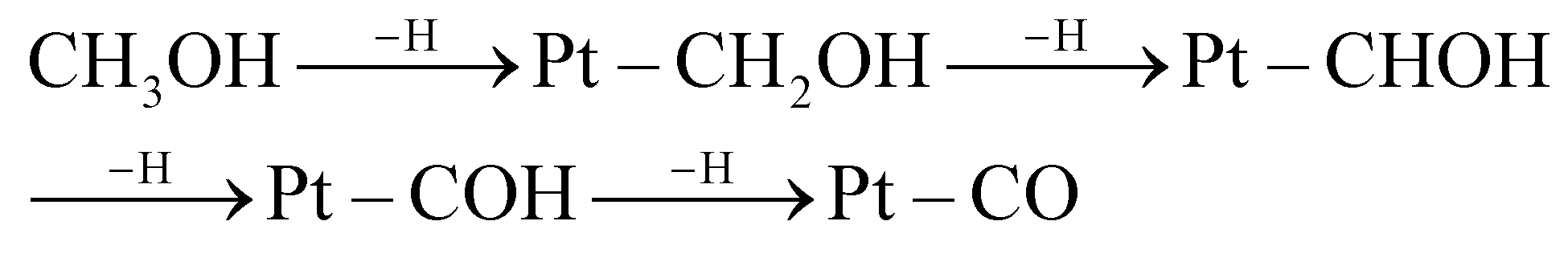DOI:
10.1039/C6NR07036A
(Paper)
Nanoscale, 2017,
9, 201-207
One-step synthesis of ultrathin PtxPb nerve-like nanowires as robust catalysts for enhanced methanol electrooxidation†
Received
6th September 2016
, Accepted 19th November 2016
First published on 21st November 2016
Abstract
Ultrathin PtxPb nerve-like nanowires (NNWs) with a diameter of only around 3.6 nm were synthesized by a one-step wet-chemical strategy, and they served as robust catalysts for greatly enhancing methanol electrooxidation both under acidic and alkaline conditions. Due to the high CO-poisoning tolerance, superior electrocatalytic activity and stability endowed by the Pt–Pb alloyed composition and the unique structure, the Pt3.5Pb NNWs showed the highest specific activity of 2.78 mA cm−2 in acidic media and 6.51 mA cm−2 in alkaline media toward the methanol oxidation reaction (MOR), which are 5.24 and 4.12 times higher than those of the commercial Pt/C catalysts, respectively. Meanwhile, the demonstrated synthetic strategy for Pt–Pb nanocrystals may stimulate more inspiration and strategies of the novel metal-based nanocrystals for promising applications in electrocatalysis.
Introduction
During the past few decades, platinum (Pt) has been used as a typical and promising catalyst in fuel cells due to its superior electrocatalytic properties.1–4 The MOR is a basic anode reaction of direct methanol fuel cells (DMFCs), and improving the electrocatalytic oxidation efficiency of the methanol molecules on the platinum catalyst is an important approach to increase the power density of fuel cells.5,6 However, the high cost, carbon monoxide (COads) poisoning and sluggish kinetics predominantly hindered the commercialization of Pt catalysts.7–9 Thus plenty of research efforts have been devoted to the precisely controlled synthesis of Pt-based intermetallic nanostructures with tailored shapes and compositions, which can not only reduce the usage of Pt but also increase the activity with the synergetic effect.10–12
With numerous Pt-based bimetallic nanocrystals being developed for the MOR, the catalytic activity and stability have been greatly promoted compared to the commercial Pt/C.13–15 But CO poisoning on the catalyst surface is still an inevitable challenge. In the process of the MOR, the Pt atom sites catalyze methanol oxidation to form adsorbed COads, and it can only be oxidized to CO2 at large overpotentials.16,17 Thus, Pt–COads can be considered as a poisoning intermediate, which hindered the further fuel oxidation on the platinum catalyst surface. Based on the poison resistance research on Pt-based bimetallic catalysts in MOR, PtRu was once thought as the best performing CO-poisoning-tolerant electrocatalyst and can be explained by the Watanabe–Motoo bifunctional mechanism: the Ru atoms provide adsorbed hydroxyl groups (OHads), which serve as the oxidant to oxidize the COads at a much lower potential.18–21 Furthermore, both DFT calculations and experiments indicate that Pt nanoparticles cooperate with some main group metals, such as Pb, Bi, Sb, and Sn, and show superior electrocatalytic properties toward the oxidation of alcohol molecules.22–27 This could be attributed to the more oxophilicity of these metals than Ru to provide oxygen-containing species that accelerate the oxidation of COads. Meanwhile, the presence of Pb (or Bi) changes the electronic structure of platinum to lower the CO adsorption energy on the catalyst surface.
Thus, we develop a one-step wet-chemical synthesis of ultrathin PtxPb nerve-like nanowires (NNWs) as robust catalysts for markedly enhanced MOR both under acidic and alkaline conditions. Different from those developed methods for nanowire networks with poly(N-vinyl-2-pyrrolidone) (PVP)-assistants,28,29 the synthesized PtxPb NNWs are inspired by the toluene–oleylamine (PhMe–OAm) system,30,31 which just uses OAm as the surfactant and structure-directing agent to accelerate the oriented attachment of nanoparticles to form the ultrathin NNWs. Deservedly, their bimetallic composition and nerve-like nanostructure provide them with high MOR electrocatalytic activity and stability.
