
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Hierarchically porous carbon supports enable efficient syngas production in electrified reactive capture†
Hengzhou
Liu‡
 a,
Heejong
Shin‡
a,
Xiao-Yan
Li‡
a,
Guangcan
Su‡
ab,
Pengfei
Ou
a,
Yong
Wang
a,
Lihaokun
Chen
c,
Jiaqi
Yu
a,
Yuanjun
Chen
a,
Rong
Xia
a,
Geonhui
Lee
d,
Kug-Seung
Lee
e,
Christine
Yu
a,
Peiying
Wang
a,
Deokjae
Choi
a,
Daojin
Zhou
a,
Cong
Tian
a,
Issam
Gereige
f,
Ammar
Alahmed
f,
Aqil
Jamal
f,
Omar K.
Farha
a,
Shannon W.
Boettcher
cg,
Jennifer B.
Dunn
b,
Ke
Xie
*a and
Edward H.
Sargent
*ah
a,
Heejong
Shin‡
a,
Xiao-Yan
Li‡
a,
Guangcan
Su‡
ab,
Pengfei
Ou
a,
Yong
Wang
a,
Lihaokun
Chen
c,
Jiaqi
Yu
a,
Yuanjun
Chen
a,
Rong
Xia
a,
Geonhui
Lee
d,
Kug-Seung
Lee
e,
Christine
Yu
a,
Peiying
Wang
a,
Deokjae
Choi
a,
Daojin
Zhou
a,
Cong
Tian
a,
Issam
Gereige
f,
Ammar
Alahmed
f,
Aqil
Jamal
f,
Omar K.
Farha
a,
Shannon W.
Boettcher
cg,
Jennifer B.
Dunn
b,
Ke
Xie
*a and
Edward H.
Sargent
*ah
aDepartment of Chemistry, Northwestern University, 2145 Sheridan Road, Evanston, IL 60208, USA. E-mail: ke-xie@northwestern.edu; ted.sargent@northwestern.edu
bDepartment of Chemical and Biological Engineering, Northwestern University, 2145 Sheridan Road, Evanston, IL 60208, USA
cDepartment of Chemistry and the Oregon Center for Electrochemistry, University of Oregon, Eugene, OR 97403, USA
dDepartment of Electrical and Computer Engineering, University of Toronto, 10 King's College Rd, Toronto, ON M5S 3G4, Canada
ePohang Accelerator Laboratory (PAL), Pohang University of Science and Technology (POSTECH), Pohang 37673, Republic of Korea
fResearch and Development Center, Saudi Aramco, Dhahran 31311, Saudi Arabia
gDepartment of Chemical & Biomolecular Engineering and Department of Chemistry, University of California, Berkeley, California 94720, USA
hDepartment of Electrical and Computer Engineering, Northwestern University, 2145 Sheridan Rd, Evanston, IL 60208, USA
First published on 29th May 2025
Abstract
Direct-air capture (DAC) of CO2 often uses alkali hydroxides (e.g. KOH) as sorbent, and relies on an energy-intensive thermal CaCO3/Ca(OH)2 step to release CO2 and regenerate the alkali hydroxide. Reactive capture instead uses alkali carbonate post-capture liquid as feedstock, seeking to convert the captured CO2 to value-added products while regenerating the capture liquid. Here we investigate the origins of low prior performance in electrochemical reactive capture systems, finding that the catalyst becomes starved of CO2 even at moderate current densities leading to a rapid decline in faradaic efficiency (FE). We then study how the catalyst support can be redesigned to tackle this problem, and construct hierarchical carbon supports featuring interconnected mesopores and micropores, our purpose to increase the interaction between in situ generated CO2, i-CO2 – the limiting reagent – and the catalyst. We find that the attachment chemistry of the catalyst to the support is critical: only when we disperse and tether the molecular catalyst do we prevent catalyst aggregation and deactivation under bias. We report as a result carbonate electrolysis at 200 mA cm−2 at 2.9 V with FE of 47 ± 1% for CO, this corresponding to an energy efficiency (EE) to 2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 syngas of 50% at 200 mA cm−2 when H2 is added using a water electrolyzer. This represents a 1.5× improvement in EE at this current density relative to the most efficient prior carbonate electrolysis reports. The CO FE remains above 40% at current densities as high as 500 mA cm−2, and all systems studied herein achieve < 1% CO2 in the outlet stream. The cradle-to-gate carbon intensity is lowered to −1.49 tonCO2 per tonsyngas as a result of the increase in EE, and a CO2-free tailgas stream is provided that minimizes separation costs.
:1 syngas of 50% at 200 mA cm−2 when H2 is added using a water electrolyzer. This represents a 1.5× improvement in EE at this current density relative to the most efficient prior carbonate electrolysis reports. The CO FE remains above 40% at current densities as high as 500 mA cm−2, and all systems studied herein achieve < 1% CO2 in the outlet stream. The cradle-to-gate carbon intensity is lowered to −1.49 tonCO2 per tonsyngas as a result of the increase in EE, and a CO2-free tailgas stream is provided that minimizes separation costs.
Broader contextOn the path to sustainable fuels – hydrocarbons having a lower carbon intensity than legacy fossil hydrocarbons – one flow of interest is direct-air capture (DAC) of CO2 followed either by solid oxide electrolysis cell (SOEC), or by reverse water–gas shift (RWGS), to syngas, readily further processed to long-chain hydrocarbons. Unfortunately, the high energy intensity of each step – both of CO2 capture-and-release, and of CO2 upgrade – leads to an energy cost of 55–65 GJ per tonsyngas. Here we pursue reactive capture, which uses alkali carbonate post-direct-air-capture liquid as feedstock, converting the captured CO2 to value-added products while regenerating the capture liquid. Prior reactive capture to syngas has been limited to 32% efficiency at 200 mA cm−2 – the result of the catalyst becoming CO2-starved. We develop hierarchical carbon supports, and this increases interaction between in situ generated CO2, i-CO2 – the limiting reagent – and the catalyst, heterogenized cobalt phthalocyanine. We report as a result carbonate electrolysis at 200 mA cm−2 having energy efficiency to 2:1 syngas of 50%. Life cycle assessment shows that – when energy is supplied using electricity having the carbon intensity of wind –CO2 emissions are reduced from today's coal-syngas of 2.3 t CO2e per t syngas to a negative emission of –1.5 t CO2e per t syngas, each cradle-to-gate. The minimum selling price (MSP) of syngas produced via the reactive capture is estimated at US$770 per t, below that for DAC-SOEC (US$1270 per t) and DAC-RWGS (US$1020 per t).
|
Main
CO2(g) capture from the atmosphere, followed by electrochemical conversion, provides an avenue to diminish the carbon-intensity of fuels and chemicals.1,2 Because CO2(g) is a weak acid and dilute in the atmosphere, strong aqueous alkali hydroxide solutions, such as KOH, are often used in direct air capture (DAC);3 however, the release from the captured state of a stream of concentrated CO2, thus regenerating hydroxide from alkaline carbonates, requires additional energy-intensive steps, these typically totaling 8–10 GJ per tonCO2 (Fig. 1a, Route 1 and Fig. 1b).3 When – in the context of carbon capture and utilization (CCU) – carbon monoxide (CO) is then produced from the captured CO2, the DAC process contributes 13–16 GJ per tonCO (GJ per tonCO2 * 44 g mol−1 CO2/28 g mol−1 CO). This is considerable when referenced to the ∼10 GJ per ton lower heating value (LHV) of CO.4 | ||
| Fig. 1 Comparison between sequential and integrated processes for DAC and CO2 upgrade. (a) Sequential process of direct air capture followed by CO2 electrolysis (Route 1) vs. an integrated process of reactive CO2 capture (Route 2). (b) Gibbs free energy comparison between Route 1 and 2. (c) A carbonate electrolyzer fed using 1.5 M K2CO3 as catholyte. Carbonate passes through the hydrophilic carbon paper, the catalyst layer, and the hydrophilic interposer layer (thickness: 135 μm) via convection and diffusion. CO32− reacts with H+ from the BPM to generate i-CO2. The i-CO2 passes back from the membrane interface and goes through the interposer layer to the catalyst layer for electroreduction to CO. (d) The transport of i-CO2 to CoPc molecules dispersed on planar (P), nanoporous (NP), and hierarchically-porous (HP) carbon supports. | ||
In reactive capture systems, CO2 capture and upgrade are instead combined into a single system (Fig. 1a, Route 2 and Fig. 1b).5–11 Reactive capture can be driven electrochemically to synthesize syngas,12,13 CO,14,15 and ethylene.11,16 For the kinetics and capital efficiency of the contactor to enable practical DAC, one may interact the pH ∼ 14 capture liquid with CO2 until it reaches the pH ∼ 12, that of carbonate; however, further reaction down to KHCO3 (pH 8.5) places too high a demand on contactor fan electricity since capture kinetics decelerate under bicarbonate conditions (Fig. S1 and Note S1, ESI†).17
Thus, in order to enable direct use of post-capture liquid, the electroreduction of carbonate post-capture liquid is seeing increased attention, and bipolar membrane (BPM) electrolyzers (Fig. S2 and Note S2, ESI†) have been limited by the overall syngas energy efficiencies (EE) of 39% at 100 mA cm−2 in light of a CO faradaic efficiency (FE) of 28% and a best prior cell voltage (Vcell) of 3.5 V at 100 mA cm−2.12 This performance further declines at higher current densities, the syngas EE reaching 32% at 200 mA cm−2.12
Mechanistic origins of low CO FE in prior carbonate-to-syngas systems
Carbonate-to-syngas systems rely on the in situ generation of CO2 (i-CO2), this is achieved by reacting carbonate with H3O+ from water dissociation in the BPM junction, the proton H+ then driven out of the cation-exchange layer (CEL).12,18,19 Unfortunately, reactive capture presents conflicting requirements: low pH at the CEL favors the release of i-CO2; but a high local pH increases the desired CO2 reduction process relative to HER.12,13,16The conflict becomes even more acute at higher current densities: prior reactive capture systems have rapidly lost FE above 100 mA cm−2. Focusing on the supply and utilization of i-CO2, the i-CO2 generation rate from the BPM at 100 mA cm−2 is ∼0.7 mL min−1 cm−2 (Note S3, ESI†), yet this is lower than the mass-transport limit of ∼5 mL min−1 cm−2 required to supply CO2 at the rate needed in gas-fed CO2 electrolyzers.20,21 Thus, the catalyst becomes starved of CO2 under high current density conditions.12 Quantitatively (Fig. S3–S5, ESI†), at 100–600 mA cm−2, the maximum i-CO2 supply rate shows a volcano trend, and is insufficient to meet i-CO2 demand for CO2-to-CO conversion.
