
Fluoroborate ionic liquids as sodium battery electrolytes†
Dale T.
Duncan
a,
Samantha L.
Piper
 a,
Maria
Forsyth
b,
Douglas R.
MacFarlane
a and
Mega
Kar
*b
a,
Maria
Forsyth
b,
Douglas R.
MacFarlane
a and
Mega
Kar
*b
aSchool of Chemistry, Monash University, Wellington Road, Clayton, VIC 3800, Australia
bInstitute of Frontier Materials, Deakin University, 221 Burwood Highway, Burwood, VIC 3125, Australia. E-mail: m.kar@deakin.edu.au
First published on 10th October 2023
Abstract
High-voltage sodium batteries are an appealing solution for economical energy storage applications. Currently available electrolyte materials have seen limited success in such applications therefore the identification of high-performing and safer alternatives is urgently required. Herein we synthesise six novel ionic liquids derived from two fluoroborate anions which have shown great promise in recent battery literature. This study reports for the first time the electrochemically applicable room-temperature ionic liquid (RTIL) N-ethyl-N,N,N-tris(2-(2-methoxyethoxy)ethyl)ammonium (tetrakis)hexafluoroisopropoxy borate ([N2(2O2O1)3][B(hfip)4]). The RTIL shows promising physical properties with a very low glass-transition at −73 °C and low viscosity. The RTIL exhibits an electrochemical window of 5.3 V on a glassy carbon substrate which enables high stability electrochemical cycling of sodium in a 3-electrode system. Of particular note is the strong passivation behaviour of [N2(2O2O1)3][B(hfip)4] on aluminium current-collector foil at potentials as high as 7 V (vs. Na+/Na) which is further improved with the addition of 50 mol% Na[FSI]. This study shows [B(hfip)4]− ionic liquids have the desired physical and electrochemical properties for high-voltage sodium electrolytes.
Introduction
Rechargeable batteries as energy storage systems have become fully integrated into many facets of modern life. Batteries provide a highly convenient energy storage solution for many applications, but their current design does not provide a sufficient safety profile, as indicated by ongoing reports of lithium-ion battery (LIB) fires.1 These fires are difficult to extinguish and produce significant quantities of toxic vapour; they have occurred in electric vehicles, mobile phones and both residential and grid-scale battery stacks.2–6 Ionic liquids (ILs) have negligible vapour pressure and low flammability which provides a safer alternative to conventional organic electrolytes (e.g. 1 M NaPF6 in ethylene carbonate (EC) and dimethyl carbonate (DMC)) which are often flammable and produce harmful species such as PF5 and HF upon combustion.7,8 The low volatility also impedes thermal runaway which greatly improves the safety of the electrolyte and of the overall device compared to organic electrolytes.9–11 The benefits of ILs expand beyond safety, also offering higher thermal stability,12 wider electrochemical windows13 and structural tunability. As such, ILs are shown to be viable electrolyte matrices in LIB and beyond-LIB technologies including sodium-ion, sodium–metal and high voltage lithium–metal batteries.14–19ILs are low melting point salts and have shown promise for a variety of applications beyond batteries including as sensors, lubricants, corrosion-inhibitors, recycling and separation processes.20 The material bulk properties are heavily influenced by the choice of cation and anion, and are a reflection of intermolecular interactions between the ions: weak and labile interactions are preferred which is reflected in high conductivity and fluidity of the bulk material. Anions offering weak and labile ion–ion interactions support high ionicity and are classified as weakly-coordinating anions (WCAs), often due to their charge-diffuse structure.21–23 Two common WCAs are hexafluorophosphate and bis(fluorosulfonyl)imide ([PF6]− and [FSI]−, respectively) of which [PF6]− is of great importance to LIBs as a component of the standard LP30 (1.0 M Li[PF6]/EC![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :DMC, 1:1 vol) electrolyte. The anion [FSI]− has lower symmetry and greater charge-delocalisation than [PF6]− and therefore exhibits weaker ion–ion interactions and better physical properties than [PF6]−.24,25 WCAs are particularly appealing for electrochemical applications as electrolytes offering high conductivity and fluidity in addition to high cathodic stability of the anion.
:DMC, 1:1 vol) electrolyte. The anion [FSI]− has lower symmetry and greater charge-delocalisation than [PF6]− and therefore exhibits weaker ion–ion interactions and better physical properties than [PF6]−.24,25 WCAs are particularly appealing for electrochemical applications as electrolytes offering high conductivity and fluidity in addition to high cathodic stability of the anion.
Identification of alternative WCAs is of research interest as currently available anions have significant drawbacks, such as the oxidative corrosion of [FSI]− towards aluminium in conventional electrolytes and the atmospheric instability of [PF6]−. Boron-containing anions such as tetrafluoroborate, difluoro(oxalato)borate and mono-closoborane ([BF4]−, [DFOB]− and [closo-CB11H12]−, respectively) are shown to be comparatively stable at high potentials and in air. Preliminary research with NaBF4 identified the salt to be an attractive material for non-flammable ionic liquid electrolytes, however it possesses limited solubility in ionic liquids which was addressed with a polyether solvent;26 the salt is also susceptible to hydrolysis. [DFOB]− has improved atmospheric stability and generates fewer hazardous decomposition products upon exposure to air when compared to [BF4]−.27 The anion also shows high oxidative stability against stainless steel with an initial decomposition potential of 5.8 V vs. Na+/Na with 1 M NaDFOB/EC:DEC 1:1 vol.28 The boron cluster [closo-CB11H12]− has also been shown to be useful as an ionic liquid anion with alkoxyammonium cations [N2(2O2O1)3]+ and [N4(2O2O1)3]+ exhibiting an electrochemical window of 4.55 V and low glass-transition (Tg) of −52 and −47 °C, respectively.29
The fluoroborate salts, sodium (tetrakis)hexafluoroisopropoxy borate (Na[B(hfip)4]·xDME, where DME = 1,2-dimethoxyethane) and sodium (tetrakis)trifluoroethoxy borate (Na[B(tfe)4]), were first reported in 2011 and 2014 respectively,30,31 however their use as battery electrolytes in organic solvents was only recently realised.32–34 From our recent publication, an electrolyte comprised of 0.5 M Na[B(hfip)4] in DME demonstrated stable electrochemical cycling over 300 cycles at 0.1 mA cm−2 in a Na‖Na symmetrical cell with very low overpotentials, below 2 mV.34 This impressive electrochemical performance was attributed to the weakly coordinating nature of [B(hfip)]− which results in high ion-dissociation of Na+. Moreover, the anion shows superior cathodic stability against aluminium, evenly passivating the surface with AlF3 and NaF.34
In other work, Roy et al. has shown 0.5 M Li[B(hfip)4]·3DME in EC:DMC (1:1 vol) to be an effective electrolyte in high-voltage Li–metal full-cells, as well as demonstrating exceptional atmospheric stability.32 Li[B(hfip)4]·3DME exposed to air for 24 hrs showed no deterioration of electrochemical performance in a full-cell when compared to an equivalent electrolyte with nonexposed salt. The promising outcomes of these studies have prompted further studies with [B(hfip)4]−, however the use of flammable organic solvents in these electrolytes has raised concerns regarding safety and high voltage stability.35 We seek to eliminate the use of such solvents by utilising the fluoroborates anions in ionic liquids. To date no electrochemically applicable ILs with [B(tfe)4]− or [B(hfip)4]− have been reported.
