
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceImproving the electrocatalytic activity of cobalt oxide with bismuth for acidic oxygen evolution reaction†
Belvin
Thomas
 a,
Bowen
Peng
b,
Xiaoxi
Huang
*c and
Tewodros
Asefa
*ab
a,
Bowen
Peng
b,
Xiaoxi
Huang
*c and
Tewodros
Asefa
*ab
aDepartment of Chemistry and Chemical Biology, Rutgers, The State University of New Jersey, 610 Taylor Road, Piscataway, NJ 08854, USA. E-mail: tasefa@chem.rutgers.edu
bDepartment of Chemical and Biochemical Engineering, Rutgers, The State University of New Jersey, 98 Brett Road, Piscataway, NJ 08854, USA
cHoffmann Institute of Advanced Materials, Shenzhen Polytechnic University, 7098 Liuxian Blvd, Nanshan District, Shenzhen, 518055, PR China. E-mail: xiaoxihuang@szpu.edu.cn
First published on 25th July 2024
Abstract
Highly durable, low-cost electrocatalysts for acidic oxygen evolution reaction (OER) are very essential for the commercial success of proton exchange membrane-based water electrolysis. The catalysts currently available for this reaction are mainly based on noble metals such as iridium (Ir) and ruthenium (Ru), which are notoriously unstable under acidic OER conditions, besides their high cost. Herein, we demonstrate that the incorporation of the main group metal bismuth (Bi) in cobalt oxide improves the latter's overall catalytic activity towards acidic OER without reducing its stability. Among the Bi-doped cobalt oxide catalysts synthesized with different Co![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :Bi ratios, the one with a Co:Bi ratio of 9:1, denoted Co9BiOx, exhibits the best performance with an overpotential of 540 mV at the current density of 10 mA cm−2 at pH 1. This catalyst is also reasonably stable for about 45 hours while driving the reaction at a current density of 5 mA cm−2. X-ray photoelectron spectroscopic studies and density functional theory-based calculations indicate that the Bi sites in these materials are catalytically active for acidic OER. In addition, the versatility of Bi in enhancing the catalytic activity of transition metal oxides towards acidic OER is demonstrated with Bi-doped iron oxides and nickel oxides.
:Bi ratios, the one with a Co:Bi ratio of 9:1, denoted Co9BiOx, exhibits the best performance with an overpotential of 540 mV at the current density of 10 mA cm−2 at pH 1. This catalyst is also reasonably stable for about 45 hours while driving the reaction at a current density of 5 mA cm−2. X-ray photoelectron spectroscopic studies and density functional theory-based calculations indicate that the Bi sites in these materials are catalytically active for acidic OER. In addition, the versatility of Bi in enhancing the catalytic activity of transition metal oxides towards acidic OER is demonstrated with Bi-doped iron oxides and nickel oxides.
1 Introduction
The rising global temperature due to the large emission of carbon dioxide and other greenhouse gases into the atmosphere has been leading to weather pattern disruptions and more frequent and intense snowstorms, rainfalls, flooding, heat waves, and forest fires around the world. These issues are further exacerbated by some unexpected events such as the recent Canadian wildfires and their environmental and societal impacts.1,2 All of these, thus, call for an urgent transition from fossil fuels to carbon-neutral, renewable energy sources for our primary energy needs.In recent decades, although large investments have been made in renewable solar and wind-based energy production methods, the intermittent nature of these energy sources has limited their full potential use. This can technically be addressed by coupling these energy sources with electrochemical water splitting to produce green hydrogen (H2). This approach has been gaining great interest as the excess off-peak energy produced by these energy sources can be stored in the form of this chemical energy (i.e., H2), which can then be transported and used to generate electricity with fuel cells or via combustion.3
One of the most promising methods for producing H2 is electrochemical water splitting in an electrolyzer via the hydrogen evolution reaction (HER) at the cathode and the oxygen evolution reaction (OER) at the anode. The HER, which is a two-electron reaction, is relatively simpler to perform compared with the OER, which involves four electrons and is thus kinetically sluggish. The latter is also the main factor limiting the overall efficiency of water electrolyzers. The overall efficiency of electrochemical water splitting can further be affected by the medium in which it is carried out. When water electrolysis in acidic medium is compared with one in alkaline medium, the former is generally a clear winner. As an example, proton exchange membrane water electrolyzers (PEMWEs), which work in acidic medium, have the advantage of having compact structures, great power density, high efficiency at low temperature, partial load range, and low gas crossovers.4–6 Additionally, the high proton concentrations in acidic media allow for faster HER while the low ohmic resistance due to the higher conductivity of hydronium ions (350 S cm2 mol−1) over hydroxide ions (198 S cm2 mol−1) creates a favorable condition for the overall process in PEMWEs.7,8 To date, the state-of-the-art OER catalysts for acidic media are noble metal oxides, namely IrO2 and RuO2; however, their high cost, instability, and scarce availability limit their large-scale application for industrial PEMWEs.9,10 So, there is an urgent need to develop noble metal-free OER electrocatalysts that can catalyze the OER in acidic media while remaining stable.
While various electrocatalysts based on first-row transition metals that are stable and show catalytic activity for OER in alkaline media have been developed, most of them are highly unstable during OER in acidic media at high oxidation potentials.11 Co- and Mn-oxides are among the few exceptions that have been the target of many studies focusing on the development of effective, acid-stable OER catalysts.12–18 But, spinel cobalt oxide (Co3O4), which can catalyze OER, still suffers from low stability under harsh acidic conditions at high oxidation potentials.19 One approach that is applied to improve the stability and activity of this promising acidic OER catalyst involves doping it with other metals. For example, the overall conductivity, stability, and electrocatalytic activity of spinel Co3O4 nanowires for OER in acid media were improved by doping them with Ag.20 Similarly, when cobalt oxide (CoOx) was doped with Fe and Pb, producing CoFePbOx, the material exhibited higher stability during OER in acidic solution than CoOx while maintaining its catalytic activity for the reaction.11 Another approach applied to improve the stability of Co3O4 is using an acid-stable protective layer, such as carbon or TiO2, to prevent the leaching of Co atoms from the material under such conditions.21–23
Other recent research reports have shown that coupling 3d transition metals (Mn/Fe/Co/Ni) with acid-stable oxides of Sb/Ti/Sn/Ge/Mo/W could yield effective OER catalysts for acidic media.24–26 For example, TiO2 was coupled with Mn/Co/Fe oxides to produce acid-stable OER catalysts.27,28 Similarly, Co2TiO4 and TiO2-incorporated MnO2 catalysts were shown to exhibit catalytic activity for OER for many hours in highly acidic solutions.13,29 Metal oxides, such as SbOx, were also demonstrated to generate acid-durable OER catalysts. For example, the incorporation of Sb into FeCoOx, which produces FeCoSbOx, gives rise to more stable and efficient electrocatalysts for OER in acidic solutions.30 (Mn,Sb)O2 possessing rutile structure, pyrochlore-type Co2Sb2O7, phase-pure CoSb2O6, and MnSb2O6 have also been shown to serve as acid-stable OER catalysts with good activity and stability.31,32
After the recent report on the stability of BiOx under acidic conditions, there has been a growing interest in this p-block metal oxide for electrocatalysis applications.33 For example, [Ag + Bi]Ox and Co–Bi–SnOx have successfully been demonstrated to catalyze the OER while remaining stable in acidic solutions.34,35 The stability of these catalysts has been attributed to the stabilizing role of the acid-stable bismuth oxide matrix. BiOx provides an added benefit in acidic OER electrocatalysis because it can sustain the reaction via the Bi3+/Bi5+ oxidation states it forms at its Bi sites, which help with the formation of the hydroxo/peroxo intermediates needed for facile OER.33 A recent study from our group also showed that Bi's oxophilic nature and ability to tune the electronic states in metal borates were helpful for stabilizing the OER intermediates and thereby enhancing the OER electrocatalysis.36
Herein we report petal-shaped Bi-doped cobalt oxides that are synthesized in situ on fluorine-doped tin oxide (FTO) substrates and their excellent electrocatalytic activity and durability during acidic OER. The catalysts are synthesized using a simple procedure wherein Co(II) and Bi(III) salts are mixed in citric acid solution, then drop-casted on FTO, and finally annealed. The catalytic active sites on the materials are comprised of thin layers of Bi-doped cobalt oxides that are deposited on FTO. Some of the catalysts remain stable while electrocatalyzing the OER in 0.1 M HClO4 solution for 45 hours at a current density of 5 mA cm−2 and for 24 hours at a current density of 10 mA cm−2. The effects of the amounts of Bi dopants in cobalt oxides on the activity and stability of the catalysts towards acidic OER are investigated by varying the Co:Bi ratios in the materials and then testing their electrocatalytic activities for hours. Bi is also found to enhance the electrocatalytic activities of iron oxide and nickel oxide for OER. These findings may encourage the exploration of other p-block metals as dopants into various transition metal oxides for acidic OER electrocatalysis.
