
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceRecent developments on microwave-assisted organic synthesis of nitrogen- and oxygen-containing preferred heterocyclic scaffolds†
Ghanshyam Tiwari
a,
Ashish Khanna
a,
Vinay Kumar Mishra
a and
Ram Sagar
 *ab
*ab
aDepartment of Chemistry, Institute of Science, Banaras Hindu University, Varanasi 221005, India
bGlycochemistry Laboratory, School of Physical Sciences, Jawaharlal Nehru University, New Delhi 110067, India. E-mail: ram.sagar@jnu.ac.in
First published on 7th November 2023
Abstract
In recent decades, the utilization of microwave energy has experienced an extraordinary surge, leading to the introduction of innovative and revolutionary applications across various fields of chemistry such as medicinal chemistry, materials science, organic synthesis and heterocyclic chemistry. Herein, we provide a comprehensive literature review on the microwave-assisted organic synthesis of selected heterocycles. We highlight the use of microwave irradiation as an effective method for constructing a diverse range of molecules with high yield and selectivity. We also emphasize the impact of microwave irradiation on the efficient synthesis of N- and O-containing heterocycles that possess bioactive properties, such as anti-cancer, anti-proliferative, and anti-tumor activities. Specific attention is given to the efficient synthesis of pyrazolopyrimidines-, coumarin-, quinoline-, and isatin-based scaffolds, which have been extensively studied for their potential in drug discovery. The article provides valuable insights into the recent synthetic protocols and trends for the development of new drugs using heterocyclic molecules.
 Ghanshyam Tiwari | Ghanshyam Tiwari completed his M.Sc. at Mahatma Gandhi Kashi Vidyapith, Varanasi, Uttar Pradesh, India. He joined the Glycochemistry lab of the Department of Chemistry at the Institute of Science, Banaras Hindu University, as a research scholar in year 2018. He completed his PhD in April 2023 under the supervision of Prof. Ram Sagar. His expertise lies in glycoscience, microwave-assisted synthesis, organic synthesis, and the development of new methods for synthesis of natural product inspired glycohybrids. |
 Ashish Khanna | Ashish Khanna completed his M.Sc. at Kumaun University, Nainital, Uttarakhand, India, in 2016. He joined the Department of Chemistry at the Institute of Science, Banaras Hindu University, as a PhD research scholar in the Glycochemistry lab in the year 2017. He completed his PhD in March 2023 under the supervision of Prof. Ram Sagar. Currently he is working as Assistant Professor in Department of Chemistry, SOPS, DIT University, Dehradun. His expertise lies in organic synthesis, the development of carbohydrate-derived bioactive molecules, natural product-inspired hybrid analogues and molecular modeling, especially protein–ligand interactions via in silico docking tools. |
 Vinay Kumar Mishra | Vinay Kumar Mishra completed his M.Sc. at Dr Ram Manohar Lohia Avadh University UP, India, in 2014. He joined the Glycochemistry lab at the Department of Chemistry, Institute of Science, Banaras Hindu University in 2019 to pursue his PhD. He completed his PhD in September 2023 under the supervision of Prof. Ram Sagar. His work is mainly focused on the development of new methods for the synthesis of carbohydrate-fused heterocyclic molecules as glycohybrids. He is also interested in the synthesis of natural product inspired bioactive scaffolds. |
 Ram Sagar | Prof. Ram Sagar received his PhD in Organic Chemistry from Central Drug Research Institute (CDRI), Lucknow, and University of Agra in 2006 under the supervision of Dr A. K. Shaw. After his PhD, he was a Research Associate under Prof. Y. D. Vankar at IIT Kanpur during 2006–2007. He pursued his first post-doctoral research at Seoul National University, South Korea, with Prof Seung Bum Park during 2007–2008. He then moved to the University of Oxford and worked with Prof Benjamin G. Davis as a BBSRC postdoctoral fellow up to August 2012. He then returned to India in August 2012 and held a faculty position at Shiv Nadar University (SNU). He moved to the Department of Chemistry, Banaras Hindu University (BHU) as an Associate Professor in February 2018 and worked there till December 2020. He subsequently moves as full professor to Jawaharlal Nehru University (JNU), New Delhi, in December 2020 and is presently working there as Professor of Chemistry in the School of Physical Sciences. His current research interests include devising new ways for the efficient chemical synthesis of natural product inspired small molecules, glycohybrids and glycopeptides implicated in various diseases, including tuberculosis and cancer. |
1. Introduction
A natural product is a substance found in the nature, and can be in the form of heterocycles or other moieties.1 Natural products have had a great influence on the developments of medicines and materials for treatment of cancer, bacterial infections, autoimmune diseases and inflammation.2 Nature is a major source for organic molecules in the form of secondary metabolites, which can be used for the development of novel therapeutics. Most of the drugs based on chemical scaffolds isolated from plants, marine organisms, microbes and other natural sources serve as treatments of various diseases. Natural products have also been used for commercial purposes such as cosmetics, dietary supplements, and many other edible products.3,4New pharmaceuticals and many effective medications were initially developed as semi-synthetic analogues of compounds found in nature.5 Natural products are very diverse in terms of three-dimensional (3D) chemical space and exhibit unique biological functions. This is inferred from the idea that virtually all natural compounds have some capability for receptor binding.6,7
Thus, the design and synthesis of natural products and natural product-inspired heterocyclic molecules have been of interest to medicinal chemists and chemical biologists because these molecules have significant roles in the drug-development process.8–10 Common heterocycles are found in natural products, especially in alkaloids, polyketals, vitamins and propanoids.11 Isatin, coumarin, quinoline, isoquinoline, pyrimidine, pyrazole, and pyrazolopyrimidine are heterocyclic motifs found widely in bioactive natural products as well as in pharmaceutical agents. Molecules derived from these scaffolds, such as coumarin derivatives, show various biological activities. Calanolide A is a coumarin based anti-HIV drug.12 Warfarin is a coumarin-based anticoagulant drug which is mainly used to prevent blood clots.13 Chloroquine is a quinoline-based antimalarial drug and recently used to treat COVID-19.14 Hydroxychloroquine is also a quinoline-based antimalarial drug.15 Nintedanib is an isatin derivative, currently used as an anticancer drug.16 The pyrazolopyrimidine derivative zaleplon is used as treatment of insomnia in humans by decreasing the activity of the brain.17 Sildenafil is a pyrazolopyrimidine-based drug mainly used in the treatment of erectile disfunction in humans18 (Fig. 1).
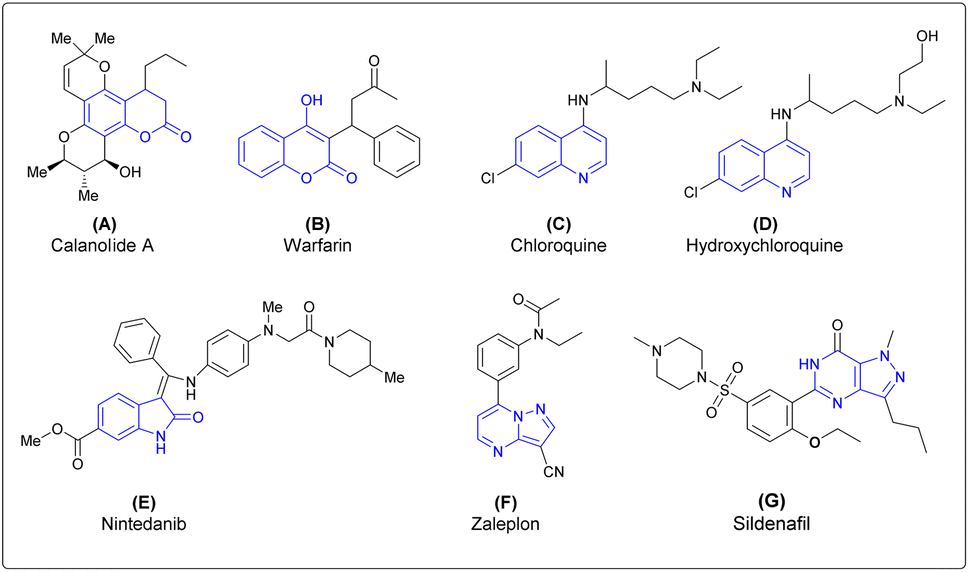 | ||
| Fig. 1 Heterocycles derived bioactive molecules (A–D): (A) calanolide A, a coumarin-based anti-HIV drug, (B) warfarin, a coumarin-based anticoagulant, (C) chloroquine, a quinoline-based antimalarial drug, (D) hydroxychloroquine, a quinoline-based antimalarial drug, (E) nintedanib, an isatin-based anticancer drug, (F) zaleplon, a pyrazolopyrimidine-based anti-insomnia drug, and (G) sildenafil, a pyrazolopyrimidine-based drug. | ||
Various techniques are available to efficiently synthesize these heterocyclic scaffolds and derivatives, including electrochemical synthesis, metal-free synthesis, photochemical synthesis, solvent-free synthesis, ultrasonication, solvothermal synthesis, ball milling, and many more.19–21 Some reactions under thermal conditions can take from a few hours to a few days.22 In recent times, various techniques and methods have enabled chemical reactions to be undertaken efficiently and in less time. As a result, a new approach has been developed to serve as an energy source for performing efficient chemical synthesis.23–26 This approach involves the use of microwave energy, which is harnessed through a microwave reactor.
A microwave reactor is an apparatus designed for carrying out microwave-assisted organic synthesis. It is important to note that this type of reactor is slightly different from the conventional household microwave oven.27 The microwave reactor comprises a microwave emitter, a pressure controller, and a rotor with safety controls that are fitted with the reactor.28 The microwave emitter in the reactor emits microwave energy, which is absorbed by the reactants in the reaction vessel, leading to a heating effect.29 The pressure controller is responsible for regulating the pressure within the reactor, ensuring that it remains at the desired level. The rotor helps to distribute the reactants evenly within the reactor, enhancing the efficiency of the reaction.30
Over the past few decades, there has been a remarkable surge in the utilization of microwave energy. This had led to the introduction of novel and revolutionary applications in various fields, such as organic synthesis, heterocycle synthesis, polymer chemistry,31,32 material sciences,33 nanotechnology, and biochemical processes.34,35 Notably, microwave irradiation has displayed the potential to significantly reduce processing time, amplify the reaction yield, as well as enhance the purity and properties of products, thereby surpassing traditional methods.36 This advancement, known as microwave-assisted organic synthesis (MAOS), relies on the efficient heat transfer achieved through dielectric heating, and is primarily dependent on the absorption capacity of the solvent or reagents for microwave energy.37
Dielectric heating provides a crucial advantage over conventional heating methods as microwaves directly interact with dipoles or ionic molecules present in the reaction mixture, allowing energy transfer to occur in less than a nanosecond (10−9 s), leading to an instantaneous temperature rise.38–40 Additionally, microwave irradiation facilitates volumetric heating by directly coupling the electromagnetic field with molecules in the reaction mixture, thereby minimizing or completely avoiding wall effects.41 This introduction outlines the promising and transformative possibilities that microwave energy offers in advancing research and development across various scientific domains.42
Overall, the microwave reactor is an essential tool for conducting MAOS, providing a reliable and efficient energy source for various chemical reactions. Therefore, this approach has proven to be useful in obtaining a diverse library of heterocyclic molecules inspired by natural products that exhibit improved pharmacological properties. Thus, microwave irradiation has been identified as a crucial technique in organic synthesis and drug-discovery processes. This review highlights recent advancements in the synthesis of quinoline, pyrazolopyrimidine, coumarin, isatin and their derivatives under MAOS.
The review covers a wide range of microwave-assisted synthetic reactions, including Suzuki–Miyaura cross-coupling, Friedlander synthesis, copper-catalyzed four-component synthesis, and the synthesis of isatin-β-thiocarbohydrazones and quinoline fused 1,4-benzodiazepines. This breadth of coverage allows readers to gain insight into the diverse applications of microwave-assisted synthesis in different chemical transformations. We provide specific examples of successful reactions, the catalysts used, reaction conditions, and isolated yields. These examples enable researchers to understand the practical aspects of microwave-assisted synthesis. It highlights the advantages of microwave-assisted synthesis over conventional heating methods. It emphasizes the significant time savings, improved yields, and higher selectivity achieved through microwave irradiation, making it a more attractive option for various chemical reactions. The review offers valuable perspectives on the potential areas of research and improvement in microwave-assisted synthesis and discusses the need for a deeper mechanistic understanding, exploration of new reactions, “green” and sustainable chemistry applications, scale-up challenges, and the combination of microwave technology with other techniques.
