
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceReactions of nickel boranyl compounds with pnictogen–carbon triple bonds†
Brady J. H. Austen,
Marissa L. Clapson and
Marcus W. Drover *
*
Department of Chemistry and Biochemistry, The University of Windsor, 401 Sunset Avenue, Windsor, ON N9B 3P4, Canada. E-mail: marcus.drover@uwindsor.ca
First published on 23rd June 2023
Abstract
The catalytic conversion of unsaturated small molecules such as nitriles into reduced products is of interest for the production of fine chemicals. In this vein, metal–ligand cooperativity has been leveraged to promote such reactivity, often conferring stability to bound substrate – a balancing act that may offer activation at the cost of turnover efficiency. This report describes the reactivity of a [(diphosphine)Ni] compound with pnictogen carbon triple bonds (R–C![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) E; E = N, P), where the diphosphine contains two pendant borane groups. For E = N, cooperative nitrile coordination is observed to afford {Ni}2 complexes displaying B–N interactions, whereas for E = P, B–P interactions are absent. This work additionally outlines a structure–activity relationship that uses nitrile dihydroboration as a model reaction to unveil the effect of SCS stabilization, employing [(diphosphine)Ni] where the diphosphine contains 0, 1, or 2 pendant Lewis acid groups.
E; E = N, P), where the diphosphine contains two pendant borane groups. For E = N, cooperative nitrile coordination is observed to afford {Ni}2 complexes displaying B–N interactions, whereas for E = P, B–P interactions are absent. This work additionally outlines a structure–activity relationship that uses nitrile dihydroboration as a model reaction to unveil the effect of SCS stabilization, employing [(diphosphine)Ni] where the diphosphine contains 0, 1, or 2 pendant Lewis acid groups.
Introduction
Metal–ligand cooperation (MLC), where both a metal and ligand participate in substrate activation has become an important tool relevant to small molecule conversion chemistry.1,2 Perhaps one of the best known examples is exemplified by compounds prepared by Milstein et al. where aromatization/dearomatization of a pyridine, fluorenyl, or acridine ligand component permits the addition of E–H bonds across both metal and ligand.3,4 Using this approach, polar substrates such as nitriles (R–CN) have been activated for hydration,5,6 as well as Michael addition-type reactivity (Fig. 1).7–10 Cooperative nitrile activation is not limited to pincer-based ligand scaffolds – Beckhaus et al. recently described a vanadium pentafulvene complex that reduces 4-chlorobenzonitrile using both a C5H5− (Cp) ring and vanadium(V) (Fig. 1).11 Despite nitrile Lewis basicity, the application of Lewis acidic ligand components to drive the activation and conversion of this substrate class remains relatively unexplored.12 A unique example is provided by Williams et al. where a ruthenium bis(pyrazolyl)borate complex is shown to promote hydride transfer, permitting selective nitrile reduction to the corresponding amine or amide (Fig. 1).13
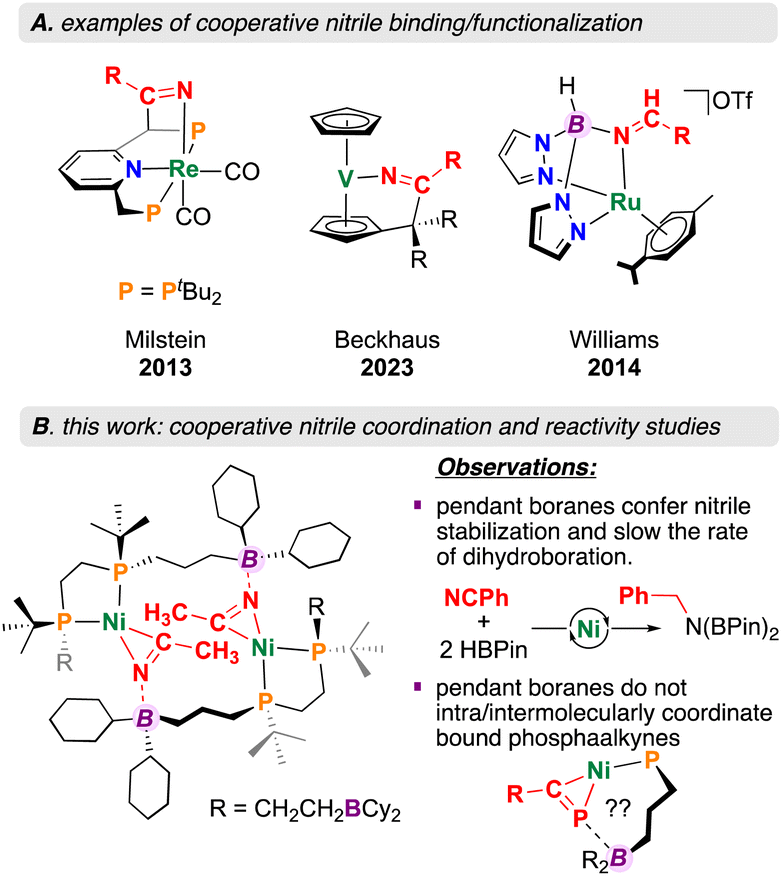 | ||
| Fig. 1 Literature precedent for cooperative nitrile coordination and this work. | ||
Incorporation of Lewis acidic groups into the secondary coordination sphere (SCS: atoms that are not directly bound to the metal) has grown in popularity over recent years.14,15 Since 2020, our group has reported on several new ligand motifs featuring SCS Lewis acidic borane groups, asking general questions relevant to the effect of Lewis acid SCS incorporation on downstream reactivity.16–19 This work has focused on expanding the “diphosphine ligand toolbox”, offering scaffolds containing four,16 two,17 or one19 Lewis acidic SCS borane groups. Of relevance here, complexation of the mono (ttbbpe: tri-tert-butylboranyldiphosphinoethane) and diboranyl (dtbbpe: di-tert-butylboranyldiphosphinoethane) diphosphine systems to Ni(0) allowed for the isolation of the monophosphine-substituted starting materials [(diphosphine)Ni(COD)] (COD = 1,4-cyclooctadiene).17,19 For the monoboranyl Ni(0) complex, [Ni(ttbbpe)(COD)], reaction with nitriles was found to form 14-membered {Ni}2 dimers, with co-stabilization being provided by both boron and nickel (Fig. 1).19
In this report, we advance our understanding of the diborane-containing ligand set, dtbbpe – studying the reactivity of (±)-rac/meso-[Ni(dtbbpe)(COD)] with nitriles to assess SCS effects on product outcome. Moreover, we expand the scope of pnictogen coordination chemistry to phosphaalkynes.20 Finally, we provide a structure activity relationship that relates the concentration (i.e., number) of SCS Lewis acid groups to reactivity by considering a model benzonitrile (PhCN) dihydroboration reaction.