2 Results and discussion
Co3O4, Bi2O3, and Bi-doped Co3O4 catalysts with different Co:Bi ratios were synthesized in situ by depositing solutions of the respective metal ions in citric acid on FTO glass slides and then annealing them, as illustrated in the schematic in Fig. 1. The Bi-doped Co3O4 catalysts obtained from the synthesis are named as CoaBibOx, such that a:b represents the mole ratios of Co(II):Bi(III) used for the synthesis of the catalysts, which are 14:1, 9:1, 2:1, and 1:2. Citric acid is added for the synthesis of these materials because the citrate ions it forms in the solution help dissolve the metal salts.37 Besides, the citrate groups are easily removable via thermal decomposition into CO2 to leave behind pristine metal oxides on the FTO. A small amount of nitric acid is also included in the solution in order to help dissolve Bi(III) nitrate and prevent it from precipitating as Bi(OH)3.38
 | ||
| Fig. 1 Schematic of the synthesis of CoaBibOx catalysts on FTO. The procedure involves: (i) mixing Co(II) nitrate and Bi(III) nitrate in citric acid solution, (ii) deposition of the solution onto FTO substrate, and (iii) drying and annealing the FTO-deposited metal salts. | ||
The catalysts synthesized on FTO are characterized first by X-ray diffraction (XRD) (Fig. 2 and S2†). The XRD pattern obtained for pristine cobalt oxide on FTO shows peaks at 2θ = 19.04, 31.26, and 37.05°, which can be ascribed to the (111), (220), and (311) crystalline planes, respectively, of Co3O4 with spinel structure (based on the Joint Committee on Powder Diffraction Standard or JCPDS no.: 42-1467).14 The XRD pattern additionally shows the diffraction peaks of the FTO substrate itself at 2θ = 26.5, 33.63, 37.72, 51.53, 61.55, and 65.51°. These peaks are also observed in all the other cases where FTO is used as a substrate. However, when the XRD patterns of Co9BiOx, Co2BiOx, and CoBi2Ox on FTO are examined, apart from the peaks corresponding to FTO, no peaks associated with spinel Co3O4 or Bi2O3 are seen on them. For example, the XRD pattern of Co14BiOx on FTO shows only a faint peak at ∼31° corresponding to the (220) plane of spinel Co3O4. This means that the XRD pattern of Co14BiOx on FTO does not show many of the peaks associated with crystalline planes of Co3O4, unlike that of pristine Co3O4 on FTO. These results indicate that incorporating Bi into Co3O4 results in materials that are largely amorphous. Additionally, a broad peak at 2θ of ∼28° is seen in the XRD pattern of CoBi2Ox due to the amorphous feature Bi2O3 in it, as Bi is the major metal in this material. It is worth noting here that the XRD pattern of pristine Bi2O3 on FTO (Fig. S1†) exhibits peaks due to α-Bi2O3 (JCPDS no.: 41-1449).39 These mean that the in situ synthesis reported herein yields amorphous catalysts in the case of Bi-doped cobalt oxides but crystalline ones in the case of the pristine Co3O4 and Bi2O3.
 | ||
| Fig. 2 XRD patterns of FTO substrate and the different catalysts synthesized in situ on FTO substrates, namely Co3O4, Co14BiOx, Co2BiOx, and CoBi2Ox. The XRD pattern of Co3O4 obtained from the JCPDS database is included for reference. | ||
The field emission scanning electron microscopy (FESEM) images of Co9BiOx, which is the best performing catalyst (vide infra), are displayed in Fig. 3a–c. The images exhibit that this material is composed of thin filament- and petal-shaped particles, just as those of the CoMnOx OER catalysts reported by Huynh et al.11 Note that this material on FTO appears black in color (see its digital image in Fig. S2†). The FESEM images of the other catalysts synthesized on FTO also exhibit similar filament- and petal-shaped morphologies (Fig. S3–S7a–c†). However, upon examining the images further, these morphologies are more pronounced when the amount of Co in the catalysts is relatively higher. For instance, the petal-like morphology is best seen in the FESEM images of Co3O4, Co14BiOx, and Co9BiOx, which have higher Co:Bi ratios (Fig. 3, S3–S4 and S6a–c†). When the catalysts are made with higher amounts of Bi, they show thicker sheet-like structures (see, for example, the FESEM images of CoBi2Ox and Bi2O3 in Fig. S5 and S7a–c†). The elemental mapping images obtained with energy dispersive X-ray spectroscopy (EDS) for Co9BiOx (Fig. 3d) show Co, Bi, and O that are distributed throughout the sample. Similarly, the EDS mapping images of all other Bi-doped cobalt oxides (Fig. S3–S6d†) show uniform distributions of Co, Bi, and O. The ratios of Co:Bi in the materials are also analyzed from these EDS mapping images, and they are found to be like the corresponding ratios of Co:Bi (in mol) used in the precursors to synthesize them (Table S1†).
 | ||
| Fig. 3 (a–c) FESEM images and (d) EDS elemental mapping images of Co9BiOx on FTO. The latter show that Co, Bi, and O are uniformly distributed on the catalyst. | ||
X-ray photoelectron spectroscopy is applied to determine the surface atomic compositions and chemical states of the catalysts present on FTO. The XPS survey spectra show peaks corresponding to Co, Bi, O, and C as well as two other peaks corresponding to Sn and Si that originate from the FTO substrate (Fig. S9†). The high-resolution Co 2p XPS spectra of all the catalysts containing Co have two main broad peaks at binding energies of 780.0 and 795.0 eV, which correspond to the Co 2p3/2 and 2p1/2 states, respectively (Fig. 4a). The high-resolution Co 2p XPS spectra show weak satellite peaks with higher binding energies around those of 2p3/2 and 2p1/2, which indicate the presence of Co2+ and Co3+ on the catalysts.40 The high-resolution Co 2p XPS spectra are deconvoluted to determine the chemical states of Co on the surfaces of the catalysts. The deconvoluted Co 2p3/2 peaks of all Co-containing catalysts are seen at binding energies of ∼779.5 and ∼780.6 eV, and they are ascribed to Co3+ and Co2+ oxidation states, respectively. In the case of pristine cobalt oxide on FTO, the ratio of Co2+:Co3+ calculated from its XPS spectrum is 3.67, although the XRD pattern indicates that this material exists in spinel structure where the Co2+:Co3+ ratio should be 0.5. This difference indicates the presence of a much larger amount of Co2+ species, and by extension, more oxygen vacancies, on the surfaces of the pristine cobalt oxide.41 This is further corroborated based on the deconvoluted high resolution O 1s spectra of the material (see below). The deconvoluted O 1s XPS survey spectrum of pristine Co3O4 on FTO (Fig. S10†) displays peaks at 529.83 eV and 531.1 eV, which correspond to Co–O bond and oxygen vacancies (Ovac) or defect sites, respectively.22,42 The O 1s XPS survey spectra of all Bi-doped cobalt oxide catalysts on FTO (Fig. S10†) also indicate the presence of oxygen vacancies, which must be formed during the decomposition of citrate groups into CO2 in the annealing step of their syntheses. The decomposition of citrate from the materials into CO2 can possibly generate the oxygen vacancies via a similar process to the one previously reported for ligand-assisted polyol synthesis of cobalt oxide using the polymer polyacrylic acid as a ligand.43
