
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicencePhotothermal catalytic oxidation of toluene over the Pt–Mn2O3/CN nanocomposite catalyst†
Xiao
Yu‡
abc,
Chuang
Zhao‡
c,
Lixia
Yang
 *c,
Jian
Zhang
*ab and
Chunlin
Chen
*ab
*c,
Jian
Zhang
*ab and
Chunlin
Chen
*ab
aNingbo Institute of Materials Technology & Engineering, Chinese Academy of Sciences, 1219 Zhongguan West Road, Ningbo 315201, Zhejiang, China
bUniversity of Chinese Academy of Sciences, Beijing 100049, China
cKey Laboratory of Jiangxi Province for Persistent Pollutants Control and Resources Recycle, Nanchang Hangkong University, Nanchang 330063, Jiangxi Province, China
First published on 20th January 2024
Abstract
The Pt–Mn2O3/CN catalyst formed through synthesis via a solvent-thermal method involves a synergistic combination of polymer CN and Pt nanoparticles loaded on Mn2O3 to catalyze the degradation of toluene. The composition incorporates Mn2O3 as the central element for photothermal conversion, CN as a uniformly dispersed matrix for Pt nanoparticles, and Pt as the catalytically active center, demonstrating significant efficacy. Particularly noteworthy is the discernible enhancement in the photothermal catalytic degradation capability of the Pt–Mn2O3/CN composite catalyst, specifically in the context of toluene. When subjected to light intensity of 300 mW cm−2 and a toluene concentration of 400 ppm, Pt–Mn2O3/CN achieves toluene conversion and CO2 mineralization rates of 99% and 80.9%, respectively. This improvement primarily stems from the Pt nanoparticles inducing a substantial presence of oxygen vacancies within the catalyst structure, thereby increasing the oxygen adsorption capacity and surface mobility. This, in turn, activates adsorbed oxygen species at the catalyst's interface. The adept utilization and conversion of solar irradiance for volatile organic compound (VOC) abatement underscore its potential as an environmentally friendly and renewable energy source.
Broader contextIn recent years, significant strides have been made in advancing photothermal catalysis – an innovative technology harnessing solar energy to enhance catalytic efficiency through synergistic photothermal effects. Toluene, classified as a volatile organic compound (VOC), poses a threat to air quality, potentially contributing to ground-level ozone and smog formation when released into the atmosphere. Exploring renewable solar power via photothermal catalysis emerges as a promising avenue to combat VOC-related environmental concerns. In our investigation, we synthesized a nanocomposite catalyst by combining polymer CN with Pt nanoparticles strategically loaded onto Mn2O3. This catalyst demonstrated remarkable efficiency in the photothermal catalytic oxidation of toluene, credited to the Pt nanoparticles inducing numerous oxygen vacancies within the catalyst structure. These vacancies notably amplified the oxygen adsorption capacity, enhanced surface fluidity, and activated adsorbed oxygen on the catalyst's surface. Our research holds the promise of significant environmental advantages, highlighting the potential of utilizing sunlight as a clean and sustainable energy source for degrading VOCs. |
Introduction
Toluene (C7H8) is commonly known as methylbenzene. Inhalation of toluene presents health risks, with prolonged exposure to its vapors causing symptoms such as headaches, dizziness, and in severe instances, damage to the central nervous system.1 Categorized as a volatile organic compound (VOC), toluene contributes to air pollution, potentially leading to the formation of ground-level ozone and smog when released into the atmosphere.2 In recent developments, photothermal catalysis has emerged as an advanced and promising technology for the removal of VOCs from the air.3 This innovative approach combines light-induced heating with catalytic processes to either eliminate or transform VOCs. Numerous studies have highlighted the superior catalytic activity of photothermal catalysis in VOC degradation. This efficacy is attributed to the synergistic interaction between photocatalysis and thermal catalysis.4 For example, research conducted by Jia's team showcased the potential of Pt–rGO–TiO2 as a highly active photothermal catalyst for VOC decomposition. This catalyst demonstrated broad light wavelength absorption and solar-thermal synergy effect.5 Additionally, they synthesized Pt-mTiO2/USY composites for the catalytic degradation of toluene under the full solar spectrum. The mTiO2/USY with increasing Pt content exhibited progressively enhanced catalytic activity, indicating the promising nature of these materials in VOC removal applications.6Catalysts based on manganese oxide are widely employed in environmental catalysis due to their affordability, abundant sources, and variable valence states.7,8 However, manganese oxide has a relatively wide band gap, which may limit its efficiency in utilizing sunlight for photothermal catalysis. To enhance catalysts’ reactivity, researchers have confirmed that heteroatom loading, which regulates the electronic structure and improves light absorption, is an effective strategy.9,10 Moreover, the lattice defects of metal oxides can be altered by loading heteroatoms, facilitating the generation of more reactive oxygen species and thereby improving catalytic activity.11 Some research also found that the dispersion of metal ions and the augmentation of the active sites are enhanced when metal oxides or noble metals are supported on flat materials.12 Polymeric carbon nitrides (CN), composed of triazine or heptazine units as building blocks, present a promising class of conjugated polymeric photocatalyst for exploring this possibility.13 Similar to graphite, CN monolayers are stacked with van der Waals force interactions, rendering them insoluble in most solvents. CN has emerged as an exceptionally attractive photocatalyst driven by visible light, distinguishing it from TiO2 materials that are responsive only to UV light (representing just 5% of the solar spectrum). With a suitable forbidden bandwidth (Eg = 2.7–2.8 eV), CN exhibits superior absorption of visible light compared to most metal oxides.14 It is particularly well-suited for the photocatalytic decomposition of water to generate hydrogen and oxygen.
Wang et al. initially reported on CN's visible light photocatalytic water decomposition for hydrogen production, marking a shift in photocatalyst research from inorganic semiconductors to diverse organic polymers.15 In a study by Zhang et al.,16 it was demonstrated that carbon nitride, characterized by excellent light-response capabilities and a layered structure, serves to prevent nanoparticle aggregation and resist chemical corrosion from acids and organic solvents. Additionally, CN can be synergistically combined with metal oxides to modulate the electronic structure of the composite catalyst. The nitrogen atoms in CN play a crucial role by providing lone pair electrons as ligands to the transition metal, contributing to the overall regulation of the catalyst's electronic structure.17 Moreover, metal nanoparticles demonstrate outstanding activity in photo-driven photothermal catalysis, often credited to the presence of hot electrons generated by the robust surface plasmon resonance (SPR) effect. Professor Zhang's research team delved into the innovative photothermal Fischer–Tropsch synthesis employing the RuCo-SAA alloy catalyst, affirming a promising approach for converting carbon monoxide into high-value chemicals.18 Additionally, their findings highlighted the beneficial role of plasmonic Cu nanoparticles in enhancing the low-temperature photo-driven water–gas shift reaction.19
Herein, we synthesized a Pt–Mn2O3/CN nanocomposite catalyst via loading the Pt and combining CN into Mn2O3 by a solvent-thermal method. This investigation focuses on utilizing Pt nanoparticles (NPs) as active sites, Mn2O3 as the center for photothermal conversion, and CN as a dispersed material to facilitate the even distribution of Pt nanoparticles. The multi-catalytic system is employed to investigate the mechanism behind the enhanced photothermal catalytic degradation of toluene by doping Pt NPs. The findings reveal that the Pt–Mn2O3/CN catalyst harnesses solar energy to activate active oxygen species on the surface of oxygen vacancies, enhancing the catalyst's stability through the robust interaction between CN and Mn2O3. This discovery holds significant potential for widespread applications in the photothermal catalytic degradation of VOCs.
