 46.4, 45.9*, 28.9, 28.8, 28.1*. HRMS (ESI) m/z [M − tBu + H]+ calcd for [C23H21ClN5O4]+ 466.1277; found 466.1287.
6a was prepared following the same procedure as for 6b, using 4a (1.00 g, 1.91 mmol). Upon completion of the reaction, the mixture was cooled to RT, diluted with DCM (30 mL), and washed with sat. aq. NH4Cl (70 mL). The aqueous phase was extracted with DCM (2 × 30 mL) and the organic layers were combined, washed with brine (100 mL), dried (MgSO4), and concentrated to a viscous liquid. The crude product (2.09 g) was purified using column chromatography on silica gel with EtOAc/hexane (20–40% gradient) as eluent to afford a white foam (1.22 g, 98%). FTIR (ATR, neat) λmax (cm−1) 3429 (N–H), 2971 (aromC–H), 1688 (CONR2). 1H NMR (600 MHz, MeOD4) δ 8.26 (s, 1H), 7.36–7.27 (m, 2H), 7.08 (dd, J = 8.1, 6.8 Hz, 1H), 6.99 (d, J = 8.3 Hz, 1H), 6.93–6.89 (m, 2H), 6.87 (d, J = 8.9 Hz, 2H), 6.85–6.81 (m, 2H), 6.43 (d, J = 2.4 Hz, 1H), 6.36 (dd, J = 8.3, 2.4 Hz, 1H), 5.59 (p, J = 8.5 Hz, 1H), 4.52 (s, 2H), 4.30 (s, 2H), 3.79–3.74 (m, 1H), 3.74 (s, 3H), 3.72–3.66 (m, 1H), 3.65 (s, 3H), 3.61–3.52 (m, 1H), 3.41–3.32 (m, 1H), 2.66–2.55 (m, 1H), 2.13–2.04 (m, 1H), 1.47 (s, 5H), 1.45 (s, 4H*). 13C NMR (151 MHz, MeOD4) δ 169.7, 162.0, 159.8, 159.0, 158.1, 156.4, 155.1, 154.2, 152.6, 142.0, 130.9, 130.3, 124.2, 121.4, 121.2, 120.2, 119.4, 109.8, 105.0, 99.2, 81.0, 57.0, 55.8, 52.9, 52.3*, 48.1, 47.9*, 46.4, 45.8*, 40.9, 29.1, 28.8, 28.2*. HRMS (ESI) m/z [M + H]+ calcd for [C36H41N6O6]+ 653.3082; found 653.3088.
7a was prepared following the same procedure as for 7b using 6a (302 mg, 0.46 mmol). The hemiaminal intermediate was obtained as a light yellow foam (315 mg) and used directly without further purification. LCMS (ESI) m/z [M + H]+ calcd for [C36H43N6O6]+ 655.3; found 655.3. The title compound 7a was prepared from the hemiaminal intermediate (250 mg, 0.38 mmol) as described for 7b using a total volume of 20% TFA (0.76 mL, 9.97 mmol). The crude product (274 mg) was purified by RP-HPLC to give the title compound as a white TFA salt (123 mg, 64%). FTIR (ATR, neat) λmax (cm−1) 3301 (broad, N–H), 3016 (broad, N–H), 2971 (aromC–H). 1H NMR (600 MHz, MeOD4) δ 8.18 (s, 1H), 7.35–7.29 (m, 2H), 7.08 (t, J = 7.4 Hz, 1H), 6.98–6.93 (m, 6H), 5.14 (br p, 1H), 3.71–3.65 (m, 3H), 3.56–3.52 (m, 2H), 3.50 (t, J = 4.8 Hz, 2H), 3.31–3.26 (m, 1H), 2.50–2.41 (m, 1H), 2.32–2.23 (m, 1H). 13C NMR (151 MHz, MeOD4) δ 159.1, 154.8, 154.3, 150.8, 146.4, 143.1, 130.8, 124.2, 123.1, 121.2, 119.4, 104.5, 58.5, 49.1, 48.4, 46.7, 44.7, 28.6. HRMS (ESI) m/z [M + H]+ calcd for [C22H25N6O]+ 389.2084; found 389.2107.