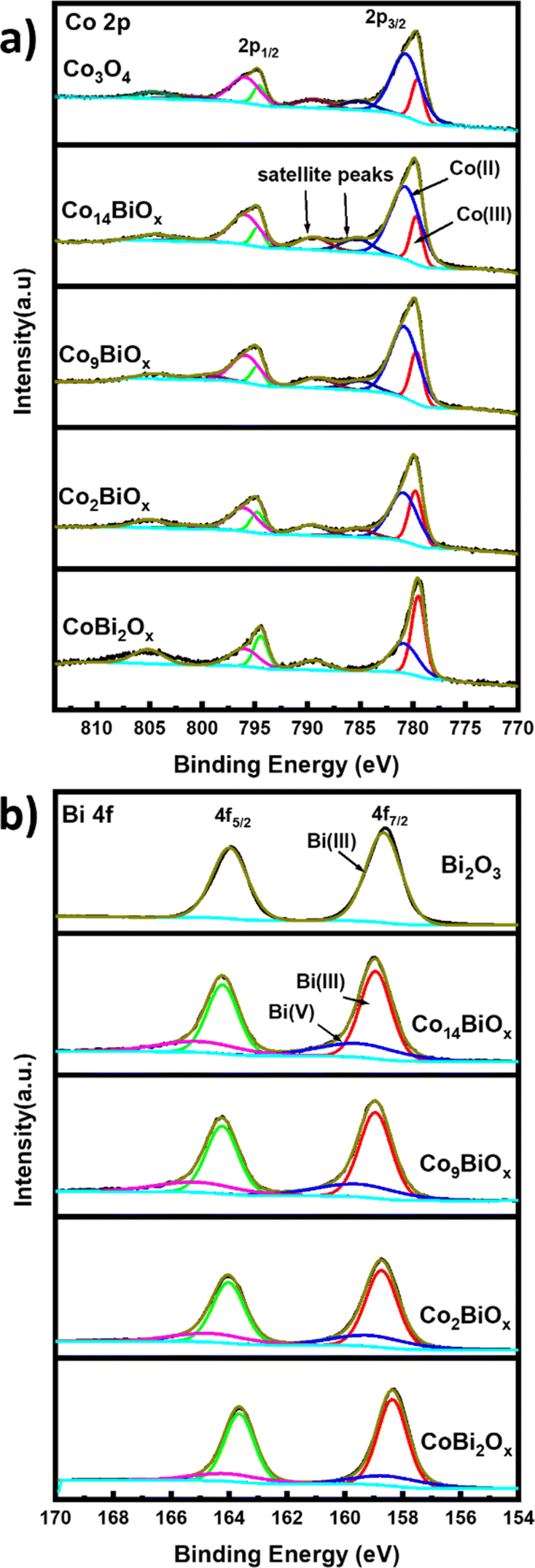 | ||
| Fig. 4 High resolution XPS spectra showing peaks corresponding to (a) Co 2p and (b) Bi 4f of Co3O4, Co14BiOx, Co9BiOx, Co2BiOx, CoBi2Ox, and Bi2O3 catalysts. | ||
Similarly, the deconvoluted Bi 4f XPS spectra of Bi in Bi-doped cobalt oxides on FTO (Fig. 4b) show two sets of peaks: the first one at binding energies of 158.9 and 164.2 eV that can be attributed to Bi 4f7/2 and Bi 4f5/2 of Bi3+, respectively, and the second one at 159.5 and 165.0 eV that can be ascribed to Bi 4f7/2 and Bi 4f5/2 of Bi5+, respectively.44 However, the high resolution Bi 4f XPS spectrum of pristine Bi2O3 on FTO shows only peaks at ca. 158.67 and ca. 163.97 eV corresponding Bi 4f7/2 and Bi 4f5/2 of Bi3+ species, respectively. These results indicate that Bi co-exists in two oxidation states (i.e., Bi3+ and Bi5+) in the Bi-doped cobalt oxides on FTO, but only as Bi3+ in Bi2O3 on FTO.
Based on the XPS results, the relative amounts of Bi3+ and Bi5+ species with respect to the total amount of Bi in Bi-doped cobalt oxides are also determined (see Table S2†). While the results indicate that the relative amount of Bi3+ is higher than that of Bi5+ in all Bi-doped cobalt oxide catalysts, the highest relative amount of Bi5+ species is obtained for Co14BiOx, which contains the lowest amount of Bi. In other words, the relative amount of Bi5+ increases as the amount of Bi in the Bi-doped cobalt oxides decreases. Additionally, the relative amounts of Co2+ or Co3+ in the Bi-doped cobalt oxides with respect to their Bi content are analyzed. In Co14BiOx, which has the lowest amount of Bi among the Bi-doped cobalt oxides, the amount of Co2+ species is 3.88 times higher than that of Co3+ species. On the other hand, when the amount of Bi in the catalysts is increased (e.g., CoBi2Ox), Co exists increasingly more as Co3+ species rather than as Co2+ species. These results show that the amounts of Bi in Bi-doped cobalt oxide catalysts affect the oxidation states of both Bi and Co as well as their relative amounts in the catalysts. Using the XPS results, the Co:Bi mole ratios on the surfaces of all Bi-doped cobalt oxide catalysts are also determined and compiled in Table S2.† The values match well with those used for the synthesis of the respective catalysts.
Raman spectroscopy is used to further confirm the presence of oxygen vacancies in the Bi-doped cobalt oxides synthesized on FTO. As shown in Fig. S11,† spinel Co3O4 shows five distinct Raman bands at 195.6, 481.5, 518.9, 615.2, and 683 cm−1, which are respectively ascribed to the F2g, Eg, F2g, F2g, and A1g Raman-active vibrational modes.45,46 Among them, the bands at 195.6 and 683 cm−1 are ascribed to the Co2+–O2− and Co3+–O2− Raman-active vibrations, respectively. In the case of Co3O4 synthesized on FTO, the A1g mode is slightly blue-shifted (to 685 cm−1), which is due to the Ovc present in Co3O4, as previously reported in the literature.45,46 This showcases that oxygen vacancies are produced during the in situ synthesis of the catalysts on FTO. Similarly, the A1g vibration mode is blue-shifted for the catalyst Co14BiOx and Co9BiOx when compared with that of spinel Co3O4. However, when the amounts of Bi in Bi-doped Co3O4 increases, the A1g vibrational mode of the catalyst is red-shifted to low vibrational frequencies. When very small amounts of Bi are doped into Co3O4, the A1g vibration mode becomes blue shifted, as a large ion such Bi3+ occupy the octahedral sites in Co3O4 by substituting Co3+ ions.47 When the amount of Bi in cobalt oxide is increased, the Co3+ ions at the octahedral sites are replaced with Bi3+ ions, resulting in a dramatic red shift of the A1g Raman-active vibration mode of the catalysts. Additionally, the Raman bands between 450 and 650 cm−1 corresponding to the Eg, F2g, and F2g modes become broad. When compared with the Raman spectra of pure Co3O4, in addition to the normal Raman bands, a new Raman band at ∼315 cm−1 is seen for the catalyst CoBi2Ox. For α-Bi2O3 synthesized on FTO, the prominent Raman bands are observed at ∼215, 317, and 451 cm−1, and they are all attributed to Bi–O stretching modes (Fig. S12†).48 However, none of these bands are seen in the Raman spectra of the Bi-doped cobalt oxides on FTO. Even in the case of CoBi2Ox, which has a large amount of Bi, except for the broad Raman band at ∼315 cm−1, no sharp Raman peak associated with α-Bi2O3 is seen in its spectrum. These results obtained with Raman spectra indirectly confirm that Bi is incorporated into the cobalt oxide structures.
Next, the electrocatalytic activities of all the materials on FTO for OER in acidic solution (0.1 M HClO4 solution with pH 1) are investigated using a three-electrode cell. In the cell, a calomel electrode is used as the reference electrode and a graphite rod is used as the counter electrode. To minimize the capacitive current, the polarization curves are obtained using a slow scan rate of 5 mV s−1. The linear sweep voltammetry (LSV) curves, which are displayed in Fig. 5a, show that the Bi-doped cobalt oxides have better electrocatalytic activities for OER in acidic solution compared with the pristine cobalt oxide. As the amount of Bi in cobalt oxide increases, its catalytic activity for OER also increases, with the highest activity being attained by the catalyst Co9BiOx (which has a Co:Bi ratio is 9:1). However, further doping of Bi in cobalt oxide beyond this amount results in lower electrocatalytic activities in the materials. The Bi-doped cobalt oxide catalyst Co9BiOx, which is found to exhibit the best performance for OER in acidic solution, does so with the lowest overpotential (540 mV at a current density of 10 mA cm−2). Meanwhile the highest overpotential (η = 671 mV) at a current density of 10 mA cm−2 is obtained for the pristine Co3O4. This means, all the Bi-doped cobalt oxide catalysts on FTO electrocatalyze the OER with lower overpotentials at a current density of 10 mA cm−2 than the pristine Co3O4 on FTO.
