
Diastereodivergent total synthesis of mosquito oviposition pheromone†
Abstract
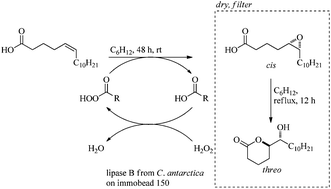
The unnatural threo-6-acetoxy-5-hexadecanolide and the natural mosquito oviposition pheromone erythro-6-acetoxy-5-hexadecanolide were synthesized in a diastereodivergent fashion in 44% and 33% overall yield respectively from 5-bromovaleric acid and undecanal. The key step utilized a chemoenzymatic epoxidation-lactonization of a naturally available fatty acid to form the 6-hydroxy-5-hexadecanolide core.
 Please wait while we load your content...
Please wait while we load your content...