Experimental
Chemicals
Platinum(II) acetylacetonate (Pt(acac)2, 98%), lead(II) acetylacetonate (Pb(acac)2, 99%) and oleylamine (OAm, >90%) were purchased from Meryer. Borane-tert-butylamine complex (BTBA, >95%), lead acetate (Pb(ac)2, 99.5%) and toluene were purchased from Aladdin Co. Commercial Pt/C catalysts (20 wt% loading of Pt on carbon black) and Nafion (5%) were purchased from Sigma Aldrich. Commercial PtRu/C catalysts (40 wt% loading of Pt and 20 wt% loading of Ru on carbon black) were purchased from Alfa Aesar. All the chemicals in the experiment were used without further purification.
Synthesis of the PtxPb NNWs
In a typical synthesis of the Pt3.5Pb NNWs, Pt(acac)2 (0.012 mmol), Pb(acac)2 (0.004 mmol) and OAm (200 μL) were added into 5.0 mL toluene, and then the mixture was ultrasonicated for around 30 min to form a homogeneous solution. The mixture solution was stirred for 2 min after BTBA (0.170 mmol) dissolved, then the solution was transferred into a Teflon-lined stainless-steel autoclave with a capacity of 10 mL and heated at 150 °C for 5 h before it was cooled to room temperature. The products collected by centrifugation were washed three times with an ethanol–cyclohexane mixture. The synthesis of the Pt3Pb NNWs and the Pt4Pb NNWs was under the same conditions except that the amount of Pb(acac)2 was altered to 0.0048 mmol and 0.0035 mmol.
Characterization
The morphology and composition of the nanoparticles were investigated using a Hitachi H-8100 EM transmission electron microscope (TEM) with an accelerating voltage of 100 kV. The HRTEM images and HAADF-STEM images were obtained with a JEM-2010 operating at 200 kV equipped with an energy dispersive spectrometer (EDS). X-ray diffraction (XRD) patterns were collected on a D8 ADVANCE (Bruker AXS, Germany) diffractometer using Cu Kα radiation. The mole ratio of nanoparticles was measured by inductively coupled plasma atomic emission spectrophotometry (ICP-AES), which was performed on a Thermo Scientific iCAP6300 (Thermo Fisher Scientific, US). X-ray photoelectron spectroscopy (XPS) measurement was conducted on an ESCALAB-MKII spectrometer (VG Co., UK) with Al Kα X-ray radiation as the X-ray source for excitation.
Catalyst preparation
The as-synthesized PtxPb NNWs were suspended in 20 mL acetic acid and stirred at 70 °C overnight to remove the OAm molecules.32 The catalysts were separated from the acid by centrifugation and washed with ethanol and deionized water, and then redispersed in a mixture of deionized water, isopropanol and Nafion (v/v/v = 4/1/0.05). Then 5 μL of the mixture (the concentration of the catalysts: 0.4 mg mL−1 for PtxPb NNWs and 1.0 mg mL−1 for the Pt/C catalysts) was cast on the working electrode and dried under ambient conditions.
Electrochemical characterization
A three-electrode cell was used to do the electrochemical measurements. An Ag/AgCl (saturated KCl) electrode served as the reference electrode, and the Pt wire served as the counter electrode. The working electrode was a glassy-carbon electrode (GCE) (diameter: 3 mm, area: 0.07065 cm2). The electrochemical active surface area (ECSA) measurement was determined by integrating the hydrogen adsorption charge on cyclic voltammetry (CV) at room temperature in 0.5 M H2SO4 solution. Methanol electrooxidation measurements under acidic and alkaline conditions were conducted in 0.5 M H2SO4 + 1 M CH3OH solution and 0.5 M KOH + 1 M CH3OH solution, respectively. All of the electrochemical measurements were performed at room temperature with the potential scan rate of 50 mV s−1.
Results and discussion
Fig. 1 shows the transmission electron microscopy (TEM) and high-angle annular dark-field scanning TEM (HAADF-STEM) images of the obtained Pt3.5Pb NNWs. The nerve-like nanocrystals are composed of ultrathin Pt–Pb wavy nanowires and connected by plentiful branches, which are similar to the veins of leaves. As the water rapidly transport in the veins, this unique structure is conducive to the electron transfer between different active sites, thus accelerating the reaction rate. The ultrathin NNWs are 3.6 ± 0.5 nm in width and hundreds of nanometers in length. The morphology and structure have no obvious change for Pt3Pb NNWs and Pt4Pb NNWs (Fig. S3†), the Pt/Pb atomic ratios in the nanocrystals were determined by EDX (Fig. S2a–c†) and ICP-AES. And the NNWs cannot be synthesized when changing the Pt2+/Pb2+ ratios of the precursors to 2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 (Fig. S3†), which may be ascribed to the tardy growth with the increase of the Pb2+ content. Therefore, it is a general synthetic strategy for the PtxPb NNWs (x ≧ 3).
