Effects of substituent groups on methane adsorption in covalent organic frameworks†
Jianfei Zhao and
Tianying Yan*
Tianjin Key Laboratory of Metal- and Molecule-Based Material Chemistry, Key Laboratory of Advanced Energy Materials Chemistry (Ministry of Education), Collaborative Innovation Center of Chemical Science and Engineering (Tianjin), Institute of New Energy Material Chemistry, College of Chemistry, Nankai University, Tianjin 300071, China. E-mail: tyan@nankai.edu.cn
First published on 20th March 2014
Abstract
Based on the structures of published three-dimensional covalent organic frameworks (COF-102, COF-103, and COF-105), we developed a sequence of modified COFs by replacing some H atoms on benzene rings with other substituent groups, including –Cl, –Br, –I, –CF3, –NH2, –CN, –OCH3, and –CH3. To explore the effects of the substituents on methane storage, we studied their adsorption properties by using Grand Canonical Monte Carlo (GCMC) simulation. The methane uptakes and isosteric heat of adsorption from 0 to 100 bar were simulated at room temperature. The results indicate that in COF-102 and COF-103, all these substituents are beneficial to methane storage at low pressure, but the advantage is weakened by the volume effect at medium and high pressure. In the case of COF-105 and its derivatives, all the isotherms are linear due to their large pore volumes. Among these groups, halogen groups (–Cl, –Br, and –I) and –NH2 are the best ones to improve methane uptake, whereas –CH3 and –OCH3 are of little help. Among all the covalent organic frameworks simulated in this study, COF-102-I (156 V(STP)/V of excess uptake and 169 V(STP)/V of methane delivery), COF-102-Br (153 V(STP)/V of excess uptake and 169 V(STP)/V of methane delivery), COF-102-Cl (148 V(STP)/V of excess uptake and 165 V(STP)/V of methane delivery), and COF-102-NH2 (143 V(STP)/V of excess uptake and 160 V(STP)/V of methane delivery) are found to be the most promising adsorbents for methane uptake.
I. Introduction
The combustion of fossil fuels has caused a series of problems. Carbon monoxide, sulfur dioxide, and nitrogen oxides are dangerous pollutants in the atmosphere. The emission of carbon dioxide is the main reason for global warming.1 Besides, the overuse of gasoline and natural gas has led to an energy crisis. Facing the serious situation, researchers are finding alternative energy resources. Methane is an ideal fuel due to its abundant resource and clean-burning characteristic. Compared with other carbon-containing compounds, methane is easier to be completely combusted and there will be less CO2 emission, attributed to its low carbon-to-hydrogen ratio. To put it into actual use, some technical difficulties need to be overcome. The premier challenge is to store methane with safety and efficiency. Until now, many ways of storage have been developed, such as liquefied natural gas (LNG), compressed natural gas (CNG), natural gas hydrate (NGH),2,3 and adsorbed natural gas (ANG).4 It has been widely accepted that the last one would be desirable, as ANG makes it possible to store methane moderately without hyperbaric or cryogenic condition. Obviously, materials with excellent physisorptive properties are essential for the application of methane storage. According to the target set by US Department of Energy (DOE),5 the capacity for methane storage has to reach 180 V(STP)/V at 35 bar, in which V(STP)/V refers to the volume of methane per volume of the adsorbent and STP is short for standard temperature and pressure (298 K and 1.01 bar).A wide range of materials can be used for storage.6 Porosity and surface area are of vital importance because they play a significant role in determining the capability of gas storage. In addition, the stability should be taken into account. If the material is too fragile to maintain its structure during the process of adsorption/desorption cycles, there would be no value for application. In the early stage, carbon nanotubes,7 activated carbon,8,9 and zeolites10,11 have been studied as methane adsorbents. An activated carbon has a surface area of 2400 m2 g−1 (ref. 12) and a disordered carbon is about 2000 m2 g−1,13 and they are among the highest ones at that time. In 1999, Yaghi and coworkers synthesized the first three-dimensional metal organic framework (3D MOF) with the combination of metal ions and organic ligands.14 This porous and crystalline structure was named MOF-5, with an appropriate pore size (11 Å) as well as a high surface area (2900 m2 g−1). After that, MOF-177 was synthesized with a surface area of 4500 m2 g−1.15 A series of studies focus on the properties of MOFs in sorption of hydrogen,16–18 methane,19,20 carbon dioxide21–23 as well as other gases,24 and in separation processes.25–27 Excitingly, some MOFs have been confirmed to surpass the DOE target of methane storage. At 35 bar and 273 K, the excess volumetric methane uptake is 220 V(STP)/V for PCN-14 (ref. 28) and 190 V(STP)/V for Ni-MOF-74.29
Inspired by the construction of MOFs, Yaghi and coworkers further designed the first two-dimensional covalent organic frameworks (2D COFs)30 in 2005, and successfully developed other 2D COFs31 and 3D COFs32,33 afterwards. Both MOFs and COFs are featured with crystalline structures. Unlike MOFs, COFs are built up merely from organic ligands without any metal elements. As a consequence, COFs always have lower densities and this could be more favorable when counting gravimetric uptake. Since being synthesized, COFs have aroused lots of interests, especially COF-102, COF-103, COF-105, and COF-108, which are reported with excellent adsorption behavior both in experimental and in theoretical research.34–39 Their building units, namely tetra(4-dihydroxyborylphenyl)methane (TBPM), tetra(4-dihydroxyborylphenyl)silane (TBPS), and triangular hexahydroxytriphenylene (HHTP) are shown in Fig. 1, and their combination patterns are listed in Table 1.
 | ||
| Fig. 1 Building units (a) TBPM or TBPS and (b) HHTP for COFs. Spheres in white, pink, cyan, and red represent hydrogen, boron, carbon, and oxygen atoms, respectively. The yellow sphere in ligand (a) denotes the central atom of TBPM or TBPS (either carbon or silicon) and the blue spheres in ligand (a) refer to the hydrogen atoms that are replaced by other groups. | ||
| Material | Combination | Central atom X |
|---|---|---|
| COF-102 | (a) | C |
| COF-103 | (a) | Si |
| COF-105 | (a) + (b) | Si |
COFs are regarded as a brand new type of adsorbent. Despite drawing so much attention, none of them has been experimentally confirmed to excel the methane storage target of 180 V(STP)/V. In order to enhance the performance of gas adsorption, they need to be modified. Some encouraging achievements have been made, and there are mainly two effective ways to increase the amount of physisorption. The first one is metal doping, especially Li, in basic COFs. Lan et al. designed Li-doping COFs and the methane uptake was almost doubled according to their computer simulation.40 Apart from the property of methane adsorption, the insertion of Li and other doping methods could also improve the performance of COFs in hydrogen storage,41,42 carbon dioxide capture,43 and gas separation.44 Another method to promote the adsorption capability of COFs is ligand modification. Mendoza-Cortés et al. reported a fantastic work,45 which modified the moiety by replacing –Hs on meta-positions with diverse alkyl groups or by changing the phenylene group into anthracene and trans-ethylene. They simulated the adsorption behavior of a variety of designed COFs and found that the delivery amount of methane in COF-102-Ant and COF-103-Eth-trans reached the goal of DOE.45 Since it is difficult to attempt every substituent in practice, computer simulation46 is a powerful tool to study the gas adsorption behavior. In this study, we explore the effects of substituent groups on the methane adsorption in COF derivatives with GCMC simulation. Without changing the topology of basic COFs, we modify their structures by introducing different substituents to the building blocks and aim to find a relatively simple and feasible way to heighten their ability of methane adsorption. Similar methodology of substitution has been attempted in a simulation study of MOFs by Wilmer et al.47 They generated 137![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 953 conceivable MOFs with 102 building blocks, which fall into inorganic, organic, and functional groups. Over 300 hypothetical MOFs were predicted as better performers for methane storage than all the known materials, among which functional groups have the most positive effect on methane uptake.
