
Twenty years of islet-on-a-chip: microfluidic tools for dissecting islet metabolism and function
Romario
Regeenes
 ab and
Jonathan V.
Rocheleau
*abc
ab and
Jonathan V.
Rocheleau
*abc
aAdvanced Diagnostics, Toronto General Hospital Research Institute, Toronto, ON, Canada
bInstitute of Biomedical Engineering, University of Toronto, Toronto, ON, Canada
cDepartments of Medicine and Physiology, University of Toronto, ON, Canada
First published on 26th January 2024
Abstract
Pancreatic islets are metabolically active micron-sized tissues responsible for controlling blood glucose through the secretion of insulin and glucagon. A loss of functional islet mass results in type 1 and 2 diabetes. Islet-on-a-chip devices are powerful microfluidic tools used to trap and study living ex vivo human and murine pancreatic islets and potentially stem cell-derived islet organoids. Devices developed over the past twenty years offer the ability to treat islets with controlled and dynamic microenvironments to mimic in vivo conditions and facilitate diabetes research. In this review, we explore the various islet-on-a-chip devices used to immobilize islets, regulate the microenvironment, and dynamically detect islet metabolism and insulin secretion. We first describe and assess the various methods used to immobilize islets including chambers, dam-walls, and hydrodynamic traps. We subsequently describe the surrounding methods used to create glucose gradients, enhance the reaggregation of dispersed islets, and control the microenvironment of stem cell-derived islet organoids. We focus on the various methods used to measure insulin secretion including capillary electrophoresis, droplet microfluidics, off-chip ELISAs, and on-chip fluorescence anisotropy immunoassays. Additionally, we delve into the various multiparametric readouts (NAD(P)H, Ca2+-activity, and O2-consumption rate) achieved primarily by adopting a microscopy-compatible optical window into the devices. By critical assessment of these advancements, we aim to inspire the development of new devices by the microfluidics community and accelerate the adoption of islet-on-a-chip devices by the wider diabetes research and clinical communities.
 Romario Regeenes | Dr. Romario Regeenes received his PhD degree in Biomedical Engineering from the University of Toronto. He currently works as a Senior Lab Manager at Sunnybrook Research Institute. His research interests are focused on measuring oxygen consumption and insulin secretion from individual pancreatic islets through islet-on-a-chip devices. In particular, he is interested in developing new microfluidic tools to understand the progression of type 1 and 2 diabetes. |
 Jonathan Rocheleau | Dr. Jonathan Rocheleau is an Associate Professor in Biomedical Engineering at the University of Toronto and a Senior Scientist at the Toronto General Hospital Research Institute of the University Health Network. His lab combines live cell imaging and bioengineering tools to tease apart mechanisms of pancreatic islet biology related to type 1 and 2 diabetes. |
1. Introduction
Pancreatic islets, found evenly distributed throughout the pancreas, are responsible for the control of blood glucose through the regulated secretion of insulin and glucagon. Islets are isolated from the pancreas using collagenase digestion for diabetes research and the treatment of diabetes through transplantation. Isolated human and mouse islets are typically 20–80 μm and 35–200 μm in diameter, respectively1 and variably comprised of a mixture of 4 endocrine cell types: α-, β-, δ-, and pancreatic polypeptide cells. The α-cells are responsible for storing and secreting the endocrine hormone glucagon, which increases blood glucose levels by stimulating liver gluconeogenesis. The β-cells, which make up ∼70–90% of mouse and ∼40–60% of human islets,1,2 are responsible for storing and secreting the endocrine hormone insulin, which decreases blood glucose levels by stimulating glucose uptake primarily by the liver and muscle cells. The δ-cells secrete somatostatin a hormone that inhibits insulin and glucagon secretion. Lastly, the pancreatic polypeptide cells secrete pancreatic polypeptide, a hormone that inhibits pancreatic secretion and gastrointestinal movement. All these cell types work together to control blood glucose levels by secreting hormones into the vascular network. Hence, islets are highly vascularized to rapidly detect and respond to changes in blood glucose levels. Typically, an islet contains 20–30 μm diameter afferent arterioles and 10–15 μm efferent venules.3 The number of arterioles and venules per islet depends on the size of the islet. Approximately 20% of the pancreatic blood supply is supplied to pancreatic islets.4 Moreover. the capillary network of islets are 5–10 times denser than the capillary network of the exocrine pancreas.5 The islet vascular architecture is comprised of endothelial cells and pericytes. When pancreatic islets are harvested for ex vivo studies, the density of intra-islet endothelial cells decreases by 50% within the first 24 hours and are almost completely gone within 96 hours.6 Furthermore, each islet contains 2 to 13 macrophages per islet7 for islet development8 and β-cell proliferation.9 Ultimately, variability in size and endocrine cell composition means that no two islets are identical, yet collectively these micro-organs maintain blood glucose homeostasis.Islet-on-a-chip devices are typically focused on measuring insulin secretion from β-cells due to their relevance to both type 1 and 2 diabetes. Type 1 diabetes (T1D) is a result of autoimmune destruction of pancreatic islets significantly decreasing the amount of insulin available to the body. CD4+ T-cells and macrophages trigger an inflammatory response in the pancreas that drives the progression of T1D.10 Type 2 diabetes (T2D) is a complex disease resulting from increased insulin resistance and β-cell dysfunction ultimately leading to poor insulin secretion. T2D is also related to inflammation where T-cells and macrophages are involved in releasing various cytokines and reactive oxygen species to promote pathogenic effects. In β-cells, glucose metabolism is rate-limited by entry through GLUT1/2 transporter and metabolism by glucokinase such that a rise in blood glucose induces a proportional increase in NAD(P)H reduction, oxidative phosphorylation (OxPhos), O2-consumption, and ATP production. The subsequent rise in ATP/ADP ratio triggers insulin secretion through the closure of ATP-sensitive K+-channels (KATP), membrane depolarization, and Ca2+ influx. Notably, glucose-stimulated insulin secretion (GSIS) occurs in two phases; a large transient first-phase spike followed by a sustained second phase of insulin secretion. Loss of first-phase insulin secretion is an early sign of both type 1 and 2 diabetes.11,12 First-phase secretion is commonly attributed to the release of docked secretory granules whereas second-phase is commonly attributed to the release of a slower ‘readily releasable’ pool of secretory granules. However, the transition between these phases also involves a metabolic shift from glycolysis to OxPhos-driven ATP production that may be ultimately involved in the loss of first-phase insulin release. The role of this metabolic shift is difficult to discern since it is transient (<10 min) and temporally variable islet-to-islet. Thus, GSIS involves ill-defined temporal dynamics in metabolism and secretion that are difficult to discern yet may lie at the heart of type 1 and 2 diabetes. We posit that islet-on-a-chip devices will be critical tools to tease apart these dynamics and the underlying mechanisms.
Most islet-on-a-chip devices have been designed to assess insulin secretion. In academic settings, these devices have aimed to provide a cost-effective and customizable platform to investigate hypothesis-driven research due to their ability to accommodate individual or multiple islets. For example, our lab is presently using islet-on-a-chip devices to measure the effects of proinflammatory cytokines, which are normally secreted by macrophages during the progression of T1D and T2D, on islet function. Excitingly, these devices also hold significant promise in clinical applications, where these devices could be used to evaluate donor islets and stem cell-derived islet organoids before transplantation for the treatment of type 1 diabetes (T1D). Islet-on-a-chip devices have also been designed for a wide range of applications beyond measuring insulin secretion. For example, they have been designed to measure other hormones secreted by the islet such as glucagon.13 They have also been designed to increase viral transduction efficiencies14 and measure O2-consumption rates (OCR).15,16 These wide-ranging applications reveal the power of islet-on-a-chip when paired with a microscope for live cell imaging. Live cell imaging of islet responses such as NAD(P)H, Ca2+-influx and OCR when coupled with insulin secretion could provide clinicians and researchers with more detailed insight into the functionality of islets.
One of the first islet-on-a-chip devices developed collected islet effluent from individual islets for insulin detection.17 This device and the many others that have followed offer many advantages over classical measures of insulin secretion including reducing assay costs using low reagent (nL–μL) and islet numbers while also offering automation, parallelization and high throughput capabilities. Considering this potential no islet-on-a-chip methods are widely used by the wider islet research community. We posit that this is likely due to the complexity and engineering expertise required to fabricate, operate, and troubleshoot the device as well as the low throughput and cost to adopt a new technique. We further posit that these tools will need to move beyond simply measuring insulin secretion from the tissue, but instead must offer unique insight such as the correlation of islet metabolism with insulin secretion. Thus, to be adopted by the wider community future devices will need to be both simple and incorporate multiparametric assays of islet function.
We recently reached the 20th anniversary of islet-on-a-chip devices.17–19 Here, we discuss the progression, current state, and future of these tools. We first review common methods to trap islets in microfluidic devices commenting on their ease of use and impact on islet physiology. We next describe islet-on-a-chip devices used to manipulate the microenvironment and to measure insulin secretion both off- and on-chip. Finally, we describe emerging devices that aim to simultaneously measure insulin secretion and metabolism, mostly through coupling to optical imaging. Throughout, we evaluate these devices based on their ease of use, temporal resolution, and sensitivity. We also highlight the need for multiparametric, single islet assays to reveal novel insight into research applications and clinical assessment of islets before transplantation.
2. Benefits of microfluidic platform
2.1. Common fabrication materials
Islet-on-a-chip devices are used to study living tissues. Therefore, the types of materials used to fabricate these devices are limited to biocompatible materials, which have been reviewed extensively elsewhere.20,21 A common material used to fabricate devices is polydimethylsiloxane (PDMS) but it presents several constraints. In our work, the high gas permeability of PDMS posed a challenge when investigating islet OCR or regulating the partial pressure of O2 and other gases to examine the effects on islets. We used PMMA for its significantly lower gas-permeability properties in comparison to PDMS to study OCR from individual islets. PDMS is also known to non-specifically adsorb hydrophobic molecules, which in turn affects the measurement of hydrophobic metabolites and proteins.22 PDMS is excellent for quick prototyping, but thermoplastic compounds are more suitable for upscaling. Thermoplastics such as PMMA, polystyrene and cyclic olefic copolymer (COC) are used to fabricate microfluidic devices due to biocompatibility, ease of fabrication and optical transparency.23 Fabrication techniques include hot embossing, laser ablation and CNC micro milling making upscaling much more feasible. Glass is also used to fabricate islet-on-a-chip devices due to superior optical clarity and high chemical resistance. The primary fabrication method for glass includes wet/dry etching. There are also many glass 3D printers available on the market with resolutions small enough to create microchannels. Unlike PDMS, thermoplastics and glass allow users to precisely control the partial pressures of gases in the chip and do not have molecule adhesion issues. However, PDMS is the easiest to work with and creates very precise microstructures without any cumbersome techniques or additional equipment. Ultimately, many different materials can be used to fabricate islet-on-a-chip devices depending on the requirements of the islet study.2.2. Different modes of immobilization
β-cell are (1) electrically coupled to each other resulting in an ensemble response,19,24 and (2) impacted by numerous ill-defined local paracrine signals. Thus, the islet research community commonly strives to preserve islet architecture during assays of metabolism and function. To assay islets inside a microfluidic chip, they first need to be immobilized in flow. Many different approaches have been developed to immobilize islets inside microfluidic devices including chambers, dam walls, and hydrodynamic traps. We here review these trapping methods based on their ease of use, compatibility with live cell imaging, and impact on islet physiology.One of the seminal islet-on-a-chip devices by Roper et al. used a 300 μm diameter Teflon chamber (∼70 nL) with an open reservoir above to trap to measure the effluent from a single islet (Fig. 1A).17 This chamber did not have any perfusion capabilities and instead relied on the diffusion of treatments injected above the chamber. The detection point was 5 cm away from the islet. This device led to the subsequent iterations of chambers perifused using an off-chip gas pressure system.26
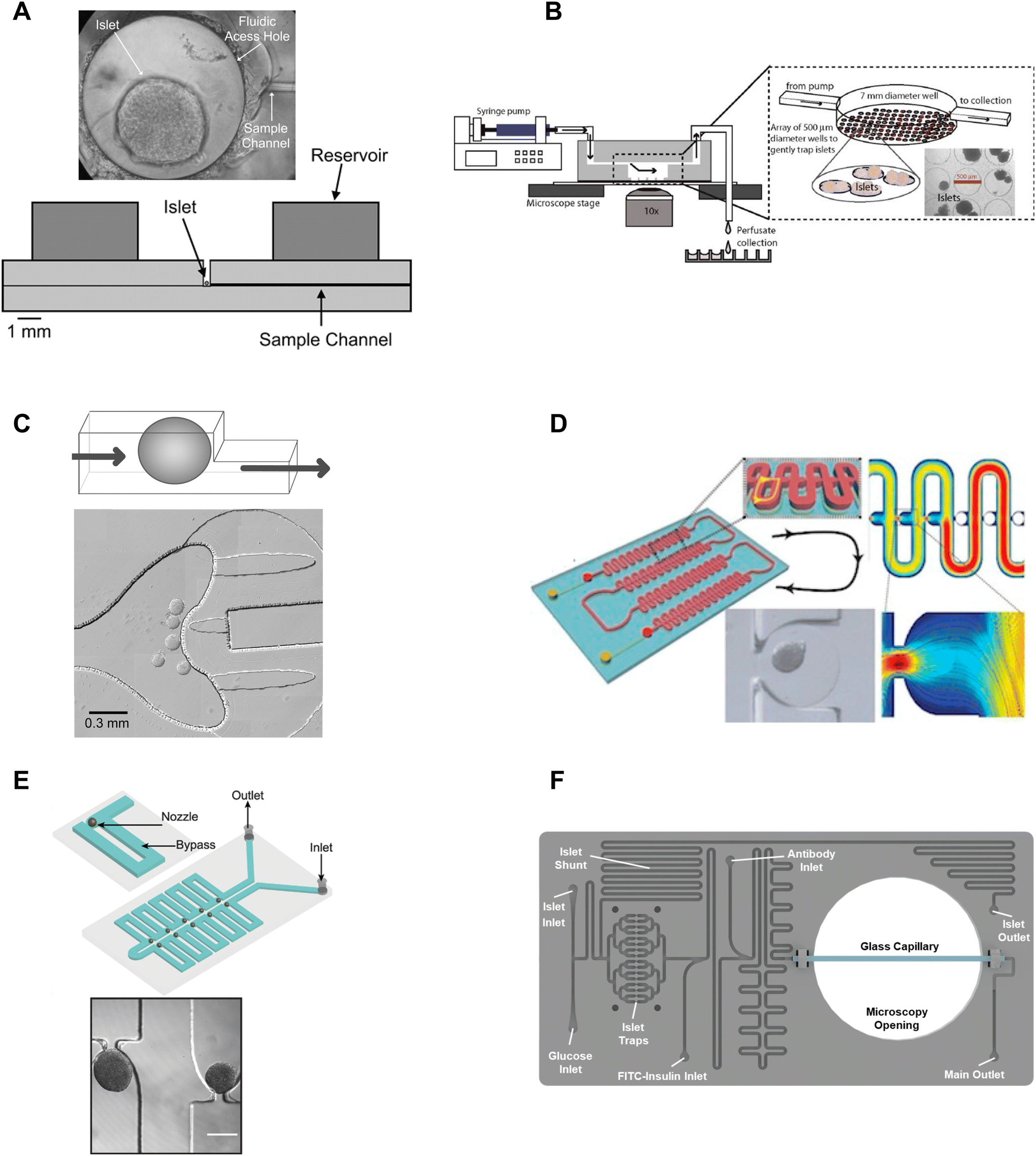 | ||
| Fig. 1 Immobilization methods used in microfluidic devices for islet entrapment. A) Open chamber loaded with a single islet for insulin secretion using capillary electrophoresis. Reprinted (adapted) with permission from ref. 17. Copyright 2003 American Chemical Society. B) Closed chamber device with an array of 500 μm wells to capture islets for simultaneous measurement of Ca2+-influx/mitochondrial membrane potential and insulin secretion. Reproduced with permission from ref. 27. C) A dam-wall device that holds islets up against a wall using controlled flow for enhanced media flow through the tissue. © 2011 Sankar et al. Reproduced with permission from ref. 19 and 30. D) A microfluidic device with an array of hydrodynamic traps to trap microencapsulated islets to explore hypoxia. Reprinted (adapted) with permission from ref. 34. Copyright 2013 American Chemical Society. E) A microfluidic device with 10 hydrodynamic traps and associated bypass in series to consecutively capture loaded islets. Reproduced with permission from ref. 31. F) A microfluidic device with 16 hydrodynamic traps in parallel with automated islet loading, stimulating and insulin secretion sensing. Reproduced with permission from ref. 36. | ||
Mohammed et al. developed a closed chamber (7 mm diameter and 3 mm deep) microfluidic device with an array of wells 500 μm in diameter and 150 μm deep to capture islets to simultaneously measure insulin secretion and Ca2+-activity or mitochondrial membrane potential (Fig. 1B).27 Ca2+ dye (Fura-2) or mitochondrial membrane potential dye (rhodamine 123) was incubated with the islets before loading and the islets were allowed to tumble into the wells of the chamber. The device was mounted on a glass slide, enabling imaging of the individual islets using an inverted microscope. Insulin secretion was simultaneously pooled from 25 mouse- or 100 human-islets, negating the ability to correlate metabolic heterogeneity to functional heterogeneity. The lower part of the chamber also had less flow than the upper chamber resulting in a nonuniform distribution of treatment throughout the chamber. This device was subsequently modified to create reproducible glucose gradients within the chamber by optimizing the inlet channel.28 Notably, this device was one of the first to report on islet functionality to predict the efficacy of islet transplantation.