Results and discussion
Structural information of the catalysts
As depicted in the XRD spectra of various catalysts presented in Fig. 1A, these catalyst formulations exhibit nearly identical diffraction patterns to the pristine Mn2O3. The distinct peaks at 66.2°, 55.3°, 49.5°, 45.5°, 38.8°, 33.1°, and 23.3° correspond to the crystallographic planes of (622), (440), (431), (332), (400), (222), and (211), respectively, all indexed to Mn2O3.10 Notably, the Pt–Mn2O3/CN catalyst shows no discernible diffraction peaks associated with the CN structure or Pt nanoparticles. This observation suggests that the catalyst predominantly consists of Mn2O3, a phenomenon likely attributable to the low Pt loading or the effective dispersion of small-size Pt NPs. However, an intriguing phenomenon is observed in the enlarged diffraction peaks at 33.1°, as illustrated in Fig. 1A (left), corresponding to the (222) crystal in Mn2O3 undergoing a negative shift. This implies an expansion of the lattice spacings along the c direction, indicating the incorporation of a fraction of CN and Pt NPs into the Mn2O3 lattice during catalyst synthesis. In this context, the thermal oxidation process's heating treatment is presumed to promote a more compact packing of CN or Pt NPs, thereby leading to an expansion of the lattice distance.20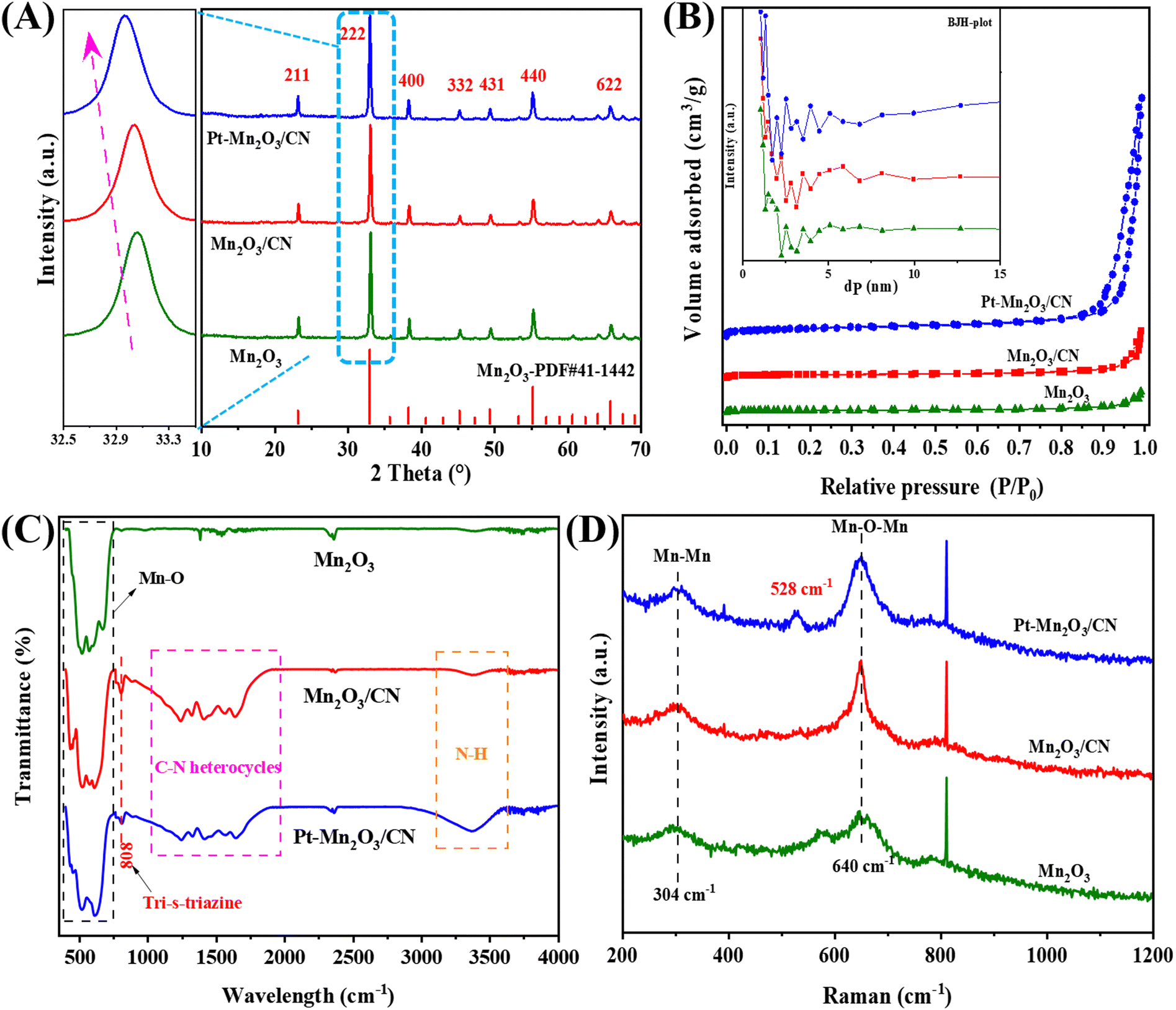 | ||
| Fig. 1 (A) XRD patterns, (B) N2 adsorption–desorption isotherm, (C) FTIR spectra, and (D) Raman spectra of different catalysts. | ||
The examination delved into the N2 adsorption–desorption isotherm and the distribution of pore sizes among diverse catalyst samples. As depicted in Fig. 1B, only Pt–Mn2O3/CN and Mn2O3/CN samples exhibit typical type IV isotherms characterized by the H3 hysteresis ring,21 signifying the existence of a mesoporous structure owing to the layering effects within CN. From the BET and BJH models, values for specific surface area, pore volume, and pore size were computed, as outlined in Table 1. As the CN content in the catalyst increased, the SBET surged from 11.01 to 17.68 m2 g−1, and notably, the SBET of Pt nanoparticles in Pt–Mn2O3/CN further escalated to 31.94 m2 g−1. This amplification can be primarily attributed to the incorporation of Mn2O3 species into the mid-layer of CN during the solvent heating process, thus fostering the creation of more medium-sized pores. Additionally, the appropriate distribution of Pt nanoparticles either evenly dispersed on the CN surface or bonded with Mn2O3 and CN further facilitates the formation of a mesoporous structure.17 The past investigation underscores that a larger specific surface area in the catalyst positively influences both the adsorption in catalytic reactions and the resultant catalytic activity.22 The pore size distribution across all samples exhibited mesoporous characteristics, showcasing variations corresponding to the varying CN and Pt content. Notably, in Pt–Mn2O3/CN, the pore volume experienced a considerable reduction to 0.231 cm3 g−1. CN possesses a π-conjugated aromatic system, a result of the sp2 hybridization involving carbon and nitrogen atoms. This configuration allows CN to adsorb and activate reactant molecules sharing a similar conjugate aromatic structure, primarily through π–π interconnection.23 Additionally, in π-delocalized electronic systems, significant delocalized p electrons become integrated with the lone pair electrons on loaded Pt atoms, thereby activating and fostering the formation of π–π interactions.24 The introduction of an appropriate quantity of Mn2O3 facilitates the opening of the interlayer structure, leading to increased adsorption of toluene molecules. Consequently, Pt–Mn2O3/CN, characterized by a larger specific surface area (SBET) and an increased π–π interaction proportionate to CN content, exhibits enhanced toluene adsorption capacity. This enhancement proves advantageous in augmenting the number of activated toluene molecules.