953 conceivable MOFs with 102 building blocks, which fall into inorganic, organic, and functional groups. Over 300 hypothetical MOFs were predicted as better performers for methane storage than all the known materials, among which functional groups have the most positive effect on methane uptake.
II. Models and computational methods
In this study, we took COF-102, COF-103, and COF-105 as basic adsorbents. Some relevant parameters about these COFs are tabulated in Table 2. The reason for choosing them is grounded on two aspects. For one thing, they are potential candidates for methane storage. For another, all of them are provided with a vast amount of benzene rings that contain many H atoms. It is convenient to explore the effects of substituents on adsorption properties by substituting H atoms with other functional groups.| Material | acell, Å | Natom | Psize, Å | Sa, Å2 | Vp, Å3 | R% | ρ, g cm−3 |
|---|---|---|---|---|---|---|---|
| a Each surface area listed above is the van der Waals (vdW) surface of the framework, which is defined as the surface intersecting with vdW radius59 of each atom.b The volume within vdW surface is considered as the occupied volume and the pore volume is calculated by subtracting the occupied volume from the total volume of unit cell. There are several methods for the calculation of surface area and pore volume in previous works40,55,56,60 and the results of COF-102, COF-103, and COF-105 listed above are comparable with those studies. | |||||||
| COF-102 | 27.1771 | 588 | 12 | 4360.18 | 14249.19 |
70.99 | 0.41 |
| COF-103 | 28.2477 | 588 | 12 | 4640.83 | 16462.88 |
73.04 | 0.38 |
| COF-105 | 44.8860 | 1020 | 19 | 8594.34 | 79606.03 |
88.03 | 0.18 |
The –Cl, –Br, –I, –CF3, –NH2, –CN, –OCH3, and –CH3 groups were chosen, and the substituted COFs were named COF-102-X, COF-103-X, and COF-105-X, in which –X refers to substituents. The choice of these eight groups is on the basis of experimental evidence. The core part of the synthesis of COF-102, COF-103, and COF-105 is condensation. COF-102 (or COF-103) is produced by the self-condensation of TBPM (or TBPS) while COF-105 by the co-condensation between TBPS and HHTP.32 A possible way to produce X-substituted COF is to link –X with boronic acid to alter the building unit and use the new ligand to finish the condensation. We did not substitute those excessive bulky groups for –Hs since this might change the tetrahedral configuration of the ligand (a) and result in a disordered framework. We did not substitute OH-containing substituents (–OH, –COOH) for –Hs because these extra –OHs might interfere in the reaction of condensation. On the grounds of these considerations, many kinds of substituents were not included in our study, such as acyl groups (–CONH2, –CONR, –NHCOR), ester groups (–COOR), aromatic groups, alkyl groups with long chains, –SO3H, –NR2, –COR, and so on.
The number of isotypes of –H on benzene rings in the fragments of ligand (a) in COF-102 is 4, the same as COF-103 and COF-105. We substituted one type of –H in each COF with –X to create derived COFs. To obtain stable configurations for subsequent calculation, we optimized the newly designed COFs by using DREIDING force field48 with conjugate gradient algorithm. The optimized coordinates as well as the cell parameters are tabulated in the ESI.† One point to be clarified is that we replaced H atoms without transforming the symmetry of COFs and all the derived frameworks still held I![[4 with combining macron]](https://www.rsc.org/images/entities/char_0034_0304.gif) 3d space group and ctn topology after the optimization. Actually, the restricted application of some covalent organic frameworks is blamed for their unstable structures. In contrast with them, the three COFs in this work are characterized by their good thermal stability. Each one is entirely constructed from C–C, C–O, B–C, and B–O covalent bonds. All of them have significant bonding energies that will contribute to the thermal stability of the whole structure. Moreover, COF-102, COF-103, and COF-105 possess carbon-nitride (ctn) topology, which is believed to be stable.49 Therefore, it would be reasonable to design new materials from the original COFs while preserving their configurations in the meantime.
3d space group and ctn topology after the optimization. Actually, the restricted application of some covalent organic frameworks is blamed for their unstable structures. In contrast with them, the three COFs in this work are characterized by their good thermal stability. Each one is entirely constructed from C–C, C–O, B–C, and B–O covalent bonds. All of them have significant bonding energies that will contribute to the thermal stability of the whole structure. Moreover, COF-102, COF-103, and COF-105 possess carbon-nitride (ctn) topology, which is believed to be stable.49 Therefore, it would be reasonable to design new materials from the original COFs while preserving their configurations in the meantime.
We employed Grand Canonical Monte Carlo (GCMC) simulation46 for our calculation of amount and isosteric heat of adsorption. The van der Waals (vdW) interactions of COF–CH4 and CH4–CH4 were simulated by Lennard-Jones potential and the parameters of each atom of COFs were obtained from DREIDING force field.48 A single-site model was adopted for CH4 molecule, whose Lennard–Jones parameters were taken from previous work50 and have been widely used in simulations.51–53 The COF models were 2 × 2 × 2 supercell for COF-102-Xs, COF-103-Xs, and 2 × 2 × 1 supercell for COF-105-Xs, which were held rigid in GCMC simulations. The temperature was set at 298 K and the pressure was 0–100 bar. The simulations were run for totally 4000000 MC steps by using a home-made code, with the first 1000000 steps for equilibrating and the following 3000000 steps for sampling.
III. Results and discussion
III.1. Comparison with experimental results
At the beginning of the work, we need to validate the COF models and the force field parameters in our simulation. For this purpose, the isotherms of methane adsorption in COF-6, COF-8, COF-10, COF-102, and COF-103 from 0 to 100 bar at 298 K are calculated. The coordinates of these five frameworks are taken from ref. 31 and 32 and we select them as they are representative COFs with different features. COF-6 is a 2D COF with small pore size (9 Å), and COF-8 and COF-10 have 2D structures but large pores (16 Å and 32 Å), while COF-102 and COF-103 are typical 3D COFs with medium pore size (12 Å). Through model testing, we compare simulation data with experimental values.37 Since the results from experiments are excess loading curves, the total number of adsorbed CH4 molecules Ntot directly acquired from GCMC needs to be converted to the excess amount of molecules Nexc. This conversion is accomplished by using eqn (1),54| Nexc = Ntot − ρVp | (1) |
Fig. 2 shows the comparison of our calculations with the experiments as well as previous simulations. In hypobaric region (below 40 bar), the simulated isotherms are pretty close to the experiments with slight deviation. As the pressure rises, the discrepancy becomes larger but the trends still remain the same, especially for COF-6, COF-8, and COF-10. From medium to hyperbaric region, there exists an overestimation in each calculated loading curve, which is more obvious in the case of COF-102 and COF-103. This disagreement can be ascribed to several factors.55 The deviation within the estimation of vdW interaction cannot be avoided and the structures of COFs may be altered under high methane uptake. Furthermore, the frameworks used for adsorption experiments might not be so perfect that some regions are inaccessible in practice. Whereas in GCMC simulation, it assumes that all the space in a COF cell can be reached, leading to an overrated methane uptake. In general, for COF-6, COF-8, COF-10, and COF-102, our calculation is basically in agreement with experimental results both in the tendencies of variation and in the values and the relative deviation is acceptable (about 5–10% in total) while for COF-103, there is an agreement at low pressure but an overestimation at high pressure. In addition to experimental work, we compare the isotherms of COF-102 and COF-103 with previous simulation studies as both of them are basic frameworks involved in subsequent discussions. For COF-102, the three groups of results are rather similar to each other; for COF-103, our isotherm approaches the one made by Lan et al.,40 while lower than that of Mendoza-Cortés et al.,56 particularly at high pressure. Apart from that, the error bars shown in Fig. 2 are quite small. Therefore, we believe that the simulation results are converged, and thus do not estimate error bars for the rest of the simulations in this study.