:1 (Fig. S3†), which may be ascribed to the tardy growth with the increase of the Pb2+ content. Therefore, it is a general synthetic strategy for the PtxPb NNWs (x ≧ 3).
 |
| | Fig. 1 Morphology and structural characterization of the Pt3.5Pb NNWs. Representative (a and b) HAADF-STEM images and (c and d) TEM images of the Pt3.5Pb NNWs. | |
Further elements and crystal structure studies of the Pt3.5Pb NNWs are shown in Fig. 2. The high resolution TEM (HRTEM) images (Fig. 2c and d) show that the lattice fringes coherently extended across the whole nanowire, indicating that the NNWs have good crystallization. X-ray diffraction (XRD) (Fig. 2h) and selected area electron diffraction (SAED) (inset in Fig. 2a) were conducted to determine the phase of the PtxPb NNWs, the positions of diffraction peaks were in accord with those of the face-center-cubic (fcc) PbPt (JCPDS no. 06-0574) and hexagonal close packed (hcp) PbPt alloy (JCPDS no. 06-0374), indicating the presence of both the PbPtx phase and PbPt phase in the Pt3.5Pb NNWs. The Pt3Pb NNWs had the PbPt phase (Fig. 2h), as well as the Pt4Pb NNWs had the PbPt phase and fcc Pt phase (JCPDS no. 04-0802) (Fig. S2d†). The SAED pattern shows concentric rings composed of bright discrete diffraction spots that can be indexed to the crystal planes of (100), (101), (111), (102) and (110), which is in conformity with the XRD pattern. The observed lattice fringes with interplanar spacings of 0.234 nm and 0.219 nm correspond respectively to the (111) and (102) planes of the Pt3.5Pb NNWs. Meanwhile, since the nerve-like structure formed from the collisions and oriented attachments between the nanoparticles, plenty of stacking faults and lattice defects by stress and Ostwald ripening between the adjacent nanoparticles were produced, which could act as highly catalytic sites.10,33 The element distribution and composition of the obtained Pt3.5Pb NNWs were analyzed using HAADF-STEM-energy dispersive X-ray spectroscopy (HAADF-STEM-EDX) and X-ray photoelectron spectroscopy (XPS). As seen in the elemental mapping images (Fig. 2e, and S1†), both Pt and Pb elements were uniformly distributed throughout the nanocrystals, confirming the homogeneous alloyed nanostructure of the NNWs. The XPS spectrum (Fig. 2f and g) shows that the Pt is mainly in the zerovalent state and the Pb contains both zerovalent and divalent states on the surface of the Pt3.5Pb NNWs. Compared to those of bulk Pt, a slightly negative shift of the Pt 4f binding energy indicates an obvious change in the electronic structure of Pt upon alloying with Pb, which can be attributed to the electron donation from Pb to Pt because of the higher electronegativity of Pt compared to that of Pb.34
 |
| | Fig. 2 (a) High-magnification TEM image and (b, c and d) HRTEM images of the Pt3.5Pb NNWs. The red dotted lines and dots marked in (c, d) indicate the crystal defects and atomic steps of the Pt3.5Pb NNWs, respectively. (e) HAADF-STEM image and EDX mapping images of the Pt3.5Pb NNWs. XPS spectra for the Pb 4f region (Pb 4f7/2 and Pb 4f5/2) (f) and Pt 4f region (Pt 4f7/2 and Pt 4f5/2) (g) of the Pt3.5Pb NNWs. (h) XRD pattern of the Pt3Pb NNWs and Pt3.5Pb NNWs. The inset image in (a) shows the corresponding SAED pattern. | |
To understand the formation mechanism of the alloyed Pt3.5Pb NNWs, the morphologies of the intermediate nanoparticles yielded at different reaction times were investigated by TEM (Fig. 3). At the initial reaction stage, the ultrathin PtPb nanoparticles, with an average size of 3.8 nm, were found after 30 minutes. After the reaction proceeded for 1 hour, the size of the nanoparticles had little change, and the oriented attachment occurred to form short wavy nanorods by connecting the adjacent nanoparticles.29 When the reaction time was increased to 2 hours, extended wavy nanowires appeared by the nose to tail connection of the nanorods, and branches began to grow on the larger nanoparticles of the wavy nanowires. After another 2 hours reaction process, there was no significant morphological change with the final product. The wavy nanowires and branches were linked together to knit an integrated vein, just like the leaf vein, the PtxPb NNWs will be very stable as a whole and the electron transfer between each active site can be quite effective through the venation channels.
 |
| | Fig. 3 TEM images of the Pt3.5Pb NNW intermediates taken at different reaction times: (a) 30 min, (b) 1 h, (c) 2 h and (d) 4 h. (e) Schematic illustration of the formation mechanism towards the Pt3.5Pb NNWs, yellow balls and green balls on behalf of platinum and lead atoms, respectively. | |
Control experiments were conducted by altering the reaction parameters to study their influence on the formation of the NNWs. As shown in Fig. S4,† NNWs could not form under the same reaction conditions on changing the volume of OAm. Compared with the typical synthesis of the Pt3.5Pb NNWs, when the OAm was decreased to 100 μL or 0 μL, it led to the formation of irregular PtPb nanoparticles, which could be attributed to the lack of OAm to induce the oriented attachment and stabilize the nanoparticles. When increasing the OAm to 500 μL or 1 mL, the nanocrystals closed together but not connected to each other, this is due to the excessive OAm wrapping the nanoparticles and blocking the attachment process. Therefore, an appropriate amount of OAm is crucial to the formation of the NNWs. Meanwhile, the precursor and reaction temperature also affect the morphology of the PtPb nanocrystals. The replacement of Pb(acac)2 with lead acetate in the synthesis resulted in the form of crosslinked PtPb networks with an irregular size (Fig. S5a–d†). And the nanocrystals conglomerated and did not form homogeneous nanowires in the absence of Pb(acac)2 (Fig. S5e and f†). With the fall and rise of the reaction temperature, the obtained products (Fig. S6†) were monodisperse nanoparticles and nanoparticle clusters, respectively. This is because the low temperature is not enough to drive the oriented attachment and high temperature is too fast for the process. Thus the typical synthetic strategy is under the optimized conditions.