Results and discussion
Reactivity with nitriles
To begin, a solution of (±)-rac/meso-[Ni(dtbbpe)(COD)] (1) was reacted with an excess of CH3CN in toluene (Fig. 2). Analysis by 31P{1H} NMR spectroscopy showed a new set of [AB]-doublets at δP = 67.3 and 70.4 ppm (2JP,P = 45.7 Hz), suggesting consumption of 1 and formation of a new nitrile-bound C1-symmetric complex 2, isolated as a yellow solid in 76% yield (Fig. 2). Consistent with nitrile coordination, the FT-IR spectrum also provided a signal at ν(CN) = 1777 cm−1 (c.f., ν(CN) = 2253 cm−1 for unbound CH3CN). Symmetry of the dtbbpe ligand warrants comment – this ligand contains two isomers – racemic (rac) and meso (differing in the presence of a mirror plane). In the instance where nitrile coordination provides an intramolecular B–N interaction, three possible isomers (two enantiomers and a meso compound) are expected - (±)-rac/meso-[Ni(dtbbpe)(η2-NCCH3)]. However, were an intermolecular B–N interaction to persist (as is the case here), matters become more complicated. In this instance, several variants could be produced, including: meso/meso-, (±)-rac/meso-, and (±)-rac/rac-pairings; these geometries all permit access to 14-membered {Ni}2 dimers. In the current instance, single-crystal X-ray diffraction data obtained on “[Ni(dtbbpe)(η2-NCCH3)]”, provides evidence for meso/meso-[Ni(dtbbpe)(η2-NCCH3)]2 (meso/meso-2), corroborating the dimeric form of these products – metrics of 2 will be discussed later.
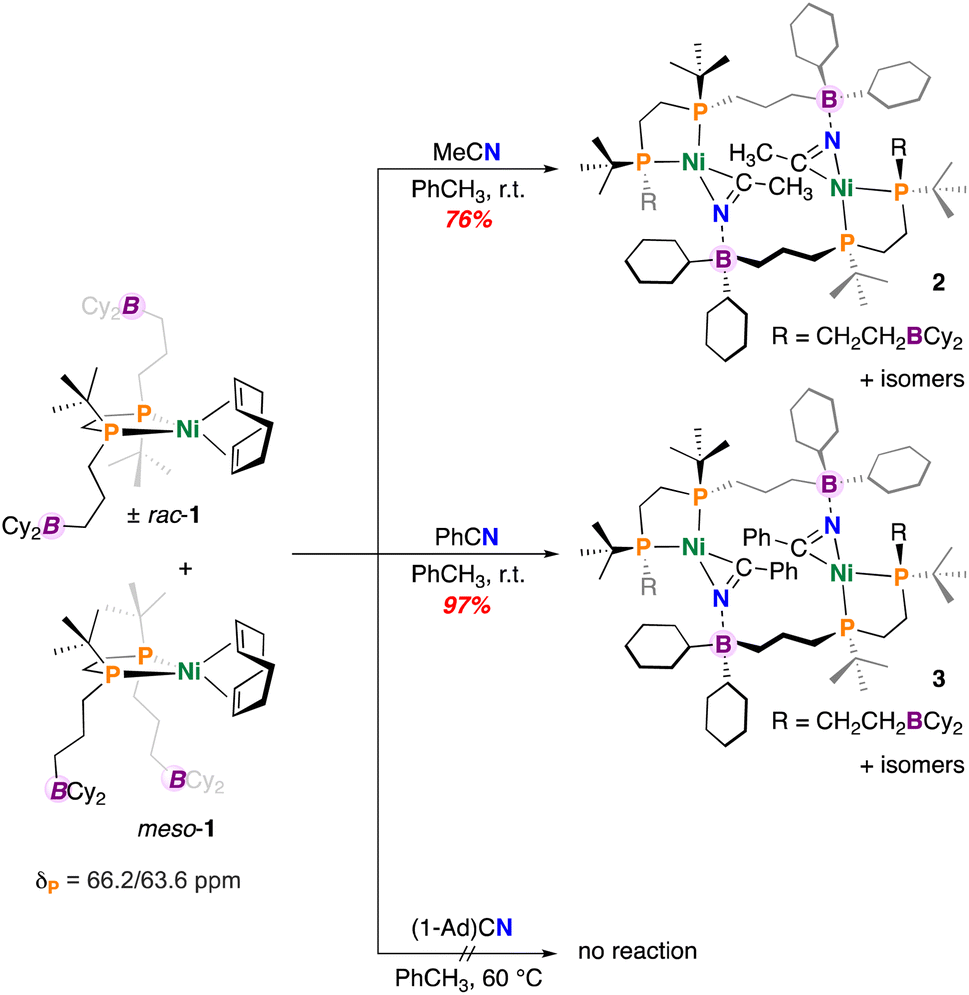 | ||
| Fig. 2 Synthesis of Ni nitrile complexes. | ||
Under similar conditions, benzonitrile (PhCN) was added to a toluene solution of 1 and allowed to stir overnight. Following work-up, the PhCN-coordinated complex [Ni(dtbbpe)(η2-NCPh)]2 (3) was isolated as a yellow-brown oil in 97% yield (Fig. 2). Analysis by 31P{1H} NMR spectroscopy provided several signatures, consistent with the stereogenic dtbbpe ligand scaffold, as introduced earlier. The major isomer features two doublets at δP = 68.2 and 67.0 ppm (2JP,P = 46.2 Hz). An additional broad signal at δP = 69.8 ppm corresponds to an additional minor isomeric component alongside a doublet at 66.7 ppm (2JP,P = 48.7 Hz). Like 2, compound 3 featured a signal at ν(CN) = 1771 cm−1 (c.f., ν(CN) = 2228 cm−1 for unbound PhCN).
