
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Next-generation CO2 electroreduction: the role of atomically precise nanoclusters and emerging catalytic strategies
Mandira
Ghosh
,
Rupa
Sarma
,
Maho
Kamiyama
,
Tokuhisa
Kawawaki
 ,
Sourav
Biswas
* and
Yuichi
Negishi
*
,
Sourav
Biswas
* and
Yuichi
Negishi
*
Institute of Multidisciplinary Research for Advanced Materials, Tohoku University, Katahira 2-1-1, Aoba-ku, Sendai 980-8577, Japan. E-mail: Sourav.biswas210@gmail.com; yuichi.negishi.a8@tohoku.ac.jp
First published on 17th April 2025
Abstract
The electrochemical reduction of carbon dioxide (CO2) is a promising approach to simultaneously mitigate greenhouse gas emissions and produce value-added carbon-based fuels and chemicals. However, sluggish reaction kinetics necessitate the development of efficient catalysts to enhance activity and selectivity. Metal nanoclusters (NCs) have emerged as highly promising candidates due to their atomically precise structures, unique electronic properties, and tunable catalytic behaviors. Despite continuous advancements in the synthesis and application of metal NCs, several challenges remain that hinder their practical deployment. Stability, scalability, and the fine control of selectivity remain critical concerns. This review systematically explores the stepwise evolution of metal NCs as catalysts for CO2 electroreduction, highlighting their key advantages while also identifying the fundamental limitations that need to be addressed. To overcome such challenges a key development is observed that leads to shift from noble-metal NCs to first-row transition-metal-based NCs, which has expanded catalytic reactivity and product selectivity. Additionally, the role of alloying in enhancing catalytic performance through synergistic interactions and electronic modifications is discussed. So, this review provides a comprehensive analysis of recent progress, with a focus on emerging NC-based electrocatalysts, and outlines future directions to address existing challenges for sustainable CO2 conversion.
 Mandira Ghosh | Mandira Ghosh is a PhD student in in Prof. Negishi's group at Tohoku University, Japan. She received her Master's in Chemistry from National Institute of Technology Durgapur (2022). Afterward she joined at Vellore Institute of Technology, Chennai (2023) as Junior Research Fellow. Her current research focuses on the synthesis of different Cu nanocluster for the electrocatalytic CO2 reduction reaction. |
 Rupa Sarma | Rupa Sarma is a PhD researcher in Prof. Negishi's group at Tohoku University, Japan. She obtained her Master's degree in Chemistry from National Institute of Technology Durgapur, India (2022). Her research focuses on synthesizing Cu nanoclusters, analysing their crystal structures, and exploring catalytic applications. |
 Maho Kamiyama | Maho Kamiyama is a masters course student at Tohoku University, Japan. She focuses her research on synthesizing diverse nanoclusters and investigating their electrocatalytic activities in Prof. Negishi's group. |
 Tokuhisa Kawawaki | Tokuhisa Kawawaki is an Associate Professor in Institute of Multidisciplinary Research for Advanced Materials (IMRAM), Tohoku University, Japan. Previously he served as junior Associate Professor at Tokyo University of Science, Japan. He received his PhD degree in applied chemistry from the University of Tokyo and worked as a Japan Society for the Promotion of Science (JSPS) postdoctoral fellow at the University of Melbourne and a JSPS super postdoctoral fellow at Kyoto University. His current research topics include synthesizing ligand-protected metal nanoparticles and nanoclusters, and their application in photoelectrochemistry as well as photocatalysis. |
 Sourav Biswas | Sourav Biswas is an Assistant Professor at IMRAM, Tohoku University, Japan. Before joining here, he served as an Assistant Professor and postdoctoral fellow at Tokyo University of Science, Japan. He obtained his PhD in Chemistry from the National Institute of Technology Durgapur, India and also worked as national post-doctoral fellow at Indian Institute of Science Education and research Thiruvananthapuram, India. His current research interests focus on the synthesis and investigation of potential applications of innovative nanoclusters. |
 Yuichi Negishi | Yuichi Negishi is a Professor at IMRAM, Tohoku University, Japan. He received his PhD degree in Chemistry from Keio University. Prior to joining Tohoku University in 2024, he was employed as a Professor at Tokyo University of Science since 2008 and before that worked as an Assistant Professor at Keio University and the Institute for Molecular Science. His research interests include the structural and functional exploration of atomically precise metal NCs, metal NC-assembled materials, and covalent organic frameworks. We are honoured to contribute to the celebration of the 10th anniversary of Nanoscale Horizons. Our association with this prestigious journal began in 2021, followed by another publication in 2023—both of which were recognized among the most popular articles of their respective years. We are deeply grateful for the continued opportunity to collaborate with such a prestigious platform. As a gesture of our appreciation and support, we are pleased to contribute once again with this article, in which we explore the potential and future directions of atomically precise metal nanoclusters for electrocatalytic carbon dioxide reduction. |
1. Introduction
One of the most pressing challenges of environmental degradation today is the global climate crisis, exacerbated by the accelerating depletion of fossil fuel resources. Among the various greenhouse gases contributing to global warming, carbon dioxide (CO2) is the most significant, with its atmospheric concentration reaching alarming levels.1–3 The rapid industrial expansion over recent decades, coupled with the continuous combustion of fossil fuels, has been a primary driver of escalating CO2 emissions.4 According to projections from the U.S. energy information administration, global CO2 emissions are expected to rise by approximately 34% by 2050 compared to 2022 levels, posing a severe threat to climate stability.5 In response to this crisis, the 29th United Nations climate change conference (COP29), held in Baku, Azerbaijan, underscored the necessity of financial support for the implementation of the Paris Agreement's long-term climate strategy.6 A central goal of this framework is to achieve carbon neutrality by 2050, necessitating a drastic reduction in net CO2 emissions. Meanwhile, the global energy sector is undergoing a transformative shift towards sustainable alternatives, including solar, wind, and hydropower, to reduce dependence on finite fossil fuel reserves and address the soaring global energy demand.7,8 However, the large-scale deployment of these renewable energy sources presents several critical challenges, such as energy intermittency, storage limitations, and geographical constraints, which hinder their ability to serve as fully reliable and independent energy solutions.9,10 As a result, achieving a sustainable energy transition requires the development of diverse alternative energy sources that can overcome existing limitations, ensuring a reliable and efficient energy supply.In the recent time, capture and conversion of CO2 into useful fuels have emerged as a game-changer for mitigating both the climate change as well as securing sustainability of energy.11–14 Among various methodologies, electrochemical CO2 reduction (ECO2R) has garnered significant attention due to its high conversion efficiency, operation under ambient conditions, and ability to generate a wide range of carbon-based fuels such as carbon monoxide (CO), formic acid (HCOOH), methanol (CH3OH), methane (CH4), ethylene (C2H4), acetaldehyde (CH3CHO), and ethanol (C2H5OH).15–19 However, the CO2 molecule exhibits high thermodynamic and kinetic stability, making its activation and subsequent reduction inherently difficult, which in turn results in sluggish reaction kinetics in ECO2R.20–22 This limitation significantly broadens the scope of catalyst development, as the efficiency and applicability of ECO2R are largely dictated by the catalytic material.23–25 The overall process is governed by the catalyst's ability to regulate reaction selectivity, activity, and stability, making catalyst design a key factor in optimizing the reaction pathway and improving product yield. Industries primarily seek high-energy-value products, such as hydrocarbons, which require multiple proton-coupled electron transfer steps, all orchestrated by the catalyst. Consequently, the design of an ideal electrocatalyst is paramount for unraveling the mechanistic pathway and enhancing product efficiency. The activation of CO2 is a crucial initial step in the reduction process, and interestingly, CO2 can adsorb onto the catalyst surface either in a linear or bent configuration. In the case of linear adsorption, the inherent stability of CO2 necessitates a significantly high negative potential to induce the formation of the *CO2˙− intermediate via single-electron capture (Scheme 1). In the subsequent step CO2 can bind to the catalyst surface either through its two oxygen atoms or via the central carbon atom, leading to the formation of distinct intermediates that ultimately dictate the final product selectivity. The HCOO* intermediate primarily form through proton-coupled electron transfer process when the O atom of *CO2˙− is weakly bind with catalyst surface, favoring the formation of HCOOH as the final product. Conversely, if the bond between the carbon of *CO2˙− and the catalyst surface is stronger, the *COOH intermediate is more likely to form.26 This intermediate further undergoes proton and electron coupling to generate *CO, a pivotal intermediate in the reaction pathway that influences the formation of both C1 and C2 products. *CO can either desorb from the catalyst surface as CO or undergo further protonation and electron transfer to form C1 products such as CH4 and CH3OH via *CHO and *COH intermediates. The formation of C2 products follows more complex mechanisms due to the distinct energy barriers associated with different intermediates. Significant C2 intermediates, including *COCO, *COCHO, and *COCOH, arise from the dimerization of *CO or its interaction with *CHO and *COH. Notably, *COCO is formed through the coupling of two *CO units, representing an additional rate-determining step in the C2 product pathway.27 Furthermore, *COCHO and *COCOH can emerge either from the protonation of *COCO or through direct reactions between *CO and *CHO or *COH. In recent years, researchers have made considerable progress in developing electrocatalysts with moderate selectivity for desired products in ECO2R, leveraging different intermediate pathways to enhance efficiency and control over product distribution. For instance, Wang et al. reported a CuBr nanoparticle (NP) with in situ restructuring ability to form Br doped Cu NP during the ECO2R process which shows faradaic efficiency (FE) of 91.6% for HCOOH formation at a current density 15.1 mA Cm−2 under the potential −0.94 V vs. reversible hydrogen electrode (RHE).28 Jiwanti et al. reported a SnO–SnO2/Ti3C2Tx NP for the ECO2R resulting HCOOH of 21.32 ppm with a FE of 94.06% using a boron doped diamond electrode.29
 | ||
| Scheme 1 Reaction mechanism of ECO2R with different products. | ||
Transition metal-based catalysts, particularly in the form of NPs, have garnered significant interest for ECO2R due to their inherent stability and high catalytic efficiency.30–33 These catalysts are crucial in enabling key reaction steps by offering active sites that enhance CO2 activation and drive its conversion into value-added products. However, despite their promising catalytic properties, several intrinsic challenges limit their overall performance and reproducibility. One of the primary concerns is the inherent heterogeneity in NP size distribution and morphology, which leads to significant variations in catalytic activity and product selectivity.33 Additionally, the presence of multiple types of active sites within these nanomaterials can introduce complex reaction pathways, often resulting in inconsistent product yields and reduced selectivity for desired products. To overcome these challenges and improve the reliability of nanomaterials for CO2 reduction, the development of catalysts with uniform particle size and well-defined active sites is crucial. In this context, atomically precise metal nanoclusters (NCs) have recently emerged as a highly promising alternative, offering precise control over size, morphology, and surface composition at the atomic level.34,35 Unlike conventional NPs, NCs consist of a discrete number of metal atoms, often with well-defined ligand environments, leading to unique electronic structures and distinct catalytic properties. This atomic-level precision enables a more systematic investigation of structure–activity relationships, allowing for the rational design of catalysts with enhanced selectivity, stability, and efficiency for ECO2R. In recent years, the ECO2R has seen remarkable progress in catalyst design, particularly with atomically precise structural architectures. Numerous studies have been published on this topic, and ongoing research continues to expand our understanding of the fundamental mechanisms and optimization strategies for catalytic performance.35–38 Although a few review articles summarize the current advancements, they often do not comprehensively cover the most recent breakthroughs, leaving gaps in consolidating the latest developments.35,39–44 Despite significant progress, a systematic and well-defined approach to identifying the optimal catalyst design—one that ensures both high selectivity and efficiency in product formation—remains lacking. Given the complexity of ECO2R, narrowing down the methodologies to determine the most effective strategies is crucial for advancing the field. Therefore, our approach focuses on addressing this gap by critically analyzing existing methodologies and establishing a well-organized framework for designing highly selective and efficient catalysts. Specifically, we aim to leverage atomically precise NCs as model systems to elucidate structure–property relationships and uncover favourable pathways for catalyst development in ECO2R.