 | ||
| Fig. 5 (a) The LSV curves obtained for OER as catalyzed by Co3O4, Co14BiOx, Co9BiOx, Co2BiOx, CoBi2Ox, and Bi2O3 in 0.1 M HClO4 solution at a scan rate of 5 mV s−1. (b) The Tafel plots of the reactions catalyzed by the materials, which are derived from their respective LSV curves. (c) Plots of the overpotential required by the catalysts to drive OER at a current density of 10 mA cm−2versus the percentage of Bi in the catalysts. (d) Plots of charging current density (obtained from the difference between the anodic and cathodic values) versus scan rate in the non-faradaic region of OER are shown. These plots are used to determine the capacitances of the catalysts. (e) Nyquist plots for Co3O4, Co14BiOx, Co9BiOx, Co2BiOx, and CoBi2Ox obtained using EIS. (f) Chronopotentiometry profiles obtained for Co9BiOx to determine its stability during electrocatalytic OER at current densities of 5 and 10 mA cm−2. | ||
To determine the kinetics of OER in acidic media over these catalysts, their Tafel plots are generated from their respective LSV curves (Fig. 5b). The lowest Tafel slope (113 mV dec−1) is obtained for the catalyst Co9BiOx, indicating that the kinetics of OER on it is better than those of all the other catalysts studied here. The lowest overpotential is also obtained for this catalyst, once again corroborating its high better electrocatalytic activity in acidic media. It is worth noting here that even the catalyst CoBi2Ox (which has a Co:Bi ratio of 1:2 or more Bi than Co) exhibits a better catalytic performance for acidic OER with a lower overpotential (η = 646 mV) at a current density of 10 mA cm−2 than the pristine Co3O4 (η = 671 mV). This suggests that the Bi sites, along with Co atoms, in the former help the acidic OER catalysis. This is despite pristine Bi2O3 on FTO is inactive for OER electrocatalysis in acidic solution, as can be seen from its LSV curve in Fig. 5a. So, although Bi2O3 does not electrocatalyze OER, the Bi incorporated into cobalt oxide substantially improves the overall electrocatalytic activity of the material for OER in acidic solution.
Next, the double-layer capacitances (Cdl) of the catalysts are determined based on the slopes of the linear plots of charging current density for the reaction over them versus scan rate (Fig. 5d and S13†). Note that the Cdl is directly proportional to the density of active sites on the surface of the catalysts exposed to the electrolyte. Among them, the value of Cdl of Co9BiOx is the highest. So, this catalyst must have the largest density of catalytically active sites on its surfaces, which may have been partly responsible for its best performance. In addition, compared with Co3O4, all the Bi-doped catalysts have higher values of Cdl, and thus larger density of electrochemically active sites to participate in the reaction. This is also in line with their better catalytic performances for OER than Co3O4. So, the incorporation of Bi in cobalt oxide increases the density of catalytically active sites on the surfaces of Co3O4, and thereby its electrocatalytic activity for OER.
We used the onset potentials and ECSA to compare our catalysts with one another, as mostly done for such catalysts. Although turnover frequencies (TOFs) can accurately reflect the intrinsic activity of catalysts, it is not easy to determine the TOFs of many types of electrocatalysts, especially multi-metallic and amorphous solid-state ones, like the ones reported here. This issue is widely discussed in a recent reference paper by Anantharaj et al.49 The main reason is that the calculation of TOF requires an accurate number of active sites to normalize the measured activities with; however, methods to precisely determine their number for many electrocatalysts, like the ones reported here, are unavailable. The two most accurate methods used to determine the active sites on electrocatalysts are underpotential deposition (UPD) and redox peak integration. While the UPD method is more accurate for noble metals, the redox peak integration is mostly applicable when the catalysts are monometallic. Additionally, since our catalysts have two active sites such as Co and Bi, precise calculation of the TOF values for each one is difficult using any available methods. To make the comparison easier, the same synthetic protocol and the same loading of catalysts on FTO substrates are employed. Furthermore, the morphologies of the catalysts obtained are almost the same, as evident from their SEM images. So, the geometrical surface areas of the catalysts should, at least, be similar for all of them. Indeed, the results of their double layer capacitance measurements (Fig. 5d) show that the catalyst Co9BiOx has the highest density of active sites. So, its highest current density (see in Fig. 5a) must be due to its highest density of active sites, as its geometrical surface area is the same as those of the other catalysts. So, unsurprisingly, the catalyst with higher ECSA is found to have a higher overall electrocatalytic activity.
In the Nyquist plots obtained with EIS measurements, the diameters of the semicircles at high frequencies typically correspond to the interfacial charge transfer resistance (Rct) of the electrocatalysts. As depicted in Fig. 5e, the smallest semicircle or Rct at higher frequencies is obtained for Co9BiOx, indicating its relatively favorable conductivity to enable more facile OER kinetics in acidic media. Additionally, the Nyquist plots show that Co3O4 has the highest Rct among all Bi-doped cobalt oxide catalysts studied here. Not surprisingly, the overpotential of the electrocatalysts to drive OER increases from Co9BiOx to Co3O4. Even the electrocatalyst CoBi2Ox, which has more Bi than Co, shows a better OER kinetics, or a lower value Rct, than Co3O4. Additionally, CoBi2Ox has more density of active sites when compared to Co3O4. So, the results obtained with EIS corroborate that the incorporation of Bi in cobalt oxide enhances the electrochemical charge transfer processes over the material during OER. This, and the higher density of catalytically active sites in CoBi2Ox, is possible only if Bi itself also contributes to them. In other words, although Bi in Bi2O3 is barely active for OER catalysis (Fig. 5a), the Bi sites in Co3O4 assist with the OER catalysis. Based on the results collectively, Co9BiOx, which has Co:Bi ratio of 9:1, is particularly found to exhibit the most facile OER kinetics, the lowest Tafel slope, the lowest interfacial charge transfer resistance, the highest density of active sites exposed on its surfaces, and the lowest overpotential to electrocatalyze the OER in acidic solution.
As one of the critical problems faced by most OER catalysts is that they are unstable under harsh acidic conditions, the stabilities of the Bi-doped cobalt oxide catalysts during the reaction are also investigated with chronopotentiometry. The chronopotentiometric curve of the best catalyst, i.e., Co9BiOx, indicates that it remains stable during the reaction at pH 1 for 45 hours at a current density of 5 mA cm−2 and for 24 hours at a current density of 10 mA cm−2 (Fig. 5f). Its stability during acidic OER is also more robust compared to many notable non-noble metal-based, acidic OER catalysts reported in the literature (see Table S4†). Similarly, Co14BiOx and Co3O4 are stable for 24 h under the same condition (Fig. S14†). However, the catalysts possessing more Bi, such as Co2BiOx and CoBi2Ox, are not as stable.
Furthermore, the catalyst recovered after 5 h-long stability test is analyzed by FESEM, EDS, and XPS. Its FESEM images (Fig. S8†) still show thin filament- and petal-shaped morphologies, indicating its structural stability during electrocatalysis in acidic solution for, at least, 5 hours. However, EDS and XPS analyses show that the ratios of Co:Bi on the surfaces of the catalyst after 5 h of electrocatalytic stability test change from 7.2:1 and 8.3:1 to 22.5:1 and 14.8:1, respectively (Tables S1 and S2†). This means that, after catalyzing the OER for 5 hours, this catalyst has a relatively larger amount of Co than Bi on its surfaces. This indicates that some of the Bi atoms on its surfaces may have leached into the electrolyte during the OER under such acidic solution, which is not uncommon for Bi. Nevertheless, the catalyst still functions reasonably well for at least 45 hours.