Overall, chambers are relatively easy to fabricate and load with islets, and the chambers can be designed to accommodate single or multiple islets. Islets in chambers are relatively immobile due to minimal flow, thus chambers have been used to hold islets during imaging and to stimulate and collect samples of effluent using channels that pass through the top of a chamber. However, care should be taken in the chamber design. Larger chamber volumes that include an access hole are easier to fabricate and load but could result in unnecessary dilution of effluent, ill-defined flow, and poor media exchange due to minimal flow.17,29 For example, it would take glucose, with a diffusion coefficient of 5 × 10−6 cm2 s−1 and the absence of flow, >250 s to reach the bottom of a 500 μm tall channel and this time increases to >800 s for molecules the size of insulin to diffuse out of the chamber. Thus, chambers with flow at the top may overly rely on diffusion for the exchange of treatment media and effluent. Moreover, increasing the flow rate to overcome this exchange problem may only further exacerbate the dilution of the effluent and would not increase diffusion rates inside the chamber. Ultimately, the ease of using chambers may be outweighed by these difficult-to-discern issues.
Dam-wall devices are relatively easy to load; however, some care is required in their design and application to avoid “ramming” the islets underneath the dam wall. This can be achieved by slowing the islets down as they approach the dam wall by design (i.e., increasing the width of the channel at the dam wall) and application (i.e., manually slowing flow as islets approach the dam wall). We originally used these traps for live cell imaging since they immobilized the islets against the glass coverslip. However, flow-induced shear stress needs to be monitored as it can lead to a loss of glucose-stimulated responses.
Nourmohammadzadeh et al. developed a serpentine channel with an array of hydrodynamic traps to hold microencapsulated islets in a single channel down the center (Fig. 1D).34 The fluidic resistance was lower through the nozzles than the bypass channels in this device until loaded with microencapsulated islets. Thus, islets loaded sequentially into each hydrodynamic trap along the serpentine channel.
We developed a device with 8 hydrodynamic traps in-series intending to limit shear-induced damage previously observed in a dam-wall device (Fig. 1E).31 A parallel arrangement was subsequently used to induce viral transduction14 and to clarify pancreatic islets.35 In each device, the bypass channels had a higher initial fluidic resistance than the U-shaped nozzle. Thus, islets were drawn into the traps rather than down the bypass channel. The islet-loaded nozzles subsequently had much higher fluidic resistance resulting in most of the flow continuing down the bypass channel. Islets in these traps still experience some flow due to a pressure drop across the tissue but at a much lower linear velocity resulting in greatly reduced shear-induced damage.
Consistent with ease of use, Glieberman et al. automated the loading of islets into hydrodynamic traps (Fig. 1F).36 The distinctive design of the hydrodynamic trap also effectively collected islet effluent beyond the nozzle and used an on-chip fluorescence anisotropy immunoassay (FAIA) to monitor insulin secretion pooled from 16 human islets. The primary objective of developing this islet-on-a-chip was to facilitate scalable manufacturing and to promote widespread adoption, through the automation of islet loading.
Overall, hydrodynamic traps are relatively easy to load with islets (e.g., unsupervised/automatic loading using gravity flow) depending on the design. Our traps were designed to ensure easy loading by setting the nozzle-to-bypass flow rate ratios at >1, meaning most of the flow goes through the nozzles until an islet fills the nozzle. We ensured this ratio was attained by adjusting the bypass channel length and width based on a simple calculation of the fluidic resistance for each element or more accurately by finite element analysis. This ensures that islets are drawn into the trap when loading (i.e., result in high loading efficiency), and ensures that as islets enter a trap, the fluidic resistance increases resulting in the islets naturally slowing down and flow continuing down the bypass channel.
2.3. Controlled microenvironment
Microfluidic devices are powerful tools to manipulate and control the microenvironment by temporally and spatially controlling the stimulation media. Islet-on-a-chip technology enables users to mimic the in vivo microenvironment of islets. By precisely controlling parameters like flow rate, temperature, and types of treatment, researchers can create customized microenvironments that mimic conditions in the body. This approach can provide a better understanding of how islets function and respond to various stimuli, as well as in the development of novel treatments for diabetes. Islet-on-a-chip devices have been used to recreate multiple islet populations37 and enhance the formation of stem cell-derived islet organoids.38–41 We now review the various islet-on-a-chip platforms developed for manipulating the microenvironment to compare various treatments,42 create glucose gradients,43 reaggregate tissue, and generate stem cell-derived islet organoids. These devices were often designed with optical windows to simultaneously measure islet morphology and function by microscopic imaging.To create two distinct islet populations, Lee et al. developed a dual microfluidic perifusion device that consisted of two chambers, each containing a perifusion and imaging zone (Fig. 2A).37 The imaging zones for each chamber were placed next to each other to facilitate simultaneous imaging of both zones using a 10× or 20× objective lens. This device provided a direct comparison of both microenvironments while imaging individual islet responses, but measured insulin secretion from the pooled islets off-chip. This device was also easy to fabricate using standard photolithography and PDMS stamping.
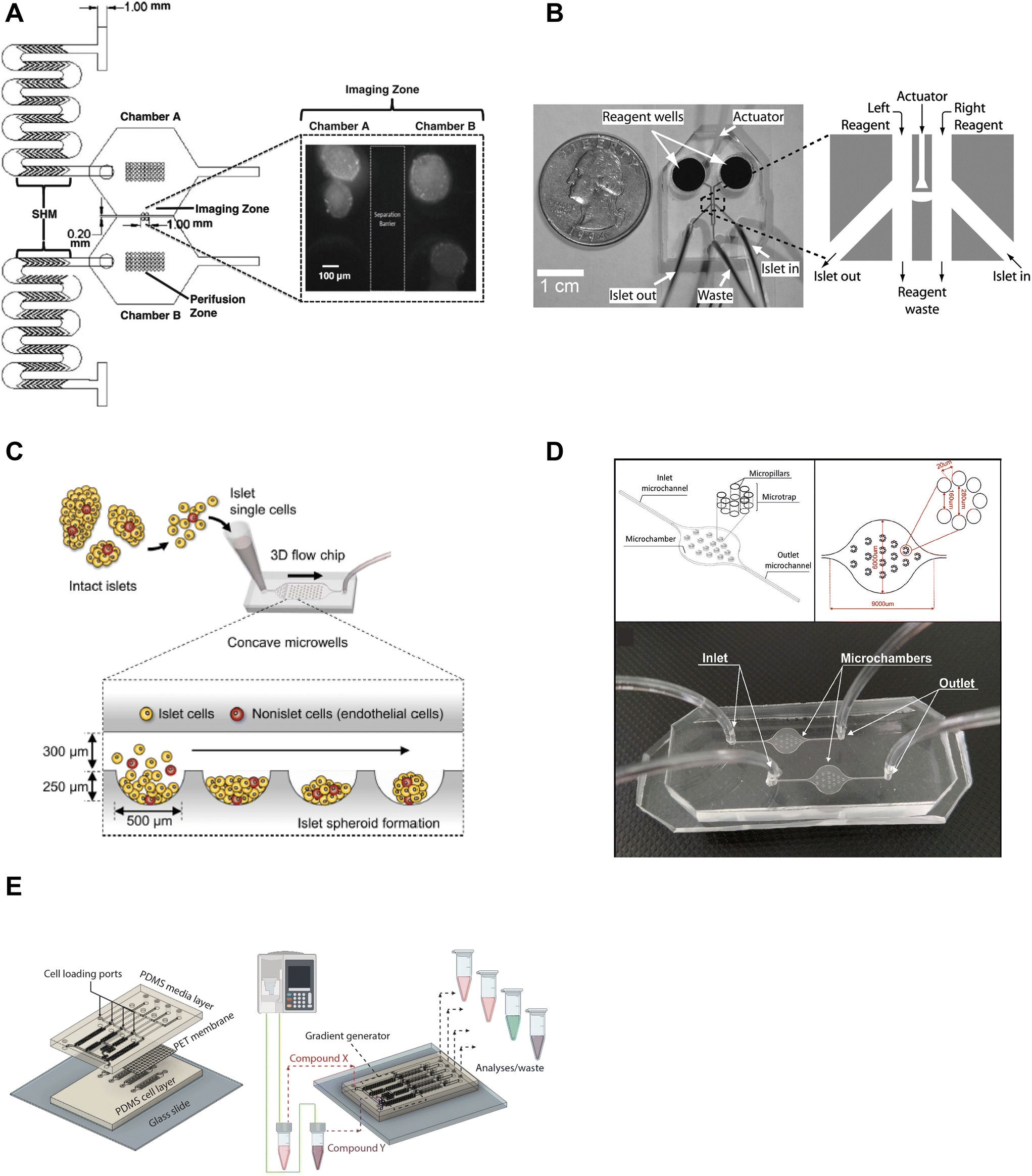 | ||
| Fig. 2 Examples of islet-on-a-chip devices designed to control the microenvironment. A) Dual perifusion chamber to compare two populations of islets or to create two different microenvironments, equipped with staggered herringbone mixers for efficient mixing. Reproduced with permission from ref. 37. B) A microfluidic device with an actuator wall to hold islets up against a PDMS wall with two on-chip wells. Copyright © 2004, the National Academy of Sciences. Reproduced with permission from ref. 19. C) A chamber device developed to reaggregate dispersed rat islets into <150 μm sized islets for better control over islet size heterogeneity using an array of microwells.45 D) An islet-on-a-chip device to coculture INS1E and aTC1-6 cells into pseudo-islets using an array of micropillars. Reproduced with permission from ref. 46. E) A 3 layered device to recapitulate the endothelial barrier to protect islets from shear stress and an on-chip gradient generator. This particular device reaggregated and cultured stem cell-derived β-cells in microwells to create islet organoids (i.e., pseudo-islets). Reproduced with permission from ref. 48. | ||
We developed an actuator device to stimulate a glucose gradient across individual islets (Fig. 2B). This device used gravity-driven flow to draw islets into a holding area that held islets between two separate channels using a thin wall that was actuated by hydraulic pressure. Islets were loaded into the device using dedicated “in” and “out” tubing while keeping the waste and reagent channels closed. Once islets were trapped, the in- and out-tubing was blocked and flow was started from the two on-chip reservoir wells to the single waste-tubing. The actuating wall pressure was controlled to accommodate different-sized islets and seal any flow across the islet. This method requires a separate pressure source to actuate the movable wall. Although mechanical stress can regulate insulin secretion,44 this design did not perturb the islet response. The left and right periphery of trapped islets were variably exposed to low (2 mM) and high (11 mM) glucose to create a glucose gradient across the tissue. The data showed that β-cells within an islet required >6.6 mM glucose locally to become Ca2+ active with neighboring cells below this threshold fully clamped. This single-layer device was easy to fabricate, but loading islets correctly into the holding chamber was not trivial and the islet throughput was low.
A group led by Sang-Hoon Lee dispersed rat islets into single cells and cultured them in a large PDMS microfluidic chamber device equipped with an array of 500 μm sized concave microwells (Fig. 2C).45 They reaggregated dispersed islet cells into <150 μm in diameter pseudo-islets and showed improved viability and function in microfluidic flow for up to 4 weeks. Here, the islets benefited from being cultured in microfluidic flow instead of traditional static culture conditions. This device was easy to fabricate using photolithography and PDMS stamping. However, the device was fully fabricated in PDMS, which due to thickness limits its useability for high-resolution fluorescence imaging.
In another example of controlling the microenvironment in forming pseudo-islets, Sokolowska et al. developed a device with micropillars that promoted 3D aggregation of β-cells by reducing the growth surface and wall shear stress (Fig. 2D).46 A reduction of the growth surface was achieved using hydrophobic PDMS micropillars. The micropillar arrangement promoted diffusion through the tissue and prevented the pseudo-islets from forming necrotic cores, a common issue culturing islets.47 They co-cultured INS1E and aTC1-6 cells to create the pseudo-islets. This device was fabricated with a borosilicate glass top to image the channels using an upright confocal microscope and the bottom mold was fabricated out of PMMA using a CNC mill.
Goswami et al. developed a device that reaggregated and cultured stem cell-derived β-cells in conjunction with a microfluidic gradient generator.48 This device consisted of three layers; a media channel, PET membrane, and a microwell cell chamber (Fig. 2E). The PET membrane protected the cultured pseudo-islets from fluid shear forces similar to endothelial cells in vivo. The diffusion properties of the PET membrane were modified to mimic the characteristics of the endothelial barrier by adjusting the pore size and density. The gradient generator increased the throughput of the device by parallelizing culture conditions, which enabled users to simultaneously explore four different conditions. This device was fabricated using standard photolithography and PDMS stamping but also attaching a PET membrane via plasma cleaning. Thus, this device requires some level of expertise to fabricate, however, the operation appears to be rather simple.
Overall, islet-on-a-chip devices offer a valuable approach to manipulating the microenvironment allowing for precise control of stimulation. In the examples provided, this control enabled temporal- and graded-glucose stimulation of islets and the formation of pseudo-islets. By attempting to mimic physiological conditions within the body, this technology enables researchers to better understand how islets respond to various stimuli, with potential implications for the development of novel treatments for diabetes. However, an aspect that is largely missing from controlled microenvironments is the immune components such as the macrophages and T-cells. Islet-on-a-chip devices do a poor job of recapitulating the immune system of pancreatic islets. This could potentially be tackled through more creative islet-on-a-chip designs and moving to pancreas-slice-on-a-chip or organ-on-a-chip devices.