| Catalysts | BET surface (m2 g−1) | Total pore volume (cm3 g−1) | Average pore diameter (nm) | Pt theoretical loading/(wt%) | ICP-Pt (wt%) | Mn4+/Mn3+ |
|---|---|---|---|---|---|---|
| Mn2O3 | 11.01 | 0.267 | 41.21 | — | 0.49 | |
| Mn2O3/CN | 17.68 | 0.282 | 51.51 | — | 0.51 | |
| Pt–Mn2O3/CN | 31.94 | 0.231 | 28.97 | 0.4 | 0.3 | 0.58 |
The FTIR spectra of the catalysts produced are presented in Fig. 1C. The infrared signal at 808 cm−1 corresponds to the distinctive breathing mode associated with triazine rings in CN.25 Peaks within the range of 1000–1950 cm−1 signify stretching vibration modes of C–N and C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N heterocycles, whereas those at 3000–3600 cm−1 indicate the stretching vibration of N–H. This is attributed to either insufficient condensed amino groups or remaining hydrogen atoms attached to the edges of graphite-like CN.16 The peaks observed at 614 cm−1 and 514 cm−1 result from the asymmetric bending of Mn–O and the stretching vibration of O–Mn–O.26 Beyond the analysis of the catalyst phase, Raman spectroscopy was employed to investigate the vibrational patterns of the catalyst's surface species. In Fig. 1D, the Raman peaks near 304 cm−1 and 640 cm−1 correspond to Mn–Mn and the Eg symmetric tensile vibration mode of Mn–O–Mn, respectively, primarily associated with the Jahn–Teller (J–T) distortion.27 In these samples, the heightened peak intensity at 640 cm−1 serves as an indicator of increased J–T distortion. Notably, Pt–Mn2O3/CN exhibits the highest peak intensity, signifying the most pronounced ordering of the Eg electron orbit.28 Raman spectra of Pt–Mn2O3/CN at 528 cm−1 are ascribed to the three Raman-active vibrations of δ (N–Pt–N), νs(Pt–N), and νas(Pt–N), respectively.29
N heterocycles, whereas those at 3000–3600 cm−1 indicate the stretching vibration of N–H. This is attributed to either insufficient condensed amino groups or remaining hydrogen atoms attached to the edges of graphite-like CN.16 The peaks observed at 614 cm−1 and 514 cm−1 result from the asymmetric bending of Mn–O and the stretching vibration of O–Mn–O.26 Beyond the analysis of the catalyst phase, Raman spectroscopy was employed to investigate the vibrational patterns of the catalyst's surface species. In Fig. 1D, the Raman peaks near 304 cm−1 and 640 cm−1 correspond to Mn–Mn and the Eg symmetric tensile vibration mode of Mn–O–Mn, respectively, primarily associated with the Jahn–Teller (J–T) distortion.27 In these samples, the heightened peak intensity at 640 cm−1 serves as an indicator of increased J–T distortion. Notably, Pt–Mn2O3/CN exhibits the highest peak intensity, signifying the most pronounced ordering of the Eg electron orbit.28 Raman spectra of Pt–Mn2O3/CN at 528 cm−1 are ascribed to the three Raman-active vibrations of δ (N–Pt–N), νs(Pt–N), and νas(Pt–N), respectively.29
To further elucidate the structure of Pt NPs, Mn2O3, and CN within Pt–Mn2O3/CN, we conducted a detailed microscopic analysis using transmission electron microscopy (TEM), displaying the findings in Fig. 2 and Fig. S3 (ESI†). Throughout the high-temperature and high-pressure treatment, the catalyst retained the layered configuration of CN, exhibiting a consistent flat and stacked sheet structure. Within this framework, Mn2O3 was found embedded, with bright white minuscule spots in Fig. 2A and G, indicating Pt NPs evenly distributed atop Mn2O3, bound to CN. The size distribution analysis of the Pt nanoparticles depicted in Fig. 2C revealed a range from 3.84 nm to 11.33 nm, with an average size of 6.37 nm. High-resolution TEM images in Fig. 2D–F revealed spherical bright spots corresponding to Pt NPs situated on the surface of Mn2O3. However, these images only exhibited lattice patterns specific to Mn2O3, with indexed lattice spacings of 0.38 nm for (211) planes and 0.19 nm for (422) planes in Mn2O3. Further examination via HRTEM (Fig. S4, ESI†) unveiled ultrafine nanoparticles adhered to CN, measuring approximately 6.0 nm in size. Enlarged sections in Fig. S4 (ESI†) delineated lattice fringe spacings of these ultrafine nanoparticles at approximately 0.22 nm, corresponding to the d-spacing observed in the (111) facets of cubic Pt.30
 | ||
| Fig. 2 (A) and (B) TEM images at different magnifications, (C) size distribution of the Pt NPs, (D) and (E) HRTEM images, (F) the shots of the selected area in the panel, (G)–(L) EDS phase mapping of the Pt (blue), O (yellow), Mn (red), N (navy), and C (green), distributions images of the Pt–Mn2O3/CN catalyst. | ||
Additionally, the elemental distribution was visualized through energy-dispersive X-ray (EDX) spectroscopy, revealing separate dispersion of Mn and Pt on CN, and some Pt dispersed on Mn2O3 particles. The actual Pt loading in Pt–Mn2O3/CN was observed to be 1% lower than the theoretical loading (refer to Fig. S5, ESI,† and Table 1). This discrepancy can be attributed to the loss of some Pt in the ethanol solution during the solvent-thermal reaction. The bonding of Pt to metal oxides typically induces the separation and migration of photo-induced carriers, thereby enhancing the photocatalytic activity. Furthermore, the interaction between Pt and metal oxides serves to augment the presence of reactive oxygen species on the catalyst, facilitating catalytic reactions. Simultaneously, the uniform distribution of Pt nanoparticles on the surface of Mn2O3 bound to CN proves advantageous in capturing and utilizing photons across a broader wavelength spectrum. In the solvent-thermal synthesis process, we also synthesized the Pt–Mn2O3 catalyst. However, TEM analysis revealed that in the absence of CN as the dispersion carrier, Pt nanoparticles tend to agglomerate, as highlighted in the red box in Fig. S6 (ESI†). These stacked and agglomerated particles are detrimental to the overall efficiency of the catalyst.
Catalytic performance