Thus, this review offers a comprehensive, well-referenced, and forward-looking perspective on microwave-assisted synthesis. It goes beyond theoretical discussions and provides concrete examples and practical benefits, making it a valuable resource for researchers and practitioners in the fields of synthetic chemistry and materials science.
2. MAOS of N- and O-containing heterocycles
Nitrogen-containing heterocyclic motifs are abundant in natural products and natural product-derived drug molecules.43,44 Therefore, chemical synthesis of these scaffolds has been of interest among organic synthesis and medicinal-chemistry research groups. Oxygen-containing molecules such as coumarins are also present in nature and display various biological functions.45–47 Chemical modification of these secondary metabolites is needed to obtain diverse derivatives that can fulfill 3D chemical space. Thus, microwave-assisted synthetic modification evolved is a powerful tool which can complete reactions in a shorter time and furnish the desired products in high yields.2.1. Quinolines and their derivatives
Quinoline is a heterocyclic organic compound consisting of a benzene ring fused to a pyridine ring. It is a fundamental building block in the synthesis of various pharmaceuticals and agrochemicals.48–50 It is found in some plants, and is also a component in the flavour of tobacco.51,52 Quinoline serves as a precursor for numerous derivatives with diverse properties. Many quinoline derivatives exhibit potent pharmacological activities,53 including anti-malarial drugs such as chloroquine54 and anti-cancer agents such as camptothecin.55 Its derivatives have been studied for their potential in treating neurodegenerative diseases because they can interact with specific receptors in the brain.56 Certain quinoline derivatives are used in agriculture as pesticides and herbicides due to their ability to control plant diseases.57 In summary, quinoline and its derivatives have pivotal roles in various fields, from medicine and chemistry to agriculture and materials science, owing to their versatile chemical properties and wide-ranging applications. Therefore, researchers have keen to synthesize quinoline-based pharmacophores for the development of new drugs. In this review, recent developments on the microwave-assisted synthesis of quinolines and their derivatives has been summarized.Ahmed and co-workers reported a one-pot, three-component domino reaction of propargylated-flavone or coumarin 1a–1b with aldehydes 3a–g and anilines 2a–e.58 The reaction was carried out under solvent-free and microwave conditions using YbCl3 as a catalyst. It resulted in an efficient and highly eco-friendly synthesis of diverse and functionalized quinolin-4-ylmethoxychromen-4-ones 4a–n (Scheme 1) and quinolin-4-ylmethoxychromen-2-ones 5a–e (Scheme 2). The desired products (4a–n and 5a–e) were obtained in excellent yields (80–95%) at 100 °C within 4 min. The reaction was also performed in an oil bath under heating conditions but it furnished a lower yield of product and longer reaction time (60 min) as compared to under microwave-assisted conditions (4 min). Hence, it is evident that the microwave approach offers several advantages, such as high yields, mild reaction conditions that are free of solvents, tolerance of functional groups, 95% atom-economy, and recyclability of the catalyst.
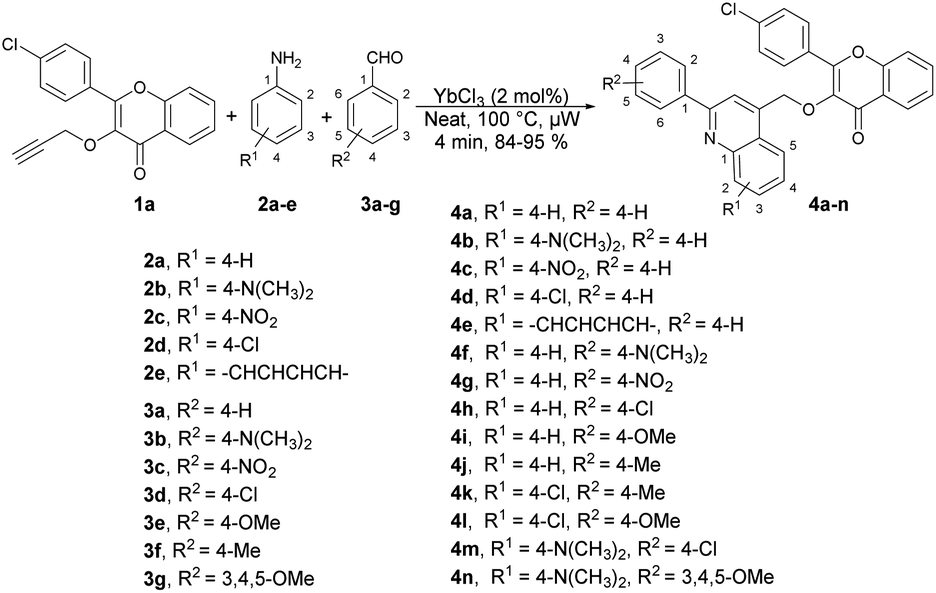 | ||
| Scheme 1 Synthesis of quinolin-4-ylmethoxychromen-4-ones 4a–n. | ||
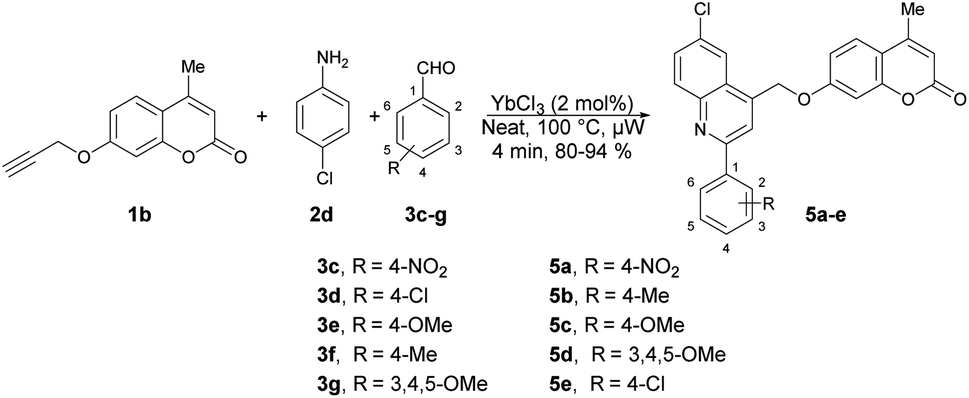 | ||
| Scheme 2 Synthesis of quinolin-4-ylmethoxychromen-2-ones 5a–e. | ||
Scheme 3 illustrates a proposed mechanism for the one-pot synthesis of quinolin-4-ylmethoxychromen-4-ones based on the aforementioned results. This one-pot three-component reaction involves a series of steps. First, it includes domino imine formation, followed by imine addition and cyclization and, finally, oxidation. The high reactivity of YbCl3 can be attributed to the smaller radius of Yb3+. Importantly, the presence of H2O in the reaction system does not deactivate or decompose the catalyst. The coordination of the hard Lewis-base site of the nitrogen lone pair with the hard Lewis-acid Yb3+ is supported by the HSAB theory. This coordination enhances the electrophilicity of the imine, leading to subsequent cyclization and aromatization. Notably, Yb3+ plays a significant role in both the imine addition and cyclization processes.
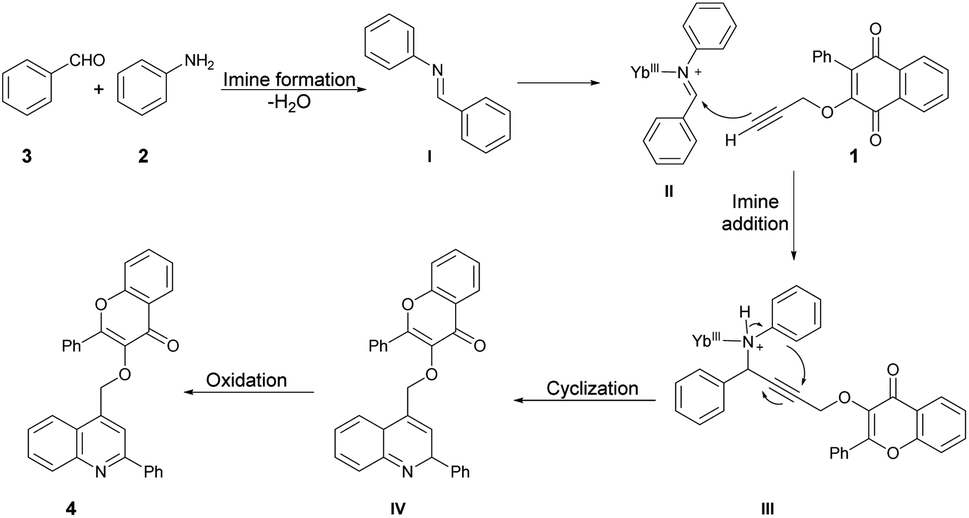 | ||
| Scheme 3 A proposed mechanism of the one-pot synthesis of quinolin-4-ylmethoxychromen-4-ones catalyzed by YbCl3. | ||
Frank and co-workers efficiently produced D- and A-ring fused novel quinolines in an estrone series.59 They synthesized the desired molecules 9a–c and 13a–g using various arylamines 8a–c and 12a–g with the corresponding β-chlorovinyl aldehydes 7a and 11a in DMF under microwave irradiation. However, they discovered that the electronic and steric characteristics of the substituents of anilines, as well as the distinct reactivities of rings D and A of the sterane skeleton, had significant impacts on the rates and yields of the one-pot, catalyst-free synthesis. They found poor-to-moderate yields (47–53%) of D-fused quinolines 9a–c applying microwave conditions at 140 °C, and A-fused quinolines (Scheme 4) 13a–g were obtained in moderate-to-good isolated yields (43–92%) at 100 °C for 2–10 min (Scheme 5). To explore the benefits of microwave irradiation, the reaction between 11a and 12b (1 equiv.) was additionally conducted using conventional heating (120 °C, DMF) but it required an extended reaction time (4 h instead of 2 min) for production of the desired quinoline 13b. Moreover, the yield achieved with conventional heating was lower, reaching 75%, as opposed to the 91% yield obtained with microwave irradiation.
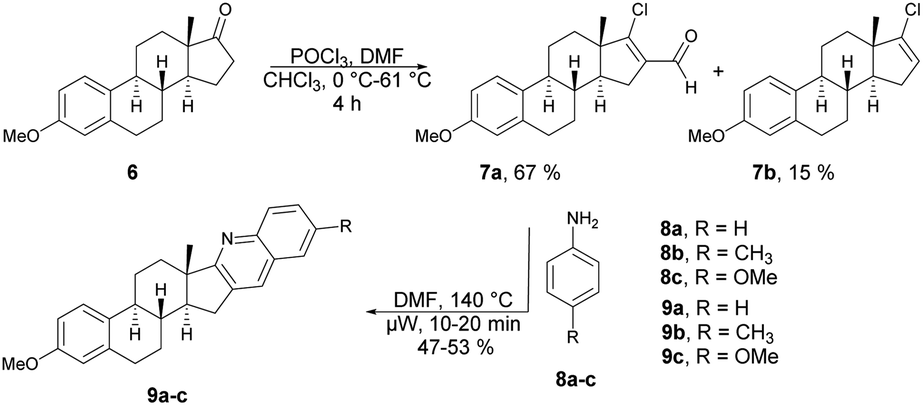 | ||
| Scheme 4 Synthesis of steroidal ring D-fused quinolines 9a–c. | ||
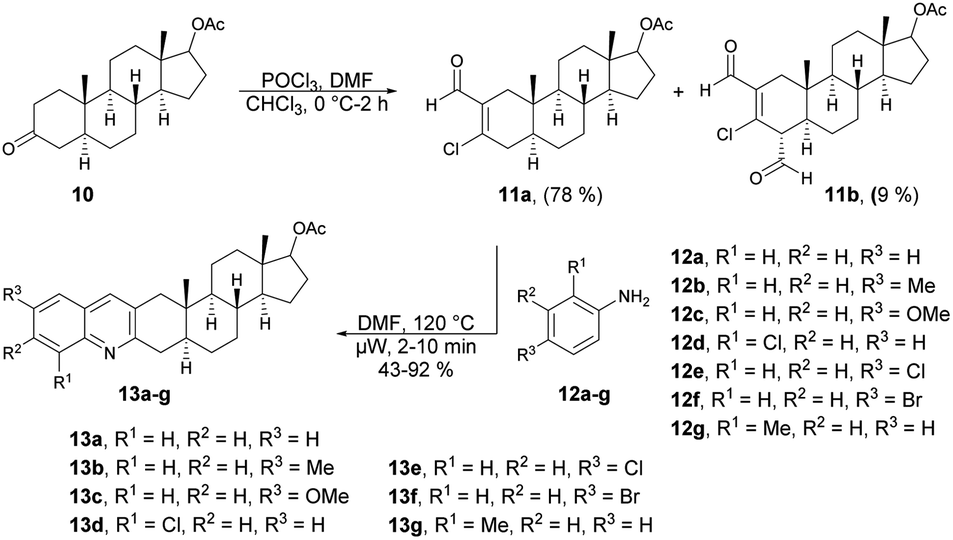 | ||
| Scheme 5 Synthesis of steroidal ring A-fused quinolines 13a–g. | ||
Fernandes and co-workers reported an efficient and environmentally friendly method to synthesize various derivatives of quinolines 17a–u using microwave irradiation.60 They optimized the reaction conditions using various solvents such as ethanol, water, and acetonitrile, as well as without a solvent. The microwave-assisted neat condition performed better for the synthesis of various derivatives of quinolines 17a–u with moderate-to-good isolated yields (40–68%). All the reactions were carried out using various anilines 14a–m, aryl aldehydes 15a–k and styrene 16 in the presence of p-sulfonic acid calix[4]arene (CX4SO3H) at neat conditions under microwave irradiation at 200 °C for 20–25 min (Scheme 6). For comparison purposes, the classical thermal method employing acetonitrile and 12 of reflux was used to obtain 17b–17h. In contrast, the utilization of microwave irradiation with various aniline substrates resulted in improved yields of quinolines, as evidenced by the results obtained for the synthesis of 17i–17u. Remarkably, microwave irradiation proved to be 11–19% more efficient than conventional heating for producing quinolines 17n, 17p and 17s. The yield of the tri-methoxy-substituted product was twice as high when using microwave irradiation compared with that using conventional heating. Additionally, reactions under microwave irradiation allowed for faster formation of the desired quinolines in comparison with using thermal heating and solvent-free conditions.