General method A – acylation with acryloyl chloride
To an oven-dried flask with a stirrer bar containing the amine intermediate was added an anhydrous solution of 3.5% Et3N in DCM (10 mL mmol−1) under argon. The solution was cooled to 0 °C before dropwise addition of 1 M acryloyl chloride in anhydrous DCM (1.3 equiv., prepared using a literature method).39 After complete addition, the reaction mixture was allowed to slowly warm up to RT. The reaction was monitored by LCMS and upon consumption of the starting material (ca. 1 hour), the reaction was diluted with 10% MeOH in DCM (10 mL) and washed with NaHCO3 (sat. aq., 10 mL). The aqueous phase was re-extracted with 10% MeOH in DCM (3 × 10 mL), and the organic layers were combined, washed with brine (30 mL), dried (MgSO4), and concentrated in vacuo.1b was prepared from 7b (128 mg, 0.25 mmol) using General method A. Purification by RP-HPLC yielded the title compound as a white lyophilised powder (73 mg, 64%). HRMS (ESI) m/z [M + H]+ calcd for [C26H29N6O2]+ 457.2347; found 457.2358.¶
1a was prepared from 7a (110 mg, 0.22 mmol) using General method A. The crude product was purified by RP-HPLC to give the title compound as a white powder (52.4 mg, 54%). HRMS (ESI) m/z [M + H]+ calcd for [C25H27N6O2]+ 443.2190; found 443.2197.¶
:1, pH = 5.3), filtered (PTFE, 0.45 μm) to remove insoluble precipitants, and lyophilised. The crude product was purified by RP-HPLC using a gradient of 20 mM ammonium acetate in H2O (pH = 5.3):20 mM ammonium acetate in MeCN/H2O (8:2, pH = 5.3) as eluent to give the amine intermediate as an acetate salt. The product was re-lyophilised with H2O/MeCN (1:1) to remove residual acetic acid (453 mg, 67%). HRMS (ESI) m/z [M + H]+ calcd for [C23H25N6O2]+ 417.2034; found 417.2041. 13b was prepared from the amine intermediate (100 mg, 0.21 mmol) using General method A to give the crude product as a beige residue (121 mg). The crude product was purified by RP-HPLC using a gradient of 20 mM ammonium acetate in H2O (pH = 5.3):20 mM ammonium acetate in MeCN/H2O (8:2, pH = 5.3) as eluent to give the title compound 13b as a white lyophilised powder. The product was re-lyophilised with H2O/MeCN (1:1) to remove residual acetic acid (49.7 mg, 50%). HRMS (ESI) m/z [M + H]+ calcd for [C26H27N6O3]+ 471.2139; found 471.2153.¶
:20 mM ammonium acetate in MeCN/H2O (8:2, pH = 5.3) as eluent gave the amine intermediate as a white acetate salt. The product was re-lyophilised with H2O/MeCN (1:1) to remove residual acetic acid (178 mg, 63%). HRMS (ESI) m/z [M + H]+ calcd for [C22H23N6O2]+ 403.1877; found 403.1891. The amine intermediate (100 mg, 0.22 mmol) was then acylated using General method A. The crude product (94 mg) was purified by RP-HPLC using a gradient of 20 mM ammonium acetate in H2O (pH = 5.3):20 mM ammonium acetate in MeCN/H2O (8:2, pH = 5.3) as eluent to give the title compound 13a as a white acetate salt. The product was re-lyophilised with H2O/MeCN (1:1) to remove residual acetic acid (36 mg, 37%). HRMS (ESI) m/z [M + H]+ calcd for [C25H25N6O3]+ 457.1983; found 457.1997.¶
Author contributions
The manuscript was written through contributions from all authors. All authors have given approval to the final version of the manuscript.Data availability
The data supporting this article have been included as part of the ESI.†Conflicts of interest
A. A. B. R. and K. S. are co-inventors on granted patents and patent applications for NLRP3 inhibitors licensed to Inflazome Ltd, subsequently acquired by Roche; these companies are developing drugs that target the NLRP3 inflammasome to address unmet clinical need in inflammatory disease. K. S. served on the Scientific Advisory Board of Inflazome (2016–2017) and Quench Bio, USA (2018–2021) and serves on a Scientific Advisory Board for Novartis, Switzerland (since 2020).Acknowledgements
This work was supported by the Australian Government Research Training Program. R.M.C. was supported by a University of Queensland Research Scholarship. S. E. is supported by the Australian Research Council (Discovery Early Career Researcher Fellowship DE220100823). K. S. is supported by the National Health and Medical Research Council of Australia (Fellowship 2009075). B. K. was funded by the Australian Research Council (ARC Laureate Fellowship FL180100109) and the National Health and Medical Research Council (NHMRC Investigator 2025931). We give our gratitude to Mr Alun Jones at the University of Queensland for mass spectrometry analysis, and Dr Grant Stuchbury at UniQuest and Shannon Jewell at the University of Queensland for assistance with biochemical assays. We thank A/Prof. Ross McGeary and A/Prof. Richard Gordon for valuable discussions.Notes and references
- F. Martinon, K. Burns and J. Tschopp, Mol. Cell, 2002, 10, 417–426 CrossRef CAS.