Anion and cation structures forming the basis of this work are shown in Fig. 1. Three cations are studied with the borate anions; N-ethyl-N,N,N-tris(2-(2-methoxyethoxy)ethyl)ammonium (1), iso-butyl(diethyl)methyl phosphonium (2) and N-methyl-N-propylpyrrolidinium ([N2(2O2O1)3]+, [P122i4]+ and [C3mpyr]+, respectively) which are of different chemical classes (ammonium, phosphonium and pyrrolidinium, respectively) and chosen based on literature merit for new electrolyte materials.29,36,37 The electrochemical stability and low Tg of [N2(2O2O1)3]+ ILs are appealing and will likely improve the physical and electrochemical properties of fluoroborate products.29,38 ILs with [P122i4]+ often display organic ionic plastic crystal (OIPC) behaviour such as [P122i4][DFOB] which exhibits this behaviour beyond −13 °C;37 the similar [P111i4]+ ILs also exhibit excellent physical and electrochemical properties.39 ILs with [C3mpyr]+ are well established in literature and have been used in SIB and Li–metal batteries.14,17,40
 | ||
| Fig. 1 Chemical structures and abbreviations of the cations N-ethyl-N,N,N-tris(2-(2-methoxyethoxy)ethyl)ammonium [N2(2O2O1)3]+ (1), iso-butyl(diethyl)methyl phosphonium [P122i4]+ (2) and N-methyl-N-propylpyrrolidinium [C3mpyr]+ (3), and anions (tetrakis)trifluoroethoxy borate [B(tfe)4]− (a) and (tetrakis)hexafluoroisopropoxy borate [B(hfip)4]− (b). | ||
In this study the novel fluoroborate ionic liquids and organic salts have been prepared and screened for useful phase behaviour as well as determination of the thermal stability and flammability hazard of the ILs. Two RTILs are identified and physically characterised against related materials to understand the effect of the charge-diffuse fluoroborate anion. The novel IL [N2(2O2O1)3][B(hfip)4] is further studied to determine its electrochemical stability, applicability to Na-electrochemistry via 3-electrode cyclic voltammetry and a thorough investigation into the underlying reason for the high voltage Al passivation behaviour via comprehensive surface analysis. [N2(2O2O1)3][B(hfip)4] is identified as a promising candidate for future research in SIB applications with the RTIL showing very high fluidity, high solubility of Na[FSI], high electrochemical cycling stability in a 3-electrode cell across 15 cycles and cathodic passivation of aluminium up to 7 V vs. Na+/Na. The high voltage passivation of Al of neat [N2(2O2O1)3][B(hfip)4] was further improved with the addition of 50 mol% Na[FSI], resulting in a surface with smooth morphology derived from [FSI]−.
Experimental
Synthetic procedures
All reagents were used as received. Na[B(OCH2CF3)4] (Na[B(tfe)4]), Li[B(OCH(CF3)2)4]·xDME (Li[B(hfip)4]·3DME) and N2(2O2O1)3Br were synthesised according to published methods.29,32,34:1.01 molar ratio) were mixed in 50 mL acetonitrile (MeCN). The mixture was stirred overnight under N2. The solvent was removed by rotovap and minimal dichloromethane (DCM) was added to dissolve the crude product, then filtered with a 0.2 μm PTFE syringe filter (25 mm ∅). The DCM was collected and centrifuged at 8000 rpm for 10 minutes. The liquid phase was collected and the sediment was discarded. The collected liquid was concentrated by rotovap and the product dried further until 8.0 × 10−3 mbar vacuum was held for two to four hours. 1H NMR: δ 3.83–3.77 ppm, 6H, t; δ 3.71–3.63 ppm, 8H, q; δ 3.63–3.58 ppm, 6H, t; δ 3.58–3.53 ppm, 6H, t; δ 3.53–3.46 ppm, 2H, q; δ 3.46–3.41 ppm, 6H, m; δ 3.26 ppm, 9H, s; δ 1.25–1.18 ppm, 3H, t. 11B NMR: 2.24, 1B, m. 19F NMR: −74.3 ppm, 12F, s.
:1.01 molar ratio) were mixed in 50 mL MeCN. The mixture was stirred overnight under N2. The solvent was removed by rotovap and minimal DCM was added to dissolve the crude product, then filtered with a 0.2 μm PFTE syringe filter (25 mm ∅). The DCM was collected and centrifuged at 8000 rpm for 10 minutes. The liquid phase was collected and the sediment was discarded. The clear liquid was then concentrated by rotovap and the product dried further until 8.0 × 10−3 mbar vacuum was held for two to four hours. 1H NMR: δ 3.73–3.61 ppm, 8H, q; δ 2.53–2.49 ppm, 2H, m; δ 2.26–2.11 ppm, 2H, m; δ 2.09–1.96 ppm, 1H, m; δ 1.85–1.79 ppm, 3H, d; δ 1.20–1.09 ppm, 6H, m; δ 1.05–1.01 ppm, 6H, d. 11B NMR: δ 2.27 ppm, 1B, s. 19F NMR: δ −74.27 ppm, 12F, s.
:1.01 molar ratio) were mixed in 50 mL MeCN. The mixture is stirred overnight under N2. The solvent was removed by rotavap and minimal DCM was added to dissolve the crude product, then filtered with a 0.2 μm PFTE syringe filter (25 mm ∅). The DCM was collected and centrifuged at 8000 rpm for 10 minutes. The liquid phase was collected and the sediment was discarded. The collected liquid was concentrated by rotovap and the product dried further until 8.0 × 10−3 mbar vacuum was held for two to four hours.1H NMR: δ 3.88–3.74 ppm, 8H, q; δ 3.53–3.37 ppm, 2H, m; δ 3.30–3.22 ppm, 4H, m; δ 2.98 ppm, 3H, s; δ 2.14–2.02 ppm, 4H, m; δ 1.78–1.65 ppm, 2H, m; δ 0.95–0.86 ppm, 3H, t. 11B NMR: δ 2.23 ppm. 19F NMR: δ −74.3 ppm.
:1.02 molar ratio) precursors were placed in a flask with 50 mL DCM. The mixture was stirred overnight under N2. The mixture was filtered and an aqueous/DCM biphasic extraction was performed 5 times on the DCM phase to remove residual Li biproduct and reactant. The DCM phase was concentrated by rotovap and the product dried further until 8.0 × 10−3 mbar vacuum was held for two to four hours. 1H NMR: δ 4.65 ppm, 4H, s; δ 3.84–3.77 ppm, 6H, t; δ 3.65–3.59 ppm, 6H, t; δ 3.59–3.54 ppm, 6H, m; δ 3.52–3.49 ppm, 2H, m; δ 3.49–3.43 ppm, 6H, m; δ 3.27 ppm, 9H, s; δ 1.28–1.23 ppm, 3H, t. 11B NMR: 1.63, 1B, m. 19F NMR: −74.6 ppm, 24F, s. MS [ES]+ = 352.28, [ES]− = 678.99.
:1.02 molar ratio) precursors were placed in a flask with 50 mL DCM. The mixture was stirred overnight under N2. The mixture was filtered and an aqueous/DCM biphasic extraction was performed 5 times on the DCM phase to remove residual Li biproduct and reactant. The DCM phase was concentrated by rotovap and the product dried further until 8.0 × 10−3 mbar vacuum was held for two to four hours. 1H NMR: δ 4.65 ppm, 4H, s; δ 2.53–2.49 ppm, 2H, m; δ 2.26–2.12 ppm, 2H, m; δ 2.09–1.95 ppm, 1H, m; δ 1.84–1.80 ppm, 3H, d; δ 1.19–1.09 ppm, 6H, m; δ 1.05–1.01 ppm, 6H, d. 11B NMR: 1.62, 1B, m. 19F NMR: −74.6 ppm, 24F, s. MS [ES]+ = 161.15, [ES]− = 678.97.