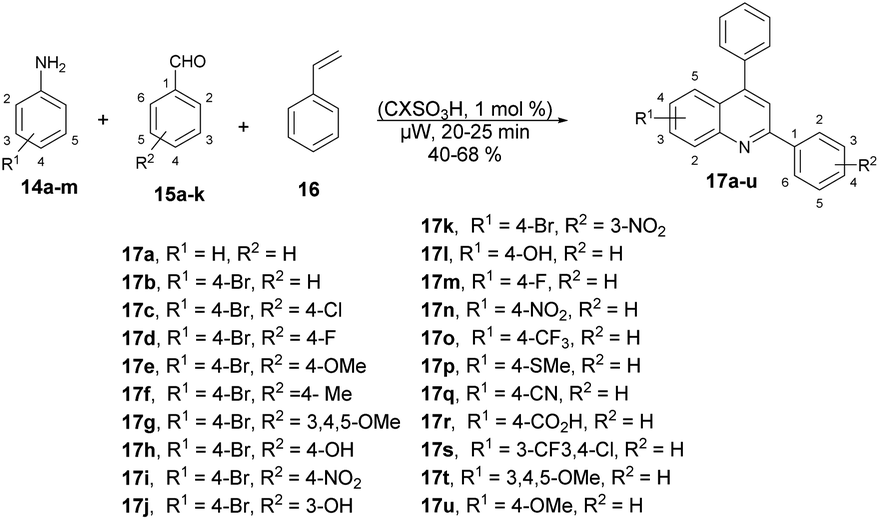 | ||
| Scheme 6 Synthesis of various derivatives of quinolines 17a–u. | ||
An elegant cascade approach was developed by Patel and co-workers to synthesize indoloquinolines. This approach enabled the synthesis of numerous valuable indoloquinoline alkaloids through the Ag2CO3-mediated cascade annulation of internal alkynes under microwave heating.61 They performed the Ag2CO3-mediated reaction with 2-(phenylethynyl)aniline 18 and phenyl isothiocynate 19 in DMSO using a conventional method, and observed the poor isolated yield (47%) of the desired product. They also carried out the reaction without using Ag2CO3 but only a trace amount of the product was formed, which indicated the significant role of Ag2CO3 in the reaction medium. Thus, to improve the yields of desired products 20a–l, all Ag2CO3-mediated reactions of 2-(phenylethynyl)anilines 18a–g with phenyl isothiocyanates 19a–f were carried out in microwave conditions at 130 °C for 20–30 min and afforded good isolated yields (64–80%) (Scheme 7). This methodology offers several benefits, including good functional-group tolerance, mild microwave conditions, and short reaction times.
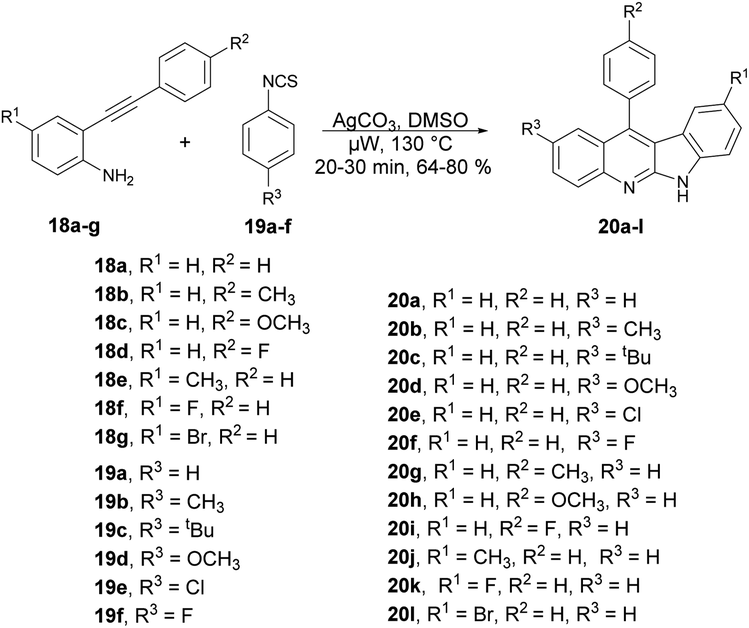 | ||
| Scheme 7 Synthesis of various indoloquinolines 20a–l. | ||
Huigens and co-workers developed a catalyst-free microwave-assisted Friedländer synthesis which provided a single-step conversion of various derivatives of 8-hydroxyquinolines with higher yield (72%) over that achieved with conventional heating (34% yield).62 Initially, they performed the reaction with generation of 2-amino-3-hydroxybenzaldehyde 21 which, upon condensation with ketones using conventional heating, afforded 8-hydroxyquinolines in low-to-moderate yields. Thus, to increase the product yields they also performed the condensation reaction with 2-amino-3-hydroxybenzaldehyde 21 and ketones 22a–g in ethanol. Using a microwave-irradiated technique, they heated the reaction mixture at 130 °C for 30–40 min to afford good isolated yields (48–84%) of designed products 23a–g (Scheme 8). They conducted additional analysis on the microwave-assisted Friedländer reaction using different substrates: significant enhancements in yields were observed for most of the quinoline library. The average yield, which was previously 34% using traditional oil-bath heating, improved to 72% with microwave irradiation.
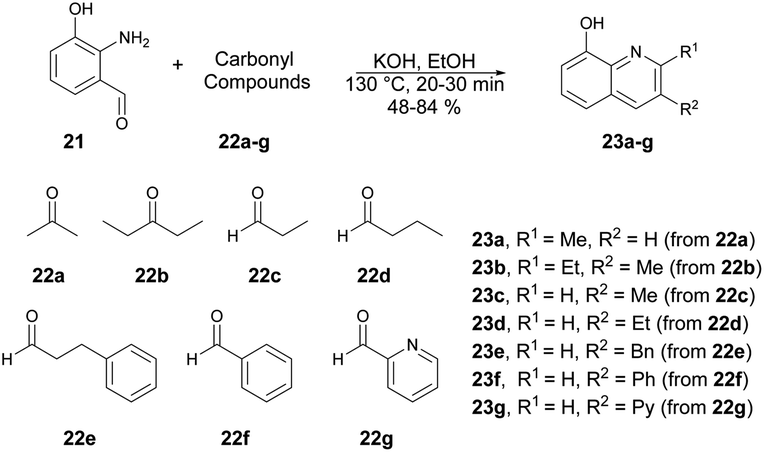 | ||
| Scheme 8 Friedländer synthesis of 8-hydroxyquinolines 23a–g. | ||
Baltork and co-workers developed an efficient and reusable catalyst to synthesize quinoline derivatives using microwave irradiation. They utilized Fe3O4-TDSN-Bi(III) as a catalyst in solvent-free conditions using microwaves for the regioselective synthesis of quinolines.63 They carried out the reaction with arylamine 24, arylaldehydes 25a–i and methyl propiolate 26 in the presence of Fe3O4-TDSN-Bi(III) as a catalyst under microwave irradiation at 80 °C to afford good-to-excellent yields (85–97%) of the designed products 27a–i (Scheme 9). They also performed the reaction without using a catalyst (Fe3O4-TDSN-Bi(III)) but there was no conversion in product, and they used a conventional method for quinoline synthesis but obtained a very low yield (40%) of the designed product 27. Thus, it was discovered that a catalyst and microwave irradiation were equally important to obtain desired products in high yields.
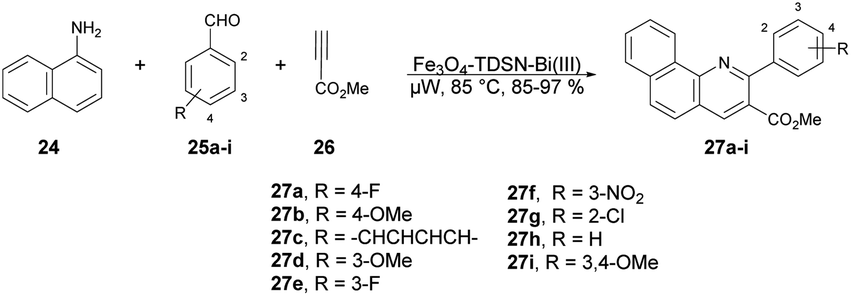 | ||
| Scheme 9 Synthesis of various quinolines 27a–i. | ||
A microwave-assisted development of a Povarov-type multicomponent reaction to synthesize quinolines was reported by Sharma and co-workers.64 They performed the reaction with anilines 28a–m, an alkyne and paraformaldehyde in the presence of (±)camphor-10-sulfonic acid (CSA; used as a promotor) and heated the reaction mixture at 90 °C for 20 min to afford poor-to-good yields (26–90%) of designed quinolines 30a–m (Scheme 10). The Povarov-type reaction involved [4 + 2] cycloaddition of imine (formed in situ with an aldehyde and aniline) and alkyne to afford various derivatives of quinolines 30a–m.
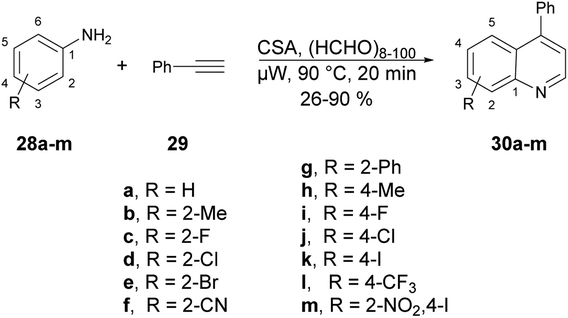 | ||
| Scheme 10 Synthesis of quinoline derivatives 30a–m. | ||
The Povarov-type multicomponent reaction could proceed through the formation of in situ-generated intermediate I from the condensation of paraformaldehyde and aniline. Cycloaddition of 29 with CSA activated imine (II) via a Povarov-type multicomponent cycloaddition reaction to afford intermediate III. The latter was converted to intermediate IV which, upon further removal of CSA and spontaneous oxidation, yielded the final product 30a (Scheme 11).