The catalyst we prepared underwent testing for its photothermal catalytic oxidation activity under the entire solar spectrum with an illumination intensity of 300 mW cm−2. In the static reaction conditions depicted in Fig. 3A and B, the photothermal removal percentage of toluene exhibited a specific order across different catalysts: Pt–Mn2O3/CN (99%) > Mn2O3/CN (53%) > Mn2O3 (45.5%) > Pt–CN (33.7%). Particularly noteworthy is the CO2 yield achieved by Pt–Mn2O3/CN, reaching an impressive 80.9%, significantly surpassing Mn2O3/CN (11.2%), Pt–CN (10.4%) and Mn2O3 (6.5%). The introduction of Pt through doping notably enhances the overall catalytic efficiency in detoxifying toluene, converting it into CO2 and H2O. This enhancement is attributed to the improved dispersion of Pt nanoparticles and the increased specific surface area of the catalyst facilitated by CN-supporting materials. The underwhelming performance of Pt–CN underscores the pivotal role of Mn2O3 in catalytic conversion. To delve into the impact of Pt loading on catalytic efficacy, various catalysts with distinct Pt loadings were scrutinized, as illustrated in Fig. S7 (ESI†). The introduction of Pt markedly enhanced the catalytic performance; however, a subsequent increase in content to 1% led to a slight reduction in performance. This diminution could be ascribed to the potential agglomeration of surface Pt NPs.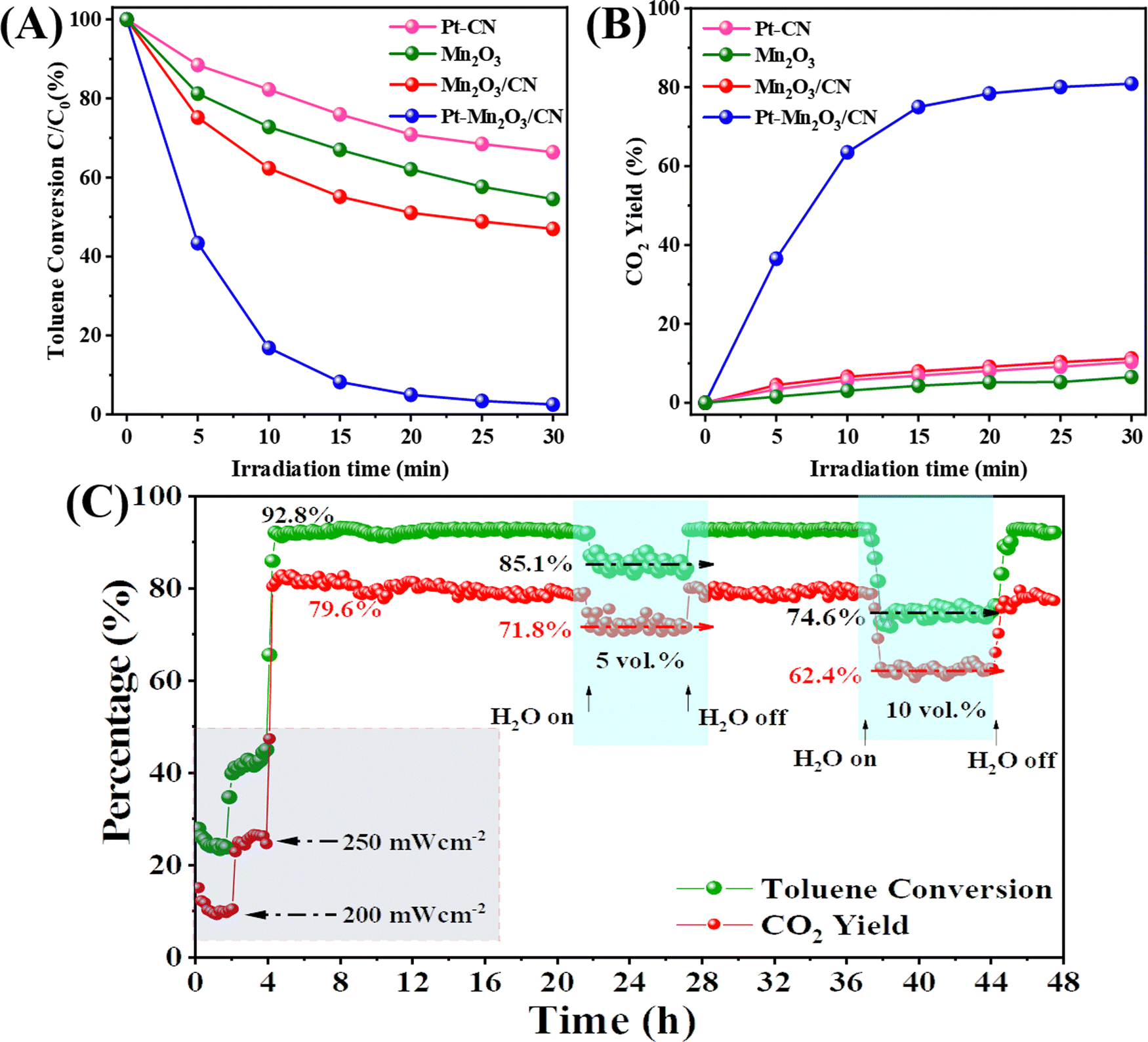 | ||
Fig. 3 Photothermal catalytic oxidation of toluene, (A) toluene conversion, and (B) CO2 yield during the static reaction with 400 ppm toluene over the different catalysts, (C) toluene conversion and CO2 yield during the continuous flow reaction over the Pt–Mn2O3/CN catalysts (full solar spectrum irradiation with 300 mW cm−2 light intensity, 200 ppm toluene, and WHSV = 30![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 mL g−1 h−1). 000 mL g−1 h−1). | ||
To assess the enduring catalytic performance and practicality of Pt–Mn2O3/CN, we employed a custom-made stainless steel fixed bed continuous flow reactor for testing its photothermal catalytic activity against toluene. The reaction conditions comprised 200 ppm toluene, 20 vol% O2, and N2. The catalyst's weight hourly space velocity (WHSV) in the reactor was controlled at 30000 mL g−1 h−1, and we investigated the impact of H2O and different light intensities on its stability. As depicted in Fig. 3C, once the reaction stabilized, the catalyst was not sufficiently activated at 200 and 250 mW cm−2 for 4 hours while the toluene conversion remained below 50%. When the light intensity was increased to 300 mW cm−2, the toluene conversion reached 92.8% and remained stable with a consistent CO2 yield of approximately 79.6%. Remarkably, even after 36 hours, the catalytic efficiency remained robust. Introducing 5 and 10 vol% H2O into the reaction system led to a decline in catalytic performance, with the toluene conversion dropping to 85.1% and 74.6% while the CO2 yield decreased to 71.8% and 62.4%. Typically, H2O and CO2 are byproducts of toluene oxidation, and hence, considering the reaction equilibrium, the introduction of water adversely affects the process.31 Noteworthy is the swift recovery of toluene conversion and CO2 yield upon the removal of water vapor, underscoring the excellent catalytic resilience of Pt–Mn2O3/CN to moisture. This underscores the catalyst's enduring performance under conditions simulating real exhaust scenarios.
Surface properties of the catalysts
The X-ray photoelectron spectroscopy (XPS) analysis of the C 1s spectra (Fig. 4A) reveals three distinct peaks at 284.8 eV, 285.8 eV, and 288.3 eV, corresponding to C–C bonds, C–NH2 species, and carbon in N–CN of the carbon nitride (CN), respectively.16 Importantly, the peaks at 285.8 eV and 288.3 eV exhibit a shift towards higher binding energy compared to the C of CN, suggesting the presence of a chemical interaction between CN and Pt–Mn2O3. To further elucidate the origin of Pt and CN electron transfer, the Mn 2p spectra were analyzed (Fig. 4B). Given the various valence states of Mn on the catalyst surface, we focused on the three most common states (Mn2+: ∼642.0 eV, Mn3+: ∼642.6 eV, and Mn4+: ∼644.1 eV).32 Evidently, the surface Mn on all catalysts predominantly exists in the Mn3+ form (Mn3+ content >40%), aligning with the XRD results indicating the presence of Mn2O3. In Table 1, the incorporation of Pt nanoparticles led to an improvement in the surface Mn4+/Mn3+ molar ratio, increasing from 0.49 to 0.58, signifying an enhanced concentration of surface Mn4+. In simpler terms, loading Pt NPs resulted in an augmented surface oxygen vacancy concentration in Mn2O3/CN. Furthermore, compared to the pure Mn2O3 sample, all Pt–Mn2O3/CN catalysts displayed a higher binding energy of the Mn 2p3/2 peak, confirming the electron transfer from CN and Mn to Pt. Consequently, the XPS spectra of C 1s and Mn 2p affirm that the electronic coupling effect at the Pt–Mn2O3/CN interfacial active sites is more pronounced than that at the active sites of Mn2O3/CN.33 This suggests that intercalating Mn2O3 and Pt nanoparticles into the lamellar CN induces structural defects, adjusts the electronic structure, and facilitates the generation of redox electric pairs of Mn4+/Mn3+.
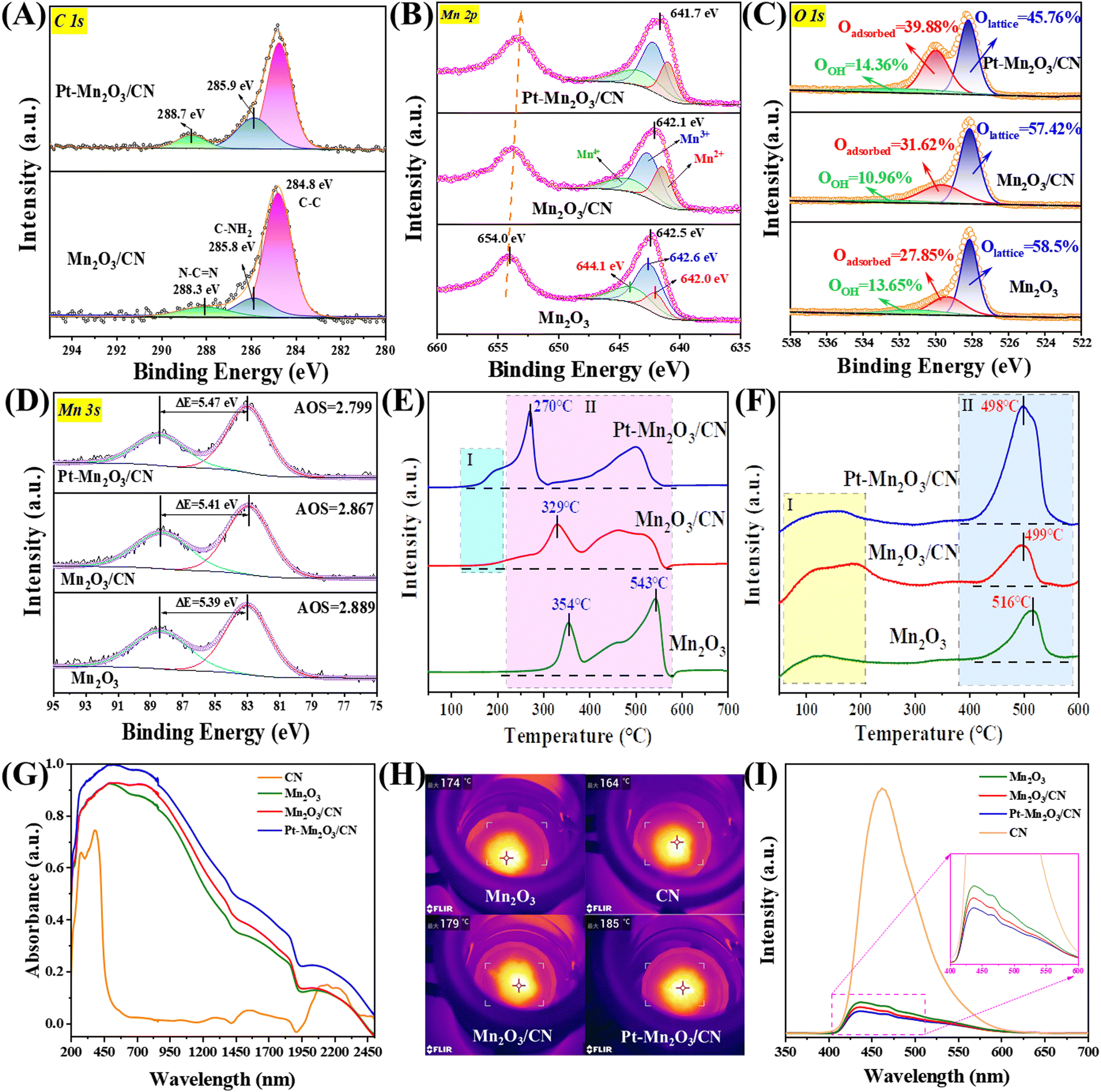 | ||
| Fig. 4 XPS survey spectra of (A) C 1s, (B) Mn 2p, (C) O 1s, and (D) Mn 3s, (E) H2-TPR, (F) O2-TPD, (G) UV-Vis-IR absorption spectrum, (H) photothermal conversion temperatures and (I) photoluminescence spectra over different catalysts. | ||
Observing the data, it becomes apparent that samples exhibiting superior activity demonstrated a higher Mn4+/Mn3+ molar ratio. The average valence state of Mn in Mn2O3 is +3. When an oxygen defect occurs, the Mn2+ content and Mn4+ content must change to maintain the overall electron balance.3,33,34 Following the incorporation of Pt NPs, a noticeable alteration in the valence distribution of various carriers occurred, aligning with the results of the activity assessment. This change followed the trend of high Mn4+ content correlating with excellent activity observed prior to loading. The outcomes of the peak separation analysis of O 1s orbitals, depicted in Fig. 4C, reveal that the asymmetrical O 1s spectrum of each sample can be deconstructed into three components at binding energies of 528.1, 529.4, and 531.4 eV. These correspond to surface lattice oxygen (Olattice), adsorbed oxygen (Oadsorbed), and adsorbed water or carbonate species (OOH).35 Notably, in Table 1, the Pt–Mn2O3/CN sample exhibited the highest Oadsorbed concentration (39.88%) among all samples, once again validating that a heightened Oadsorbed concentration promotes toluene combustion. It is observed that carriers with higher Mn4+ content also displayed a higher oxygen content on their surface, consistent with the peak separation results of Mn 2p orbitals. Within this context, Oadsorbed species play a pivotal role in catalytic degradation. Their effectiveness hinges on the presence of oxygen vacancies (Ovs) implanted in the catalysts. In essence, an increased number of oxygen vacancies translates to more surface-adsorbed active oxygen, ultimately leading to higher final degradation efficiency.