To evaluate the performance of the PtxPb NNWs, the electrocatalytic properties toward the MOR were investigated. Cyclic voltammetry (CV) (Fig. S8†) was used to evaluate the electrochemically active surface areas (ECSAs) of the Pt4Pb NNWs, Pt3.5Pb NNWs, Pt3Pb NNWs, commercial PtRu/C catalysts and Pt/C catalysts. The CV curves of these catalysts were recorded in N2-saturated 0.5 M H2SO4 solution at a sweep rate of 50 mV s−1. The ECSAs were calculated to be 45.1 m2 g−1 for Pt4Pb NNWs, 42.2 m2 g−1 for Pt3.5Pb NNWs, 38.6 m2 g−1 for Pt3Pb NNWs, 50.6 m2 g−1 for PtRu/C catalysts and 54.8 m2 g−1 for Pt/C catalysts. Fig. 4a shows the CV of MOR catalyzed by these catalysts, which were measured in 0.5 M aqueous H2SO4 solution with 1 M CH3OH at a sweep rate of 50 mV s−1. The methanol oxidation current density on the PtxPb catalysts is much higher than that on the Pt/C catalysts, and the PtxPb catalysts exhibit much higher If (If is the forward current density) to Ib (Ib is the backward current density) ratio. The If/Ib ratios of the Pt4Pb NNWs, Pt3.5Pb NNWs and Pt3Pb NNWs were 1.09, 1.18 and 1.14, respectively, compared to 1.08 of the Pt/C catalysts. It implies that the methanol molecules could be more effectively oxidized on the PtxPb NNW catalysts during the forward potential scan as well as generating relatively less poisoning species and higher tolerance toward CO poisoning compared to the Pt/C catalysts.9,18 Moreover, significant negative shifts of the onset potential of the MOR can be observed on the PtxPb NNWs (Fig. 4b), which indicate that the electrooxidation will be inclined to start at a lower potential on the Pt3.5Pb NNWs. As shown in Fig. 4c, the MOR current densities were normalized with the loading amount of Pt and the ECSA to compare the mass and specific activities. The Pt3.5Pb NNWs exhibited a mass activity of 1.18 A mgPt−1 at 0.65 V, which was 1.55, 1.14, 2.21 and 4.10 times greater than that of the Pt4Pb NNWs (0.74 A mgPt−1), Pt3Pb NNWs (1.01 A mgPt−1), the PtRu/C catalysts (0.46 A mgPt−1) and the Pt/C catalysts (0.29 A mgPt−1), respectively. Meanwhile, the Pt3.5Pb NNWs exhibit a specific activity of 2.78 mA cm−2, which is 5.24 times higher than that of the Pt/C catalysts (0.53 mA cm−2). In order to further understand the high electrocatalytic activity of the PtxPb nanocrystals, we must analyze the elementary reactions of methanol molecules oxidized at the anode. Bagotzky et al. described the methanol oxidation processes in acidic media as the stepwise H subtraction of methanol (CH3OH) as shown in eqn (1) and (2).35
| | | CH3OH + H2O → CO2 + 6H+ + 6e− | (1) |
| | 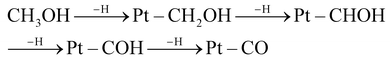 | (2) |
 |
| | Fig. 4 Electrocatalytic properties of the Pt4Pb NNWs, Pt3.5Pb NNWs, Pt3Pb NNWs, PtRu/C catalysts and Pt/C catalysts. (a) CV, (b) onset potential–current curves and (c) histogram of mass and specific activities of different catalysts for MOR in 0.5 M H2SO4 + 1 M CH3OH solution. (d) CV of the Pt3.5Pb NNWs, PtRu/C catalysts and Pt/C catalysts before and after accelerated durability test (ADT). (e) Potential–time curves measured by chronopotentiometry at a constant current of 1 mA cm−2 in 0.5 M H2SO4 + 1 M CH3OH solution. (f) Current–time curves recorded at 0.6 V, the inset is the magnified image marked by the square. | |
Although Pt is considered as the best catalyst for breaking C–H and O–H bonds in methanol molecules, the strong adsorption and high oxidation potential of CO on Pt atoms result in the large occupation of the active sites. The pathways of COads oxidation on the Pt catalyst and Pt–Pb catalyst are described by eqn (3) and (4) and eqn (5) and (6), respectively.35,36
| | | Pt + H2O → Pt–OHads + H+ + e− | (3) |
| | | Pt–COads + Pt–OHads → CO2 + 2Pt + H+ + e− | (4) |
| | | Pb + H2O → Pb–OHads + H+ + e− | (5) |
| | | Pt–COads + Pb–OHads → CO2 + Pt + Pb + H+ + e− | (6) |
The Pt (or Pb) atoms provide adsorbed hydroxyl groups (OHads), which served as the oxidant to oxidize the COads at a lower potential. However, the adsorption of oxygen-like species on Pt (eqn (3)) will not occur to any appreciable extent below −0.7 V, while it could occur on Pb at a lower potential (eqn (5)).37 Furthermore, the COads oxidation to CO2 must occur on the M–OHads catalytic surface, while the Pt–Pb bimetallic alloy structure can effectively provide OHads to adjacent Pt–COads. Therefore, the PtxPb nanocrystals showed a high electrocatalytic activity for MOR in acidic media. By analyzing the CV of MOR catalyzed by these catalysts, the forward current density rapidly increased with the potential rising from 0.4 V to 0.7 V, this might be attributed to the direct methanol molecules oxidation process. Then the current density tobogganed after the peak potential, this is because of the strong electro-adsorption of COads on Pt atoms that led to the great reduction of the MOR efficiency. When the potential exceeded 0.9 V, the catalysts reactivated due to the COads oxidation at high potential. In the backward scan process, the low current density continued to 0.5 V, and then gradually increased. This might be due to the readsorption of the poisoning intermediates from 0.9 V to 0.5 V, and desorption from the catalyst surface under low potential, thus the Pt catalyst became reactivated for MOR.