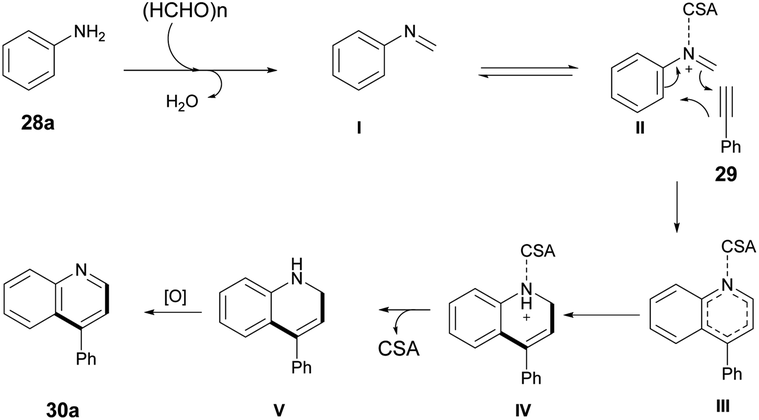 | ||
| Scheme 11 A plausible mechanism for quinoline synthesis. | ||
Kamble and co-workers reported an efficient microwave-assisted protocol for the synthesis of quinoline fused 1,4-benzodiazepine.65 They performed the reduction reaction with 2-chloro,6,7,8-substituted quinoline-3-carbaldehydes 31a–j to afford compounds 32a–j which, on further bromination and condensation with 1,2-phenylenediamine, afforded the final products quinoline-fused 1,4-benzodiazepines 34a–j. The condensation reaction was performed using brominated products 6,7,8-substituted 3-bromomethyl-2-chloro-quinolines 33a–j in a microwave reactor at 80 °C to afford excellent isolated yields (92–97%) of quinoline-fused 1,4-benzodiazepines 34a–j (Scheme 12). To compare the conventional and microwave irradiation methods, substituted quinoline derivatives were synthesized using a conventional method, resulting in final products 33a–j with satisfactory yields of 62–65%. Researchers reported a microwave-assisted synthesis of benzimidazoles using amides and observed improved yields. This observation prompted them to investigate further by reacting substituted 3-bromomethyl-2-chloroquinolines (33a–j) with 1,2-phenylenediamine under microwave conditions to obtain 33a–j. Remarkably, they achieved a significant increase in yield, ranging from 92% to 97%, within a very short time duration of 5 min using 150 W of microwave irradiation. Hence, microwaves produced good results in the form of higher yields, less time as well as atom-economy as compared with the traditional heating method.
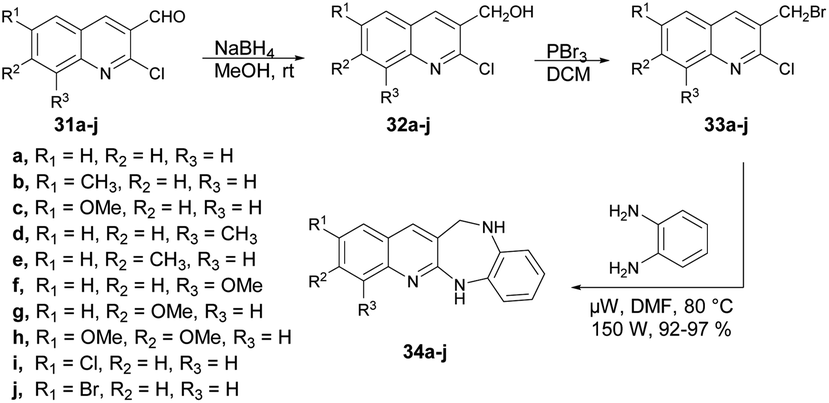 | ||
| Scheme 12 Synthesis of quinoline-fused 1,4-benzodiazepines 34a–j. | ||
Munir and co-workers reported a microwave-assisted synthesis of 2-((6/8-methyl-2-(piperidin-1-yl)quinolin-3-yl)methylene)hydrazinecarbothioamides.66 They developed a new series of piperidine-containing quinolinyl thiosemicarbazones using commercially available anilines. Initially, they performed the acetylation of anilines 35a–l, produced acetanilides 36a–l which, on further Vilsmeier–Haack formylation using dimethylformamide and phosphoryl chloride, afforded 2-chloroquinoline-3-carbaldehydes 37a–l. Subsequently, cetyltrimethylammonium bromide (CTAB)-catalyzed nucleophilic aromatic substitution of 37a–l with piperidine in polyethylene glycol-400 (PEG-400) produced 2-(piperidin-1-yl)quinoline-3-carbaldehydes 38a–l in 97–98% yields. Finally, microwave-assisted condensation of 38a–l with thiosemicarbazides afforded the desired hybrid compounds 39a–l in excellent yields (89–98%) within 3–5 min (Scheme 13). They investigated the reactions for comparative study between conventional and microwave irradiation and found a 10% fall in the yield using the conventional method instead of microwave irradiation.
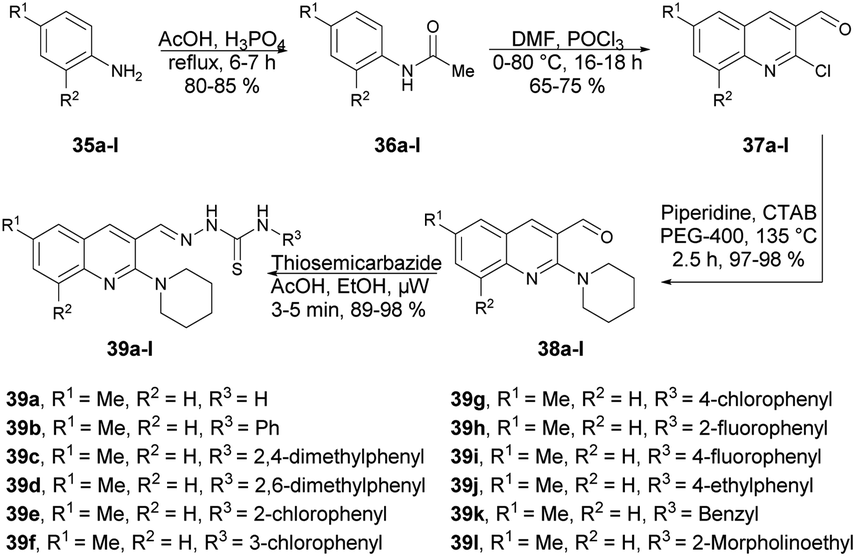 | ||
| Scheme 13 Synthesis of quinolinyl-hydrazinecarbothioamides 39a–l. | ||
Jonnalagadda reported a new method for synthesizing pyrazolo-[3,4-b]-quinolines in an eco-friendly, fast, and convenient way using a microwave-assisted multi-component/one-pot protocol.67 This method was conducted in aqueous ethanol at 50 °C for 5 min. This process resulted in a new series of pyrazolo-[3,4-b]-quinoline derivatives 43a–r with excellent yields ranging from 91% to 98%. These derivatives were produced by the reaction with aryl aldehydes 40a–r, dimedone 41, and 5-amino-3-methyl-1-phenylpyrazole 42 applying microwave conditions (Scheme 14). Furthermore, during optimization of the reaction, they observed a significant decrease in the yield (15%) when varying the reaction temperature from 50 °C to 30 °C. They also noticed that increasing the reaction temperature resulted in a lower product yield. This result provided evidence that maintaining a constant temperature was crucial for this reaction, making microwave heating a superior choice compared with traditional heating methods.
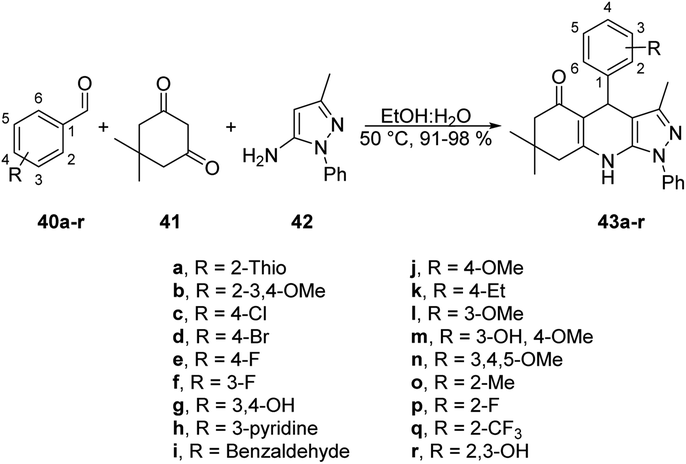 | ||
| Scheme 14 Synthesis of pyrazolo-[3,4-b]-quinoline derivatives 43a–r. | ||
Scheme 15 illustrates a plausible mechanism for the synthetic reaction. Initially, there is a Knoevenagel condensation reaction between dimedone 41 and an aryl aldehyde 40, leading to the formation of adduct A. The adduct A undergoes dehydration to produce intermediate B. Subsequently, a Michael-type addition reaction occurs between 5-amino-3-methyl-1-phenylpyrazole 42 and intermediate B, yielding a new intermediate, C. This intermediate C then undergoes tautomerisation to form an intermediate D. Finally, through cyclization and dehydration reactions, the product 43 is obtained.
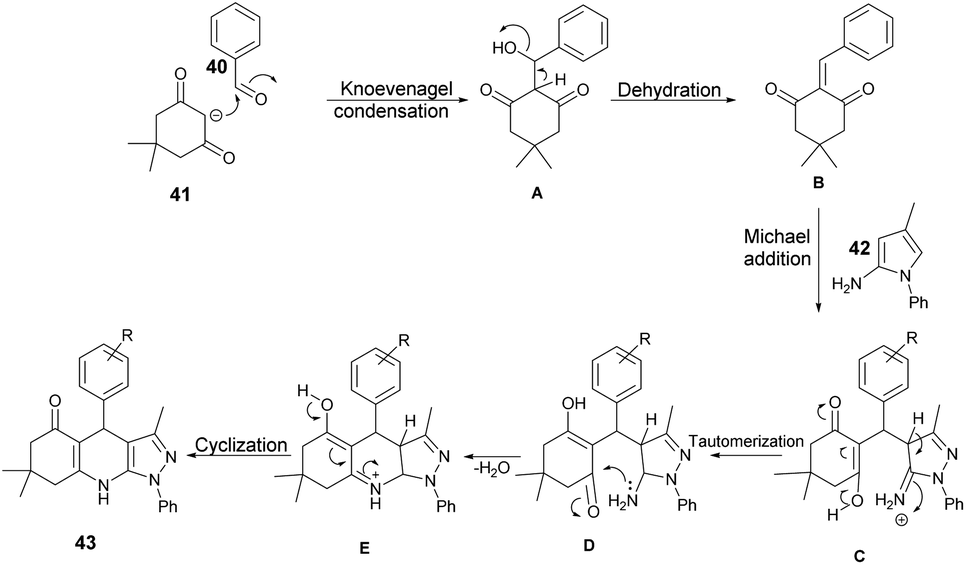 | ||
| Scheme 15 Plausible mechanism for the synthesis of pyrazolo-[3,4-b]-quinoline derivatives. | ||
Jonnalagadda and co-workers reported an efficient and environmentally benign approach for the synthesis of substituted 1,4-dihydroquinolines.68 The one-pot multicomponent reaction was performed using resorcinol 44, aromatic aldehydes 45a–j, ammonium acetate 46 and acetoacetanilide 47 under microwave conditions (100 W) at 110 °C for 8–10 min to afford good isolated yields (88–96%) of compounds 48a–j. The reaction proceeded through the Knoevenagel condensation, Michael addition followed by intramolecular cyclization (Scheme 16). To validate the advantage of microwaves, the reaction was conducted by heating a simple mixture of resorcinol, benzaldehyde, acetoacetanilide, and ammonium acetate in ethanol under reflux conditions, which led to completion of the reaction in 6 h and provided 48a at a yield of 79%. Recognizing the advantages associated with microwave irradiation for conducting reactions, researchers investigated the same reaction under microwave irradiation at 100 W and 110 °C in ethanol. Surprisingly, this microwave-assisted reaction reached completion in just 10 min, resulting in a higher yield of 48a (92%) compared with that using the traditional thermal reaction. Thus, microwaves are a better choice to proceed this type of reaction and obtain a maximum yield in a shorter time.
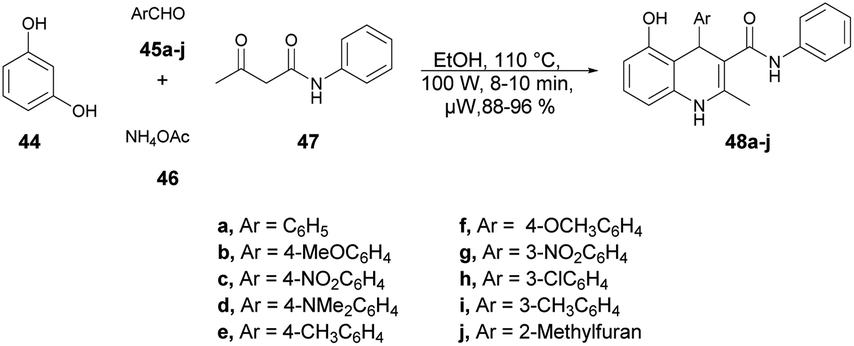 | ||
| Scheme 16 Synthesis of 1,4-dihydroquinolines 48a–j. | ||
Abonia and co-workers developed a catalyst-free and one-pot three-component protocol to synthesize dihydropyrido[2,3-d]pyrimidines and dihydro-1H-pyrazolo[3,4-b]pyridines containing a quinoline pharmacophore.69 They performed the microwave-assisted synthesis with formyl-quinoline derivatives 49a–f, primary heterocyclic amine 50 and cyclic 1,3-diketone 51 in DMF at 150 °C for 8 min, which afforded the good isolated yields (68–82%) of target compounds 52a–f (Scheme 17). The reaction proceeded via aldol condensation, Michael addition and intramolecular addition of an amine to a carbonyl group.