- F. Martinon, V. Pétrilli, A. Mayor, A. Tardivel and J. Tschopp, Nature, 2006, 440, 237 CrossRef CAS PubMed.
- Y. Yang, H. Wang, M. Kouadir, H. Song and F. Shi, Cell Death Dis., 2019, 10, 128 CrossRef PubMed.
- T. Próchnicki and E. Latz, Cell Metab., 2017, 26, 71–93 CrossRef PubMed.
- M. S. J. Mangan, E. J. Olhava, W. R. Roush, H. M. Seidel, G. D. Glick and E. Latz, Nat. Rev. Drug Discovery, 2018, 17, 588 CrossRef CAS PubMed.
- A. J. Mohamed, B. F. Nore, B. Christensson and C. I. Smith, Scand. J. Immunol., 2002, 49, 113–118 CrossRef PubMed.
- A. N. R. Weber, Z. Bittner, X. Liu, T.-M. Dang, M. P. Radsak and C. Brunner, Front. Immunol., 2017, 8, 1454 CrossRef.
- A. T. Bender, A. Gardberg, A. Pereira, T. Johnson, Y. Wu, R. Grenningloh, J. Head, F. Morandi, P. Haselmayer and L. Liu-Bujalski, Mol. Pharmacol., 2017, 91, 208–219 CrossRef CAS PubMed.
- M. Ito, T. Shichita, M. Okada, R. Komine, Y. Noguchi, A. Yoshimura and R. Morita, Nat. Commun., 2015, 6, 7360 CrossRef PubMed.
- X. Liu, T. Pichulik, O.-O. Wolz, T.-M. Dang, A. Stutz, C. Dillen, M. Delmiro Garcia, H. Kraus, S. Dickhöfer, E. Daiber, L. Münzenmayer, S. Wahl, N. Rieber, J. Kümmerle-Deschner, A. Yazdi, M. Franz-Wachtel, B. Macek, M. Radsak, S. Vogel, B. Schulte, J. S. Walz, D. Hartl, E. Latz, S. Stilgenbauer, B. Grimbacher, L. Miller, C. Brunner, C. Wolz and A. N. R. Weber, J. Allergy Clin. Immunol., 2017, 140, 1054–1067 CrossRef CAS PubMed.
- Z. A. Bittner, X. Liu, M. Mateo Tortola, A. Tapia-Abellán, S. Shankar, L. Andreeva, M. Mangan, M. Spalinger, H. Kalbacher, P. Düwell, M. Lovotti, K. Bosch, S. Dickhöfer, A. Marcu, S. Stevanović, F. Herster, Y. Cardona Gloria, T.-H. Chang, F. Bork, C. L. Greve, M. W. Löffler, O.-O. Wolz, N. A. Schilling, J. B. Kümmerle-Deschner, S. Wagner, A. Delor, B. Grimbacher, O. Hantschel, M. Scharl, H. Wu, E. Latz and A. N. R. Weber, J. Exp. Med., 2021, 218, e20201656 CrossRef CAS PubMed.
- D. Rozkiewicz, J. M. Hermanowicz, I. Kwiatkowska, A. Krupa and D. Pawlak, Molecules, 2023, 28, 2400 CrossRef CAS PubMed.
- J. Krämer, A. Bar-Or, T. J. Turner and H. Wiendl, Nat. Rev. Neurol., 2023, 19, 289–304 CrossRef.
- G. S. D. Purvis, M. Collino, H. Aranda-Tavio, F. Chiazza, C. E. O'Riordan, L. Zeboudj, S. Mohammad, D. Collotta, R. Verta, N. E. S. Guisot, P. Bunyard, M. M. Yaqoob, D. R. Greaves and C. Thiemermann, Br. J. Pharmacol., 2020, 177, 4416–4432 CrossRef CAS PubMed.
- V. Asati, A. Anant, P. Patel, K. Kaur and G. D. Gupta, Eur. J. Med. Chem., 2021, 225, 113781 CrossRef CAS.