The XPS spectrum of the best catalyst (i.e., Co9BiOx) recovered after the 5-h long chronopotentiometric OER test is further analyzed to probe its active sites. Just like the deconvoluted Co 2p XPS spectra of pristine Co9BiOx, those of Co9BiOx after OER show peaks corresponding to Co2+ and Co3+ species (Fig. S15a and Table S3†). However, the relative ratio of Co2+:Co3+ species on its surfaces increases from 3.14 to 3.44 after the OER, indicating that Co with an oxidation state of +2 is more favored during the OER (Table S1†). Similarly, the Bi 4f XPS spectra of Co9BiOx before and after OER show that the relative ratio of Bi3+:Bi5+ species changes from 2.39 to 3.42 after the OER (Fig. S15b and Tables S1 and S3†). This indicates that some of the Bi3+ sites become Bi5+ species during the OER. This change in the relative ratio of the oxidation state of Bi also indirectly indicates Bi's participation during the electrocatalytic OER under such conditions. The deconvoluted O 1s XPS spectra of Co9BiOx after OER show peaks corresponding to oxygen vacancies (at a binding energy of 530.69 eV), lattice oxygen (at a binding energy of 529.95 eV), and hydroxyl species (at a binding energy of 531.48 eV) (Fig. S15c and Table S3†). As the OER is performed in acidic solution, the latter must have formed due to the protonation of the surfaces of the catalyst, as such species form as intermediates during acidic OER.7
Since oxides of cobalt show reasonably good electrocatalytic activity towards OER, extensive studies have been conducted to determine the oxidation state of cobalt, i.e., Co2+ or Co3+, that is more responsible for it.50–53 In alkaline conditions, the studies showed that having more Co3+ in the catalyst improves the catalyst's activity for OER.53,54 However, in acidic conditions, Co2+-rich Co3O4 has been found to do better as an OER catalytic site.52 A recent report also corroborated that (i) the high activity of Co2+ site towards acidic OER was due to the transformation of the Co2+–O sites to reducible Co3+–O sites in situ during the reaction and (ii) the latter species would facilitate the deprotonation of water and the release of dioxygen.55 Note also that these reducible Co3+–O species forming in situ during OER have better activities for OER than the originally present Co3+–O species on the catalyst. Additionally, studies have shown that the oxygen vacancies present on such catalysts can help with the in situ transformation of Co2+–O species to Co3+–O species.55,56 Based on these XPS results, the density of Co2+ species on the catalyst increases after the OER. This means that the catalyst Co9BiOx becomes Co2+-rich, which can translate to more oxygen vacancies and more favorable catalytically active sites on its surfaces for the OER. Furthermore, the oxygen vacancies can generate new bandgap states in the catalyst, which improve its overall conductivity and thereby its catalytic performance for OER.22
Since the densities of Bi3+ and Bi5+ species in the catalyst Co9BiOx have changed after OER, we believe that Bi in cobalt oxides also acts as the active site by forming Bi3+/Bi5+ redox couple, as proposed for related materials in the literature.33 Additional evidence on Bi acting as active site is found from electrochemical studies of CoBi2Ox catalyst. In this catalyst, the density of Co2+ species is less than that of Co3+ species, and the total amount of Bi (present in the form of both Bi3+ and Bi5+) is more than the total amount of Co (present in the form of both Co2+ and Co3+). However, CoBi2Ox requires a lower overpotential and has a higher density of active sites than Co3O4, which has the highest density of Co2+ species. So, the improved catalytic performance exhibited by CoBi2Ox toward OER is possible only if Bi itself in the material catalyzes OER with an activity that is even better than that of Co. Pristine Bi2O3 is OER active only at potentials greater than ∼2.0 V vs. RHE33 whereas CoBi2Ox is OER active at potentials greater than ∼1.6 V vs. RHE. This indicates that the Bi sites in CoBi2Ox are OER active at potentials below 2.0 V vs. RHE. This enhancement in OER activity of Bi in CoBi2Ox than Bi in Bi2O3 can thus be attributed to the synergistic interaction between the Co and Bi species present in CoBi2Ox. Based on our studies, while all Bi-doped cobalt oxides show higher electrocatalytic activities for OER in acidic condition, the optimal composition that does so the best is Co:Bi ratio of 9:1 (or the material Co9BiOx). Bi's ability to improve the acidic OER activity of first-row transition metal oxides is apparently not limited to Co oxides, as our preliminary studies showed that Bi-doped iron oxides and nickel oxides also do the same. A detailed discussion on these two materials and their electrocatalytic properties are available in the ESI.†
Since the electrochemical studies point towards the possibility of Bi atoms contributing to the OER catalysis in acidic solutions, density functional theory (DFT) calculations are performed for the Bi-doped cobalt oxide catalysts. To do so, two catalyst slab models, namely Co3O4(100) surface and Bi-doped Co3O4(100) surface, are built as displayed in Fig. 6a and b. Their optimized structures with surface-adsorbed *OH, *O, and *OOH intermediates are depicted in Fig. 6c and d. Their calculated free energy changes are shown in diagram presented in Fig. 7. The results show that the (H+ + e−) transfer step in each case, which involves the largest free energy change and which is represented with a bold line, is the potential determination step (PDS). The PDS of OER on Co3O4(100) surface involves the conversion from *O to *OOH, with an overpotential of 0.509 V. In contrast, the PDS of OER on Bi-doped Co3O4(100) surface involves the conversion of *OH to *O, which requires a calculated overpotential of 0.396 V. The energetic gap between *OH and *OOH is generally constant (which is 3.2 ± 0.2 eV) due to the scaling relationship among different intermediates. This applies to both metal oxide and metallic catalysts.8,57,58 Based on the theoretical studies, when Bi in Bi-doped cobalt oxides serves as the active site for OER, the energy difference between the binding energies of *OH and *OOH on it is 2.56 eV, which is much lower than 3.2 eV. This shows that, the presence of Bi in cobalt oxides enables the scaling relationship between *OH and *OOH to be broken, making these materials require lower overpotentials to electrocatalyze the OER. In other words, the incorporation of Bi in cobalt oxide catalysts is beneficial for promoting the electrocatalysis of OER on them in acidic solutions.
