DOI:
10.1039/C6RA15140J
(Paper)
RSC Adv., 2016,
6, 75651-75663
Discovery of a new class of 16-membered (2Z,11Z)-3,11-di(aryl/naphthyl)-1,13-dioxa-5,9-dithia-2,12-diazacyclohexadeca-2,11-dienes as anti-tumor agents†
Received
10th June 2016
, Accepted 3rd August 2016
First published on 3rd August 2016
Abstract
A series of new 16-membered small macrocyclic compounds, (2Z,11Z)-3,11-di(aryl/naphthyl)-1,13-dioxa-5,9-dithia-2,12-diazacyclohexadeca-2,11-dienes (1a–k) were designed and developed by a simple and practical synthetic route from readily available substrates using simple organic transformations. Evaluation of in vitro anti-tumor activities on human triple negative breast cancer cells MDAMB-231 cell lines reveal that the macrocycles, 1a, 1f, 1g, 1i and 1k are promising anti-tumor compounds as evidenced from inhibition of cell migration and proliferation, upregulation of anti-tumor genes p53, MDA7 and TRAIL. The anti-proliferative effect of macrocycles is specific to cancer cells but no cytotoxic effect on normal breast epithelial cells has been observed (MCF10A). The developed synthetic route is free from metals, protecting groups and air-free techniques. The structure of macrocycle (1e) is confirmed by single crystal XRD studies.
Introduction
Design, synthesis and development of new anti-tumor agents has always been a challenging task for the scientific community as cancer is a major public health problem in many parts of the world1–4 particularly in developing countries increasingly, adopting a cancer-associated lifestyle options including smoking, westernized diets and lack of adequate exercise.5 Globally every 8th death is caused by cancer.6 Many of the available anti-tumor drugs have adverse effects on human health, therefore, there is a great need to develop new class of drugs for the treatment of cancer that are cost effective and chemically stable with low toxicity to normal cells.
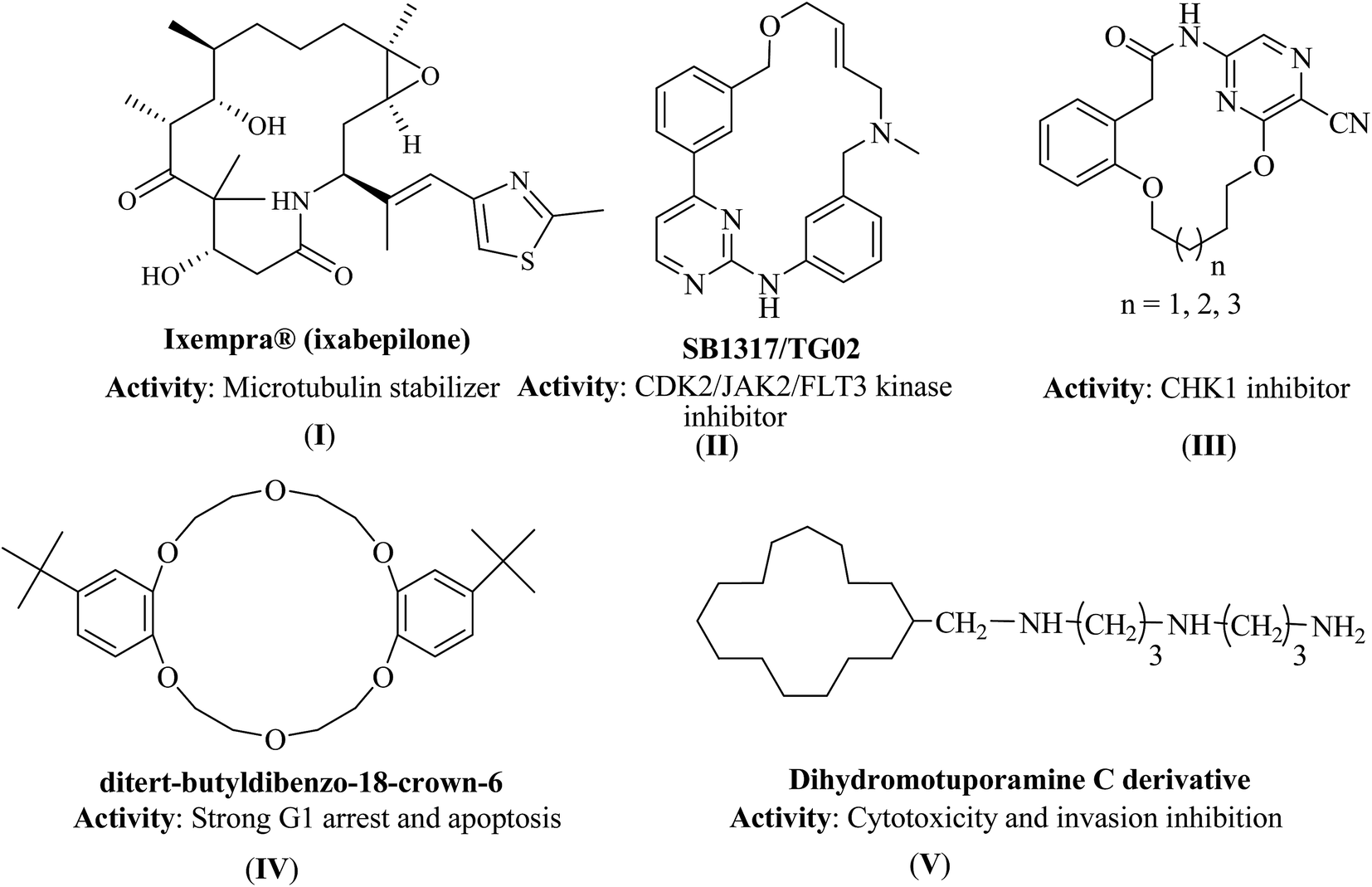
Macrocyclic molecules played a key role in drug discovery as evidenced from natural products.7 Among the macrocycles, the hetero-atoms incorporated macrocyclic molecules7 might be the best option as they possess unusual characteristics such as (i) structural complexity, regiogenicity, stereogenicity and rigidity, (ii) potential hydrogen bonding to interact with biomolecules such as RNA, DNA, proteins (enzymes),8,9 (iii) high degree of structural pre-organization which leads to the interaction of the key functional groups across extended protein's active site and (iv) ‘drug-like’ physico-chemical as well as pharmacokinetic properties.10,11 For instance, small macrocyclic molecules derived from natural products (I)12 and many synthetic macrocycles (II–IV)13–15 displayed apoptosis and 15-membered macrocycles (V)16 exhibited the highest levels of cytotoxicity (Fig. 1). Further, macrocyclic molecules exhibited various pharmacological properties which include anti-bacterial, anti-fungal, anti-tumor and anti-HIV activities.17
 |
| | Fig. 1 Representative examples of biologically active macrocyclic molecules. | |