2. Nanoclusters and their effects
Metal NCs consist of a few to a few hundred metal atoms held together predominantly by metallophilic interactions within a sub-3 nm length scale.45–47 Due to their ultra-small dimensions, quantum confinement effects play a crucial role in defining their electronic structures, setting them apart from their bulk and NP counterparts. Unlike metal NPs, where electronic bands are continuous, NCs exhibit discrete, quantized energy levels, which impart them with unique molecular-like properties.48–50 In addition, metal NCs exhibits strong structure–property correlation, wherein precise tuning of their electronic charge states necessitates controlled modifications in their structural framework. This fine modulation is predominantly governed by metal–ligand interactions, which dictate both the stability and reactivity of the NCs.51–58 Consequently, optimizing these interactions is critical for tailoring the physicochemical properties of NCs for specific applications.59–64The molecular-like nature of NCs, with their well-defined geometric and electronic structures, presents new opportunities for enhancing catalytic performance. Through electronic and geometric modifications, their catalytic activity can be systematically tuned to achieve superior efficiency. This advantage is particularly significant in electrocatalysis, where NCs provide a promising platform for designing next-generation catalysts with enhanced activity, selectivity, and stability. In the context of ECO2R, NCs are emerging as advanced catalysts due to their precisely defined active sites and tuneable charge states.34,35,65 Their structural specificity allows for in-depth mechanistic investigations, facilitating the identification of optimal active sites and reaction pathways. This mechanistic understanding is crucial for the rational design of highly efficient catalysts for CO2 conversion, thereby contributing to the advancement of sustainable energy solutions. As a result, metal NCs are at the forefront of contemporary electrocatalysis research, offering an exciting avenue for the development of innovative catalytic systems with superior performance for ECO2R and other important transformations.
2.1. Effect of noble metal NCs
The application of NCs in ECO2R initially focused on noble metal catalysts, specifically gold (Au) and silver (Ag) NCs, due to their superior stability compared to other transition metal NCs.34 Among these, Au NCs demonstrated remarkable catalytic performance in CO2 reduction. In a pioneering study conducted in 2012, Kauffman et al. investigated the catalytic efficiency of previously synthesized atomically precise [Au25(PET)18]− (PET: phenylethylthiolate) [Au25]− NCs (Fig. 1a) in ECO2R and compared their performance with Au NPs and bulk Au surfaces.66,67 Their findings revealed that [Au25]− NC (1.2 nm) exhibited an exceptionally high FECO, reaching ≈100% at an applied potential of −0.973 V vs. RHE. This efficiency was significantly enhanced compared to other Au-based catalysts: approximately 1.2 times higher than 2 nm Au NPs, 10 times greater than 5 nm Au NPs, and nearly 80 times superior to bulk Au (Fig. 1b). The remarkable catalytic performance of [Au25]− NC was attributed to its well-defined molecular structure, which facilitated binding interactions with CO2 molecules at the active catalytic site, where O atom of CO2 interacts with the three associated S atoms in the shell, partially driven by electrostatic attraction (inset Fig. 1b). These interactions facilitated selective activation of the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O bond and promoted the formation of surface-adsorbed hydrogen species, which are crucial intermediates in the CO2 reduction process. In contrast, such precise catalytic sites and interactions were either absent or significantly less effective in larger Au NPs and bulk Au surfaces, underscoring the advantages of atomically precise NCs in ECO2R. Although this study achieved the highest FECO using [Au25]− NCs, it also paved the way for further exploration of other NCs for ECO2R. This breakthrough also inspired researchers to investigate the influence of structural architecture, electronic charge, and ligand environment on the catalytic performance of NCs. Building on their initial work, the same research group conducted an in-depth study on the role of NC charge in dictating the mechanistic pathway of ECO2R.68 They discovered that the charge state of the NC significantly impacted catalytic reactivity by stabilizing the reactant or key reaction intermediates, ultimately influencing the final product yield. Their study concluded that anionic NCs exhibited superior catalytic performance in terms of FECO compared to neutral or cationic NCs (Fig. 1c). This enhancement was attributed to the negatively charged NCs, which facilitated CO2 adsorption and stabilized key intermediates, thereby improving overall reaction efficiency (Fig. 1d). This was determined through the reaction turnover frequency measured at −1 V vs. RHE in the presence of both acidic and alkaline electrolytes (Fig. 1e and f). Subsequent research efforts led to modifications in the electrochemical setup, transitioning from conventional H-type cells to gas diffusion electrode-based flow electrolyzers which change the mechanistic pathway of the reduction process and the stabilization of the key intermediates, however the final yield is CO.69
O bond and promoted the formation of surface-adsorbed hydrogen species, which are crucial intermediates in the CO2 reduction process. In contrast, such precise catalytic sites and interactions were either absent or significantly less effective in larger Au NPs and bulk Au surfaces, underscoring the advantages of atomically precise NCs in ECO2R. Although this study achieved the highest FECO using [Au25]− NCs, it also paved the way for further exploration of other NCs for ECO2R. This breakthrough also inspired researchers to investigate the influence of structural architecture, electronic charge, and ligand environment on the catalytic performance of NCs. Building on their initial work, the same research group conducted an in-depth study on the role of NC charge in dictating the mechanistic pathway of ECO2R.68 They discovered that the charge state of the NC significantly impacted catalytic reactivity by stabilizing the reactant or key reaction intermediates, ultimately influencing the final product yield. Their study concluded that anionic NCs exhibited superior catalytic performance in terms of FECO compared to neutral or cationic NCs (Fig. 1c). This enhancement was attributed to the negatively charged NCs, which facilitated CO2 adsorption and stabilized key intermediates, thereby improving overall reaction efficiency (Fig. 1d). This was determined through the reaction turnover frequency measured at −1 V vs. RHE in the presence of both acidic and alkaline electrolytes (Fig. 1e and f). Subsequent research efforts led to modifications in the electrochemical setup, transitioning from conventional H-type cells to gas diffusion electrode-based flow electrolyzers which change the mechanistic pathway of the reduction process and the stabilization of the key intermediates, however the final yield is CO.69
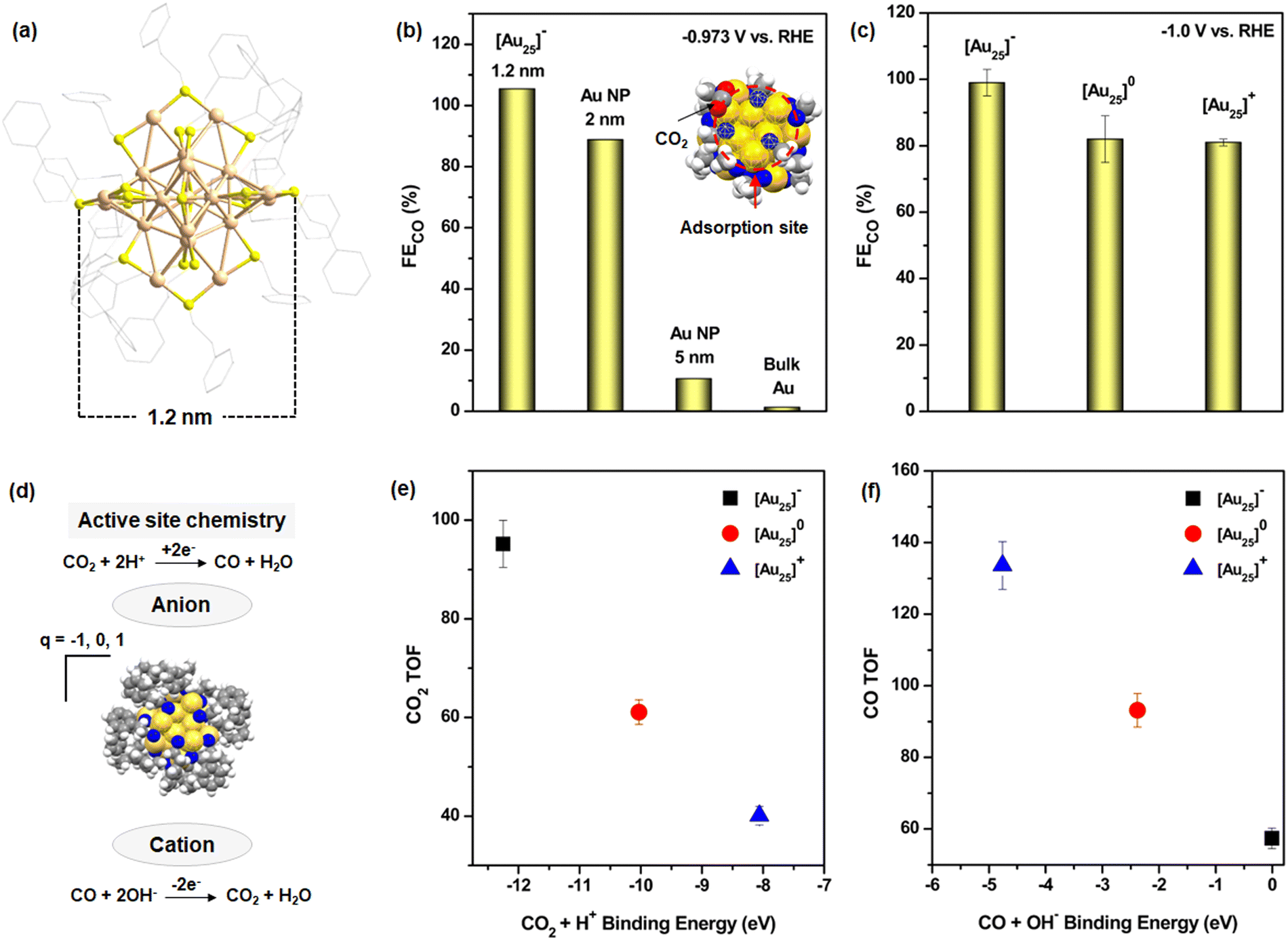 | ||
| Fig. 1 (a) Structural architecture of [Au25]− NCs. H atoms are omitted for the clarity. Color legend: Au, tan; S, yellow; and C, gray. (b) Comparision of FECO among of [Au25]− NC, Au NPs and bulk Au surface. Inset showing the active sites for CO2 adsorption in the theoretically model structure. Color legend: Au, gold; S, blue; C, gray; H, white; and O, red. (c) Comparison of FECO in [Au25] NCs depending on their charge. (d) Active site chemistry of [Au25]− NC depending on their change. (e) and (f) Binding energy vs. reaction TOF for (e) CO2 reduction at −1 V vs. RHE and (f) CO oxidation at +0.89 V vs. RHE. | ||
Further investigations delved into the influence of NC size and geometric structure while ensuring that the ligand environment remained consistent. A series of Au NCs from Au25(SC6H13)18, Au38(SC6H13)24, and Au144(SC6H13)60—previously synthesized, were systematically examined to assess their catalytic performance.70–73 The results demonstrated that as the Au NC size increased, catalytic activity improved, leading to higher TOF values (Fig. 2a). This suggests a size-dependent influence on reaction kinetics or active site availability. Despite these variations in nuclearity and geometric arrangement, all three NCs ultimately facilitated the reduction reaction, yielding CO as the final product, consistent with previous findings. Subsequent research investigated the influence of surface-protecting ligands in ECO2R while preserving a consistent NC structural framework. This study focused on three Au25 NCs, each stabilized by distinct ligands: PET, 1-naphthalenethiolate (Nap), and benzoselenolate (SePh).74 It has been observed that modifications in the carbon tail of the ligands had minimal influence on the reaction outcome, provided that the overall NC framework remained unchanged. However, altering the connecting atoms between the ligand and the NC core had a pronounced effect on product selectivity. For instance, when sulfur (S) was used as the anchoring atom, the reduction reaction predominantly yielded CO as the final product. In contrast, substituting S with selenium (Se) significantly altered the reaction pathway, favoring the competitive hydrogen evolution reaction (HER) (Fig. 2b). Additional studies revealed that modifying surface-protecting ligands could be an effective strategy for fine-tuning the electronic structure of Au NCs, especially by controlling the number of available valence electrons, as illustrated in two separate studies involving Au28 NCs.75,76 This electronic modulation, in turn, had a direct impact on both the selectivity and efficiency of the ECO2R. By varying ligand composition, one could influence the electron density around the NC core, thereby modifying the adsorption strength of CO2 intermediates and steering the reaction pathway toward specific products. Additionally, it was discovered that steric effects introduced by bulky ligands imposed spatial constraints on active sites, adversely affecting catalytic performance by hindering substrate accessibility and electron transfer efficiency.77 These findings underscored the dual role of ligand modifications in both electronic and steric regulation, offering valuable insights for designing high-performance NC-based catalysts. Furthermore, even when the overall