Moreover, the oxygen vacancy concentration can be represented by the average oxidation state (AOS) of Mn 3s in the Mn2O3 samples. The principle here is that a lower AOS value corresponds to a higher oxygen vacancy concentration.36 This calculation is derived from the multiplet splitting of Mn 3s orbitals, and it follows the formula outlined below:
| AOS = 8.98–1.13ΔEs (eV) |
The ΔEs, denoting the disparity in binding energy between the primary peak and its associated satellite arising from the multiplet splitting within Mn 3s orbitals, play a crucial role. The determined ΔEs values for Mn2O3, Mn2O3/CN, and Pt–Mn2O3/CN stand at 5.39, 5.41, and 5.47 eV, respectively (refer to Fig. 4D). Notably, the Pt–Mn2O3/CN catalyst exhibits the lowest Average Oxidation State (AOS) among all counterparts, registering at 2.78, indicative of its heightened Ovs content, which is in line with EPR analysis (Fig. S8, ESI†). The escalation in oxygen vacancy concentration aligns with enhanced oxygen migration capability.37 Within this spectrum, Pt–Mn2O3/CN manifests a substantial increase in oxygen vacancies, primarily attributed to the pronounced influence of strong metal–support interactions (SMSI) on the Mn–O bond within the carrier. This phenomenon results in the formation of additional oxygen vacancies, emerging as the principal factor contributing to the marked enhancement in its catalytic activity.34
The reduction performance of the catalyst was explored through H2 temperature-programmed reduction (H2-TPR). In Fig. 4E, the reduction spectrum of manganese oxides with diverse morphologies is depicted. The peaks around 354 °C signify the reduction from Mn4+ to Mn3+, while those at approximately 543 °C indicate the reduction from Mn3+ to Mn2+. The remaining high-temperature peaks are attributed to the partial reduction of the surface phase.38 Typically, the temperature at which Mn2O3 transforms from Mn4+ to Mn3+ is employed to assess the reduction capacity of Mn2O3.39 It is evident from the analysis that the Pt-loaded sample exhibits superior low-temperature reducibility. Region I represents the reduction absorption peak of Pt–Mn2O3/CN and surface-adsorbed oxygen, while Region II corresponds to the Mn2O3 reduction absorption peak. Despite the extremely low Pt loading, even if all Pt oxides exist in the form of PtO2, it remains significantly lower than the hydrogen consumption in Region I. Consequently, the absorption peak in Region I is predominantly ascribed to the hydrogen overflow effect induced by strong metal–support interaction (SMSI). This prompts the Mn2O3/CN surface oxygen species near Pt NPs to react with H2.34 The absorption peak in Region II reveals that the Mn4+ → Mn3+ reduction peak intensity of the Pt–Mn2O3/CN sample is the highest, with the lowest reduced temperature. This suggests that Pt–Mn2O3/CN exhibits superior low-temperature oxidizability, heightened surface oxygen mobility, an abundance of reactive oxygen species, and a greater ability to accept electrons compared to Mn2O3.
Oxygen temperature-programmed desorption (O2-TPD) analysis was conducted to discern the oxygen species within Mn2O3 and investigate potential factors influencing its oxidation properties. The results of the analysis are depicted in Fig. 4F. The desorption peak in the range of 50–200 °C was ascribed to the release of surface-adsorbed oxygen (peroxide species, O2−), while the peak at 400–600 °C was associated with the desorption of surface lattice oxygen (monatomic species, O−). Furthermore, the desorption peak exceeding 600 °C was attributed to the release of bulk lattice oxygen (O2− species).10 Considerable attention has been directed towards the first two peaks, as lattice oxygen species (O2− species, >550 °C) tend to become inert during toluene oxidation at low temperatures.40 The diagram illustrates that the order of surface active lattice oxygen content among the three samples is Pt–Mn2O3/CN > Mn2O3/CN > Mn2O3. Notably, the content of surface lattice oxygen (monatomic species, O−) has a pronounced positive impact on high efficiency and low-temperature oxidation. This observation is attributed to the catalyst's surface lattice oxygen being the primary substance adsorbed by the activated carrier during low-temperature oxidation. The migration of oxygen to replenish the consumed surface lattice oxygen after oxygen vacancy activation can further mineralize the activated toluene, indicating superior oxygen mobility.
Photothermal catalysis involves the utilization of light energy to transform it into heat energy during a chemical reaction. This process enhances the efficiency and speed of catalytic reactions by absorbing and converting light energy into heat.41 The initial step in photothermal catalysis is light absorption, a crucial determinant of how much light energy the catalyst absorbs. Catalysts that can absorb light across a broader range of wavelengths generally exhibit superior catalytic performance because they can harness a more extensive portion of the solar energy spectrum. The Pt–Mn2O3/CN sample, as depicted in Fig. 4G, displays a notable photoresponse in the ultraviolet to visible and near-infrared (IR) spectral regions (up to 2500 nm), confirming its exceptional solar absorption capacity. Comparing the absorption spectra intensities of different samples, the order is Pt–Mn2O3/CN > Mn2O3/CN > Mn2O3 > CN. Furthermore, the photothermal conversion temperature is critical, as it denotes the temperature at which the heat energy generated by light absorption elevates the temperature of both the catalyst and the reactants. The increase in temperature can accelerate the reaction rate, improve the selectivity of the reaction, or enable the reaction to take place under milder conditions. Infrared thermography was employed to assess the equilibrium temperatures of various catalysts under light irradiation, revealing that the Pt–Mn2O3/CN sample exhibited the best catalytic performance with the highest surface temperature at 185 °C (Fig. 4H). Kirchhoff's law posits that the infrared (IR) emissivity is equal to the IR absorptivity of a material in thermodynamic equilibrium. Consequently, the Pt–Mn2O3/CN's IR absorption results in robust IR radiation, leading to intense thermal radiation diffusion.42 Moreover, Pt nanoparticles uniformly dispersed on the catalyst surface produce a plasmonic resonance effect under light, which enhances the light absorption and photothermal conversion efficiency.43
Under 280 nm excitation light, the singular Mn2O3 demonstrates a pronounced photoluminescence (PL) intensity attributable to substantial photogenerated charge recombination. In contrast, the Pt–Mn2O3/CN composite exhibits a markedly diminished PL intensity (Fig. 4I), suggesting the timely migration of photogenerated electrons from Mn2O3 to Pt nanoparticles, effectively suppressing charge recombination. Likewise, Mn2O3/CN displays a reduction in fluorescence intensity, albeit less pronounced than the Pt–Mn2O3/CN catalyst scenario. This disparity may stem from the lower Mn2O3 decoration content on CN, making the migration of photogenerated holes comparatively more challenging than that of electrons. These outcomes underscore the superior efficiency of the Pt–Mn2O3/CN co-catalyst and heterojunction synergy over single modifications, aligning with the overall enhancement observed in photothermal catalytic performance.