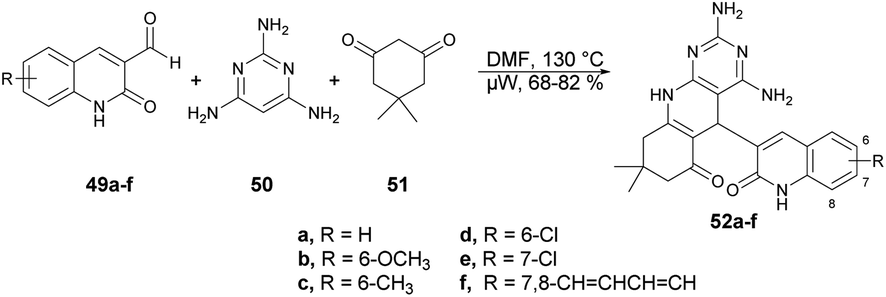 | ||
| Scheme 17 Synthesis of dihydropyrido[2,3-d]pyrimidines 52a–f. | ||
A microwave-assisted, temperature-variable, metal-free and acid-mediated procedure to prepare spiro-indolenines and quinolines was established by Eycken and co-workers.70 They discovered that indolyl-ynones 53a–p treated with TFA and continuous stirring at room temperature for 15 min afforded spiro-indolenines 54a–p in good isolated yields (41–99%) whereas, upon heating the reaction mixture at 100 °C for 30 min under microwave conditions (100 W) afforded the quinolines 55a–p in excellent yields (95–99%). For the synthesis of spiro-indolenine, the reaction proceeded through acid-catalyzed intramolecular Michael addition and double-protonation of the spiro-indolenine, rearrangement with the formation of cyclopropane ring, followed by expansion and restoration of aromaticity (Scheme 18). Thus, in this protocol, two types of product were noticed while using the same reactant in both cases. When the reaction was performed at room temperature using the conventional method, it generated spiro-indolenines and applying microwave conditions led to quinoline being formed as a major product. Hence, in some cases, microwave-assisted synthesis produces different products compared with those using traditional heating methods.
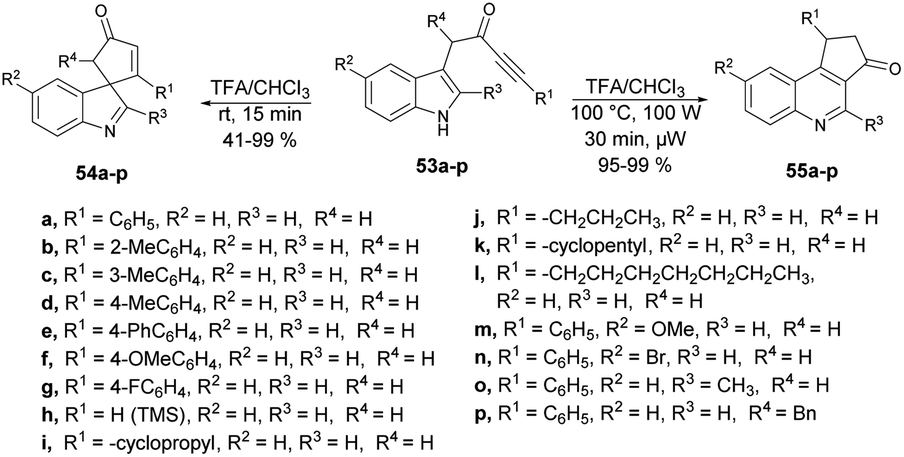 | ||
| Scheme 18 Synthesis of spiro-indolenines 54a–p and quinoline analogs 55a–p. | ||
Scheme 19 illustrates a plausible mechanism. Initially, an intramolecular Michael addition is catalyzed by a Brønsted acid, involving the C-3 carbon of the indole and the ynone, resulting in the formation of spiro-indolenine 54. The activation energy for the formation of 3a is notably higher than that for 54, as evidenced by the requirement of a higher temperature for the rearrangement process. Under elevated temperature conditions, the second step involves double-protonation of the spiro-indolenine, leading to ketone tautomerization and the generation of an electrophilic iminium ion. This iminium ion is then attacked by the enol, followed by a rearrangement that forms a cyclopropane ring, leading to ring expansion and the restoration of aromaticity. Consequently, this series of reactions culminates in the formation of the quinoline core 55.
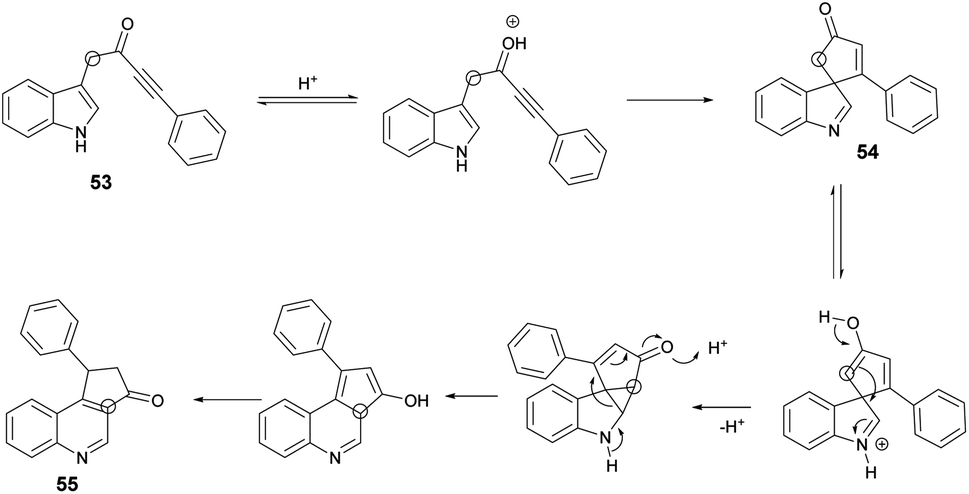 | ||
| Scheme 19 Plausible mechanism for the synthesis of spiro-indolenines and quinolines. | ||
Kumar and co-workers reported a microwave-assisted synthesis of a series of quinoline derivatives having antitubercular properties.71 To synthesize 4-aminoquinoline-phthalimides 58a–x, they performed the reaction with various phthalic anhydrides 56a–f and 4-aminoquinoline-diamines 57a–e in DMSO, heated at 160 °C for 2 min in microwave obtained good isolated yields (81–92%) (Scheme 20). They have also evaluated the antitubercular potential of synthesized compounds against mc26230 strain of Mycobacterium tuberculosis and compound 58d found the most potent (MIC99 value of 13.7 μM) among all the derived compounds.
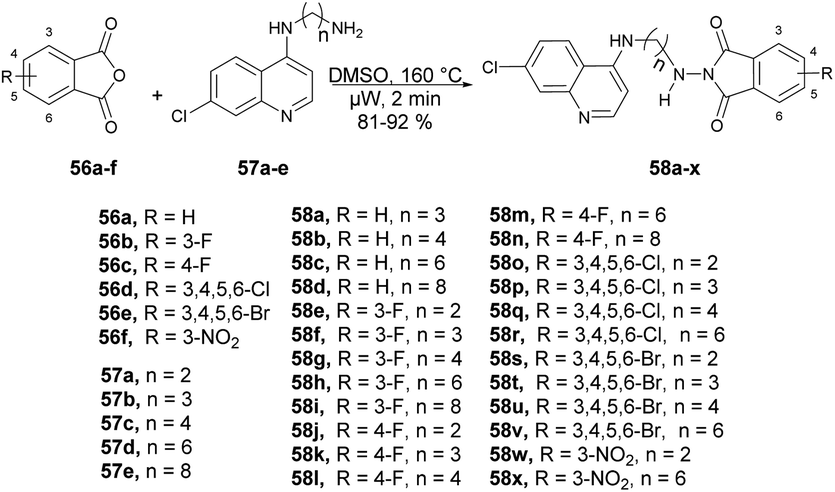 | ||
| Scheme 20 Synthesis of 4-aminoquinoline-phthalimides 58a–x. | ||
Jonnalagadda and co-workers developed a series of quinoline derivatives in ethanol under microwave conditions.72 They performed the TEA-catalyzed reaction between 5,5-dimethylcyclohexane-1,3-dione, aromatic aldehydes, malononitrile and ammonium acetate in ethanolic solvent under microwave conditions at 100 W for 12 min to afford excellent yields (85–98%) of quinoline derivatives 63a–h (Scheme 21).
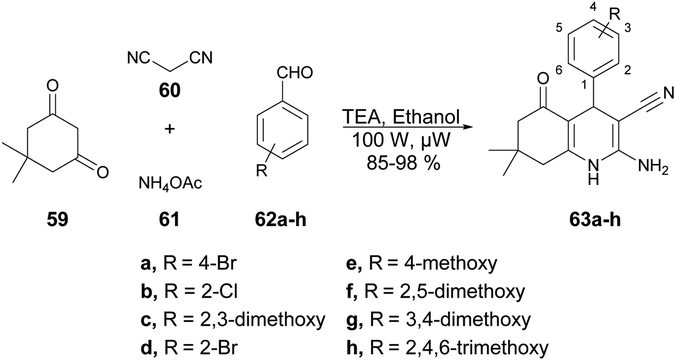 | ||
| Scheme 21 Synthesis of quinoline derivatives 63a–h. | ||
Yu and co-workers reported a microwave-assisted development of polyheterocyclic-fused quinoline-2-thiones through catalyst-free annulation of various hetero anilines and carbon disulphide using green solvent.73 In order to synthesize imidazopyridine-fused quinoline-2-thiones 66a–r, they performed the reaction with various substituted 2-(imidazo[1,2-a]pyridin-2-yl)anilines 64a–r and carbon disulfide 65, heated at 140 °C for 30 min in water using microwave and obtained good isolated yields (51–99%). The reaction proceeds through 6π-electrocyclization followed by the aromatization of the intermediate (Scheme 22). This represents the first example of synthesizing polyheterocyclic-fused quinoline-2-thiones, showcasing several advantageous aspects, such as a catalyst-free process, a short reaction duration, a reusable and environmentally friendly solvent system, and easy isolation via simple filtration, thereby avoiding the need for column chromatography.
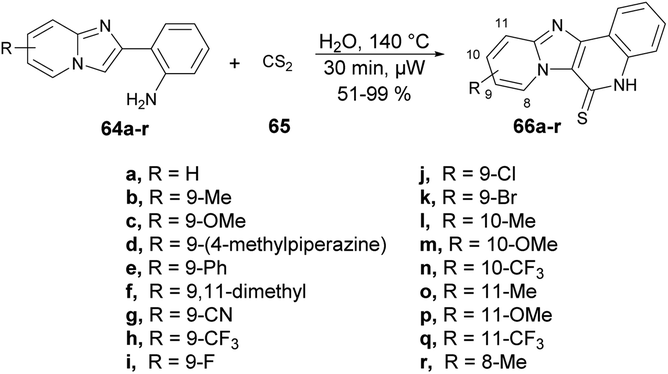 | ||
| Scheme 22 Synthesis of imidazopyridine fused quinoline-2-thiones 66a–r. | ||
Patel and co-workers developed a green and efficient approach to synthesize a series of quinoline-4-carboxylic derivatives using an organocatalyst under microwave irradiation.74 They carried out the three-component reaction of aromatic aldehydes 67a–c, substituted anilines 68a–g, and pyruvic acid 69 in ethanol using p-TSA as a catalyst under microwave conditions at 80 °C for 3 min, which furnished good isolated yields (50–80%) of quinoline derivatives 70a–p (Scheme 23). They also performed the reaction using a conventional method but it required longer (3 h to overnight) instead of 3–4 min under microwave conditions. They also observed a lower yield in the conventional method as compared with the microwave method.