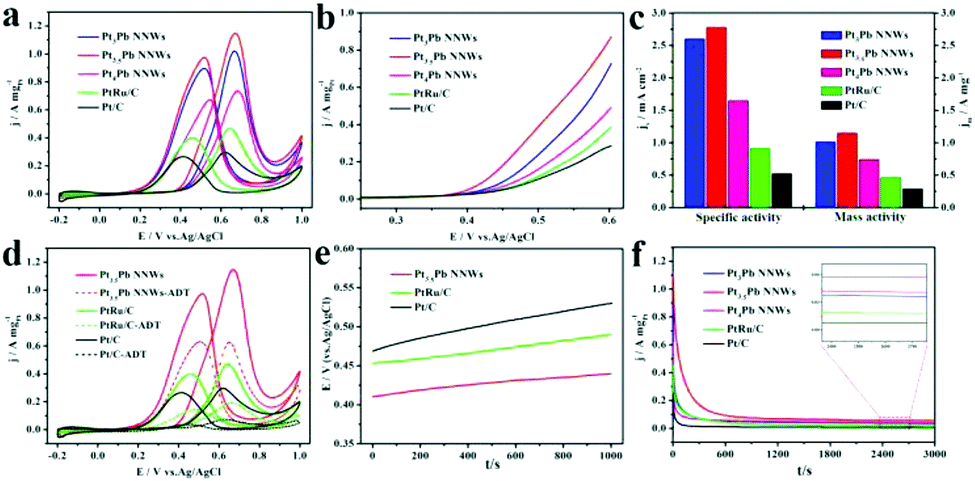
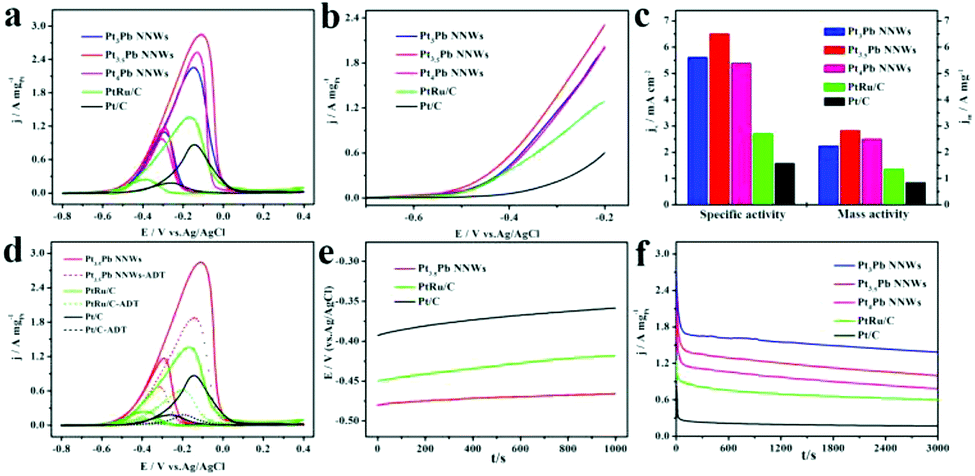
Similarly, we also investigated their electrocatalytic performance towards methanol oxidation under alkaline conditions. The MOR was carried out in an aqueous solution containing 0.5 M KOH and 1 M CH3OH at a sweep rate of 50 mV s−1. As shown in Fig. 6a–c, all of the catalysts had a higher If/Ib ratio and current density compared to themselves in acidic media. And the Pt3.5Pb NNWs exhibited the lowest onset potential and highest mass activity (2.84 A mgPt−1), which was 1.14, 1.26, 2.10 and 3.45 times higher than that of the Pt4Pb NNWs (2.25 A mgPt−1), Pt3Pb NNWs (2.50 A mgPt−1), PtRu/C catalysts (1.35 A mgPt−1) and Pt/C catalysts (0.83 A mgPt−1), respectively. Meanwhile, the Pt3.5Pb NNWs showed a high specific activity of 6.51 mA cm−2, which was 4.12 times higher than that of the Pt/C catalysts (1.58 mA cm−2). The higher reaction kinetics and current density of the MOR in alkaline media could be ascribed to the different mechanistic pathways of the methanol molecules oxidation as follows (eqn (7)–(12)).38
| | | CH3OH + 6OH− → CO2 + 5H2O + 6e− | (7) |
| | | Pb + OH− → Pb–OHads + e− | (8) |
| | | 2Pt + CH3OH → Pt–H + Pt–(CH3O)ads | (9) |
| | | Pt–(CH3O)ads + xPb–OHads → Pt–(CH3−xO)ads–Pbx + xH2O | (10) |
| | | Pt–COads–Pb + Pb–OHads → Pt–(COOH)ads + 2Pb | (11) |
| | | Pt–(COOH)ads + Pb–OHads → CO2 + H2O + Pt + Pb | (12) |
In alkaline media, abundant hydroxyl ions will accelerate the H subtraction process of methanol (eqn (8)–(12)),38 thus the MOR has lower overpotential and onset potential. Meanwhile, the different pathways of MOR lead to the complete Pt–(CH3O)ads oxidation process under the forward scan and avoid producing much COads absorbed on the catalyst surface. Therefore, the catalysts possessed a higher current density and catalytic activity of the MOR in alkaline media. Fig. 5, S10† and Fig. 7 show the CV plots of MOR on these catalysts at different scan rates under acidic and alkaline conditions, respectively, ranging from 10 mV s−1 to 100 mV s−1. With an increase of the scan rate, the peak current potential shifted positively, and the current density also increased correspondingly. A linear relationship was found between the square root of the scan rate (v1/2) and the forward peak current density (jm). The results indicate that the MOR on these catalysts follows a diffusion-controlled process. The higher slope values of the PtxPb NNWs relative to that of the Pt/C catalysts suggest the improved electrooxidation kinetics of the PtxPb NNWs.39
 |
| | Fig. 5 CV of MOR on the Pt3.5Pb NNWs at different scan rates under acidic conditions (a) and the corresponding plot of forward peak current (jm) versus the square root of the scan rate (v1/2) (b). CV of MOR on the Pt/C catalyst modified electrodes at different scan rates (c) and the corresponding plot of jmversus the v1/2 (d). | |
 |
| | Fig. 6 Electrocatalytic properties of the Pt4Pb NNWs, Pt3.5Pb NNWs, Pt3Pb NNWs, PtRu/C catalysts and Pt/C catalysts under alkaline conditions. (a) CV, (b) onset potential–current curves and (c) histogram of mass and specific activities of different catalysts for MOR in 0.5 M KOH + 1 M CH3OH solution. (d) CV of the Pt3Pb NNWs, PtRu/C catalysts and Pt/C catalysts before and after the accelerated durability test (ADT). (e) Potential–time curves measured by chronopotentiometry at a constant current of 1 mA cm−2 in 0.5 M KOH + 1 M CH3OH solution. (f) Current–time curves recorded at −0.1 V. | |
 |