The probable mechanism involved in the photothermal degradation of toluene
An in-depth exploration of the mechanisms underlying toluene oxidation led us to conduct in situ diffuse reflection infrared Fourier transform spectroscopy (DRIFTS) measurements under specific conditions. Employing the typical reaction setup, Fig. 5A presents in situ DRIFTS spectra recorded at different temperatures. Prior to the measurement, the catalyst underwent pretreatment under an N2 flow (30 mL min−1) at 300 °C for 30 minutes. Subsequently, the system was cooled down to 100 °C, and the background spectra were collected.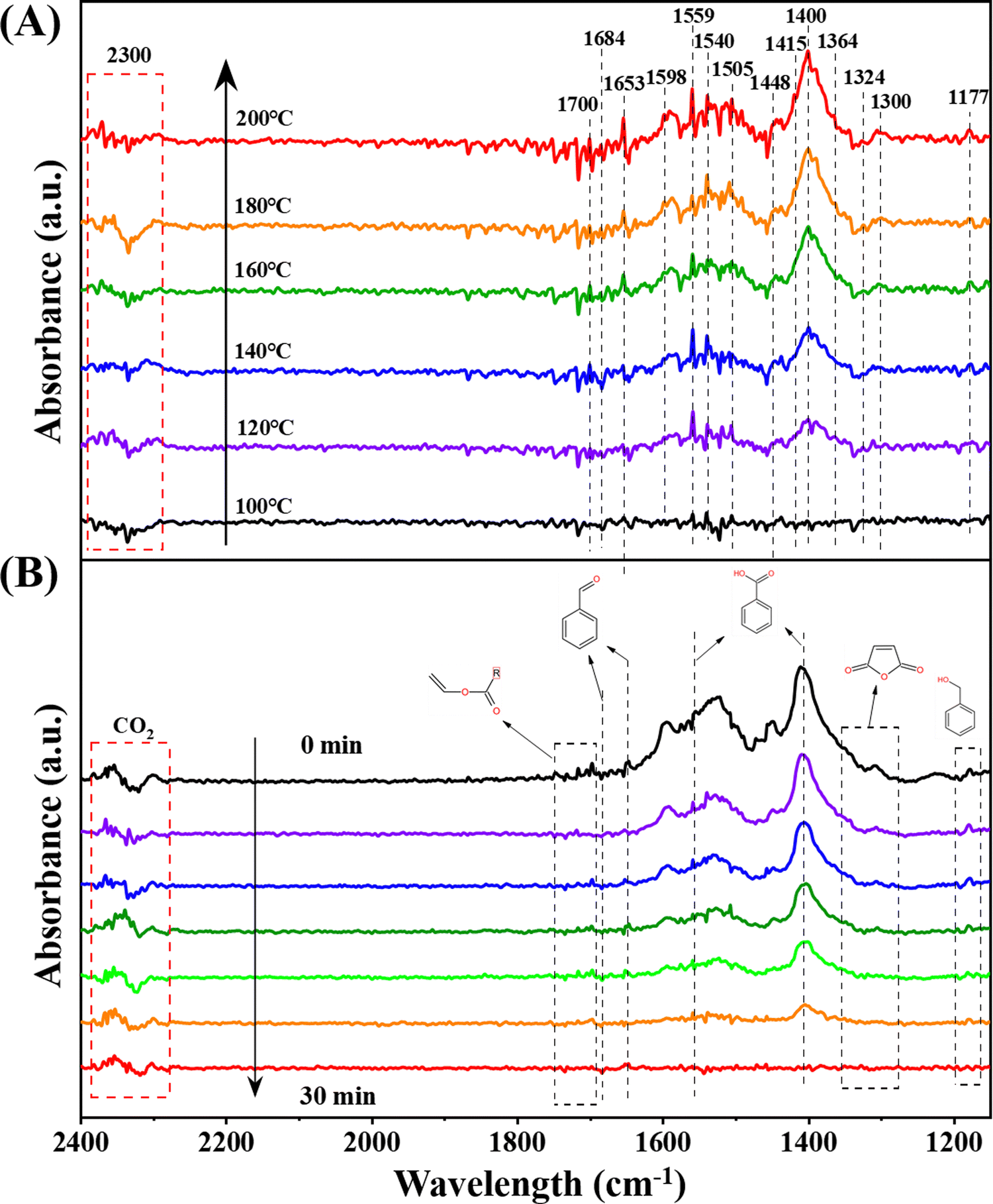 | ||
| Fig. 5 In situ DRIFTS spectra of (A) toluene adsorption at different temperatures and (B) toluene oxidation (1000 ppm toluene + O2 + N2 feed gas) over Pt–Mn2O3/CN. | ||
The peak at 1177 cm−1 is unequivocally identified as a C–O stretching vibration mode distinctive of alkoxide species. This implies that the adsorbed toluene can engage with the catalyst by cleaving the C–H bond of the methyl group (–CH4) to generate benzyl alcohol.16 The vibrational band at 1300 cm−1 is assigned to the maleic anhydride species, while the emergence of the band at 1364 cm−1, due to the symmetrical deformation vibration of the methyl group, signifies the formation of acetone.44 The peaks at 1448 cm−1 and 1598 cm−1 are ascribed to the vibrational mode of CC in the benzene ring.45 Furthermore, the vibrational bands at 1415 cm−1 and 1540 cm−1 are segregated into the C–O symmetry of benzoate and antisymmetry modes.17 The bands at 1559 cm−1 and 1400 cm−1 are indicative of carboxylic acid groups (1559 cm−1 representing asymmetric –COO− tensile vibration, and 1400 cm−1 representing symmetric –COO− tensile vibration), suggesting the formation of benzoate species.46 Bands situated at 1598 cm−1 and 1505 cm−1 are associated with the in-plane skeleton vibration of the aromatic ring.34 Additionally, bands at 1684 cm−1 and 1653 cm−1 are attributed to the CO stretching vibration,47 while the band at 1324 cm−1 corresponds to an aldehyde group consisting of skeletal C–C stretching vibration, suggesting the formation of benzaldehyde species.7 The multiple signal peaks in the range of 1668–1700 cm−1 can be categorized as CC tensile vibration in vinyl acetate. Notably, characteristic bands (2300–2340 cm−1) of adsorbed CO2 are also discerned, and their intensities escalate with an increase in the reaction temperature.46
Examining the spectra reveals a discernible trend where intermediate species are progressively amassed and depleted during the adsorption and oxidation of toluene. In the course of toluene heating and adsorption, the Pt–Mn2O3/CN catalyst exhibited peaks of intermediate species on the surface, and the peak intensity demonstrated an increase. This suggests the involvement of surface lattice oxygen from the catalyst in the oxidation process. When toluene adsorption was halted, and only oxygen was introduced (Fig. 5B), the intermediate species on the catalyst surface underwent continuous oxidation, leading to a gradual reduction in peak intensity. This phenomenon can be attributed to the activation of surface O2 molecules, facilitating the profound oxidation of intermediates by reactive oxygen species. Nevertheless, the examination of in situ infrared spectra for pristine Mn2O3 reveals only a marginal alteration in the signal intensity of benzoic acid with no discernible progression to subsequent stages (Fig. S9, ESI†). This observation underscores the difficulty of activating reactive oxygen species on the surface of Mn2O3.
In the context of incomplete toluene combustion, the Pt–Mn2O3/CN catalyst surface displayed peaks corresponding to benzyl alcohol, benzaldehyde, benzoic acid, vinyl acetate, and maleic anhydride. Analyzing in situ DRIFTS spectrum data, the degradation pathway of toluene on Pt–Mn2O3/CN aligns with the illustrated route in Fig. 6. A hydrogen atom within the methyl group of toluene is eliminated, giving rise to benzyl alcohol. Subsequently, benzyl alcohol is subject to additional oxidation mediated by reactive oxygen species, leading to the sequential formation of benzaldehyde and then benzoic acid. This oxidative process further disrupts the structural stability of the benzene ring. Following this, the activated benzene ring and the carboxyl group undergo an attack, culminating in the generation of maleic anhydride. Ultimately, maleic anhydride undergoes conversion into carbon dioxide (CO2) and water (H2O).