The design of porous carbon supports for i-CO2 transport to catalyst sites
We sought strategies to resolve this conflict in requirements, and considered how engineering of the porosity of the catalyst support can offer a degree of freedom for this purpose. A planar support (Fig. 1d, left) offers little opportunity to enhance the availability of active catalytic sites to interact with inbound i-CO2. Nanoporous carbon (Fig. 1d, middle), with a typical pore diameter of <2 nm, increases surface area, but the tight pores may inhibit diffusion of i-CO2.In contrast, hierarchical carbon supports – having a first, larger, pore radius (mesopore radius ∼2–50 nm, Fig. 1d, right), and a second length scale (micropore <2 nm diameter) – may potentially offer the combination of enhanced catalyst surface area, accompanied by better transport of i-CO2. We term the control and candidate supports planar (P), nanoporous (NP), and hierarchically porous (HP: an admixture of mesopores further interpenetrated by micropores).
Brunauer–Emmett–Teller (BET) analysis showed specific surface areas of 300 (P), 920 (NP), and 1960 (HP) m2 g−1 supports (Fig. S6, ESI†). BET also enables estimation of effective pore sizes and their distributions (Fig. S6b, ESI†), and is consistent, in the case of HP, with prominent porosity components at both the mesopore and the micropore length scales. HP (after optimizing the meso-to-micro ratio) exhibits an 11× higher volumetric density of mesopores than does P. The meso-to-micro pore volume ratio in HP is 2× that of NP (Fig. S7, ESI†). Scanning transmission electron microscopy (STEM) analysis (Fig. S8, ESI†) shows (more qualitatively) the same trend as seen in the Fourier domain via BET.
We then examined reactive capture performance in BPM-based electrolyzers (Fig. S9, ESI†) using each support. Unfortunately, and to our surprise, the HP support exhibited a peak FECO of 34% at 200 mA cm−2. Its performance declined further with increasing current density, dropping to <30% at current densities >300 mA cm−2 (Fig. 2a).
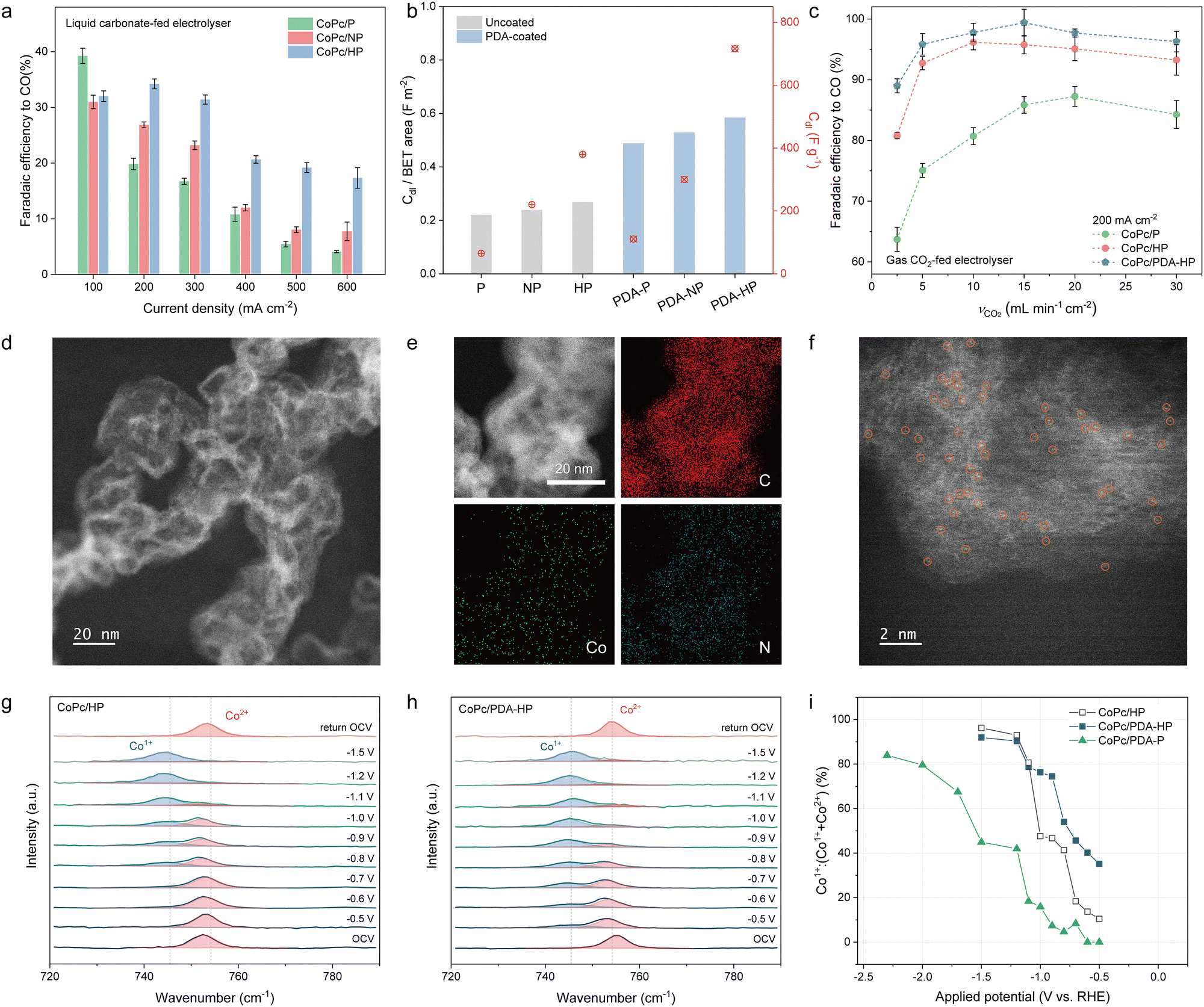 | ||
| Fig. 2 Investigation of i-CO2 transport and molecular catalyst dispersion for uncoated vs. PDA-coated porous carbon supports. (a) Faradaic efficiency to CO for liquid carbonate-fed electrolyzer using CoPc catalyst loaded on different carbon supports. We used the optimum catalyst loading of 2 mg cm−2 (CoPc + carbon). The catalyst contains 40 wt% carbon by mass. (b) Cdl/BET ratio on uncoated and PDA-coated carbon. (c) Faradaic efficiency to CO for gas-CO2-fed electrolyzer as a function of CO2 flow rate at 200 mA cm−2. 1.5 M K2CO3 was used as catholyte and anolyte. The optimal meso-to-micro ratio of HP was used for electrochemical testing in Figure (a)–(c). (d) Low-magnification STEM image, (e) HAADF-STEM-EDS mapping, and (f) Atomic-resolution HAADF-STEM images of CoPc/PDA-HP. The individual bright dots marked by orange circles are associated with the Co centres of CoPc molecules. (g) and (h) In situ Raman spectra of CoPc/HP and CoPc/PDA-HP catalysts in 1.5 M K2CO3. The in situ analysis was performed in a flow cell with three compartments. CoPc/PDA-HP was coated on the carbon paper substrate as the cathode, and Pt/C as the anode. (i) Co1+:(Co1+ + Co2+) ratio as a function of applied potential for CoPc catalysts loaded on a variety of carbon supports: HP, PDA-HP, and PDA-P. | ||
Electrochemical impedance spectroscopy (EIS) allows us to estimate total capacitance (Cdl), related to electrochemically active surface area accessible to the reaction (Fig. S10 and Note S4, ESI†).22,23 By normalizing Cdl to the BET surface area, we estimate the fraction of active sites participating in the electrochemical reaction, referred to as active site utilization. We observed that each class of carbon support had a low Cdl/BET value in the 0.2–0.3 range (Fig. 2b). Thus more than half of CoPc molecules are inactive.