To investigate the effect of nitrile steric bulk on dimer formation, complex 1 was reacted with adamantyl nitrile (1-Ad)CN (Fig. 2). However, no reaction was concluded between 1 and (1-Ad)CN by 31P{1H} NMR spectroscopy. Additional heating of the reaction mixture at 60 °C over 48 h likewise provided no evidence for (1-Ad)CN coordination or even decay of 1, suggesting that the 1-Ad group inhibits nitrile binding, which is contrasted with the facile coordination observed for CH3CN and PhCN.
Yellow crystals of complex 2, suitable for X-ray crystallography, were grown from a saturated toluene solution layered with hexane at −35 °C overnight (Fig. 3). For the ttbbpe system discussed previously, structural data was only available for the PhCN adduct – this represents the first crystallographically characterized CH3CN derivative. Complex 2 features two intermolecular B–N interactions resulting in a 14-membered {Ni}2 ring system with a Ni⋯Ni separation of 6.484(8) Å. The average C–N bond length is 1.236(2) Å (c.f., ∼1.13 Å for a free nitrile).21 Two hybridizations, sp2 and sp3, are assigned to the borane moieties, one for the non-interacting (ΣC–B–C = 360°) and one for the interacting (ΣC–B–C = 339°), respectively. This notion is also reflected in the 11B{1H} NMR spectrum, which features two resonances at δB = 83.7 (non-interacting) and 1.42 ppm (interacting); the B–N bond length for the interacting borane was observed to be 1.629(2) Å.
 | ||
| Fig. 3 (A) Molecular structure of 2; (B) top view; (C) side view. Thermal ellipsoids are shown at the 50% probability level. Hydrogen atoms have been omitted for clarity. Selected bond distances (Å): N1–C1 1.236(2), N2–C2 1.236(2), Ni1–Ni2 6.484(8), B1–N2 1.629(2), B3–N1 1.629(2), Ni1–N1 1.907(14), Ni2–N2 1.907(14), Ni1–C1 1.856(17), Ni2–C2 1.856(17). | ||
Reactivity with 1-adamantylphosphaethyne
The controlled activation and transformation of phosphaalkynes, congeners of nitriles, but more closely resembling alkynes, is of interest for the synthesis of organophosphorus compounds.20 Despite this, isolated examples of η2-bound R–CP units are far less common than their nitrile counterparts.22–29 To investigate the possibility of inter/intramolecular P⋯B interactions and inspired by some elegant [(diphosphine)Ni(η2-PCR)] chemistry from Jones and et al.,28 we accordingly sought to examine the reactivity of 1 with a commercially available phosphaalkyne, 1-adamantylphosphaethyne ((1-Ad)CP), a heavier analogue of (1-Ad)CN, described above. However, unlike in the above case of (1-Ad)CN, reaction of 1 with (1-Ad)CP in toluene resulted in an immediate colour change from light yellow to deep brown. Following work-up, complex (±)-rac/meso-[Ni(dtbbpe)(η2-PC(1-Ad))] (4) was isolated as a brown oily solid in 46% yield (Fig. 4). Analysis by 31P{1H} NMR spectroscopy corroborated loss of signals corresponding to 1 and the appearance of six additional resonances with doublet of doublet multiplicity, attributable to a mixture of meso- and (±)-rac-isomers. For the coordinated phosphaalkyne, resonances were observed at δP = 133.3 and 135.0 ppm for the minor and major isomers, respectively in a roughly 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2 ratio, significantly downfield-shifted when compared to free (1-Ad)CP (δP = −67.9 ppm). These values are consistent with previously reported η2-coordinated phosphaalkyne complexes.26,29 For the diphosphine ligand scaffold, signals at δP = 76.8 ppm (major/minor overlapping), 69.4 (minor), and 68.8 (major) ppm are consistent with two compounds. The presence of two (1-Ad)C
:2 ratio, significantly downfield-shifted when compared to free (1-Ad)CP (δP = −67.9 ppm). These values are consistent with previously reported η2-coordinated phosphaalkyne complexes.26,29 For the diphosphine ligand scaffold, signals at δP = 76.8 ppm (major/minor overlapping), 69.4 (minor), and 68.8 (major) ppm are consistent with two compounds. The presence of two (1-Ad)C![[P with combining low line]](https://www.rsc.org/images/entities/char_0050_0332.gif) resonances clearly suggests the formation of a monomeric species, rather than a dimer, which would produce a more convoluted spectrum. Moreover, the lack of an upfield shifted boron resonance (δB = 83.7 ppm only), suggests the absence of a persistent B⋯P interaction, which might be due to the lower electronegativity of P as compared to N.30 Apparently, the difference in reactivity between 1 and (1-Ad)CN/(1-Ad)CP is not attributable to sterics. With complexes 2, 3, and 4 in-hand, we next turned to density functional theory (DFT) to understand the thermodynamics associated with borane co-coordination in such [(diphosphine)Ni(η2-ECR)]n (E = N, P) compounds.