| | Fig. 7 CV of MOR on the Pt3.5Pb NNWs at different scan rates under alkaline conditions (a) and the corresponding plot of jmversus the v1/2 (b). CV of MOR on the Pt/C catalyst modified electrodes at different scan rates (c) and the corresponding plot of jmversus the v1/2 (d). | |
To further evaluate the durability of different catalysts, we investigated the long-term stability of the catalysts. The decrease of the ECSA and current density were evaluated by potential cycling for 600 cycles at room temperature. The variation of ECSA was carefully calculated as shown in Fig. S7–9,† in comparison to the PtRu/C catalysts and Pt/C catalysts, the PtxPb NNWs showed much higher stability. After 600 cycles, the ECSA of the Pt4Pb NNWs, Pt3.5Pb NNWs and Pt3Pb NNWs still remained 92.2%, 93.9% and 90.1%, respectively, while the Pt/C catalysts and PtRu/C catalysts just remained 72.1% and 59.5% of the ECSA, respectively. After 600 sweeping cycles in 0.5 M H2SO4 + 1 M CH3OH (Fig. 4d and S11a and b†) and 0.5 M KOH + 1 M CH3OH (Fig. 6d and S11c and d†), the Pt3.5Pb NNWs show 54.8% and 66.4% of the initial catalytic activity, much better than those of the PtRu/C catalysts (41.5% and 44.1%) and the Pt/C catalysts (24.3% and 21.5%), respectively. Fig. 4e and 6e show the potential–time plots measured by chronopotentiometry at a constant current of 1 mA cm−2 in acidic and alkaline media, respectively. The onset potentials on Pt3.5Pb NNWs are more negative than that of the Pt/C catalysts, which indicates that the Pt3.5Pb NNWs have higher catalytic activity relative to Pt/C catalysts. The potential change due to surface poisoning and variation of morphology in 1000 s are 29 mV, 38 mV and 61 mV for the Pt3.5Pb NNWs, PtRu/C catalysts and Pt/C catalysts in acidic media, 15 mV, 27 mV and 33 mV for the Pt3.5Pb NNWs, PtRu/C catalysts and Pt/C catalysts in alkaline media, respectively. Obviously, the Pt3.5Pb NNWs exhibit lower voltage degradation rates than PtRu/C catalysts and Pt/C catalysts, indicating that the Pt3.5Pb NNWs are more durable than PtRu/C catalysts and Pt/C catalysts.13Fig. 4f and 6f show the current–time curves recorded in acidic and alkaline media, respectively. Over the entire 3000 s test, the currents remaining on the Pt3.5Pb NNWs were still higher than that of the PtRu/C catalysts and Pt/C catalysts, which further confirmed the high durability of the Pt3.5Pb NNWs. The morphological changes of the catalysts after the durability tests were characterized by TEM. There was almost no change of the nerve-like structure in the Pt3.5Pb NNWs (Fig. S12†), while obvious aggregation occurred at the Pt/C catalysts after the durability test (Fig. S13†). Uniform dispersed Pt nanoparticles disappeared from the supported carbon and aggregated into large irregular nanoparticles. Fig. S14† shows the TEM images of the PtRu/C catalysts. Overall, the electrocatalytic activity and stability of the Pt3.5Pb NNWs toward the MOR both under acidic and alkaline conditions had been greatly promoted compared to the Pt/C catalysts and various reported electrocatalysts (Table S1†). The enhanced electrocatalytic activity and stability of the Pt3.5Pb NNWs can be attributed to the following aspects: (i) the Pt–Pb bimetallic composition and the alloyed nanostructure resulted in that the Pt atoms on the surface of the catalysts will be stable and hard to dissolve. Moreover, the electronic structure of Pt was changed through charge transfer from Pb to Pt, due to the higher electronegativity of Pt. And the adsorption energy (Eads) for the adsorbed PtxPb–CO was lower than the corresponding Pt–CO under the same conditions. Thus, the catalysts can effectively avoid COads-poisoning at the