However, disinclination to investigate and develop macrocycles as new drug candidates in the academic and pharmaceutical industry was observed owing to poor yields during macrocyclization, difficulties in analog synthesis and unproductive side reactions during the joining of ends of a linear precursor molecule.18 Later, metal template methods were developed,19 but these methods were also associated with few disadvantages such as difficulties in choosing the correct template metal ion and its removal.20 Other methods were non-template, but required high dilution conditions21 which lead to huge organic waste generation along with low yields of desired product and also these methods require lengthy separations to remove oligomeric by-products. Later, ring closing metathesis,22 Glaser–Eglinton–Hay-type sp–sp coupling23 and head to tail macrocyclizations (build/couple/pair strategies)24 methods have emerged. But most of these methods suffer from one or more disadvantages as discussed above. Keeping in view of various pharmacological and biomedical applications and difficulties in the synthesis of macrocycles, we aimed to design and synthesize a new class of non-peptidic 16-membered macrocycles i.e. (2Z,11Z)-3,11-di(aryl/naphthyl)-1,13-dioxa-5,9-dithia-2,12-diazacyclohexadeca-2,11-dienes (1) from readily available substrates using simple organic transformations. The following factors formed the basis for the present design and synthesis: (i) high probability of cyclic oxime-ethers in the synthesis of biologically active synthetic molecules25 and (ii) accessibility of diverse topologies26 by the presence of sulfur atoms which were amiable to disrupt protein–protein interactions (PPIs).27
Retro-synthetic analysis
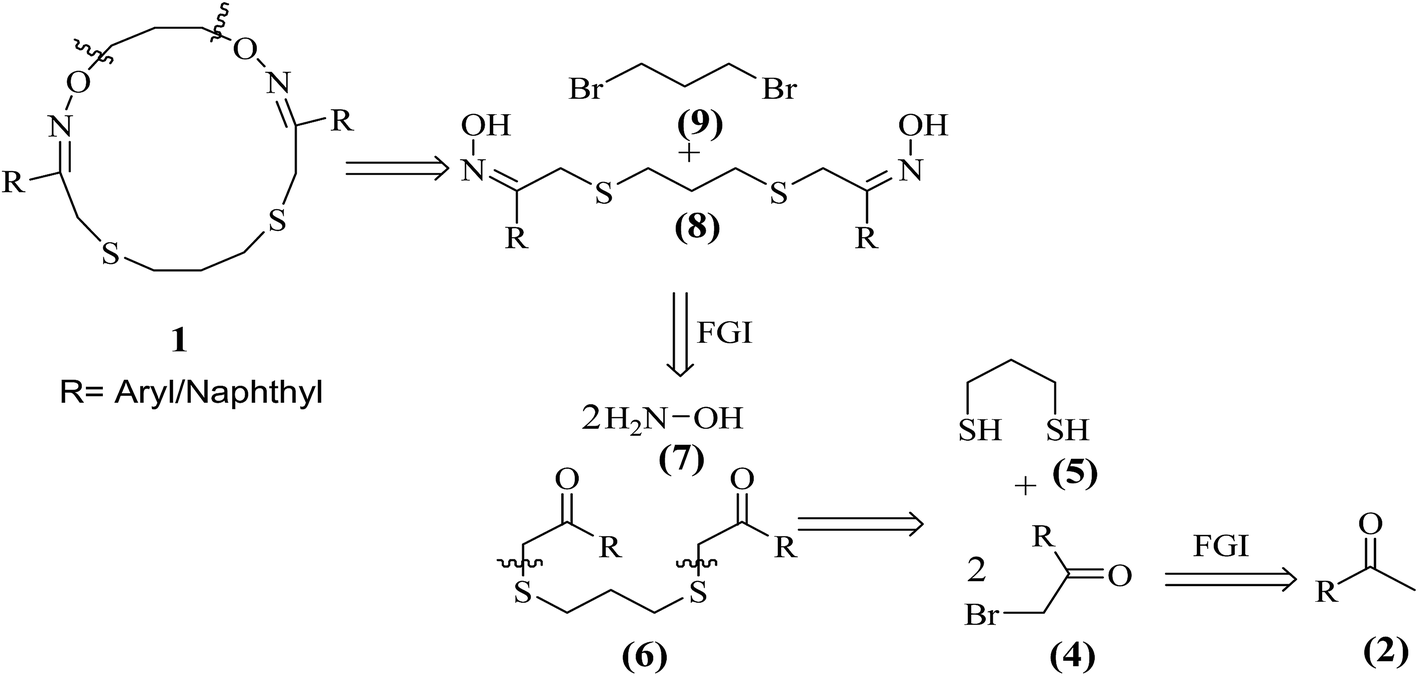
The retrosynthetic analysis of target 16-membered (2Z,11Z)-3,11-di(aryl/naphthyl)-1,13-dioxa-5,9-dithia-2,12-diazacyclohexadeca-2,11-dienes (1) is outlined in Scheme 1.
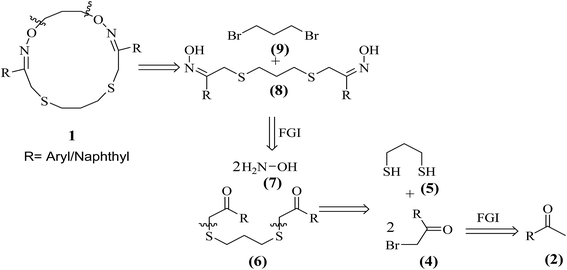 |
| | Scheme 1 Retrosynthetic analysis for the synthesis of (2Z,11Z)-3,11-di(aryl/naphthyl)-1,13-dioxa-5,9-dithia-2,12-diazacyclohexadeca-2,11-dienes (1). | |
Herein, a simple, efficient and practical synthetic route is developed for the synthesis of structurally diverse non-peptidic 16-membered (2Z,11Z)-3,11-di(aryl/naphthyl)-1,13-dioxa-5,9-dithia-2,12-diazacyclohexadeca-2,11-dienes (1) (1a–k) from readily available substrates such as aralkyl ketones (2a–k), N-bromosuccinimide (3), 1,3-propanedithiol (5), NH2OH·HCl (7) and 1,3-dibromopropane (9) using simple organic transformations involving α-bromination, C–S bond construction, oxime formation followed by macrocyclization (C–O bond formation) as shown in Scheme 2. Further, the synthesized macrocyclic molecules (1a–k) are well characterized by 1H, 13C NMR, HRMS and single crystal XRD studies and evaluation of their anti-tumor activity is duly undertaken.
 |
| | Scheme 2 Synthesis of (2Z,11Z)-3,11-di(aryl/naphthyl)-1,13-dioxa-5,9-dithia-2,12-diaza cyclohexadeca-2,11-dienes (1). Reaction conditions: (i) 10% (w/w) silicagel, NBS (3), MeOH, reflux, 15–20 min; (ii) 1,3-propanedithiol (5), K2CO3, acetonitrile, 15–20 °C, 85–95 min, (iii) NH2OH·HCl (7), NaOAc, reflux, 2 h; (iv) 1,3-dibromopropane (9), KOH in a mixture of H2O–DMSO (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :5 ratio), 5–10 °C, 25–35 min. :5 ratio), 5–10 °C, 25–35 min. | |
Results and discussion
Chemistry