resonances clearly suggests the formation of a monomeric species, rather than a dimer, which would produce a more convoluted spectrum. Moreover, the lack of an upfield shifted boron resonance (δB = 83.7 ppm only), suggests the absence of a persistent B⋯P interaction, which might be due to the lower electronegativity of P as compared to N.30 Apparently, the difference in reactivity between 1 and (1-Ad)CN/(1-Ad)CP is not attributable to sterics. With complexes 2, 3, and 4 in-hand, we next turned to density functional theory (DFT) to understand the thermodynamics associated with borane co-coordination in such [(diphosphine)Ni(η2-ECR)]n (E = N, P) compounds.
 | ||
| Fig. 4 Synthesis of phosphaalkyne adduct, 4. | ||
Theoretical considerations
DFT analysis utilizing the BP86-D3(BJ)/def2-TZVP level of theory provided optimized structures for nickel complexes having bound CH3CN and CH3CP, as models (Fig. 5 and 6). Examination of the non-interacting geometries 2_none and 4_none provided stark differences in η2-bonding (Fig. 5). While in both cases, Ni–C bond lengths are similar (1.90 vs. 1.92 Å), the Ni–P bond length is 0.25 Å longer than that the corresponding Ni–N bond length (1.97 vs. 2.22 Å). The pnictogen–carbon multiple bond (ECR) lengths are also elongated due to Ni(dπ)-to-ECR(π*) back-bonding, giving values of 1.23 vs. 1.65 Å for E = N and P, respectively.
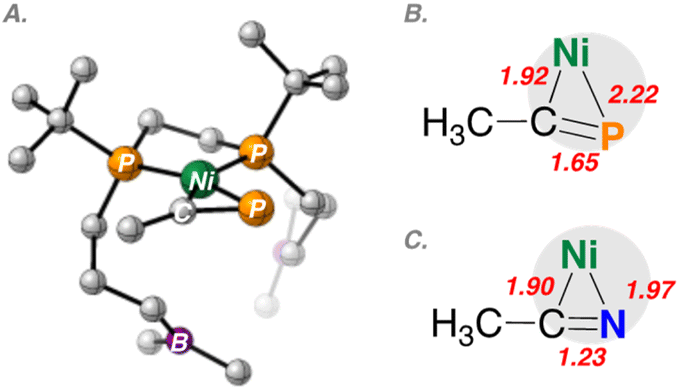 | ||
| Fig. 5 DFT-optimized structures for CH3CN/P-coordinated Ni compounds 2_none and 4_none showing differences in geometric parameters of bound substrate. Bond distances (Å) and angles (deg). (A) [Ni(dtbbpe)(η2-PCCH3)]. (B) Core of [Ni(dtbbpe)(η2-PCCH3)]: <C–Ni–P 46.2, <Ni–P–C 76.7, <Ni–C–P 57.1. (C) Core of [Ni(dtbbpe)(η2-NCCH3)]: <C–Ni–N 37.0, <Ni–N–C 68.4, <Ni–C–N 74.5. | ||
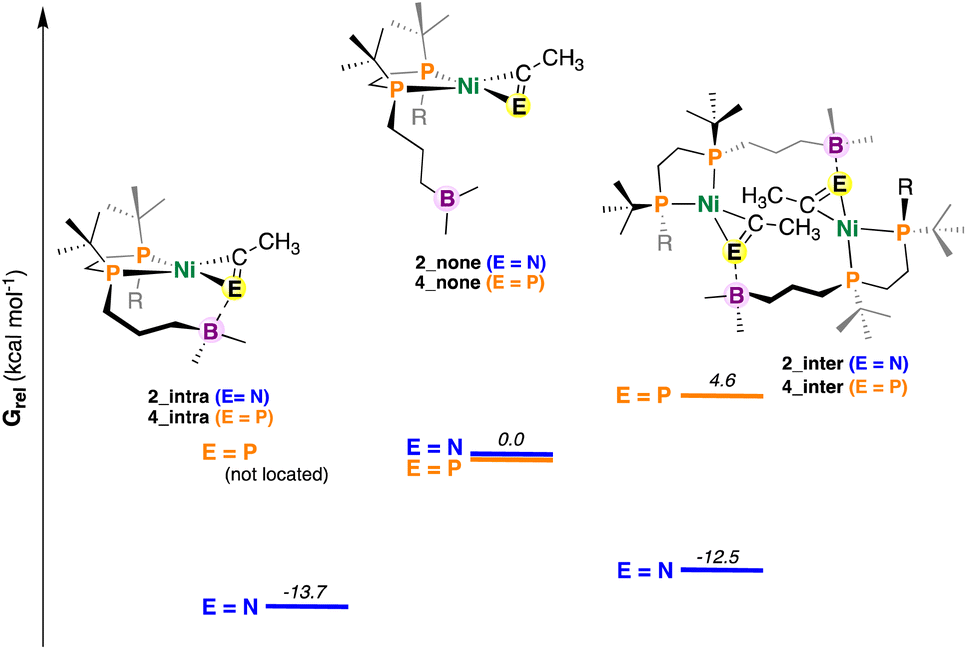 | ||
| Fig. 6 DFT-optimized structures of 2 and 4 with differing ECR (E = N or P) coordination modes. Values in kcal mol−1. Energies have been calculated from DFT-optimized structures using DLPNO-CCSD(T) (see ESI†). Note that for simplicity E = N and P have been placed on the same graph – these energies cannot be directly compared. | ||
For the nitrile system, it was determined that intramolecular stabilization of the coordinated nitrile (2_intra) was favoured by 13.7 kcal mol−1 (Fig. 6). Dimer formation via intermolecular B–N coordination to give 2_inter was also found to be favorable (−12.5 kcal mol−1); the difference between them (ΔGrel = 1.2 kcal mol−1) being close to the margin of calculation error. By contrast, an intramolecular variant of 4 (e.g., 4_intra) could not be located; a potential energy surface scan that varied the B–P distance from roughly 7.8 to 1.4 Å (see ESI†) provided no energy minimum. A dimeric complex 4_inter was located, albeit this compound was found to be uphill (Grel = +4.6 kcal mol−1), consistent with experimental results.