2.4. Insulin secretion detection methods
Most islet-on-a-chip devices have been designed to measure islet insulin secretion. These devices mainly aimed to lower the costs compared to commercially available perifusion setups by reducing the reagent use and islet number. These devices used various methods to measure insulin including capillary electrophoresis, droplet microfluidics, off-chip ELISA, and on-chip fluorescence anisotropy immunoassay. We review here the associated islet-on-a-chip designs for each insulin detection method and discuss how these devices have become more user-friendly. We evaluate each for ease of use, cost, and temporal resolution. We note that most papers reported the fastest sampling time or speed. However, temporal resolution, defined as the shortest period of insulin pulses that can be resolved by the device, is critical to measuring the dynamics of first-phase insulin secretion and second-phase insulin pulses yet was often not reported perhaps due to the difficulty in measuring it. This oversight needs to be addressed by the field as we aim to measure insulin dynamics. Finally, we comment on the ability of each method to measure insulin secretion from individual islets, which is necessary to reveal islet heterogeneity.The Kennedy group specialized in electrophoresis-based immunoassays for continuous insulin monitoring of individual islets.17,26,49–52 A significant number of contributions from this group helped develop a better understanding of insulin secretion from individual islets. Tao et al. developed a capillary device designed to perfuse individual islets with various treatments and collect the effluent for online mixing and insulin detection.18 Their device used a CE-based competitive immunoassay, enabling a 9 s temporal resolution and revealing high-frequency oscillations within the islets. They also showed first and second-phase insulin secretion in rat islets.
Roper et al. translated the capillary design into an islet-on-a-chip capable of on-chip electrophoresis-based immunoassay.17 Individual islets were placed in a Teflon chamber and perfused with different treatments (Fig. 1A). Chamber effluent was sampled via electroosmotic flow for on-chip mixing with FITC-labelled insulin and antibody prior to separation and detection by a photon counting detector (Fig. 3A). They achieved a temporal resolution of 30 s based on a step change in insulin. However, the assay required 50 s for the FITC-insulin, antibody and secreted insulin to travel down the reaction channel before being separated for detection. This duration likely resulted in some pulse broadening, although it was not explicitly tested. Furthermore, this device did not perfuse the islet chamber, relying instead on the diffusion of treatments. This led to the development of devices coupled with gas pressure systems (Fig. 3B)26 capable of measuring insulin secretion from 4 individual islets (Fig. 3C)49 and eventually 15 individual islets in parallel with a temporal resolution of 20 s (Fig. 3D).51
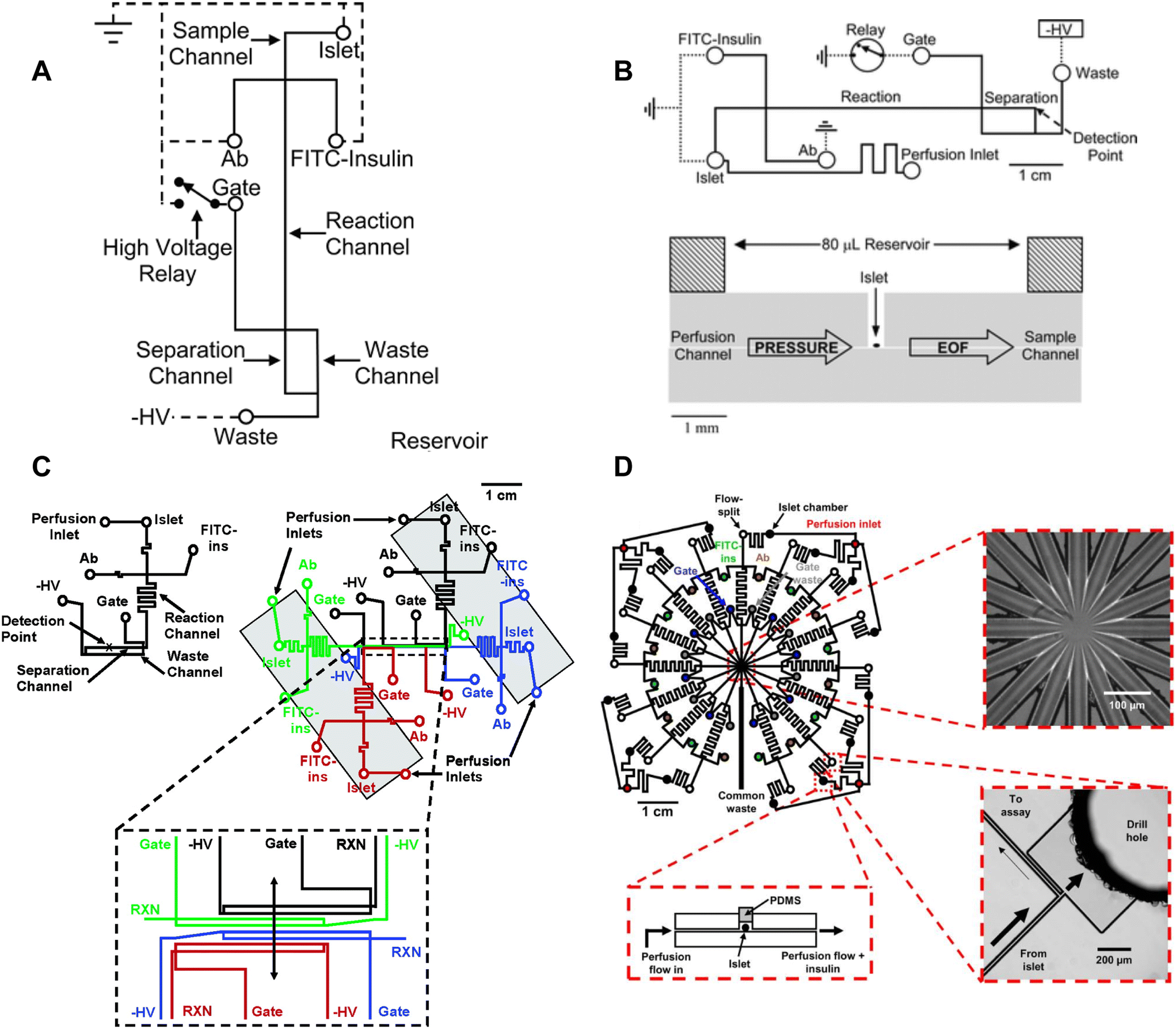 | ||
| Fig. 3 Examples of CE-based immunoassays to measure insulin secretion on-chip from individual islets. A) Online competitive immunoassay with a single islet. Islet effluent is mixed with FITC-insulin and antibody until it reaches the reactor capillary where it is injected into the separation capillary to be separated and detected. Reproduced with permission from ref. 18. B) An islet-on-a-chip capable of on-chip electrophoresis-based competitive immunoassay. Top: The islet effluent is mixed with FITC-insulin and antibody (Ab) downstream from the islet and loaded into a separation channel using a relay for separation and detection. Bottom: The islet chamber was continuously perfused in a well with a large reservoir above it and sampled using electroosmotic flow (EOF). Reproduced with permission from ref. 17. C) A microfluidic device capable of measuring insulin secretion from four individual islets. Each channel network works independently of one another. However, the separation and detection channels run parallel to one another allowing for laser detection. Reprinted (adapted) with permission from ref. 49. Copyright 2007 American Chemical Society. D) A parallel network of channels dedicated to measuring insulin secretion from 15 individual islets using CE-based immunoassays on-chip. This device was fabricated out of glass. Reprinted (adapted) with permission from ref. 51. Copyright 2009 American Chemical Society. | ||
Overall, CE-based competition immunoassays show high temporal resolution and very low detection limits making quantification of the relative and absolute insulin secretion better than some ELISAs. CE-based competition immunoassays on-chip have translated to multiple research groups. However, this method is not yet widely used by the islet community likely due to the relatively low throughput, specialized fabrication of glass microfluidic devices, and the complexity of using flow gate controllers and electrodes.
Insulin is stored as a crystal with Zn2+ in secretory granules, thus Zn2+ release has been used as a surrogate for insulin secretion. The Piston lab measured Zn2+-release from islets using FluoZin-3, a dye that shows a 50-fold increase in fluorescence upon chelating free Zn2+. This dye is normally used as an intracellular stain but was adapted here to reveal the dynamics of insulin release. A droplet-based microfluidic was used to sample at a frequency of 1.09 s.56 The experimental setup involved perfusing the islets with various treatments in conjunction with Fluozin-3 (Fig. 4A). Downstream from the islets, droplets formed from effluent and Fluozin-3 media were mixed, enabling precise insulin measurements. Unfortunately, the limited number of secretion traces suggests low throughput and the data shown provides no evidence of first and second-phase insulin secretion.
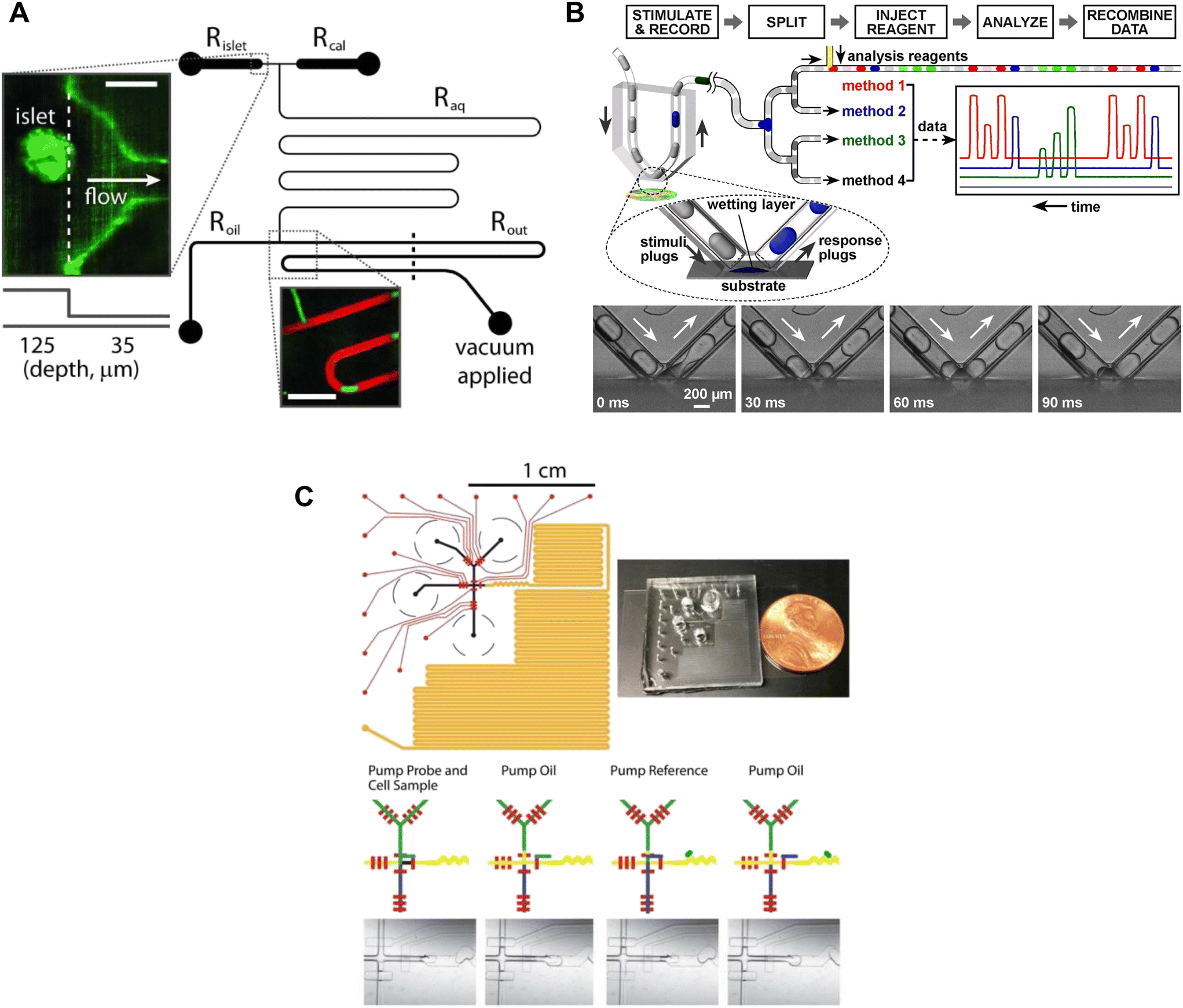 | ||
| Fig. 4 Examples of droplet microfluidic devices to measure insulin secretion from individual islets. A) Islets were continuously perfused with treatment and FluoZin-3 through Rislet. Droplets of islet effluent with FluoZin-3 are formed downstream of Roil and sequentially stored for analysis in Rout. Rcal inlet was primarily used as a vacuum inlet to introduce stimulation or for calibration measurements of FluoZin-3. Reproduced with permission from ref. 56. B) A droplet microfluidic device that used surface wetting of droplets to sample islet effluent. The droplets were then injected with immunoassay reagents for subsequent insulin quantification by FCS measurement. Islets sat in a stimulant buffer within a MatTek dish. © 2008 by the National Academy of Sciences of the USA. Reproduced with permission from ref. 57. C) A droplet microfluidic device with a long incubation channel (orange) to store and image droplets after incubation time has been met. The black circles represent the reservoirs for the islet, oil, probe and reference. The red channels represent the pneumatic control channels that automate the chip operation. Reproduced with permission from ref. 59. | ||
The collection of droplets for later offline analysis is a strategy to reduce dispersion and maintain the temporal response while benefiting from off-chip quantitation. Chen et al. developed a droplet-based microfluidic device capable of forming droplets from a hydrophilic surface.57 Islets were loaded into a coverslip-bottomed dish and the tip of a V-shaped tube, termed a chemistrode, trapped single islets while isolating them from the bulk solution (Fig. 4B). Plugs of solution with varying glucose concentrations travelled down the chemistrode to mix with the islet solution and simultaneously carry away the released insulin. Downstream from the chemistrode tip, droplets were injected with immunoassay reagents for insulin quantification of insulin concentrations by using fluorescence correlation spectroscopy (FCS). The chemistrode sampled islet secretion at a sampling frequency of 1.5 s, but could go as fast as ∼50 ms at higher flow rates. This device and the surrounding methods (e.g., FCS) are highly complex and based on the limited number of traces presented this method also suffers from low throughput. Thus, it is perhaps not surprising that this advanced method has not been more widely adopted.
Capturing homogenous immunoassays within droplets of islet effluent is another effective method to sample insulin secretion.58,59 Li et al. developed an automated droplet-generating microfluidic device that mixed and read immunoassays to measure insulin secretion from individual islets.59 The device consists of several channels such as a Y-channel for islet reagent and sample mixing, a T-junction for droplet formation, serpentine channels for droplet mixing, a long channel for droplet storage, and pneumatic control channels (Fig. 4C). All these channels worked together to achieve a sampling resolution of 15 s. The assay probe mixture and islet effluent were captured in a droplet and mixed as they travelled down the channel, and a fluorescent image was taken of each droplet. The assay probe mixture was from a highly sensitive and user-friendly human insulin FRET-PINCER assay kit, enabling accurate quantification of insulin secretion.
Overall, droplet microfluidics are a powerful approach to limit dispersion and enhance the temporal resolution of insulin secretion measurements. Each droplet captured a timepoint that could be accessed later, providing valuable data. The control of temporal resolution was achieved by increasing the frequency of droplet formation. Nonetheless, the complexity and labour-intensive nature of current droplet microfluidic devices, involving multiple inlets, outlets, and pumps, may restrict their application to non-specialized labs. The development of simpler systems is critically necessary to facilitate the adoption of droplet microfluidics in everyday research conducted by non-specialist labs.