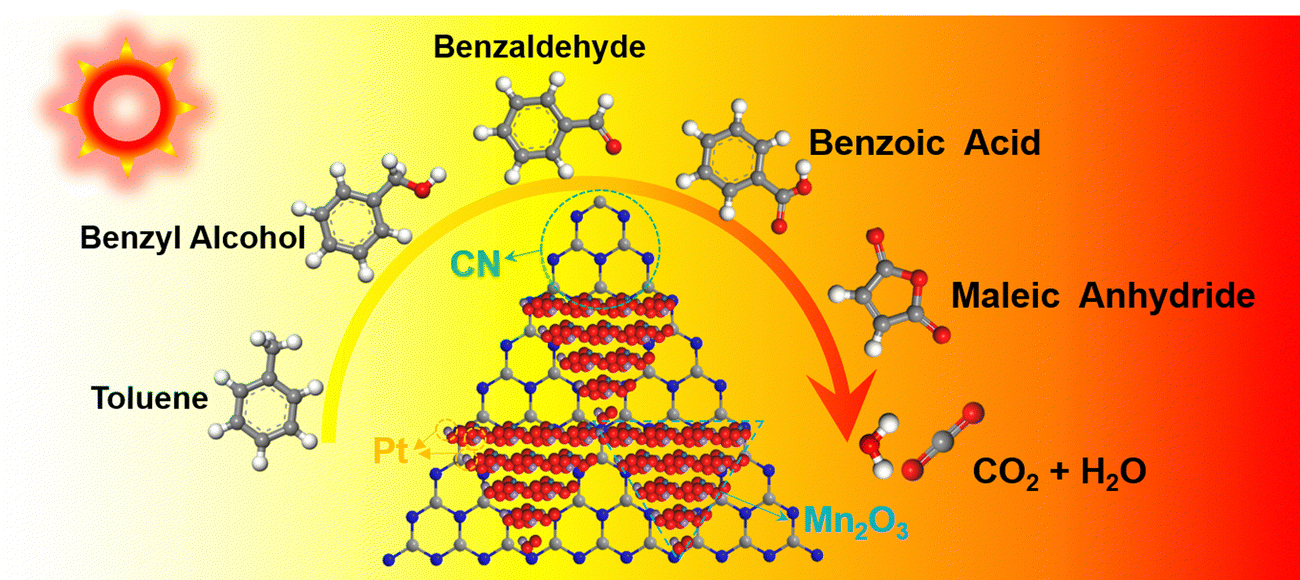 | ||
| Fig. 6 Scheme of toluene oxidation over Pt–Mn2O3/CN. | ||
Conclusion
In summary, harnessing renewable solar energy through photothermal catalysis presents a promising alternative to remediating VOCs. The approach involving Mn2O3 as the photothermal conversion center, CN as a uniformly dispersed medium for Pt nanoparticles, and Pt as the active site has proven successful. The loading of an appropriate proportion of Pt nanoparticles significantly enhanced the photothermal catalytic degradation activity of the Pt–Mn2O3/CN nanocomposite catalyst on toluene. This improvement is primarily attributed to the Pt nanoparticles inducing numerous oxygen vacancies in the catalyst, enhancing the oxygen adsorption content and surface fluidity, and activating the absorbed oxygen on the catalyst's surface. The effective utilization of sunlight for VOC degradation highlights its potential as a clean and renewable energy source, offering substantial environmental benefits.Experimental
Chemical reagents
Manganous acetate tetrahydrate (C4H6MnO4·4H2O), and platinum tetra ammonia nitric acid (H12N6O6Pt) were purchased from Aladdin Reagents (Shanghai) Co., Ltd. Melamine (C3H6N6) and glycol (C2H6O2) were purchased from Sinopharm Chemical Reagent Co., Ltd. All these chemicals were of analytical grade and used without further purification.Catalyst preparation
In parallel, Mn2O3 and Mn2O3/CN were synthesized using the same aforementioned procedure, omitting H12N6O6Pt.
Characterization
The X-ray diffraction (XRD) patterns of the catalysts were acquired employing a Bruker D8 Advance X-ray diffractometer (Bruker, Germany) with Cu Kα radiation (λ = 1.542 Å). BET adsorption and desorption isotherms were conducted using a Belsorp-Mini II analyzer (Japan). Fourier transform infrared spectroscopy (FTIR) data were obtained using a Nicolet Nexus 410 spectrometer. The Laser Raman spectrum was measured using an Ar ion laser as the excitation source, covering a frequency range of about 50–1400 cm−1 (HORIBA Jobin Yvon S.A.S.). X-ray photoelectron spectroscopy (XPS) analyses were executed within an ultrahigh vacuum chamber at room temperature, maintaining a pressure of 2 × 10−9 bar (Thermo Fisher Scientific, ESCALAB 250). The morphology and microstructures of the catalyst were scrutinized through transmission electron microscopy (TEM) (FEI Talos F200X) and scanning electron microscopy (SEM) (Hitachi, model SU1510). Gas chromatography (GC, Agilent Technologies 7890A) was employed to monitor the concentration of toluene and CO2 during the catalytic process, while gas chromatography–mass spectrometry (GC-MS, Agilent Technologies 7890A-5975C) was utilized to identify specific components in the reactor. Furthermore, H2-TPR and O2 temperature-programmed desorption (O2-TPD) analyses were conducted on the MicrotracBEL BELCAT-II chemical adsorption analyzer. EPR spectra were obtained through an electron paramagnetic resonance spectrometer (Bruker A300). A UV-Vis-IR spectrophotometer (UV-3600 plus) was employed to record the full-spectrum light absorption on the catalysts. The photoluminescence (PL) spectra were measured using a Hitachi F4500 fluorescence spectrophotometer with an excitation wavelength of 280 nm at room temperature.In situ DRIFTS experiments were conducted using a Nicolet 50 spectrometer (Thermo Fisher, USA) equipped with a Harrick DRIFTS cell and an MCT/A detector. To perform time-dependent in situ DRIFTS, the catalyst underwent pretreatment under a nitrogen (N2) flow at a rate of 30 mL min−1, maintaining a temperature of 300 °C for 30 minutes. Subsequently, the system was cooled to 200 °C, and the background spectrum was recorded. Introducing 1000 ppm of toluene (at a flow rate of 10 mL min−1) into the system occurred over 1 hour, followed by N2 purging (at a flow rate of 30 mL min−1) for 30 minutes to eliminate weakly adsorbed toluene. The sample was then subjected to a reaction at 200 °C under an air flow containing 21% O2 in N2. Concurrently, spectral data were collected throughout the entire process.
Photothermal catalytic activity
The photothermal catalytic oxidation of toluene was systematically investigated through both static and continuous flow reaction modes. The static reaction test, chosen for its capacity to elucidate the temporal evolution of intermediates, provides valuable insights into the mechanistic intricacies of the process. Concurrently, the continuous flow reaction test serves as a comprehensive evaluation of the catalyst's stability and practical efficacy under dynamic conditions.For a detailed exposition of the catalytic performance experiments conducted on the aforementioned catalysts, we direct the reader to the ESI,† accompanying this manuscript.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors gratefully acknowledge the financial support from the National Natural Science Foundation of China (22072170, U23A20125), the Zhejiang Provincial Key Research and Development Program (2021C03170), and the Ningbo Science and Technology Bureau (2019B10096).References
- Y. Yang, S. H. Zhao, L. F. Cui, F. K. Bi, Y. N. Zhang, N. Liu, Y. X. Wang, F. D. Liu, C. He and X. D. Zhang, Green Energy Environ., 2023, 8, 654–672 CrossRef CAS.
- B. Liu, J. Ji, B. Zhang, W. Huang, Y. Gan, D. Y. C. Leung and H. Huang, J. Hazard. Mater., 2022, 422, 126847 CrossRef CAS PubMed.
- X. Yu, B. H. Deng, L. X. Yang, M. L. Zou, Z. L. Chen, Y. X. Fan, Z. H. Wei, K. Chen, M. X. Lu, T. T. Ying, J. Zhang and W. L. Dai, J. Environ. Chem. Eng., 2022, 10(5), 108496 CrossRef CAS.
- C. Song, Z. Wang, Z. Yin, D. Xiao and D. Ma, Chem. Catal., 2022, 2, 52–83 CrossRef CAS.
- J.-J. Li, S.-C. Cai, E.-Q. Yu, B. Weng, X. Chen, J. Chen, H.-P. Jia and Y.-J. Xu, Appl. Catal., B, 2018, 233, 260–271 CrossRef CAS.
- E. A. Elimian, M. Zhang, J. Chen, H. Jia, Y. Sun and J. He, Appl. Catal., B, 2022, 307, 121203 CrossRef CAS.
- M. Zhang, G. Li, Q. Li, J. Chen, E. A. Elimian, H. Jia and H. He, Environ. Sci. Technol., 2023, 57, 4286–4297 CrossRef CAS PubMed.
- X. Li, Y. Wang, D. Chen, N. Li, Q. Xu, H. Li, J. He and J. Lu, Green Energy Environ., 2023, 8, 538–547 CrossRef CAS.