We posited that this inactivity could be the result of CoPc being poorly-dispersed on support. CoPc molecules have strong π-stacking interactions and are known to form micron-sized aggregates.24,25 These are expected to reduce catalyst conductivity, hinder reactant access to the catalytic centers, and thus diminish turnover frequency.26,27
Chemical strategies to disperse CoPc inside hierarchical porous supports
We sought surface treatments that would better disperse and immobilize CoPc on each carbon support. Polydopamine (PDA) coatings have previously been found to enable the deposition of adherent and uniform layers on a variety of substrates.28 We deposited PDA using self polymerization (Methods) and found using elemental analysis a nitrogen content of ∼2% by weight (Table S2, ESI†), with TEM, XPS, EDX, and FTIR indicating PDA coating on each class of carbon support (Fig. S11–S15, ESI†). The pore size and distribution did not exhibit significant change following PDA coating (Fig. S16, ESI†).When we employed PDA-coating, the Cdl/BET values increased for each choice of carbon support compared to the uncoated case, with HP exhibiting a 3× increase (Fig. 2b). HRTEM and SEM-EDS (Fig. 2d–f and Fig. S17 and S18, ESI†) suggest that PDA-coated carbon (i.e., CoPc/PDA-HP) led to a more spatially-uniform dispersion of CoPc. Without PDA, CoPc aggregates form (Fig. S17 and S18, ESI†). An accompanying study (Fig. S19, ESI†) of optical absorption spectra vs. concentration25 shows that PDA militates against aggregate formation.
The transition in the oxidation state of the active site, under the action of reductive bias, from inactive Co2+Pc to active Co1+Pc,29,30 reports on whether CoPc has been successfully dispersed and electrically connected to the conductive support. From in situ Raman (Fig. 2g–i and Fig. S20 and S21, ESI†) we found that PDA coated HP led to an earlier onset potential for, and to a more complete transition form, inactive to active states (Fig. 2i). PDA-coated HP shows a significantly higher Co1+ ratio (35%) at –0.5 VRHE, along with a faster and more complete Co2+Pc-to-Co1+Pc transformation, compared to the reference carbon supports. At –1.2 VRHE, over 90% of Co is in the Co1+Pc state for PDA-HP. The axial coordination31 of PDA to the cobalt center, the aromatic π–π stacking interactions between the polymer and the Pc, along with the hydrophilic nature of PDA, which aids in CoPc stabilization, have all been shown to enhance the dispersion of CoPc molecules.
The impact of porous supports on reactant transport
To study the transport of CO2, we first sought a test of whether CO2 is capable of reaching the catalytic sites inside each catalyst. Turning away for the moment from the carbonate system, we studied CO2-gas-fed systems (Note S5 (ESI†) and Fig. 2c) and measured, in a flow cell, FECO at low CO2 gas feed rate (vCO2) and low partial pressure (PCO2). The HP support with PDA, when functionalized using CoPc, maintained high FE (>80%) even when the vCO2 was slowed to 2.5 mL min−1 cm−2 (Fig. 2c) and PCO2 was reduced to 20% (Fig. S22 and S23, ESI†). In anion exchange membrane (AEM)-based MEA electrolyzers with gas CO2 feed, the single pass conversion (SPC) was 48% (close to the 50% upper bound for the case of neutral CO2-to-CO32) and the stoichiometric CO2 consumption ratio (λstoic) was 1.3 (Fig. S24 and S25 and Table S3, ESI†) in the case of the optimized HP PDA-treated support. In sum, the optimized support enables CO2 diffusion and adsorption on the electrode surface even when CO2 supply is severely curtailed, a finding in accord with adsorption isotherm analysis (Fig. S26 and Note S6, ESI†).We then used EIS (Fig. S27, ESI†) to study mass transfer in porous structures, focusing on the interaction time of ions/reactants inside the pores.22,23 This we accomplished by measuring both capacitance and relaxation time constant (τo). The capacitance increases 10× moving from CoPc/PDA-P to CoPc/PDA-HP, while τo increases by a factor of 3×. The HP porous carbon support thus appears to enrich reactants within its pores, prolonging reactant retention and facilitating transport deep within pores.
Efficient i-CO2 supply in the carbonate system also relies on the effective transport of CO32− anions from the bulk electrolyte to the CEL/electrolyte interface. Again using EIS and cyclic voltammetry (CV) (Fig. S28 and S32 and Note S7, ESI†), we saw evidence of increased binding affinity and interaction between PDA-carbon and carbonate.
Simply increasing the BET surface area by raising the micropore ratio in NP is not effective in achieving the needed reactant transport (Fig. S33–S36 and Note S8, ESI†); instead, we found that an optimized proportion of mesopores in HP is crucial. These mesopores serve as reservoirs and channels, reducing the required length of tortuous diffusion pathways, and ensuring thereby access to active sites within the micropores.33,34 Control experiments (Fig. S37–S39, ESI†) exclude catalyst layer thickness and hydrophilicity as principal factors in transport behavior.
Electronic tuning effect between catalyst and support
We further examined, using X-ray absorption near edge structure (XANES) (Fig. 3a), how PDA influenced the electronic interaction between the Co center and the PDA on the support. XANES shows an upshift of the white line to higher binding energy and an increase in intensity on CoPc/PDA-HP compared to the unsupported CoPc molecules (Fig. 3a and Fig. S40, ESI†). This indicates charge transfer between Co and PDA, and the partial oxidation of Co centers. CV shows a positive shift of the Co2+/Co1+ redox wave after coating with PDA, consistent with the presence of more-electron deficient CoPc (Fig. 3b and Fig. S41–S44, ESI†). These results are in agreement with conclusions from XPS on Co 2p (Fig. S14, ESI†).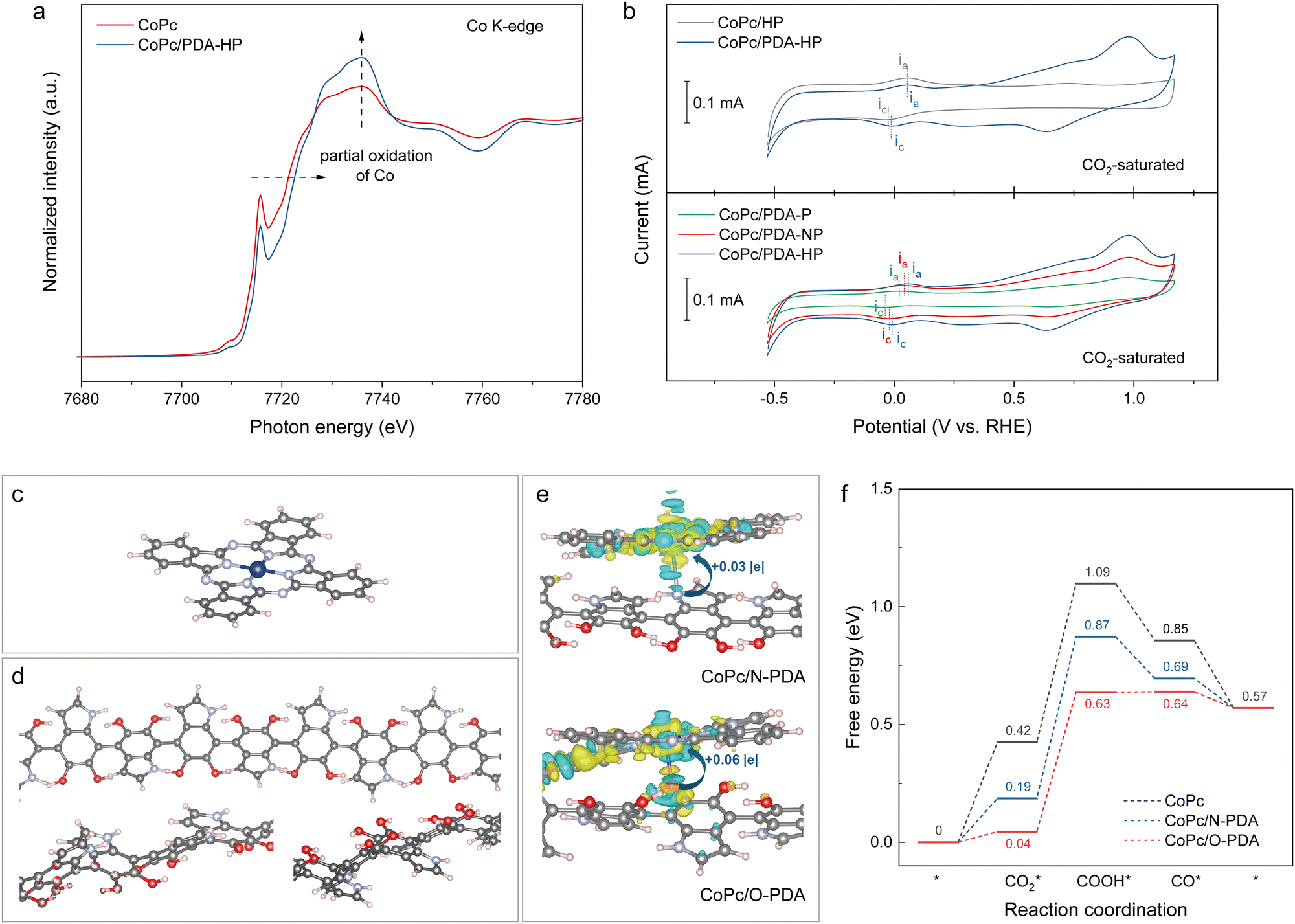 | ||
| Fig. 3 Electronic tuning effect between catalyst and support. (a) XANES spectra in the Co K-edge spectrum, with vs. without PDA. (b) CV analysis in Ar-saturated 1.5 M K2CO3 solution. CV was performed on RDE electrode with different CoPc-based catalysts coated on the surface at a scan rate of 500 mV s−1. Similar positive shifts of Co redox positions were observed on PDA-P (+20 mV) and PDA-NP (+30 mV) compared to their counterparts loaded on pristine supports. (c) Molecular diagram of CoPc, with Co center coordinated with four N. (d) Molecular diagram of PDA. In (c) and (d), different colors represent different atoms: blue, Co; red, O; silver, N; pink, H; grey, C. N-PDA and O-PDA denote the N and O terminations from PDA, respectively. (e) Difference of charge density between CoPc and PDA considering two different loaded sites with an isosurface value of 0.001 e Bohr−3. Cyan, losing electrons; yellow, gaining electrons. From Bader charge analysis, the Co site possesses more positive charge on CoPc/N-PDA (+1.04 |e|) and CoPc/O-PDA (+1.07 |e|) compared to pristine CoPc (+1.01 |e|). (f) Free energy diagrams for CO2 reduction to CO. The adsorption energies of CO2* on CoPc/N-PDA and CoPc/O-PDA were 0.19 eV and 0.04 eV, lower than the 0.42 eV on pristine CoPc. The formation energies of COOH* from CO2* were 0.58 eV and 0.59 eV for CoPc/N-PDA and CoPc/O-PDA, respectively, both less than the 0.67 eV for pristine CoPc. | ||
In accompanying density functional theory (DFT) studies, calculated electron-density-difference plots (Fig. 3c–e) show that CoPc loses electrons (blue), while PDA gains electrons (yellow), in the N- and O-terminations of PDA (N-PDA and O-PDA). Also from DFT, the adsorption energies of CO2* and the formation energies of COOH* on CoPc/N-PDA and CoPc/O-PDA are lower than those for the case of pristine CoPc (Fig. 3f and Fig. S45–S47, ESI†). The combined experimental and computational results suggest that the modified electronic structure of CoPc facilitates the binding and activation of CO2, resulting in a lower energy barrier for its conversion into CO.