same time improving the catalytic activity.23,34 (ii) The nerve-like structure consists of ultrathin wavy nanowires and branches could serve as an integrated electrocatalytic reaction region, which would be quite effective for the electron transfer between each active site. Furthermore, the unique structure makes it have a high contact area with the electrode surface and difficult to aggregate, because it has both the properties of one-dimensional and two-dimensional nanomaterials. (iii) The formation mechanism of the PtxPb NNWs led to plentiful distinct defects on the surface of the catalysts, such as atomic steps and crystal face defects, which could act as highly catalytic sites and may serve as particular COads rapid oxidation sites. Because the PtxPb NNWs come from the collisions and oriented attachments between the nanoparticles at high temperatures, the nanocrystals went through the Ostwald ripening and aggregation processes, and finally reached a stable structure. In general, the unique nanostructure and the Pt–Pb bimetallic synergistic effect led to the electrocatalytic activity and stability of the Pt3.5Pb NNWs toward the MOR, greatly promoted both under acidic and alkaline conditions.
Conclusions
In summary, we demonstrate a one-step synthetic strategy for ultrathin PtxPb nerve-like nanowires and they served as robust catalysts for greatly enhancing methanol electrooxidation both under acidic and alkaline conditions. In acidic media, the mass and specific activities of the Pt3.5Pb NNWs reach 1.18 A mgPt−1 and 2.78 mA cm−2, which are 4.10 and 5.24 times higher than Pt/C catalysts, respectively. And in alkaline media, the mass and specific activities of the Pt3.5Pb NNWs are 2.84 A mgPt−1 and 6.51 mA cm−2, respectively, 3.45 and 4.12 times higher than Pt/C catalysts. The Pt–Pb alloyed composition and the nerve-like structure endowed the catalysts with high CO-poisoning tolerance, superior electrocatalytic activity and stability toward the MOR. And the demonstrated synthetic strategy of the ultrathin PtxPb nerve-like nanowires will exploit more inspiration and strategies for the rational design of novel metal nanocrystals for various electrocatalytic applications.
Acknowledgements
This work was supported by the National Natural Science Foundation of China (no. 21375123 and 21675151) and the Ministry of Science and Technology of China (no. 2013YQ170585 and 2016YFA0203201).
Notes and references
- J. Wu and H. Yang, Acc. Chem. Res., 2013, 46, 1848–1857 CrossRef CAS PubMed.
- N. Jung, D. Y. Chung, J. Ryu, S. J. Yoo and Y.-E. Sung, Nano Today, 2014, 9, 433–456 CrossRef CAS.
- H. L. Liu, F. Nosheen and X. Wang, Chem. Soc. Rev., 2015, 44, 3056–3078 RSC.
- J. Lai, W. Niu, R. Luque and G. Xu, Nano Today, 2015, 10, 240–267 CrossRef CAS.
- G. A. Olah, Angew. Chem., Int. Ed., 2013, 52, 104–107 CrossRef CAS PubMed.
- B. C. Steele and A. Heinzel, Nature, 2001, 414, 345–352 CrossRef CAS PubMed.
- H. A. Gasteiger and N. M. Markovic, Science, 2009, 324, 48–49 CrossRef CAS PubMed.
- F. A. de Bruijn, V. A. T. Dam and G. J. M. Janssen, Fuel Cells, 2008, 8, 3–22 CrossRef CAS.
- Y. X. Chen, A. Miki, S. Ye, H. Sakai and M. Osawa, J. Am. Chem. Soc., 2003, 125, 3680–3681 CrossRef CAS PubMed.
- B. Lim, M. Jiang, P. H. Camargo, E. C. Cho, J. Tao, X. Lu, Y. Zhu and Y. Xia, Science, 2009, 324, 1302–1305 CrossRef CAS PubMed.
- C. Chen, Y. Kang, Z. Huo, Z. Zhu, W. Huang, H. L. Xin, J. D. Snyder, D. Li, J. A. Herron, M. Mavrikakis, M. Chi, K. L. More, Y. Li, N. M. Markovic, G. A. Somorjai, P. Yang and V. R. Stamenkovic, Science, 2014, 343, 1339–1343 CrossRef CAS PubMed.