The synthetic route of title compounds i.e. (2Z,11Z)-3,11-di(aryl/naphthyl)-1,13-dioxa-5,9-dithia-2,12-diazacyclohexadeca-2,11-dienes (1) was accomplished via a four-step process as shown in Scheme 2. Accordingly, first 2-bromo-1-(4-chlorophenyl)ethanone (4a) was prepared from 4-chloroacetophenone (2a) by α-bromination using NBS (3) in MeOH in the presence of silica gel under reflux condition in high yield (91%) within a short period of time.28 The same procedure was applied for the preparation of other α-bromoaralkyl ketones (4b–k) from various acetophenones and acetonaphthones (2a–k) and the obtained results were presented in Scheme 3.
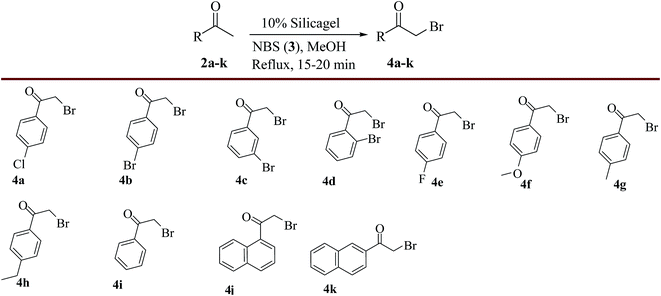 |
| | Scheme 3 Synthesis of α-bromoaralkyl ketones (4). Reagents and conditions: aralkyl ketones, 2a–k (10.0 mmol), NBS (12.0 mmol) (3), 10% (w/w) silica gel, MeOH, reflux, 15–20 min; isolated yields: 4a (91%), 4b (87%), 4c (81%), 4d (74%), 4e (94%), 4f (87%), 4g (92%), 4h (88%), 4i (95%), 4j (86%), 4k (91%). | |
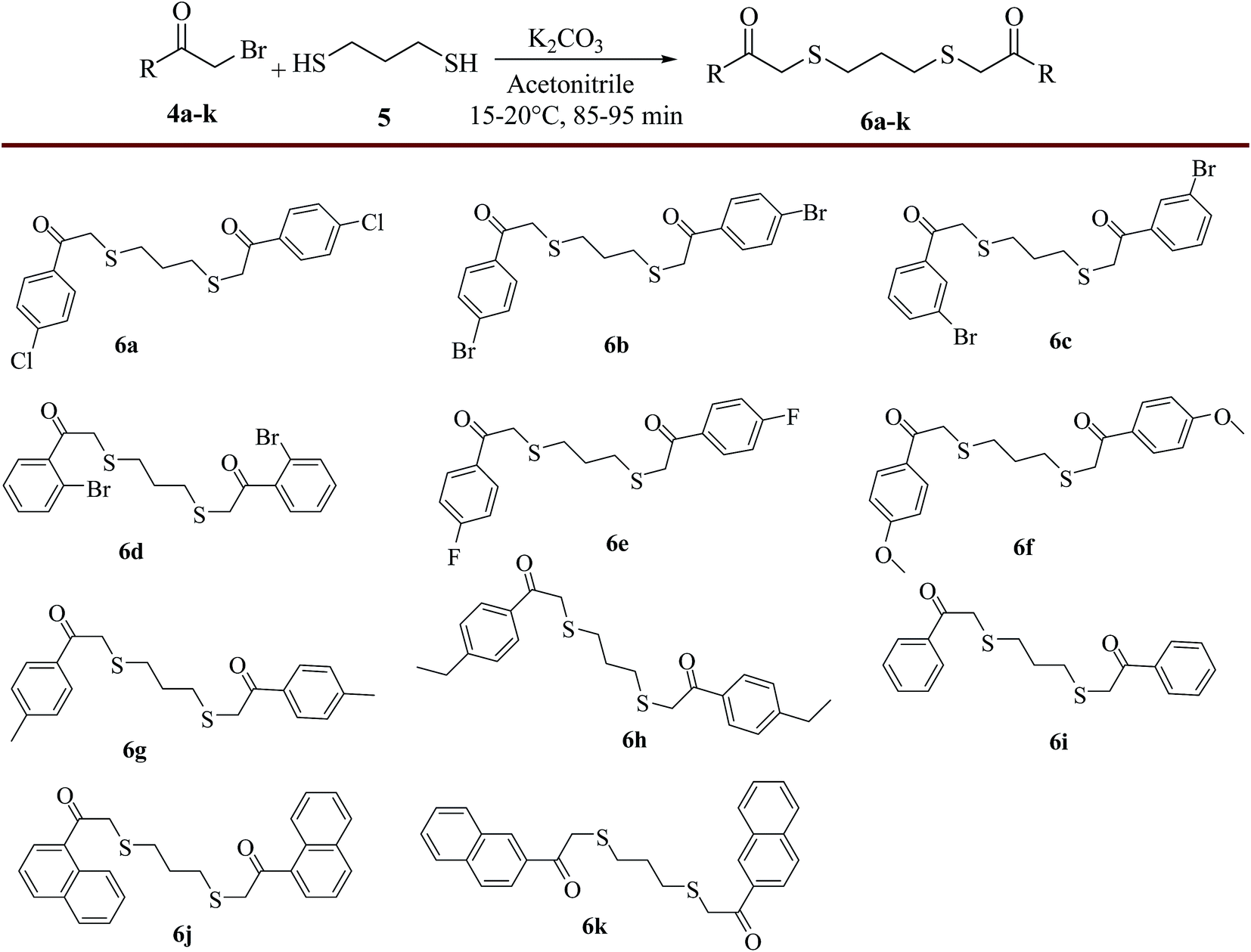
Later, the reaction conditions for the synthesis of 2,2′-(propane-1,3-diylbis(sulfanediyl))bis(1-(4-chlorophenyl)ethanone) (6a) were optimized. Towards this direction, firstly, 1,3-propanedithiol was added drop wise to the α-bromo compound, 2-bromo-1-(4-chlorophenyl)ethanone (4a) in presence of Na2CO3 (3.0 equiv.) in ethanol at 20–25 °C and obtained 45% yield of the product (6a). To improve the yield further, the effect of various solvents such as dichloromethane, methanol, acetonitrile, THF, DMF and DMSO was investigated and 42%, 54%, 65%, 49%, 58% and 53% yields of product (6a) were obtained respectively. Preliminary screening of solvent study revealed that acetonitrile was the best option for maximum conversion of product (6a). To improve the yield of the product (6a) further, various bases such as K2CO3 (3.0 equiv.), Et3N (3.0 equiv.) and pyridine (3.0 equiv.) have been employed in acetonitrile which resulted in 85%, 65% and 55% yields of the product (6a) respectively. Finally, the effect of temperature on the course of C–S bond formation was also studied at various temperatures ranging from 5 to 25 °C. The study indicated that 15–20 °C was the optimum temperature for maximum yield of product 6a (98%). With the help of optimized reaction conditions, the substrate scope was tested for the synthesis of various 2,2′-(propane-1,3-diylbis(sulfanediyl))-bis(1-(aryl/naphthyl)ethanone) derivatives (6b–k) using α-bromoketones (4b–k) as substrates and the obtained results were presented in Scheme 4.
 |
| | Scheme 4 Synthesis of various 2,2′-(propane-1,3-diylbis(sulfanediyl))bis(1-aryl/naphthyl ethanone) derivatives (6). Reagents and conditions: α-bromoaralkyl ketones, 4a–k (10.0 mmol), 1,3-propanedithiol (5.0 mmol) (5), K2CO3, acetonitrile, 15–20 °C, 85–95 min; isolated yields: 6a (98%), 6b (96%), 6c (93%), 6d (86%), 6e (97%), 6f (95%), 6g (96%), 6h (95%), 6i (95%), 6j (94%), 6k (98%). | |
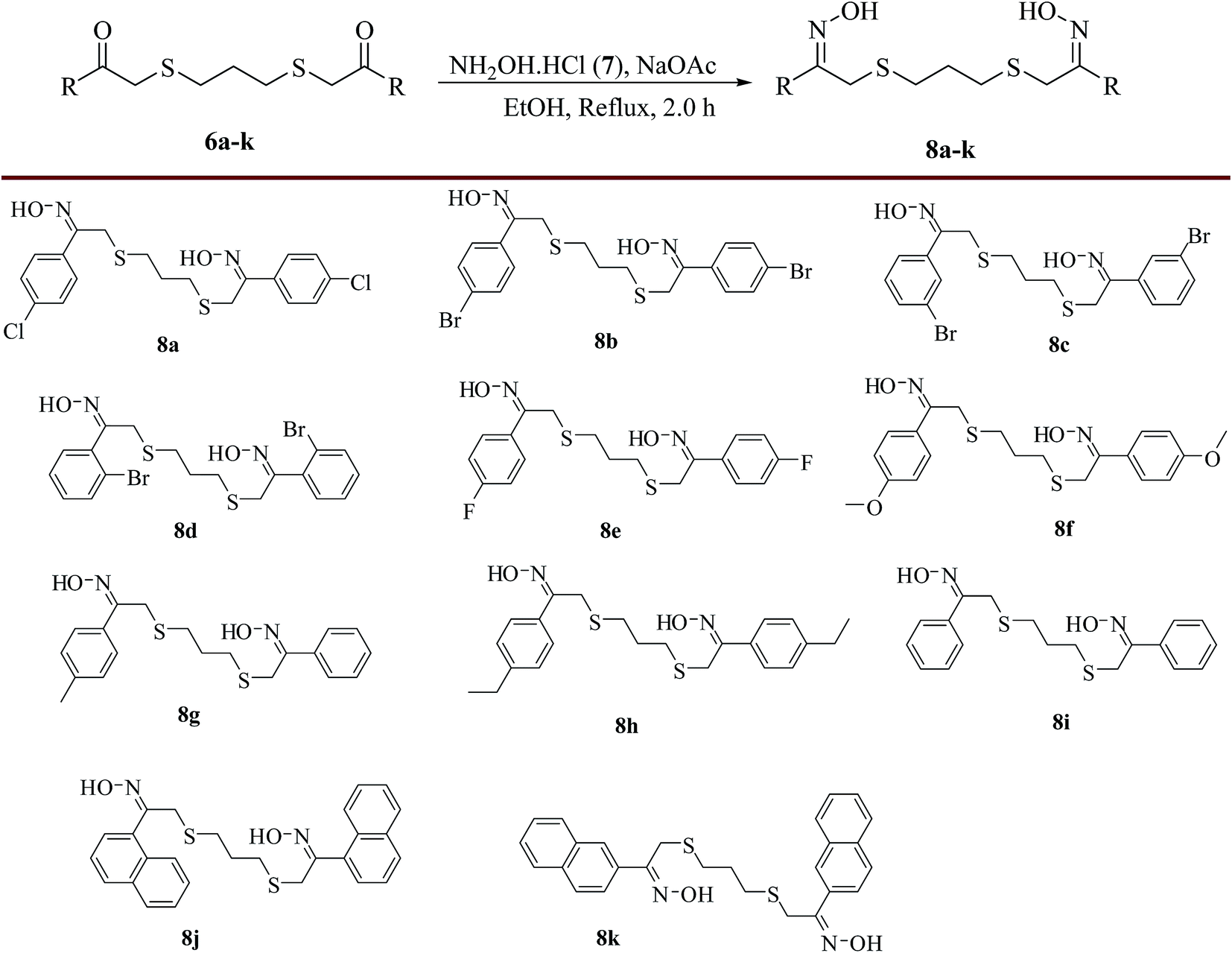
The third step was the oxime formation step which involves, the reaction between compound (6a) and NH2OH·HCl (7) in the presence of NaOAc in ethanol under reflux conditions29 to produce the precursor of the pharmacophore, (1Z,1′Z)-1-(4-chlorophenyl)-2-(3-((Z)-2-(4-chlorophenyl)-2-(hydroxyimino)-ethylthio)propylthio)ethanone oxime (8a) in excellent yield (98%). The same procedure was followed for the preparation of (1Z,1′Z)-1-(aryl/naphthyl)-2-(3-((Z)-2-(aryl/naphthyl)-2-(hydroxyimino)-ethylthio)propylthio)ethanone oximes (8b–k) using various substrates, 6b–k and the obtained results were presented in Scheme 5.