Islet perifusion systems that capture effluent from several hundred islets are commonly used to measure the dynamics of first- and second-phase insulin secretion.60–68 These perifusion systems have a large footprint and do not provide an easy optical window for simultaneous imaging. Nevertheless, these devices have been extensively used and built upon. Sweet et al. developed a perifusion system capable of collecting effluent into a fraction collector for insulin ELISA quantification with a temporal resolution of ∼2–3 minutes.60 The perifusion system was not limited to hormone secretion measurements due to the perifusion column being accessible by optical fibers69 or a microscope61 enabling the quantification of metabolism by autofluorescence64 and Ca2+-activity in dye-loaded islets.61 The Sweet group has also successfully integrated O2 sensors into the perfusion column with temporal resolutions ranging from ∼2–5 minutes.16,61,62,69 Unfortunately, these perifusion systems have a relatively low temporal resolution, and a large footprint (Fig. 5A). These perifusion systems also use >100 islets per channel leading to high animal costs and obfuscating any heterogeneity in secretion.
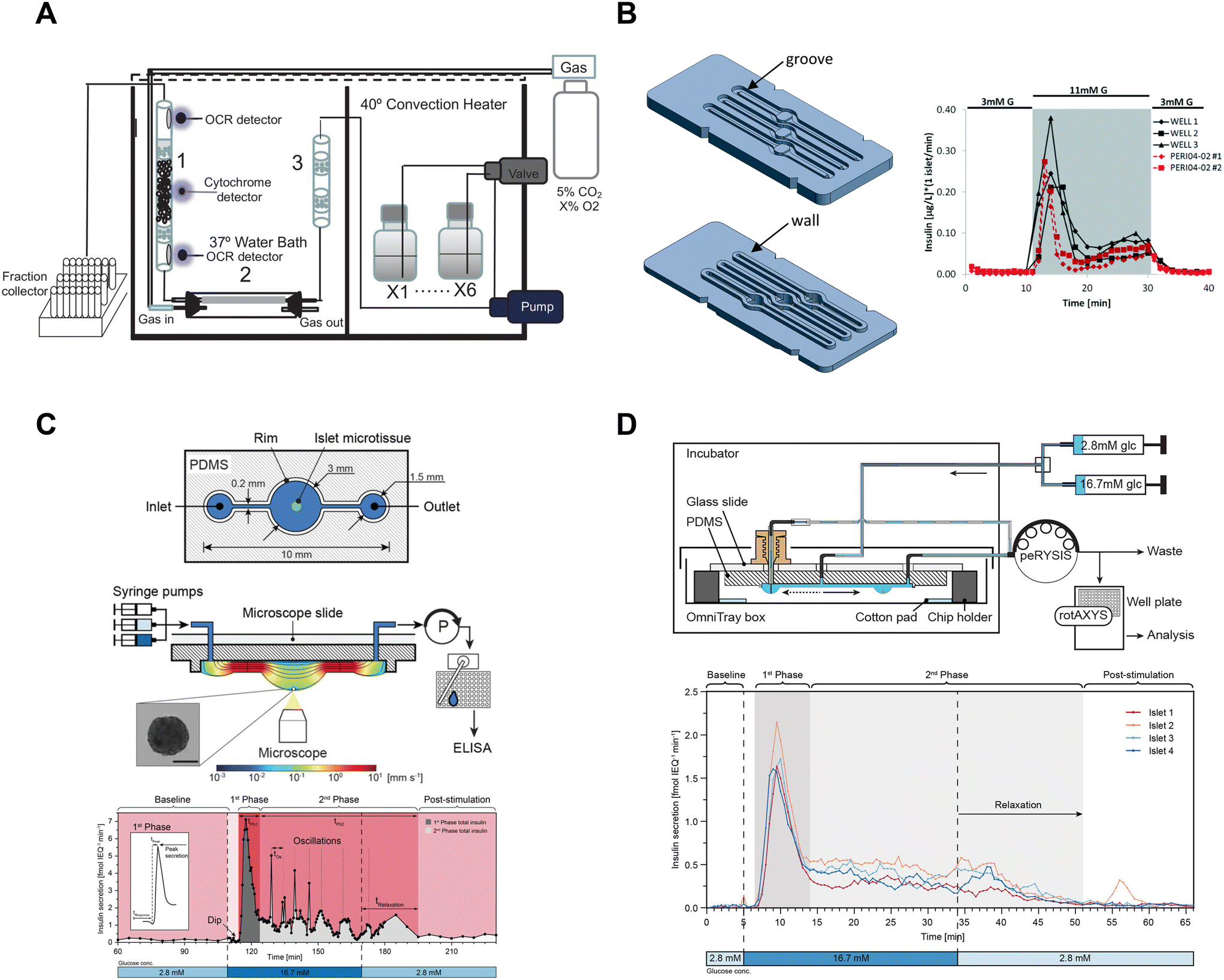 | ||
| Fig. 5 Examples of fluidic systems that collect islet effluent off-chip for insulin ELISA quantification. A) A perfusion system that loads islets into a column where flow is controlled by a peristaltic pump. OCR measurements are made before and after the islets, cytochrome c measurements are made at the islet level and insulin secretion is collected in bulk using a fraction collector. This device requires 50 islets for insulin secretion and Ca2+ measurements but requires 900 islets for OCR measurements. Reproduced with permission from ref. 16. B) A resealable acrylic microfluidic device to measure insulin secretion and image pancreatic islets. The top layer (left-top) consists of grooves for the walls of the bottom layer (left-bottom) to sit in and a gasket to seal the liquid within the device. The three chambers have dedicated inlets and outlets attached to a PERI4-02 perifusion machine. A representative GSIS trace from all 3 wells compared to the PERI4-02 instrument (right). Reproduced with permission from ref. 72. C) A hanging droplet islet-on-a-chip device to measure GSIS from individual islets (top). A cross-sectional view of an operating device where an islet is situated at the bottom of the hanging droplet (middle). This device uses a peRYSIS peristaltic pump to withdraw effluent into a 384-well plate that is controlled by a rotAXYS sampling arm. A representative GSIS trace from a single islet displaying first- and second-phase insulin secretion (bottom). Reproduced with permission from ref. 70. D) An improved hanging droplet islet-on-a-chip device to measure GSIS from 4 individual islets (top). This device uses a peRYSIS peristaltic pump to withdraw effluent into a 384-well plate that is controlled by a rotAXYS sampling arm. A representative GSIS trace from 4 individual islets showing first- and second-phase insulin secretion (bottom). Copyright © 2021 Wu Jin, Rousset, Hierlemann and Misun. Reproduced with permission from ref. 71. | ||
To improve the temporal resolution, automated sampling systems designed for multi-well plates have been developed in conjunction with perfusion systems that decrease the sampling time to ≤1 minute.66–68,70,71 However, these devices still require multiple islets per well (confounding heterogeneity) and whether these devices improve temporal resolution to ≤1 min is unclear. Finally, these devices still suffer from a general inability to simultaneously image the islets during the assay and the cost of off-chip analysis using ELISAs.
Islet-on-a-chip devices have also been designed that offer miniaturization of the perifusion and a microscope-friendly optical window. Lenguito et al. reported a PDMS-free fluidic platform to collect insulin for off-chip ELISA while being paired with a microscope.72 This acrylic device consisted of 3 holding chambers (4 mm diameter and 1.5 mm height), each capable of trapping 50 islets, and a top and bottom layer constructed with a gasket and grooves (Fig. 5B, left). A perfusion system called PERI4-02 controlled the treatment switching and fractionation collection from each holding chamber into a 96-well plate for offline insulin quantification using an ELISA kit. The sampling frequency of the system was reported to be 2 min with the microfluidic device and 1 min with the PERI4-02 vial chamber alone (Fig. 5B, right). They also showed the benefits of simultaneous microscopy by imaging live–dead stained islets and performing optogenetic experiments. The device was also used without perifusion for acute imaging experiments. Although this microfluidic device allows simultaneous imaging with a microscope, it lacks the temporal resolution and sensitivity required to explore the heterogeneity of islets.
To increase temporal resolution and sensitivity, Misun et al. developed a microfluidic device that holds single islets in hanging-drops formed using surface tension and capillary action for fluid flow (Fig. 5C, top).70 The effluent from this device was collected into a 384 well plate for offline ELISA using a rotAXYS system (Fig. 5C, middle). This innovative approach combines perifusion and microscopy, achieving a sampling resolution of 30 s (Fig. 5C, bottom). The subsequent iteration of the device improved throughput to 4 islets (Fig. 5D), reduced ELISA assay costs and eliminated the need for a microscope.71 However, the sampling resolution was not improved, and the system lacked a microscope for real-time monitoring of the islets. Nevertheless, the use of ultra-sensitive insulin ELISA kits made it possible to measure insulin secretion from individual islets, challenging the conventional requirement of using perifusion systems with hundreds of islets.
These devices are examples of innovative approaches used to explore the dynamics of islet insulin secretion. However, their reliance on automated sampling systems and/or perfusion systems for off-chip ELISA analysis may make them less accessible to research labs without these resources. Simplifying these systems would increase adoption by the wider islet community. Additionally, the use of ELISA kits adds to the ongoing cost and slows turnaround times restricting experimental throughput. These methods notably reduced the number of islets per treatment and improved sampling resolution and are moving to single islet sampling. However, these methods each need to be more critically assessed to determine the temporal resolution, which depends on dispersion by the system and is much more difficult to experimentally determine.
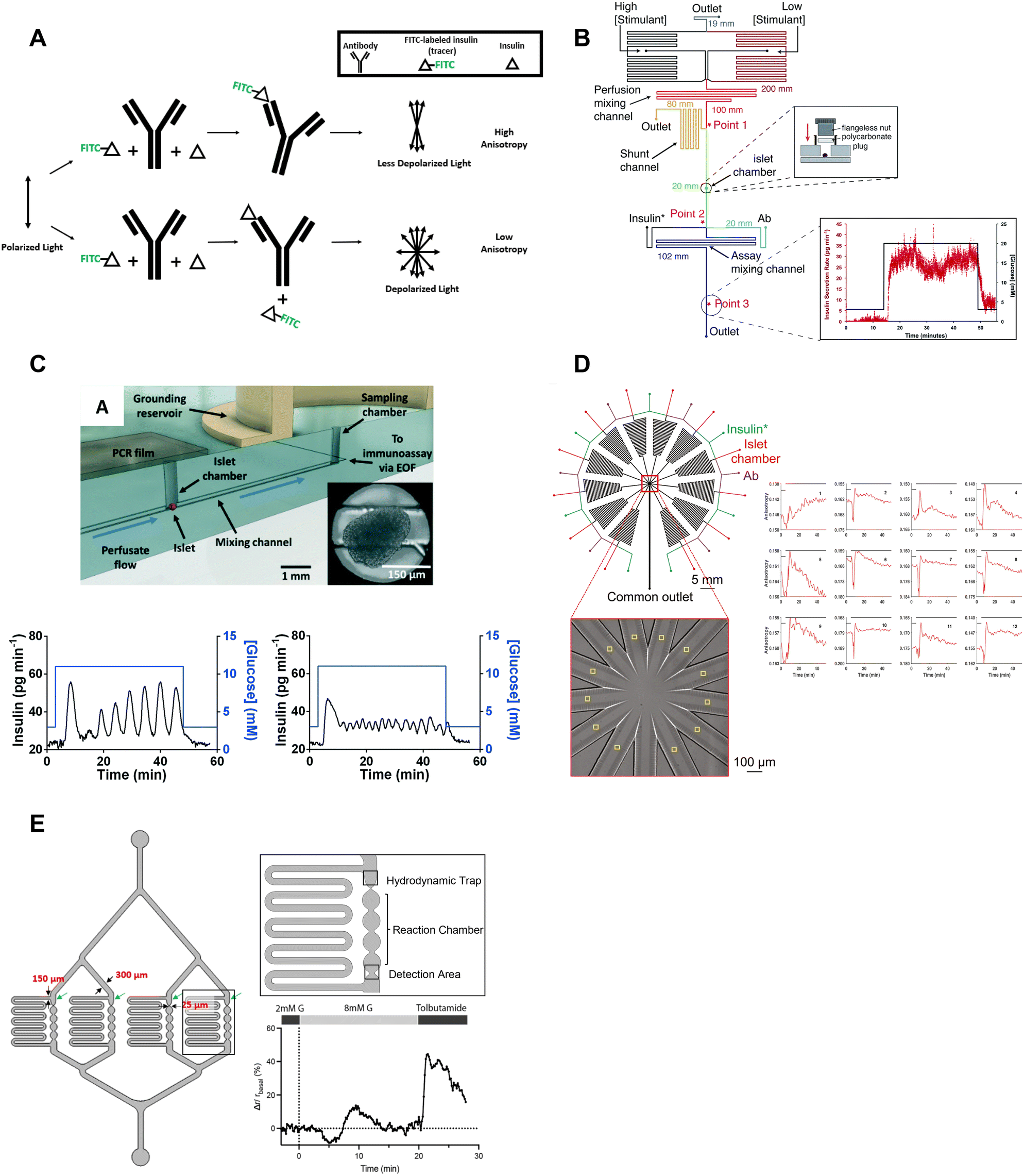 | ||
| Fig. 6 Examples of islet-on-a-chips with on-chip FAIAs to measure insulin secretion from individual and pooled islets. A) A schematic showing direct (top) and competitive (bottom) binding events that result in varying anisotropy levels. B) An islet-on-a-chip equipped with long on-chip mixing channels to measure insulin secretion from individual islets. A fraction of the mixed high- and low-glucose is sent to the islet chamber where the islet chamber is stimulated to release insulin. The released insulin mixes with insulin* and antibody while traveling down the assay mixing channel. Anisotropy measurements of the mixed product are continuously made at point 3 and converted to insulin concentration (right). Reproduced with permission from ref. 73. C) An islet-on-a-chip with an electrically grounded sampling chamber that allowed the perfusate to be continuously sampled using electroosmotic flow (top). Bottom: A representative trace of slow (left) and fast (right) GSIS traces using the device. Reproduced with permission from ref. 74. D) A high throughput microfluidic system that measures insulin secretion (right) from 12 groups of 5 or more islets. This design only has one detection area that encompasses all 12 readouts in one location. Reproduced with permission from ref. 80. E) An islet-on-a-chip developed to measure insulin secretion from 4 individual islets. Each reaction channel has a hydrodynamic trap and a short reaction chamber that makes the detection area very close to the islet permitting multiparametric readouts. A representative trace of glucose-stimulated c-peptide secretion from an individual islet (right). Changes in anisotropy were normalized to the anisotropy at 2 mM glucose. Reproduced with permission from ref. 77. | ||
The Roper group developed an FAIA that can be easily adopted by laboratories with access to fluorescence microscopes modified for steady-state fluorescence anisotropy imaging. This device mixes islet effluent with an antibody-bound tracer solution to measure insulin secretion on-chip from individual islets (Fig. 6B).73 This on-chip assay eliminates the need for expensive ELISAs. They subsequently built upon this microfluidic device to explore the acute and chronic effects of 5-palmitic acid hydroxy stearic acid on healthy and diabetic human islets (Fig. 6C).74 They measured slow and fast oscillations in insulin secretion demonstrating the high temporal resolution and power of individual islet measurements (Fig. 6C, bottom). They also revealed fatty acid esters of hydroxy fatty acids improved oscillatory dynamics and increased GSIS in both healthy and T2D human islets. This device used an electrically grounded sampling chamber that allowed continuous sampling into the immunoassay. This led to a microfluidic device designed for scalable manufacturing that increased islet throughput by arranging the islets in parallel hydrodynamics traps.36 Insulin secretion was pooled from 8 islets since the device only had a single location for anisotropy measurements (Fig. 1F). More recently, Wang et al. increased the throughput to measure insulin secretion from 12 groups (Fig. 6D, left) of 5 or more islets with a temporal resolution of 0.8 min (Fig. 6D, right). While these devices show promise in investigating heterogeneity in GSIS within islets, their current limitations include the pooling of secretion from multiple islets and the inability to integrate with other islet sensors, as the detection location is distant from the islet holding area. Potential improvements could include fabrication using a glass coverslip to improve the optical window and a motorized microscope stage to sequentially image islet and FAIA locations to correlate islet metabolism and insulin secretion, respectively.
