
p-Aminobenzoic acid polymorphs under high pressures†
Abstract
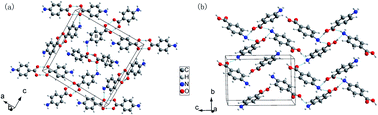
The effect of high pressure on two forms (α, β) of p-aminobenzoic acids (PABA) is studied in a diamond anvil cell using in situ Raman spectroscopy. Previous research showed that α-PABA undergoes a phase transition, and β-PABA is transformed to α-PABA at high temperatures. In the present study, we investigate whether a new polymorph or a transformation between the two polymorphs occurs upon the application of pressure. Experimental results reveal that the two forms remain stable up to ∼13 GPa. Ab initio calculations are performed to account for the changes in unit cell parameters, molecular arrangements, and hydrogen bonds. Polymerization is observed in type B molecules of α-PABA through the calculated geometric parameters of hydrogen bonds. Based on a systematic comparison of the subtle structural changes, anisotropic characteristic, and various interactions of the two polymorphs, we propose that the stability of α-form crystals is associated with the special dimer structure. The stability of the β-form is attributed to hydrogen-bonded networks with four-membered ring construction.
 Please wait while we load your content...
Please wait while we load your content...