Catalytic nitrile hydroboration
Catalytic hydroboration, the addition of a B–H unit across an unsaturated bond, is a useful tool for access to reduced products.31–33 Hypothesizing that co-coordination would be deleterious to turnover, we wished to quantize the effect of stabilization on nitrile reduction. Accordingly, 10 mol% [Ni(COD)2], a diphosphine ligand, PhCN, and 2 equivs. of HBPin in C6D6 were reacted over the period of 72 h (Fig. 7); three diphosphine ligands were tested having 0 (dtbpe: 1,2-bis(di-tert-butylphosphino)ethane), 1 (ttbbpe), and 2 (dttbbpe) pendant borane groups. The all-alkyl ligand, dtbpe provided the diborylated amine in 97% yield (TOF = 0.15 h−1), ttbbpe resulted in 80% (TOF = 0.1 h−1), and dtbbpe resulted in 40% (TOF = 0.05 h−1). These TOFs are lower than those found for related nickel-based systems. For example, Shimada et al. reported a nickel(II) bis(acetylacetonato) catalyst for the hydroboration of arylnitriles with catecholborane (HBcat) (catalyst loading: 0.5 mol%, RT, 12 h, >99% yield),34 while Baker et al. reported a [Ni(II)(2,5-Me2-pyrrolyl)(κ3-NNN)] complex for nitrile dihydroboration (catalyst loading: 2.0 mol%, RT, 10–18 h, >99% yield).35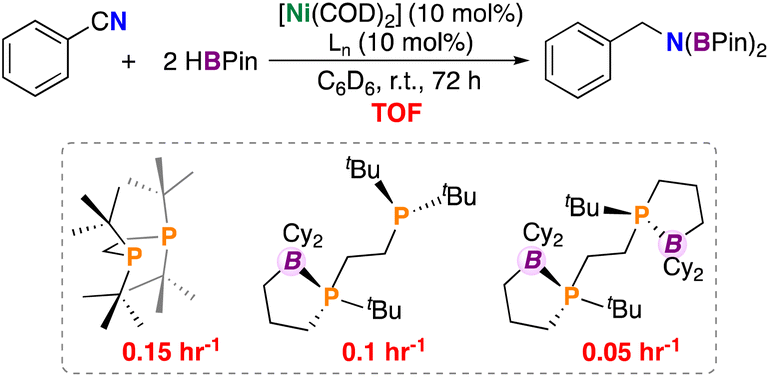 | ||
| Fig. 7 Nitrile dihydroboration using a series of borane-containing ligands. Values in red are turnover frequencies after 72 h (h−1). dtbbpe is used as a mixture of isomers. | ||
Conclusion
This study has explored the reactivity of (±)-rac/meso-[(dtbbpe)Ni(COD)] (1) with two nitriles (PhCN and CH3CN) and a phosphaalkyne ((1-Ad)CP) to understand boron SCS effects on R–CE (E = N, P) coordination and reduction chemistry (for E = N). We found that a Lewis acidic SCS can be used for the co-coordination of nitriles, providing 14-membered ring systems. Moreover, nickel coordination was found to depend on the nature of “E”: compound 1 does not react with (1-Ad)CN, but readily reacts with (1-Ad)CP. For the phosphorus analogue, the SCS borane groups do not engage with the nickel-bound (1-Ad)CP unit, as shown by NMR spectroscopy and as supported by DFT calculations. Finally, this work shows that Ni/B-nitrile co-coordination comes at the cost of catalytic dihydroboration efficiency – speaking to the balance that must be achieved between coordination/stabilization and achieving turnover.