Our lab also adopted FAIA to measure insulin secretion from individual islets.77 Our goal was to achieve the highest temporal resolution to monitor the dynamics of first- and second-phase insulin secretion while simultaneously imaging islet metabolism. We also aimed for this device to be as simple as possible to facilitate the throughput necessary for live cell imaging. To ensure high temporal dynamics, we first chose to flow the antibody bound-tracer past the islets to reduce the need to mix the effluent and tracer solution. We also chose to sense insulin c-peptide as a surrogate for insulin realizing insulin is secreted as a hexamer crystal that requires time to dissolve prior to assay. These design choices enabled measuring insulin secretion from 4 individual islets with a temporal resolution of ≤7 s (Fig. 6E, left). The device was designed to be compatible with a fluorescence anisotropy microscope with a motorized stage allowing the users to image 4 separate locations for anisotropy measurements (Fig. 6E, right). This work revealed new details in the first-phase response. More specifically, the data showed a glucose-dependent double-peak in first-phase GSIS that would normally be buried in a bulk perifusion assay. The device also allowed us to simultaneously image mitochondrial membrane potential, Ca2+-influx, and ATP with insulin secretion to reveal a transition from glycolytic to OxPhos-driven ATP production at the nadir between double-peak of insulin. Thus, this device could be used to explore islet heterogeneity and the associated metabolic dynamics. It is also relatively easy to use (requiring a single pump) but could be improved further by increasing the throughput. The device also does a poor job of measuring absolute secretion by individual islets due to its limited ability to control/measure flow rates.
Overall, on-chip FAIAs are versatile islet-on-a-chip methods that could be used for measuring many secreted peptides, extending beyond insulin secretion from pancreatic islets. In particular, we envision the potential to measure glucagon secretion. The successful development of each on-chip FAIA relies on understanding the kinetics of the competition. By determining the equilibrium time, the device design needs to account for the efficient mixing of reagents and equilibrium on the chip. As a result, each assay requires the device to be somewhat tailored to the specific kinetics. We posit that a major advantage of on-chip FAIA is the ability to enable simultaneous measurements when paired with a microscope, which we hope to use to reveal the underlying metabolism driving insulin secretion.
2.5. Multiparametric readouts
Islet-on-a-chip devices have the potential to screen islets prior to transplantation for the treatment of type 1 diabetes. However, islet-on-a-chip devices are not commonly used by clinicians for this purpose. Instead, batch assays of islet function (≥50 islets) are done using commercial instruments including insulin secretion using perifusion assays and OCR analysis using the Seahorse Analyzer. Islet-on-a-chip assays are not presently used in the clinic likely due to their complexity and that they presently do not significantly increase the speed and/or quality of the assay (e.g., many rely on the same underlying enzyme-linked immunosorbent assays (ELISA)). We posit that islet-on-a-chip devices that simply measure insulin secretion will never be clinically adopted since GSIS alone (1) can already be easily measured from small batches of islets and (2) is a poor prognostic of islet transplantation outcomes.81 Instead, islet-on-a-chip devices will need to provide novel insight such as the correlation of insulin secretion with islet metabolism. This may include revealing the timing of the metabolic transition between first and second-phase insulin secretion, which will require simultaneous measurements with high temporal resolution. This type of information is lost using typical bulk perifusion systems. Ultimately, islet-on-a-chip devices hold promise to screen islets before transplantation if they are made simple and reliable enough to be used in a clinical setting and provide unique insight (i.e., temporal and/or functional variability of islets).Over the past two decades, significant advancements have been made in the development of multiparametric islet-on-a-chip devices that provide readouts beyond insulin secretion. These innovative platforms enable NAD(P)H and FAD autofluorescence, Ca2+ activity, and O2-consumption rates (OCR) primarily by immobilizing islets for live cell imaging of autofluorescence, fluorescent dyes, and biosensors. These devices also show great research spinoff potential, such as organ- and tumour-on-a-chip due to their ability to effectively capture and perifuse 3D tissue. In the following section, we highlight examples of islet-on-a-chip devices used to investigate and measure islet metabolism.
Our lab has leveraged both our dam-wall and hydrodynamic traps to immobilize islets during live cell two-photon NAD(P)H imaging. These devices were fabricated by bonding the PDMS-formed channel to a no. 1.5 glass coverslip. The channel height was ∼125 mm tall where the islet was imaged resulting in the islets being pressed against the coverslip. A dam-wall device (Fig. 1C) was used to measure NAD(P)H and mitochondrial membrane potential responses in FGF21-treated islets.82 In contrast, a hydrodynamic trap device (Fig. 7A, left) was used to increase viral transduction within the core of the tissue and the two-photon NAD(P)H response was used to determine the impact on islet metabolism (Fig. 7A, right).14 Both devices offered subcellular resolution by immobilizing the tissue, allowing us to track the same regions of interest (i.e., same cells) between treatments.
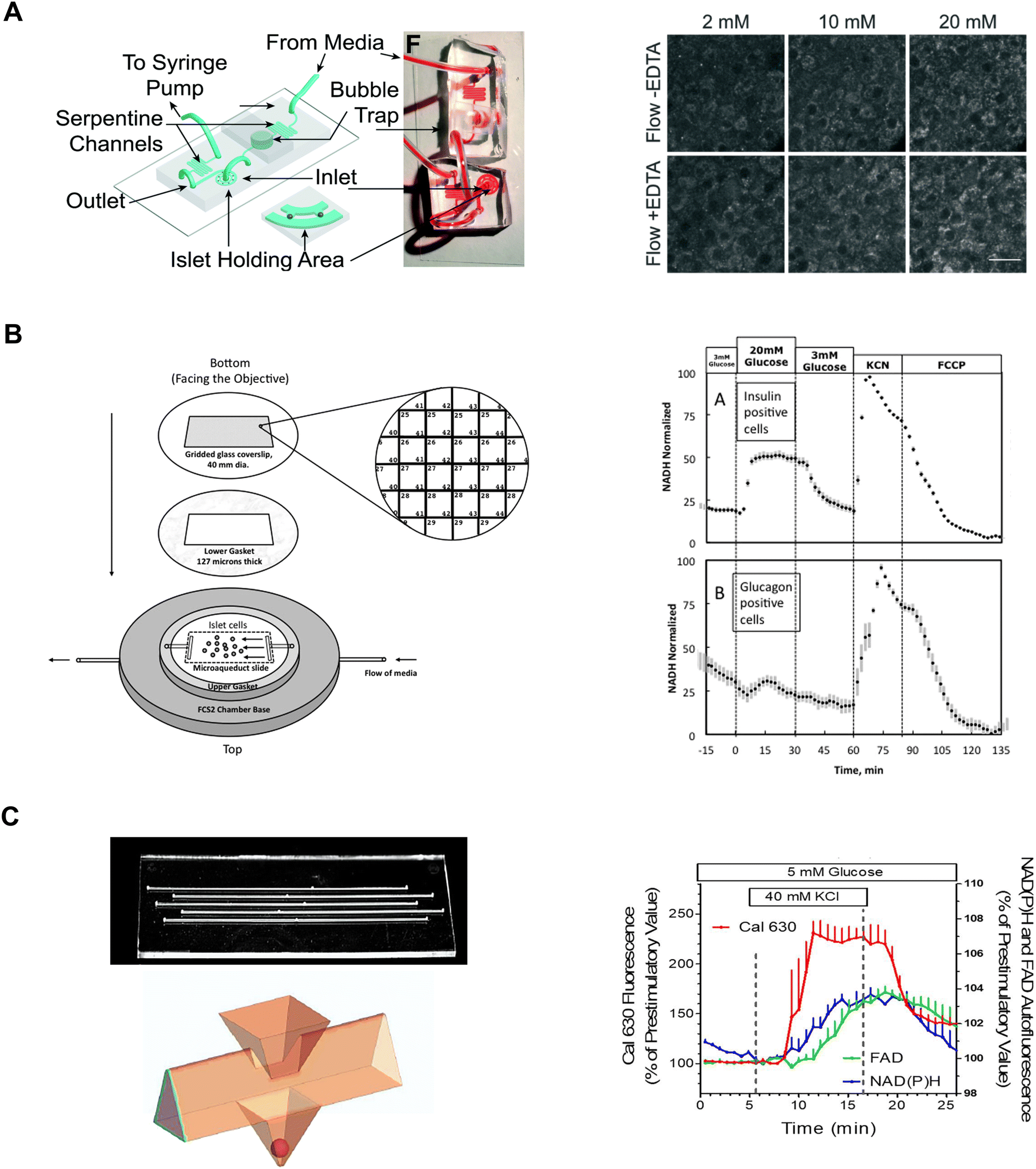 | ||
| Fig. 7 Examples of different microfluidic platforms used to explore NAD(P)H autofluorescence. A) Highly efficient adenoviral transduction (HEAT) on a chip. A parallel hydrodynamic trap device used to increase viral transduction efficiencies throughout the islet (left). This technique uses EDTA to transiently expand the tissue such that virus flow can penetrate deeper into the tissue. NAD(P)H measurements (right) reveal EDTA had no effect on islet health. Reproduced with permission from ref. 14. B) A custom gridded glass-coverslip to perifuse dispersed islet cells within the FCS2 chamber while also identifying cell locations (left). NAD(P)H measurements (right) show normal β- (inset A) and α-cell (inset B) responses to changes in glucose, KCN and FCCP. Reproduced with permission from ref. 84. C) An islet-on-a-chip designed to perifuse 5 single islets in parallel in separate channels (left). The islets are placed in a pyramidal well (below) that have a dedicated loading window above the well. This device can be used for multiparametric readouts that include NAD(P)H and FAD autofluorescence, and Ca2+-activity (right). Reproduced with permission from ref. 85. | ||
Neal et al. designed a custom-gridded glass coverslip for the commercially available FCS2 fluidics chamber.84 Rat pancreatic islets were dispersed and the cells were seeded onto the coverslip for real-time NAD(P)H imaging and Ca2+-influx imaging (Fig. 7B, left). The grids on the coverslip were numbered allowing users to identify cell locations based on the spatial map. The authors used the fluidic chamber to perifuse the cells with low and high glucose to determine the metabolic responses between α- and β-cells using NAD(P)H imaging. The cell type was confirmed by subsequent immunostaining and identification facilitated by the spatial map. β-cells showed an increase in NAD(P)H levels whereas a-cells showed no change (Fig. 7B, right). This device brought perifusion and microscopy together while allowing users to control flow rate and temperature. The FCS2 chamber can be used like a microfluidic device if cells are immobilized on the coverslip. However, the chamber can only accommodate coverslips limiting the chamber to cells rather than islets.
Schulze et al. developed an islet-on-a-chip device fabricated in glass to simultaneously measure NAD(P)H, FAD and Ca2+-influx from 5 individual islets.85 The device had 5 channels running in parallel with separate inlets and outlets (Fig. 7C, left-top). Islets were immobilized in 500 μm deep pyramidal-shaped chambers and perifused with channels at the top of the chambers (Fig. 7C, left-bottom). The authors used the device to show a glucose-stimulated rise in NAD(P)H autofluorescence after Ca2+-influx (Fig. 7C, right). Effluent from each islet was also collected to measure insulin secretion, but this suffered from relatively low temporal resolution likely due to dispersion and off-chip analysis. A major advantage of this device lies in its glass composition, which enables the seamless integration of additional sensors such as O2 sensors. However, this device has a relatively long (∼20 min) fluid exchange time and according to their simulations, a narrow size range of islets (i.e., 250 μm) should be used to facilitate a faster exchange process for a more homogenous exposure. This device requires expertise to fabricate, but it looks relatively straightforward to operate.
Overall, the platforms we reviewed demonstrate how NAD(P)H and FAD measurements are used in islet-on-a-chip devices to investigate normal or perturbed islet metabolism. These innovative microfluidic platforms immobilized islets or cells in an optical window, which allowed for autofluorescence measurements when used with a microscope. NAD(P)H and FAD autofluorescence imaging offered insight into islet health without adding dyes as the sensors are intrinsic. Consequently, this makes NAD(P)H and FAD imaging accessible methods to determine islet health while preserving intrinsic biology. Unlike other measurements that require incubation with dyes, autofluorescence imaging offers a quick and effective readout of islet health.
Tao et al. developed an islet-on-a-chip platform to engineer human induced-pluripotent stem cell-derived (iPSC) islet organoids.88 Human iPSCs were seeded into a microfluidic device and left overnight to form embryoid bodies in microwells in either static or dynamic conditions (Fig. 8A, left). In the static condition, the devices were placed in a tissue culture dish containing media. In the dynamic condition, the microwells were perifused with media. Following the formation of embryoid bodies, they were exposed to chemical factors to induce endoderm formation and promote islet differentiation. On day 23, the islet organoids from both static and dynamic conditions were retrieved to compare their Ca2+-activity using Fluo-4 imaging. The Ca2+ measurements showed perifused tissue was more responsive to changes in glucose than islet organoids derived in static conditions (Fig. 8A, right). Islet organoids from the static condition showed delayed response to glucose and an inability to clamp Ca2+ activity when switching from high to low glucose.