:1.02 molar ratio) precursors were placed in a flask with 50 mL DCM. The mixture was stirred overnight under N2. The mixture was filtered and an aqueous/DCM biphasic extraction was performed 5 times on the DCM phase to remove residual Li biproduct and reactant. The DCM phase was concentrated by rotovap and the product dried further until 8.0 × 10−3 mbar vacuum was held for two to four hours. 1H NMR: δ 4.65 ppm, 4H, s; δ 3.51–3.37 ppm, 2H, m; δ 3.28–3.22 ppm, 4H, m; δ 2.98 ppm, 3H, s; δ 2.10–2.05 ppm, 4H, m; δ 1.78–1.65 ppm, 2H, m; δ 0.96–0.89 ppm, 3H, t. 11B NMR: 1.62, 1B, m. 19F NMR: −74.6 ppm, 24F, s. MS [ES]+ = 128.11, [ES]− = 678.99.
Crystal structure and refinement
Single crystal X-ray crystallographic data was collected on a Rigaku Xtalab Synergy Dualflex using a monochromator equipped with Cu-Kα (λ = 1.5418 Å) radiation, at 123 K. Data was processed using proprietary software CrysAlisPro.41 The structures were solved and refined using the SHELX software suite42,43 and refined against F2 using Olex244 as a graphical interface. Non-hydrogen atoms were refined with anisotropic displacement parameters. Alkyl hydrogen atoms were included in calculated positions (riding model).Thermal properties
Phase transition temperatures (melting Tm, solid–solid Tss) and enthalpies were determined using a DSC TA Q200 calorimeter (TA Instruments). The equipment was calibrated using standard indium (TA Instruments, Tm = 156.6 °C, ΔHf = 28.47 J g−1) and cyclohexane (Sigma-Aldrich, Tm = 8 °C). Measurements were performed under a N2 atmosphere with a sample size of 3–5 mg and a scanning rate of 10 °C min−1. Presented values were taken from the second heating cycle unless otherwise specified. Tm was considered as the peak maximum and ΔHf as the area of the peak.Thermal stability was determined by thermogravimetric analysis (TGA) using Mettler Toledo TGA/DSC 1 STARe system. Samples were heated with a dynamic heating rate of 10 °C min−1 over 40 to 450 °C under N2. Material flammability was determined by igniting approximately 0.25 g or 0.25 mL of IL on Al-foil with a butane torch. The sample was exposed to the flame for 2 seconds to ensure combustion of the sample; the material was deemed flammable if combustion was sustained for one second or more following removal of the ignition source.
Physical properties
Density and viscosity measurements were conducted in 5 °C increments from 20 to 90 °C. For density measurements, 1.8 mL electrolyte was injected into a Mettler Toledo DM40 density meter. For viscosity measurements, a 1.59 mm ∅ steel rolling-ball capillary tube was prepared and inserted into an Anton Paar Lovis 2000ME viscosity meter at 30° incline. Conductivity measurements for liquid samples were conducted with electrical impedance spectroscopy (EIS) on a Biologic MTZ-35 instrument with an air-tight dip-cell fitted with Pt electrodes, designed in-house. For solid-state conductivity, pellets (13 mm ∅) were prepared with a hydraulic press to yield pellets of 0.5 to 0.8 mm thickness. The pellets were analysed in a barrel cell with stainless-steel electrode, the reported value is the average of two measurements. The frequency range of analysis was 7.0 × 107 to 1 Hz, with 10 points per decade and 10 cycles per measurement. Instrument temperature was controlled by a Eurotherm 2204e thermostat with analysis in 5 °C increments from 25 °C to 90 °C. The conductivity of the electrolytes was determined by relating the touch-down point of the Nyquist Plot against a 0.01 M KCl/H2O.Electrochemical characterisation
All electrolytes were stored and prepared in an Ar glovebox (<0.1 ppm H2O, <0.1 ppm O2). The Na[FSI]:1b electrolyte was prepared by measuring equimolar quantities before dissolution at 50 °C with stirring over 48 hours.Three-electrode cells were prepared with 0.5 mL electrolyte. The electrochemical window was determined with a glassy carbon working electrode and Pt wires as the counter electrode and pseudo reference electrode; the cell was referenced to the Fc+/Fc redox. The oxidative and reductive stability were determined independently with a clean cell and electrodes. The cells were cycled in a cyclic voltammetry protocol at 25 mV s−1 from open-circuit voltage (OCV), incrementally increasing the scan range until run-away current (>0.2 mA cm−2). Na electrochemistry was explored on a Cu working electrode, Na-metal reference electrode and a Pt wire counter electrode; all electrodes were cleaned prior to use and the Cu electrode was polished with 0.3 um beta-alumina and water slurry. The cell was cycled from OCV to −1.4 V to 2 V at 100 mV s−1 at 50 °C with ZIR correction determined by the x-intercept of the first touch-down on the Nyquist plot.
The Al electrodes were punched from a roll of battery-grade Al-foil with 19 mm diameter hollow punch. Sodium metal counter electrodes were prepared by rolling-out a Na metal chunk into a foil, first placed within a plastic bag before rolling on a glass pane. 18 mm diameter disks of Na metal were punched from the foil. The 2032 coin-cells were fabricated in an Ar-glovebox with a Solupor PE separator and 50 μL electrolyte. At either RT or 50 °C, the cells were at OCV for 12 h before cyclic voltammetry (OCV start) from 3 V to 7 V with a scan rate of 1 mV s−1 on a Biologic VMP2 or VMP3e potentiostat. Following the CV, the cell was held at OCV for 1 minute before the LSV to 7 V at 1 mV s−1. The CA at 7 V for 12 h commenced immediately after the LSV sweep.
Surface characterisation
The materials were investigated by a scanning electron microscope (JEOL JSM-7001F FEG-SEM) at an accelerating voltage (HT) of 15 kV and EDS that is attached to the microscope. The samples were attached on the SEM specimen holder with supporting stubs with double-side conducting carbon tape.X-ray photoelectron spectroscopic (XPS) analysis was performed using a Nexsa Surface Analysis System (Thermo Fisher Scientific). Analysis was undertaken with a monochromated Al Kα source at a power of 180 W (15 kV × 12 mA) and a hemispherical analyser operating in a fixed analyser transmission mode. The total pressure in the main vacuum chamber during the analysis was typically between 10−9 and 10−8 mbar. Survey spectra were acquired at a pass energy of 200 eV and a step size of 1 eV. High-resolution spectra were recorded from individual peaks at 50 eV pass energy using 0.1 eV step size, typically yielding a full width at half-maximum (FWHM) of 0.8–0.9 eV for the Ag 3d5/2 peak and <1.0 eV for the ester peak in poly(ethylene terephthalate) (PET) during performance tests. Samples were analysed at a nominal photoelectron emission angle of 0° with respect to the surface normal. Since the actual emission angle is ill-defined in the case of rough surfaces (ranging from 0 to 90°), the sampling depth may range from 0 to approximately 10 nm. Samples were mounted in a manner excluding any electrical contact of the analysed conductive area with the instrument ground. Data processing was performed using the ThermoScientific Avantage processing software version 5.9925. Binding energies were referenced to the C 1s peak at 284.8 eV (aliphatic hydrocarbon).