 |
| | Scheme 5 Preparation of various (1Z,1′Z)-1-(aryl/naphthyl)-2-(3-((Z)-2-(aryl/naphthyl)-2-(hydroxyimino)ethylthio)propylthio)ethanone oxime (8). Reagents and conditions: substrates, 6a–k (10.0 mmol), NH2OH·HCl (40.0 mmol) (7), NaOAc (30.0 mmol w. r. t. NH2OH·HCl), ethanol, reflux, 2 h; isolated yields: 8a (98%), 8b (97%), 8c (95%), 8d (88%), 8e (98%), 8f (97%), 8g (98%), 8h (96%), 8i (97%), 8j (96%), 8k (98%). | |
Then, the attention was focused on macrocyclization step to synthesize the title compound, (2Z,11Z)-3,11-bis(4-chlorophenyl)-1,13-dioxa-5,9-dithia-2,12-diazacyclohexadeca-2,11-diene (1a) by the reaction of pharmacophore, (1Z,1′Z)-1-(4-chlorophenyl)-2-(3-((Z)-2-(4-chlorophenyl)-2-(hydroxyimino)-ethylthio)propylthio)ethanoneoxime (8a) with linker i.e. 1,3-dibromopropane (9). Towards this direction, initially, a reaction was carried out by using the substrate, 8a and 1,3-dibromopropane (9) in MeOH in the presence of NaOH at 20–25 °C and lower yield (15%) of product, 1a was obtained. To improve the yield, the reaction conditions for the preparation of target 16-membered macrocycle 1a have been optimized using different solvents and bases. Initially, the effect of solvent or mixtures of solvents was studied for their impact on the course of macrocyclization. It was found that the solvents like MeOH, EtOH, acetonitrile, DMSO and DMF were provided 15%, 20%, 10%, 50% and 35% yields, respectively. From this study, it was found that DMSO provided considerable yield (50%) of the product 1a. For the improvement of the yield of the product 1a further, the effect of mixture of solvents was investigated. Towards this direction, a mixture of H2O and DMSO was selected and the study revealed that 1:5 ratio of H2O and DMSO provided good yield (75%) of product 1a. Further, the effect of temperature was also studied and found that 5–10 °C was the optimum temperature for maximum yield of product 6a (86%). The effect of various bases such as Na2CO3, K2CO3, NaOH, KOH, Et3N and pyridine on the course of macrocyclization was studied. From this study, it was concluded that KOH was the best in obtaining maximum isolated yield (94%) of the desired product 1a in presence of 1:5 ratio of H2O and DMSO at 5–10 °C. Encouraged by the productive results the same procedure was applied for the synthesis of a series of non-peptidic 16-membered (2Z,11Z)-3,11-di(aryl/dinaphthyl)-1,13-dioxa-5,9-dithia-2,12-diazacyclohexadeca-2,11-diene derivatives (1b–k) from various oximes (8b–k) and 1,3-dibromopropane (9) and the yields obtained were shown in Scheme 6.
 |
| | Scheme 6 Synthesis of (2Z,11Z)-3,11-di(aryl/dinaphthyl)-1,13-dioxa-5,9-dithia-2,12-diazacyclohexadeca-2,11-diene derivatives (1). Reagents and conditions: substrates, 8a–k (10.0 mmol), 1,3-dibromopropane (10.0 mmol) (9), KOH (20.0 mmol) in a mixture of H2O–DMSO (1:5 ratio), 5–10 °C, 25–35 min; isolated yields: 1a (94%), 1b (94%), 1c (91%), 1d (86%), 1e (95%), 1f (92%), 1g (94%), 1h (94%), 1i (95%), 1j (89%), 1k (95%); overall yield: (%): 82, 76, 65, 48, 85, 74, 81, 75, 83, 69 and 83. | |
The synthesized compounds were characterized by 1H-NMR, 13C-NMR and HRMS data and the structure of one of the macrocyclic compound, (2Z,11Z)-3,11-bis(4-fluorophenyl)-1,13-dioxa-5,9-dithia-2,12-diazacyclohexadeca-2,11-diene (1e) was confirmed by single crystal X-ray diffraction studies as shown in Fig. 2.
 |
| | Fig. 2 ORTEP view of (2Z,11Z)-3,11-bis(4-fluorophenyl)-1,13-dioxa-5,9-dithia-2,12-diazacyclohexadeca-2,11-diene (1e). | |
Anti-tumor activity studies
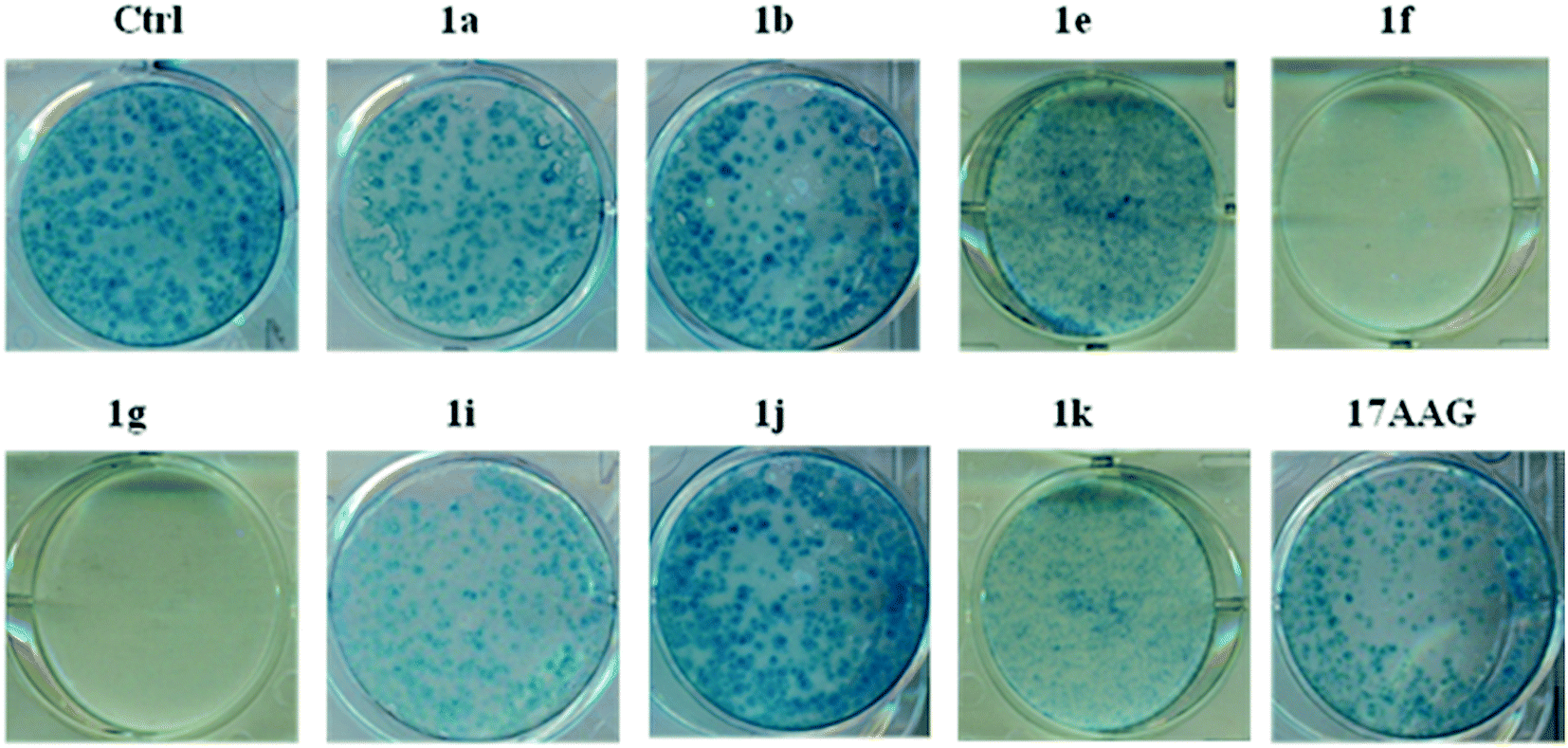
The anti-tumor activity of macrocyclic compounds (1a–k) was evaluated in vitro using triple negative breast cancer cells (TNBC) MDAMB-231. The anti-tumor effects of macrocyclic compounds were evaluated using standard anti-tumor assays, including wound healing assays (Fig. 3) and colonogenic assay (Fig. 4), depicting effect on cancer cells metastatic and cell division potential, respectively. For understanding the underlying molecular mechanism involved in anti-tumor effects of macrocyclic compounds (1a–k), transcription levels of key anti-tumor genes was measured by quantitative (q) RT-PCR (Fig. 5). Fragmentation of genomic DNA representing induction of apoptosis was analyzed by DNA laddering (Fig. 6) in agarose gel electrophoresis. The cytotoxic effects of these compounds (1a–k) were measured on normal breast epithelial cells (MCF10A) (Fig. 7).