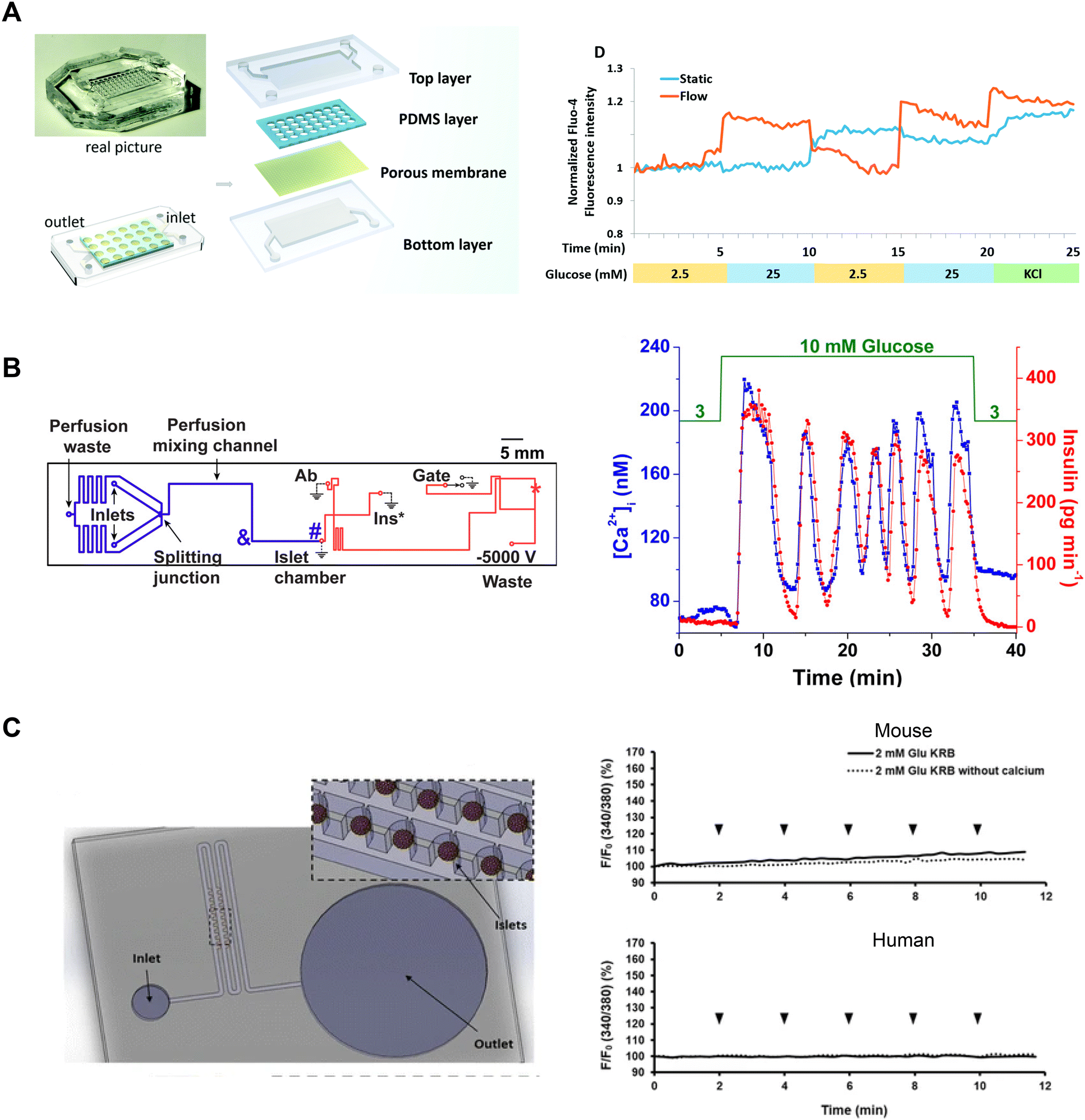 | ||
| Fig. 8 Examples of Ca2+-activity measurements in microfluidic devices designed for varying purposes. A) A microfluidic device developed to culture and differentiate human iPSC into islet organoids (top). Glucose-stimulated Ca2+-activity shows islet organoids generated under flow conditions respond better than islet organoids generated in static conditions. Reproduced with permission from ref. 88. B) A microfluidic device designed to be compatible with a dual detection system for Ca2+-activity and insulin secretion (left). The blue line represents the perfusion channel used to deliver glucose to the islet. The red lines represent the electroosmotic flow channel used to sample insulin secretion from the islet chamber. The islet-on-a-chip shows synchronous glucose-stimulated Ca2+-activity and insulin secretion (right). Reprinted (adapted) with permission from ref. 89. Copyright 2016 American Chemical Society. C) A pumpless microfluidic device driven by surface tension with an array of 20 hydrodynamic traps (left). 10 μL drops were placed at the inlet to achieve a flow rate of 20 μL min−1. Ca2+-activity measurements revealed shear stress was minimal within the device (right). Reproduced with permission from ref. 90. | ||
Yi et al. developed an islet-on-a-chip to integrate imaging Ca2+-activity with automated insulin detection by electrophoretic immunoassay (Fig. 8B, left).89 In their design, a single islet was placed in a chamber away from a detection channel separation system. This allowed simultaneous Ca2+-imaging in the chamber and detection of insulin secretion by using a dual objective system. This setup involved a xenon lamp for Fura-2 excitation with a CMOS camera for detection, and a 635 nm laser for excitation of an insulin tracer with a PMT for detection. Ca2+ and insulin traces were aligned by correcting for time delays introduced by the flow rate. The authors showed oscillations in glucose stimulation entrained Ca2+ activity and insulin secretion consistent with previous work (Fig. 8B, right). The temporal resolution of this device (∼80 s) was not quite sufficient to resolve fast Ca2+ oscillations (i.e., <60 s). This device also requires a dual-objective microscope, which will restrict easy translation to other research groups.
Xing et al. designed a pumpless microfluidic device with an array of hydrodynamic traps and flow driven by surface tension.90 This device achieved a stable flow rate of 20 μL min−1 by placing a 10 μL droplet at the inlet. The device was used to image 20 individual islets and pooled insulin secretion for off-chip analysis (Fig. 8C, left). The authors presented Ca2+, mitochondrial membrane potential, and insulin secretion from both mouse and human islets, indicating that their device accommodated a range of islet sizes. The authors used Ca2+-influx in low glucose as an indicator of shear stress during perifusion. To investigate the shear stress effects of fluid flow on the islets, they introduced continuous drops of 2 mM glucose, with and without Ca2+, at the inlet. They showed continuous drops did not elevate Ca2+-activity, suggesting changes in fluid flow did not elevate shear stress on mouse and human islets (Fig. 8C, right). This device has a simple design and holds great potential for easy adaptation by other research groups due to its pumpless operation, eliminating the need for specialized expertise. Potential limitations of this device are that it requires users to constantly top up the inlet and only operates at one flow rate.
Overall, Ca2+-activity is directly related to first- and second-phase insulin secretion, and thus has been a crucial tool in measuring the glucose-stimulated response of islets. Islet-on-a-chip platforms offer a controlled microenvironment to maintain islet health and elicit healthy responses. Thus, the devices we reviewed were developed for varying purposes yet share a common utilization of Ca2+ as a reporter of islet health. Furthermore, these islet-on-a-chip devices are compatible with microscopy, meaning these devices are immediately adaptable to a wide range of quantitative live cell imaging.
Our lab developed on-chip O2 microsensors to simultaneously image OCR and Ca2+-activity of individual pancreatic islets.15 This device was based on a dam-wall device fabricated out of PMMA with micron-sized O2-sensitive sensors on the glass slide (Fig. 9A, left). We used PMMA instead of typically used O2-permeable PDMS to avoid O2 saturating our measurements. To calibrate the microsensors, phosphorescence was measured while flowing ddH2O and 10% sodium sulfite for 100% air saturated and 0% O2 water, respectively. The microwells were fabricated in 10 μm tall SU-8, which digitized the sensor response while maintaining optical clarity. The temporal resolution of the O2-microsensors was <15 s. This device used an on-chip reservoir making it easy to load islets and exchange treatments. This design allowed simultaneous imaging of the glucose-stimulated OCR and Ca2+ responses to reveal a biphasic respiratory response that is independent of Ca2+ influx. Furthermore, we used this device to conduct a series of mitochondrial respiration treatments to show that individual islet responses in the device were consistent with normally measured bulk islet responses (Fig. 9A, right). Thus, this device can be used to measure the fast dynamics of islet OCR response to glucose as well as classic assays of islet function.
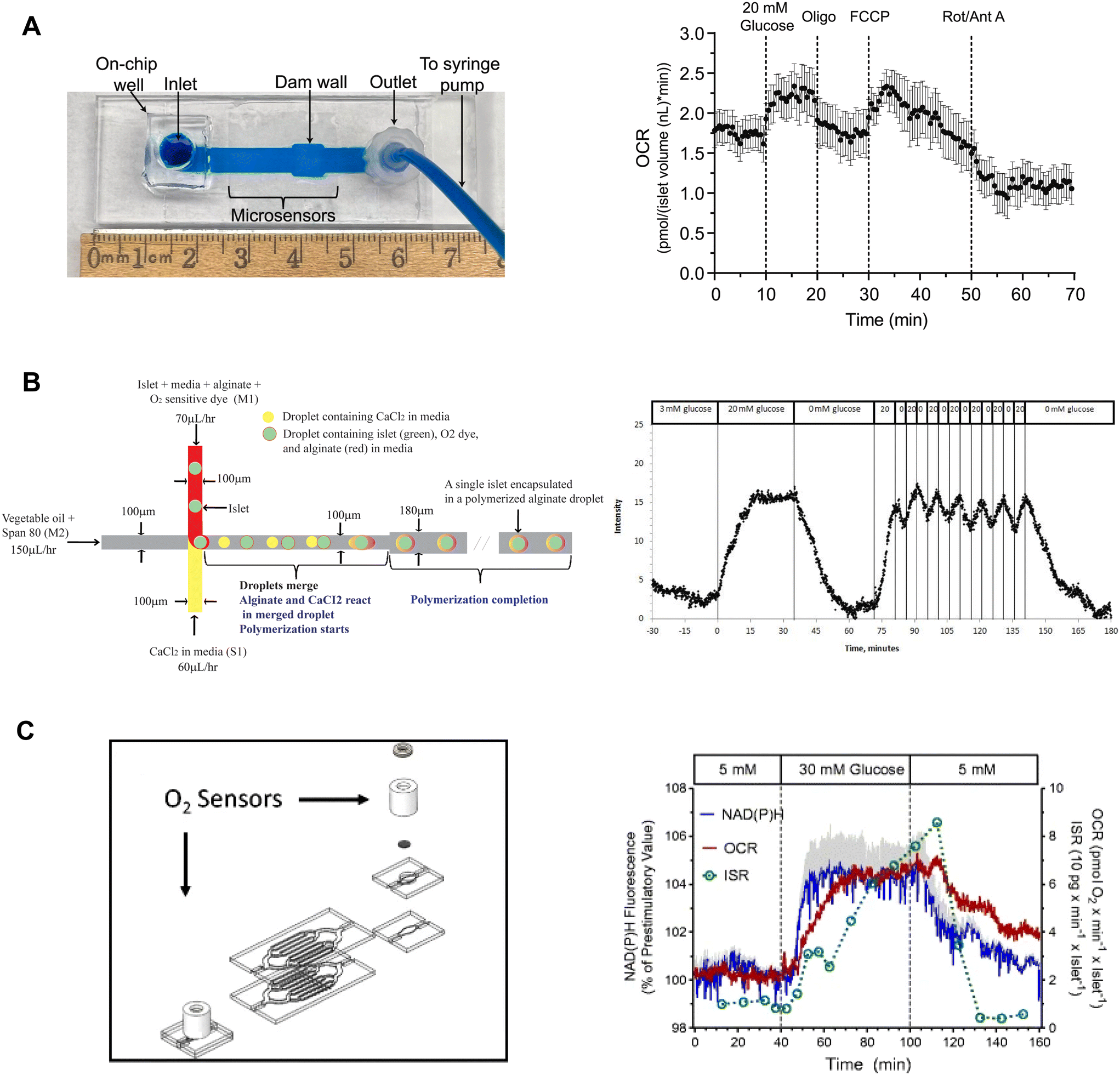 | ||
| Fig. 9 Examples of islet-on-a-chip devices capable of OCR measurements. A) A dam wall device with O2-sensitive microwells on the glass slide (left). Pancreatic islets sit on top of the microsensors when pushed up against the dam wall. Mean OCR of individual islets treated with respiratory inhibitors (right). Reproduced with permission from ref. 15. B) A T-junction device that encapsulates islets in alginate droplets with O2-sensitive dye (left). The device uses vegetable oil as the carrier fluid and injects islets, media, alginate and O2-sensitive dye from one inlet and CaCl2 from the other. The CaCl2 merges with the alginate downstream causing the alginate to polymerize encapsulating the islet and O2-sensitive dye. Oscillations in glucose treatment revealed glucose-induced oscillations in islet O2 (right). © 2012 Chen et al. Reproduced with permission from ref. 98. C) A microfluidic device designed for multiparametric imaging of OCR, NAD(P)H autofluorescence, Ca2+-influx and insulin secretion from 50 islets. O2-sensor spots (Pst3 sensors) are strategically positioned at both the inlet and outlet of the device to accurately measure the changes in O2 levels resulting from islet respiration (left). Representative traces of simultaneous measurements of NAD(P)H, OCR and insulin secretion (right). NAD(P)H autofluorescence averaged from 3 islets in three different wells. The device effluent was collected using a fraction collector, and subsequently, the insulin concentration of each fraction was quantified using an ELISA. Reproduced with permission from ref. 98. | ||
Chen et al. used droplet microfluidics to capture islets in thin alginate droplets with an O2-sensitive fluorescent dye.98 This T-junction device used vegetable oil as the carrier fluid to create the droplets (Fig. 9B, left). The other two inlets were injected with CaCl2-containing media and islets with the O2-sensitive dye. The CaCl2 induced alginate cross-linking thereby trapping islets with the O2-sensor. The islets still secreted insulin showing that encapsulation did not alter GSIS. This method had a sampling time of 30 s and required some expertise to operate. To calibrate the O2-sensor, fluorescence was measured at varying levels of dissolved O2 while suppressing islet respiration by using KCN. The authors showed a glucose-stimulated first-phase response and second-phase oscillations of individual islets (Fig. 9B, right).
Schulze et al. developed a glass islet-on-a-chip device capable of multiparametric imaging of OCR, NAD(P)H autofluorescence, and Ca2+-influx with insulin secretion pooled from 50 islets.99 The device consists of 8 branched channels in parallel with 7 wells that are progressively deeper from the inlet to the outlet to protect islets from shear stress and still have good media exchange. To measure OCR, O2 sensor (Pst3 sensors) was placed at the inlet and outlet of the device (Fig. 9C, left). The device required 50 islets to achieve a stable bulk OCR measurement. Islet effluent was collected from the device using a fractionator to perform an off-chip insulin ELISA. The authors showed simultaneous measurements of NAD(P)H, OCR and insulin secretion as well as OCR and Ca2+-activity (Fig. 9C, right). The authors did not report the temporal resolution of the OCR and insulin measurements, albeit the difference in O2 between the outlet and inlet sensor was continuously monitored. This device measures OCR from at least 50 islets, which negates using it to explore islet heterogeneity. This device appears to be easy to operate but does require some specialized equipment and expertise to simultaneously assay insulin secretion.
In summary, OCR measurements are widely used to explore the mitochondrial metabolism of islets during GSIS. Most methods developed so far rely on bulk islet responses, limiting the ability to gain insight into the metabolic heterogeneity of islets. Bulk responses are also inadequate for capturing rapid changes in metabolism due to the temporal variability of islet responses. Fortunately, the development of more advanced islet-on-a-chip devices is underway to address these limitations. This includes the integration of optical O2-sensors into the device.100 These sensors can allow for live cell imaging if the sensor is selected to avoid spectral overlap with other sensors and/or the position of the sensors is away from the islet to mitigate any potential spectral overlap. We postulate that these devices will become much more widely used by the islet community once their ease of use is improved and once the unique temporal insight they provide is further revealed.