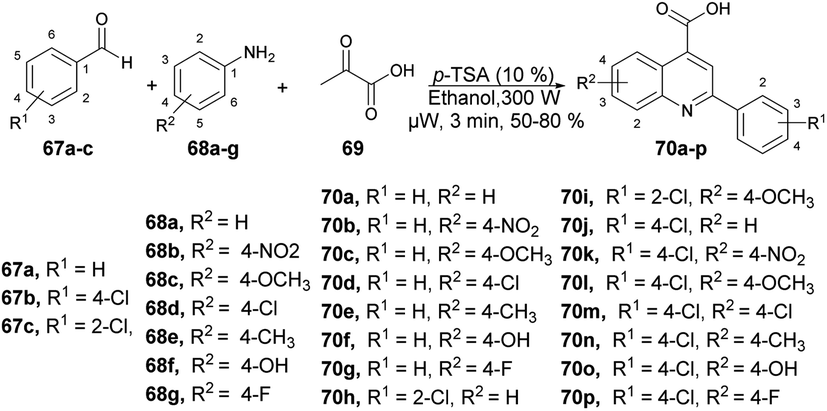 | ||
| Scheme 23 Synthesis of quinoline derivatives 70a–p. | ||
Wang and co-workers discovered a novel, environmentally friendly synthetic route for the Friedländer quinoline synthesis of various substituted quinolines.75 They performed the reaction with 2-aminobenzophenone 71 and various dicarbonyl moieties 72a–k using Nafion NR50 as a catalyst in ethanolic solvent under microwave conditions at 200 °C for 60 min to afford good isolated yields (43–95%) of polysubstituted quinolines 73a–k (Scheme 24).
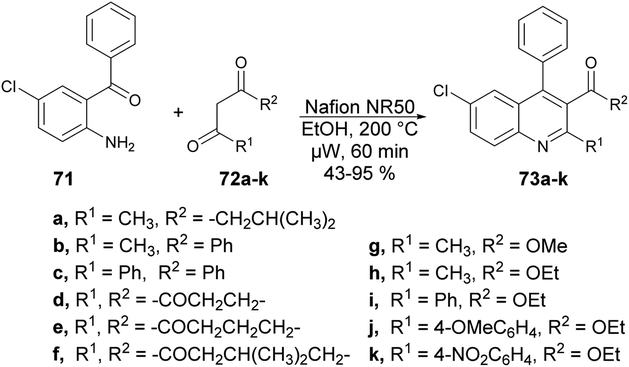 | ||
| Scheme 24 Synthesis of polysubstituted quinolines 73a–k. | ||
Andrade and co-workers reported a new method to synthesize poly-functionalized 2-quinolinones from N-(o-ethynylaryl)-acrylamides (1,7-enynes).76 They discovered the microwave-assisted synthesis of 2-quinolinone-fused γ-lactones using Fenton's reagent within 10 s. They performed the reaction with various derivatives of compound 74a–o and tBuOH/formamide using Fenton's reagent heated in microwave-irradiated conditions at 130 °C for 10 s to afford the desired compounds 75a–o in isolated yields of 33–45% (Scheme 25). A similar reaction was also performed using a conventional method, but took (4 h) instead of 10 s in microwave conditions.
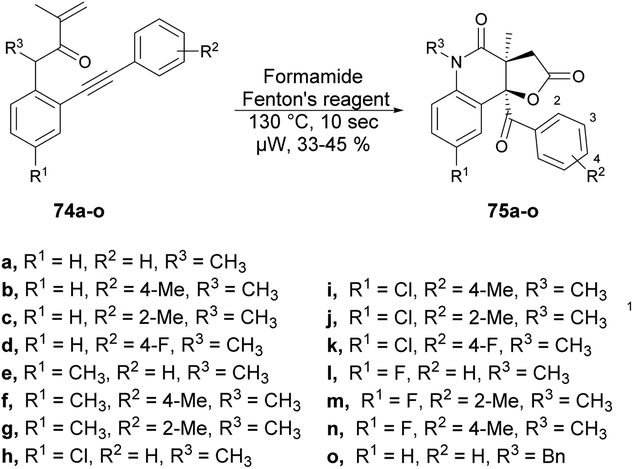 | ||
| Scheme 25 Synthesis of tetrahydrofuro quinoline-diones 75a–o. | ||
Scheme 26 illustrates the possible mechanism for the synthesis of 2-quinolinone-fused γ-lactones 75a–o. The initial phase involves the generation of hydroxyl radicals using Fenton's reaction. In an acidic environment, 6H2O2 and Fe(II) facilitate the production of hydroxyl radicals, water, and Fe(III). The reaction also allows for the regeneration of Fe(II) from the reaction between Fe(III) and H2O2. Subsequently, formamide is transformed into the carbamoyl radical by the action of the hydroxyl radical. This carbamoyl radical then attacks the carbon–carbon double bond of the acrylic amide moiety and undergoes cyclization with the carbon–carbon triple bond, resulting in the formation of vinyl radical II. The recombination of hydroxyl and vinyl radicals II leads to the formation of enol III. Enol III is then converted into racemic epoxide IV through an epoxidation reaction. Studies have shown that peroxides, in the presence of iron, can oxidize C–C double bonds that are conjugated to aromatic rings, resulting in the formation of epoxides. The pathway for the construction of lactone involves a selective epoxide-opening reaction via an SN2-type mechanism, which explains the observed maximum yield (50%) in this process. Once intermediate IV is formed with the appropriate stereochemistry (anti), 5-exo-tet cyclization can take place. This involves a nucleophilic attack of the amide group from the backside of the epoxide through an SN2-type mechanism, leading to the formation of intermediate V (geminal diol). After simple hydrolysis and dehydration of V, product 75a–o is obtained with the release of ammonia.
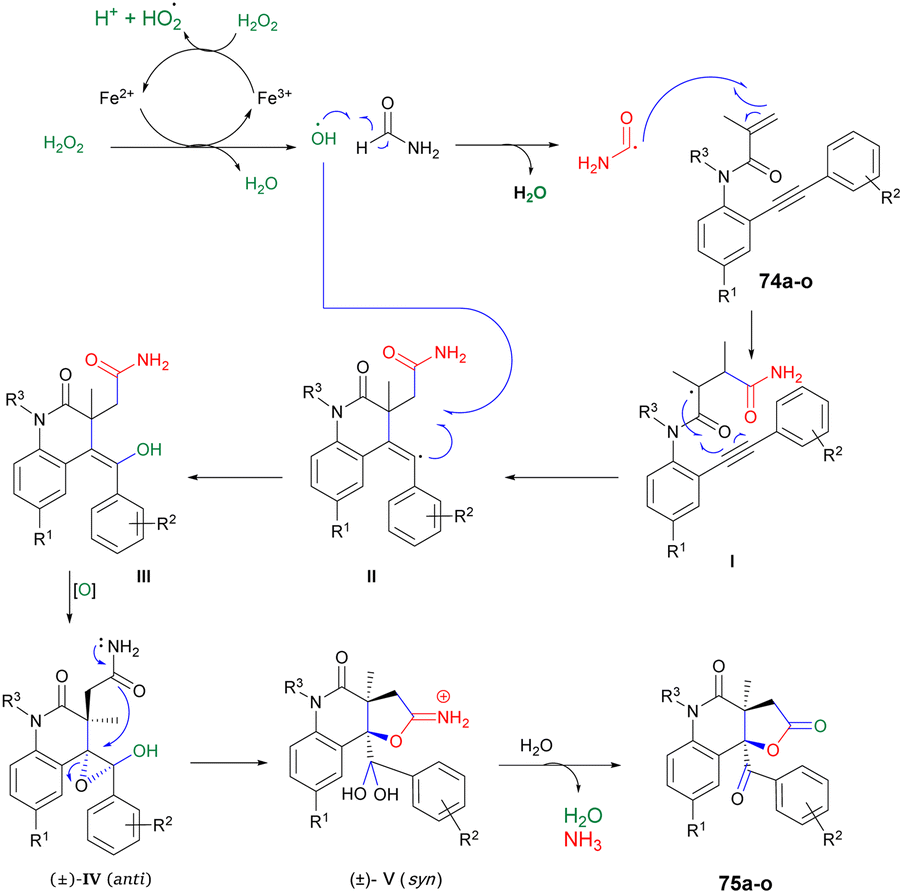 | ||
| Scheme 26 Proposed mechanism for the synthesis of 2-quinolinone-fused γ-lactones 75a–75o. | ||
2.2. Pyrazolopyrimidines and their derivatives
The pyrazolopyrimidine scaffold is an important class of N-heterocycles in the realm of drug discovery.77 It comprises fusion between a pyrazole ring and a six-membered pyrimidine ring. Pyrazolopyrimidines exist in several isomeric forms, including pyrazolo [3,4-d]pyrimidines, pyrazolo [4,3-d]pyrimidines, pyrazolo [5,1-b]pyrimidines, and pyrazolo [1,5-a]pyrimidines, all of which hold scientific importance.78 The pyrazolopyrimidine structure is particularly alluring due to its synthetic feasibility and profound pharmacological significance. Several well-known drugs in the market incorporate the pyrazolopyrimidine nucleus, including zaleplon,79 indiplon,80 allopurinol,81 dinaciclib,82 ocinaplon,83 pyrazophos,84 and sildenafil.85 Pyrazolopyrimidines showcase a diverse range of pharmacological properties, serving as cyclin-dependent kinase (CDK) inhibitors,86 antibacterial agents,87 antifungal agents,88 anti-proliferative compounds,89 as well as anti-leishmanial90 and antiviral agents.91 Therefore, the synthesis of these types of scaffolds and investigation of their biological potential are of keen interest to researchers. Herein, we have compiled the microwave-assisted synthetic developments on pyrazolopyrimidines and their derivatives.An acid-promoted one-pot development of 6-amino-pyrazolopyrimidines was reported by Wong and co-workers.92 The reaction was carried out with 1H-pyrazol-5-yl-N,N-dimethylformamidines or 5-amino-1H-pyrazole-4-carbaldehydes 76a–o and cyanamide (NH2CN) in an acid-mediated solution applying microwave conditions at 55 °C to afford good isolated yields (63–86%) of desired compounds 77a–o (Scheme 27). The overall reaction involves deprotection, imination, acid promoted heterocyclization and aromatization. For comparative experiments, conventional heating was conducted using a standard hot plate/magnetic stirrer. However, this method resulted in a lower yield compared with that obtained using microwave irradiation.
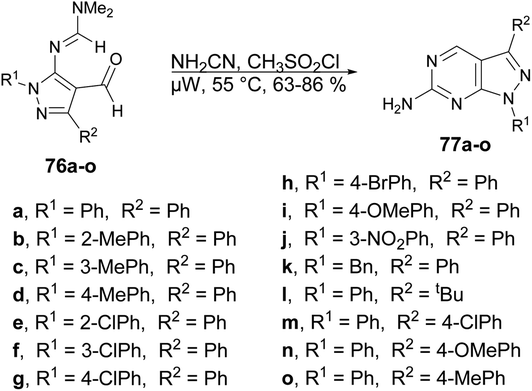 | ||
| Scheme 27 Synthesis of 6-amino-pyrazolopyrimidines 77a–o. | ||
A protocol for microwave-assisted Suzuki–Miyaura cross-coupling reaction of 3-bromo pyrazolo[1,5-a]pyrimidin-5(4H)-one was reported by Ababri and co-workers.93 They optimized arylation reactions using various aryl and heteroaryl boronic acids 79a–n with a catalyst XPhosPdG2/XPhos to avoid the debromination reaction. The improved conditions were effectively utilized to synthesize various arylated pyrazolo[1,5-a]pyrimidin-5-ones 80a–n from their respective brominated starting material 78. In addition, the Suzuki–Miyaura cross-coupling conditions were used to perform the second C-5 arylation of C-3-arylated pyrazolo[1,5-a]pyrimidin-5-ones. By utilizing microwave irradiation at 135 °C instead of conventional thermal heating, only the coupled product 80a–n was generated, with a yield of 67–89% (Scheme 28). The most noteworthy aspect was that the reaction duration was shortened from 12 h to 40 min, and there was no indication of the debrominated product in the crude material.