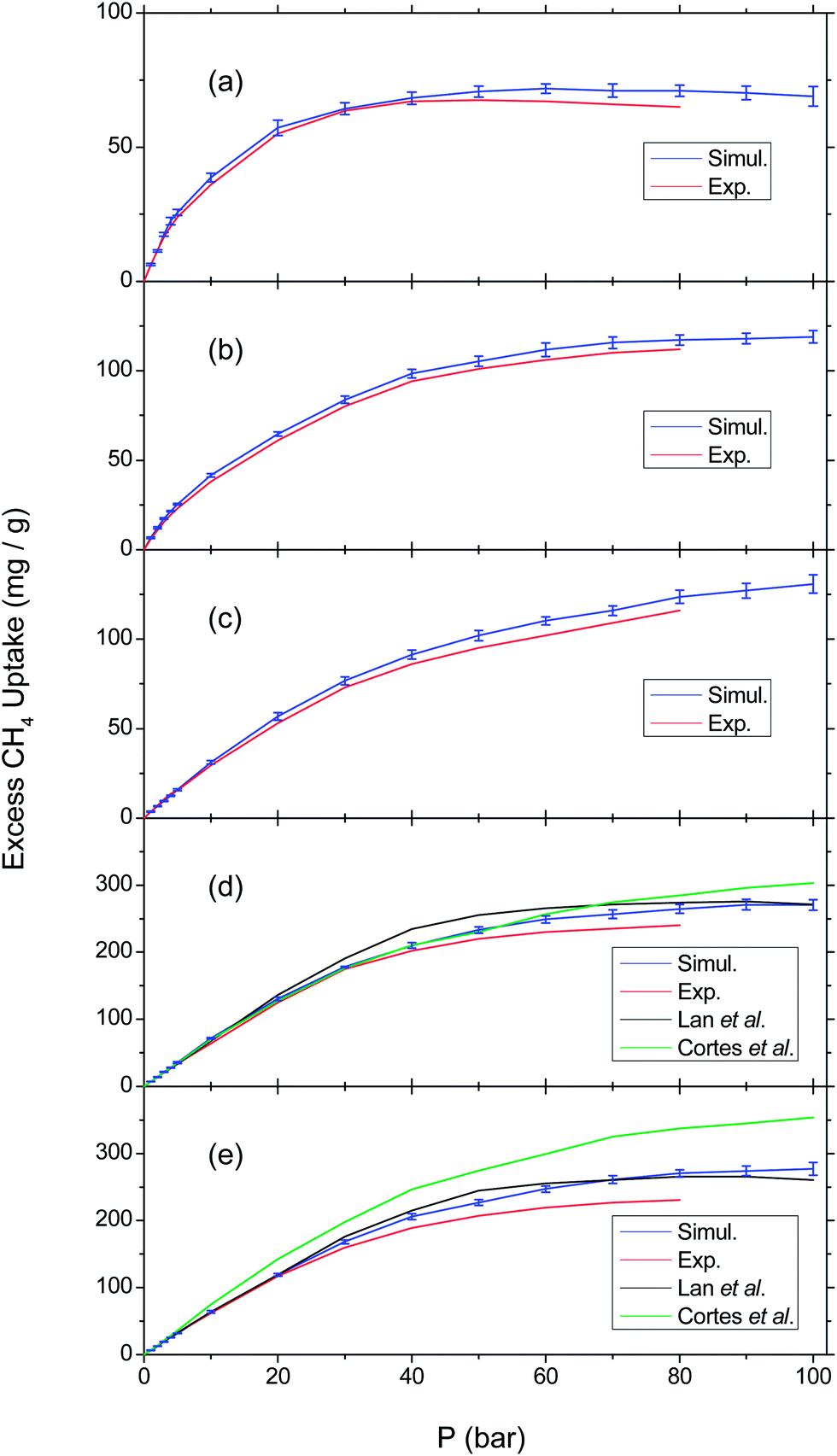 | ||
| Fig. 2 Comparison of CH4 adsorption isotherms between our results and experiments37 and also previous simulations:40,56 (a) COF-6; (b) COF-8; (c) COF-10; (d) COF-102; (e) COF-103. The blue lines are the simulation results in this work, with error bars estimated via the standard deviation of five independent GCMC simulations at each pressure. The red lines are experimental results.37 The black and green lines refer to the simulation studies by Lan et al.40 and by Mendoza-Cortés et al.,56 respectively. | ||
III.2. Methane uptake in basic COFs and substituted COFs
The total volumetric CH4 uptakes of the three COFs at 298 K are shown in Fig. 3. Fig. 3(a) displays the isotherms of COF-102 and its derivatives COF-102-Xs. From 0 to 20 bar, the uptakes nearly grow with linearity. In hypobaric coverage, the absolute amount of absorbed methane molecules is limited so that the free space in covalent organic framework is plentiful. Thus, CH4 can enter the pores without too much constraint. At higher pressure, the uptakes diverge. The variations are affected by their abilities of CH4 capture, attributed to the difference in the interactions with the substituents. Because the linkage pattern and the organic ligands almost remain unchanged, the substituents should be the single factor that affects the adsorption. Thus, each substitution group is beneficial for the elevation of CH4 storage due to the stronger interaction between CH4 molecule and the substitution groups. When the pressure is higher than 20 bar, the growth becomes slow because of the volume effect. More gas molecules are adsorbed so the free space in COF-102-X decreases. When a CH4 enters the pore, it will be subject to an attraction force as well as a growing repulsion force from other molecules. In the case of low and intermediate pressure, the former one would be in a dominant position while the latter one would impede the adsorption process. At high pressure, the gentle curves imply that the methane uptake is gradually reaching its limit. The pores are crowded and the repulsion lessens the inclination of adsorbing more methane. The faster the isotherm flattens, the more significant the volume effect is.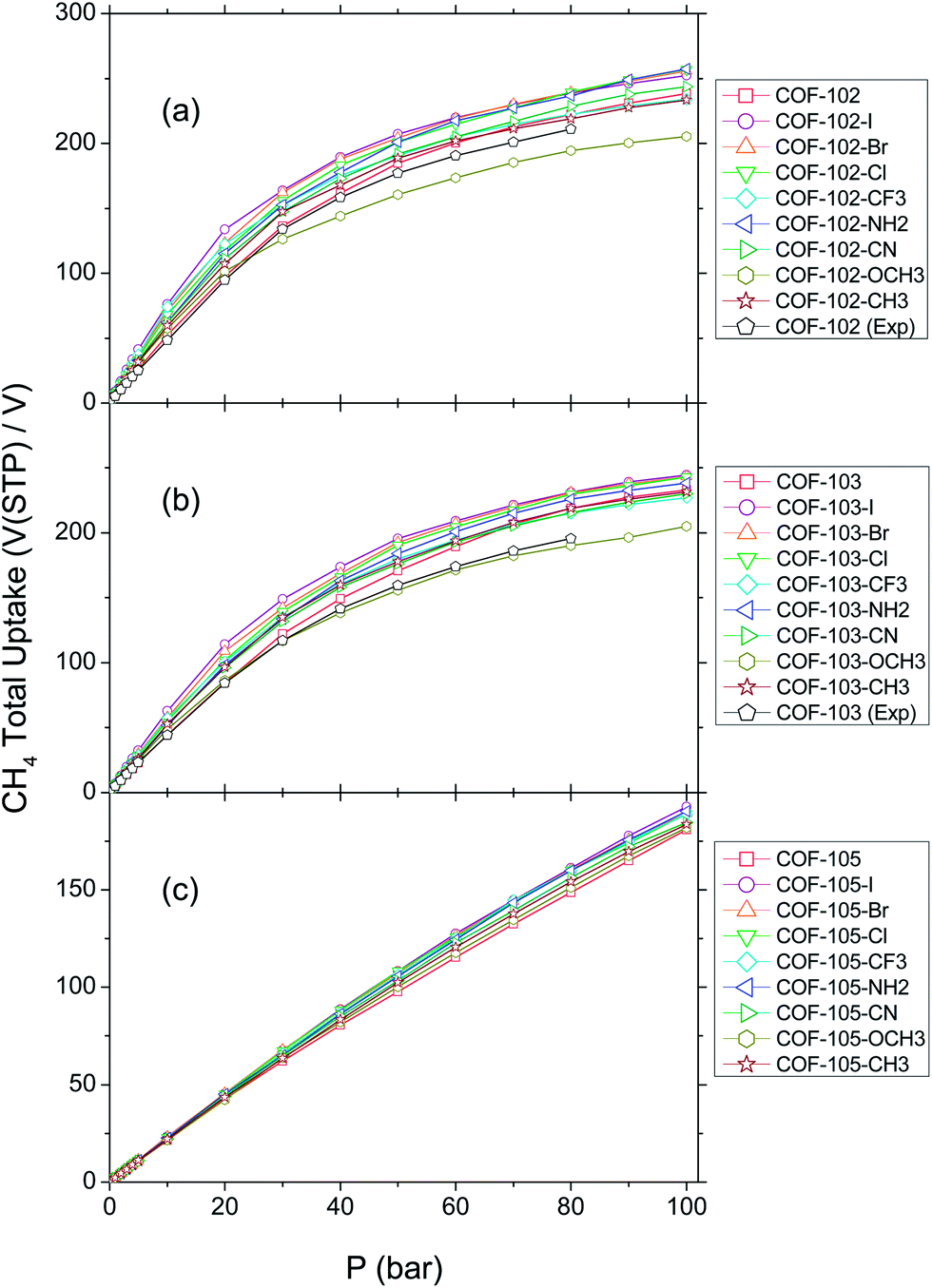 | ||
| Fig. 3 Isotherms of total volumetric methane uptake at 298 K. (a) COF-102-Xs; (b) COF-103-Xs; (c) COF-105-Xs. | ||
Among COF-102-Xs, all the substituted COFs except COF-102-OCH3 are predicted with advances in CH4 uptake and the top adsorbents at 35 bar are COF-102-Cl (169 V(STP)/V), COF-102-Br (175 V(STP)/V), and COF-102-I (176 V(STP)/V). The increments between their uptakes and COF-102 (149 V(STP)/V) are 13.4%, 17.4%, and 18.1%, respectively. The volumetric uptake of COF-102-I, namely the highest one, is at least 12.5% more than COF-102 from 0 to 50 bar. Their remarkable adsorption capabilities reveal the strong interactions between halogen groups and methane. Given that we simply substitute one H atom on each benzene ring, such amplification is quite impressive. To explain this idea further, we compare the isodensity graph of CH4 in basic COF-102 and COF-102-Cl in Fig. 4, where the density of CH4 is 5 times higher than their individual average densities, as depicted as the gray clouds. It can be seen in Fig. 4(a) that the distribution of CH4 is intermittent in COF-102. By contrast, in Fig. 4(b), there are consecutive rings around the fragments of ligand (a), indicating that –Cl groups improve the amount of adsorption mainly in this area. Besides the attraction with CH4, the substitution of –Cl also enlarges the spacing between benzene moieties, due to the Cl–Cl repulsion. As a consequence, the spacing, which is not filled out by CH4 in basic