Experimental data
General considerations
The storage and manipulation of all compounds was carried out under an atmosphere of dry nitrogen either in an MBraun glovebox or employing standard Schlenk techniques under an atmosphere of dry nitrogen. Dried solvents were retrieved from a solvent purification system supplied by PPT, LLC. and stored over molecular sieves. Benzene-d6 was dried over molecular sieves and degassed by three freeze–pump–thaw cycles prior to use. HBCy2,36 1,2-bis(di-tert-butylphosphino)ethane (dtbpe),37 di-tert-butylboranyldiphosphinoethane (dtbbpe),17 tri-tert-butylboranyldiphosphinoethane (ttbbpe),19 and compound 1 (ref. 17) were prepared following literature procedures. All other reagents were purchased from commercial vendors and used without further purification unless otherwise stated.Physical methods
NMR spectra were collected on a Bruker Avance III 500 (BBFO probe, TOPSPIN 3.5). 1H NMR spectra are reported in parts per million (ppm) and are referenced to residual solvent e.g., 1H(C6D6): δ 7.16; 13C(C6D6): 128.06; coupling constants are reported in Hz. 13C, 11B, and 31P NMR spectra were performed as proton-decoupled experiments and are reported in ppm. Infrared spectra were collected on a Bruker Alpha II FT-IR spectrophotometer with ATR module.Preparation of compounds
Note: all compounds were isolated as mixtures of isomers. Where appropriate, data is listed in terms of major and minor isomers; ratios were determined by 31P{1H} NMR spectroscopy.[Ni(dtbbpe)(η2-MeCN)]2 [2: C84H162B4N2Ni2P4, Mw = 1484.8 g mol−1]: to a 20 mL scintillation vial equipped with a stir bar was added 1 (0.046 g, 0.057 mmol) and dissolved in 4 mL toluene. To the solution was added MeCN (5 mL). The mixture was stirred for 12 h and toluene removed in vacuo. The resulting residue was dissolved in hexane and filtered through Celite®. Hexane was removed under vacuum and the product redissolved in minimal toluene, layered with hexane, and allowed to crystallize at −35 °C, giving 2 (as the majority meso/meso-isomer) as yellow blocks in a 76% yield (0.032 g, 0.015 mmol). 1H NMR (500 MHz, tol-d8, 298 K, select signals for major isomer only): δH = 2.46 (d, 4JP,H = 4.7 Hz, 6H; C![[H with combining low line]](https://www.rsc.org/images/entities/char_0048_0332.gif) 3CN), 0.99 (d, 3JP,H = 12.7 Hz, 18H; t-Bu), 0.91 (d, 3JP,H = 13.3 Hz, 18H; t-Bu). 13C{1H} NMR (125.8 MHz, tol-d8, 298 K, select signal for major isomer only): δC = 164.5 (dd, 2JP–C = 46.2 Hz, 2JP–C = 6.3 Hz; N
3CN), 0.99 (d, 3JP,H = 12.7 Hz, 18H; t-Bu), 0.91 (d, 3JP,H = 13.3 Hz, 18H; t-Bu). 13C{1H} NMR (125.8 MHz, tol-d8, 298 K, select signal for major isomer only): δC = 164.5 (dd, 2JP–C = 46.2 Hz, 2JP–C = 6.3 Hz; N![[C with combining low line]](https://www.rsc.org/images/entities/char_0043_0332.gif) -CH3). 31P{1H} NMR (202.5 MHz, tol-d8, 298 K): δP = 70.4 (d, 2JP–P = 45.7 Hz), 67.3 (d, 2JP–P = 45.7 Hz). 11B{1H} NMR (160.5 MHz, tol-d8, 298 K): δB = 83.7 (Δ1/2 = 1002 Hz; non-interacting), 1.42 (interacting). FT-IR: ν(CN): 1777 cm−1.
-CH3). 31P{1H} NMR (202.5 MHz, tol-d8, 298 K): δP = 70.4 (d, 2JP–P = 45.7 Hz), 67.3 (d, 2JP–P = 45.7 Hz). 11B{1H} NMR (160.5 MHz, tol-d8, 298 K): δB = 83.7 (Δ1/2 = 1002 Hz; non-interacting), 1.42 (interacting). FT-IR: ν(CN): 1777 cm−1.
[Ni(dtbbpe)(η2-PhCN)]2 (3: C94H166B4N2Ni2P4, Mw = 1608.9 g mol−1): to a 20 mL scintillation vial equipped with a stir bar was added 1 (0.028 g, 0.035 mmol) and dissolved in 4 mL toluene. PhCN (0.004 g, 0.035 mmol) was added to the reaction mixture and was stirred for 12 h before toluene was removed in vacuo. The resulting crude product was dissolved in hexane and filtered through Celite®, giving 3 (as a mixture of isomers) as a brown oil in 97% yield (0.027 g, 0.017 mmol). 1H NMR (500 MHz, tol-d8, 298 K, select signals for major isomer only): δH = 7.52 (d, 3JH–H = 7.6 Hz, 4H; 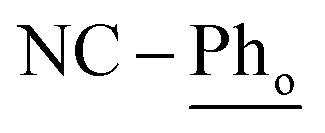 ), 7.21 (t, 3JH–H = 7.6 Hz, 2H;
), 7.21 (t, 3JH–H = 7.6 Hz, 2H; 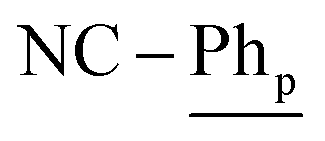 ), 7.15 (t, 3JH–H = 7.6 Hz, 4H;
), 7.15 (t, 3JH–H = 7.6 Hz, 4H; 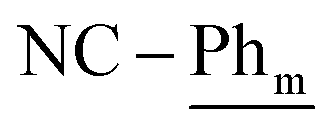 ), 1.05 (d, 3JP,H = 12.4 Hz, 18H; t-Bu), 0.81 (d, 3JP,H = 13.3 Hz, 18H; t-Bu). 13C{1H} NMR (125.8 MHz, tol-d8, 298 K, select signals for major isomer only): δC = 168.8 (dd, 2JP–C = 42.7 Hz, 2JP–C = 7.2 Hz; N-Ph). 31P{1H} NMR (202.5 MHz, tol-d8, 298 K): δP = 69.8 (br; minor isomer), 68.2 (d, 2JP–P = 46.2 Hz; major isomer), 67.0 (d, 2JP-P = 46.2 Hz; major isomer), 66.7 (d, 2JP–P = 48.7 Hz; minor isomer). [Major]:[Minor] = [1.00]:[0.41]. 11B{1H} NMR (160.5 MHz, tol-d8, 298 K): δB = 83.7 (Δ1/2 = 1367 Hz; non-interacting), 2.85 (interacting). FT-IR: ν(CN): 1771 cm−1.