Reactive capture system performance and its optimization for energy efficiency
We then returned to reactive capture systems and their optimization, focusing now on the HP polydopamine-coated carbon support. We varied the CoPc to PDA-HP ratio (Fig. S48, ESI†), meso-to-micro ratio (Fig. S49 and S50, ESI†), catalyst layer thickness (Fig. S31, ESI†), and interposer pore size (Fig. S51–S54 and Note S9, ESI†), and in this way obtained an FECO of 47% at 100 and 200 mA cm−2 (Fig. 4a, left). The FE on CoPc/PDA-HP remained above 37% at current densities as high as 600 mA cm−2. This contrasts with lower-porosity and with uncoated supports which – at 600 mA cm−2 – decline to FE < 30% (Fig. 2a and Fig. 4a, right). | ||
| Fig. 4 Electrochemical generation of syngas from carbonate liquid as a function of catalyst. (a) Comparison of faradaic efficiency to CO as a function of carbon-based support: (left) PDA coating on different porous supports and (right) with and without PDA coating on HP. Electrolysis was carried out in the current density range of 100–600 mA cm−2. (b) Cell voltage (left Y-axis) and energy efficiency (right Y-axis) for carbonate electrolysis (1.5 M K2CO3) at different cell configurations. The energy efficiency is calculated using experimental data for a system that employed the optimized BPM and a NiFeOx anode. (c) Cell voltage (left Y-axis) and faradaic efficiency to CO (right Y-axis) for 40 hours of electrolysis at 200 mA cm−2 using the CoPc/PDA-HP catalyst and homemade SnO2-BPM. The interposer layer was replaced after 15 hours and 30 hours. (d) Comparison of energy consumption for the generation of 1 ton syngas for different processes. In order, top to bottom, these are: carbonate electrolysis reported in this work; carbonate best prior performance; direct air capture (DAC) coupled with bicarbonate electrolysis; DAC coupled with reverse water gas shift (RWGS); and DAC coupled with a solid oxide electrolysis cell (SOEC). | ||
We also evaluated whether the carbonate electrolyzer could regenerate the solution all the way to basic conditions, i.e. to the pH needed such that it can be returned to the contactor for the next capture cycle – all this while still efficiently converting carbonate to CO. Through the analysis in Note S10 (ESI†), we estimate that ∼ each electron utilized for carbon-to-CO conversion generates one OH−. Our results indicate that after 24 hours of electrolysis at 100 mA, the pH of the regenerated post-capture liquid reached 13.2, corresponding to an OH− regeneration efficiency of ∼ 35%, a figure in accord with the time-averaged FECO. A total of 0.7 g of CO2 had thus been extracted and converted into CO from the post-capture liquid. This result indicates the system's ability to circulate the capture liquid and enable repeated cycles of CO2 capture and conversion. Regenerating a higher pH capture liquid (KOH + K2CO3) requires a greater selectivity of i-CO2 to CO, particularly under conditions where K2CO3 is gradually consumed.
We then sought to optimize the carbonate electrolyzer for energy efficiency (EE). To study the distribution of voltages, we constructed an analytic BPM electrolyzer that would allow us to monitor, operando, the voltage difference across each electrode/membrane element (Fig. S56, ESI†).35,36 At 200 mA cm−2, the voltage across the BPM accounted for 1.8 V, fully 42% of Vcell. We then replaced the BPM with one incorporating a nanoparticle P25-TiO2 WD catalyst (Fig. S57 and S58, ESI†),37 after which, at 200 mA cm−2, VBPM decreased to 1 V. We also coated the Ni foam anode with Ni(Fe)Ox catalyst, reducing the anode overvoltage by ∼200 mV.
Thus, in energy efficiency to 2:1 syngas, the improved support and system achieved 50% at 200 mA cm−2 (Fig. 4b), compared to 32% at the same current density in the highest-performing previously-reported BPM-based electrified carbonate reactive capture systems (details in Table S4, ESI†). In estimating a projected syngas EE, we assumed that an efficient water electrolyzer (EE to H2 = 65%) will be employed in parallel to provide the H2 missing to make up the 2:1 H2:CO syngas.10 Of the overall improvement in EE, 1.3× comes from reduced Vcell, and 1.2× from increased FECO at the 200 mA cm−2 current density, corresponding to the 1.3 × 1.2 = 1.56× improvement in EE from 32% to 50%.
To query operating stability, we ran the system at 200 mA cm−2 for 40 hours of electrolysis (Fig. 4c). We observed that the systems maintained a cell voltage (Vcell) within the range of 2.8 to 3.3 V. The FECO gradually declined from >40% to ∼32–35% during each 15-hour interval, primarily due to the gradual increase in electrolyte pH and the dissolution of the interposer under electrolysis conditions. The FECO >40% can be recovered by periodically replacing the interposer. The mixed cellulose ester (MCE) in the interposer is a weak link: in future it will need to be made using a more long-lived material, presumably with similar hydrophobicity and porosity, to extend operating lifetime.
The CoPc/PDA-HP catalyst itself remained stable: Co K-edge X-ray absorption spectra after operation indicate that CoPc was unchanged (Fig. S59, ESI†). In contrast, without a PDA layer, and when loading CoPc in a non-porous support (CoPc/P), the CoPc molecules aggregated rapidly and became inactive (FECO < 10% within 20 hours) (Fig. S60, ESI†). SEM-EDS and inductively coupled plasma mass spectrometry (ICP-MS) show that CoPc/PDA-HP exhibited significantly lower aggregation and leaching rates compared to CoPc/P, 4 ppb h−1versus 9 ppb h−1 (Fig. S60c and d, ESI†).
Carbon utilization is defined as:
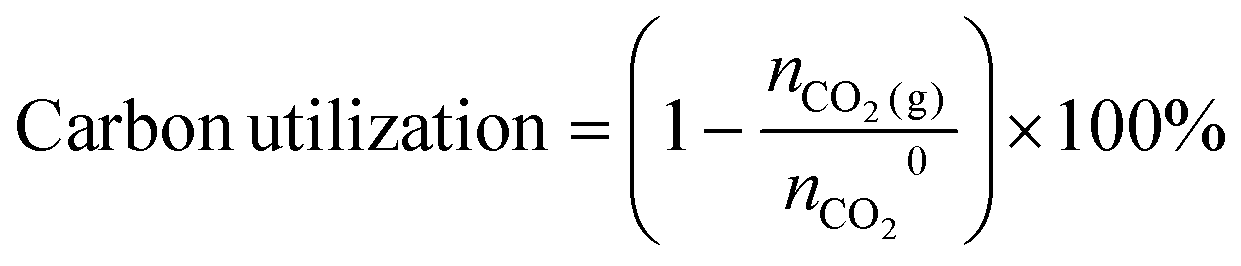
We then used a membrane contactor (Fig. S63–S66 and Note S11, ESI†) with a 3 M KOH capture medium to perform direct air capture of laboratory air to produce the liquid for carbonate electrolysis. When DAC had been run for a duration such that the pH of the capture liquid had declined to ∼12.7, we used this post-capture solution for electrolysis, and achieved on CoPc/PDA-HP a 40% FECO across 100–300 mA cm−2.
We compare estimated full system energy requirements (Fig. 4d, Note S12, Fig. S68, and Table S6, ESI†) for: (i) sequential DAC capture-and-release followed by gas-fed electrochemical CO2 upgrade, such as solid oxide electrolyzer cell (SOEC); (ii) sequential DAC capture-and-release followed by reverse water–gas shift (RWGS) using H2 from an efficient water electrolyzer; (iii) the integrated reactive capture approach studied herein, again with missing H2 filled in using the same water electrolyzer. The improved reactive capture system provides 44 GJ per tonsyngas, compared to 61 GJ for the most efficient prior electrified reactive capture systems, and compared to 65 GJ for DAC + RWGS and 55 GJ for DAC + SOEC. An advantage from the reactive capture system derives from the avoidance of the CO2 regeneration step in sequential approaches.