Au NC size remained constant, differences in ligand isomerism resulted in variations in ECO2R efficiency.78 These variations were primarily attributed to subtle rearrangements in the structural architecture of NC, which altered the stabilization energies of key reaction intermediates. Consequently, these changes influenced the reaction kinetics and product distribution, demonstrating the critical role of ligand orientation in modulating catalytic performance. Despite these changes in catalytic performance, the final reduction products consistently remained CO and the competitive HER, highlighting the inherent limitations imposed by the Au NC. Liu et al. modified the surface microenvironment of the nanocluster by incorporating two 2-thiouracil-5-carboxylic acid ligands into the pocket-like cavity of [Au25(p-MBA)18]− (p-MBA: para-mercaptobenzoic acid) NC, significantly enhancing the interaction between nitrogen and CO2 molecules and thereby improving the FECO activity compared to the model NC.79 Later, structural morphology has been identified as a critical factor in determining the ECO2R activity of Au NCs. Comparative studies between different structural configurations have revealed significant variations in catalytic performance. For instance, the spherical [Au25(PET)18]− NC exhibited a markedly higher FECO than the rod-shaped [Au25(PPh3)10(PET)5Cl2]2+ NC (Fig. 2c).80 This disparity was attributed to the more favourable interaction dynamics between negatively charged reaction intermediates and the active catalytic sites in the spherical NC, facilitating enhanced reaction efficiency. Beyond structural morphology, recent investigations have also emphasized the critical role of hydrides in Au NCs in dictating ECO2R efficiency.81 The presence and accessibility of these hydrides significantly influence electron transfer dynamics and intermediate stabilization, thereby affecting the overall reaction pathway. Studies on bridged structures have provided further insights into their catalytic contributions, shedding light on their potential role in optimizing selectivity and efficiency.82 The presence of alkali metal ions in the electrocatalyst has also been identified as a stimulative factor for ECO2R when using model Au25 NC, attributed to the cation-coupled electron transfer step.83 Despite significant progress in understanding the complex interplay between morphology, ligand effects, and both the geometrical and electronic structures of Au NCs, the ECO2R using these NCs has largely been limited to CO as the primary reduction product. One of the key challenges hindering further advancement is the persistent competition with HER, which remains a dominant side reaction. This competition effectively limits the efficiency and selectivity of CO2 reduction processes. Therefore, there is a critical need for the development of other materials that can enhance the selectivity for desired products, improve catalytic efficiency, and minimize the impact of side reactions like HER.
 | ||
| Fig. 2 (a) TOF comparison in −0.56 V vs. RHE among Au25(SC6H13)18, Au38(SC6H13)24, and Au144(SC6H13)60 NCs. (b) Comparision of FECO among of [Au25] NCs with ligand variation. (c) Comparison of FECO in [Au25] NCs depending on their morphology. | ||
Researchers then shifted their focus toward Ag NCs as potential catalysts for ECO2R.40 Compared to their larger Ag NP counterparts, Ag NCs demonstrated significantly enhanced ECO2R performance, making them promising candidates for improving reaction efficiency and selectivity. Experimental studies revealed that Ag NCs could achieve nearly 20 times higher CO mass production than Ag NPs, emphasizing their superior catalytic activity in ECO2R.84 However, when comparing with Au NCs containing the same number of metal atoms, such as [Au25]− and [Ag25(SPhMe2)18]− (Ag25) (SPhMe2: 2,4-dimethylbenzenethiolate) NCs (Fig. 3a), a significant disparity in CO production efficiency is evident.85,86 Experimental studies have shown that the Au25 NC exhibits a much higher FECO and current density than its Ag25 counterpart, despite both NCs possessing structurally similar active catalytic sites (Fig. 3b). The variation in the onset potential (η) is also visible. This variation in catalytic performance is primarily attributed to differences in their electronic structures, which influence the required limiting potential for CO2 reduction. Specifically, the Au25 NC requires a lower overpotential to activate CO2via *COOH intermediate stabilization compared to the Ag25 NC. The stronger stabilization of the *COOH intermediate in Au25 facilitates a more efficient reaction pathway, leading to improved CO2 conversion rates. In contrast, the Ag25 NC required a higher overpotential to achieve the same level of activation, which in turn reduced its catalytic efficiency. These findings highlight the superior reactivity of Au NCs over Ag NCs in ECO2R, emphasizing the critical role of electronic structure and intermediate stabilization in determining catalytic performance. While Ag NCs offer advantages over conventional Ag NPs, Au NCs remains more efficient catalysts for selective CO2 reduction, necessitating further optimization of Ag-based NCs to bridge this performance gap. As previously discussed in the case of Au NCs, ligand chemistry plays a pivotal role in modulating both the selectivity and efficiency of the ECO2R. A similar influence is observed in Ag NCs, where the nature of the surface-protecting ligands profoundly affects catalytic performance by altering electronic properties, active site accessibility, and intermediate stabilization. In Au NCs, thiolate ligands have been shown to impact CO selectivity by tuning the electronic structure and binding affinity of key intermediates. However, Ag NCs protected by alkynyl ligands have been reported to exhibit an exceptionally high FECO, consistently reaching approximately 95% at an applied potential of −0.6 V vs. RHE.87 A detailed reaction kinetics analysis further revealed that the formation of *COOH serves as the rate-determining step in Ag NC-mediated ECO2R. The efficiency of CO production is therefore directly linked to the ability of the NC to stabilize this key intermediate. Ligands that enhance the stabilization of *COOH facilitate more efficient CO2 reduction, thereby increasing the overall CO production rate. The nature of the ligands is also appeared as a decisive factor in this process, even when the core structure and number of metal atoms in the NCs remain unchanged. A striking contrast is observed between Au NCs and Ag NCs regarding the role of ligands in ECO2R performance. In Au NCs, thiolate ligands are known to enhance the FECO by fine-tuning the electronic environment around the active catalytic sites and stabilizing crucial intermediates. However, in Ag NCs, thiolate-protected species do not exhibit similarly high CO2 reduction efficiency. Instead, Ag NCs demonstrate significantly superior ECO2R performance when protected by alkynyl ligands rather than thiolate ligands (Fig. 3c).88 The underlying reaction mechanism suggests that alkynyl ligands, in particular, play a crucial role in facilitating the formation of key reactive intermediates, thereby influencing catalytic efficacy. These ligands are believed to alter the electronic density around the NC core, modulating the adsorption and stabilization of *COOH, which is widely recognized as the rate-determining intermediate in CO2 reduction. By promoting a more favourable energy landscape for *COOH formation and desorption, alkynyl ligands enhance the overall catalytic efficiency of Ag NCs, leading to improved CO selectivity and higher reaction rates. Recently, Chen et al. identified the supportive role of inner-sphere Na+ ions in alkynyl-protected Ag15 NC, which enhances FECO activity by promoting CO2 activation and facilitating proton transfer through the formation of a Na+–CO2(*COOH) complex.89
 | ||
| Fig. 3 (a) Structural architecture of Ag25 NC. H atoms are omitted for the clarity. Color legend: Ag, aqua; S, yellow; and C, gray. (b) Comparison of ECO2R data between [Au25]− and Ag25 NCs. Reproduced with permission form ref. 85. Copyright 2022 Americal Chemical Society. (c) Structural architecture of alkynyl-protected Ag32 NC. H atoms are omitted for the clarity. Color legend: Ag, aqua; F, green; and C, gray. | ||
Despite extensive research on Au and Ag NCs for ECO2R, a fundamental limitation persists: CO remains the predominant final reduction product, and the reaction is largely confined to a two-electron transfer pathway. While theoretical studies suggest that deeper reductions involving multi-electron transfer processes could lead to value-added products such as hydrocarbons and alcohols, the weak binding stability of key reaction intermediates—such as *CO, *CHO, and *CH2—limits further progression along the reduction pathway. As a result, although noble metal NC-based catalysts have demonstrated exceptional efficiency in selectively converting CO2 to CO, their practical application remains constrained by the inability to achieve more complex reductions. To overcome these limitations and move toward practical implementation, future research must focus on developing alternative catalyst materials capable of stabilizing crucial reaction intermediates. Among the emerging candidates, Cu-based NCs have shown the potential to break the current selectivity bottleneck and unlock new catalytic pathways for CO2 utilization. Unlike Au and Ag NCs, which primarily facilitate CO evolution, Cu NCs have demonstrated the ability to promote C–C coupling, leading to the formation of multi-carbon products.90 Despite their promising catalytic activity, Cu NCs face challenges related to stability under electrochemical conditions. However, due to their superior catalytic potential in driving multi-electron CO2 reduction, Cu NCs are gaining significant attention in the field. Given the rapid development of Cu NCs for ECO2R, there is now a pressing need to systematically understanding and summarize the various Cu-based NC catalysts that have been utilized for this specific purpose. A comprehensive analysis of their catalytic activity, structural stability, and key influencing parameters—such as ligand effects, electronic properties, and surface morphology—will be essential for understanding their performance trends and optimizing their design. This will not only facilitate the development of next-generation Cu NC catalysts but also contribute to the broader goal of achieving sustainable and selective CO2 conversion to value-added products.