Results and discussion
Synthesis of the fluoroborate ionic liquids
The synthetic procedures to yield the six ionic liquids (ILs) are shown in Scheme 1 (see Fig. 1 for structures and Table S1, ESI† for abbreviations). Both products incorporating cation 1 were found to be room-temperature ionic liquids which is typical of ILs formed by the alkoxyammonium cation;29 the four remaining ILs are solids at room temperature. Purification of the [B(hfip)4]− ILs is possible via an aqueous biphasic extraction which achieves sufficient purification for the electrochemistry studied here. Unfortunately, the same procedure is not suitable for the [B(tfe)4]− ILs due to the tendency of the product to favour the aqueous phase. Purification was therefore limited to filtration and centrifuging techniques which prohibits the complete removal of Na+ species in the products. Inductively coupled plasma optical emission spectroscopy (ICP-OES) measurements determined residual alkali ions was present at trace quantities (<1000 ppm) for all products excluding 1a and 3a (Table S2, ESI†). Unfortunately, the measured concentration of Na+ reflects the maximum solubility of Na+ which is probably a mixture of Na[B(tfe)4] and NaBr. Both 1a and 3a measured a high Br− content by ICP-OES (Table S2, ESI†) which was expected to limit electrochemical investigations. The synthesis of 1b instead used the tosylate (i.e. [CH3(C6H4)SO3]−) pathway instead of Br−, however the sodium tosylate by-product was detected by NMR. The 1H-NMR spectra of the purified materials synthesised are shown in Fig. S2–S7 (ESI†).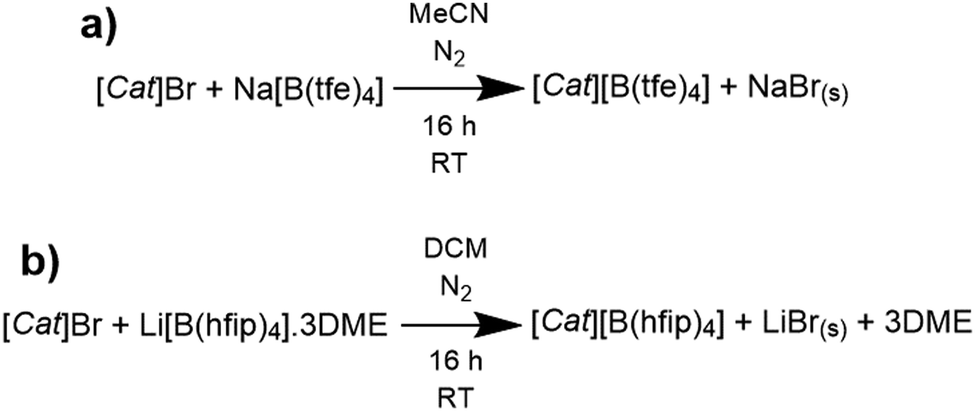 | ||
| Scheme 1 Generalised reaction schemes of for the metathesis of the (a) [B(tfe)4]− and (b) [B(hfip)4]− ILs where [Cat]+ represents either [N2(2O2O1)3]+, [P122i4]+ or [C3mpyr]+. Note: for [P122i4]+, the tosylate ([H3C(C6H4)SO3]−) salt is used instead of Br−. MeCN is acetonitrile and DCM is dichloromethane. | ||
Single-crystal X-ray diffraction (SC-XRD) crystal structures of 2a, 3a and 3b were obtained from crystals grown in a DCM/n-hexane slow diffusion crystallisation set-up, with DCM as the solvent and hexane as the anti-solvent. The vials were stored at room-temperature and crystals were collected after one week. A summary of the crystal structure data is available in Table S3 (ESI†) and the structures of 2a, 3a and 3b are available in Fig. S7–S9 (ESI†). Attempts to obtain crystals of 2b of suitable quality for SC-XRD analysis were unsuccessful. At room-temperature (RT), crystals of 2b were found to be malleable and tacky. Notably, this is characteristic plastic behaviour and explains the poor-quality diffraction observed for these crystals.
Physical characterisation
 | ||
| Fig. 2 Differential scanning calorimetry (DSC) traces of all synthesised compounds from −120 °C to Tm at 10 °C min−1. The first heating cycle is shown for all compounds excluding 2b where the 3rd cycle is shown to include the melt. | ||
The low entropies of fusion (ΔSf) of 2b and 3b (17 and 18 J mol−1 K−1, respectively), which are calculated from the first law of thermodynamics (ΔS = ΔH/Tm), suggest that these materials exhibit organic ionic plastic crystal (OIPC) behaviour. OIPCs typically exhibit only a small (<20 J mol−1 K−1) ΔSf upon melting,45 due to their highly disordered “plastic” phases that exist prior to melting; this disorder contributes to high mobility in these solid phases.46,47
All materials analysed here undergo solid-state transitions prior to melting and the solid-state phases are denoted here by IV, III, II and I, where I is the highest temperature solid phase. These transitions are typically associated with the onset of translational, rotational or orientational motions of the ions, or crystallographic changes in the material.48,492b and 3b exhibit several solid–solid transitions prior to the melt and the calculation of ΔS (Table 1) shows both materials undergo low entropy melting transitions; these compounds therefore likely have high entropy in the solid phases. Furthermore, 2b exhibits the disordered phase I (ΔSIII-I = 46 J mol−1 K−1, ΔSf = 17 J mol−1 K−1) at room temperature (RT). On cooling, two exotherms are observed with a combined entropy of 46 J mol−1 K−1, indicating the endothermic transition observed at 9 °C is likely a two-step process or a convoluted peak representing both the III–II and II–I transitions, hence the lower temperature phase is denoted III. Materials which display OIPC behaviour at RT are of strong research interest for a variety of applications which include atmospheric gas separation and solid-state electrolytes.49–52 The broad temperature range of the phase I of 2b (9 to 180 °C) is ideal as it provides the opportunity to utilise the unique properties of the plastic phase over a large temperature range.48,49
| Glass transition | Solid–solid transitions | Melt | |||||||
|---|---|---|---|---|---|---|---|---|---|
| T g | Phase IV–III | Phase III–II | Phase II–I | Phase I–melt | |||||
| T g ± 1/°C | T ss ± 1/°C | ΔS ± 10%/J K−1 mol−1 | T ss ± 1/°C | ΔS ± 10%/J K−1 mol−1 | T ss ± 1/°C | ΔS ± 10%/J K−1 mol−1 | T m ± 1/°C | ΔSf ± 10%/J K−1 mol−1 | |
| 1a | −74 | ||||||||
| 1b | −73 | ||||||||
| 2a | 70 | 10 | 100 | 47 | |||||
| 2b | 9 | 46 | — | — | 189 | 17 | |||
| 3a | 90 | 5.0 | 113 | — | 128 | 38 | |||
| 3b | 47 | 6.5 | 108 | 25 | 142 | 8.2 | 158 | 18 | |
The Tm of the [B(tfe)4]− (a) compounds were lower for both [P122i4]+ and [C3mpyr]+, when compared to their respective [B(hfip)4]− salts (Table 1). For example, the Tm of 2a is 100 °C whereas the Tm of 2b is 188 °C. The difference in melting point is attributed to the additional molar mass of [B(hfip)4]−.