), 1.05 (d, 3JP,H = 12.4 Hz, 18H; t-Bu), 0.81 (d, 3JP,H = 13.3 Hz, 18H; t-Bu). 13C{1H} NMR (125.8 MHz, tol-d8, 298 K, select signals for major isomer only): δC = 168.8 (dd, 2JP–C = 42.7 Hz, 2JP–C = 7.2 Hz; N-Ph). 31P{1H} NMR (202.5 MHz, tol-d8, 298 K): δP = 69.8 (br; minor isomer), 68.2 (d, 2JP–P = 46.2 Hz; major isomer), 67.0 (d, 2JP-P = 46.2 Hz; major isomer), 66.7 (d, 2JP–P = 48.7 Hz; minor isomer). [Major]:[Minor] = [1.00]:[0.41]. 11B{1H} NMR (160.5 MHz, tol-d8, 298 K): δB = 83.7 (Δ1/2 = 1367 Hz; non-interacting), 2.85 (interacting). FT-IR: ν(CN): 1771 cm−1.
(±)-rac/meso-[Ni(dtbbpe)(η2-(1-Ad)CP)] ((±)-rac/meso-4: C51H93B2NiP3, Mw = 879.6 g mol−1): To a 20 mL scintillation vial equipped with a stir bar was added 1 (0.038 g, 0.047 mmol) and (1-Ad)CP (0.084 mg, 0.047 mmol) and dissolved in 4 mL toluene. This mixture was stirred for 12 h and toluene removed in vacuo. The resulting crude product was dissolved in hexane and filtered through Celite®, giving (±)-rac/meso-4 as a brown oil in 46% yield (0.019 g, 0.01 mmol). 1H{31P} NMR (500 MHz, tol-d8, 298 K, select signals only): δH = 1.21 (s, t-Bu, major isomer), 1.16 (s, t-Bu, minor isomer), 1.13 (s, t-Bu, minor isomer), 1.12 (s, t-Bu, major isomer). 13C{1H} NMR (125.8 MHz, tol-d8, 298 K, select signals only): δC = 255.0 (from 1H–13C HMBC; P-Ad), 48.0 (br, 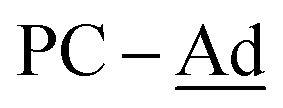 ), 43.6 (br,
), 43.6 (br, 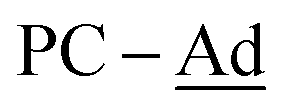 ), 37.6 (br,
), 37.6 (br, 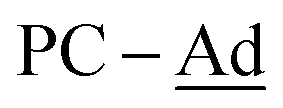 ), 30.3 (br,
), 30.3 (br, 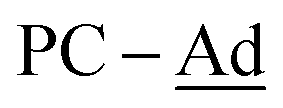 ). 31P{1H} NMR (202.5 MHz, tol-d8, 298 K): δP = 135.1 (br, PC-Ad; minor isomer), 133.5 (br, PC-Ad; major isomer), 77.09 (d, 2JP–P = 34.9 Hz; minor isomer), 76.87 (d, 2JP–P = 33.3 Hz; major isomer), 69.5 (dd, 2JP–P = 34.9 Hz, 2JP–P = 16.0 Hz; minor isomer), 68.9 (dd, 2JP–P = 33.3 Hz, 2JP–P = 13.5 Hz; major isomer). [Major]:[Minor] = [1.00]:[0.47]. 11B{1H} NMR (160.5 MHz, tol-d8, 298 K): δB = 83.7 (Δ1/2 = 1397 Hz).
). 31P{1H} NMR (202.5 MHz, tol-d8, 298 K): δP = 135.1 (br, PC-Ad; minor isomer), 133.5 (br, PC-Ad; major isomer), 77.09 (d, 2JP–P = 34.9 Hz; minor isomer), 76.87 (d, 2JP–P = 33.3 Hz; major isomer), 69.5 (dd, 2JP–P = 34.9 Hz, 2JP–P = 16.0 Hz; minor isomer), 68.9 (dd, 2JP–P = 33.3 Hz, 2JP–P = 13.5 Hz; major isomer). [Major]:[Minor] = [1.00]:[0.47]. 11B{1H} NMR (160.5 MHz, tol-d8, 298 K): δB = 83.7 (Δ1/2 = 1397 Hz).
Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors are grateful to the University of Windsor, the Council of Ontario Universities for a John C. Polanyi award to M. W. D., the Natural Sciences and Engineering Research Council of Canada (Discovery Grant, RGPIN-2020-04480, Discovery Launch Supplement, DGECR-2020-00183, and PGS-D to B. J. H. A.), and donors of the American Chemical Society Petroleum Research Fund (PRF #62284-ND).Notes and references
- M. R. Elsby and R. T. Baker, Chem. Soc. Rev., 2020, 49, 8933–8987 RSC.
- J. R. Khusnutdinova and D. Milstein, Angew. Chem., Int. Ed., 2015, 54, 12236–12273 CrossRef CAS PubMed.
- J. Zhang, G. Leitus, Y. Ben-David and D. Milstein, J. Am. Chem. Soc., 2005, 127, 10840–10841 CrossRef CAS PubMed.
- C. Gunanathan and D. Milstein, Acc. Chem. Res., 2011, 44, 588–602 CrossRef CAS PubMed.
- B. Guo, J. G. de Vries and E. Otten, Chem. Sci., 2019, 10, 10647–10652 RSC.
- Q.-Q. Zhou, Y.-Q. Zou, S. Kar, Y. Diskin-Posner, Y. Ben-David and D. Milstein, ACS Catal., 2021, 11, 10239–10245 CrossRef CAS PubMed.