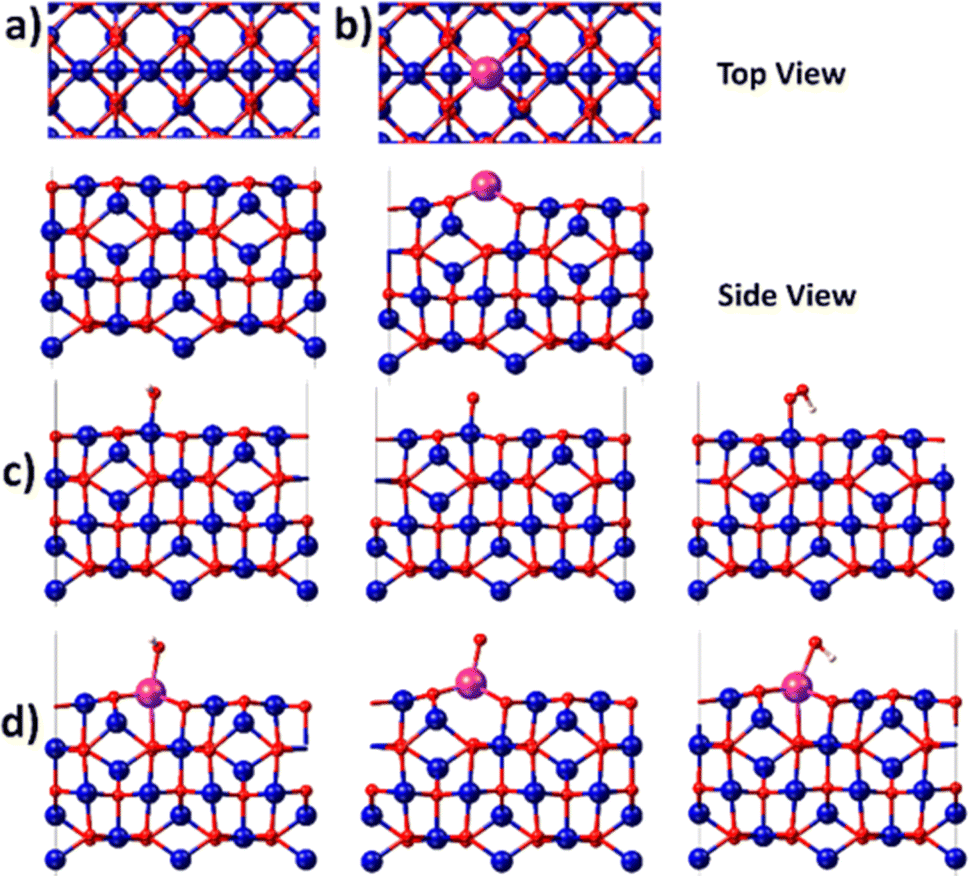 | ||
| Fig. 6 Top view and side view images of (a) Co3O4(100) surface and (b) Bi-doped Co3O4(100) surface. Red: oxygen, blue: cobalt, and purple: bismuth. Optimized structures of (c) Co3O4(100) surface and (d) Bi-doped Co3O4(100) surface, with both showing surface-adsorbed *OH, *O, and *OOH intermediates. | ||
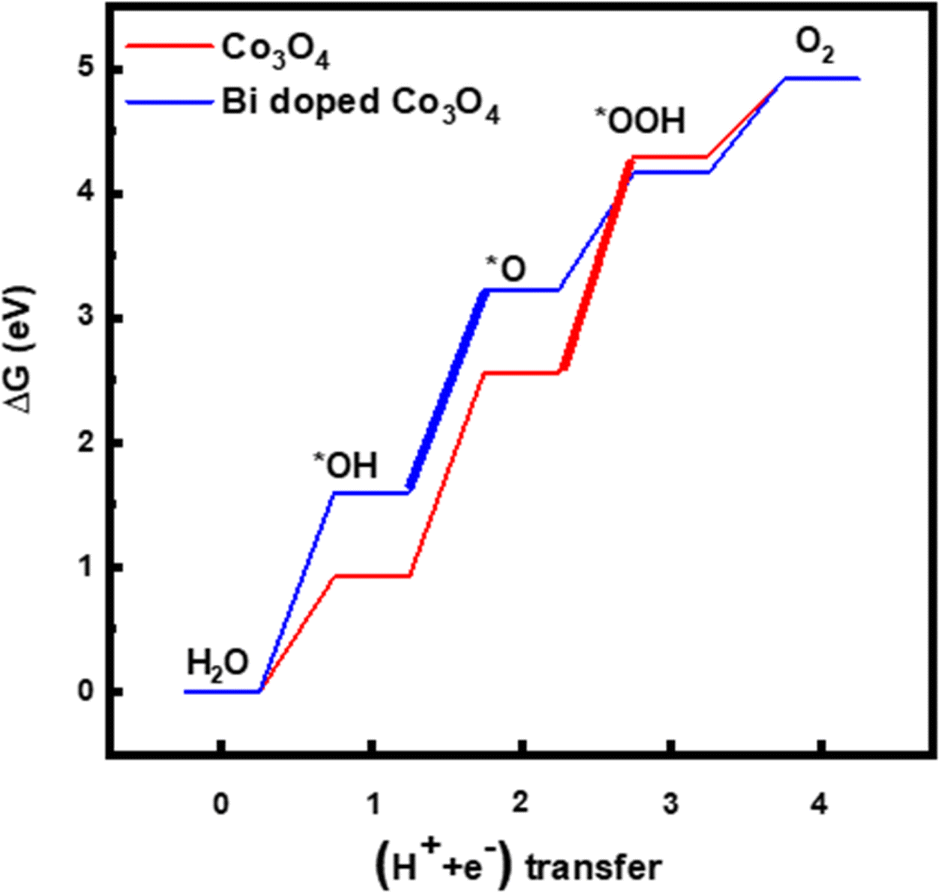 | ||
| Fig. 7 Diagrams showing calculated free energy changes of OER on the surface of Co3O4(100) and Bi-doped Co3O4. | ||
As a final note, Bi is also found to improve the electrocatalytic activities of iron oxides and nickel oxides for OER in acidic solution. However, the stabilities of these materials are much inferior to the Bi-doped cobalt oxides. The results and the pertinent discussions on these materials are presented in the ESI and Fig. S16–S19.†
3 Conclusions
In summary, cobalt oxides doped with the p-block metal Bi in different ratios of Co:Bi were synthesized in situ on FTO substrates using a simple procedure. The materials had thin filament- and petal-like morphologies and showed high durability and electrocatalytic activity for acidic OER. Upon investigating the catalysts synthesized with different Co:Bi ratios, the one with Co:Bi ratio of 9:1 (Co9BiOx) exhibited the best performance for the reaction, with an overpotential of 540 mV at the current density of 10 mA cm−2. This catalyst was also found to be stable for about 45 h while driving the reaction at a current density of 5 mA cm−2. XRD patterns showed that the addition of Bi into cobalt oxide made the materials to have more amorphous structures. The SEM images of the Bi-doped cobalt oxides revealed that their morphologies would change from thin filament- and petal-like morphologies to sheet-like structures as the amount of Bi in them was increased. XPS spectra of the catalysts before and after OER indicated that the relative ratios of the metallic species, namely, Co2+:Co3+ and Bi3+:Bi5+, on the surfaces of the catalysts changed, indirectly suggesting that both Co and Bi could may have participated as active sites for the OER. DFT calculations also corroborated that Bi incorporated into cobalt oxide would help with the catalytic activity of the material for OER, lowering the overpotential of the reaction. In addition, Bi was found to enhance the electrocatalytic activities of iron oxides and nickel oxides for OER in acidic solutions. These results collectively show that the p-block metal Bi, which is underexplored for electrocatalysis, holds a great potential for producing stable and effective acidic OER electrocatalysts. We believe that our findings will further inspire more research on main group metals towards the development of acidic OER catalysts.
Data availability
Data for this article, including materials characterizations, electrochemical and electrocatalytic test results, and DFT calculations, are available the authors' computers and university instrumentation computers. Some of the data supporting this article have been included as part of the ESI.†Author contributions
B. T. and B. P. contributed equally to the work. B. T. and B. P. conducted most of the experiments and the synthesis and characterization of the catalysts, and X. H. conducted the DFT studies on the catalysts. B. T., B. P., and T. A. conceived the study. T. A. supervised the study and provided the resources. B. T., T. A., and X. H. wrote the draft paper. B. T. and T. A. reviewed and revised the paper. T. A. submitted the paper. All authors discussed the results and commented on the manuscript.Conflicts of interest
There are no conflicts to declare in this work and report.Acknowledgements
We thank Ann Mary Jose and Prof. Ashutosh Goel for their assistance with elemental analysis to cross-check some of the samples in this work, the results of which ultimately ended up not relevant and thus not included.Notes and references
- K. Chen, Y. Ma, M. L. Bell and W. Yang, Canadian Wildfire Smoke and Asthma Syndrome Emergency Department Visits in New York City, J. Am. Med. Assoc., 2023, 220(14), 1385–1387 CrossRef PubMed.
- Z. Wang, Z. Wang, Z. Zou, X. Chen, H. Wu, W. Wang, H. Su, F. Li, W. Xu, Z. Liu and J. Zhu, Severe Global Environmental Issues Caused by Canada’s Record-Breaking Wildfires in 2023, Adv. Atmos. Sci., 2024, 41, 565–571 CrossRef CAS.
- M. Chatenet, B. G. Pollet, D. R. Dekel, F. Dionigi, J. Deseure, P. Millet, R. D. Braatz, M. Z. Bazant, M. Eikerling, I. Staffell, P. Balcombe, Y. Shao-Horn and H. Schäfer, Water electrolysis: from textbook knowledge to the latest scientific strategies and industrial developments, Chem. Soc. Rev., 2022, 51(11), 4583–4762 RSC.
- J. Gao, H. Tao and B. Liu, Progress of Nonprecious-Metal-Based Electrocatalysts for Oxygen Evolution in Acidic Media, Adv. Mater., 2021, 33(31), 2003786 CrossRef CAS PubMed.
- S. A. Grigoriev, V. N. Fateev, D. G. Bessarabov and P. Millet, Current status, research trends, and challenges in water electrolysis science and technology, Int. J. Hydrogen Energy, 2020, 45(49), 26036–26058 CrossRef CAS.
- Q. Feng, X. Z. Yuan, G. Liu, B. Wei, Z. Zhang, H. Li and H. Wang, A review of proton exchange membrane water electrolysis on degradation mechanisms and mitigation strategies, J. Power Sources, 2017, 366, 33–55 CrossRef CAS.
- L. Li, P. Wang, Q. Shao and X. Huang, Recent Progress in Advanced Electrocatalyst Design for Acidic Oxygen Evolution Reaction, Adv. Mater., 2021, 33(50), 2004243 CrossRef CAS PubMed.
- L. An, C. Wei, M. Lu, H. Liu, Y. Chen, G. G. Scherer, A. C. Fisher, P. Xi, Z. J. Xu and C. H. Yan, Recent Development of Oxygen Evolution Electrocatalysts in Acidic Environment, Adv. Mater., 2021, 33(20), e2006328 CrossRef PubMed.