Life cycle assessment and techno-economic analysis
A life-cycle assessment (LCA) was conducted to evaluate the prospective environmental impacts of the reactive capture system (Fig. 5a and Note S13, Fig. S69 and S70 and Tables S7 and S8, ESI†). In both coelectrolysis and reactive capture approaches, the greenhouse gas (GHG) emissions are dominated by the carbon footprint of electric energy. Using the U.S. grid, the reactive capture process emits 0.6 t CO2e per t syngas less than the DAC-SOEC process and is comparable to the DAC-RWGS process. As the carbon intensity (CI) of electricity declines over the coming half-decade to 280 g CO2e per kW h, the reactive capture method begins to outperform syngas production methods such as natural gas reforming (NG-syngas: 1.26 t CO2e per t syngas) and coal gasification (coal-syngas: 2.32 t CO2e per t syngas).38 According to the International Energy Agency (IEA), these grid carbon intensity targets could be reached in the U.S. before 2030.39 Eventually, when wind-generated electricity is used (Fig. 5a), the reactive capture process achieves a global warming potential (GWP) of –1.49 t CO2e per t syngas, 0.78 t CO2e per t syngas lower than the DAC-SOEC and DAC-RWGS processes. | ||
| Fig. 5 Environmental and economic analysis. (a) Cradle-to-gate GWP values of different syngas production routes using three different electricity sources: US mix, US 2030, and wind energy. US 2030 electricity CI (160 g kW−1 h−1) is obtained from IEA report.39 Each color in the legend indicates the GHG emission in various stages or carbon credits due to biogenic CO2 and byproduct of oxygen. The gray bar is the net GWP value. (b) Breakdown of the MSP of syngas production routes. The green bar is the MSP, and the gray bar is the MSP after considering the social cost of carbon. (c) Effects of Faraday efficiency and cell voltage on the MSP. The white line is the commercial syngas price obtained from previous study.40 In this figure, we included and compared the performance metrics, FE and Vcell, from our work with the best prior results. | ||
A techno-economic analysis (TEA) was also performed (Note S14 and Fig. S71 and Tables S9–S11, ESI†). The minimum selling price (MSP) of syngas produced via the reactive capture is estimated at US$770 per t, below that for DAC-SOEC (US$1270 per t) and DAC-RWGS (US$1020 per t), and just slightly (by a margin of ∼US$120 per t) higher than commercial syngas (Fig. 5b). The reactive capture system avoids the need for a CO2 regeneration unit, CO2 circulation system, and RWGS reactor (these latter requiring ∼800 °C operation). Incorporating the social cost of carbon into TEA results brings parity earlier: a social cost of carbon of US$190 per t CO2,41 decreases the net/effective cost of reactive capture-syngas to US$490 per t (Fig. 5b grey bar), lower than the fossil-syngas of US$900 per t–US$1100 per t (Fig. 5b dash line). These costs employed based on LCA results achieved when electricity comes from wind energy.
Conclusions
In this work, we used alkali carbonate post-direct-air-capture liquid as feedstock, converting the captured CO2 to pure syngas while regenerating the capture liquid. We developed hierarchical carbon supports, their pores coated internally first with PDA and then with catalyst, that improve reactant mass transport and that molecularly disperse the heterogenized molecular catalyst, also tuning the electronic environment of the Co center. By improving the determinants of syngas production in reactive capture (Fig. 5c), FECO and Vcell, we achieved an EE to 2:1 syngas of 50% at 200 mA cm−2. The higher CO2 utilization efficiency and lower energy consumption surpass those of previous reactive capture and gas-CO2 electrolysis systems (Tables S4 and S5, ESI†). In future work, developing robust interposer materials or transitioning to an interposer-free design, will be essential to further enhance system stability. To improve OH− regeneration efficiency, it is also critical to increase the selectivity for i-CO2-to-CO conversion, particularly under conditions of gradual K2CO3 depletion. Additionally, continued advancements in catalyst design and system integration are essential to reduce energy consumption and progress toward a fully closed carbon cycle for air-to-product conversion—that is, producing carbon products from air-derived capture liquids while efficiently regenerating the capture media.
Methods
Electrode preparation
All reagents used in this work were purchased from suppliers without further purification. The planar carbon (Vulcan XC 72R), nanoporous carbon (Ketjenblack EC-300J), and highly porous carbon (Ketjenblack EC-600JD) were all purchased from Fuel Cell Store. To tune the mesopore-to-micropore structures of the carbon support of HP, we employed a CO2-activation method. Ketjenblack EC-600JD was heated in a tube furnace up to 1000 °C at a ramp rate of 5 °C min−1 under continuous argon gas flow (500 sccm). Once the temperature reached 1000 °C, the gas supply was switched from argon to CO2 at the same flow rate (500 sccm) and maintained for a certain duration (1 hour or 2.5 hours), followed by natural cooling under continuous argon flow (500 sccm). Optimal performance in HP was achieved with a 2.5-hour CO2 treatment. The polydopamine (PDA) coating on carbon materials were carried out using a solution of dopamine hydrochloride (1 mg mL−1, Sigma-Aldrich) with pH 8.5 Tris–HCl buffer (10 mM Trizma® base, Sigma-Aldrich and hydrochloric acid, 37%, Sigma-Aldrich), using a previously-reported method.28 Briefly, carbon materials (0.5 mg mL−1) were added into the solution, and the thickness of the polydopamine coating is roughly proportional to the coating time. The optimal PDA coating duration is 30 min for P and NP, and 15 min for HP (with the optimum meso-to-micro ratio by CO2-activation of 2.5 hours). The coated sample was washed using distilled water several times to remove unreacted precursors. After filtering, the samples were dried in a vacuum oven (80 °C) overnight. The catalyst ink was prepared by dispersing CoPc (Sigma-Aldrich) and polydopamine-treated carbons in 2-propanol with added Nafion ionomer by ultrasonication. The ink needs to be well-sonicated for a good dispersion of catalyst. The mass ratio of the powders (CoPc and carbons) and ionomer was 9:1. The ink was then airbrushed onto the hydrophilic carbon substrate (Freudenberg H23, Fuel cell store) to the final loading of ∼2 mg cm−2 (CoPc + carbon). The mass loading of carbon was optimized and described in the supplementary figures. Unless otherwise specified, a carbon to CoPc ratio of 4:6 is used for all electrochemical measurements.
NiFeOx electrode was prepared from a modified method in the literature.42 Ni foam was first cleaned by 6 M HCl and DI-water for 15 min under sonication. Then, a 40 mL solution with 4 mmol NH4F, 10 mmol urea, 2 mmol Ni(NO3)2·6H2O, and 2 mmol Fe(NO3)3·9H2O was prepared and transferred to a 50 mL Teflon-lined stainless steel autoclave. The hydrothermal growth of the hydroxides on Ni foam was performed at 120 °C for 6 hours with a heating rate of 3 °C min−1, followed by sonication in DI-water and drying in the oven at 80 °C.
Electrochemical measurements
The carbonate electrolyzer contains two stainless steel flow-field plates with serpentine channels, PTFE and silicone gaskets, and the membrane electrode assembly (MEA), which contains two electrodes and a membrane, and was formed after assembling the cell hardware. The catholyte and anolyte were circulated by peristaltic pumps (INTLLAB) at 15 mL min−1. The current was controlled by an Autolab potentiostat/galvanostat. The membrane used to separate catholyte and anolyte was a commercial (Fumasep BPM, Fuel Cell Store) or custom-designed BPM. The CoPc-based catalyst was used as the cathode. A piece of filter membrane was inserted as the interposer layer between cathode and cation exchange layer (CEL). The thickness of the interposer layer was controlled as 135 μm. The pore size of the hydrophilic interposer layer was controlled at 1.2, 3, 5, and 8 μm for MCE membrane, 30 μm and 160 μm for Nylon membrane. The catholyte was 1.5 M K2CO3, and the anolyte was 1 M KOH. For stability measurements in homemade BPM system, a large tank (1 L) of catholyte and anolyte was used, and the refresh of electrolytes was conducted at each 15-hour interval. When we performed carbonate electrolysis in cation exchange membrane (CEM)-based electrolyzer, Nafion 117 (H+ transport) was used as the membrane, 0.5 M H2SO4 was used as the anolyte, and the IrO2/Ti felt (US Research Nanomaterials, Inc., IrO2 loading: ∼1 mg cm−2) was applied as the anode. All experiments were performed at room temperature.For gas CO2 reduction reaction in three-compartment flow cells, Ag/AgCl (4 M KCl) and a piece of Ni foam were employed as the reference electrode and counter electrode, respectively. The cathode catalysts were airbrushed onto hydrophobic carbon paper (Freudenberg H23C3, Fuel cell store). The anion-exchange membrane (PiperION, 40 microns) was used as the membrane to separate the cathode and anode chambers. 1.5 M K2CO3 was used for both catholyte and anolyte. In experiments with varying CO2 gas flow rate, the CO2 flow was regulated using a mass flow controller (Alicat Scientific). For the gas CO2 reduction with various concentrations, N2 was used as a balance gas to adjust the CO2 partial pressure, and a tee-type connector was employed to mix the gases before purging them into the flow cell. The total gas flow rates were measured using a gas flow meter (Thermo Fisher Scientific), and the product concentrations at the catholyte outlet were quantified by gas chromatography (GC). A detailed analysis and description of the experimental setup can be found in Supplementary Note S5 (ESI†).