 |
| | Fig. 3 Effect of macrocyclic compounds in cell migration: macrocyclic compounds (1a, 1b, 1e, 1f, 1g, 1i, 1j and 1k) differentially inhibit the migration of TNBC MDAMB-231 in the wound. Compounds 1a, 1f, 1g, 1i and 1k possess substantial anti-proliferative properties. | |
 |
| | Fig. 4 Effect of macrocyclic compounds on colonogenic survival of tumor-cells: macrocyclic compounds (1a, 1b, 1e, 1f, 1g, 1i, 1j and 1k) possess differential effect on colonogenic survival and proliferation of MDAMB-231 cells. Compounds 1e, 1f, 1g, and 1k substantially inhibited colonogenic survival of tumor cells. | |
 |
| | Fig. 5 Effect of macrocyclic compounds on expression of anti-tumor genes: macrocyclic compounds 1a, 1i and 1k robustly induced the transcription of p53, MDA7 and TRAIL, while 1f and 1g have moderate effect, however 1b and 1j fail to induce anti-tumor genes. Expression of genes is represented in fold change where as compared solvent only treated control groups and were measured after 24 h treatment with respective compounds. | |
 |
| | Fig. 6 Effect of macrocyclic compounds on integrity of genomic DNA: macrocyclic compounds 1a and 1i induce genomic DNA fragmentation in MDAMB-231 cells as evidenced from observed DNA ladder like impression of genomic DNA in agarose gel. | |
 |
| | Fig. 7 Cytotoxic activity studies on primary human breast MCF10A cells: investigated macrocyclic compounds 1a, 1b, 1e, 1f, 1g, 1i, 1j and 1k have no cytotoxic effect on normal human breast epithelial cells (MCF10A). | |
Macrocycles inhibit migration of cancer cells. Tumor cells possess the property of cell migration and invasion that can be measured by standard wound healing assay measuring effect of anti-tumor drugs on migration cells in cancer cell line ex vivo.30 The inhibitory effect of macrocyclic compounds was evaluated in comparison to known anti-cancer drug 17-N-allylamino-17-demthoxygeldanamycin (17AAG). The macrocyclic compounds 1a, 1f, 1g and 1i strongly reduced migration of TNBC MDAMB-231 and healing of the wound delayed (Fig. 3), compounds 1b, 1e and 1j showed negligible effects on cell migration, while compound 1k had moderate effect on migration of cancer cells (Fig. 3). These results suggested that macrocycles 1a, 1f, 1g, 1i and 1k are anti-tumor in their nature.
Macrocycles hampers proliferation of cancer cells. The cell division potential of cancer cells was measured by colonogenic assay besides measuring the effect of anti-tumor drug on survival and proliferation of cancer cells in ex vivo system. The anti-proliferative effect of macrocyclic compounds (1a, 1b, 1e, 1f, 1g, 1i, 1j and 1k) was evaluated on MDAMB-231 cells. Except 1b and 1j all the tested macrocycles possess varying degree of anti-proliferative potential (Fig. 4), compared with positive controls 17AAG and hydroxyl urea (HU). Macrocycles 1a, 1e, 1f, 1g, 1i and 1k effectively inhibited colonogenic survival of MDAMB-231 cells. Similar results were observed with IMR32 cells (data not shown). In comparison to macrocycles 1e, 1f, 1g and 1k, macrocycles 1b and 1j showed least response on colonogenic survival of cancer cells, while macrocycles 1a and 1i had intermediate effect. These results suggest that macrocycles 1a, 1e, 1f, 1g, 1i and 1k are anti-tumor in their nature and the results are identical to wound healing assay.
Macrocyclic compounds induces transcription of anti-tumor genes. The anti-tumor agents are known to induce array of tumor suppressor genes such as p53, TRAIL, MDA7, STAT-3 etc. These genes exert anti-tumor activity by induction of apoptosis through variety of mechanisms, upregulation of tumor suppressor protein have direct impact on tumor cells survival.31 Transcriptional upregulation of p53, TRAIL and MDA7 was observed for MDAMB-231 cells, treated with macrocyclic compounds 1a, 1i, 1f, 1g and 1k. Compounds 1b and 1j did not show significant induction of anti-tumor genes. Previous studies clearly establish the role of TRAIL mediated induction of caspase dependent apoptotic cell death of tumor cells.32,33 Macrocycles compounds 1a, 1f, 1g, 1i and 1k, upregulate transcription of TRAIL might be leading to activation of caspase dependent apoptotic cell death of MDAMB-231 cells. Macrocycles 1a, 1i and 1k synergistically induce transcripts for TRAIL, MDA7 and p53 (Fig. 5) genes, higher than observed with compounds 1f and 1g. The tumor suppressor gene p53 regulates transcription of TRAIL,34 also regulates MDA7 activities.35 The MDA7 is an anti-cancer gene, which can be induced by retroviral mediated restriction of tumor growth, both in vivo and in vitro.35,36 Among 8 tested macrocycles, compounds 1a, 1f, 1g, 1i and 1k induced transcription of MDA7. Both MDA7 and TRAIL belong to IL-10 family of cytokines which are known to induce apoptosis in tumor cells in caspase-3 dependent manner that has been considered as model genes for studying anti-tumor activities and gene therapy.37 Earlier report suggests that MDA7 induces apoptotic cells death of tumor cells in type – I interferon dependent manner,31b however, we have not found any induction of IFN-β gene (data not shown). The study suggests that anti-cancer activities might be interferon independent. Further it establishes that the down regulation of pro-cancer gene STAT3 concurrently elevates TRAIL mediated-induction of cell death38 and two compounds 1a and 1i follow similar trend38 (Fig. 5). Induction of STAT3 in 1f, 1g and 1k treatment groups might be antagonizing TRAIL effect and that might be the reason for the absence of DNA laddering in groups treated with these compounds (Fig. 6).
Macrocycles induces apoptosis in tumor cells. Among the investigated macrocyclic compounds (1a, 1b, 1e, 1f, 1g, 1i, 1j and 1k), only two compounds 1a and 1i effectively induced DNA laddering in MDAMB-231 cells (Fig. 6). The ladder like impression of genomic DNA in agarose gel represents fragmentation of DNA into nucleosome monomers (consisting of 146 bp DNA) and oligomers, a caspase-3 dependent cellular event, observed during induction of apoptosis. These results suggest that compounds 1a and 1i induces caspase-3 mediated activation of apoptosis.
Macrocyclic compounds have no observed cytotoxic effect on normal breast cells. None of the investigated macrocyclic compounds 1a, 1b, 1e, 1f, 1g, 1i, 1j and 1k exerted cytotoxic effect on breast epithelia cells (MCF10A) (Fig. 7) were observed till 72 hours. These results suggest that macrocyclic compounds 1a, 1f, 1g, 1i, and 1k, which restrict proliferation of tumor cells (Fig. 4) have no cytotoxic effect on primary cells and can be considered for further validation in animal system.
Conclusion
We designed and synthesized a new class of small macrocyclic molecules, (2Z,11Z)-3,11-di(aryl/naphthyl)-1,13-dioxa-5,9-dithia-2,12-diazacyclohexadeca-2,11-dienes (1a–k) from readily available substrates using simple organic transformations. These macrocycles were evaluated for their anti-tumor effect on triple negative breast cancer cell line, MDAMB-231. Compounds 1a, 1f, 1g, 1i and 1k restrict cell invasion, compounds 1e, 1f, 1g, and 1k restrict cell proliferation and colonogenic survival, compounds 1a, 1i and 1k induces TRAIL, MDA7 and p53 anti-tumor genes. None of the tested compound was found to be cytotoxic for normal breast epithelial cells. Further, it would be of great interest to evaluate these macrocyclic compounds in animal system, especially the compounds 1a and 1i to establish its effect under physiological condition. Other notable advantages are (i) accessibility of diverse topologies due to presence of two ‘S’ atoms that are amiable to disrupt protein–protein interactions (PPIs) and (ii) the presence of pharmacophore cyclic oxime-ether functionalities (C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N–O–) which are suitable for further ‘FG’ transformations holds the potential to access lead analogs other than the present reported macrocyclic molecules.