2.2. Effect of Cu NCs
Cu is unique among metal electrocatalysts in its ability to drive ECO2R toward a diverse range of C1 and C2 products, with over 16 different reduction products identified beyond CO.91–95 This exceptional versatility stems from Cu's ability to bind CO (*CO) on its surface with an optimal balance—strong enough to enable further reduction but not so strong as to hinder desorption—while simultaneously stabilizing key reaction intermediates essential for multi-electron transfer processes. In contrast, metals such as Au and Ag exhibit weak CO binding affinities, which favor the evolution of CO as the predominant product rather than its further reduction.95 On the other hand, metals like nickel (Ni) and platinum (Pt) bind CO too strongly, leading to CO poisoning that deactivates catalytic sites and severely limits their effectiveness in CO2 reduction. One of the critical factors that set Cu apart is its ability to exist in multiple oxidation states—Cu0, Cu+, and Cu2+—which dynamically change under reaction conditions and significantly influence product selectivity. Despite these advantages, a major challenge in utilizing Cu NCs for ECO2R lies in their low reduction potential, which affects both their stability and the controlled synthesis of well-defined nanostructures. In addition, Cu NCs are highly susceptible to oxidation, aggregation, and surface reconstruction during electrochemical cycling, leading to structural instability and loss of catalytic activity over time. Additionally, their low reduction potential complicates the formation of NCs with precise size and shape control, making it difficult to achieve uniform catalytic behavior. However, significant progress has been made in addressing these limitations through advances in ligand engineering, electrolyte optimization, and surface modification strategies.The synthesis of ligand-protected stable Cu NCs is typically achieved through the reduction of Cu precursor salts in the presence of surface-protecting ligands, which play a crucial role in stabilizing the resulting NCs.96,97 However, during the reduction process, Cu enables efficient interaction with a wide range of reaction intermediates that are crucial for converting CO2 into valuable multi-carbon products.98 Additionally, Cu nanoclusters (NCs) exhibit a tendency to bind hydride species, which can be introduced through specific reduction methods, potentially influencing the reaction pathway and product selectivity. These hydrides can be accommodated either in interstitial positions within the NC core or as lattice-bound species, both of which significantly alter the overall electronic structure of the NCs. The presence of these hydrides has been shown to have a profound impact on the catalytic efficiency of Cu NCs in ECO2R, influencing key reaction intermediates, charge transfer dynamics, and product selectivity. Theoretical studies have provided valuable insights into the role of hydrides in Cu NCs, revealing that these species can modulate the adsorption strength of CO2-derived intermediates and directly affect reaction pathways. Given these observations, the subsequent discussion categorizes Cu NCs into two broad groups—hydride-containing Cu NCs and hydride-free Cu NCs—and systematically evaluates their comparative effectiveness in ECO2R. Our discussion also aims to provide an in-depth exploration of the intricate mechanisms governing Cu NC-based ECO2R, with a particular emphasis on the role of hydride species in modulating reaction selectivity and influencing the structural evolution of the NCs.
 | ||
| Fig. 4 (a) Structural architecture of Cu32 NC. H atoms are omitted partially for the clarity. Color legend: Cu, brown; S, yellow; P, magenta stick; O, red stick; C, gray stick; and H, white. (b) Theoretical Free energy calculations for CO and HCOOH formation by Cu32 NC. (c) Mechanistic pathway for HCOOH formation form CO2 reduction. Reproduced with permission form ref. 100. Copyright 2017 Americal Chemical Society. | ||
To further investigate the role of hydrides in Cu NCs and their impact on ECO2R selectivity, Li et al. conducted a comprehensive theoretical study.101 Their work focused on the [Cu25H22(PPh3)12]Cl (Cu25) NC, which contains a higher number of lattice hydrides compared to previously studied systems. This NC features a well-defined Cu13 icosahedral core that is further stabilized by four Cu3 triangular units, which cap the tetrahedral sites of the core. Unlike the previously discussed Cu32 NC, where hydrides were distributed between both interstitial and lattice positions, in Cu25 NC, all twenty-two hydrides are exclusively located in lattice positions, each adopting distinct coordination modes. DFT calculations revealed that, similar to earlier findings, the most negatively charged lattice hydrides play a crucial role in facilitating the initial adsorption of CO2. The study identified that these lattice hydrides preferentially promote the formation of the HCOO* intermediate, which subsequently leads to the production of HCOOH. In contrast, the formation of the *COOH intermediate, a key precursor for CO production, was found to be thermodynamically unfavorable in the presence of lattice hydrides. This computational insight aligns well with previous experimental findings, reinforcing the idea that lattice-bound hydrides serve as active catalytic sites that influence product selectivity in ECO2RR. Li et al. synthesized a unique thiol-free [Cu26H11(DPPE)3(CF3CO2)8(CH3O)2(tBuC![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) C)4]+ (DPPE: 1,2-bis(diphenylphosphino)ethane) (Cu26) NC which features a distinctive nested structure composed of a tetrahedral Cu4 core encapsulated by Cu6 and Cu16 shells (Fig. 5a).102 The precise positions of the hydride atoms within the NC were determined using theoretical calculations, which confirmed that all eleven hydrides are located in interstitial positions, rather than lattice sites. This structural characteristic significantly influences the catalytic behavior of Cu26 NC in ECO2R, as the absence of accessible lattice-bound hydrides limits their direct participation in the reaction. Due to the lack of available lattice hydrides, the adsorption and subsequent reduction of CO2 follow an alternative mechanistic pathway in Cu26 NC, which ultimately leads to the stabilization of the *COOH intermediate rather than the HCOO*. This shift in intermediate formation dictates the final product selectivity, favoring CO evolution over HCOOH. Experimental results demonstrated that Cu26 NC achieves a FE of 81% for CO production at an applied potential of −0.8 V vs. RHE, highlighting its strong selectivity toward CO2-to-CO conversion. To further investigate the role of hydrides in ECO2R, a comparative theoretical study was performed on the optimized structure of Cu26, both with and without hydrides. The results revealed that in the as-synthesized Cu26 NC, the energy barrier for the formation of the *COOH intermediate was 1.35 eV, whereas the energy barrier for the competing HER was significantly lower at 0.16 eV. This energy difference suggests that in the presence of interstitial hydrides, HER is more favorable than CO2 reduction. However, when hydrides were removed from the NC, the energy barrier for *COOH formation drastically decreased from 1.35 eV to 0.14 eV, indicating a substantial enhancement in CO2 activation. Additionally, the absence of hydrides shifted the rate-determining step to *CO formation, and the energy barrier for HER increased to 0.72 eV. This increase in HER energy barrier suggests that without hydrides, CO2 reduction becomes the dominant process, effectively suppressing hydrogen evolution.
C)4]+ (DPPE: 1,2-bis(diphenylphosphino)ethane) (Cu26) NC which features a distinctive nested structure composed of a tetrahedral Cu4 core encapsulated by Cu6 and Cu16 shells (Fig. 5a).102 The precise positions of the hydride atoms within the NC were determined using theoretical calculations, which confirmed that all eleven hydrides are located in interstitial positions, rather than lattice sites. This structural characteristic significantly influences the catalytic behavior of Cu26 NC in ECO2R, as the absence of accessible lattice-bound hydrides limits their direct participation in the reaction. Due to the lack of available lattice hydrides, the adsorption and subsequent reduction of CO2 follow an alternative mechanistic pathway in Cu26 NC, which ultimately leads to the stabilization of the *COOH intermediate rather than the HCOO*. This shift in intermediate formation dictates the final product selectivity, favoring CO evolution over HCOOH. Experimental results demonstrated that Cu26 NC achieves a FE of 81% for CO production at an applied potential of −0.8 V vs. RHE, highlighting its strong selectivity toward CO2-to-CO conversion. To further investigate the role of hydrides in ECO2R, a comparative theoretical study was performed on the optimized structure of Cu26, both with and without hydrides. The results revealed that in the as-synthesized Cu26 NC, the energy barrier for the formation of the *COOH intermediate was 1.35 eV, whereas the energy barrier for the competing HER was significantly lower at 0.16 eV. This energy difference suggests that in the presence of interstitial hydrides, HER is more favorable than CO2 reduction. However, when hydrides were removed from the NC, the energy barrier for *COOH formation drastically decreased from 1.35 eV to 0.14 eV, indicating a substantial enhancement in CO2 activation. Additionally, the absence of hydrides shifted the rate-determining step to *CO formation, and the energy barrier for HER increased to 0.72 eV. This increase in HER energy barrier suggests that without hydrides, CO2 reduction becomes the dominant process, effectively suppressing hydrogen evolution.
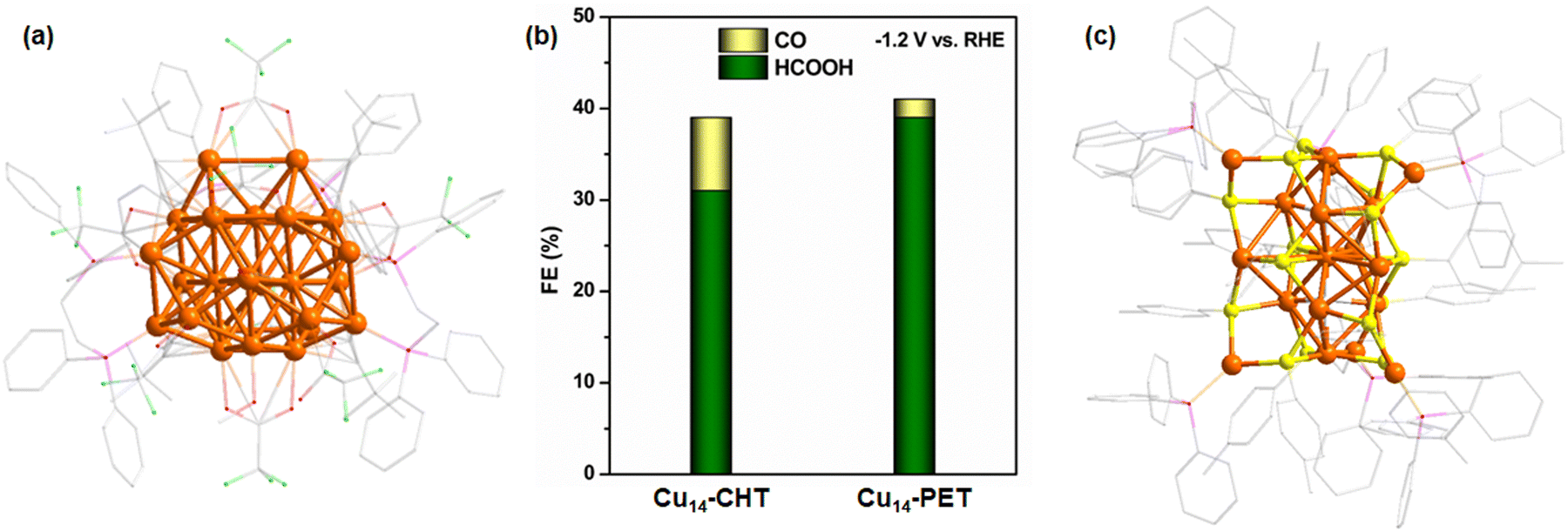 | ||
| Fig. 5 (a) Structural architecture of Cu26 NC. H atoms are omitted for the clarity. Color legend: Cu, brown; P, magenta stick; Cl, green stick; O, red stick; and C, gray stick. (b) Comparison of FE between Cu14–CHT and Cu14–PET NCs. (c) Structural architecture of Cu26 NC. H atoms are partially omitted for the clarity. Color legend: Cu, brown; P, magenta stick; C, gray stick; and H, white. | ||
Our research group has also made significant contributions to this field by synthesizing a variety of hydride-containing Cu NCs and evaluating their catalytic performance in ECO2R. Recently, we conducted a comparative study on two structurally similar yet distinct hydride-containing Cu14 NCs: [Cu14(PET)3(PPh3)8H10]+ (Cu14–PET) and [Cu14(CHT)3(PPh3)8H10]+ (CHT: cyclohexanethiolate) (Cu14–CHT) NCs.103 Although these NCs were confirmed to contain hydrides via electrospray ionization mass spectrometry (ESI-MS), their precise locations could not be determined through single-crystal X-ray diffraction (SCXRD). However, the experimental findings revealed that Cu14–CHT exhibited a maximum FE of 31% for HCOOH and 8% for CO at an applied potential of −1.2 V vs. RHE, whereas Cu14–PET demonstrated a slightly enhanced FE of 39% for HCOOH and a lower FE of 2% for CO under the same reaction conditions (Fig. 5b). This difference in selectivity highlights the influence of ligand environment and electronic structure on catalytic behavior. Our analysis suggests that the stronger cuprophilic (Cu⋯Cu) interactions in Cu14–PET lead to a more compact NC structure, which may contribute to greater stability of key reaction intermediates, ultimately favoring HCOOH formation over CO. Building upon this understanding, we also investigated the catalytic performance of a larger hydride-containing Cu NC, [Cu18H2(PTT)15(PPh3)6] (PTT: p-toluenethiolate) (Cu18PTT) NC (Fig. 5c).104 The ECO2RR experiments demonstrated that Cu18-PTT achieved a maximum FE of ∼35% for HCOOH and ∼4% for CO at −1.2 V vs. RHE. Notably, in this NC, the two hydrides were confirmed to be positioned at interstitial sites rather than lattice positions, meaning they were not directly involved in the proton transfer process during CO2 reduction. Despite this, Cu18-PTT NC still exhibited a preference for HCOOH production over CO, further reinforcing the hypothesis that factors beyond the presence of lattice hydrides influence catalytic selectivity. A deeper structural analysis of Cu18-PTT revealed that its fused-core architecture, featuring a central Cu(0) atom surrounded by strong cuprophilic interactions, played a pivotal role in determining product selectivity. The presence of this metallophilic interaction is believed to modulate the electronic structure of the NC, stabilizing the HCOO* intermediate and promoting HCOOH formation. This finding is particularly significant as it suggests that HCOOH selectivity in Cu NCs is not solely dictated by the presence of lattice hydrides; rather, metallophilic interactions also play a crucial role in tuning catalytic performance. The influence of cuprophilic interactions has also been demonstrated in other studies, where stronger cuprophilic interactions were found to play a crucial role in enhancing catalytic selectivity.105 Specifically, these interactions have been shown to favor CH4 formation by stabilizing key intermediates and facilitating the reaction pathways leading to CH4 production.