3. Discussion
Based on searches in PubMed, ∼210 publications over the last two decades used the term “islet on a chip” (Fig. 10A) and 164 used the terms “islet and microfluidic” (Fig. 10B). These numbers highlight that this technology is still niche in nature primarily being used by bioengineers who possess the necessary equipment and expertise to fabricate and operate these devices. We postulate that islet-on-a-chip devices will be embraced by the larger islet/diabetes community when (1) they reveal unique insights into islet metabolism and function, (2) they are simpler than current commercial perfusion systems, and (3) they are significantly cheaper (i.e., use fewer islets and expensive kits/reagents) to run. Within the publications, notable advancements have been made to develop more user-friendly techniques for investigating the dynamic metabolism of islets.15,77,80 While some of the devices reviewed here may be limited to specialized laboratories, many of them are relatively straightforward to use once a lab is properly outfitted. They typically require a minimum of premade treatments that are added to a reservoir, a pump or vacuum-based pressure controller to regulate flow rate, tubing for inlet and/or outlet, and a standard operating procedure (SOP) for user guidance.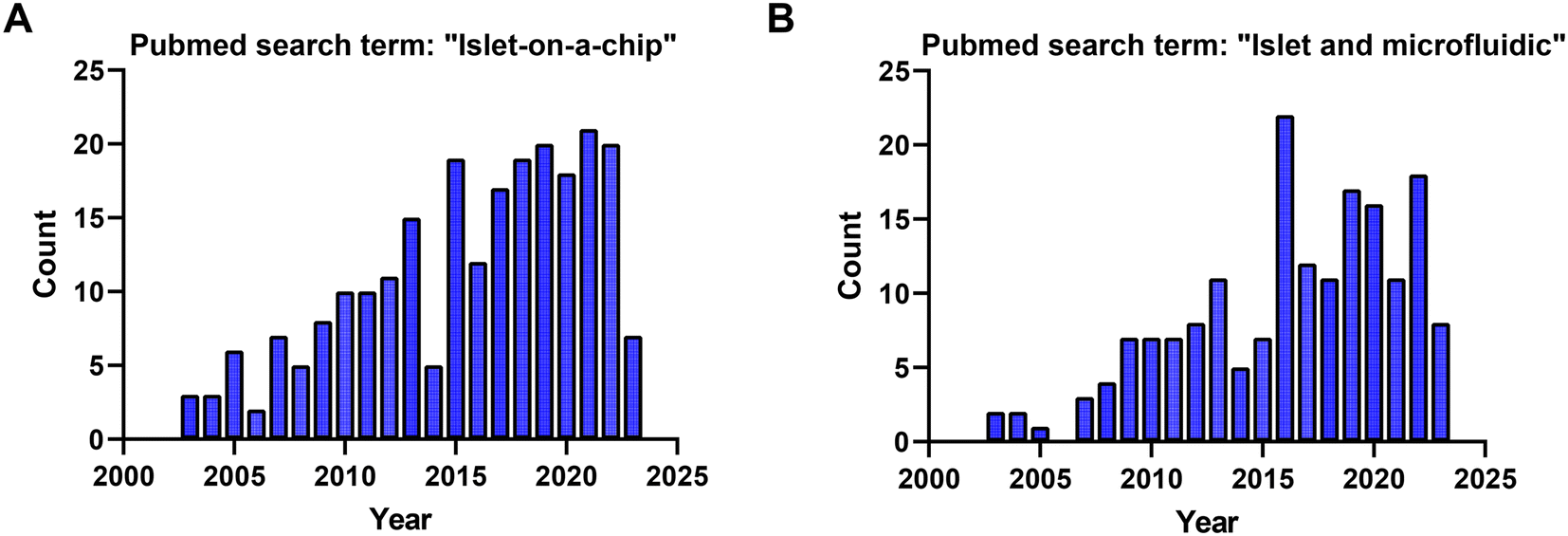 | ||
| Fig. 10 PubMed search term results for the studies used in this review from 2003 to 2023. A) 210 PubMed search results for the search term: “islet on a chip”. B) 164 PubMed search results for the search term: “islet and microfluidic”. | ||
A large majority of islet-on-a-chip devices used the device to fractionate the secretion for off-chip assay of secretion using ELISAs. Effluent from these devices is collected using sampling systems, often with an islet holding area positioned away from the outlet. These devices often work on a lower number of islets per treatment but still rely on relatively expensive ELISA kits. These devices often offer increased sampling frequency. However, it should be recognized that as islet effluent travels down the channel and off-chip, the insulin is dispersed, diluted, and possibly adsorbed to the walls of the device. Thus, although these devices may offer high sampling frequency, they may do little to increase the temporal resolution (i.e., the ability to resolve two closely spaced pulses of insulin).
Islet-on-a-chip devices often offer the opportunity to simultaneously image glucose-stimulated metabolism using a host of fluorescence sensors. For example, our lab has made extensive use of sensors for mitochondrial membrane potential, ATP, and Ca2+, with each providing insight into the temporal dynamics of mitochondrial metabolism and voltage-gated channel opening. Often, microfluidic devices are optically clear, but not optimized for live cell fluorescence microscopy. Live cell fluorescence microscopy methods use high NA objective lenses, which are commonly designed to image cells seeded onto a no. 1.5 glass coverslip with a specified thickness and index of refraction. However, many devices are fabricated using much thicker glass, plastic, and PDMS. Moving away from the standard coverslip results in spherical aberrations and loss of working distance. Islets are also unique in that they are not attached to the glass surface. This results in only a few cells being placed up against the coverslip when depending on gravity alone. Thus, our lab has generally relied on dam walls and hydrodynamic traps with channel heights that push the tissue against the coverslip rather than chamber/wells that rely simply on gravity to immobilize the islet.
Islet-on-a-chip devices are being used for a wide range of applications beyond measuring islet insulin secretion including 1) treating and enhancing tissue viability through flow, 2) modulating stem cell development, 3) imaging subcellular features, 4) simultaneously measuring metabolites specific to the tissue and 5) disease modelling in vitro. However, we believe these devices offer a much wider range of applications. In the future, we anticipate these devices being applied to culture and enhance stem cell-derived tissue as a potential disease treatment. Similarly, ex vivo tissue also could be cultured in an islet-on-a-chip to be preserved for a longer culture period under flow conditions.45 We anticipate these devices will show greater application to live cell imaging with subcellular resolution by taking advantage of the islet-on-a-chip to immobilize the tissue. This resolution could help reveal how β-cell heterogeneity determines islet function. We also anticipate more applications using multiple channels for different culturing conditions to increase the detail and throughput of disease models.37 Islet-on-a-chip devices undoubtedly open a new door to measuring the functional heterogeneity of stem cell-derived islet organoids. We anticipate the future application of islet-on-a-chip devices to screen stem cell-derived islet organoids to determine how well they resemble native islets and/or to screen the tissue prior to transplantation. Thus far, islet-on-a-chip devices have also shown limited application to measuring the impact of endothelial and immune cells on islet function. Endothelial cells and the extracellular matrix support GSIS but are lost after isolation.101,102 The stem cell-derived islet organoids lack endothelial cells and macrophages which are normally present in islets. These cell types need to be incorporated into the tissues we study to have a better understanding of T1D and T2D. Regrettably, these aspects have been overlooked by the community during pseudo-islet development. This could be attributed to the main focus on developing stem cell-derived β-cells that secrete insulin in a pulsatile fashion. In the meantime, our lab and others are indirectly exploring the effects of immune cells by treating healthy islets with proinflammatory cytokines. To fully explore how immune cells and islets interact with one another ex vivo will require organ-on-a-chip devices where the immune cells are somehow incorporated and/or pancreas-slice-on-a-chip where macrophages and T-cells are already present in the tissue.103
The future of islet-on-a-chip devices relies on their accessibility to the broader islet biology community. The simpler, less complex, and more facile these devices become for live cell imaging will lead to adoption in islet research. Additionally, these devices hold potential for use in studying other tissues, such as 3D aggregates, spheroids, and tumors that are ellipsoids or spheres. This includes stem-cell-derived 3D aggregates, spheroids derived from adipose stem cells, liver cells, and lung spheroids, among many others. These microfluidic platforms offer immobilization for imaging, a customizable controlled microenvironment, and the possibility of detecting secreted metabolites such as VEGF from adipose derived stem cell spheroids104 or albumin from liver spheroids.105 However, it is important to note that islet-on-a-chip devices designed for detecting secreted metabolites are tailored based on the reaction kinetics of the tracer molecule and antibody. Consequently, different metabolites require new devices specifically designed for those assays. This limitation makes most devices originally designed for insulin secretion less adaptable to studying other metabolites. Nevertheless, configurations like capillary electrophoresis devices, droplet microfluidics, off-chip, and on-chip devices can be utilized to explore other metabolites and address this challenge.
4. Conclusion
Islet-on-a-chip technology currently offers users many ways to explore islet metabolism and insulin secretion. These microfluidic devices serve to immobilize islets, enabling precise control of media exchange, while also providing a means to monitor islet function through the temporal assay of insulin secretion. We believe the true power of islet-on-a-chip devices lies in their compatibility with microscopy. Firstly, this allows users to explore morphological differences between islets, before and after treatments, including size-related measurements. Secondly, microscopy can be used with a plethora of fluorescent dyes and genetically encoded sensors to explore metabolic and electrical responses behind islet secretion. Lastly, microscopy maintains access to other techniques such as capillary electrophoresis-based immunoassays, droplet microfluidics, on-chip FAIA, and multiparametric readouts.A large majority of islet-on-a-chip devices have been designed to measure secretion from multiple islets. This strategy has resulted in using fewer islets compared to classic perifusion methods, but it is not clear whether these devices have significantly improved the resolution of the detection and reduced costs since many still rely on off-chip ELISA kits. There is also growing evidence that islet heterogeneity is critical to normal physiology and pathophysiology of both type 1 and type 2 diabetes.106–112 Loss of first-phase insulin secretion is also a hallmark of both diseases. Thus, we suggest that future islet-on-a-chip devices should be focused on measuring insulin secretion from individual islets with high temporal resolution. Ultimately, this would result in tools that provide unique insight into islet heterogeneity, which will be critical to our understanding of islet dysfunction in T2D and as the field advances toward screening islets for T1D treatment. In this regard, the development of high-throughput devices capable of simultaneously measuring insulin secretion and metabolism (e.g., OCR) of individual islets holds great promise for clinical applications.
Author contributions
Romario Regeenes: conceptualization, data curation, investigation, project administration, writing – original draft, writing – review & editing, visualization. Jonathan V. Rocheleau: conceptualization, project administration, writing – review & editing, supervision.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors gratefully acknowledge funding (RGPIN-2022-04454, JVR) and stipend support (PGSD-535059-2019, RR) from NSERC.References
- A. Kim, K. Miller, J. Jo, G. Kilimnik, P. Wojcik and M. Hara, Islets, 2009, 1, 129 CrossRef PubMed.
- O. Cabrera, D. M. Berman, N. S. Kenyon, C. Ricordi, P. O. Berggren and A. Caicedo, Proc. Natl. Acad. Sci. U. S. A., 2006, 103, 2334–2339 CrossRef CAS PubMed.
- L. Jansson, A. Barbu, B. Bodin, C. J. Drott, D. Espes, X. Gao, L. Grapensparr, Ö. Källskog, J. Lau, H. Liljebäck, F. Palm, M. Quach, M. Sandberg, V. Strömberg, S. Ullsten and P. O. Carlsson, Upsala J. Med. Sci., 2016, 121, 81 CrossRef PubMed.
- L. Jansson and C. Hellerstrom, Am. J. Physiol., 1986, 251, E644–E647 CAS.
- Y. El-Gohary, S. Sims-Lucas, N. Lath, S. Tulachan, P. Guo, X. Xiao, C. Welsh, J. Paredes, J. Wiersch, K. Prasadan, C. Shiota and G. K. Gittes, Anat. Rec., 2012, 295, 1473–1481 CrossRef CAS PubMed.
- D. Nyqvist, M. Köhler, H. Wahlstedt and P. O. Berggren, Diabetes, 2005, 54, 2287–2293 CrossRef CAS PubMed.
- H. C. Denroche, S. Miard, S. Sallé-Lefort, F. Picard and C. B. Verchere, Immun. Ageing, 2021, 18(1), 1–13 CrossRef PubMed.
- M. Brissova, K. Aamodt, P. Brahmachary, N. Prasad, J. Y. Hong, C. Dai, M. Mellati, A. Shostak, G. Poffenberger, R. Aramandla, S. E. Levy and A. C. Powers, Cell Metab., 2014, 19, 498 CrossRef CAS PubMed.
- X. Xiao, I. Gaffar, P. Guo, J. Wiersch, S. Fischbach, L. Peirish, Z. Song, Y. El-Gohary, K. Prasadan, C. Shiota and G. K. Gittes, Proc. Natl. Acad. Sci. U. S. A., 2014, 111, E1211 CAS.
- C. Li, Q. Gao, H. Jiang, C. Liu, Y. Du and L. Li, Sci. Rep., 2022, 12, 1–9 CrossRef PubMed.
- G. C. Weir and S. Bonner-Weir, J. Clin. Invest., 2021, 131, 10–13 CrossRef PubMed.
- J. E. Gerich, Diabetes, 2002, 51, S117–S121 CrossRef CAS PubMed.
- W. J. Eaton and M. G. Roper, Anal. Methods, 2021, 13, 3614–3619 RSC.
- P. N. Silva, Z. Atto, R. Regeenes, U. Tufa, Y. Y. Chen, W. C. W. Chan, A. Volchuk, D. M. Kilkenny and J. V. Rocheleau, Lab Chip, 2016, 16, 2921–2934 RSC.
- R. Regeenes, Y. Wang, A. Piro, A. Au, C. M. Yip, M. B. Wheeler and J. V. Rocheleau, Biosens. Bioelectron.: X, 2023, 13, 100285 CAS.
- V. Kamat, B. M. Robbings, S. R. Jung, J. Kelly, J. B. Hurley, K. P. Bube and I. R. Sweet, eLife, 2021, 10, 1–37 CrossRef PubMed.
- M. G. Roper, J. G. Shackman, G. M. Dahlgren and R. T. Kennedy, Anal. Chem., 2003, 75, 4711–4717 CrossRef CAS PubMed.
- L. Tao, C. A. Aspinwall and R. T. Kennedy, Electrophoresis, 1998, 19, 403–408 CrossRef CAS PubMed.
- J. V. Rocheleau, G. M. Walker, W. S. Head, O. P. McGuinness and D. W. Piston, Proc. Natl. Acad. Sci. U. S. A., 2004, 101, 12899–12903 CrossRef CAS PubMed.
- I. Miranda, A. Souza, P. Sousa, J. Ribeiro, E. M. S. Castanheira, R. Lima and G. Minas, J. Funct. Biomater., 2022, 13(1), 1–20 Search PubMed.
- E. Berthier, E. W. K. Young and D. Beebe, Lab Chip, 2012, 12, 1224–1237 RSC.
- M. W. Toepke and D. J. Beebe, Lab Chip, 2006, 6, 1484–1486 RSC.
- E. Roy, J. C. Galas and T. Veres, Lab Chip, 2011, 11, 3193–3196 RSC.
- J. V. Rocheleau, M. S. Remedi, B. Granada, W. S. Head, J. C. Koster, C. G. Nichols and D. W. Piston, PLoS Biol., 2006, 4, e26 CrossRef PubMed.
- L. A. Godwin, M. E. Pilkerton, K. S. Deal, D. Wanders, R. L. Judd and C. J. Easley, Anal. Chem., 2011, 83, 7166–7172 CrossRef CAS PubMed.
- J. G. Shackman, G. M. Dahlgren, J. L. Peters and R. T. Kennedy, Lab Chip, 2005, 5, 56–63 RSC.
- J. S. Mohammed, Y. Wang, T. A. Harvat, J. Oberholzer and D. T. Eddington, Lab Chip, 2009, 9, 97 RSC.
- A. F. Adewola, D. Lee, T. Harvat, J. Mohammed, D. T. Eddington, J. Oberholzer and Y. Wang, Biomed. Microdevices, 2010, 12, 409–417 CrossRef PubMed.
- J. T. Walker, R. Haliyur, H. A. Nelson, M. Ishahak, G. Poffenberger, R. Aramandla, C. Reihsmann, J. R. Luchsinger, D. C. Saunders, P. Wang, A. Garcia-Ocaña, R. Bottino, A. Agarwal, A. C. Powers and M. Brissova, JCI Insight, 2020, 5, e137017 CrossRef PubMed.
- K. S. Sankar, B. J. Green, A. R. Crocker, J. E. Verity, S. M. Altamentova and J. V. Rocheleau, PLoS One, 2011, 6, 1–11 CrossRef PubMed.
- P. N. Silva, B. J. Green, S. M. Altamentova and J. V. Rocheleau, Lab Chip, 2013, 13, 4374–4384 RSC.
- W. H. Tan and S. Takeuchi, Proc. Natl. Acad. Sci. U. S. A., 2007, 104, 1146 CrossRef CAS PubMed.
- L. Y. Wu, D. Di Carlo and L. P. Lee, Biomed. Microdevices, 2008, 10, 197–202 CrossRef CAS PubMed.
- M. Nourmohammadzadeh, J. F. Lo, M. Bochenek, J. E. Mendoza-Elias, Q. Wang, Z. Li, L. Zeng, M. Qi, D. T. Eddington, J. Oberholzer and Y. Wang, Anal. Chem., 2013, 85, 11240 CrossRef CAS PubMed.