N–O–) which are suitable for further ‘FG’ transformations holds the potential to access lead analogs other than the present reported macrocyclic molecules.
Materials and methods
General information
All commercially available starting materials, reagents and solvents were purchased from Sigma Aldrich, Acros organics and Merck were used without further purification. Melting points of various obtained products were determined and uncorrected. NMR spectra were recorded on a Varian, Jeol/AL and Bruker 400 MHz. Chemical shifts were expressed in parts per million (ppm), coupling constants were expressed in Hertz (Hz). Splitting patterns describe apparent multiplicities and were designated as s (singlet), br s (broad singlet), d (doublet), t (triplet), q (quartet), quintet, m (multiplet). High-resolution mass spectra (MS) and compound purity data were acquired on a Waters LCT premier XE TOF HRMS and single quadrupole system equipped with electro spray ionization (ESI) source and Exactive orbitrap mass spectrometer (Thermo Scientific). Single crystal XRD analysis was carried out on a Bruker Apex ii diffractometer with CCD detector MoKα radiation (λ = 0.71069 Å) operating ω–2θ scanning mode at room temperature. Thin-layer chromatography was performed on 0.25 mm Merck silica gel plates and visualized with UV light. Column chromatography was performed on silica gel. The structures of isolated compounds were elucidated on the basis of spectroscopic data (1H & 13C-NMR, HRMS) and single crystal XRD analysis.
Cell culture
Human triple negative breast cancer (TNBC) MDAMB231-cells (ATCC; cat no. HTB-26™) and neuroblastoma cell, IMR-32 were grown in 10% fetal bovine supplemented (FBS) (GIBCO, Life technologies, USA) complete Dulbecco's modified Eagles medium (cDMEM) (Sigma Aldrich, USA). The primary human breast epithelial cells (MCF10) were cultured in MEBM (Lonza/clonetics MEGM Kit cat# CC-3150), supplemented with 100 ng mL−1 cholera toxin, 2 mL per liter bovine pituitary extract (cat# CC4009G), 0.5 mL per liter gentamicin sulfate (cat# 4081G), 0.5 mL per liter recombinant growth factor human (cat# CC-4017G), recombinant insulin human (cat# CC-4021G), 0.5 mL per liter hypo cortisone (cat# CC-4031G). All the culture were maintained at 37 °C in humidified chamber (Thermo Fisher Scientific USA), supplied with supplied with 5% CO2. For subculture and seeding, TNBC MDAMB-231, IMR-32 cells were harvested with 0.01% trypsin-EDTA and MCF10A with 0.05% trypsin-EDTA, setting the cells density as per experimental requirements.
Wound healing assay
For wound healing assay TNBC MDAMB-231 cells were seeded in six well plate, at density of 1 × 106 cells per well in cDMEM. The 95% monolayer of cells carefully wounded using 20–200 μL micro tips. Cells were washed with DMEM for removing cell debris. Wounded monolayer of cells was treated with 1a, 1b, 1e, 1f, 1g, 1i, 1j and 1k (5 μM each) for 24 h 17AAG (5 μM) and solvent only treated cells were used as positive and negative control. Migration of cells into wounded area was captured by phase contrast microscope (Zeiss Axiovert model) at 0 h and 24 h time points.
Colonogenic assay
The anti-proliferative effect of macrocycles (1a, 1b, 1e, 1f, 1g, 1i, 1j and 1k) were tested by this assay. TNBC MDAMB-231 cells were seeded in six well plates at density of 1 × 103 cells per well and left in culture for 10 days for formation of colonies. Than cells were treated with test macrocyclic compounds 1a, 1b, 1e, 1f, 1g, 1i, 1j and 1k (5 μM each), positive control 17AAG (5 μM) or hydroxyl urea (1 mM) for 24 h. Followed by cells were washed with 1×-PBS and fixed using 70:30 solution of ethanol acetic acid for 10 minutes. Fixed cells were stained with 1% methylene blue for 15 min. Extra stain was drained by washing with water. Dried plates were scanned for representation of effect of macrocycles on colonogenic survival of cells.
Quantitative-PCR analysis
The MDAMB-231 cells were seeded in six well plate at density of 0.2 × 106. Next day, cells were treated with macrocycles 1a, 1b, 1e, 1f, 1g, 1i, 1j and 1k (5 μM each) for 24 h. The cells were washed with 1×-PBS and total RNA was extracted TRIzol reagent (Invitrogen USA) and first strand cDNA was synthesized for 1 μg RNA, using iScript-cDNA synthesis kit (BIORAD USA), according to manufacturers instruction. Relative quantification of anti-tumor genes (p53, MDA7, and TRAIL) and transcription factor STAT3 was performed with SYBR green master mix, using 18S as housekeeping control. Following, initial denaturation step at 95 °C for 10 min, 40 cycles of PCR amplification were performed with denaturation for 15 s and annealing at 60 °C for 1 min. In order to confirm amplification as single specific product, melt curve analysis was done as 95 °C for 15 s, 60 °C for 1 min, 95 °C for 15 s. The sequences of primer used in RNA quantification is given is Table 1.
Table 1 Primer used for quantitative PCR
| Primer |
Direction |
Sequence |
Size bp |
Amplicon |
| S18 |
Forward |
ATCACCATTATGCAGAATCCACG |
23 |
93 bp |
| Reverse |
GACCTGGCTGTATTTTCCATCC |
22 |
| P53 |
Forward |
GCCATCTACAAGCAGTCACAG |
21 |
143 bp |
| Reverse |
TCATCCAAATACTCCACACGC |
21 |
| TRAIL |
Forward |
AGCAATGCCACTTTTGGAGT |
20 |
120 bp |
| Reverse |
TTCACAGTGCTCCTGCAGTC |
20 |
| STAT3 |
Forward |
TTTGTCAGCGATGGAGTACG |
20 |
168 bp |
| Reverse |
TGTTGACGGGTCTGAAGTTG |
20 |
| MDA7 |
Forward |
GAGGAACACGAGACTGAGAG |
20 |
116 bp |
| Reverse |
TCCAGAGAAGCAGGGTAAAAC |
21 |
DNA fragmentation assay
Human TNBC MDAMB-231 and IMR32 cells were seeded in 6 well culture dish at density of 0.2 × 106 cells per well. Next day cells were treated with macrocyclic compounds 1a, 1b, 1e, 1f, 1g, 1i, 1j and 1k. Followed to 48 h incubation, cells were harvested by trypsin-EDTA and pellet down at 200 g for 2 min. Cells washed with 1×-PBS and re-suspended lysis buffer (10 mM Tris-Cl: pH 8.0, 10 mM: EDTA, 0.5% Triton X-100) and nuclei pellet was collected at 13000 g for 20 min at 4 °C. Obtained nuclei pellet, re-suspended in lysis buffer and treated DNAse-free RNAse (0.1 mg mL−1) for 1 h at 37 °C. Then proteinase K (0.2 mg mL−1) and SDS (final concentration of 1%) were added and a resultant mixture was incubated further for 2 h at 50 °C. DNA from these samples was extracted using phenol chloroform isoamyl alcohol (25:24:1). DNA precipitation was done using 3.5 volume of 100% ethanol and 0.1 volume of CH3COOK (pH 5.2) at −80 °C. The DNA pellet was resuspended in 40 μL of MQ and equal quantity of DNA was analyzed in 2% agarose gel.
Cell cytotoxicity assay
The cytotoxic effect of newly synthesized macrocyclic compounds 1a, 1b, 1e, 1f, 1g, 1i, 1j and 1k was analyzed on human breast epithelial cells MCF10A. Cells were seeded in 24 well plates at density of 0.5 × 106 cells per well, cultured till >90% cell density and treated with macrocyclic compounds 1a, 1b, 1e, 1f, 1g, 1i, 1j and 1k (200 μM each) for 24 h. Cells micrographs were taken before and after treatment at 0 h and 24 h time points using phase contrast microscopy.
General experimental procedure
Synthesis of 2-bromo-1-(4-chlorophenyl)ethanone (4a). In a 100 mL two necked round-bottom flask, 1-(4-chlorophenyl)ethanone (2a) (10.0 mmol), N-bromosuccinimide (3) (12.0 mmol), 10% (w/w) silica gel in methanol (20 mL) were stirred under reflux conditions for 19 min. The progress of the reaction was monitored by TLC. After completion of the reaction, the stirring was stopped and cooled to RT. The solvent was removed under reduced pressure using rotary evaporator. To the crude product dichloromethane and distilled water were added. The organic layer was collected and again washed with water (2 × 50 mL) twice. The layers were separated and the organic layer was collected and it was dried using anhydrous Na2SO4. Again, the solvent was evaporated using rotary evaporator. The crude was purified over column chromatography using silica gel (99:1 ratio of n-hexane and EtOAc) and pure product 3a was obtained in 91% yield. The same experimental procedure was adopted for the preparation of other α-bromoketones (4b–k).