COF-102, is extended in COF-102-Cl and is readily to accommodate more CH4. Additionally, it should be noticed in Fig. 4(b) that CH4 molecules are also likely to gather on one side of the hexatomic ring B3O3, and the distribution looks like a triangle. On the three moieties of ligand (a) that are connected to the same B3O3 ring, there will be three Cl atoms at three sides of this triangle. The powerful attraction of these Cl atoms makes their inner region as a favored area for methane adsorption, causing the enhanced distribution of CH4. However, the triangle area cannot be seen on the other side of B3O3 because there will not be such Cl atoms.
 | ||
| Fig. 4 Isodensity graphs of methane in (a) COF-102 and (b) COF-102-Cl at 10 bar; (c) COF-103-CN and (d) COF-103-CF3 at 30, 60, 80 bar. The sticks refer to the structures of basic COFs, and the spheres in green, blue, and yellow highlight Cl, N, and F atoms. Clouds in gray denote methane with a density of 5 times higher than individual average density. | ||
It is also notable that COF-102-NH2 also performs well, particularly at high pressure. Indeed, its isotherm almost overlaps with COF-102-Cl, COF-102-Br, and COF-102-I from 80 to 100 bar. In comparison with halogen groups, COF-102-NH2 benefits more from its relatively large Vp (cf. Table 3) rather than from the attraction ability of –NH2. As one of the smallest ones in these eight substituents, –NH2 group occupies less space while leaving more for accommodating CH4 molecules. By contrast, COF-102-OCH3 is the modified COF with the lowest uptake. Above 25 bar, it is the only substituted COF whose loading curve is beneath COF-102. With the pressure rising, the uptake of COF-102-OCH3 falls far behind COF-102, which may be caused by the volume effect of –OCH3 since it occupies much more space than –H. Though –OCH3 substitution extends the surface (Sa) and provides substantial sites for adsorption, which could promote the uptake before 30 bar, the increment is marginal. This promotion is overwhelmed by volume effect at high pressure, causing the restriction of CH4 uptake. In the rest of COF-102-X, the isotherms of COF-102-CN and COF-102-CF3 are better than COF-102 throughout the coverage. Though weaker than –Cl, –CN still has notable contribution to the adsorbing efficiency of basic COF-102 structure, possibly ascribed to the similarity of –CN and –Cl, as –CN is known as a pseudohalogen group. COF-102-CF3 possesses ultra high adsorption amount at low pressure. Actually, its isotherm almost overlaps COF-102-Br below 30 bar, only lower than COF-102-I. –CF3 groups offer extensive surface area together with sites and F atoms strengthen the affinity to CH4, both enhancing the methane uptake to a high level. However, –CF3 is so bulky that the little Vp makes the curve flatten very quickly, even lower than COF-102 after 80 bar. COF-102-CH3 displays an increase at first but is left behind by COF-102 over 65 bar. Taking the large Vp of COF-102-CH3 (in Table 3) into account, its loading curve suggests a weak inclination of methane physisorption. The disparity between the curves of COF-102-I and COF-102-CH3 reveals that alkyl group is not an advisable choice for the improvement of CH4 storage in COFs. It may seem to be a little strange as Wilmer et al. point out that methyl, ethyl, and propyl groups occur most frequently in those MOFs with top values of methane uptake.47 However, the premise of their conclusion is that the absolute CH4 adsorption should be at least 180 V(STP)/V at 35 bar. If the uptake is around 160–170 V(STP)/V (the values in this paper), chlorine would be the functional group with the most frequent occurrence, which is corresponded with the performance of halogen groups in our study.