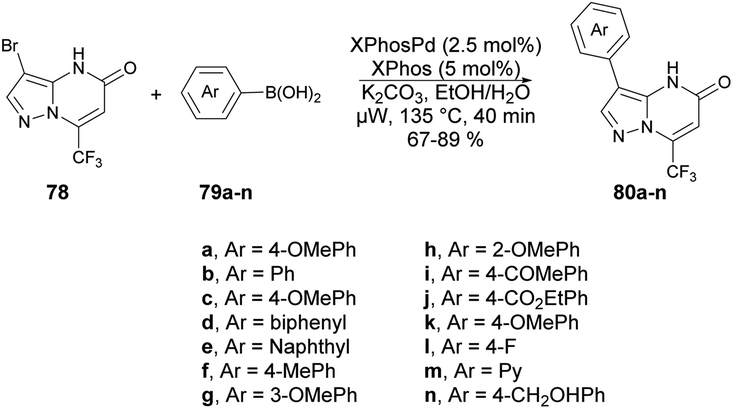 | ||
| Scheme 28 Synthesis of arylated pyrazolo[1,5-a]pyrimidin-5-ones 80a–n. | ||
Peterson and co-workers reported a straightforward synthesis of 3-substituted pyrazolo[1,5-a]pyrimidines using microwave-assisted synthesis.94 The synthesis of pyrazolo[1,5-a]pyrimidines 84a–g involved sequential treatment of commercially available acetonitrile derivative 81 with DMF-dimethylacetal (120 °C, 20 min), followed by treatment with NH2NH2·HBr (120 °C, 20 min), and 1,1,3,3-tetramethoxypropane or 2-aryl-substituted malondialdehdyes (120 °C, 20 min) (Scheme 29).
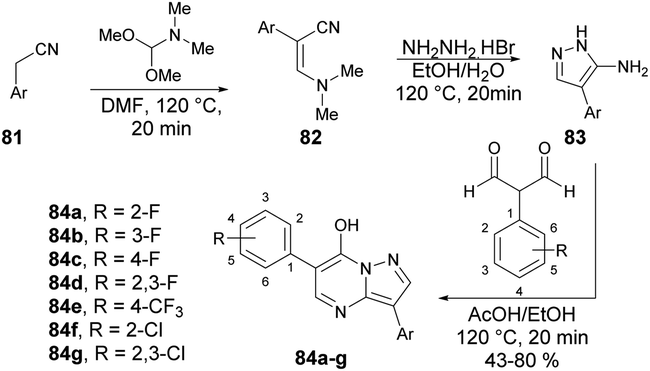 | ||
| Scheme 29 Synthesis of 3-substituted pyrazolo[1,5-a]pyrimidines 84a–g. | ||
Dolzhenko and co-workers developed a microwave-assisted three-component protocol for the synthesis of pyrazolo[3,4-d]pyrimidin-4-ones 87a–o.95 They carried out the microwave-assisted one-pot reaction using methyl 5-aminopyrazole-4-carboxylates 85a–g, trimethyl orthoformate and primary amines 86a–h to afford poor-to-good isolated yields of desired products 87a–o. The reaction mixture was heated at 160 °C for 55 min in ethanolic conditions (Scheme 30). The method that was optimized for benzylamine proved to be effective in producing pyrazolo[3,4-d]pyrimidin-4-ones with substituted benzylamines and their analogues, resulting in yields of 60–85%. Nevertheless, when aromatic amines were utilized as substrates, the yields dropped to 21–53%. To compare conventional and microwave methods, the reaction was also attempted under conventional heating with reflux in EtOH. However, even after 3 days of heating, the desired product was not obtained. Thus, the developed microwave protocol offers several advantages, including a shorter reaction time, efficient utilization of materials and steps, and the convenience of isolating the product without the need for chromatography.
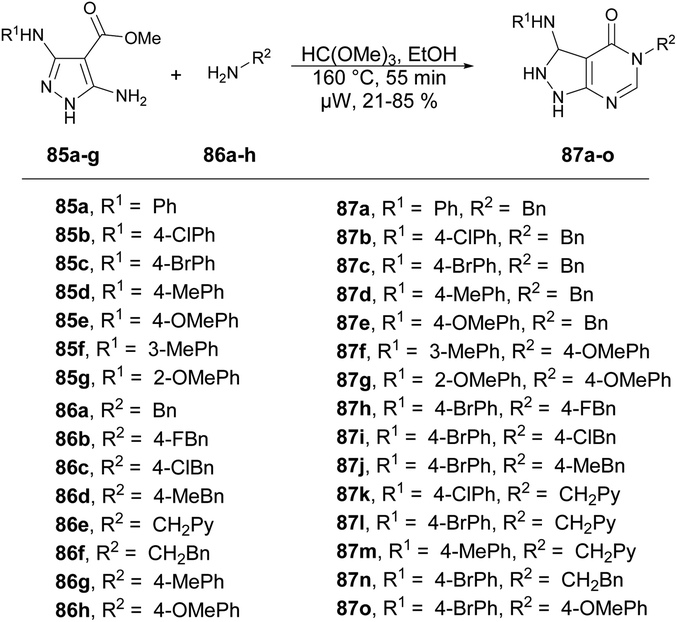 | ||
| Scheme 30 Synthesis of pyrazolo[3,4-d]pyrimidin-4-ones 87a–o. | ||
A palladium-catalyzed microwave-assisted development was reported by Portilla and co-workers for the synthesis of functionalized 6-(aryldiazenyl)pyrazolo[1,5-a]pyrimidin-7-amines 90a–q from the cyclization of 3-oxo-2-(2-arylhydrazinylidene)butanenitriles and 5-amino-1H-pyrazoles in neat conditions.96 They performed the reaction with 2-arylhydrazinyliden-3-oxobutanenitriles 88a–c and various substituted 5-amino-1H-pyrazoles 89a–h without a solvent under microwave conditions at 180 °C for 4 min to afford yields of 70–94% of 6-(aryldiazenyl)pyrazolo[1,5-a]pyrimidin-7-amines 90a–q (Scheme 31). In order to validate the efficiency and advantage of microwaves, the reaction was also carried out using a conventional heating method with variation of solvent and temperature. When the reaction was carried out in ethanol or toluene, it did not favour the desired product. Interestingly, they observed that a high-boiling-point solvent such as DMSO and DMF favoured the product at high temperature, but the yield was not satisfactory. Thus, to retain the high temperature with low-boiling-point solvent was not possible, which is why microwaves were the best choice to perform the reaction in ethanol while using high temperature and pressure.
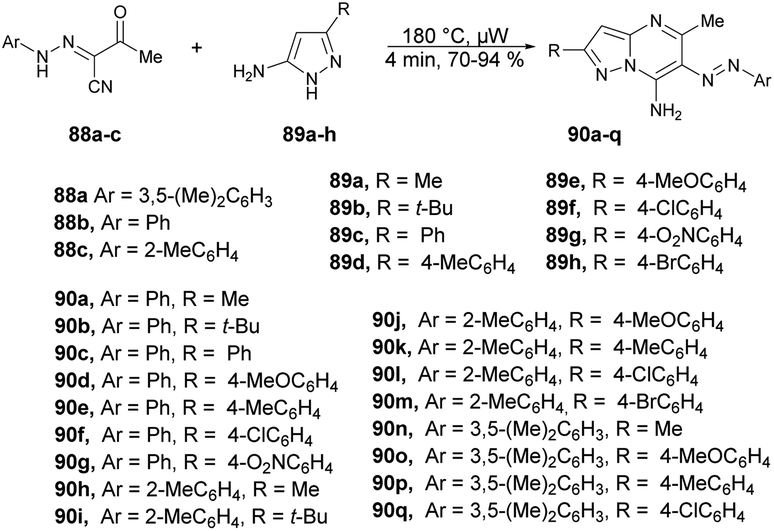 | ||
| Scheme 31 Synthesis of 6-(aryldiazenyl)pyrazolo[1,5-a]pyrimidin-7-amines 90a–q. | ||
Raboin and co-workers reported microwave-assisted developments of 7-substituted pyrazolo[1,5-a]pyrimidines. They developed a one-pot two-step reaction through palladium-catalyzed CH-activation and saponification–decarboxylation reaction.97 The reaction was carried out between 7-ethoxycarbonylmethyl-5-methyl-2-phenylpyrazolo[1,5-a]pyrimidine 91, aryl or heteroaryl halides 92a–l and 2-dicyclohexylphosphino-20-(N,N-dimethylamino) biphenyl (DavePhos) in the presence of a palladium catalyst and toluene as a solvent under microwave irradiation at 150 °C for 30 min which, upon further saponification–decarboxylation, afforded moderate-to-good isolated yields (48–80%) of 7-substituted pyrazolo[1,5-a]pyrimidines 93a–l (Scheme 32). In contrast to the classical method, this novel approach offers several consistent advantages, such as achieving good yields, shorter reaction times, a reduced number of steps, and the utilization of commercially available and relatively non-toxic compounds.
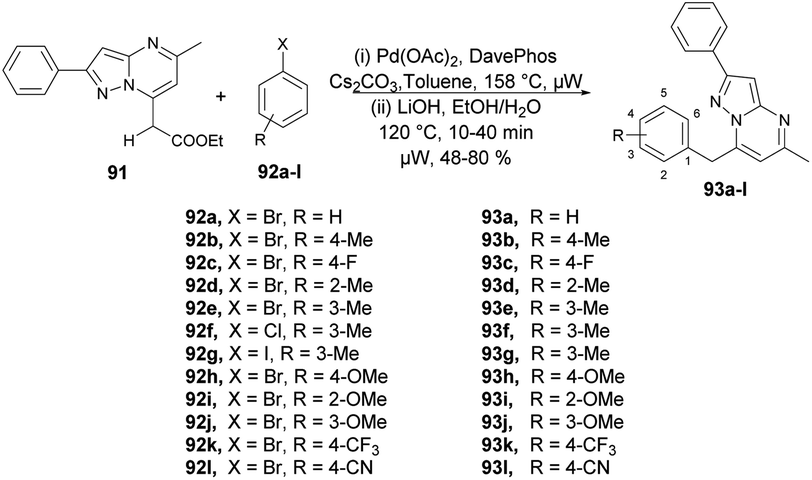 | ||
| Scheme 32 Synthesis of 7-substituted pyrazolo[1,5-a]pyrimidines 93a–l. | ||
Zhang and co-workers reported a microwave-assisted chemoselective synthesis of 5,6-pyrazolo[1,5-a]pyrimidines 96a–l and their antifungal properties against Cytospora sp., Colletotrichum gloeosporioides, Botrytis cinerea, Alternaria solani, and Fusarium solani.98 They performed the reaction between various isoflavones 94a–k and substituted 3-aminopyrazole 95a–b in DMSO using potassium tert-butoxide as a base under microwave conditions at 100 °C for 15 min to yield 60–85% isolated yields of 5,6-pyrazolo[1,5-a]pyrimidines 96a–l (Scheme 33).
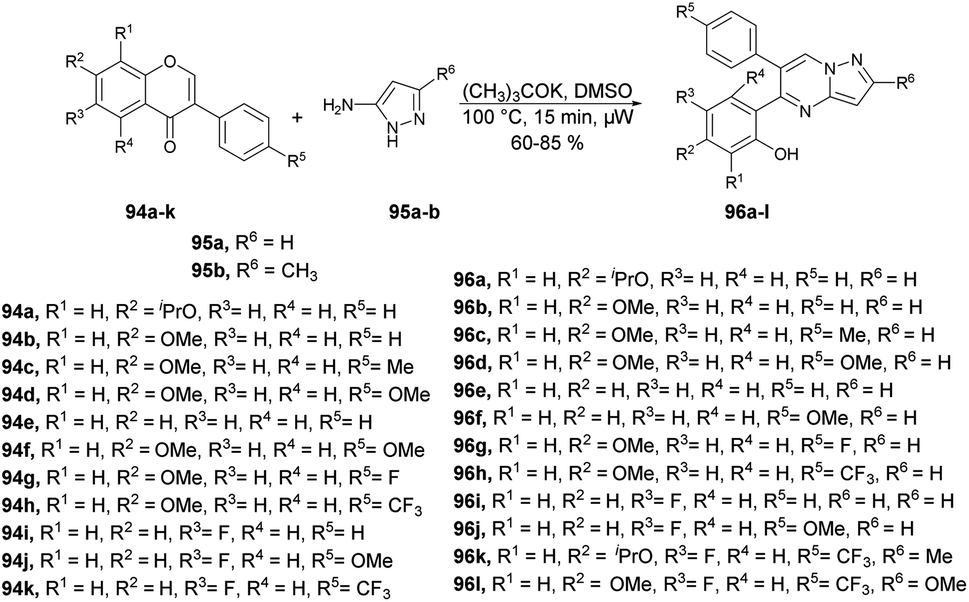 | ||
| Scheme 33 Synthesis of 5,6-pyrazolo[1,5-a]pyrimidines 96a–l. | ||
An efficient, microwave-assisted route to synthesize various substituted 3-halo and 3-nitropyrazolo[1,5-a]pyrimidines was reported by Portilla and co-workers.99 They performed the reaction between various β-enaminones 97a–d and NH-5-aminopyrazoles 98a–e under microwave conditions at 180 °C and 300 W for 2 min to afford good isolated yields (85–97%) of 3-halo and 3-nitropyrazolo[1,5-a]pyrimidines 99a–p (Scheme 34). The reaction proceeds through condensation of β-enaminones and NH-5-aminopyrazole followed by regioselective electrophilic substitution using electrophilic reagents. During the optimization process, the researchers explored various factors such as temperature, solvent, and the impact of conventional heating versus microwave irradiation. Refluxing the reaction in ethanol or toluene for 12 h did not result in the formation of the desired product. When they attempted the reaction in DMF at a higher temperature, the yield of the desired product remained poor. It became evident that higher temperatures were necessary for completion of the product. Consequently, the conventional heating method was not sufficient to achieve the maximum yield in high-temperature reactions. Microwave irradiation proved to be a crucial technique in attaining higher yields, especially when using high-boiling-point solvents.