- B. Nammalwar and R. A. Bunce, Pharmaceuticals, 2024, 17, 104 CrossRef CAS PubMed.
- A. Daina, O. Michielin and V. Zoete, Sci. Rep., 2017, 7, 42717 CrossRef PubMed.
- F. Lovering, J. Bikker and C. Humblet, J. Med. Chem., 2009, 52, 6752–6756 CrossRef CAS PubMed.
- O. Trott and A. J. Olson, J. Comput. Chem., 2009, 455–461, DOI:10.1002/jcc.21334.
- E. C. Taylor and W. R. Sherman, J. Am. Chem. Soc., 1959, 81, 2464–2471 CrossRef CAS.
- R. G. Wilde, DuPont Merck Pharmaceutical Company, WO9744038A1, 1997.
- A. R. Heald and D. A. Morley, AstraZeneca AB, WO2007148064A1, 2007.
- S. W. Bailey, R. Y. Chandrasekaran and J. E. Ayling, J. Org. Chem., 1992, 57, 4470–4477 CrossRef CAS.
- R. Suzuki, A. Hiramatsu, M. Tateyama and H. Sasahara, Ono Pharmaceutical Co., Ltd., US10239879B2, 2019.
- C. M. Starks, C. L. Liotta and M. Halpern, Phase-transfer catalysis: fundamentals, applications, and industrial perspectives, Springer Netherlands, Dordrecht, 1994 Search PubMed.
- G. Li, X. Wang, C. Tian, T. Zhang, Z. Zhang and J. Liu, Tetrahedron Lett., 2012, 53, 5193–5196 CrossRef CAS.
- K. Sukata, Bull. Chem. Soc. Jpn., 1983, 56, 280–284 CrossRef CAS.
- C. Rieux, S. Goffinont, F. Coste, Z. Tber, J. Cros, V. Roy, M. Guérin, V. Gaudon, S. Bourg, A. Biela, V. Aucagne, L. Agrofoglio, N. Garnier and B. Castaing, Int. J. Mol. Sci., 2020, 21, 2058 CrossRef CAS PubMed.
- N. Nisha, M. C. Sharma, R. Kumar and Y. Kumar, ChemistrySelect, 2018, 3, 4822–4826 CrossRef.
- N. Desai and R. Shah, Synthesis, 2006, 3275–3279, DOI:10.1055/s-2006-950206.
- J. D. Oslob, J. Zhu, K. Barr, J. Crossrow, B. Raimundo and H. Tanaka, Sunesis Pharmaceuticals, Inc., WO2006065703A1, 2006.
- T. Nowak, AstraZeneca AB, WO2008093075A2, 2008.
- D. A. Burnette, M. G. Bursavich and A. J. Mcriner, WO2015066697A1, 2015.
- C. G. Bashore, I. J. Samardjiev, J. Bordner and J. W. Coe, J. Am. Chem. Soc., 2003, 125, 3268–3272 CrossRef CAS PubMed.
- Y. Sun, Y.-L. Li and D. M. Burns, Incyte Corporation, WO2016183071, 2016.
- J. Gu, B. X. Xiao, Y. R. Chen, W. Du and Y. C. Chen, Adv. Synth. Catal., 2016, 358, 296–302 CrossRef CAS.
- Z. Pan, H. Scheerens, S.-J. Li, B. E. Schultz, P. A. Sprengeler, L. C. Burrill, R. V. Mendonca, M. D. Sweeney, K. C. K. Scott, P. G. Grothaus, D. A. Jeffery, J. M. Spoerke, L. A. Honigberg, P. R. Young, S. A. Dalrymple and J. T. Palmer, ChemMedChem, 2007, 2, 58–61 CrossRef CAS PubMed.
- A. G. Baldwin, D. Brough and S. Freeman, J. Med. Chem., 2016, 59, 1691–1710 CrossRef CAS PubMed.
- G. H. Stempel, R. P. Cross and R. P. Mariella, J. Am. Chem. Soc., 1950, 72, 2299–2300 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available: Details on biochemical and synthetic procedures and compound spectral characterisation and assignment. See DOI: https://doi.org/10.1039/d4ob01453g |
| ‡ Present address: IP Australia – 47 Bowes St., Phillip, ACT, 2606, Australia. |
| § See the ESI† for Scifinder® search results. |
| ¶ See the ESI† for additional synthetic procedures, NMR characterisation data, and NMR spectral assignments. |
| This journal is © The Royal Society of Chemistry 2025 |