| Materials | Sa, Å2 | Vp, Å3 | Ratio of pore volume% |
|---|---|---|---|
| a Each surface area listed above is the van der Waals (vdW) surface of the framework, which is defined as the surface intersecting with vdW radius59 of each atom.b The volume within vdW surface is considered as the occupied volume and the pore volume is calculated by subtracting the occupied volume from the total volume of unit cell. | |||
| COF-102-Cl | 4794.29 | 13814.35 |
67.49 |
| COF-102-Br | 4946.74 | 13541.99 |
66.14 |
| COF-102-I | 5168.01 | 13173.09 |
64.14 |
| COF-102-CF3 | 5239.03 | 13139.52 |
63.24 |
| COF-102-NH2 | 4643.79 | 14108.47 |
68.95 |
| COF-102-CN | 4985.50 | 13725.13 |
66.81 |
| COF-102-OCH3 | 5745.56 | 12887.93 |
63.45 |
| COF-102-CH3 | 4928.99 | 13949.70 |
67.38 |
| COF-103-Cl | 4905.23 | 14672.14 |
68.40 |
| COF-103-Br | 5046.22 | 14401.88 |
67.13 |
| COF-103-I | 5276.21 | 14037.36 |
65.20 |
| COF-103-CF3 | 5356.80 | 14070.84 |
64.58 |
| COF-103-NH2 | 4764.75 | 14960.90 |
69.77 |
| COF-103-CN | 5137.60 | 14641.27 |
67.76 |
| COF-103-OCH3 | 5864.83 | 13767.39 |
64.59 |
| COF-103-CH3 | 4954.02 | 14777.89 |
68.29 |
| COF-105-Cl | 8612.66 | 73083.91 |
86.65 |
| COF-105-Br | 8756.27 | 72841.10 |
86.33 |
| COF-105-I | 8988.41 | 72586.06 |
85.84 |
| COF-105-CF3 | 9093.29 | 72883.89 |
85.64 |
| COF-105-NH2 | 8473.92 | 73352.61 |
86.99 |
| COF-105-CN | 8841.17 | 73239.28 |
86.48 |
| COF-105-OCH3 | 9574.65 | 71935.39 |
85.67 |
| COF-105-CH3 | 8660.37 | 73386.40 |
86.60 |
Fig. 3(b) displays the isotherms of COF-103 and its derivatives COF-103-X. The curves of COF-103-Xs stay linear in 0–20 bar and the data are chiefly affected by their ability of sorption. After then, the volume effect competes with the adsorbing interaction at medium pressure. The latter one is the controlling factor but the former one becomes distinct over 60 bar, making the growth rate slow down. The overall uptake and the variation of COF-103-X follow the trend of COF-102-X in Fig. 3(a). The comparable adsorption properties may be attributed to the similar structures of these two COFs. As shown in Fig. 1, the configuration, the linkage pattern, and even the atom number of COF-103 resemble those of COF-102, with the only distinction lying in the central atom of the organic ligand (a). COF-102 is C while COF-103 is Si, and yet they are both the 4th main group elements in periodic table. It seems peculiar that the uptake of COF-103-X is less than corresponding COF-102-X while the pore volume (Vp) of COF-103-X (∼14500 Å3 per cell) is higher than COF-102-X (∼13500 Å3 per cell). In fact, the absolute adsorbed CH4 molecules in COF-103-Xs rival those in COF-102-Xs; however, the larger total volume of COF-103-Xs causes the smaller volumetric uptake. Like COF-102-I, the total uptake of COF-103-I at 35 bar (163 V(STP)/V) exceeds other COF-103-Xs, with 19% higher than that of COF-103 (137 V(STP)/V), followed by COF-103-Br (157 V(STP)/V), COF-103-Cl (155 V(STP)/V), and COF-103-NH2 (151 V(STP)/V). Each of these four COF-103-Xs retains a decided advantage over COF-103 through 0–100 bar. Again, the cross point of COF-103-OCH3 and COF-103 is about 25 bar, indicating that –OCH3 is the least suitable one for improving adsorption.
Nevertheless, COF-103-Xs differ from COF-102-Xs in some details. Unlike COF-102, the performance of –CN and –CF3 is not ideal when combined with COF-103. The isotherm of COF-103-CF3 just holds a tiny advantage over COF-103-CH3 and COF-103-CN is even below COF-103-CH3. Even worse, the total absorbed CH4 molecules in either COF-103-CN or COF-103-CF3 are less than COF-103 above 80 bar. This could be ascribed to the gap of pore volumes between COF-103 and COF-103-CN (or COF-103-CF3), which is much larger than the gap between COF-102 and COF-102-CN (or COF-102-CF3). According to Tables 2 and 3, the difference of Vp is 1000 Å3 per cell between COF-102 and COF-102-CF3 and is 500 Å3 per cell between COF-102 and COF-102-CN. However, the Vp of COF-103-CN and COF-103-CF3 could be ∼2000 Å3 per cell less than COF-103. Such volume effect in COF-103-Xs is more important at high pressure, and thus COF-103-CN and COF-103-CF3 flatten more quickly when compared with the corresponding COF-102 substituted materials. Considering that both –CF3 and –CN are powerful substituents for methane capture, there would be another explanation for their poor values of Ntot, except for the Vp. It is possible that the most preferable sites for CH4 adsorption have already been occupied below 80 bar. As reflected in Fig. 4(c), there are mainly two preferable sites for methane storage in COF-103-CN: ring-like distribution around the moiety of ligand (a) and triangle distribution at one side of B3O3. The graph seems like the one of COF-102-Cl because –CN has similar property as –Cl group. As the pressure comes to 60 bar and then to 80 bar, the rings are still clear in COF-103-CN but the triangles become sparse, especially at their centers. It demonstrates that CH4 will gradually lose the inclination to be adsorbed at some favored sites when the pressure goes up to 80 bar. The situation of COF-103-CF3 in Fig. 4(d) resembles that of COF-103-CN, except that there are ‘clover’ areas instead of triangles above the B3O3 rings. This kind of distribution means that CH4 will gather inward to enjoy stronger attraction. It is quite evident that the ‘clover’ will transfer from a solid one to a hollow one with the pressure rising from 30 to 80 bar.
The isotherms of COF-105 and its derivatives COF-105-X in Fig. 3(c) extremely vary from Fig. 3(a) and (b), in the sense that every isotherm approximately keeps a linear relationship with pressure. The uptake of COF-105-X primarily relies on the ability of adsorption but is barely affected by volume effect, as can be seen from the fact that all the isotherms of COF-105-Xs are above COF-105. As recorded in Table 3, the pore volumes of COF-105-Xs are much bigger than COF-102-Xs and COF-103-Xs. Thus, there is enough room in COF-105-Xs, and the pores are scarcely saturated with the adsorbed CH4, making the adsorption proceed smoothly. As expected, halogen substituents (–Cl, –Br, and –I) are the most helpful candidates for adsorption, followed by –CF3, –NH2, and –CN. The fact that COF-105-CF3 outperforms COF-105-NH2 illustrates that –CF3 will have better adsorption efficiency over –NH2 without the interference from volume effect. At this time, the isotherm of COF-105-OCH3 is still beneath the other COF-105-Xs, though slightly higher than COF-105. Hence, it can be inferred that –OCH3 is an inferior choice for CH4 capture when comparing with other substituents in this work. It is noteworthy that the distribution of the isotherms is very narrow. Even the maximum difference between COF-105-I and COF-105 from 0 to 100 bar is within 11%, due to the small percentage of modification. In the cell of COF-105, there are merely 4.7% of all atoms (48 H atoms out of 1020 atoms) replaced by other substituents, whereas in COF-102 or COF-103, the ratio is almost doubled to 8.2% (48 out of 588).