The first heating cycle is shown for all compounds, except 2b, where the 3rd cycle is shown instead to include the high Tm; the material was not originally cycled to this temperature due to decomposition concerns around 200 °C. Both 3a and 3b displayed evidence of decomposition after their first melting transitions. The second cycle for 3a and 3b (Fig. S15 and S16, ESI,† respectively) was altered compared to the first with phase II to I and the melting temperature merging to a single peak which implies partial decomposition of the analysed material (as decomposition products serve as impurities which can broaden transitions and decrease transition temperatures).53
| Compound | T m/°C | T d (−5%)/°C | Flammable? | σ (50 °C)/S cm−1 | ρ (50 °C)/g cm−3 | η (50 °C)/mPa s | ECW (WE: GC)/V | |
|---|---|---|---|---|---|---|---|---|
| vs. Fc+/Fc | vs. Na+/Na | |||||||
| a T g observed instead of Tm. | ||||||||
| 1a | −74a | 132 | Yes | 9.4 × 10−4 | 1.25 | 40.1 | Up to 1.0 | Up to 4.1 |
| 1b | −73a | 207 | No | 1.6 × 10−3 | 1.39 | 21.1 | −3.1 to 2.2 | 0.0 to 5.3 |
| 2a | 100 | 168 | Yes | 5.1 × 10−4 | — | — | — | |
| 2b | 189 | 210 | No | 3.7 × 10−6 | — | — | — | |
| 3a | 128 | 200 | Yes | 2.4 × 10−4 | — | — | — | |
| 3b | 158 | 185 | No | 1.4 × 10−4 | — | — | — | |
| [C3mpyr][FSI] | −9 | 319 | No | 1.6 × 10−2 | 1.32 | 20.1 | −3.0 to 2.325 | 0.1 to 5.4 |
| [N2(2O2O1)3] [TFSI]38,58 | −68a | 300 | — | 3.5 × 10−3 | 1.28 | 25.7 | Up to 1.3 | Up to 4.4 |
| [N2(2O2O1)3] [closo-CB11H12]29 | −47a | 300 | — | 3.9 × 10−4 | 1.01 | 269 | Up to 1.7 | Up to 4.7 |
Ionic liquids are often described as being non-flammable, despite combustion being a complex and unpredictable process therefore materials should be judged on a case-by-case basis.11,57 In the flammability test, a small sample is exposed to an ignition source until combustion, after which the ignition source is removed and the sample is observed for self-sustaining behaviour.10 Still images and experimental observations from video recordings of the tests are available in the ESI† (Fig. S18–S24) with a brief discussion of experimental observations; a summary of the results are shown in Table 2. The image in Fig. 3a shows the material 1a after 2 s of combustion with an applied ignition source, which sustained combustion for 9 s following ignition source removal (Fig. 3b); the sample was entirely consumed and left minimal residue. TGA was concordant with this observation by also recording 100% mass loss.
 | ||
| Fig. 3 Still images at key time-points of the flammability experiments for 1a at (a) 2 s and (b) 12 s following ignition showing the sustained combustion of 1a. Stills at (c) 5 s and (d) 7 s of 1b show non-sustained combustion following removal of the ignition source. The (e) self-extinguishing times of all analysed samples; safe ILs are indicated by a self-extinguishing time of <2 s (shown by the green region). | ||
The suspected source of the flammability is attributed to disassociation of the anion into known highly volatile and flammable compounds B(tfe)3 and Na[tfe] (tfe = –OCH2CF3).8,30 In contrast, Fig. 3c shows the combustion of 1b which quickly extinguished upon removal of the ignition source, as shown in Fig. 3d. The self-extinguishing time following removal of the ignition source for all samples is shown in Fig. 3e. [C3mpyr][FSI] was used as an experimental benchmark; this was found to be combustible but not self-sustaining, therefore non-flammable. 1b behaved identically to the [C3mpyr][FSI] and therefore possesses a comparably low flammability hazard profile. Overall, [B(hfip)4]− salts all displayed safety profiles equivalent to the experimental benchmark making them highly appealing for large-scale energy storage applications.
Solid-state conductivity measurements were made to determine the ionic mobility of the organic salts. Such physicochemical properties are crucial in electrochemical applications such as batteries in determining the transport of charged ions through the electrolyte; the results are summarised in Table 2 and plotted in Fig. S25 (ESI†) along with some benchmark data from the literature. The conductivity of the ILs 1a and 1b is shown in Fig. 4a. The measured values of 1a and 1b at 50 °C (9.4 × 10−4 and 1.6 × 10−3 S cm−1, respectively, Table 2) are lower in comparison to [N2(2O2O1)3][TFSI] (3.5 × 10−3 S cm−1), due to the smaller anion volume of [TFSI]− relative to the fluoroborates. Conductivity for 1a and 1b was higher than [N2(2O2O1)3][closo-CB11H12] (3.9 × 10−4 S cm−1) which implies fewer charge-neutral ion aggregates which cannot contribute to bulk conductivity.
 | ||
| Fig. 4 (a) Conductivity, (b) viscosity, (c) density and (d) Walden analysis of the RTILs 1a and 1b. Data for [N2(2O2O1)3][closo-CB11H12] and [N2(2O2O1)3][TFSI] were obtained from literature.29,38 The solid and dashed grey lines on the Walden plot represent the Ideal KCl and 10% ionicity lines, respectively. | ||
The measured conductivity of all of the solid ILs particularly 2b (1.0 × 10−9 S cm−1 at 30 °C) was lower than published data for [N111,1O1][FSI] which exhibits an impressive conductivity of 9.9 × 10−6 S cm−1 at 30 °C. The low conductivity for the newly designed materials is thought to be a consequence of the bulky fluoroborate anion. The steric bulk of the fluoroborate anions inherently hinders diffusion of the anion which counteracts the reduced electrostatic pairing. This is evident when comparing solid-state conductivities of [B(tfe)4]− and [B(hfip)4]− salts where the smaller [B(tfe)4]− anion exhibits higher conductivity with either cation.
| σ = σ0exp[−kσ/(T − Tσ)] | (1) |
T 0 is often referred to as the ideal glass transition temperature and often lies 20–50 °C below experimental data, as observed for 1a and 2b; DSC experiments have determined the Tg to be 200 and 201 K respectively. [C3mpyr][FSI] measured a higher conductivity than both 1a and 1b which is reflected in the σ0 values however the difference of kσ between 1b and [C3mpyr][FSI] was not significant, given the large standard deviation in the value for [C3mpyr][FSI].
Viscosity data was also fitted to a VTF type equation (eqn (2))
| η = η0exp[kη/(T − Tη)] | (2) |
Both fluoroborate ILs have shown potential for electrochemical applications, which is further investigated in the following section. The electrochemical window is initially investigated, followed by Na-cycling and cathodic passivation of Al of the best material.
Electrochemical characterisation
Electrochemical stability for 1b was first investigated via a three-electrode cell against a glassy carbon (GC) working electrode at RT under argon (Ar) atmosphere. 1a was not studied electrochemically due to the known [Br]− content. Fig. 5a shows the 5.3 V ECW of 1b against a GC electrode. During the experiment the maximum potential for each sweep was gradually increased until runaway current is observed. The sixth and eighth cycle display the decreasing currents as the native GC surface is passivated; the mechanism for passivation is currently unknown. The ninth cycle appears to be the 2.2 V (vs. Fc+/Fc, 5.3 V vs. Na+/Na) oxidative limit of 1b. This work improves upon the 5 V (vs. Li+/Li) window of 0.5 M Li[B(hfip)4]·3DME/EC:DMC (1:1 vol), despite the ether ammonium cation which often exhibits lower stabilities.29,59 The oxidative stability of 2.2 V (vs. Fc+/Fc, 5.3 V vs. Na+/Na) for 1b is higher than the 2.1 V limit of [P111i4][FSI], one the most electrochemically stable ILs known.39 It substantially improves upon the stability of [N2(2O2O1)3]+ ILs with alternative anions such as [closo-CB11H12]− or [TFSI]− which display stabilities of 4.4 and 4.7 V (vs. Na+/Na), respectively.29,59 This suggests that 1b does not oxidise easily at high oxidative potentials up to 5.3 V (vs. Na+/Na). This makes it an excellent candidate as a battery electrolyte with high-voltage cathodes which function at high operating voltages (i.e. >4.0 V vs. Na+/Na).64
 | ||
| Fig. 5 Three-electrode CV traces of the (a) ECWa of 1b, earlier sweeps are shown with the dotted and dashed traces with the passivation indicated by the black arrow (WE: GC, CE: Pt, RE: Pt) at RT. Cell potential was referenced to a ferrocenium/ferrocene redox-couple. (b) CV trace of the Na+/Na redox with 50 mol% Na[FSI] in 1b (WE: Cu, CE: Pt, RE: Na) in the potential range of −1.4 to 2.0 V at 100 mV s−1 at 50 °C. All 15 cycles are shown where black is the first cycle and subsequent cycles in blue. aoxidative and reductive stability were measured separately and plotted together. | ||
To confirm the applicability of 1b in a Na electrochemical system, a three-electrode cell with a copper working electrode was prepared. The cyclic voltammetry trace with Na[FSI]:1b (at 1:1 molar ratio) is shown in Fig. 5b. In the first cycle (black) a coulombic efficiency of 72% and a maximum deposition current density (Jmax) of 1.4 mA cm−2 at −1.4 V vs. Na+/Na was achieved. The overpotential for Na deposition onset was measured at −80 mV vs. Na+/Na which implied the energy barrier for deposition is low. The Jmax of Na[FSI]:1b was lower than the similar Na[FSI]:[N2(2O2O1)3][FSI] system (Jmax = 12 mA cm−2) and is believed to result from the lower molar Na+ concentration (1.2 M) of Na[FSI]:1b which is a consequence of the bulky anion of 1b.59 The Na[FSI]:1b electrolyte outperforms other alkoxyammonium RTILs such as [N2(2O2O1)3][TFSI] with 2.0 mol kg−1 Na[TFSI] which measured a Jmax of 0.18 mA cm−2 (10 mV s−1, 50 °C).60 The cell cycling behaviour over 15 cycles with Na[FSI]:1b shows consistent deposition and stripping currents which implies stable electrochemical cycling of Na. In the first cycle (black trace), solid–electrolyte interface formation on the native Cu surface can be seen by the elevated current from ∼1.5 V to the potential of Na0 deposition. Currents for Na0 deposition were very stable across the 15 cycles where two convoluted peaks were observed which suggests two or more oxidative mechanisms occurring. Elucidation of these processes was not attempted as in-depth studies outside of the scope of this work would be required.