- A. Nerush, M. Vogt, U. Gellrich, G. Leitus, Y. Ben-David and D. Milstein, J. Am. Chem. Soc., 2016, 138, 6985–6997 CrossRef CAS PubMed.
- L. E. Eijsink, S. C. P. Perdriau, J. G. de Vries and E. Otten, Dalton Trans., 2016, 45, 16033–16039 RSC.
- M. Vogt, A. Nerush, M. A. Iron, G. Leitus, Y. Diskin-Posner, L. J. W. Shimon, Y. Ben-David and D. Milstein, J. Am. Chem. Soc., 2013, 135, 17004–17018 CrossRef CAS PubMed.
- S. Perdriau, D. S. Zijlstra, H. J. Heeres, J. G. de Vries and E. Otten, Angew. Chem., Int. Ed., 2015, 54, 4236–4240 CrossRef CAS PubMed.
- S. de Graaff, K. Schwitalla, H. Thye, Z. Yusufzadeh, M. Willms, M. Schmidtmann and R. Beckhaus, Chem. - Eur. J., 2023, e202203846 CrossRef CAS PubMed.
- S. Das, J. Maity and T. K. Panda, Chem. Rec., 2022, 22, e202200192 CAS.
- Z. Lu and T. J. Williams, Chem. Commun., 2014, 50, 5391–5393 RSC.
- M. W. Drover, Chem. Soc. Rev., 2022, 51, 1861–1880 RSC.
- J. A. Zurakowski, B. J. H. Austen and M. W. Drover, Trends Chem., 2022, 4, 331–346 CrossRef CAS.
- J. A. Zurakowski, M. Bhattacharyya, D. M. Spasyuk and M. W. Drover, Inorg. Chem., 2021, 60, 37–41 CrossRef CAS PubMed.
- B. J. H. Austen, H. Sharma, J. A. Zurakowski and M. W. Drover, Organometallics, 2022, 41, 2709–2715 CrossRef CAS.
- J. A. Zurakowski, B. J. H. Austen, K. R. Brown and M. W. Drover, Chem. Commun., 2022, 58, 2500–2503 RSC.
- M. L. Clapson, H. Sharma, J. A. Zurakowski and M. W. Drover, Chem. - Eur. J., 2023, 29, e202203763 CrossRef CAS PubMed.
- A. Chirila, R. Wolf, J. Chris Slootweg and K. Lammertsma, Coord. Chem. Rev., 2014, 270–271, 57–74 CrossRef CAS.
- R. J. Boyd and S. C. Choi, Chem. Phys. Lett., 1985, 120, 80–85 CrossRef CAS.
- M. F. Meidine, M. A. N. D. A. Lemos, A. J. L. Pombeiro, J. F. Nixon and P. B. Hitchcock, J. Chem. Soc., Dalton Trans., 1998, 3319–3324 RSC.
- R. B. Bedford, A. F. Hill, J. D. E. T. Wilton-Ely, M. D. Francis and C. Jones, Inorg. Chem., 1997, 36, 5142–5144 CrossRef CAS.
- T. Schaub and U. Radius, Z. Anorg. Allg. Chem., 2006, 632, 981–984 CrossRef CAS.
- A. D. Burrows, A. Dransfeld, M. Green, J. C. Jeffery, C. Jones, J. M. Lynam and M. T. Nguyen, Angew. Chem., 2001, 113, 3321–3324 CrossRef.
- P. Binger, B. Biedenbach, A. T. Herrmann, F. Langhauser, P. Betz, R. Goddard and C. Krüger, Chem. Ber., 1990, 123, 1617–1623 CrossRef CAS.
- J. C. T. R. B.-S. Laurent, P. B. Hitchcock, H. W. Kroto and J. F. Nixon, J. Chem. Soc., Chem. Commun., 1981, 1141–1143 RSC.
- T. Görlich, D. S. Frost, N. Boback, N. T. Coles, B. Dittrich, P. Müller, W. D. Jones and C. Müller, J. Am. Chem. Soc., 2021, 143, 19365–19373 CrossRef PubMed.
- M. Trincado, A. J. Rosenthal, M. Vogt and H. Grützmacher, Eur. J. Inorg. Chem., 2014, 10, 1599–1604 CrossRef.
- C. Tantardini and A. R. Oganov, Nat. Commun., 2021, 12, 2087 CrossRef CAS PubMed.
- A. Rezaei Bazkiaei, M. Findlater and A. E. V. Gorden, Org. Biomol. Chem., 2022, 20, 3675–3702 RSC.
- J. B. Geri and N. K. Szymczak, J. Am. Chem. Soc., 2015, 137, 12808–12814 CrossRef CAS PubMed.
- A. Kaithal, B. Chatterjee and C. Gunanathan, J. Org. Chem., 2016, 81, 11153–11161 CrossRef CAS PubMed.
- G. Nakamura, Y. Nakajima, K. Matsumoto, V. Srinivas and S. Shimada, Catal. Sci. Technol., 2017, 7, 3196–3199 RSC.
- S. Ataie and R. T. Baker, Inorg. Chem., 2022, 61, 19998–20007 CrossRef CAS PubMed.
- A. Abiko, Org. Synth., 2003, 79, 103 Search PubMed.
- A. A. Del Paggio, R. A. Andersen and E. L. Muetterties, Organometallics, 1987, 6, 1260–1267 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Invited contribution for the Emerging Investigators 2023 Special Issue 1H, 13C{1H}, 31P{1H}, and 11B NMR spectra for all complexes. XYZ coordinates for DFT calculations. CCDC 2251021. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d3ra02797j |
| This journal is © The Royal Society of Chemistry 2023 |