- M. Blasco-Ahicart, J. Soriano-López, J. J. Carbó, J. M. Poblet and J. R. Galan-Mascaros, Polyoxometalate electrocatalysts based on earth-abundant metals for efficient water oxidation in acidic media, Nat. Chem., 2018, 10(1), 24–30 CrossRef CAS PubMed.
- S. Niu, X.-P. Kong, S. Li, Y. Zhang, J. Wu, W. Zhao and P. Xu, Low Ru loading RuO2/(Co,Mn)3O4 nanocomposite with modulated electronic structure for efficient oxygen evolution reaction in acid, Appl. Catal., B, 2021, 297, 120442 CrossRef CAS.
- M. Huynh, T. Ozel, C. Liu, E. C. Lau and D. G. Nocera, Design of template-stabilized active and earth-abundant oxygen evolution catalysts in acid, Chem. Sci., 2017, 8(7), 4779–4794 RSC.
- A. Li, H. Ooka, N. Bonnet, T. Hayashi, Y. Sun, Q. Jiang, C. Li, H. Han and R. Nakamura, Stable Potential Windows for Long-Term Electrocatalysis by Manganese Oxides Under Acidic Conditions, Angew. Chem., Int. Ed., 2019, 58(15), 5054–5058 CrossRef CAS PubMed.
- R. Frydendal, E. A. Paoli, I. Chorkendorff, J. Rossmeisl and I. E. L. Stephens, Toward an Active and Stable Catalyst for Oxygen Evolution in Acidic Media: Ti-Stabilized MnO2, Adv. Energy Mater., 2015, 5(22), 1500991 CrossRef.
- W. Zhao, F. Xu, J. Yang, X. Hu and B. Weng, Ce Single-Atom Incorporation Enhances the Oxygen Evolution Reaction of Co3O4 in Acid, Inorg. Chem., 2024, 63(4), 1947–1953 CrossRef CAS PubMed.
- R.-Y. Fan, H.-Y. Zhao, Y.-N. Zhen, F.-G. Wang, H. Hu, Y.-M. Chai and B. Dong, Mn-induced strengthening hybridization effect of Co–O bond for stable oxygen evolution in acidic media, Fuel, 2023, 333, 126361 CrossRef CAS.
- M. Etzi Coller Pascuzzi, M. van Velzen, J. P. Hofmann and E. J. M. Hensen, On the Stability of Co3O4 Oxygen Evolution Electrocatalysts in Acid, ChemCatChem, 2021, 13(1), 459–467 CrossRef CAS.
- A. Li, S. Kong, C. Guo, H. Ooka, K. Adachi, D. Hashizume, Q. Jiang, H. Han, J. Xiao and R. Nakamura, Enhancing the stability of cobalt spinel oxide towards sustainable oxygen evolution in acid, Nat. Catal., 2022, 5(2), 109–118 CrossRef CAS.
- L. Sun, M. Feng, Y. Peng, X. Zhao, Y. Shao, X. Yue and S. Huang, Constructing oxygen vacancies by doping Mo into spinel Co3O4 to trigger a fast oxide path mechanism for acidic oxygen evolution reaction, J. Mater. Chem. A, 2024, 12, 8796–8804 RSC.
- Y. Zhu, J. Wang, T. Koketsu, M. Kroschel, J.-M. Chen, S.-Y. Hsu, G. Henkelman, Z. Hu, P. Strasser and J. Ma, Iridium single atoms incorporated in Co3O4 efficiently catalyze the oxygen evolution in acidic conditions, Nat. Commun., 2022, 13(1), 7754 CrossRef CAS PubMed.
- K.-L. Yan, J.-Q. Chi, J.-Y. Xie, B. Dong, Z.-Z. Liu, W.-K. Gao, J.-H. Lin, Y.-M. Chai and C.-G. Liu, Mesoporous Ag-doped Co3O4 nanowire arrays supported on FTO as efficient electrocatalysts for oxygen evolution reaction in acidic media, Renewable Energy, 2018, 119, 54–61 CrossRef CAS.
- X. Yang, H. Li, A.-Y. Lu, S. Min, Z. Idriss, M. N. Hedhili, K.-W. Huang, H. Idriss and L.-J. Li, Highly acid-durable carbon coated Co3O4 nanoarrays as efficient oxygen evolution electrocatalysts, Nano Energy, 2016, 25, 42–50 CrossRef CAS.
- Q. Lai, V. Vediyappan, K.-F. Aguey-Zinsou and H. Matsumoto, One-Step Synthesis of Carbon-Protected Co3O4 Nanoparticles toward Long-Term Water Oxidation in Acidic Media, Adv. Energy Sustainability Res., 2021, 2(11), 2100086 CrossRef CAS.
- T. Tran-Phu, H. Chen, R. Daiyan, M. Chatti, B. Liu, R. Amal, Y. Liu, D. R. Macfarlane, A. N. Simonov and A. Tricoli, Nanoscale TiO2 Coatings Improve the Stability of an Earth-Abundant Cobalt Oxide Catalyst during Acidic Water Oxidation, ACS Appl. Mater. Interfaces, 2022, 14(29), 33130–33140 CrossRef CAS PubMed.
- Z. Wang, Y.-R. Zheng, I. Chorkendorff and J. K. Nørskov, Acid-Stable Oxides for Oxygen Electrocatalysis, ACS Energy Lett., 2020, 5(9), 2905–2908 CrossRef CAS.
- B. Han, M. Risch, S. Belden, S. Lee, D. Bayer, E. Mutoro and Y. Shao-Horn, Screening Oxide Support Materials for OER Catalysts in Acid, J. Electrochem. Soc., 2018, 165(10), F813 CrossRef CAS.
- G. T. K. K. Gunasooriya and J. K. Nørskov, Analysis of Acid-Stable and Active Oxides for the Oxygen Evolution Reaction, ACS Energy Lett., 2020, 5(12), 3778–3787 CrossRef CAS.
- L. Vallez, S. Jimenez-Villegas, A. T. Garcia-Esparza, Y. Jiang, S. Park, Q. Wu, T. M. Gill, D. Sokaras, S. Siahrostami and X. Zheng, Effect of doping TiO2 with Mn for electrocatalytic oxidation in acid and alkaline electrolytes, Energy Adv., 2022, 1(6), 357–366 RSC.
- L. Zhao, Q. Cao, A. Wang, J. Duan, W. Zhou, Y. Sang and H. Liu, Iron oxide embedded titania nanowires – an active and stable electrocatalyst for oxygen evolution in acidic media, Nano Energy, 2018, 45, 118–126 CrossRef CAS.
- S. Anantharaj, K. Karthick and S. Kundu, Spinel Cobalt Titanium Binary Oxide as an All-Non-Precious Water Oxidation Electrocatalyst in Acid, Inorg. Chem., 2019, 58(13), 8570–8576 CrossRef CAS PubMed.
- J. Wang, H. Kim, H. Lee, Y.-J. Ko, M. H. Han, W. Kim, J. M. Baik, J.-Y. Choi, H.-S. Oh and W. H. Lee, Sb incorporated into oxides enhances stability in acid during the oxygen evolution reaction by inhibiting structural distortion, Nano Energy, 2023, 110, 108355 CrossRef CAS.
- T. A. Evans and K.-S. Choi, Electrochemical Synthesis and Investigation of Stoichiometric, Phase-Pure CoSb2O6 and MnSb2O6 Electrodes for the Oxygen Evolution Reaction in Acidic Media, ACS Appl. Energy Mater., 2020, 3(6), 5563–5571 CrossRef CAS.
- Y. Wang, H. Zhao, Y. Guo, J. Wu, X. Lu and X. Tang, Pyrochlore-type cobalt and manganese antimonate electrocatalysts with excellent activity and stability for OER in acidic solution, Nanoscale, 2023, 15(21), 9413–9422 RSC.
- A. E. Thorarinsdottir, C. Costentin, S. S. Veroneau and D. G. Nocera, -Block Metal Oxide Noninnocence in the Oxygen Evolution Reaction in Acid: The Case of Bismuth Oxide, Chem. Mater., 2022, 34(2), 826–835 CrossRef CAS.
- D. Simondson, M. Chatti, J. L. Gardiner, B. V. Kerr, D. A. Hoogeveen, P. V. Cherepanov, I. C. Kuschnerus, T. D. Nguyen, B. Johannessen, S. L. Y. Chang, D. R. MacFarlane, R. K. Hocking and A. N. Simonov, Mixed Silver–Bismuth Oxides: A Robust Oxygen Evolution Catalyst Operating at Low pH and Elevated Temperatures, ACS Catal., 2022, 12(20), 12912–12926 CrossRef CAS.