As a combination of ligand (a) and (b), COF-105-X contains a much greater amount of benzene rings since it has extensive conjugated systems besides p-phenylene. With considerable Vp and Sa and massive adsorption sites offered by extra aromatic rings, the absolute amount of Ntot is higher in comparison with COF-102-X or COF-103-X. Unfortunately, this amount of increment is not commensurate with its sizable unit cell, which leads to disappointing low volumetric uptakes. The uptake of COF-105-I, also the highest one among COF-105-Xs, is merely 78 V(STP)/V at 35 bar, far below the commercial requirement. This suggests that the involvement of polycyclic aromatic structure does not serve as a prerequisite heading to outstanding volumetric storage capacity due to the exorbitant pore size (Psize) and void fraction it brings about. According to Wilmer et al.,47 0.8 seems to be the cusp of optimal void fraction since going past this point could only worsen the storage property of MOFs. As listed in Table 3, the ratio of pore volume (void fraction) of each COF-105-X oversteps that cusp, thereby lessening the capability for methane uptake. The Psize of COF-105-X is around 19 Å, and therefore the central part of the pores (excessively distant from those adsorption sites of COFs) could hardly enjoy strong interactions from the framework. As a result, the central part would suffer from a kind of spatial waste with relatively few methane molecules stored in this area.
III.3. Isosteric heat of adsorption
Apart from the uptakes, isosteric heat of adsorption, Qst, also functions to interpret the effects of substituents on methane adsorption. Qst can be calculated from the following equation,
 | (2) |
 | ||
| Fig. 5 Isosteric heat of adsorption at 298 K. (a) COF-102-Xs; (b) COF-103-Xs; (c) COF-105-Xs. | ||
As shown in Fig. 5, all the derived COFs display higher isosteric heats than the basic COF. This can be reflected in the isotherms in Fig. 3, where substituted COFs exceed the corresponding basic COFs in their total amount of adsorbed molecules and have larger slopes at low pressure. To be specific, COFs with –Cl, –Br, –I, and –CF3 groups commonly have higher Qst values, which demonstrate that they possess stronger adsorptive capabilities than the others. Nonetheless, the property of a physisorptive material is by no means simply determined by Qst, since it can just serve as a reference for the analysis of adsorption. To further illustrate this point, we take –CF3, –OCH3, and –NH2 as examples. The former two maintain higher Qst compared to the latter one; however, the sequence of their volumetric uptakes is almost inverse. The uptakes of COFs combined with –CF3 or –OCH3 are restricted as a result of low Vp, as can be seen in COF-102-Xs and COF-103-Xs at high pressure in Fig. 3(a) and (b). On the contrary, COF-102-NH2 and COF-103-NH2 are featured with moderate Qst, but their pore volumes ensure the adsorption of large amount of CH4. Furthermore, exceedingly high Qst may bring some undesired outcomes in practice.57 For materials like activated carbon, if the Qst reaches about 20 kJ mol−1, both the rates of adsorption and desorption of methane will slow down. Other than this, as an exothermal process, there might be an increase in temperature during the adsorption of methane if the heat cannot be dispersed effectively, which will negatively influence or even prohibit the adsorption in return. This correlation between high heat of adsorption and low uptake has been demonstrated by a recent theoretical study by Ihm et al.58
III.4. Potential for application
From the total uptake of COF-102-Xs and COF-103-Xs, we calculated their excess uptakes with eqn (1), and the results are depicted in Fig. 6. Here we just show –Cl, –Br, –I, and –NH2 substituted COFs because they are the best substitution groups for the enhancement of methane storage, as shown in Section III.2. Complete results are provided in the ESI,† where excess CH4 isotherms of all the COF-102-Xs and COF-103-Xs are reported. In this part, we do not include COF-105-Xs because the highest total uptake among them at 35 bar (COF-105-I) cannot attain 80 V(STP)/V, meaning that their excess uptake will definitely be rather poor. Hence, COF-105-Xs can hardly be put into practical use for CH4 uptakes.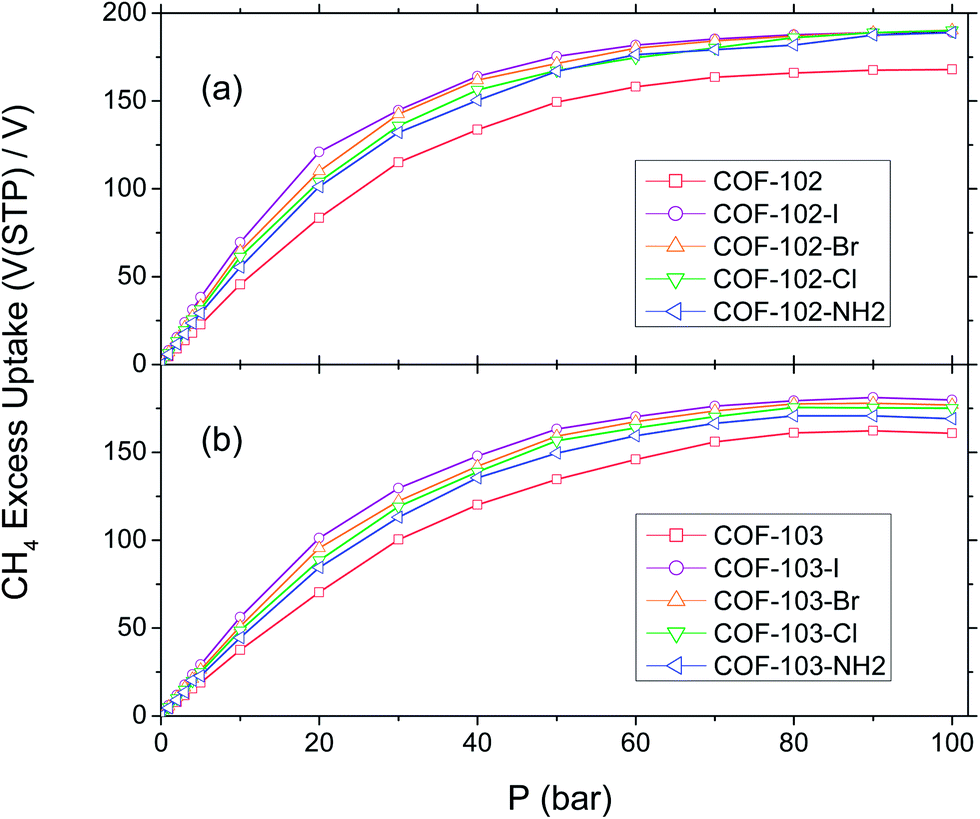 | ||
| Fig. 6 Isotherms of excess volumetric methane uptake at 298 K. (a) COF-102-Xs and (b) COF-103-Xs. | ||
According to Fig. 6(a), the excess uptake at 35 bar is 156 V(STP)/V for COF-102-I, 153 V(STP)/V for COF-102-Br, 148 V(STP)/V for COF-102-Cl, and 143 V(STP)/V for COF-102-NH2. These might not be able to parallel the best MOF (PCN-14),28 and there still exists a gap between our results and the top performers in carbonaceous materials such as monolithic carbon4 and carbon nanotubes7 predicted by simulations (∼210 V(STP)/V). Nevertheless, these COFs have already exhibited distinct improvements. In comparison with COF-102 (125 V(STP)/V), the percentage of promotion of these four derivatives ranges from 14.4% to 24.8%, higher than that in their total uptakes (13.4–18.1%), which could be attributed to their smaller Vp. Based on eqn (1), a large Ntot together with a low Vp will lead to high Nexc and thus, it is reasonable that the disparity between the Nexc of COF-102-Xs and COF-102 is expanded. The trends are similar in COF-103-Xs, though the values are slightly lower. The excess uptakes for COF-103-I, COF-103-Br, COF-103-Cl, and COF-103-NH2 are 138 V(STP)/V, 133 V(STP)/V, 130 V(STP)/V, and 125 V(STP)/V, 12.6–24.3% higher than COF-103 (111 V(STP)/V).