For gas CO2 reduction reaction in MEA-type electrolyzer, the setup involved sequential assembly of the cathode GDE (4 cm2 geometric area), PiperION AEM (40 microns), and anode (IrO2-GDE, 4 cm2, Dioxide Materials). Both electrodes were secured within polytetrafluoroethylene gaskets, with a window size of 4 cm2. The entire cathode/membrane/anode assembly was then compressed between the electrolyzer plates, ensuring proper sealing, electron conductivity, and ionic transport within the MEA. The humidified CO2 gas, controlled by a mass flow controller (Alicat Scientific), is directed into the cathode flow field for the reaction. After the reaction, the product stream is mixed with a 2.5 mL min−1 N2 stream for GC analysis. In the anode chamber, 0.1 M KHCO3 solution is circulated at a flow rate of 20 mL min−1. In MEA configurations for electrolysis, especially at large current densities, significant CO2 loss for (bi)carbonate formation can occur, the mixed N2 is considered as an internal standard for accuracy. A detailed analysis and description of the experimental setup can be found in Supplementary Note S5 (ESI†).
CV was conducted by a general three electrode configuration from −1.2 to 0.3 V (vs. Ag/AgCl) at scan rate from 100 to 500 mV s−1 in Ar-saturated 1.5 M K2CO3 electrolyte to observe the redox behavior of cobalt sites. EIS analysis was conducted in the same cell configuration as the CV experiments, using a VIONIC (Metrohm) instrument. The measurements were performed under Ar-saturated conditions in 1.5 M K2CO3 (carbonate) and 1.5 M KOH (non-carbonate) solutions. Before the EIS measurement, the electrode was equilibrated around 20 min in open circuit voltage (OCV). The measurement was conducted at OCV in the frequency range from 50 mHz to 100 kHz with an AC amplitude of 5 mV.
Bipolar membrane fabrication and measurements
The homemade BPM electrolyzer was assembled by incorporating P25-TiO2 and SnO2 as water dissociation (WD) catalysts. P25-TiO2 was purchased from Thermo Fisher, and SnO2 was synthesized according to a previously reported method.43 Nafion 212 (50 μm, Ion Power) and PiperION-A40-HCO3 (40 μm, Versogen) were used as the cation exchange layer (CEL) and anion exchange layer (AEL), respectively. The CEL, purchased in a pre-protonated state, was soaked and stored in DI H2O, while the AEL was soaked in 0.50 M KOH for more than 1 hour, stored in fresh 0.50 M KOH, and rinsed with 18.2 MΩ water before use. Both membrane layers were cut into 2 cm × 2 cm squares.The WD catalysts were spin-coated onto the CEL at 3000 rpm for 30 seconds. The WD catalysts were dispersed in a water/IPA mixture (1:1 by weight) to create an ink with different wt% solids. The edges of the CEL were taped onto a glass slide, and the ink was applied until the surface was fully covered, followed by spin-coating to form a uniform thin layer. The final concentration of WD catalysts (0.2 wt%) was diluted from a 2 wt% mother solution.
Voltage breakdown measurements for both the commercial and homemade BPM were conducted using a membrane-potential-sensing setup. The detailed step-by-step assembly of the voltage distribution setup in a MEA configuration was described in previous literature.37 The setup included two reference electrodes (Ag/AgCl and Hg/HgO, Pine Research), sensing strips, gaskets, and O-rings. Measurements were performed using a potentiostat (Bio-Logic VSP 300).
Product analysis
The gas products (H2 and CO) were quantified by GC (Shimadzu2014, PerkinElmer Clarus 580) equipped with a thermal conductivity detector (TCD) and a flame ionization detector (FID) equipped with a Methanizer. The calibration curve was established by analyzing the standard calibration gases with different concentrations (10–10000 ppm). Argon (100 mL min−1) was purged as the carrier gas to carry the gas products out of the system for quantification.
The rate of H2/CO generation (r, mol s−1) for each cycle was calculated by the following equation:
r = c × 10−6 × [P![[V with combining dot above]](https://www.rsc.org/images/entities/i_char_0056_0307.gif) × 10−6/(RT)] × 10−6/(RT)] |
is the volumetric flow rate of the inlet gas (100 mL min−1); p is the ambient pressure (p = 1.013 × 105 Pa); R is the gas constant (R = 8.314 J mol−1 K−1); T is the room temperature (293.15 K). The total amount of gas (mol) was calculated by integrating the plot of H2/CO production rate (mol s−1) vs. reaction time (s).
The faradaic efficiency (FEi) can be calculated by equations as follows:
 485 C mol−1); Q is the total charge passed through the electrolytic cell.
485 C mol−1); Q is the total charge passed through the electrolytic cell.
Carbon utilization is defined as 1 minus the normalized ratio of CO2 gas detected in the outlet of the carbonate system to the theoretically produced i-CO2 at the BPM/electrolyte interface. The carbon utilization can be calculated by the equations as follows:
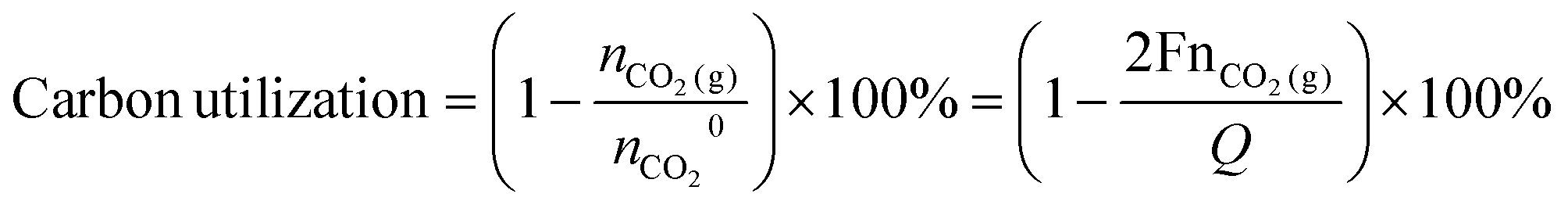 485 C mol−1); Q is the total charge passed through the electrolytic cell. With a sealed catholyte tank and Ar as the carrier gas (gas flow rate: 50 mL min−1), gas products were collected using a gas-tight needle. The carrier gas flow rate was regulated by a mass flow controller (Alicat Scientific), and the total gas flow rate (products + carrier gas) was measured using a flow meter (Thermo Fisher Scientific). The amount of gas products (H2, CO, and CO2) were subsequently quantified and calculated through GC analysis.
485 C mol−1); Q is the total charge passed through the electrolytic cell. With a sealed catholyte tank and Ar as the carrier gas (gas flow rate: 50 mL min−1), gas products were collected using a gas-tight needle. The carrier gas flow rate was regulated by a mass flow controller (Alicat Scientific), and the total gas flow rate (products + carrier gas) was measured using a flow meter (Thermo Fisher Scientific). The amount of gas products (H2, CO, and CO2) were subsequently quantified and calculated through GC analysis.
Materials characterization
X-ray photoelectron spectroscopy (XPS) was carried out on a Thermo Scientific NEXSA G2 XPS spectrometer, equipped with an Al K alpha radiation source and electron flood-gun, at a pressure of 8 × 10−8 mbar with a pass energy of 50 eV. All spectra were calibrated with the C 1s peak at 284.8 eV. Scanning Electron Microscopy-Energy Dispersive X-ray Spectroscopy (SEM-EDS) was conducted by JEOL JSM-7900FLV SEM at an accelerating voltage of 10 kV with backscattered electron detection, which is equipped with a light-element X-ray detector and an Oxford Aztec energy-dispersive X-ray analysis system. Aberration corrected scanning transmission electron microscopy (STEM) images and energy-dispersive X-ray spectroscopy (EDS) mappings were taken using JEOL ARM200CF TEM equipped with dual SDD EDS detector. Transmission electron microscopy (TEM) samples were prepared by dispersing powder samples in ethanol followed by drop-casting on the grid. N2 and CO2 isotherms were conducted by Micromeritics 3Flex instrument, samples were dried at 373 K under dynamic vacuum for 12 h until the pressure stabilized below 5 × 10−6 bar. Brunauer–Emmett–Teller (BET) method was used to calculate surface area from N2 adsorption isotherm. Fourier-transform infrared (FTIR) measurements were carried out using a Nicolet iS50 FTIR spectrometer equipped with a Harrick Scientific Praying Mantis DRIFTS accessory. X-ray absorption fine structure (XAFS) measurements were made at the 8C nano-probe XAFS beamline (BL8C) of Pohang Light Source (PLS-II) in the 3.0GeV storage ring, with a ring current of 250mA. The X-ray beam was monochromated by a Si (111) double crystal where the beam intensity was reduced by 30% to eliminate higher order harmonics. The X-ray beam was then delivered to a secondary source aperture where the beam size was adjusted to 0.3mm (v) × 1mm (h). A high voltage (3000V) was applied to ionization chambers filled with N2/Ar mixture gases to detect X-ray intensity. XAFS spectra were collected in both transmission and fluorescence modes.