Synthesis of 2,2′-(propane-1,3-diylbis(sulfanediyl))bis(1-(4-chlorophenyl)ethanone) (6a). In a 100 mL two necked round bottom flask, 2-bromo-1-(4-chlorophenyl)ethanone (4a) [10.0 mmol] in acetonitrile (15 mL) was taken. The temperature of the reaction mixture was cooled to 15–20 °C and then K2CO3 (30 mmol) was added. Then, the propane-1,3-dithiol (5) [5.0 mmol] was taken in acetonitrile (15 mL) and it was added drop wise using dropping funnel at 15–20 °C. The reaction mixture was stirred for 90 min at the same temperature up to the disappearance of the substrate (4a). The completion of the reaction was confirmed by TLC. Then, the reaction mass was filtered and then the filtrate was concentrated under reduced pressure. The obtained crude product (6a) was purified by column chromatography using silica gel (4:1 ratio of n-hexane and ethyl acetate). The same experimental procedure was adopted for the preparation of other 2,2′-(propane-1,3-diylbis(sulfanediyl))bis(1-aryl/naphthyl)ethanone derivatives (6b–k).
Synthesis of (1Z,1′Z)-1-(4-chlorophenyl)-2-(3-((Z)-2-(4-chlorophenyl)-2-(hydroxyimino)ethylthio)propylthio)ethanoneoxime (8a). In a 100 mL two necked round bottom flask, substrate (6a) [10.0 mmol] and hydroxyl ammonium chloride (NH2OH·HCl) (7) [40.0 mmol] were dissolved in ethanol (30 mL) and then NaOAc (30.0 mmol w.r.t NH2OH·HCl) was added. Then, the reaction mixture was stirred at reflux temperature for 2 h. The progress of the reaction was monitored by TLC. After completion of the reaction, reaction mixture was cooled to RT and then filtered, washed thoroughly with ethanol. The filtrate was collected and the solvent was removed under reduced pressure. To the crude reaction mass, water (50 mL) was added and the product (8a) was extracted with EtOAc (2 × 25 mL). Then organic layer was collected and washed with water (2 × 25 mL) twice. Again, the organic layer was collected and dried over anhydrous Na2SO4 and the solvent was removed under reduced pressure. The crude product (8a) was recrystallized from the ethanol (10 mL) and n-hexane (30 mL) mixture. The same experimental procedure was followed for the preparation of other ketoxime derivatives (8b–k).
Synthesis of (2Z,11Z)-3,11-bis(4-chlorophenyl)-1,13-dioxa-5,9-dithia-2,12-diazacyclohexadeca-2,11-diene (1a). In a 100 mL two necked round bottom flask, compound 8a (10.0 mmol) was dissolved in DMSO (15 mL). The reaction mass was cooled to 5 to 10 °C. At this temperature, aqueous KOH solution (20.0 mmol of KOH was dissolved in 5 mL of water) was added slowly and stirred at 5 to 10 °C for 15 min. TLC was checked to confirm the disappearance of substrate (it convert to dipotassium salt form). Then, 1,3-dibromopropane (9) [10.0 mmol] was taken in 10 mL of DMSO and was added slowly at 5 to 10 °C and the reaction mass was stirred at the same temperature for 10 min. The reaction progress was monitored by TLC. After completion of the reaction, the reaction mixture was quenched in crushed ice (100 mL) and then the product (1a) was extracted with diethyl ether (2 × 50 mL) twice and the solvent was evaporated under reduced pressure. The obtained crude product (1a) was purified by column chromatography using silica gel (99:1 ratio of n-hexane and ethyl acetate). The same experimental procedure has been followed for the preparation of other macrocyclic compounds (1b–k). The synthesized compounds were characterized by 1H, 13C NMR and HRMS spectral data and the structure of compound 1e was confirmed by single crystal XRD data.
Acknowledgements
The authors acknowledge the financial support for this work from the Council of Scientific and Industrial Research (CSIR), New Delhi, Government of India under a major research project (No. 01 (2391)/10/EMR-II) and Department of Atomic Energy-Board of Research in Nuclear Sciences (DAE-BRNS), Mumbai, Government of India through a major research project (No. 2011/37C/52/BRNS/2264). The study related to the analysis of biological activities is also supported by research grants number SR/S2/RJN-55/2009, BT/PR6009/GBD/27/382/2012 from Department of Science and Technology (DST) and Department of Biotechnology (DBT), Government of India respectively. This study was partly supported by the Intramural Research Grant of IISER, Bhopal, India and Grant for Joint Research Program of the Institute for Genetic Medicine, Hokkaido University, Japan to HK. Authors also thank Dr R.V.J. Kashyap, Dept. of English, Yogi Vemana University, for critical reading of the manuscript to check the linguistic corrections.
Notes and references
-
(a) R. Siegel, J. Ma, Z. Zou and A. Jemal, Cancer statistics, 2014, Ca-Cancer J. Clin., 2014, 64, 9–29 CrossRef PubMed;
(b) C. E. DeSantis, C. C. Lin, A. B. Mariotto, R. L. Siegel, K. D. Stein, J. L. Kramer, R. Alteri, A. S. Robbins and A. Jemal, Cancer Treatment and Survivorship Statistics, 2014, Ca-Cancer J. Clin., 2014, 64, 252–271 CrossRef PubMed;
(c) C. DeSantis, J. Ma, L. Bryan and A. Jemal, Breast Cancer Statistics, 2013, Ca-Cancer J. Clin., 2014, 64, 52–62 CrossRef PubMed.
- World Cancer Report 2014, World Health Organization, 2014, ch. 1.1., ISBN 9283204298 Search PubMed.
- The Top 10 Causes of Death Fact Sheet No 310, World Health Organization (http://www.who.int/mediacentre/factsheets/fs310/en/), May 2014 Search PubMed.
- World Cancer Report 2014, World Health Organization, 2014, ch. 1.3., ISBN 9283204298 Search PubMed.
- A. Jemal, F. Bray, M. M. Center, J. Ferlay, E. Ward and D. Forman, Global Cancer Statistics, Ca-Cancer J. Clin., 2011, 61, 69–90 CrossRef PubMed.
- R. L. Siegel, K. D. Miller and A. Jemal, Cancer statistics, 2015, Ca-Cancer J. Clin., 2015, 65, 5–29 CrossRef PubMed.
-
(a) A. R. Shareef, D. H. Sherman and J. Montgomery, Chem. Sci., 2012, 3, 892–895 RSC;
(b) R. M. Wilson and S. J. Danishefsky, Chem. Soc. Rev., 2007, 36, 1207–1226 RSC;
(c) E. Marsault and M. L. Peterson, J. Med. Chem., 2011, 54, 1961–2004 CrossRef CAS PubMed.
-
(a) T. E. Rose, K. V. Lawson and P. G. Harran, Chem. Sci., 2015, 6, 2219–2223 RSC;
(b) D. F. Veber, S. R. Johnson, H. Y. Cheng, B. R. Smith, K. W. Ward and K. D. Kopple, J. Med. Chem., 2002, 45, 2615–2623 CrossRef CAS PubMed;
(c) T. Rezai, B. Yu, G. L. Millhauser, M. P. Jacobsen and R. S. Lokey, J. Am. Chem. Soc., 2006, 128, 2510–2511 CrossRef CAS PubMed.
-
(a) R. Tripier, O. Siri, F. Rabiet, F. Denat and R. Guilard, Tetrahedron Lett., 1999, 40, 79–82 CrossRef CAS;
(b) C. Mcfarland, D. A. Vivic and A. V. Debnath, Synthesis, 2006, 5, 807–812 Search PubMed.
- E. M. Driggers, S. P. Hale, J. Lee and N. K. Terrett, Nat. Rev. Drug Discovery, 2008, 7, 608–624 CrossRef CAS PubMed.
-
(a) A. K. Yudin, Chem. Sci., 2015, 6, 30–49 RSC;
(b) J. Levin, Macrocycles in Drug Discovery, RSC Drug Discovery, The Royal Society of Chemistry, 2015 Search PubMed;
(c) C. A. Lipinski, F. Lombardo, B. W. Dominy and P. J. Feeney, Adv. Drug Delivery Rev., 1997, 23, 3–25 CrossRef CAS.
-
(a) V. Takiar, E. A. Strom, D. P. Baumann, F. Meric-Bernstam, R. H. Alvarez and A. M. Gonzalez-Angulo, Oncologist, 2013, 18, 265–270 CrossRef CAS PubMed;
(b) C. C. Rohena and S. L. Mooberry, Nat. Prod. Rep., 2014, 31, 335–355 RSC.