In evaluating the potential of materials for CH4 storage, another factor is also considered to be critical. Methane delivery is defined as the volume of CH4 released from the adsorbent at room temperature when the pressure is reduced from 35 to 1 bar. This concept has been paid much attention in carbonaceous materials and introduced in COFs.45,56 Fig. 7(a) and (b) display the CH4 delivery, calculated by subtracting the total uptake at 1 bar from those above 1 bar. Again, we merely include the best ones among COF-102-Xs and COF-103-Xs, and the complete results can be found in the ESI.† COF-105-Xs are not involved for the reason that their CH4 deliveries must be lower than total uptakes.
 | ||
| Fig. 7 Isotherms of methane delivery at 298 K. (a) COF-102-Xs and (b) COF-103-Xs. | ||
In contrast with Fig. 6, the difference of deliveries between COF-102-Xs (or COF-103-Xs) and COF-102 (or COF-103) shown in Fig. 7 is smaller. However, the methane deliveries are higher than excess uptakes and even close to the DOE target at 35 bar. The methane deliveries of –Cl, –Br, –NH2, and –I in COF-103 vary from 145 V(STP)/V to 156 V(STP)/V at 35 bar. Inspiringly, the values of COF-102-Cl, COF-102-Br, COF-102-I, and COF-102-NH2 are 165 V(STP)/V, 169 V(STP)/V, 169 V(STP)/V, and 160 V(STP)/V respectively. From a perspective of application, COF-102-I and COF-102-Br have good potentials as they are approaching the goal of 180 V(STP)/V.
IV. Conclusion
In this study, 24 covalent organic frameworks are designed on the basis of 3 published COFs (COF-102, COF-103, and COF-105). The original structures are modified by replacing one H atom on each benzene ring in the fragments of ligand (a) in Fig. 1(a) with eight substituent groups, namely –Cl, –Br, –I, –CF3, –NH2, –CN, –OCH3, and –CH3. After acquiring the optimized stable configurations, we study their methane adsorption properties at room temperature by using GCMC simulation. To validate our methodology and models, we choose 5 representative COFs (COF-6, COF-8, COF-10, COF-102, and COF-103) and simulate their excess uptakes from 0 to 100 bar, which are comparable with experimental results.As shown in the total uptake isotherms, all of them maintain good linearity at the beginning. Within this area, the values and the variation trends of the curves are controlled by the attraction ability of adsorbents. All the substituted COFs are better than original COFs below 20 bar, attributed to their stronger interactions with CH4. When the pressure is above 20 bar, the isotherms of COF-102-Xs, COF-103-Xs, and COF-105-Xs have different tendencies. For COF-102-Xs and COF-103-Xs, the slopes start to decrease and the uptakes display approaches to saturation, due to the volume effect. For COF-105-Xs, their large Psize and Vp guarantee that the isotherms would increase constantly and scarcely be filled with CH4. It can be inferred from the three sets of materials that it is the reasonable combination of adsorption sites (related to surface area), pore volume, and attraction ability (related to isosteric heat) that results in an ideal uptake of methane storage.
In general, the uptakes and Qst of COF-102-Xs, COF-103-Xs, and COF-105-Xs suggest that among these eight groups, –NH2 and halogen-containing groups, such as –Cl, –Br, –I, and –CF3 have excellent abilities to ameliorate adsorption. Specifically, their advantages are not the same. Single-site groups such as –Cl, –Br, and –I benefit from their intrinsic strong interactions with methane. The isodensity graphs of methane in COF-102 and COF-102-Cl imply that the region with greatest improvement is around the fragments of ligand (a), since consecutive ring-like distribution of CH4 can be seen in COF-102-Cl while there are only discontinuous pieces in basic COF-102. Multi-sites group, –CF3, contributes to the uptake by adding large amount of sites to interact with CH4 and by providing F atoms to enhance the attraction with CH4. Unlike halogen-containing groups with high Qst values, –NH2 enjoys the balance between moderate adsorptive ability and small occupied volume, which leaves plenty of space available for CH4 molecules. In contrast, –CN achieves nice performance in COF-102 but bad in COF-103 and COF-105 while –OCH3 and –CH3 are confirmed as poorer choices.
Overall, based on the data of total uptakes, excess uptakes, deliveries, and isosteric heats, –Cl, –Br, –I, and –NH2 are found to be promising substituents to strengthen the capability of methane storage, where COF-102-I and COF-102-Br are most potential for practical application. Further study on double substitution is in progress.
Acknowledgements
This work is supported by NSFC (21373118, 21073097), NCET-10-0512, and the MOE Innovation Team (IRT13022) of China.References
- J. P. Kennett, K. G. Cannariato, I. L. Henry and R. J. Behl, Science, 2000, 288, 128–133 CrossRef CAS PubMed.
- S. Watanabe, K. Saito and R. Ohmura, Cryst. Growth Des., 2011, 11, 3235–3242 CAS.
- S. Takeya, A. Yoneyama, K. Ueda, H. Mimachi, M. Takahashi, K. Sano, K. Hyodo, T. Takeda and Y. Gotoh, J. Phys. Chem. C, 2012, 116, 13842–13848 CAS.
- K. R. Matranga, A. L. Myers and E. D. Glandt, Chem. Eng. Sci., 1992, 47, 1569–1579 CrossRef CAS.
- T. Burchell and M. Rogers, SAE Tech. Pap. Ser., 2000, 01, 2205 Search PubMed.
- V. C. Menon and S. Komarneni, J. Porous Mater., 1998, 5, 43–58 CrossRef CAS.
- D. Cao, X. Zhang, J. Chen, W. Wang and J. Yun, J. Phys. Chem. B, 2003, 107, 13286–13292 CrossRef CAS.
- J. Alcañiz-Monge, M. A. de la Casa-Lillo, D. Cazorla-Amorós and A. Linares-Solano, Carbon, 1997, 35, 291–297 CrossRef.
- D. Lozano-Castelló, D. Cazorla-Amorós, A. Linares-Solano and D. F. Quinn, Carbon, 2002, 40, 2817–2825 CrossRef.
- A. K. Nowak, C. J. J. Den Ouden, S. D. Pickett, B. Smit, A. K. Cheetham, M. F. M. Post and J. M. Thomas, J. Phys. Chem., 1991, 95, 848–854 CrossRef CAS.
- S. Zhang, O. Talu and D. T. Hayhurst, J. Phys. Chem., 1991, 95, 1722–1726 CrossRef CAS.
- N. Texier-Mandoki, J. Dentzer, T. Piquero, S. Saadallah, P. David and C. Vix-Guterl, Carbon, 2004, 42, 2744–2747 CrossRef CAS.
- M. G. Nijkamp, J. E. M. J. Raaymakers, A. J. van Dillen and K. P. de Jong, Appl. Phys. A, 2001, 72, 619–623 CrossRef CAS.
- H. Li, M. Eddaoudi, M. O'Keeffe and O. M. Yaghi, Nature, 1999, 402, 276–279 CrossRef CAS.
- H. K. Chae, D. Y. Siberio-Perez, J. Kim, Y. Go, M. Eddaoudi, A. J. Matzger, M. O'Keeffe and O. M. Yaghi, Nature, 2004, 427, 523–527 CrossRef CAS PubMed.