Later we explored the impact of structural defects on the catalytic performance of Cu NCs, particularly when interstitial hydrides were present.106 To systematically investigate this, we synthesized and compared three closely related Cu58 NCs: a regular cubic [Cu58H20(SPr)36(PPh3)8]2+ (SPr: propanethiolate) (Cu58-I) NC a surface–ligand–vacant distorted [Cu58H20(SPr)36(PPh3)7]2+ (Cu58-II), and a further distorted variant [Cu58H20(SEt)36(PPh3)6]2+ (SEt: ethanethiolate) (Cu58-III) NCs (Fig. 6a–c). These NCs provided an ideal platform to examine how structural distortions, ligand vacancies, and cuprophilic interactions influence ECO2R product selectivity. In the Cu58-I NC, the cubic framework was well-preserved, maintaining a relatively moderate cuprophilic interaction. The interstitial hydrides, though present, were not directly accessible during the catalytic process, which limited its CO2 reduction activity. As a result, this NC predominantly facilitated CO evolution, with no significant formation of other products (Fig. 6d). However, when we introduced a surface-ligand vacancy by removing one PPh3 ligand from the Cu58-I structure, we obtained the Cu58-II NC, which featured a partial dislocation of Cu atoms at one of the cube's vertices. This structural modification created an open site that subtly altered the cuprophilic interaction, leading to a distorted geometry. The distortion appeared to stabilize key reaction intermediates, particularly those favoring further proton-coupled electron transfer steps. Consequently, Cu58-II NC yielded CH3OH as the major ECO2R product, with a FE of ∼44% at −0.9 V vs. RHE. This result highlighted the importance of structural flexibility and ligand vacancy effects in facilitating deeper CO2 reduction pathways with different stabilization of intermediates (Fig. 6e). Further structural modification in Cu58-III NC, achieved by introducing additional Cu atom dislocations at the diagonal vertex of the cube, dramatically influenced the cluster's reactivity. This alteration reoriented the cuprophilic interactions in such a way that it favored competitive HER over CO2 reduction with very low HCOOH production. This shift suggests that excessive structural distortion and loss of electronic coherence among Cu centers can significantly alter the reaction pathway, ultimately reducing the efficiency of CO2 reduction and promoting HER. So, these findings provide valuable insights into the delicate balance between NC symmetry, ligand environment, hydride accessibility, and electronic structure in governing ECO2R selectivity.
 | ||
| Fig. 6 Structural architecture of (a) Cu58-I, (b) Cu58-II, (c) Cu58-III NCs. H atoms and ligands are partially omitted for the clarity. Color legend: Cu, brown; S, yellow; P, magenta; and C, gray stick. (d) Comparison of FE among Cu58-I, Cu58-II, Cu58-III NCs. (e) Mechanistic insights of the reaction free energy diagram for CO2 reduction process. Reproduced with permission form ref. 106. Copyright 2025 John Wiley & Sons, Inc. | ||
 | ||
| Fig. 7 (a) Structural architecture of [Cu6(MBD)6] NC and FE. H atoms are omitted for the clarity. Color legend: Cu, brown; S, yellow; N, blue; and C, gray. (b) Theoretical reaction mechanism through free enrgy profile. Reproduced with permission form ref. 107. Copyright 2023 John Wiley & Sons, Inc. (c) Structural architecture of Cu8-I, Cu8-II, Cu8-III, NCs along with their FEHCOOH. H atoms are partially omitted for the clarity. Color legend: Cu, brown; S, yellow; N, blue; O, red. | ||
Mu et al. investigated the role of anion-templated Cu NCs in ECO2R. They synthesized two structurally distinct Cu NC variants by employing thiacalix[4]arene and alkynyl ligands as protective frameworks.109 The first variant was a discrete Cu38 NC (Fig. 8a), while the second was a hierarchical assembly composed of forty three Cu atoms. From a structural standpoint, their study revealed that the Cu38 NC exhibited greater intrinsic stability than the Cu43 NC, which in turn enhanced its applicability for ECO2R. Electrochemical studies demonstrated that the Cu38 NC achieved a maximum FE of 62.01% for hydrocarbon production at an applied potential of −1.57 V vs. RHE (Fig. 8a). The hydrocarbon product distribution consisted of 34.03% C2H4 and 27.98% CH4. The theoretical study revealed that the energy barrier for the *CO → *COH transition was significantly higher compared to the *CO → *OCCO step (Fig. 8b). This energy difference favored the formation of the *OCCO intermediate, which subsequently facilitated C–C coupling and C2H4 production. Guan et al. reported the synthesis and catalytic performance of hydrophobically stabilized Cu4 NCs supported on multi-walled carbon nanotubes (MWCNTs) for ECO2R.110 They systematically compared the catalytic behavior of these Cu4 NCs with molecular Cu species embedded in MWCNTs to evaluate their efficiency and product selectivity. Their study revealed that the chemically anchored Cu4 NCs formed stable catalytic sites on the MWCNT surface through strong interactions, which contributed to significantly enhanced selectivity for hydrocarbon products. Notably, they observed a crucial shift in product distribution when the Cu active site changed from a single Cu atom to a tetra-nuclear Cu4 NC (Fig. 8c). While single Cu atoms predominantly favored CH4 production, the transition to Cu4 NCs altered the reaction pathway, leading to a preference for C2H4 formation. This shift in selectivity was attributed to the increased catalytic surface area of the Cu4 NCs, which promoted the aggregation of active sites. The enhanced surface area facilitated *CO dimerization, a key intermediate step in C–C bond formation, thereby favoring C2H4 production over CH4. Theoretical studies establish that Cu(100) favors C–C coupling and ethylene formation, while Cu(111) preferentially yields methane due to facet-dependent adsorption energies of key intermediates.111 This effect is more pronounced in NPs and extensively studied in the literature.112–114 Thus, these findings highlight the critical role of nuclearity in modulating catalytic performance and selectivity in ECO2R reactions for Cu NCs.
 | ||
| Fig. 8 (a) Structural architecture of Cu38 NC and FE. H atoms are omitted for the clarity. Color legend: Cu, brown; S, yellow; O, red; and C, gray. (b) Theoretical reaction mechanism through free enrgy profile. Reproduced with permission form ref. 109. Copyright 2024 Americal Chemical Society. (c) Comparison in ECO2R product when Cu NCs embedded on MWCNT. Reproduced with permission form ref. 110. Copyright 2021 Americal Chemical Society. | ||
2.3. Effect of alloy NCs
It is evident from the previous discussion that Cu NCs exhibit superior catalytic activity and product selectivity beyond CO compared to overcrowded noble metal NCs. However, further catalyst engineering is necessary to fine-tune the CO2 reduction pathway, which can be achieved by doping foreign metal atoms into the NCs. Bimetallic or trimetallic catalysts often exhibit superior performance compared to their monometallic counterparts, owing to synergistic effects arising from the interaction between distinct active catalytic centers.115–117 A pioneering study by Li et al. compared the electrocatalytic activity of Pd1Au24 NCs with the well-established Au25 NC model system.118 Their results revealed that Pd doping significantly suppressed the competing HER at high currents while maintaining a remarkable 100% FE for CO production over a broad potential range (−0.6 V to −1.2 V vs. RHE). In contrast, Au25 NCs exhibited a decline in CO selectivity beyond −0.9 V vs. RHE. Theoretical calculations revealed that doping with Pd significantly increases the thermodynamic barrier for the desorption of thiolate ligands from the nanocluster surface. This enhancement in ligand binding strength leads to greater structural stability of the nanocluster, particularly by preserving the coordination environment around the Au active sites. As a result, the Pd-doped system exhibits improved resilience under electrochemical conditions, which in turn enhances its selectivity for ECO2R, especially at higher potentials. Subsequent studies explored the effects of Cd doping in Au NCs, revealing that modifications in surface geometry and electronic structure through narrowing the energy gap altered intermediate binding energies, leading to enhanced CO selectivity in high-potential regions.119–121 However, despite these advances, both homometallic and bimetallic noble metal NCs predominantly yield CO as the main product, limiting their practical applicability. In contrast, Cu-based catalysts have a unique ability to generate a wider range of products in ECO2R, making them more versatile for real-world applications. Given this distinction, the following section will explore the effectiveness of Cu-based doped NCs in ECO2R and their potential for optimizing product selectivity. Ma et al. reported a series of alkynyl-protected M15 alloy NCs—Au7Ag8, Ag9Cu6, and Au2Ag8Cu5—synthesized using a site-specific metal exchange approach to investigate the role of different metal atoms in the ECO2R.122 Structural analysis revealed that all three NCs adopt a similar core–shell-shell configuration with a well-defined Mcore@Mcube@Moctahedron geometry. However, while the central core position remains fixed with Au, variations in the shell composition due to the presence of different metal atoms lead to distinct electronic effects, altering ligand coordination and modifying the active surface motifs. This structural diversification significantly influences catalytic performance in ECO2R. The Au7Ag8 NC demonstrated exceptional CO selectivity, achieving a maximum FE of 98.1% at −0.49 V vs. RHE (Fig. 9a). In contrast, Ag9Cu6 and Au2Ag8Cu5 NCs predominantly facilitated the production of HCOOH at more negative potentials. These findings highlight the crucial role of Cu in favoring HCOOH formation over CO production. Notably, Ag9Cu6 exhibited a higher FE for HCOOH compared to the trimetallic Au2Ag8Cu5 NC, which was attributed to the enhanced stabilization of the HCOO* intermediate at the Ag–Cu active sites. Deng et al. reported the effect of Cu doping in Ag NCs by synthesizing a core–shell alkynyl-protected [Ag15Cu6(CCR)18(DPPE)2]− (HCCR: 3,5-bis(trifluoromethyl)phenylacetylene) (Ag15Cu6) NC.123 This NC features a body-centered cubic (bcc) Ag11Cu4 core, which follows the Mcore@Mcube@Moctahedron structural motif, with an atomic arrangement of Ag@Ag8@Ag2Cu4. The core is further stabilized by two Cu atoms, two Ag2DPPE motifs, and eighteen alkynyl ligands, contributing to its well-defined structural integrity. ECO2R studies revealed that Ag15Cu6 NC exhibited superior catalytic efficiency and selectivity, achieving a high FECO production (FECO ∼ 91.3% at −0.81 V vs. RHE). This performance was significantly better compared to a structurally similar bcc-type Ag9Cu6 NC, which displayed a much lower FECO of 48.5% under similar conditions. They identified that ligand removal from Ag9Cu6 NC led to a deterioration in catalytic performance, whereas the Ag15Cu6 NC maintained its ligand environment more effectively, preserving its reactivity and selectivity. The free energy profile diagram signifies the stability of the intermediate on