- H.-L. Du, M. Chatti, B. Kerr, C. K. Nguyen, T. Tran-Phu, D. A. Hoogeveen, P. V. Cherepanov, A. S. R. Chesman, B. Johannessen, A. Tricoli, R. K. Hocking, D. R. MacFarlane and A. N. Simonov, Durable Electrooxidation of Acidic Water Catalysed by a Cobalt-Bismuth-based Oxide Composite: An Unexpected Role of the F-doped SnO2 Substrate, ChemCatChem, 2022, 14(11), e202200013 CrossRef CAS.
- B. Thomas, C. Tang, M. Ramírez-Hernández and T. Asefa, Incorporation of Bismuth Increases the Electrocatalytic Activity of Cobalt Borates for Oxygen Evolution Reaction, ChemPlusChem, 2023, 88(5), e202300104 CrossRef CAS PubMed.
- H.-Y. Wang, S.-F. Hung, H.-Y. Chen, T.-S. Chan, H. M. Chen and B. Liu, Operando Identification of Geometrical-Site-Dependent Water Oxidation Activity of Spinel Co3O4, J. Am. Chem. Soc., 2016, 138(1), 36–39 CrossRef CAS PubMed.
- T. Ollevier, A. Jalba and H. Keipour, Bismuth(III) Nitrate Pentahydrate, in Encyclopedia of Reagents for Organic Synthesis (EROS), 2016, pp. 1–9 Search PubMed.
- R. Irmawati, M. N. Noorfarizan Nasriah, Y. H. Taufiq-Yap and S. B. Abdul Hamid, Characterization of bismuth oxide catalysts prepared from bismuth trinitrate pentahydrate: influence of bismuth concentration, Catal. Today, 2004, 93–95, 701–709 CrossRef CAS.
- A. Younis, D. Chu, X. Lin, J. Lee and S. Li, Bipolar resistive switching in p-type Co3O4 nanosheets prepared by electrochemical deposition, Nanoscale Res. Lett., 2013, 8(1), 36 CrossRef PubMed.
- Q. Zhang, P. Yang, H. Zhang, J. Zhao, H. Shi, Y. Huang and H. Yang, Oxygen vacancies in Co3O4 promote CO2 photoreduction, Appl. Catal., B, 2022, 300, 120729 CrossRef CAS.
- J. Wang, R. Gao, D. Zhou, Z. Chen, Z. Wu, G. Schumacher, Z. Hu and X. Liu, Boosting the Electrocatalytic Activity of Co3O4 Nanosheets for a Li–O2 Battery through Modulating Inner Oxygen Vacancy and Exterior Co3+/Co2+ Ratio, ACS Catal., 2017, 7(10), 6533–6541 CrossRef CAS.
- W. Xu, F. Lyu, Y. Bai, A. Gao, J. Feng, Z. Cai and Y. Yin, Porous cobalt oxide nanoplates enriched with oxygen vacancies for oxygen evolution reaction, Nano Energy, 2018, 43, 110–116 CrossRef CAS.
- S. Gautam, V. Aggarwal, B. Singh, V. P. S. Awana, R. Ganesan and S. S. Kushvaha, Signature of weak-antilocalization in sputtered topological insulator Bi2Se3 thin films with varying thickness, Sci. Rep., 2022, 12(1), 9770 CrossRef CAS PubMed.
- Y. Zhang, L. Feng, W. Zhan, S. Li, Y. Li, X. Ren, P. Zhang and L. Sun, Co3O4 Hollow Porous Nanospheres with Oxygen Vacancies for Enhanced Li–O2 Batteries, ACS Appl. Energy Mater., 2020, 3(4), 4014–4022 CrossRef CAS.
- Z. Wang, W. Wang, L. Zhang and D. Jiang, Surface oxygen vacancies on Co3O4 mediated catalytic formaldehyde oxidation at room temperature, Catal. Sci. Technol., 2016, 6(11), 3845–3853 RSC.
- Y. Wang, Y.-Q. Zhu, Z. Xie, S.-M. Xu, M. Xu, Z. Li, L. Ma, R. Ge, H. Zhou, Z. Li, X. Kong, L. Zheng, J. Zhou and H. Duan, Efficient Electrocatalytic Oxidation of Glycerol via Promoted OH* Generation over Single-Atom-Bismuth-Doped Spinel Co3O4, ACS Catal., 2022, 12(19), 12432–12443 CrossRef CAS.
- X. Wang, W. He, J. Shi, J. R. C. Junqueira, J. Zhang, S. Dieckhöfer, S. Seisel, D. Das and W. Schuhmann, Ag-induced Phase Transition of Bi2O3 Nanofibers for Enhanced Energy Conversion Efficiency towards Formate in CO2 Electroreduction, Chem.–Asian J., 2023, 18(2), e202201165 CrossRef CAS PubMed.
- S. Anantharaj, P. E. Karthik and S. Noda, The Significance of Properly Reporting Turnover Frequency in Electrocatalysis Research, Angew. Chem., Int. Ed., 2021, 60(43), 23051–23067 CrossRef CAS PubMed.
- M. Ya, J. Wang, G. Li, G. Gao, X. Zhao, J. Cui, H. Wu and L. Li, Surface Engineering in MgCo3O4 Spinel Oxide for an Improved Oxygen Evolution Reaction, ACS Sustain. Chem. Eng., 2023, 11(2), 744–750 CrossRef CAS.
- Y. Xu, F. Zhang, T. Sheng, T. Ye, D. Yi, Y. Yang, S. Liu, X. Wang and J. Yao, Clarifying the controversial catalytic active sites of Co3O4 for the oxygen evolution reaction, J. Mater. Chem. A, 2019, 7(40), 23191–23198 RSC.
- K.-L. Yan, J.-F. Qin, J.-H. Lin, B. Dong, J.-Q. Chi, Z.-Z. Liu, F.-N. Dai, Y.-M. Chai and C.-G. Liu, Probing the active sites of Co3O4 for the acidic oxygen evolution reaction by modulating the Co2+/Co3+ ratio, J. Mater. Chem. A, 2018, 6(14), 5678–5686 RSC.
- X. Han, G. He, Y. He, J. Zhang, X. Zheng, L. Li, C. Zhong, W. Hu, Y. Deng and T.-Y. Ma, Engineering Catalytic Active Sites on Cobalt Oxide Surface for Enhanced Oxygen Electrocatalysis, Adv. Energy Mater., 2018, 8(10), 1702222 CrossRef.
- C. Alex, S. C. Sarma, S. C. Peter and N. S. John, Competing Effect of Co3+ Reducibility and Oxygen-Deficient Defects Toward High Oxygen Evolution Activity in Co3O4 Systems in Alkaline Medium, ACS Appl. Energy Mater., 2020, 3(6), 5439–5447 CrossRef CAS.
- J. Liu, T. Wang, X. Liu, H. Shi, S. Li, L. Xie, Z. Cai, J. Han, Y. Huang, G. Wang and Q. Li, Reducible Co3+–O Sites of Co–Ni–P–Ox on CeO2 Nanorods Boost Acidic Water Oxidation via Interfacial Charge Transfer-Promoted Surface Reconstruction, ACS Catal., 2023, 13(8), 5194–5204 CrossRef CAS.
- A. Bergmann, T. E. Jones, E. Martinez Moreno, D. Teschner, P. Chernev, M. Gliech, T. Reier, H. Dau and P. Strasser, Unified structural motifs of the catalytically active state of Co(oxyhydr)oxides during the electrochemical oxygen evolution reaction, Nat. Catal., 2018, 1(9), 711–719 CrossRef CAS.
- J. Rossmeisl, Z. W. Qu, H. Zhu, G. J. Kroes and J. K. Nørskov, Electrolysis of water on oxide surfaces, J. Electroanal. Chem., 2007, 607(1), 83–89 CrossRef CAS.
- I. C. Man, H.-Y. Su, F. Calle-Vallejo, H. A. Hansen, J. I. Martínez, N. G. Inoglu, J. Kitchin, T. F. Jaramillo, J. K. Nørskov and J. Rossmeisl, Universality in Oxygen Evolution Electrocatalysis on Oxide Surfaces, ChemCatChem, 2011, 3(7), 1159–1165 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4ta02845g |
| This journal is © The Royal Society of Chemistry 2024 |