 | ||
| Fig. 6 The first, third and sixth voltammetry sweeps (currents decreasing with cycles) of 1b and Na[FSI]:1b electrolytes in an Al‖Na coin-cell (WE: Al, RE/CE: Na) and (b) CA following the sixth voltammetry sweep at 50 °C. | ||
Notably, the electrochemical data implies the passivation behaviour was substantially improved with the addition of Na[FSI] seen by the lower currents; the exact role of [FSI]− is further understood in the surface analysis in the following section. The Na[FSI]:1b electrolyte displayed an impressively low current of 0.037 μA cm−2 after 12 h of polarisation at 7 V this being representative of the leakage current of the Al current-collector. Compared to Na[FSI]:1b, neat 1b at 50 °C was less stable and exhibited a temporary increase in current at 2 h (from 1.5 to 1.6 μA cm−2) in the CA experiment (Fig. 6b), which is suspected to be caused by Na+ ions from the CE reaction becoming involved in the WE process which compromises the passivation layer. Dissolution is often enhanced with increasing temperatures which may explain the worse passivation at 50 °C with neat 1b.
The electrochemical data for the anodisation of Al at the very high potential of 7 V (vs. Na+/Na) is promising for all systems however each system showed a varying degree of passivation effectiveness. To further understand the passivation behaviour between electrolytes, the foils were recovered after the electrochemisty shown and analysed to investigate the morphology and chemical composition of the anodised foils.
 | ||
| Fig. 7 SEM micrographs of (a) pristine Al current-collector foil (top-down), (b) Al-foil anodised with Na[FSI]:1b (top-down) and (c) cross-section of the same foil to show surface smoothness. SEM micrographs of anodised foils with neat 1b at (d) 50 °C (top-down) and (e) RT (top-down), and the (f) cross-section of the RT anodisation to show surface roughness. | ||
The results and discussion of the EDX data can be found in the ESI† below Table S5. The EDX data (Fig. S30–S33, ESI†) shows Na[FSI]:1b at 50 °C exhibits a smaller amount of electrolyte decomposition than 1b, measuring the highest abundance of Al (87.4 and 75.9 atomic%, respectively).
The X-ray photoelectron spectroscopy (XPS) analysis of Al 2p environments on the anodised foils (Fig. 8a) show significant alteration of all surfaces when compared to the pristine Al. The surface of the pristine Al foil was as expected, with the bulk Al0 and overlying Al2O3 signals at 73 and 74 eV, respectively.68,69 Surprisingly the sample produced from 1b at RT showed no Al 2p environment which implies an Al-free passivation layer, which is thicker than the penetration depth (∼10 nm) of XPS. Al 2p was detected for 1b at 50 °C although the weak Al0 response implies a thickening of the passivation layer which is weighted heavily towards inorganic Al species (76–74 eV).68,69 The Al 2p in the Na[FSI]:1b sample was also heavily weighted towards inorganic Al rather than Al0 which suggests thickening of the passivation layer with additional inorganic Al compounds such as AlF3 (76 eV) and AlFO2 (75 eV).69 Neat 1b at 50 °C also fluorinated the native Al2O3 layer however AlF3 was in lower abundance and an outcome of the inferior passivation.
 | ||
| Fig. 8 X-ray photoelectron spectroscopy of the (a) Al 2p, (b) Na 1s, (c) F 1s, and (d) B 1s chemical environments detected on the Al foil surface. Important chemical species are indicated above each figure. Data for all samples is normalised and low abundance regions (i.e. noisy) are reduced until noise is comparable to other samples. | ||
The S 2p spectra are shown in Fig. S34 (ESI†) where Na[FSI]:1b, the only S-containing system, produced a S 2p response at 170.5 eV (FO2S–) which is representative of the [FSI]− and/or decomposition products;70 Na[FSI] is assumed to be removed by washing with DMC. In organic systems, [FSI]− is well-known to corrode Al foil at potentials greater than 4.3 V vs. Na+/Na, which involves the formation of Al[FSI]3 and/or [FSI]− decomposition products on the Al surface. Al[FSI]3 exhibits high solubility in organic solvents and therefore dissolves into the electrolyte to reveal underlying Al2O3 or Al0 which is continually attacked by [FSI]−.71Fig. 8b shows the Na 1s environments and confirms the presence of NaF (1071.5 eV) on the surface after anodisation with Na[FSI]:1b; higher binding energy species were detected but their identity could not be determined and are likely complex products of electrolyte decomposition.
The dominating signals in the F 1s spectra are shown in Fig. 8c, with the 1b at RT sample producing AlF3 (687.7 eV) whereas for the Na[FSI]:1b sample, NaF and [FSI]− were the major species present.14,32,34 The –CF3 moiety detected for 1b at 50 °C is attributed to residual electrolyte in the porous Al surface which can be seen in the SEM micrograph in Fig. S35 (ESI†). The figure is accompanied by a short discussion regarding the inferior passivation of 1b at 50 °C. The C 1s spectra in Fig. S36 (ESI†) detected abundant –CF3 for 1b at 50 °C from the residual electrolyte. Ethereal C was also detected the surface for all anodised samples which is probably from cation decomposition products. The residual electrolyte from 1b at 50 °C can also be observed in the nitrogen (N) 1s spectra in Fig. S37 (ESI†) at 401.6 eV which corresponds to an alkyl ammonium environment.72 The same environment was observed for Na[FSI]:1b additional to the N–SO2 environment (399.8 eV) which confirms the presence of Al[FSI]3, or [FSI]− decomposition products.14 The B 1s spectra in Fig. 8d shows B was a significant component only for 1b at RT, all other samples detected negligible B. The B 1s environment detected is attributed to [B4O7]− and confirms its role in passivation of the Al foil. The O 1s spectra in Fig. S38 (ESI†) supports the presence of tetraborate ([B4O7]−) (192.4 eV) after anodisation with 1b at RT.73
Conclusions
This work reports novel borate ionic liquids and studies of their thermal, physical, and electrochemical properties for a broad scope of applications. All ILs prepared with [B(hfip)4]− show intriguing thermal properties such as the plastic and liquid behaviour. The best material uncovered was [N2(2O2O1)3][B(hfip)4] (1b), found to be a room-temperature ionic liquid which exhibits a 5.3 V electrochemical window and passivation of Al shown up to 7 V (vs. Na+/Na). Electrochemical cycling of Na was shown with an electrolyte of an equimolar mixture of Na[FSI] and 1b where high stability was observed over 15 cycles. Additional to the electrochemical properties, 1b was found to exhibit good ionicity and non-flammability. The properties discovered in this work show 1b to be a promising material for high-voltage electrochemical applications.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors acknowledge the Australian Research Council funded StorEnergy Industrial Transformation Training Centre (IC180100049) for financial support. The authors also thank Darcy Simondson-Tammer and Dr Cuong Nguyen for their assistance with analysis.References
- L. Wong, Crews battle Tesla battery fire at Moorabool, near Geelong – ABC News, https://www.abc.net.au/news/2021-07-30/tesla-battery-fire-moorabool-geelong/100337488, (accessed 10 December 2022).