the surface of the Ag15Cu6 NC which leads to the formation of CO (Fig. 9b). Further, they extended their synthetic strategy to incorporate Cu doping into Au NCs and successfully synthesized [Au15Cu4(DPPM)6Cl4(CCR)1]2+ (DPPM: bis(diphenylphosphino)methane) NC which exhibited a significantly higher FECO compared to its undoped counterpart, Au18 NC.124 Theoretical calculations revealed that Cu doping induces strong synergistic effects within the NC framework, fundamentally altering the electronic structure and catalytic behavior. In particular, the study suggested that the presence of exposed Au–Cu dual sites played a crucial role in initiating the ECO2R process. These active dual sites facilitated electron transfer, optimizing the adsorption and activation of CO2 molecules. Additionally, Cu incorporation led to a synergistic d-state shift between Au and Cu, which fine-tuned the binding strength of key reaction intermediates, thereby enhancing catalytic efficiency and selectivity. Su et al. conducted a comparative study using a model M9 NC system, in which partially thiol-capped Au NCs were doped with Cu and Ag to evaluate their effectiveness in ECO2R effectivity.125 Electrochemical studies demonstrated that Cu doping significantly enhanced the catalytic activity, with [Au8Cu1(SAdm)4(Dppm)3Cl]2+ (SAdm: adamantanethiolate) NC achieving a higher FECO (∼82.2%) whereas its Ag-doped counterparts [Au8Ag1(SAdm)4(Dppm)3Cl]2+ NC exhibited much lower FECO (∼33.1%) (Fig. 9c). Theoretical calculations were performed, revealing that the Au–Cu active surface effectively lowers the energy barrier for CO2 reduction and provides enhanced stability to key reaction intermediates. Shi et al. recently conducted a theoretical investigation into the catalytic performance of Cu NCs, focusing on the role of surrounding atoms in modulating electrocatalytic activity.126 They selected Cu13 and Cu55 as model NCs, considering their magic-number-associated symmetrical stability, which provides well-defined electronic and geometric structures. To explore the influence of transition metal doping, they systematically substituted the corner atoms of these NCs with widely available transition metals—Fe, Co, and Ni—leading to the formation of a crown jewel structure. Their theoretical findings revealed that multi-atom alloy NCs exhibit significantly enhanced catalytic activity by supressing the energy barrier compared to conventional Cu (211) surfaces, which are often considered active sites for CO2 reduction on bulk Cu catalysts. Among the different dopants studied, Co incorporation in Cu-based clusters demonstrated the lowest free energy barrier (0.33 eV) for CO2 reduction, indicating a highly favorable reaction pathway. Furthermore, their analysis indicated that catalytic performance is largely governed by the relative proportion of metallic dopants, which influences local coordination environments. In particular, a coordination number of six—formed by specific dopant arrangements—was found to be a key structural feature enabling the efficient reduction of CO2 to CH4. This study underscores the potential of precise atomic doping strategies in optimizing Cu-based NCs for ECO2R, offering fundamental insights into the design of highly efficient and selective electrocatalysts.
 | ||
| Fig. 9 (a) Comparison of FE series of among alkynyl-protected M15 alloy NCs. (b) Theoretical reaction mechanism through free enrgy profile by using Ag15Cu6 NC as catalyst. Reproduced with permission form ref. 123. Copyright 2023 Americal Chemical Society. (c) Comparison of FE with different M9 alloy NCs. | ||
3. Conclusions and outlook
This article provides a comprehensive analysis of recent advancements in atomically precise metal NC-based catalysts for ECO2R. While noble metal NCs have demonstrated the highest FE for CO2 reduction, their catalytic performance is largely limited to the formation of CO, with little diversification in product selectivity. This constraint arises from the intrinsic properties of noble metals, which do not effectively stabilize intermediates required for the formation of more complex reduction products. To overcome this limitation, researchers have explored various factors that influence catalytic activity, including ligand effects, electronic structures, and the introduction of external stimuli. However, despite these efforts, the primary product of CO2 reduction using noble metal NCs remains CO, highlighting the need for alternative strategies to expand product selectivity. In contrast, Cu NCs and Cu-based alloy NCs have emerged as promising candidates for broadening the scope of ECO2R products. Due to their unique electronic structures and intermediate stabilization capabilities, Cu NCs facilitate multi-step reduction processes, leading to the formation of high-value products such as HCOOH, hydrocarbons, and oxygenates. However, the synthesis of Cu NCs poses significant challenges due to their inherent instability. Unlike noble metal NCs, Cu NCs tend to undergo structural degradation, limiting their practical applicability. One promising approach to address this issue involves the incorporation of hydrides, which not only enhance the stability of Cu NCs but also influence product selectivity, particularly favoring the formation of HCOOH. Furthermore, the absence or limited availability of hydrides in the catalytic system has been found to promote deeper reduction pathways, yielding more complex and energy-dense products. In addition to hydride incorporation, various external and internal factors—such as ligand effects, support interactions, and electrochemical conditions—have been studied to optimize Cu NC-based catalysts. The incorporation of Cu into alloy NCs has also proven beneficial, extending the product selectivity window while maintaining high FE. Thus, this review article systematically outlines the stepwise evolution of NC-based catalyst design, detailing the interplay between structural modifications and catalytic performance. By elucidating the structure–property correlations of NC catalysts, this work provides valuable insights into the rational design of efficient catalysts tailored for selective CO2 reduction.Looking ahead, several key challenges and opportunities remain in the development of NC-based catalysts for ECO2R. Although Cu-based NCs exhibit considerable catalytic potential influenced by their size, the limited availability of stable high-nuclear Cu NCs continues to hinder the comprehensive exploration of their catalytic properties. This limitation primarily stems from challenges related to their stability. Future research should therefore focus on developing innovative synthetic strategies to obtain high-nuclear Cu NCs with precisely controlled compositions—including both hydride-rich and hydride-free variants—as the presence of hydrides has also been shown to play a crucial role in influencing catalytic activity. Additionally, while Cu is currently the most promising transition metal for selective CO2 reduction, exploring other transition metal NCs may uncover alternative catalytic pathways, potentially altering intermediate binding affinities and enabling new reaction mechanisms. Furthermore, the scope of alloy-based NC catalysts remains underexplored, particularly in the context of Cu alloying strategies. The integration of additional transition metals into Cu NCs may lead to synergistic effects that enhance stability, selectivity, and catalytic efficiency. Future research should prioritize the rational design of alloy NCs with tunable electronic structures to achieve optimal CO2 reduction performance. In addition to catalyst design, the configuration of the electrolyzer plays a crucial role in determining the overall efficiency, selectivity, and stability of ECO2R. Factors such as electrode architecture, ion transport pathways, and electrolyte composition can significantly influence the reaction environment and, consequently, the catalytic performance. Despite its importance, this aspect remains relatively underexplored, particularly in the context of employing NCs as catalysts. Most studies to date have focused primarily on material development, with limited attention given to optimizing the integration of NCs within practical electrolyzer setups. This presents a valuable opportunity for future research to bridge the gap by systematically investigating the interplay between nanocluster-based catalysts and electrolyzer design, potentially unlocking new avenues for enhancing ECO2R efficiency at scale. Ultimately, advancing the field of NC-based electrocatalysis holds immense promise for sustainable and environmentally friendly CO2 conversion technologies. As research progresses, the continued development of atomically precise NC catalysts will pave the way for transformative breakthroughs in catalytic science, offering new opportunities for industrial and environmental applications. By addressing current challenges and leveraging innovative synthesis strategies, we are poised to unlock the full potential of metal NCs for next-generation CO2 reduction processes.
Data availability
No primary research results, software or code have been included and no new data were generated or analysed as part of this review.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the JSPS KAKENHI (grant no. 23H00289 and 22K19012), Scientific Research on Innovative Areas “Aquatic Functional Materials” (grant no. 22H04562), the Yazaki Memorial Foundation for Science and Technology, Advanced Technology Institute Research Grants, Takahashi Industrial and Economic Research Foundation, and the Kumagai Foundation for Science and Technology and the Ogasawara Foundation for the Promotion of Science and Engineering.Notes and references
- S. J. Davis, K. Caldeira and H. D. Matthews, Science, 2010, 329, 1330–1333 CrossRef CAS.
- J. Hansen, M. Sato, R. Ruedy, A. Lacis and V. Oinas, Proc. Natl. Acad. Sci. U. S. A., 2000, 97, 9875–9880 CrossRef CAS.
- M. Meinshausen, N. Meinshausen, W. Hare, S. C. Raper, K. Frieler, R. Knutti, D. J. Frame and M. R. Allen, Nature, 2009, 458, 1158–1162 CrossRef CAS PubMed.
- G. A. Olah, G. S. Prakash and A. Goeppert, J. Am. Chem. Soc., 2011, 133, 12881–12898 CrossRef CAS PubMed.
- K. B. Keerthana, S.-W. Wu, M.-E. Wu and T. Kokulnathan, Sustainability, 2023, 15, 7932 CrossRef CAS.
- S. C. Obiora, O. Bamisile, Y. Hu, D. U. Ozsahin and H. Adun, Heliyon, 2024, 10, e28770 CrossRef CAS.
- P. A. Østergaard, N. Duic, Y. Noorollahi and S. Kalogirou, Renewable Energy, 2022, 199, 1145–1152 CrossRef.
- I. E. Agency, World energy outlook, OECD/IEA Paris, 2009 Search PubMed.
- D. Gielen, F. Boshell, D. Saygin, M. D. Bazilian, N. Wagner and R. Gorini, Energy Strategy Rev., 2019, 24, 38–50 CrossRef.
- W. Strielkowski, L. Civín, E. Tarkhanova, M. Tvaronavičienė and Y. Petrenko, Energies, 2021, 14, 8240 CrossRef CAS.
- S. M. Kim, P. M. Abdala, M. Broda, D. Hosseini, C. Copéret and C. Müller, ACS Catal., 2018, 8, 2815–2823 CrossRef CAS.
- K. M. K. Yu, I. Curcic, J. Gabriel and S. C. E. Tsang, ChemSusChem, 2008, 1, 893–899 CrossRef CAS PubMed.
- W. D. Jones, J. Am. Chem. Soc., 2020, 142, 4955–4957 CrossRef CAS PubMed.
- T. P. Senftle and E. A. Carter, Acc. Chem. Res., 2017, 50, 472–475 CrossRef CAS PubMed.