In situ Raman analysis was performed using a Renishaw inVia Raman spectrometer with a custom in situ cell and a ×50 water immersion lens. The CoP-based catalysts, supported on hydrophobic carbon paper (Freudenberg H23C3), were used as the cathode in a 1.5 M K2CO3 electrolyte, with N2 purged from the backside. Pt/C on carbon cloth and Ag/AgCl were used as the counter and reference electrodes, respectively. A piece of CEM (Nafion 212) was used as the membrane. The detailed experimental set-up and discussion was shown in the Fig. S20 and S21 (ESI†).
For CoPc dispersion experiments, ultrasmall PDA particles were prepared as dispersion additives. 180 mg of dopamine hydrochloride was dissolved in 90 mL of deionized water, and 840 μL of 1 M NaOH was added at 60 °C with vigorous stirring for 5 hours. The solution color changed from pale yellow to dark brown. The product was collected by centrifugation (16000 rpm for 20 minutes) and washed with deionized water three times, followed by freeze-drying to obtain black PDA solids. 20 mg of PDA nanoparticles were dissolved in 10 mL of 0.1 M NaOH. The pH was adjusted to 7.0 by adding 0.1 M HCl under sonication (150 W for 8 minutes). The particles were then retrieved using a centrifugal filter (MWCO = 30 kDa) at 8000 rpm for 8 minutes, washed with deionized water, and freeze-dried to obtain ultrasmall PDA particles. Stock solutions of CoPc and ultrasmall PDA in DMF were prepared at concentrations of 5.0 × 10−6 M for CoPc and 5.0 × 10−6 M for PDA. For targeted solution concentration, these stock solutions were diluted. The solutions were sonicated until ready for UV-vis measurement. Contact angle measurements were performed using a Biolin Optical Tensiometer under ambient conditions (22 °C). Using the sessile drop method, a 5–7 μL water droplet was placed on the catalyst layer substrate, and the contact angle was measured within five seconds. The dissolution rate of cobalt during the stability test was tracked by calculating the cobalt content in the running electrolyte using an ICP-MS (Thermo iCap Q).
DFT calculations
All spin unrestricted DFT calculations were performed by using the Vienna Ab initio Simulation Package (VASP).44,45 For all geometric optimizations,46 we adopted Perdew–Burke–Ernzerhof formulation of the generalized gradient approximation (GGA-PBE) to determine the exchange–correlation energy and projector augmented wave (PAW) to construct the plane wave.47 The cutoff energy was set to 450 eV.48 The DFT-D3 method of Grimme was derived from the long-range dispersion correction.49 All atomic coordinates were allowed to relax until the self-consistent energy and force converge to below 10−5 eV and 0.05 eV Å−1, respectively. The simulated crystal-cell (30 Å × 26.16 Å × 30 Å) was constructed periodically, and 1 × 1 × 1 k-point mesh was generated by Monkhorst–Pack scheme in the Brillouin scheme.50 The hydrated potassium ion clusters containing six water molecules and one potassium ion near the Co site on the CoPc was used to simply simulate the local solvation effect and field effect in the electro-CO2RR.51 Therefore, the CO2RR for CO includes four elementary steps, as follows.

| COOH* + H+ + e− → CO* + H2O(l) |
| CO* ↔ CO(g) + * |
Here, Gibbs free energy (G) was calculated and corrected by the following equation:

 , and TS are the contribution form electronic energy directly calculating by DFT, zero-point energy, temperature enthalpic, and entropic correction (T = 300 K), respectively. Using the computational hydrogen electrode model, the Gibbs free energy for a proton/electron [G(H+ + e−)] in the electrolyte was treated by the half of the Gibbs free energy of molecule H2 [0.5G(H2)].52
, and TS are the contribution form electronic energy directly calculating by DFT, zero-point energy, temperature enthalpic, and entropic correction (T = 300 K), respectively. Using the computational hydrogen electrode model, the Gibbs free energy for a proton/electron [G(H+ + e−)] in the electrolyte was treated by the half of the Gibbs free energy of molecule H2 [0.5G(H2)].52
Environmental and economic assessment
We conducted a cradle-to-gate life cycle assessment to evaluate the GHG emissions of syngas production from various routes, such as DAC-SOEC, DAC-RWGS, and carbonate electrolysis proposed in this study. The functional unit for this assessment is 1 t of syngas with 2:1 H2:CO molar ratio. We utilized three electricity sources: US mix, US2030, and wind energy. Emission factors for these electricity sources are derived from the Greenhouse Regulated Emissions and Energy use in Technologies (GREET 2023) model.38 We analyzed a medium-sized syngas plant with an annual production capacity of 365000 t, with a production rate of 1000 t per day. Detailed assumptions and calculations are provided in the ESI.†
Author contributions
E. H. S. and K. X. supervised the project. H. L. and H. S. performed electrochemical measurements, materials synthesis, and characterization. X. L. and P. O. carried out the DFT calculations. G. S. and J. B. D conducted the environmental and economic assessment. L. C. contributed to the synthesis of BPM. J. Y. and Y. W performed HRTEM characterization. K. L. performed the XAFS analysis. I. G., A. A., and A. J., supervised the project and commented on the TEA. All authors discussed the results and contributed to the manuscript writing and editing.Data availability
The data supporting the findings of this study are available within the article and its ESI.†Conflicts of interest
The authors declare no competing interests.Acknowledgements
This work received financial support from Saudi Aramco Technologies Company under the Agreement No. SATC-2022-016. We thank Drs. Tirzah Abbott and Krysten Lauren Villalon from Northwestern University's NUANCE Center for their assistance with cross-sectional SEM analysis. This work was supported by the National Research Foundation of Korea (NRF) grant funded by the Korea government (MSIT) (RS-2023-00253850). The XAFS analysis was conducted with the support of the grant NRF-2019M3D1A1079309. Theoretical calculations of this work were supported through computational resources and staff contributions provided for the Quest high-performance computing facility at Northwestern University which is jointly supported by the Office of the Provost, the Office of Research, and Northwestern University Information Technology.References
- S. Nitopi, E. Bertheussen, S. B. Scott, X. Liu, A. K. Engstfeld and S. Horch, et al., Progress and perspectives of electrochemical CO2 reduction on copper in aqueous electrolyte, Chem. Rev., 2019, 119(12), 7610–7672 CrossRef CAS PubMed.
- P. De Luna, C. Hahn, D. Higgins, S. A. Jaffer, T. F. Jaramillo and E. H. Sargent, What would it take for renewably powered electrosynthesis to displace petrochemical processes?, Science, 2019, 364(6438), eaav3506 CrossRef CAS PubMed.
- D. W. Keith, G. Holmes, D. S. Angelo and K. Heidel, A process for capturing CO2 from the atmosphere, Joule, 2018, 2(8), 1573–1594 CrossRef CAS.
- M. Fasihi, O. Efimova and C. Breyer, Techno-economic assessment of CO2 direct air capture plants, J. Cleaner Prod., 2019, 224, 957–980 CrossRef CAS.
- A. J. Welch, E. Dunn, J. S. DuChene and H. A. Atwater, Bicarbonate or carbonate processes for coupling carbon dioxide capture and electrochemical conversion, ACS Energy Lett., 2020, 5(3), 940–945 CrossRef CAS.
- H. Liu, Y. Chen, J. Lee, S. Gu and W. Li, Ammonia-Mediated CO2 Capture and Direct Electroreduction to Formate, ACS Energy Lett., 2022, 7(12), 4483–4489 CrossRef CAS.
- L. Chen, F. Li, Y. Zhang, C. L. Bentley, M. Horne and A. M. Bond, et al., Electrochemical reduction of carbon dioxide in a monoethanolamine capture medium, ChemSusChem, 2017, 10(20), 4109–4118 CrossRef CAS PubMed.
- R. E. Siegel, S. Pattanayak and L. A. Berben, Reactive capture of CO2: opportunities and challenges, ACS Catal., 2022, 13(1), 766–784 CrossRef.
- Q. Xia, K. Zhang, T. Zheng, L. An, C. Xia and X. Zhang, Integration of CO2 Capture and Electrochemical Conversion: Focus Review, ACS Energy Lett., 2023, 8, 2840–2857 CrossRef CAS.
- P. Debergh, O. Gutierrez-Sanchez, M. N. Khan, Y. Y. Birdja, D. Pant and M. Bulut, The Economics of Electrochemical Syngas Production via Direct Air Capture, ACS Energy Lett., 2023, 8, 3398–3403 CrossRef CAS.
- J. Lee, H. Liu and W. Li, Bicarbonate Electroreduction to Multicarbon Products Enabled by Cu/Ag Bilayer Electrodes and Tailored Microenviroments, ChemSusChem, 2022, 15(22), e202201329 CrossRef CAS PubMed.
- Y. C. Li, G. Lee, T. Yuan, Y. Wang, D.-H. Nam and Z. Wang, et al., CO2 electroreduction from carbonate electrolyte, ACS Energy Lett., 2019, 4(6), 1427–1431 CrossRef CAS.
- Y. C. Xiao, C. M. Gabardo, S. Liu, G. Lee, Y. Zhao and C. P. O’Brien, et al., Direct carbonate electrolysis into pure syngas, EES Catal., 2023, 1(1), 54–61 RSC.
- Y. Kim, E. W. Lees and C. P. Berlinguette, Permeability Matters When Reducing CO2 in an Electrochemical Flow Cell, ACS Energy Lett., 2022, 7(7), 2382–2387 CrossRef CAS.
- G. Lee, Y. C. Li, J.-Y. Kim, T. Peng, D.-H. Nam and A. Sedighian Rasouli, et al., Electrochemical upgrade of CO2 from amine capture solution, Nat. Energy, 2021, 6(1), 46–53 CrossRef CAS.