- Y. Park, V. N. Pham, K. Lee and H. Lee, Inorg. Chem., 2023, 62, 13428–13434 CrossRef CAS PubMed.
- X. Yu, M. Shi, Y. Fan, L. Yang, J. Zhang, W. Liu, W. Dai, S. Zhang, L. Zhou, X. Luo and S. Luo, Appl. Catal., B, 2022, 309, 121236 CrossRef CAS.
- W. Zhao, M. Adeel, P. Zhang, P. Zhou, L. Huang, Y. Zhao, M. A. Ahmad, N. Shakoor, B. Lou, Y. Jiang, I. Lynch and Y. Rui, Environ. Sci.: Nano, 2022, 9, 61–80 RSC.
- T. Pan, H. Deng, Y. Lu, J. Ma, L. Wang, C. Zhang and H. He, Environ. Sci. Technol., 2023, 57, 1123–1133 CrossRef CAS PubMed.
- G. Chen, F. Wei, Z. Zhou, B. Su, C. Yang, X. F. Lu, S. Wang and X. Wang, Sustainable Energy Fuels, 2023, 7, 381–388 RSC.
- Q. Wang, G. Zhang, W. Xing, Z. Pan, D. Zheng, S. Wang, Y. Hou and X. Wang, Angew. Chem., Int. Ed., 2023, 62(25), e202303486 CrossRef PubMed.
- M. Liu, G. Zhang, X. Liang, Z. Pan, D. Zheng, S. Wang, Z. Yu, Y. Hou and X. Wang, Angew. Chem., Int. Ed., 2023, 62(11), e202217448 CrossRef PubMed.
- M. Zhang, S. Cai, J. Li, E. A. Elimian, J. Chen and H. Jia, J. Hazard. Mater., 2021, 412, 125266 CrossRef CAS PubMed.
- M. Zhang, H. Gao, J. Chen, E. A. Elimian and H. Jia, Appl. Catal., B, 2022, 307, 121208 CrossRef CAS.
- J. Zhao, J. Liu, Z. Li, K. Wang, R. Shi, P. Wang, Q. Wang, G. I. N. Waterhouse, X. Wen and T. Zhang, Nat. Commun., 2023, 14(1), 1909 CrossRef CAS PubMed.
- J. Zhao, Y. Bai, Z. Li, J. Liu, W. Wang, P. Wang, B. Yang, R. Shi, G. I. N. Waterhouse, X. D. Wen, Q. Dai and T. Zhang, Angew. Chem., Int. Ed., 2023, 62(13), e202219299 CrossRef CAS PubMed.
- P. Niu, L. Zhang, G. Liu and H.-M. Cheng, Adv. Funct. Mater., 2012, 22, 4763–4770 CrossRef CAS.
- X. Zhang, X. Lv, F. Bi, G. Lu and Y. Wang, Mol. Catal., 2020, 482, 110701 CrossRef CAS.
- C. Kaewbuddee, P. Chirawatkul, K. Kamonsuangkasem, N. Chanlek and K. Wantala, Chem. Eng. Commun., 2021, 209, 512–528 CrossRef.
- P. Yang, H. Ou, Y. Fang and X. Wang, Angew. Chem., Int. Ed., 2017, 56, 3992–3996 CrossRef CAS PubMed.
- I. A. Abdelhafeez, J. Chen and X. Zhou, Sep. Purif. Technol., 2020, 250, 117085 CrossRef CAS.
- W. Zhao, J. Wang, R. Yin, B. Li, X. Huang, L. Zhao and L. Qian, J. Colloid Interface Sci., 2020, 564, 28–36 CrossRef CAS PubMed.
- Q. Huang, X. Cai, M. Chen, Q. Yang, S. Fan, Y. Zhang, H. Hu, T. Gan and Z. Huang, Chem. Eng. J., 2022, 436, 135258 CrossRef CAS.
- Z. Xiao, F. Xia, L. Xu, X. Wang, J. Meng, H. Wang, X. Zhang, L. Geng, J. Wu and L. Mai, Adv. Funct. Mater., 2021, 32(14), 2108244 CrossRef.
- X. Liu, J. Mi, L. Shi, H. Liu, J. Liu, Y. Ding, J. Shi, M. He, Z. Wang, S. Xiong, Q. Zhang, Y. Liu, Z. S. Wu, J. Chen and J. Li, Angew. Chem., Int. Ed., 2021, 60, 26747–26754 CrossRef CAS PubMed.
- M. S. Brogan, T. J. Dines and J. A. Cairns, J. Chem. Soc., Faraday Trans., 1994, 90, 1461–1466 RSC.
- J. Xiao, C. Chen, S. Chen, H. Liu and T. Peng, Chem. Eng. J., 2021, 425, 131627 CrossRef CAS.
- J. Li, M. Zhang, E. A. Elimian, X. Lv, J. Chen and H. Jia, Chem. Eng. J., 2021, 412, 128560 CrossRef CAS.
- D. Zhang, J. Cao, X. Zhang, N. Insin, S. Wang, J. Han, Y. Zhao, J. Qin and Y. Huang, Adv. Funct. Mater., 2021, 31(14), 2009412 CrossRef CAS.
- H. Yan, S. Yao, J. Wang, S. Zhao, Y. Sun, M. Liu, X. Zhou, G. Zhang, X. Jin, X. Feng, Y. Liu, X. Chen, D. Chen and C. Yang, Appl. Catal., B, 2021, 284, 119803 CrossRef CAS.
- Z. Xu, S. Mo, Y. Li, Y. Zhang, J. Wu, M. Fu, X. Niu, Y. Hu and D. Ye, Chemosphere, 2022, 286, 131754 CrossRef CAS PubMed.
- X. Fu, Y. Liu, J. Deng, L. Jing, X. Zhang, K. Zhang, Z. Han, X. Jiang and H. Dai, Appl. Catal., A, 2020, 595, 117509 CrossRef CAS.
- S. Fang, Y. Li, Y. Yang, J. Chen, H. Liu and X. Zhao, Environ. Sci.: Nano, 2017, 4, 1798–1807 RSC.
- C. Feng, F. Jiang, G. Xiong, C. Chen, Z. Wang, Y. Pan, Z. Fei, Y. Lu, X. Li, R. Zhang and Y. Liu, Chem. Eng. J., 2023, 451, 138868 CrossRef CAS.
- Z. Xie, Z. Lyu, J. Wang, A. Li and P. François-Xavier Corvini, Chem. Eng. J., 2022, 429, 132299 CrossRef CAS.
- H. Yan, Q. Shen, Y. Sun, S. Zhao, R. Lu, M. Gong, Y. Liu, X. Zhou, X. Jin, X. Feng, X. Chen, D. Chen and C. Yang, ACS Catal., 2021, 11, 6371–6383 CrossRef CAS.
- X. Yu, Z. Chen, Z. Wei, K. Chen, Y. Fan, M. Zou, X. Si, B. Deng, L. Yang and J. Zhang, Funct. Mater. Lett., 2022, 15(05), 2251042 CrossRef CAS.
- B. Su, Y. Kong, S. Wang, S. Zuo, W. Lin, Y. Fang, Y. Hou, G. Zhang, H. Zhang and X. Wang, J. Am. Chem. Soc., 2023, 145, 27415–27423 CrossRef CAS PubMed.
- Y. Li, J. Hao, H. Song, F. Zhang, X. Bai, X. Meng, H. Zhang, S. Wang, Y. Hu and J. Ye, Nat. Commun., 2019, 10, 2359 CrossRef PubMed.
- S. C. Cai, J. J. Li, E. Q. Yu, X. Chen, J. Chen and H. P. Jia, ACS Appl. Nano Mater., 2018, 1, 6368–6377 CrossRef CAS.
- Z. Wang, H. Yang, R. Liu, S. Xie, Y. Liu, H. Dai, H. Huang and J. Deng, J. Hazard. Mater., 2020, 392, 122258 CrossRef CAS PubMed.
- E. Yu, J. Li, J. Chen, J. Chen, Z. Hong and H. Jia, J. Hazard. Mater., 2020, 388, 121800 CrossRef CAS PubMed.
- Z. Wang, S. Xie, Y. Feng, P. Ma, K. Zheng, E. Duan, Y. Liu, H. Dai and J. Deng, Appl. Catal., B, 2021, 298, 120612 CrossRef CAS.
- Y. Shen, J. Deng, L. Han, W. Ren and D. Zhang, Environ. Sci. Technol., 2022, 56, 10433–10441 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3ey00298e |
| ‡ These authors contributed equally. |
| This journal is © The Royal Society of Chemistry 2024 |