- H. Furukawa, M. A. Miller and O. M. Yaghi, J. Mater. Chem., 2007, 17, 3197–3204 RSC.
- W. Zhou, H. Wu, M. R. Hartman and T. Yildirim, J. Phys. Chem. C, 2007, 111, 16131–16137 CAS.
- J. L. C. Rowsell, A. R. Millward, K. S. Park and O. M. Yaghi, J. Am. Chem. Soc., 2004, 126, 5666–5667 CrossRef CAS PubMed.
- M. Eddaoudi, J. Kim, N. Rosi, D. Vodak, J. Wachter, M. O'Keeffe and O. M. Yaghi, Science, 2002, 295, 469–472 CrossRef CAS PubMed.
- T. Düren, L. Sarkisov, O. M. Yaghi and R. Q. Snurr, Langmuir, 2004, 20, 2683–2689 CrossRef.
- T. Düren, Y. S. Bae and R. Q. Snurr, Chem. Soc. Rev., 2009, 38, 1237–1247 RSC.
- X. Zhou, Z. Zhang, B. Li, F. Yang, Y. Peng, G. Li, Z. Shi, S. Feng and J. Li, New J. Chem., 2013, 37, 425–430 RSC.
- Z. Zhang, Y. Zhao, Q. Gong, Z. Li and J. Li, Chem. Commun., 2013, 49, 653–661 RSC.
- J. L. C. Rowsell, E. C. Spencer, J. Eckert, J. A. K. Howard and O. M. Yaghi, Science, 2005, 309, 1350–1354 CrossRef CAS PubMed.
- R. Q. Snurr, J. T. Hupp and S. T. Nguyen, AIChE J., 2004, 50, 1090–1095 CrossRef CAS.
- B. Liu, Q. Yang, C. Xue, C. Zhong, B. Chen and B. Smit, J. Phys. Chem. C, 2008, 112, 9854–9860 CAS.
- J. J. Gutierrez-Sevillano, A. Caro-Perez, D. Dubbeldam and S. Calero, Phys. Chem. Chem. Phys., 2011, 13, 20453–20460 RSC.
- S. Ma, D. Sun, J. M. Simmons, C. D. Collier, D. Yuan and H. Zhou, J. Am. Chem. Soc., 2008, 130, 1012–1016 CrossRef CAS PubMed.
- H. Wu, W. Zhou and T. Yildirim, J. Am. Chem. Soc., 2009, 131, 4995–5000 CrossRef CAS PubMed.
- A. P. Cote, A. I. Benin, N. W. Ockwig, M. O'Keeffe, A. J. Matzger and O. M. Yaghi, Science, 2005, 310, 1166–1170 CrossRef CAS PubMed.
- A. P. Cote, H. M. El-Kaderi, H. Furukawa, J. R. Hunt and O. M. Yaghi, J. Am. Chem. Soc., 2007, 129, 12914–12915 CrossRef CAS PubMed.
- H. M. El-Kaderi, J. R. Hunt, J. L. Mendoza-Cortés, A. P. Cote, R. E. Taylor, M. O'Keeffe and O. M. Yaghi, Science, 2007, 316, 268–272 CrossRef CAS PubMed.
- J. R. Hunt, C. J. Doonan, J. D. LeVangie, A. P. Cote and O. M. Yaghi, J. Am. Chem. Soc., 2008, 130, 11872–11873 CrossRef CAS PubMed.
- R. Babarao and J. Jiang, Energy Environ. Sci., 2008, 1, 139–143 CAS.
- G. Garberoglio and R. Vallauri, Microporous Mesoporous Mater., 2008, 116, 540–547 CrossRef CAS.
- S. S. Han, H. Furukawa, O. M. Yaghi and W. A. Goddard, J. Am. Chem. Soc., 2008, 130, 11580–11581 CrossRef CAS PubMed.
- H. Furukawa and O. M. Yaghi, J. Am. Chem. Soc., 2009, 131, 8875–8883 CrossRef CAS PubMed.
- E. Klontzas, E. Tylianakis and G. E. Froudakis, J. Phys. Chem. C, 2008, 112, 9095–9098 CAS.
- E. Tylianakis, E. Klontzas and G. E. Froudakis, Nanoscale, 2011, 3, 856–869 RSC.
- J. Lan, D. Cao and W. Wang, Langmuir, 2010, 26, 220–226 CrossRef CAS PubMed.
- J. Lan, D. Cao and W. Wang, J. Phys. Chem. C, 2010, 114, 3108–3114 CAS.
- F. Li, J. Zhao, B. Johansson and L. Sun, Int. J. Hydrogen Energy, 2010, 35, 266–271 CrossRef CAS.
- J. Lan, D. Cao, W. Wang and B. Smit, ACS Nano, 2010, 4, 4225–4237 CrossRef CAS PubMed.
- Z. Yang and D. Cao, J. Phys. Chem. C, 2012, 116, 12591–12598 CAS.
- J. L. Mendoza-Cortés, T. A. Pascal and W. A. Goddard, J. Phys. Chem. A, 2011, 115, 13852–13857 CrossRef PubMed.
- D. Frenkel and B. Smit, Understanding Molecular Simulation: From Algorithms to Applications (Computational Science), Academic Press, 2nd edn, 2002 Search PubMed.
- C. E. Wilmer, M. Leaf, C. Y. Lee, O. K. Farha, B. G. Hauser, J. T. Hupp and R. Q. Snurr, Nat. Chem., 2012, 4, 83–89 CrossRef CAS PubMed.
- S. L. Mayo, B. D. Olafson and W. A. Goddard, J. Phys. Chem., 1990, 94, 8897–8909 CrossRef CAS.
- R. Schmid and M. Tafipolsky, J. Am. Chem. Soc., 2008, 130, 12600–12601 CrossRef CAS PubMed.
- S. J. Goodbody, K. Watanabe, D. MacGowan, J. P. R. B. Walton and N. Quirke, J. Chem. Soc., Faraday Trans., 1991, 87, 1951–1958 RSC.
- E. Atci, I. Erucar and S. Keskin, J. Phys. Chem. C, 2011, 115, 6833–6840 CAS.
- L. Chen, L. Grajciar, P. Nachtigall and T. Düren, J. Phys. Chem. C, 2011, 115, 23074–23080 CAS.
- S. Wang, Energy Fuels, 2007, 21, 953–956 CrossRef CAS.
- A. L. Myers, AIChE J., 2002, 48, 145–160 CrossRef CAS.
- G. Garberoglio, Langmuir, 2007, 23, 12154–12158 CrossRef CAS PubMed.
- J. L. Mendoza-Cortés, S. S. Han, H. Furukawa, O. M. Yaghi and W. A. Goddard, J. Phys. Chem. A, 2010, 114, 10824–10833 CrossRef PubMed.
- D. Lozano-Castelló, J. Alcañiz-Monge, M. A. de la Casa-Lillo, D. Cazorla-Amorós and A. Linares-Solano, Fuel, 2002, 81, 1777–1803 CrossRef.
- Y. Ihm, V. R. Cooper, N. C. Gallego, C. I. Contescu and J. R. Morris, J. Chem. Theory Comput., 2013, 10, 1–4 CrossRef.
- A. Bondi, J. Phys. Chem., 1964, 68, 441–451 CrossRef CAS.
- B. Assfour and G. Seifert, Microporous Mesoporous Mater., 2010, 133, 59–65 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: The coordinates of published and optimized covalent organic frameworks, and the complete graphs of CH4 excess uptake and CH4 delivery for COF-102-Xs and COF-103-Xs. See DOI: 10.1039/c3ra47429a |
| This journal is © The Royal Society of Chemistry 2014 |