- G. Lee, A. S. Rasouli, B.-H. Lee, J. Zhang, Y. C. Xiao and J. P. Edwards, et al., CO2 electroreduction to multicarbon products from carbonate capture liquid., Joule, 2023, 7(6), 1277–1288 CrossRef CAS.
- H. M. Almajed, R. Kas, P. Brimley, A. M. Crow, A. Somoza-Tornos and B. M. Hodge, et al., Closing the Loop: Unexamined Performance Trade-Offs of Integrating Direct Air Capture with (Bi)carbonate Electrolysis, ACS Energy Lett., 2024, 9(5), 2472–2483 CrossRef CAS PubMed.
- L. Chen, Q. Xu, S. Z. Oener, K. Fabrizio and S. W. Boettcher, Design principles for water dissociation catalysts in high-performance bipolar membranes, Nat. Commun., 2022, 13(1), 3846 CrossRef CAS PubMed.
- S. Z. Oener, M. J. Foster and S. W. Boettcher, Accelerating water dissociation in bipolar membranes and for electrocatalysis, Science, 2020, 369(6507), 1099–1103 CrossRef CAS PubMed.
- D. Kim, W. Choi, H. W. Lee, S. Y. Lee, Y. Choi and D. K. Lee, et al., Electrocatalytic reduction of low concentrations of CO2 gas in a membrane electrode assembly electrolyzer, ACS Energy Lett., 2021, 6(10), 3488–3495 Search PubMed.
- Y. Zhao, L. Hao, A. Ozden, S. Liu, R. K. Miao and P. Ou, et al., Conversion of CO2 to multicarbon products in strong acid by controlling the catalyst microenvironment, Nat. Synth., 2023, 1–10 Search PubMed.
- S. H. Lee, J. Kim, D. Y. Chung, J. M. Yoo, H. S. Lee and M. J. Kim, et al., Design principle of Fe–N–C electrocatalysts: how to optimize multimodal porous structures?, J. Am. Chem. Soc., 2019, 141(5), 2035–2045 Search PubMed.
- J. H. Jang and S. M. Oh, Complex capacitance analysis of porous carbon electrodes for electric double-layer capacitors, J. Electrochem. Soc., 2004, 151(4), A571 Search PubMed.
- M. Zhu, R. Ye, K. Jin, N. Lazouski and K. Manthiram, Elucidating the reactivity and mechanism of CO2 electroreduction at highly dispersed cobalt phthalocyanine, ACS Energy Lett., 2018, 3(6), 1381–1386 Search PubMed.
- W. S. Dean, T. L. Soucy, K. E. Rivera-Cruz, L. L. Filien, B. D. Terry and C. C. McCrory, Mitigating Cobalt Phthalocyanine Aggregation in Electrocatalyst Films through Codeposition with an Axially Coordinating Polymer, Small, 2024, 2402293 Search PubMed.
- S. Anantharaj and S. Noda, iR drop correction in electrocatalysis: everything one needs to know!, J. Mater. Chem. A, 2022, 10(17), 9348–9354 Search PubMed.
- Y. J. Son, R. A. Marquez, K. Kawashima, L. A. Smith, C. E. Chukwuneke and J. Babauta, et al., Navigating iR compensation: practical considerations for accurate study of oxygen evolution catalytic electrodes, ACS Energy Lett., 2023, 8(10), 4323–4329 CrossRef CAS.
- H. Lee, S. M. Dellatore, W. M. Miller and P. B. Messersmith, Mussel-inspired surface chemistry for multifunctional coatings, Science, 2007, 318(5849), 426–430 Search PubMed.
- S. Ren, E. W. Lees, C. Hunt, A. Jewlal, Y. Kim and Z. Zhang, et al., Catalyst aggregation matters for immobilized molecular CO2RR electrocatalysts, J. Am. Chem. Soc., 2023, 145(8), 4414–4420 Search PubMed.
- D. Masheder and K. Williams, Raman spectro-electrochemistry. II. In situ Raman spectroscopic studies of the electrochemical reduction of CO2 at cobalt (II) phthalocyanine-impregnated PTFE-bonded carbon gas diffusion electrodes, J. Raman Spectrosc., 1987, 18(6), 391–398 Search PubMed.
- T. L. Soucy, W. S. Dean, J. Zhou, K. E. Rivera Cruz and C. C. McCrory, Considering the influence of polymer–catalyst interactions on the chemical microenvironment of electrocatalysts for the CO2 reduction reaction., Acc. Chem. Res., 2022, 55(3), 252–261 Search PubMed.
- S. Brückner, Q. Feng, W. Ju, D. Galliani, A. Testolin and M. Klingenhof, et al., Design and Diagnosis of high-performance CO2-to-CO electrolyzer cells, Nat. Chem. Eng., 2024, 1(3), 229–239 CrossRef.
- V. Yarlagadda, M. K. Carpenter, T. E. Moylan, R. S. Kukreja, R. Koestner and W. Gu, et al., Boosting fuel cell performance with accessible carbon mesopores, ACS Energy Lett., 2018, 3(3), 618–621 Search PubMed.
- H. T. Chung, D. A. Cullen, D. Higgins, B. T. Sneed, E. F. Holby and K. L. More, et al., Direct atomic-level insight into the active sites of a high-performance PGM-free ORR catalyst, Science, 2017, 357(6350), 479–484 Search PubMed.
- D. Salvatore and C. P. Berlinguette, Voltage matters when reducing CO2 in an electrochemical flow cell, ACS Energy Lett., 2019, 5(1), 215–220 CrossRef.
- K. U. Hansen, L. H. Cherniack and F. Jiao, Voltage Loss Diagnosis in CO2 Electrolyzers Using Five-Electrode Technique, ACS Energy Lett., 2022, 7(12), 4504–4511 CrossRef CAS.
- L. Chen, Q. Xu and S. W. Boettcher, Kinetics and mechanism of heterogeneous voltage-driven water-dissociation catalysis, Joule, 2023, 7(8), 1867–1886 CrossRef CAS.
- GREET® Model: the Greenhouse Gases, Regulated Emissions, and Energy Use in Transportation (GREET) Model [Internet]. 2023. Available from: https://greet.es.anl.gov/.
- IEA. Carbon intensity of electricity generation in selected regions in the Sustainable Development Scenario, 2000-2040. 2020 2020/06/02/.
- H. M. Almajed, O. J. Guerra, W. A. Smith, B.-M. Hodge and A. Somoza-Tornos, Evaluating the techno-economic potential of defossilized air-to-syngas pathways, Energy Environ. Sci., 2023, 16(12), 6127–6146 Search PubMed.
- EPA. Report on the Social Cost of Greenhouse Gases: Estimates Incorporating Recent Scientific Advances. 2023 2023/12/02.
- B. Zhang, Y. H. Lui, H. Ni and S. Hu, Bimetallic (FexNi1−x)2P nanoarrays as exceptionally efficient electrocatalysts for oxygen evolution in alkaline and neutral media, Nano Energy, 2017, 38, 553–560 Search PubMed.
- S. Sasmal, L. Chen, P. V. Sarma, O. T. Vulpin, C. R. Simons and K. M. Wells, et al., Materials descriptors for advanced water dissociation catalysts in bipolar membranes, Nat. Mater., 2024, 1–7 Search PubMed.
- G. Kresse and J. Hafner, Ab initio molecular dynamics for liquid metals, Phys. Rev. B: Condens. Matter Mater. Phys., 1993, 47(1), 558 CrossRef CAS PubMed.
- G. Kresse and J. Furthmüller, Efficient iterative schemes for ab initio total-energy calculations using a plane-wave basis set, Phys. Rev. B: Condens. Matter Mater. Phys., 1996, 54(16), 11169 CrossRef CAS PubMed.
- G. Kresse and D. Joubert, From ultrasoft pseudopotentials to the projector augmented-wave method, Phys. Rev. B: Condens. Matter Mater. Phys., 1999, 59(3), 1758 CrossRef CAS.
- J. P. Perdew, K. Burke and M. Ernzerhof, Generalized gradient approximation made simple, Phys. Rev. Lett., 1996, 77(18), 3865 Search PubMed.
- P. E. Blöchl, O. Jepsen and O. K. Andersen, Improved tetrahedron method for Brillouin-zone integrations, Phys. Rev. B: Condens. Matter Mater. Phys., 1994, 49(23), 16223 CrossRef PubMed.
- S. Grimme, Semiempirical GGA-type density functional constructed with a long-range dispersion correction, J. Comput. Chem., 2006, 27(15), 1787–1799 Search PubMed.
- H. J. Monkhorst and J. D. Pack, Special points for Brillouin-zone integrations, Phys. Rev. B, 1976, 13(12), 5188 Search PubMed.
- L. Han, P. Ou, W. Liu, X. Wang, H.-T. Wang and R. Zhang, et al., Design of Ru-Ni diatomic sites for efficient alkaline hydrogen oxidation, Sci. Adv., 2022, 8(22), eabm3779 CrossRef CAS PubMed.
- J. K. Nørskov, J. Rossmeisl, A. Logadottir, L. Lindqvist, J. R. Kitchin and T. Bligaard, et al., Origin of the overpotential for oxygen reduction at a fuel-cell cathode, J. Phys. Chem. B, 2004, 108(46), 17886–17892 CrossRef PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d5ee00094g |
| ‡ These authors contributed equally. |
| This journal is © The Royal Society of Chemistry 2025 |