- J. Sun, J. Li, T. Zhou, K. Yang, S. Wei, N. Tang, N. Dang, H. Li, X. Qiu and L. Chen, Nano Energy, 2016, 27, 313–319 CrossRef CAS.
- F. Larsson, P. Andersson, P. Blomqvist and B. E. Mellander, Sci. Rep., 2017, 7, 1–13 CrossRef CAS PubMed.
- M. Li, J. Lu, Z. Chen and K. Amine, Adv. Mater., 2018, 30, 1800561 CrossRef PubMed.
- Q. Wang, B. Mao, S. I. Stoliarov and J. Sun, Prog. Energy Combust. Sci., 2019, 73, 95–131 CrossRef.
- Q. Wang, P. Ping, X. Zhao, G. Chu, J. Sun and C. Chen, J. Power Sources, 2012, 208, 210–224 CrossRef CAS.
- P. Barnes, K. Smith, R. Parrish, C. Jones, P. Skinner, E. Storch, Q. White, C. Deng, D. Karsann, M. L. Lau, J. J. Dumais, E. J. Dufek and H. Xiong, J. Power Sources, 2020, 447, 227370 CrossRef.
- K. Tasaki, K. Kanda, S. Nakamura and M. Ue, J. Electrochem. Soc., 2003, 150, A1628 CrossRef CAS.
- H. Sun, G. Zhu, X. Xu, M. Liao, Y. Y. Li, M. Angell, M. Gu, Y. Zhu, W. H. Hung, J. Li, Y. Kuang, Y. Meng, M. C. Lin, H. Peng and H. Dai, Nat. Commun., 2019, 10, 1–11 CrossRef PubMed.
- C. Arbizzani, G. Gabrielli and M. Mastragostino, J. Power Sources, 2011, 196, 4801–4805 CrossRef CAS.
- D. M. Fox, J. W. Gilman, A. B. Morgan, J. R. Shields, P. H. Maupin, R. E. Lyon, H. C. De Long and P. C. Trulove, Ind. Eng. Chem. Res., 2008, 47, 6327–6332 CrossRef CAS.
- Y. Cao and T. Mu, Ind. Eng. Chem. Res., 2014, 53, 8651–8664 CrossRef CAS.
- M. Armand, F. Endres, D. R. MacFarlane, H. Ohno and B. Scrosati, Nat. Mater., 2009, 8, 621–629 CrossRef CAS PubMed.
- J. Sun, L. A. O’Dell, M. Armand, P. C. Howlett and M. Forsyth, ACS Energy Lett., 2021, 6, 2481–2490 CrossRef CAS.
- S. Aminah, M. Noor, P. C. Howlett, D. R. Macfarlane and M. Forsyth, Electrochim. Acta, 2013, 114, 766–771 CrossRef.
- S. Brutti, E. Simonetti, M. De Francesco, A. Sarra, A. Paolone, O. Palumbo, S. Fantini, R. Lin, A. Falgayrat, H. Choi, M. Kuenzel, S. Passerini and G. B. Appetecchi, J. Power Sources, 2020, 479, 228791 CrossRef CAS.
- M. Hasanpoor, D. Saurel, R. C. Barreno, K. Fraysse, M. Echeverría, M. Jáuregui, F. Bonilla, G. W. Greene, R. Kerr, M. Forsyth and P. C. Howlett, ACS Appl. Mater. Interfaces, 2022, 14, 13196–13205 CrossRef CAS PubMed.
- J. Sun, D. Rakov, J. Wang, Y. Hora, M. Laghaei, N. Byrne, X. Wang, P. C. Howlett and M. Forsyth, ChemElectroChem, 2022, 9, e202200382 CrossRef CAS.
- F. Makhlooghiazad, L. A. O’Dell, L. Porcarelli, C. Forsyth, N. Quazi, M. Asadi, O. Hutt, D. Mecerreyes, M. Forsyth and J. M. Pringle, Nat. Mater., 2022, 21, 228–236 CrossRef CAS PubMed.
- N. V. Plechkova and K. R. Seddon, Chem. Soc. Rev., 2007, 37, 123–150 RSC.
- W. Xu and C. A. Angell, Science, 2003, 302, 422–425 CrossRef CAS PubMed.
- M. Yoshizawa, W. Xu and C. A. Angell, J. Am. Chem. Soc., 2003, 125, 15411–15419 CrossRef CAS PubMed.
- W. Xu, E. I. Cooper and C. A. Angell, J. Phys. Chem. B, 2003, 107, 6170–6178 CrossRef CAS.
- J. Golding, N. Hamid, D. R. MacFarlane, M. Forsyth, C. Forsyth, C. Collins and J. Huang, Chem. Mater., 2001, 13, 558–564 CrossRef CAS.
- H. Matsumoto, H. Sakaebe, K. Tatsumi, M. Kikuta, E. Ishiko and M. Kono, J. Power Sources, 2006, 160, 1308–1313 CrossRef CAS.
- M. Egashira, T. Asai, N. Yoshimoto and M. Morita, Electrochim. Acta, 2011, 58, 95–98 CrossRef CAS.
- J. L. Allen, D. W. McOwen, S. A. Delp, E. T. Fox, J. S. Dickmann, S. D. Han, Z. Bin Zhou, T. R. Jow and W. A. Henderson, J. Power Sources, 2013, 237, 104–111 CrossRef CAS.
- J. Chen, Z. Huang, C. Wang, S. Porter, B. Wang, W. Lie and H. K. Liu, Chem. Commun., 2015, 51, 9809–9812 RSC.
- M. Kar, O. Tutusaus, D. R. MacFarlane and R. Mohtadi, Energy Environ. Sci., 2019, 12, 566–571 RSC.
- S. Bulut, P. Klose and I. Krossing, Dalton Trans., 2011, 40, 8114 RSC.
- A. B. A. Rupp, P. Klose, H. Scherer and I. Krossing, ChemPhysChem, 2014, 15, 3729–3731 CrossRef CAS PubMed.
- B. Roy, P. Cherepanov, C. Nguyen, C. Forsyth, U. Pal, T. C. Mendes, P. Howlett, M. Forsyth, D. MacFarlane and M. Kar, Adv. Energy Mater., 2021, 11, 2101422 CrossRef CAS.
- D. M. C. Ould, S. Menkin, H. E. Smith, V. Riesgo-Gonzalez, E. Jónsson, C. A. O’Keefe, F. Coowar, J. Barker, A. D. Bond, C. P. Grey and D. S. Wright, Angew. Chem., 2022, 134, e202202133 CrossRef.
- D. T. Duncan, B. Roy, S. L. Piper, C. Nguyen, P. Howlett, M. Forsyth, D. R. MacFarlane, J. Sun and M. Kar, J. Phys. Chem. C, 2022, 126, 18918–18930 CrossRef CAS.