- S. Guo, S. Zhang, D. Su and S. Sun, J. Am. Chem. Soc., 2013, 135, 13879–13884 CrossRef CAS PubMed.
- Z. Cui, H. Chen, M. Zhao, D. Marshall, Y. Yu, H. Abruna and F. J. DiSalvo, J. Am. Chem. Soc., 2014, 136, 10206–10209 CrossRef CAS PubMed.
- J. Zheng, D. A. Cullen, R. V. Forest, J. A. Wittkopf, Z. Zhuang, W. Sheng, J. G. Chen and Y. Yan, ACS Catal., 2015, 5, 1468–1474 CrossRef CAS.
- Q. Chen, Y. Yang, Z. Cao, Q. Kuang, G. Du, Y. Jiang, Z. Xie and L. Zheng, Angew. Chem., Int. Ed., 2016, 128, 9167–9171 CrossRef.
- N. V. Long, C. M. Thi, Y. Yong, M. Nogami and M. Ohtaki, J. Nanosci. Nanotechnol., 2013, 13, 4799–4824 CrossRef CAS PubMed.
- W. T. Cahyanto, W. Widanarto, M. Effendi, M. R. Hamdi and H. Kasai, AIP Conf. Proc., 2016, 1712, 050023 CrossRef.
- T. Yajima, H. Uchida and M. Watanabe, J. Phys. Chem. B, 2004, 108, 2654–2659 CrossRef CAS.
- F. Taufany, C. J. Pan, F. J. Lai, H. L. Chou, L. S. Sarma, J. Rick, J. M. Lin, J. F. Lee, M. T. Tang and B. J. Hwang, Chem. – Eur. J., 2013, 19, 905–915 CrossRef CAS PubMed.
- D.-J. Chen and Y. J. Tong, Angew. Chem., Int. Ed., 2015, 127, 9526–9530 CrossRef.
- C. Zhu, Q. Shi, S. Fu, J. Song, H. Xia, D. Du and Y. Lin, Adv. Mater., 2016, 28, 8779–8783 CrossRef CAS PubMed.
- X. Yu and P. G. Pickup, Electrochim. Acta, 2010, 55, 7354–7361 CrossRef CAS.
- Q. Jiang, L. Jiang, J. Qi, S. Wang and G. Sun, Electrochim. Acta, 2011, 56, 6431–6440 CrossRef CAS.
- S. Maksimuk, S. Yang, Z. Peng and H. Yang, J. Am. Chem. Soc., 2007, 129, 8684–8685 CrossRef CAS PubMed.
- Y. Kang, L. Qi, M. Li, R. E. Diaz, D. Su, R. R. Adzic, E. Stach, J. Li and C. B. Murray, ACS Nano, 2012, 6, 2818–2825 CrossRef CAS PubMed.
- K. Jiang, L. Bu, P. Wang, S. Guo and X. Huang, ACS Appl. Mater. Interfaces, 2015, 7, 15061–15067 Search PubMed.
- N. Zhang, S. Guo, X. Zhu, J. Guo and X. Huang, Chem. Mater., 2016, 28, 4447–4452 CrossRef CAS.
- W. Hong, C. Shang, J. Wang and E. Wang, Energy Environ. Sci., 2015, 8, 2910–2915 Search PubMed.
- Y. Feng, L. Bu, S. Guo, J. Guo and X. Huang, Small, 2016, 12, 4464–4470 CrossRef CAS PubMed.
- X. Teng, W.-Q. Han, W. Ku and M. Hücker, Angew. Chem., Int. Ed., 2008, 120, 2085–2088 CrossRef.
- X. Yu, D. Wang, Q. Peng and Y. Li, Chem. – Eur. J., 2013, 19, 233–239 CrossRef CAS PubMed.
- V. Mazumder and S. Sun, J. Am. Chem. Soc., 2009, 131, 4588–4589 CrossRef CAS PubMed.
- N. Tian, Z. Y. Zhou, S. G. Sun, Y. Ding and Z. L. Wang, Science, 2007, 316, 732–735 CrossRef CAS PubMed.
- A. Ferre-Vilaplana, J. V. c. Perales-Rondón, J. M. Feliu and E. Herrero, ACS Catal., 2015, 5, 645–654 CrossRef CAS.
- V. S. Bagotzky, Y. B. Vassiliev and O. A. Khazova, J. Electroanal. Chem., 1977, 81, 229–238 CrossRef.
- C. Lamy, A. Lima, V. LeRhun, F. Delime, C. Coutanceau and J.-M. Léger, J. Power Sources, 2002, 105, 283–296 CrossRef CAS.
- E. Casado-Rivera, D. J. Volpe, L. Alden, C. Lind, C. Downie, T. Vazquez-Alvarez, A. C. Angelo, F. J. DiSalvo and H. D. Abruna, J. Am. Chem. Soc., 2004, 126, 4043–4049 CrossRef CAS PubMed.
- R. Manoharan and J. Prabhuram, J. Power Sources, 2001, 96, 220–225 CrossRef CAS.
- Y. Wang, B. Wu, Y. Gao, Y. Tang, T. Lu, W. Xing and C. Liu, J. Power Sources, 2009, 192, 372–375 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Fig. S1–S14. See DOI: 10.1039/c6nr07036a |
|
| This journal is © The Royal Society of Chemistry 2017 |
Click here to see how this site uses Cookies. View our privacy policy here.