
A novel hybrid self-assembly process for synthesising stratified polyethylene–organoclay films

Abstract
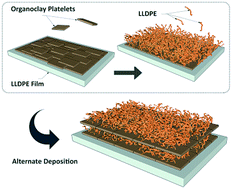
This study reports the first effort to synthesize a new type of PE–organoclay multilayer film by starting from an uncharged apolar polymer substrate and successively depositing apolar organoclay and uncharged apolar polyethylene (PE) layers with subsequent repeating depositions. The alternate variation of contact angle (85° average for organoclay and 107° for PE layers) confirmed the profilometry and the scanning electron microscopy results as well as the linear growth pattern, i.e. the successful highly stratified assembly of repetitive bilayers comprised of 450 nm organoclays and 2.25 μm PE layers. The self-assembly of organoclays on PE surfaces was driven by solvophobic molecular construction involving hydrophobic interactions between the organic parts of the organoclay tactoids dispersed in an organic solvent and the PE hydrophobic surface. The deposition of PE molecules on the organoclay layers was the result of a dip-coating process involving physical sorption of a highly viscous PE solution on the surface of the organoclay layers.
 Please wait while we load your content...
Please wait while we load your content...