- R. Gond, W. van Ekeren, R. Mogensen, A. J. Naylor and R. Younesi, Mater. Horiz., 2021, 8, 2913 RSC.
- Y. Wang, R. Jiang, Y. Liu, H. Zheng, W. Fang, X. Liang, Y. Sun, R. Zhou and H. Xiang, ACS Appl. Energy Mater., 2021, 4, 7376–7384 CrossRef CAS.
- C. S. M. Kang, O. E. Hutt and J. M. Pringle, ChemPhysChem, 2022, 23, e202200115 CrossRef CAS PubMed.
- M. Hilder, M. Gras, C. R. Pope, M. Kar, D. R. MacFarlane, M. Forsyth and L. A. O’Dell, Phys. Chem. Chem. Phys., 2017, 19, 17461–17468 RSC.
- G. M. A. Girard, M. Hilder, H. Zhu, D. Nucciarone, K. Whitbread, S. Zavorine, M. Moser, M. Forsyth, D. R. Macfarlane and P. C. Howlett, Phys. Chem. Chem. Phys., 2015, 17, 8706–8713 RSC.
- H. Yoon, H. Zhu, A. Hervault, M. Armand, D. R. MacFarlane and M. Forsyth, Phys. Chem. Chem. Phys., 2014, 16, 12350–12355 RSC.
- CrysAlisPRO, Oxford Diffr./Agilent Technol. UK Ltd.
- G. M. Sheldrick, Acta Crystallogr., Sect. C: Struct. Chem., 2015, 71, 3–8 Search PubMed.
- G. M. Sheldrick, Acta Crystallogr., Sect. A: Found. Crystallogr., 2015, 71, 3–8 CrossRef PubMed.
- O. V. Dolomanov, L. J. Bourhis, R. J. Gildea, J. A. K. Howard and H. Puschmann, J. Appl. Crystallogr., 2009, 42, 339–341 CrossRef CAS.
- J. Timmermans, J. Phys. Chem. Solids, 1961, 18, 1–8 CrossRef CAS.
- U. A. Rana, M. Forsyth, D. R. Macfarlane and J. M. Pringle, Electrochim. Acta, 2012, 84, 213–222 CrossRef CAS.
- J. M. Pringle, Phys. Chem. Chem. Phys., 2013, 15, 1339–1351 RSC.
- H. Zhu, D. R. MacFarlane, J. M. Pringle and M. Forsyth, Trends Chem., 2019, 1, 126–140 CrossRef CAS.
- D. R. MacFarlane, M. Forsyth, P. C. Howlett, M. Kar, S. Passerini, J. M. Pringle, H. Ohno, M. Watanabe, F. Yan, W. Zheng, S. Zhang and J. Zhang, Nat. Rev. Mater., 2016, 1, 15005 CrossRef CAS.
- N. Sirigiri, F. Chen, C. M. Forsyth, R. Yunis, L. O’Dell, J. M. Pringle and M. Forsyth, Mater. Today Phys., 2022, 22, 100603 CrossRef CAS.
- A. Basile, M. Hilder, F. Makhlooghiazad, C. Pozo-Gonzalo, D. R. MacFarlane, P. C. Howlett and M. Forsyth, Adv. Energy Mater., 2018, 8, 1703491 CrossRef.
- R. Yunis, D. Al-Masri, A. F. Hollenkamp, C. M. Doherty, H. Zhu and J. M. Pringle, J. Electrochem. Soc., 2020, 167, 070529 CrossRef CAS.
- M. Kerner, N. Plylahan, J. Scheers and P. Johansson, RSC Adv., 2016, 6, 23327–23334 RSC.
- F. U. Shah, O. I. Gnezdilov and A. Filippov, Phys. Chem. Chem. Phys., 2017, 19, 16721–16730 RSC.
- M. Taher, F. U. Shah, A. Filippov, P. De Baets, S. Glavatskih and O. N. Antzutkin, RSC Adv., 2014, 4, 30617–30623 RSC.
- N. Wongittharom, T. C. Lee, C. H. Wang, Y. C. Wang and J. K. Chang, J. Mater. Chem. A, 2014, 2, 5655–5661 RSC.
- M. Smiglak, W. M. Reichert, J. D. Holbrey, J. S. Wilkes, L. Sun, J. S. Thrasher, K. Kirichenko, S. Singh, A. R. Katritzky and R. D. Rogers, Chem. Commun., 2006, 2554 RSC.
- M. Kar, B. Winther-Jensen, M. Armand, T. J. Simons, O. Winther-Jensen, M. Forsyth and D. R. MacFarlane, Electrochim. Acta, 2016, 188, 461–471 CrossRef CAS.
- M. Hilder, P. C. Howlett, D. Saurel, E. Gonzalo, A. Basile, M. Armand, T. Rojo, M. Kar, D. R. MacFarlane and M. Forsyth, Electrochim. Acta, 2018, 268, 94–100 CrossRef CAS.
- C. R. Pope, M. Kar, D. R. MacFarlane, M. Armand, M. Forsyth and L. A. O’Dell, ChemPhysChem, 2016, 17, 3187–3195 CrossRef CAS PubMed.
- D. Morales, L. Gomes Chagas, D. Paterno, S. Greenbaum, S. Passerini and S. Suarez, Electrochim. Acta, 2021, 377, 138062 CrossRef CAS.
- K. Matsumoto, Y. Okamoto, T. Nohira and R. Hagiwara, J. Phys. Chem. C, 2015, 119, 7648–7655 CrossRef CAS.
- S. A. M. Noor, N. C. Su, L. T. Khoon, N. S. Mohamed, A. Ahmad, M. Z. A. Yahya, H. Zhu, M. Forsyth and D. R. MacFarlane, Electrochim. Acta, 2017, 247, 983–993 CrossRef CAS.
- H. Gao, I. D. Seymour, S. Xin, L. Xue, G. Henkelman and J. B. Goodenough, J. Am. Chem. Soc., 2018, 140, 18192–18199 CrossRef CAS PubMed.
- S. T. Myung, Y. Hitoshi and Y. K. Sun, J. Mater. Chem., 2011, 21, 9891–9911 RSC.
- E. Cho, J. Mun, O. B. Chae, O. M. Kwon, H.-T. Kim, J. H. Ryu, Y. G. Kim and S. M. Oh, Electrochem. Commun., 2012, 22, 1–3 CrossRef CAS.
- Z. Hou, X. Zhang, H. Ao, M. Liu, Y. Zhu and Y. Qian, Mater. Today Energy, 2019, 14, 100337 CrossRef.
- S. Theivaprakasam, G. Girard, P. Howlett, M. Forsyth, S. Mitra and D. MacFarlane, npj Mater. Degrad., 2018, 2, 13 CrossRef.
- R. Ramos, G. Cunge, B. Pelissier and O. Joubert, Plasma Sources Sci. Technol., 2007, 16, 711 CrossRef CAS.
- A. Lahiri, M. Olschewski, R. Gustus, N. Borisenko and F. Endres, Phys. Chem. Chem. Phys., 2016, 18, 14782–14786 RSC.
- L. Qiao, U. Oteo, M. Martinez-Ibañez, A. Santiago, R. Cid, E. Sanchez-Diez, E. Lobato, L. Meabe, M. Armand and H. Zhang, Nat. Mater., 2022, 21, 455–462 CrossRef CAS PubMed.
- Y. Nakayama, T. Takahagi, F. Soeda, A. Ishitani, M. Shimomura and T. Kunitake, J. Colloid Interface Sci., 1988, 122, 464–474 CrossRef CAS.
- A. Y. Kuznetsov, A. V. Kruzhalov, I. N. Ogorodnikov, A. B. Sobolev and L. I. Isaenko, Phys. Solid State, 1999, 41, 48–50 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 2246728–2246730. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d3cp03694d |
| This journal is © the Owner Societies 2023 |