- A. D. William, A. C. H. Lee, K. C. Goh, S. Blanchard, A. Poulsen, E. L. Teo, H. Nagaraj, C. P. Lee, H. Wang, M. Williams, E. T. Sun, C. Hu, R. Jayaraman, M. K. Pasha, K. Ethirajulu, J. M. Wood and B. W. Dymock, J. Med. Chem., 2012, 55, 169–196 CrossRef CAS PubMed.
- Z. F. Tao, L. Wang, K. D. Stewart, Z. Chen, W. Gu, M.-H. Bui, P. Merta, H. Zhang, P. Kovar, E. Johnson, C. Park, R. Judge, S. Rosenberg, T. Sowin and N. H. Lin, J. Med. Chem., 2007, 50, 1514–1527 CrossRef CAS PubMed.
- M. Marjanovic, M. Kralj, F. Supek, L. Frkanec, I. Piantanida, T. Sÿmuc and L. Tusek-Bozic, J. Med. Chem., 2007, 50, 1007–1018 CrossRef CAS PubMed.
- A. Muth, V. Pandey, N. Kaur, M. Wason, C. Baker, X. Han, T. R. Johnson, D. A. Altomare and O. Phanstiel, J. Med. Chem., 2014, 57, 4023–4034 CrossRef CAS PubMed.
-
(a) P. Rajakumar, N. Venkatesan and G. Mohanraj, RSC Adv., 2014, 4, 21190–21194 RSC;
(b) M. Altendorfer, A. Raja, F. Sasse, H. Irschik and D. Menche, Org. Biomol. Chem., 2013, 11, 2116–2139 RSC;
(c) J. Choi, J. Kim, K. Kim, S. T. Yang, J. Il Kim and S. Jon, Chem. Commun., 2007, 1151–1153 RSC;
(d) Y. X. Tan and F. E. Romesberg, Med. Chem. Commun., 2012, 3, 916–925 RSC;
(e) J. M. Wilson, F. Giordani, L. J. Farrugia, M. P. Barrett, D. J. Robins and A. Sutherland, Org. Biomol. Chem., 2007, 5, 3651–3656 RSC;
(f) E. L. Dodd and D. S. Bohle, Chem. Commun., 2014, 50, 13765–13768 RSC;
(g) T. Takizawa, K. Watanabe, K. Narita, T. Oguchi, H. Abe and T. Katoh, Chem. Commun., 2008, 1677–1679 RSC;
(h) T. L. Newkirk, A. A. Bowers and R. M. Williams, Nat. Prod. Rep., 2009, 26, 1293–1320 RSC;
(i) W. M. Kazmierski, R. L. Jarvest, J. J. Plattner and X. Li, Linear and Macrocyclic Hepatitis C Virus Protease Inhibitors: Inhibitor Design and Macrocyclization Strategies for HCV Protease and Related Targets, Macrocycles in Drug Discovery, ch. 7, 2015 Search PubMedB. L. Dutton, R. R. A. Kitson, S. Parry-Morris, S. M. Roe, C. Prodromou and C. J. Moody, Org. Biomol. Chem., 2014, 12, 1328–1340 CAS; J. G. Rudick, M. M. Laakso, A. C. Schloss and W. F. DeGrado, Org. Biomol. Chem., 2013, 11, 7096–7100 Search PubMed.
-
(a) D. Parker, Macrocycle Synthesis: A Practical Approach, Oxford University Press, Oxford, 1996 Search PubMed;
(b) S. Vogel, K. Rohr, O. Dahl and J. Wengel, Chem. Commun., 2003, 1006–1007 RSC.
-
(a) A. Noor, S. C. Moratti and J. D. Crowley, Chem. Sci., 2014, 5, 4283–4290 RSC;
(b) Comprehensive Supramolecular Chemistry, ed. J. L. Atwood, J. E. D. Davies, D. D. MacNicol and F. Vögtle, Pregamon Press, NewYork, 1996, vol. 1–10 Search PubMed;
(c) H. W. Gibson, H. Wang, K. Bonrad, J. W. Jones, C. Slebodnick, L. N. Zackharov, A. L. Rheingold, B. Habenicht, P. Lobue and A. E. Ratliff, Org.
Biomol. Chem., 2005, 3, 2114–2121 CAS.
- K. E. Krakoviak, J. S. Bradshaw, W. Jiang, N. K. Dalley, G. Wu and R. M. Izatt, J. Org. Chem., 1991, 56, 2675–2680 CrossRef.
- F. Sancenon, R. Martinez-Manez and J. Soto, Angew. Chem., Int. Ed., 2002, 41, 1416–1419 CrossRef CAS.
-
(a) S. Dasgupta and J. Wu, Org. Biomol. Chem., 2011, 9, 3504–3515 RSC;
(b) S. A. Dietrich, L. Banfi, A. Basso, G. Damonte, G. Guanti and R. Riva, Org. Biomol. Chem., 2005, 3, 97–106 RSC.
- Naveen, S. A. Babu, G. Kaur, N. A. Aslam and M. Karanam, RSC Adv., 2014, 4, 18904–18916 RSC.
- M. E. Fitzgerald, C. A. Mulrooney, J. R. Duvall, J. Wei, B. C. Suh, L. B. Akella, A. Vrcic and L. A. Marcaurelle, ACS Comb. Sci., 2012, 14, 89–96 CrossRef PubMed.
-
(a) A. Y. Sukhorukov and S. L. Ioffe, Chem. Rev., 2011, 111, 5004–5041 CrossRef CAS PubMed and references cited therein;
(b) H.-U. Reissig and R. Zimmer, Science of Synthesis, ed. G. A. Molander, Thieme, Stuttgart, 2006, vol. 33, p. 371 Search PubMed.
-
(a) J. M. Smith, N. C. Hill, P. J. Krasniak and R. Fasan, Org. Biomol. Chem., 2014, 12, 1135–1142 RSC;
(b) J. Y. Lee, H. J. Kim, J. H. Jung, W. Sim and S. S. Lee, J. Am. Chem. Soc., 2008, 130, 13838–13839 CrossRef CAS PubMed.
- J. A. Robinson, ChemBioChem, 2009, 10, 971–973 CrossRef CAS PubMed.
- B. Mohan Reddy, V. V. Ramana Kumar, N. C. Gangi Reddy and S. Mahender Rao, Chin. Chem. Lett., 2014, 25, 179–182 CrossRef.
- B. S. Furniss, A. J. Hannaford, P. W. G. Smith and A. R. Tatchell, Vogel's text book of organic chemistry, Longmann Scientific & Technical and John Wiley & Sons, Inc., New York, 5th edn, 1989 Search PubMed.
- K. I. Hulkower and R. L. Herber, Pharmaceutics, 2011, 3, 107–124 CrossRef CAS PubMed.
-
(a) U. Ramp, E. Caliskan, C. Mahotka, A. Krieg, S. Heikaus, H. E. Gabbert and C. D. Gerharz, Br. J. Cancer, 2003, 88, 1800–1807 CrossRef CAS PubMed;
(b) S. Ekmekcioglu, J. B. Mumm, M. Udtha, S. Chada and E. A. Grimm, Cytokine, 2008, 43, 34–44 CrossRef CAS PubMed.
- A. Ashkenazi and V. M. Dixit, Science, 1998, 281, 1305–1308 CrossRef CAS PubMed.
- G. S. Wu, Cancer Lett., 2009, 285, 1–5 CrossRef CAS PubMed.
- A. Ventura, D. G. Kirsch, M. E. McLaughlin, D. A. Tuveson, J. Grimm, L. Lintault, J. Newman, E. E. Reczek, R. Weissleder and T. Jacks, Nature, 2007, 445, 661–665 CrossRef CAS PubMed.
- S. Chanda, A. M. Mhashilkar, Y. Liu, T. Nishikawa, D. Bocangel, M. Zheng, S. A. Vorburger, A. Pataer, S. G. Swisher, R. Ramesh, K. Kawase, R. E. Meyn and K. K. Hunt, Cancer Gene Ther., 2006, 13, 490–502 CrossRef PubMed.
- W. Zhu, L. Wei, H. Zhang, J. Chen and X. Qin, J. Exp. Clin. Cancer Res., 2012, 31, 51 CrossRef CAS PubMed.
- M. H. Mirzaei and A. Esmaeilzadeh, J. Med. Hypotheses Ideas, 2014, 8, 7–13 CrossRef.
- S. Huang and F. A. Sinicrope, Mol. Cancer Ther., 2010, 9, 742–750 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Physical and spectral characterization data and single crystal XRD analysis data. CCDC 1046587. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c6ra15140j |
|
| This journal is © The Royal Society of Chemistry 2016 |
Click here to see how this site uses Cookies. View our privacy policy here.