- G. A. Ozin, Adv. Mater., 2015, 27, 1957–1963 CrossRef CAS PubMed.
- D.-H. Nam, P. De Luna, A. Rosas-Hernández, A. Thevenon, F. Li, T. Agapie, J. C. Peters, O. Shekhah, M. Eddaoudi and E. H. Sargent, Nat. Mater., 2020, 19, 266–276 CrossRef CAS.
- J. Wu, Y. Huang, W. Ye and Y. Li, Adv. Sci., 2017, 4, 1700194 CrossRef PubMed.
- W. Zhang, Y. Hu, L. Ma, G. Zhu, Y. Wang, X. Xue, R. Chen, S. Yang and Z. Jin, Adv. Sci., 2018, 5, 1700275 CrossRef PubMed.
- S. Nitopi, E. Bertheussen, S. B. Scott, X. Liu, A. K. Engstfeld, S. Horch, B. Seger, I. E. L. Stephens, K. Chan, C. Hahn, J. K. Nørskov, T. F. Jaramillo and I. Chorkendorff, Chem. Rev., 2019, 119, 7610–7672 CrossRef CAS PubMed.
- F. Dattila, R. R. Seemakurthi, Y. Zhou and N. López, Chem. Rev., 2022, 122, 11085–11130 CrossRef CAS.
- X. She, Y. Wang, H. Xu, S. Chi Edman Tsang and S. Ping Lau, Angew. Chem., Int. Ed., 2022, 61, e202211396 CrossRef CAS PubMed.
- J. Han, X. Bai, X. Xu, A. Husile, S. Zhang, L. Qi and J. Guan, Chem. Sci., 2024, 15, 7870–7907 RSC.
- S. Garg, M. Li, A. Z. Weber, L. Ge, L. Li, V. Rudolph, G. Wang and T. E. Rufford, J. Mater. Chem. A, 2020, 8, 1511–1544 RSC.
- S. Zhang, Q. Fan, R. Xia and T. J. Meyer, Acc. Chem. Res., 2020, 53, 255–264 CrossRef CAS.
- C. S. Diercks, Y. Liu, K. E. Cordova and O. M. Yaghi, Nat. Mater., 2018, 17, 301–307 CrossRef CAS.
- J. Yu, J. Wang, Y. Ma, J. Zhou, Y. Wang, P. Lu, J. Yin, R. Ye, Z. Zhu and Z. Fan, Adv. Funct. Mater., 2021, 31, 2102151 CrossRef CAS.
- W. Zhang, Y. Yang, D. Guo and L. Liu, J. Energy Chem., 2024, 88, 10–27 CrossRef CAS.
- X. Wang, A. Guo, Y. Wang, Z. Chen, Y. Guo, H. Xie, W. Shan and J. Zhang, J. Colloid Interface Sci., 2024, 653, 238–245 CrossRef CAS.
- P. K. Jiwanti, M. A. Jazuli, D. K. A. Sukardi, G. T. M. Kadja, M. Tomisaki, A. Purwaningsih and Y. Einaga, J. Solid State Electrochem., 2025, 29, 401–411 CrossRef CAS.
- F. Franco, C. Rettenmaier, H. S. Jeon and B. R. Cuenya, Chem. Soc. Rev., 2020, 49, 6884–6946 RSC.
- M. Li, S. Garg, X. Chang, L. Ge, L. Li, M. Konarova, T. E. Rufford, V. Rudolph and G. Wang, Small Methods, 2020, 4, 2000033 CrossRef CAS.
- D. Johnson, Z. Qiao and A. Djire, ACS Appl. Energy Mater., 2021, 4, 8661–8684 CrossRef CAS.
- T. Zheng, K. Jiang and H. Wang, Adv. Mater., 2018, 30, 1802066 CrossRef PubMed.
- S. Biswas, Y. Shingyouchi, M. Ogami, M. Kamiyama, T. Kawawaki and Y. Negishi, EcoEnergy, 2024, 2, 400–418 CrossRef CAS.
- D. Yang, J. Wang, Q. Wang, Z. Yuan, Y. Dai, C. Zhou, X. Wan, Q. Zhang and Y. Yang, ACS Nano, 2022, 16, 15681–15704 CrossRef CAS.
- K. Kwak and D. Lee, Acc. Chem. Res., 2019, 52, 12–22 CrossRef CAS PubMed.
- J. Huang, N. Hörmann, E. Oveisi, A. Loiudice, G. L. De Gregorio, O. Andreussi, N. Marzari and R. Buonsanti, Nat. Commun., 2018, 9, 3117 CrossRef PubMed.
- T. Liu and H. Yabu, EcoEnergy, 2024, 2, 419–432 CrossRef.
- Y. Liu, J. Yu, Y. Lun, Y. Wang, Y. Wang and S. Song, Adv. Funct. Mater., 2023, 33, 2304184 CrossRef CAS.
- G. Ma, L. Qin, Y. Liu, H. Fan, L. Qiao, C. Yu and Z. Tang, Surf. Interfaces, 2023, 36, 102555 CrossRef CAS.
- S. Zhao, R. Jin and R. Jin, ACS Energy Lett., 2018, 3, 452–462 CrossRef CAS.
- T. Kawawaki, T. Okada, D. Hirayama and Y. Negishi, Green Chem., 2024, 26, 122–163 RSC.
- M. Wu, F. Dong, Y. Yang, X. Cui, X. Liu, Y. Zhu, D. Li, S. Omanovic, S. Sun and G. Zhang, Electrochem. Energy Rev., 2024, 7, 10 CrossRef CAS.
- L. Chen, L. Wang, Q. Shen, Y. Liu and Z. Tang, Mater. Chem. Front., 2023, 7, 1482–1495 RSC.
- R. Jin, C. Zeng, M. Zhou and Y. Chen, Chem. Rev., 2016, 116, 10346–10413 CrossRef CAS PubMed.
- S. Biswas, S. Das and Y. Negishi, Coord. Chem. Rev., 2023, 492, 215255 CrossRef CAS.
- I. Chakraborty and T. Pradeep, Chem. Rev., 2017, 117, 8208–8271 CrossRef CAS.
- Y. Du, H. Sheng, D. Astruc and M. Zhu, Chem. Rev., 2020, 120, 526–622 CrossRef CAS PubMed.
- X. Kang and M. Zhu, Chem. Soc. Rev., 2019, 48, 2422–2457 RSC.
- S. Biswas and Y. Negishi, J. Phys. Chem. Lett., 2024, 15, 947–958 CrossRef CAS PubMed.
- H. Hirai, S. Ito, S. Takano, K. Koyasu and T. Tsukuda, Chem. Sci., 2020, 11, 12233–12248 RSC.
- Y. Jin, C. Zhang, X.-Y. Dong, S.-Q. Zang and T. C. Mak, Chem. Soc. Rev., 2021, 50, 2297–2319 RSC.
- S. Biswas, A. K. Das and S. Mandal, Acc. Chem. Res., 2023, 56, 1838–1849 CrossRef CAS PubMed.
- S. Biswas, A. K. Das, S. S. Manna, B. Pathak and S. Mandal, Chem. Sci., 2022, 13, 11394–11404 RSC.
- S. Biswas, A. K. Das, A. C. Reber, S. Biswas, S. Bhandary, V. B. Kamble, S. N. Khanna and S. Mandal, Nano Lett., 2022, 22, 3721–3727 CrossRef CAS.
- R. K. Gupta, Z. Wang, B. Mohan, C. H. Tung and D. Sun, Adv. Mater., 2024, 36, 2410054 CrossRef CAS PubMed.
- Y.-Z. Huang, R. K. Gupta, G.-G. Luo, Q.-C. Zhang and D. Sun, Coord. Chem. Rev., 2024, 499, 215508 CrossRef CAS.
- W.-D. Tian, C. Zhang, S. Paul, W.-D. Si, Z. Wang, P.-P. Sun, A. Anoop, C.-H. Tung and D. Sun, Angew. Chem., Int. Ed., 2025, e202421656 CAS.
- X. Liu and D. Astruc, Coord. Chem. Rev., 2018, 359, 112–126 CrossRef CAS.
- Z. Wang, B. Chen and A. L. Rogach, Nanoscale Horiz., 2017, 2, 135–146 RSC.
- S. Maity, D. Bain and A. Patra, Nanoscale, 2019, 11, 22685–22723 RSC.
- H. Lin, X. Song, O. J. H. Chai, Q. Yao, H. Yang and J. Xie, Adv. Mater., 2024, 36, 2401002 Search PubMed.
- A. K. Das, S. Biswas, S. S. Manna, B. Pathak and S. Mandal, Chem. Sci., 2022, 13, 8355–8364 Search PubMed.
- S. Biswas, A. Pal, M. K. Jena, S. Hossain, J. Sakai, S. Das, B. Sahoo, B. Pathak and Y. Negishi, J. Am. Chem. Soc., 2024, 146, 20937–20944 Search PubMed.
- R. K. Raju, P. Rodriguez and E. N. Brothers, Phys. Chem. Chem. Phys., 2023, 25, 11630–11652 Search PubMed.
- D. R. Kauffman, D. Alfonso, C. Matranga, H. Qian and R. Jin, J. Am. Chem. Soc., 2012, 134, 10237–10243 Search PubMed.
- M. Zhu, C. M. Aikens, F. J. Hollander, G. C. Schatz and R. Jin, J. Am. Chem. Soc., 2008, 130, 5883–5885 Search PubMed.
- D. R. Kauffman, D. Alfonso, C. Matranga, P. Ohodnicki, X. Deng, R. C. Siva, C. Zeng and R. Jin, Chem. Sci., 2014, 5, 3151–3157 RSC.
- B. Kim, H. Seong, J. T. Song, K. Kwak, H. Song, Y. C. Tan, G. Park, D. Lee and J. Oh, ACS Energy Lett., 2020, 5, 749–757 Search PubMed.
- H. Seong, V. Efremov, G. Park, H. Kim, J. S. Yoo and D. Lee, Angew. Chem., Int. Ed., 2021, 60, 14563–14570 CrossRef CAS.
- K. Kwak, Q. Tang, M. Kim, D.-E. Jiang and D. Lee, J. Am. Chem. Soc., 2015, 137, 10833–10840 CrossRef CAS PubMed.
- H. Qian, Y. Zhu and R. Jin, ACS Nano, 2009, 3, 3795–3803 CrossRef CAS.
- H. Qian and R. Jin, Chem. Mater., 2011, 23, 2209–2217 CrossRef CAS.
- S. Li, A. V. Nagarajan, Y. Li, D. R. Kauffman, G. Mpourmpakis and R. Jin, Nanoscale, 2021, 13, 2333–2337 RSC.
- J. Wang, F. Xu, Z. Y. Wang, S. Q. Zang and T. C. Mak, Angew. Chem., Int. Ed., 2022, 61, e202207492 CrossRef CAS.
- S.-F. Yuan, R.-L. He, X.-S. Han, J.-Q. Wang, Z.-J. Guan and Q.-M. Wang, Angew. Chem., Int. Ed., 2021, 60, 14345–14349 CrossRef CAS PubMed.
- M. R. Narouz, K. M. Osten, P. J. Unsworth, R. W. Y. Man, K. Salorinne, S. Takano, R. Tomihara, S. Kaappa, S. Malola, C.-T. Dinh, J. D. Padmos, K. Ayoo, P. J. Garrett, M. Nambo, J. H. Horton, E. H. Sargent, H. Häkkinen, T. Tsukuda and C. M. Crudden, Nat. Chem., 2019, 11, 419–425 CrossRef CAS PubMed.