- Y. Y. Chen, P. N. Silva, A. M. Syed, S. Sindhwani, J. V. Rocheleau and W. C. W. Chan, Proc. Natl. Acad. Sci. U. S. A., 2016, 113, 14915–14920 CrossRef CAS PubMed.
- A. L. Glieberman, B. D. Pope, J. F. Zimmerman, Q. Liu, J. P. Ferrier, J. H. R. Kenty, A. M. Schrell, N. Mukhitov, K. L. Shores, A. B. Tepole, D. A. Melton, M. G. Roper and K. K. Parker, Lab Chip, 2019, 19, 2993–3010 RSC.
- D. Lee, Y. Wang, J. E. Mendoza-Elias, A. F. Adewola, T. A. Harvat, K. Kinzer, D. Gutierrez, M. Qi, D. T. Eddington and J. Oberholzer, Biomed. Microdevices, 2012, 14, 7–16 CrossRef CAS PubMed.
- S. H. Lee, S. G. Hong, J. Song, B. Cho, E. J. Han, S. Kondapavulur, D. Kim and L. P. Lee, Adv. Healthcare Mater., 2018, 7, 1701111 CrossRef PubMed.
- A. Essaouiba, T. Okitsu, R. Jellali, M. Shinohara, M. Danoy, Y. Tauran, C. Legallais, Y. Sakai and E. Leclerc, Mol. Cell. Endocrinol., 2020, 514, 110892 CrossRef CAS PubMed.
- Y. Jun, A. R. Kang, J. S. Lee, S. J. Park, D. Y. Lee, S. H. Moon and S. H. Lee, Biomaterials, 2014, 35, 4815–4826 CrossRef CAS PubMed.
- A. Tevlek, S. Kecili, O. S. Ozcelik, H. Kulah and H. C. Tekin, ACS Omega, 2022, 8, 3630–3649 CrossRef PubMed.
- C. Quintard, E. Tubbs, J. L. Achard, F. Navarro, X. Gidrol and Y. Fouillet, Biosens. Bioelectron., 2022, 202, 113967 CrossRef CAS PubMed.
- X. Zhang and M. G. Roper, Anal. Chem., 2009, 81, 1162–1168 CrossRef CAS PubMed.
- J. Kim, D. Bilder and T. P. Neufeld, Genes Dev., 2018, 32, 156–164 CrossRef CAS PubMed.
- Y. Jun, J. Lee, S. Choi, J. H. Yang, M. Sander, S. Chung and S.-H. Lee, Sci. Adv., 2019, 5, eaax4520 CrossRef CAS PubMed.
- P. Sokolowska, K. Zukowski, J. Janikiewicz, E. Jastrzebska, A. Dobrzyn and Z. Brzozka, Biosens. Bioelectron., 2021, 183, 113215 CrossRef CAS PubMed.
- S. J. Williams, H. H. Huang, K. Kover, W. Moore, C. Berkland, M. Singh, I. V. Smirnova, R. MacGregor and L. Stehno-Bittel, Organogenesis, 2010, 6, 115 CrossRef PubMed.
- I. Goswami, E. de Klerk, P. Carnese, M. Hebrok and K. E. Healy, Lab Chip, 2022, 22, 4430–4442 RSC.
- J. F. Dishinger and R. T. Kennedy, Anal. Chem., 2006, 79, 947–954 CrossRef PubMed.
- K. R. Reid and R. T. Kennedy, Anal. Chem., 2009, 81, 6837–6842 CrossRef CAS PubMed.
- J. F. Dishinger, K. R. Reid and R. T. Kennedy, Anal. Chem., 2010, 81, 3119–3127 CrossRef PubMed.
- L. Tao and R. T. Kennedy, Anal. Chem., 1996, 68, 3899–3906 CrossRef CAS PubMed.
- K. S. Elvira, F. Gielen, S. S. H. Tsai and A. M. Nightingale, Lab Chip, 2022, 22, 859–875 RSC.
- Q. Chen, J. Li, Y. Song, B. Chen, D. M. Christopher and X. Li, Int. J. Multiphase Flow, 2021, 140, 103648 CrossRef CAS.
- T. S. Kaminski and P. Garstecki, Chem. Soc. Rev., 2017, 46, 6210–6226 RSC.
- C. J. Easley, J. V. Rocheleau, W. S. Head and D. W. Piston, Anal. Chem., 2009, 81, 9086–9095 CrossRef CAS PubMed.
- D. Chen, W. Du, Y. Liu, W. Liu, A. Kuznetsov, F. E. Mendez, L. H. Philipson and R. F. Ismagilov, Proc. Natl. Acad. Sci. U. S. A., 2008, 105, 16843–16848 CrossRef CAS PubMed.
- X. Li, J. C. Brooks, J. Hu, K. I. Ford and C. J. Easley, Lab Chip, 2017, 17, 341–349 RSC.
- X. Li, J. Hu and C. J. Easley, Lab Chip, 2018, 18, 2926–2935 RSC.
- I. R. Sweet, D. L. Cook, R. W. Wiseman, C. J. Greenbaum, A. Lernmark, S. Matsumoto, J. C. Teague and K. A. Krohn, Diabetes Technol. Ther., 2002, 4, 67–76 CrossRef CAS PubMed.
- I. R. Sweet and M. Gilbert, Diabetes, 2006, 55, 3509–3519 CrossRef CAS PubMed.
- M. Gilbert, S. R. Jung, B. J. Reed and I. R. Sweet, J. Biol. Chem., 2008, 283, 24334–24342 CrossRef CAS PubMed.
- S. R. Jung, B. J. Reed and I. R. Sweet, Am. J. Physiol., 2009, 297, 717–727 Search PubMed.
- S. R. Jung, I. T. D. Kuok, D. Couron, N. Rizzo, D. H. Margineantu, D. M. Hockenbery, F. Kim and I. R. Sweet, J. Biol. Chem., 2011, 286, 17422–17434 CrossRef CAS PubMed.
- I. T. Kuok, A. M. Rountree, S. R. Jung and I. R. Sweet, Islets, 2019, 11, 51–64 CrossRef CAS PubMed.
- K. Bentsi-Barnes, M. E. Doyle, D. Abad, F. Kandeel and I. H. Al-Abdullah, Islets, 2011, 3, 284–290 CrossRef PubMed.
- O. Alcazar and P. Buchwald, Front. Endocrinol., 2019, 10, 680 CrossRef PubMed.
- O. Cabrera, M. C. Jacques-Silva, D. M. Berman, A. Fachado, F. Echeverri, R. Poo, A. Khan, N. S. Kenyon, C. Ricordi, P. O. Berggren and A. Caicedo, Cell Transplant., 2008, 16, 1039 Search PubMed.
- I. R. Sweet, G. Khalil, A. R. Wallen, M. Steedman, K. A. Schenkman, J. A. Reems, S. E. Kahn and J. B. Callis, Diabetes Technol. Ther., 2002, 4, 661–672 CrossRef PubMed.
- P. M. Misun, B. Yesildag, F. Forschler, A. Neelakandhan, N. Rousset, A. Biernath, A. Hierlemann and O. Frey, Adv. Biosyst., 2020, 4, 1–14 Search PubMed.
- P. Wu Jin, N. Rousset, A. Hierlemann and P. M. Misun, Front. Bioeng. Biotechnol., 2021, 9, 674431 CrossRef PubMed.
- G. Lenguito, D. Chaimov, J. R. Weitz, R. Rodriguez-Diaz, S. A. K. Rawal, A. Tamayo-Garcia, A. Caicedo, C. L. Stabler, P. Buchwald and A. Agarwal, Lab Chip, 2017, 17, 772–781 RSC.
- A. M. Schrell, N. Mukhitov, L. Yi, J. E. Adablah, J. Menezes and M. G. Roper, Anal. Methods, 2017, 9, 38–45 RSC.
- B. Bandak, L. Yi and M. G. Roper, Lab Chip, 2018, 18, 2873–2882 RSC.
- J. E. Adablah, Y. Wang, M. Donohue and M. G. Roper, Anal. Chem., 2020, 92, 8464–8471 CrossRef CAS PubMed.
- L. Wang, W. Liu, Y. Wang, J. J. C. Wang, Q. Tu, R. Liu and J. J. C. Wang, Lab Chip, 2013, 13, 695–705 RSC.
- Y. Wang, R. Regeenes, M. Memon and J. V. Rocheleau, Cells Rep. Methods, 2023, 3(10), 100602 CrossRef CAS PubMed.
- M. D. Hall, A. Yasgar, T. Peryea, J. C. Braisted, A. Jadhav, A. Simeonov and N. P. Coussens, Methods Appl. Fluoresc., 2016, 4, 022001 CrossRef PubMed.
- M. H. A. Roehrl, J. Y. Wang and G. Wagner, Biochemistry, 2004, 43, 16056–16066 CrossRef CAS PubMed.
- Y. Wang, D. I. Adeoye, Y. J. Wang and M. G. Roper, Anal. Chim. Acta, 2022, 1212, 339942 CrossRef CAS PubMed.
- K. K. Papas, T. M. Suszynski and C. K. Colton, Curr. Opin. Organ Transplant., 2009, 14, 674–682 CrossRef PubMed.
- M. Y. Sun, E. Yoo, B. J. Green, S. M. Altamentova, D. M. Kilkenny and J. V. Rocheleau, Biophys. J., 2012, 103, 2379 CrossRef CAS PubMed.
- A. K. Lam, P. N. Silva, S. M. Altamentova and J. V. Rocheleau, Integr. Biol., 2012, 4, 838–846 CrossRef CAS PubMed.
- A. S. Neal, A. M. Rountree, J. R. Radtke, J. Yin, M. W. Schwartz, C. S. Hampe, J. D. Posner, V. Cirulli and I. R. Sweet, Sci. Rep., 2016, 6, 1–10 CrossRef PubMed.
- T. Schulze, K. Mattern, P. Erfle, D. Brüning, S. Scherneck, A. Dietzel and I. Rustenbeck, Front. Bioeng. Biotechnol., 2021, 9, 615639 CrossRef PubMed.
- N. I. Mourad, M. Nenquin and J. C. Henquin, Am. J. Physiol., 2010, 299, 389–398 CrossRef PubMed.
- J. Xu, N. Wijesekara, R. Regeenes, D. Al Rijjal, A. L. Piro, Y. Song, A. Wu, A. Bhattacharjee, Y. Liu, L. Marzban, J. V. Rocheleau, P. E. Fraser, F. F. Dai, C. Hu and M. B. Wheeler, JCI Insight, 2021, 6, e143037 CrossRef PubMed.
- T. Tao, Y. Wang, W. Chen, Z. Li, W. Su, Y. Guo, P. Deng and J. Qin, Lab Chip, 2019, 19, 948–958 RSC.
- L. Yi, B. Bandak, X. Wang, R. Bertram and M. G. Roper, Anal. Chem., 2016, 88, 10368–10373 CrossRef CAS PubMed.
- Y. Xing, M. Nourmohammadzadeh, J. E. M. Elias, M. Chan, Z. Chen, J. J. McGarrigle, J. Oberholzer and Y. Wang, Biomed. Microdevices, 2016, 18, 1–9 CrossRef CAS PubMed.
- P. C. Hinkle, M. A. Kumar, A. Resetar and D. L. Harris, Biochemistry, 1991, 30, 3576–3582 CrossRef CAS PubMed.
- I. G. Darvey, Biochem. Educ., 1998, 26, 22–23 CrossRef CAS.
- S. K. Jung, L. M. Kauri, W. J. Qian and R. T. Kennedy, J. Biol. Chem., 2000, 275, 6642–6650 CrossRef CAS PubMed.
- K. K. Papas, C. K. Colton, R. A. Nelson, P. R. Rozak, E. S. Avgoustiniatos, W. E. Scott, G. M. Wildey, A. Pisania, G. C. Weir and B. J. Hering, Am. J. Transplant., 2007, 7, 707 CrossRef CAS PubMed.
- J. P. Kitzmann, D. O'Gorman, T. Kin, A. C. Gruessner, P. Senior, S. Imes, R. W. Gruessner, A. M. J. Shapiro and K. K. Papas, Transplant. Proc., 2014, 46, 1985 CrossRef CAS PubMed.
- K. K. Papas, M. D. Bellin, D. E. R. Sutherland, T. M. Suszynski, J. P. Kitzmann, E. S. Avgoustiniatos, A. C. Gruessner, K. R. Mueller, G. J. Beilman, A. N. Balamurugan, G. Loganathan, C. K. Colton, M. Koulmanda, G. C. Weir, J. J. Wilhelm, D. Qian, J. C. Niland and B. J. Hering, PLoS One, 2015, 10, e0134428 CrossRef PubMed.
- I. Chareyron, S. Christen, S. Moco, A. Valsesia, S. Lassueur, L. Dayon, C. B. Wollheim, J. Santo Domingo and A. Wiederkehr, Diabetologia, 2020, 63, 2628–2640 CrossRef CAS PubMed.
- W. Chen, M. Lisowski, G. Khalil, I. R. Sweet and A. Q. Shen, PLoS One, 2012, 7, 1–10 Search PubMed.
- T. Schulze, K. Mattern, E. Früh, L. Hecht, I. Rustenbeck and A. Dietzel, Biomed. Microdevices, 2017, 19, 1–11 CrossRef CAS PubMed.
- S. M. Grist, L. Chrostowski and K. C. Cheung, Sensors, 2010, 10, 9286–9316 CrossRef CAS PubMed.
- A. Zbinden, M. Urbanczyk, S. L. Layland, L. Becker, J. Marzi, M. Bosch, P. Loskill, G. P. Duffy and K. Schenke-Layland, Tissue Eng., Part A, 2021, 27, 977–991 CrossRef CAS PubMed.
- L. M. Weber, K. N. Hayda and K. S. Anseth, Tissue Eng., Part A, 2008, 14, 1959–1968 CrossRef CAS PubMed.
- N. Rafiei, M. G. Moghadam, A. Au, R. Regeenes, S. Chidambaram, T. Liang, Y. Wang, C. M. Yip, H. Gaisano and J. V. Rocheleau, Biofabrication, 2022, 14, 041001 CrossRef PubMed.
- A. B. Di Stefano, V. Urrata, M. Trapani, F. Moschella, A. Cordova and F. Toia, J. Cell. Physiol., 2022, 237, 4397 CrossRef CAS PubMed.
- A. A. Banaeiyan, J. Theobald, J. Paukštyte, S. Wölfl, C. B. Adiels and M. Goksör, Biofabrication, 2017, 9, 015014 CrossRef PubMed.
- R. K. P. Benninger, C. Dorrell, D. J. Hodson and G. A. Rutter, Curr. Diabetes Rep., 2018, 18, 112 CrossRef PubMed.
- M. A. Miranda, J. F. Macias-Velasco and H. A. Lawson, Am. J. Physiol., 2021, 320, E716–E731 CAS.
- G. Da Silva Xavier and G. A. Rutter, J. Mol. Biol., 2020, 432, 1395–1406 CrossRef CAS PubMed.
- D. Avrahami, A. Klochendler, Y. Dor and B. Glaser, Diabetologia, 2017, 60, 1363–1369 CrossRef CAS PubMed.
- D. G. Pipeleers, Diabetes, 1992, 41, 777–781 CrossRef CAS PubMed.
- D. Pipeleers, R. Kiekens, Z. Ling, A. Wilikens and F. Schuit, Diabetologia, 1994, 37, S57–64 CrossRef PubMed.
- C. Dorrell, J. Schug, P. S. Canaday, H. A. Russ, B. D. Tarlow, M. T. Grompe, T. Horton, M. Hebrok, P. R. Streeter, K. H. Kaestner and M. Grompe, Nat. Commun., 2016, 7, 1–9 Search PubMed.
| This journal is © The Royal Society of Chemistry 2024 |