- S. Li, A. V. Nagarajan, X. Du, Y. Li, Z. Liu, D. R. Kauffman, G. Mpourmpakis and R. Jin, Angew. Chem., Int. Ed., 2022, 61, e202211771 CrossRef CAS PubMed.
- Z. Liu, J. Chen, B. Li, D.-E. Jiang, L. Wang, Q. Yao and J. Xie, J. Am. Chem. Soc., 2024, 146, 11773–11781 CrossRef CAS PubMed.
- S. Zhao, N. Austin, M. Li, Y. Song, S. D. House, S. Bernhard, J. C. Yang, G. Mpourmpakis and R. Jin, ACS Catal., 2018, 8, 4996–5001 CrossRef CAS.
- V. K. Kulkarni, B. N. Khiarak, S. Takano, S. Malola, E. L. Albright, T. I. Levchenko, M. D. Aloisio, C.-T. Dinh, T. Tsukuda, H. Häkkinen and C. M. Crudden, J. Am. Chem. Soc., 2022, 144, 9000–9006 CrossRef CAS.
- Z.-H. Gao, K. Wei, T. Wu, J. Dong, D.-E. Jiang, S. Sun and L.-S. Wang, J. Am. Chem. Soc., 2022, 144, 5258–5262 CrossRef CAS.
- S. M. Han, M. Park, J. Kim and D. Lee, Angew. Chem., Int. Ed., 2024, 63, e202404387 CrossRef CAS PubMed.
- Z. Wang, T. Li, Q. Wang, A. Guan, N. Cao, A. M. Al-Enizi, L. Zhang, L. Qian and G. Zheng, J. Power Sources, 2020, 476, 228705 CrossRef CAS.
- H. Seong, M. Choi, S. Park, H.-W. Kim, J. Kim, W. Kim, J. S. Yoo and D. Lee, ACS Energy Lett., 2022, 7, 4177–4184 CrossRef CAS.
- C. P. Joshi, M. S. Bootharaju, M. J. Alhilaly and O. M. Bakr, J. Am. Chem. Soc., 2015, 137, 11578–11581 CrossRef CAS PubMed.
- L. Qin, F. Sun, X. Ma, G. Ma, Y. Tang, L. Wang, Q. Tang, R. Jin and Z. Tang, Angew. Chem., Int. Ed., 2021, 60, 26136–26141 CrossRef CAS.
- L. Chen, F. Sun, Q. Shen, L. Qin, Y. Liu, L. Qiao, Q. Tang, L. Wang and Z. Tang, Nano Res., 2022, 15, 8908–8913 CrossRef CAS.
- Y. Chen, X. Zhou, X. Liu, Z. Tang, L. Wang and Q. Tang, J. Am. Chem. Soc., 2025, 147, 2699–2713 CrossRef CAS PubMed.
- S. Biswas, S. Das and Y. Negishi, Nanoscale Horiz., 2023, 8, 1509–1522 RSC.
- J. Huang, X. Zhang, J. Yang, J. Yu, Q. Chen and L. Peng, Adv. Sci., 2024, 11, 2309865 CrossRef CAS.
- Y. Ma, M. Sun, H. Xu, Q. Zhang, J. Lv, W. Guo, F. Hao, W. Cui, Y. Wang, J. Yin, H. Wen, P. Lu, G. Wang, J. Zhou, J. Yu, C. Ye, L. Gan, D. Zhang, S. Chu, L. Gu, M. Shao, B. Huang and Z. Fan, Adv. Mater., 2024, 2402979 CrossRef CAS.
- J. Wang, M. Sun, H. Xu, F. Hao, Q. Wa, J. Su, J. Zhou, Y. Wang, J. Yu, P. Zhang, R. Ye, S. Chu, B. Huang, M. Shao and Z. Fan, ACS Nano, 2024, 18, 7192–7203 CrossRef CAS PubMed.
- L. Guo, J. Zhou, F. Liu, X. Meng, Y. Ma, F. Hao, Y. Xiong and Z. Fan, ACS Nano, 2024, 18, 9823–9851 CrossRef CAS.
- Y. Ma, J. Yu, M. Sun, B. Chen, X. Zhou, C. Ye, Z. Guan, W. Guo, G. Wang, S. Lu, D. Xia, Y. Wang, Z. He, L. Zheng, Q. Yun, L. Wang, J. Zhou, P. Lu, J. Yin, Y. Zhao, Z. Luo, L. Zhai, L. Liao, Z. Zhu, R. Ye, Y. Chen, Y. Lu, S. Xi, B. Huang, C.-S. Lee and Z. Fan, Adv. Mater., 2022, 34, 2110607 CrossRef CAS.
- S. Biswas and Y. Negishi, Dalton Trans., 2024, 53, 9657–9663 RSC.
- A. Pal, S. Biswas, B. Sahoo and Y. Negishi, Synlett, 2025 DOI:10.1055/a-2504-3530.
- M. Kamiyama, Y. Shingyouchi, R. Sarma, M. Ghosh, T. Kawawaki, S. Biswas and Y. Negishi, Chem. Commun., 2025, 61, 1048–1062 RSC.
- R. S. Dhayal, J.-H. Liao, S. Kahlal, X. Wang, Y.-C. Liu, M.-H. Chiang, W. E. van Zyl, J.-Y. Saillard and C. W. Liu, Chem. – Eur. J., 2015, 21, 8369–8374 CrossRef CAS PubMed.
- Q. Tang, Y. Lee, D.-Y. Li, W. Choi, C. Liu, D. Lee and D.-E. Jiang, J. Am. Chem. Soc., 2017, 139, 9728–9736 CrossRef CAS PubMed.
- F. Li and Q. Tang, J. Catal., 2020, 387, 95–101 CrossRef CAS.
- S. Li, X. Yan, J. Tang, D. Cao, X. Sun, G. Tian, X. Tang, H. Guo, Q. Wu, J. Sun, J. He and H. Shen, Chem. Mater., 2023, 35, 6123–6132 CrossRef CAS.
- Y. Shingyouchi, M. Ogami, S. Biswas, T. Tanaka, M. Kamiyama, K. Ikeda, S. Hossain, Y. Yoshigoe, D. J. Osborn, G. F. Metha, T. Kawawaki and Y. Negishi, Small, 2024, 2409910 Search PubMed.
- S. Biswas, Y. Shingyouchi, M. Kamiyama, M. K. Jana, M. Ogami, T. Kawawaki, B. Pathak and Y. Negishi, Small, 2025, 2500302 CrossRef.
- L. Zhang, X.-X. Li, Z.-L. Lang, Y. Liu, J. Liu, L. Yuan, W.-Y. Lu, Y.-S. Xia, L.-Z. Dong and D.-Q. Yuan, J. Am. Chem. Soc., 2021, 143, 3808–3816 CrossRef CAS PubMed.
- S. Biswas, T. Tanaka, H. Song, M. Ogami, Y. Shingyouchi, S. Hossian, M. Kamiyama, T. Kosaka, R. Nakatani, Y. Niihori, S. Das, T. Kawawaki, D.-E. Jiang and Y. Negishi, Small Sci., 2025, 5, 2400465 CrossRef PubMed.
- Q.-J. Wu, D.-H. Si, P.-P. Sun, Y.-L. Dong, S. Zheng, Q. Chen, S.-H. Ye, D. Sun, R. Cao and Y.-B. Huang, Angew. Chem., Int. Ed., 2023, 62, e202306822 CrossRef CAS.
- L. J. Liu, Z. Y. Wang, Z. Y. Wang, R. Wang, S. Q. Zang and T. C. Mak, Angew. Chem., Int. Ed., 2022, 61, e202205626 CrossRef CAS.
- W.-L. Mu, L. Li, X.-Z. Cong, X. Chen, P. Xia, Q. Liu, L. Wang, J. Yan and C. Liu, J. Am. Chem. Soc., 2024, 146, 28131–28140 CAS.
- A. Guan, C. Yang, Q. Wang, L. Qian, J. Cao, L. Zhang, L. Wu and G. Zheng, ACS Sustainable Chem. Eng., 2021, 9, 13536–13544 CrossRef CAS.
- M. Salehi, H. Al-Mahayni, A. Farzi, M. McKee, S. Kaviani, E. Pajootan, R. Lin, N. Kornienko and A. Seifitokaldani, Appl. Catal., B, 2024, 353, 124061 CrossRef CAS.
- P. Iyengar, J. Huang, G. L. De Gregorio, C. Gadiyar and R. Buonsanti, Chem. Commun., 2019, 55, 8796–8799 RSC.
- D. Cheng, Z.-J. Zhao, G. Zhang, P. Yang, L. Li, H. Gao, S. Liu, X. Chang, S. Chen, T. Wang, G. A. Ozin, Z. Liu and J. Gong, Nat. Commun., 2021, 12, 395 CrossRef CAS PubMed.
- F. Chang, M. Xiao, R. Miao, Y. Liu, M. Ren, Z. Jia, D. Han, Y. Yuan, Z. Bai and L. Yang, Electrochem. Energy Rev., 2022, 5, 4 CrossRef CAS.
- J. He, N. J. Johnson, A. Huang and C. P. Berlinguette, ChemSusChem, 2018, 11, 48–57 CrossRef CAS PubMed.
- L. Liu, H. Akhoundzadeh, M. Li and H. Huang, Small Methods, 2023, 7, 2300482 CrossRef CAS PubMed.
- X. Kang, Y. Li, M. Zhu and R. Jin, Chem. Soc. Rev., 2020, 49, 6443–6514 RSC.
- S. Li, D. Alfonso, A. V. Nagarajan, S. D. House, J. C. Yang, D. R. Kauffman, G. Mpourmpakis and R. Jin, ACS Catal., 2020, 10, 12011–12016 CrossRef CAS.
- S. Zhuang, D. Chen, L. Liao, Y. Zhao, N. Xia, W. Zhang, C. Wang, J. Yang and Z. Wu, Angew. Chem., Int. Ed., 2020, 59, 3073–3077 CrossRef CAS.
- S. Li, A. V. Nagarajan, D. R. Alfonso, M. Sun, D. R. Kauffman, G. Mpourmpakis and R. Jin, Angew. Chem., Int. Ed., 2021, 60, 6351–6356 CrossRef CAS.
- Y. Sun, X. Liu, K. Xiao, Y. Zhu and M. Chen, ACS Catal., 2021, 11, 11551–11560 CrossRef CAS.
- X. Ma, F. Sun, L. Qin, Y. Liu, X. Kang, L. Wang, D.-E. Jiang, Q. Tang and Z. Tang, Chem. Sci., 2022, 13, 10149–10158 RSC.
- G. Deng, J. Kim, M. S. Bootharaju, F. Sun, K. Lee, Q. Tang, Y. J. Hwang and T. Hyeon, J. Am. Chem. Soc., 2023, 145, 3401–3407 CrossRef CAS.
- G. Deng, H. Yun, M. S. Bootharaju, F. Sun, K. Lee, X. Liu, S. Yoo, Q. Tang, Y. J. Hwang and T. Hyeon, J. Am. Chem. Soc., 2023, 145, 27407–27414 CrossRef CAS PubMed.
- S. Su, Y. Zhou, L. Xiong, S. Jin, Y. Du and M. Zhu, Angew. Chem., Int. Ed., 2024, 63, e202404629 CrossRef CAS PubMed.
- L. Shi, H. Wu, W. Xu, W. Fu, X. Wang, Z. Gu, X. Zhang, J. Chen, Y. Ma and J. Zhao, Sci. Chin. Mater., 2024, 67, 3602–3608 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2025 |