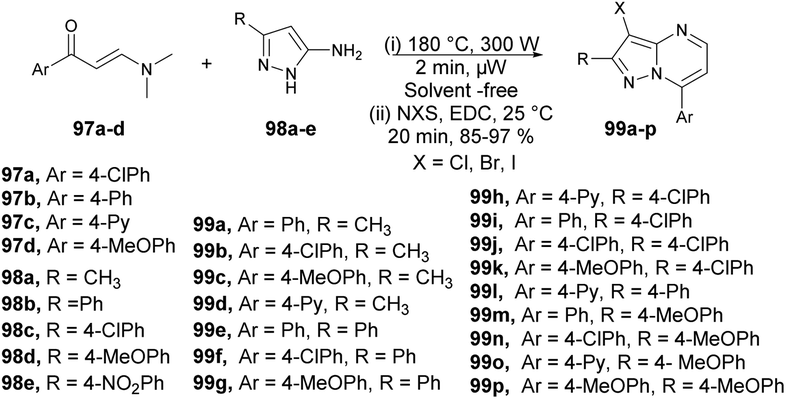 | ||
| Scheme 34 Synthesis of 3-substituted pyrazolo[1,5-a]pyrimidines 99a–p. | ||
Portilla and co-workers developed a new microwave-assisted route for the regioselective development of 3-formylpyrazolo[1,5-a]pyrimidines.100 They carried out the cyclocondensation reaction between β-enaminones 100a–d and NH-3-aminopyrazoles 101a–e under microwave conditions at 180 °C for 2 min to afford an intermediate 102, which was not isolated and reaction was continued by the formylation of an iminium salt using Vilsmeier–Haack reagent under microwave conditions at 80 °C for 10 min to afford good to excellent isolated yields (72–91%) of 3-formylpyrazolo[1,5-a]pyrimidines 103a–j (Scheme 35).
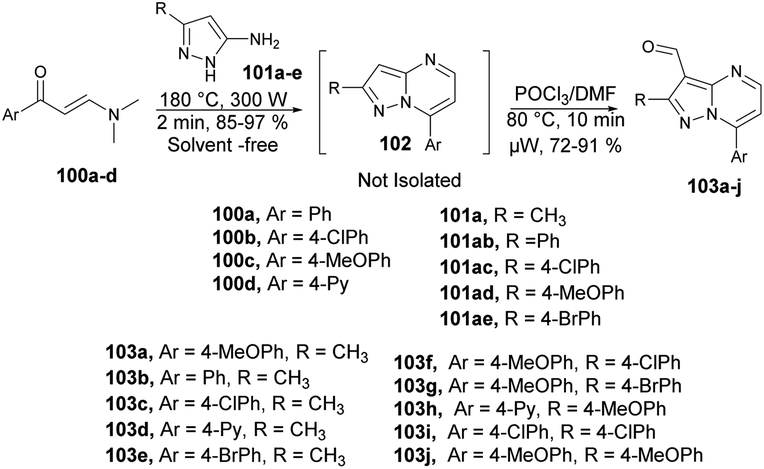 | ||
| Scheme 35 Synthesis of diverse 3-formylpyrazolo[1,5-a]pyrimidines 103a–j. | ||
To investigate the specific effect of microwave heating compared with conventional heating on reaction acceleration, a preheated sand bath was employed as a heat source for comparative experiments. Encouragingly, the heteroaryl aldehyde was obtained using conventional heating at 80 °C for 60 min. However, this method was not entirely satisfactory because there was a loss of intermediate mass during the transfer from the microwave tube to the round-bottom flask. The validity of their hypothesis was confirmed by performing a reliable cyclocondensation-formylation sequence directly in the same microwave tube without the need for further isolation steps.
Ameta and co-workers reported an efficient and environmentally friendly method to synthesize pyrazolo[3,4-d]pyrimidine-3-ones using microwave irradiation.101 The reaction was carried out between 2,4-dinitrophenylhydrazine 104 and diethyl malonate in 1,3-dibutylimidazolium bromide ionic liquid under microwave irradiation for 5–8 min to afford compound 105 which, on further treatment with base and 1,3-dibutylimidazolium bromide ionic liquid under microwave irradiation for 7–8 min, afforded compound 106a–e. The latter, on further reaction with urea under similar conditions, afforded desired compounds 107a–e in moderate-to-good isolated yields (65–85%) (Scheme 36).
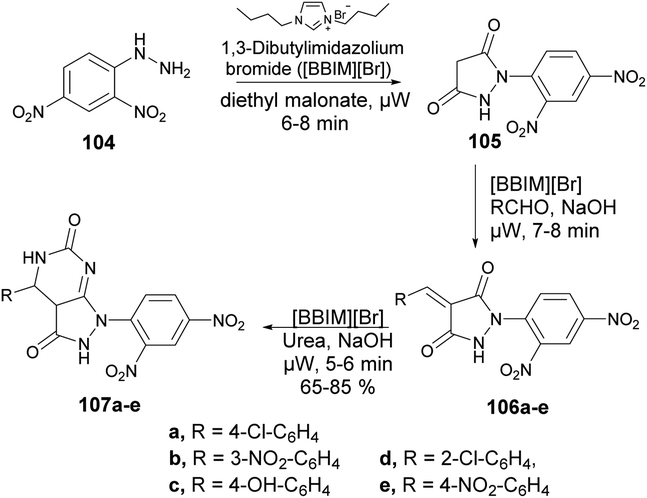 | ||
| Scheme 36 Synthesis of pyrazolo[3,4-d]pyrimidin-3-ones 107a–e. | ||
A microwave-assisted synthesis was reported by Shaaban and co-workers for the preparation of a series of pyrazolo[1,5-a]pyrimidines through the reaction of 2-(phenylsulfonyl)-1-(4-(phenylsulfonyl)phenyl)ethan-1-one, 2-benzenesulfonyl-1-(4-benzenesulfonyl-phenyl)-3-dimethylamino-propenone and 3-(dimethylamino)-1-(4-(phenylsulfonyl)phenyl)prop-2-en-1-one.102 They performed the reaction between a mixture of enaminosulfone 108 with arylazodiaminopyrazole 109a–g in acetic acid under microwave conditions for 15 min to afford good isolated yields (90–95%) of pyrazolo[1,5-a]pyrimidines 110a–g (Scheme 37). Furthermore, a comparative study between conventional and microwave methods was carried out, revealing a 10% decrease in yield when using conventional heating as opposed to microwave heating. Consequently, in order to achieve a superior yield with fewer impurities, the researchers opted to employ microwave heating for all reactions.
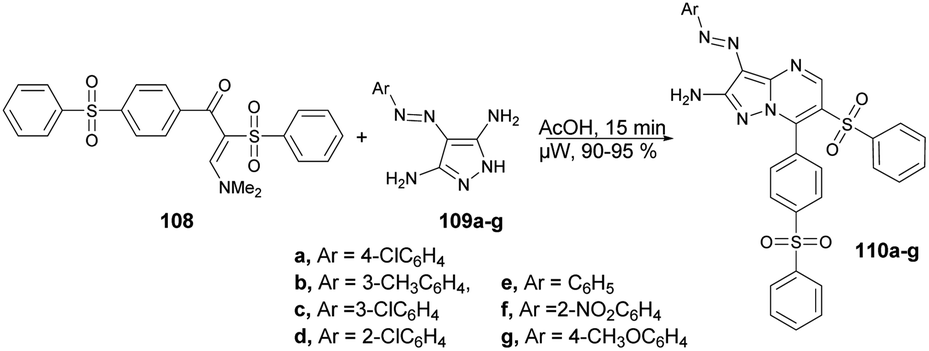 | ||
| Scheme 37 Synthesis of pyrazolo[1,5-a]pyrimidines 110a–g. | ||
From a mechanistic perspective, it was anticipated that the multi-component synthesis of compounds 110a–g would occur through Michael-type addition under acidic conditions. Scheme 38 illustrates the process leading to the formation of pyrazolo[1,5-a]pyrimidine derivatives 110a–g. This process involves Michael-type addition, resulting in the formation of a non-isolable Michael adduct IV, which then undergoes cyclocondensation with the loss of dimethylamine, leading to the formation of three additional non-isolable intermediates V–VII. Subsequently, a water molecule is eliminated to yield the corresponding pyrazolo[1,5-a]pyrimidine derivatives 110a–g.
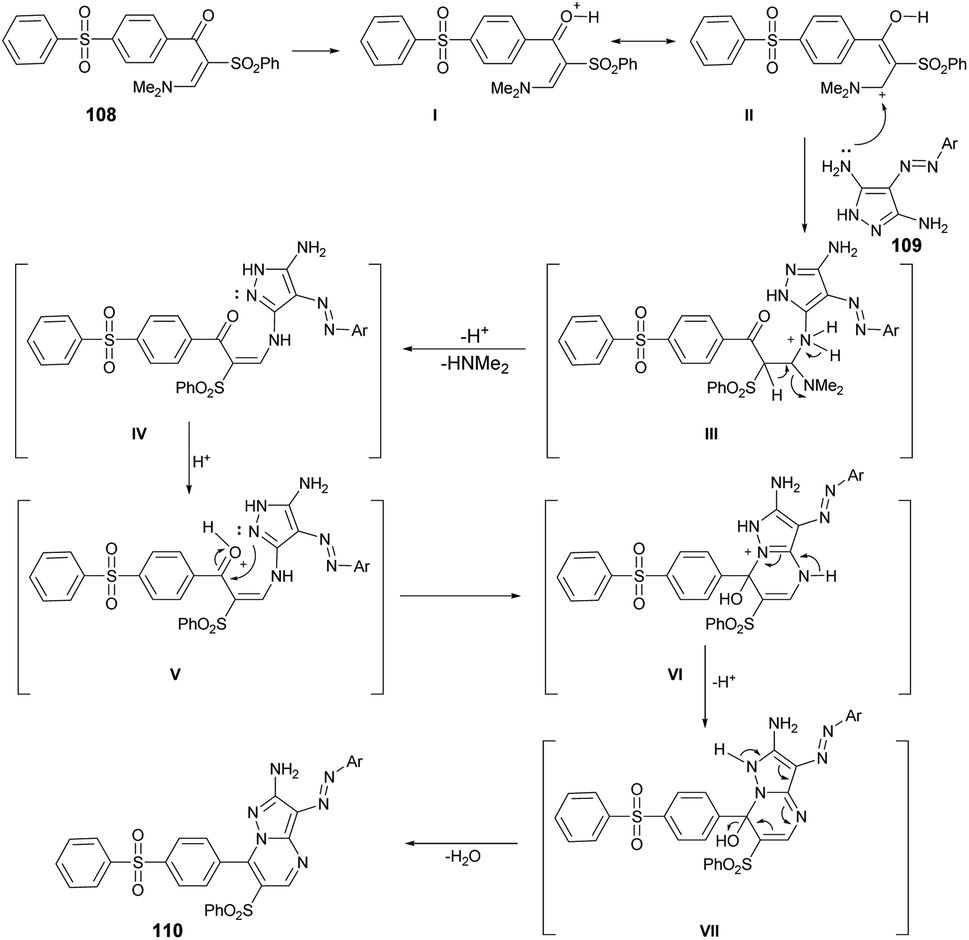 | ||
| Scheme 38 General mechanism for the synthesis of compound 110a–g. | ||
Abbas and co-workers reported a microwave-assisted procedure for the synthesis of a series of pyrazolo[1,5-a]pyrimidine derivatives. They developed various symmetrical and asymmetrical 3,6-diarylazo-2,5,7-triaminopyrazolo[1,5-a]pyrimidines by applying microwave conditions.103 The reaction was carried out between hydrazone 111 and hydrazine hydrate in ethanol/pyridine, heated under microwave conditions at 140 °C for 2 min to afford a good isolated yield (71–78%) of desired compounds 112a–b (Scheme 39). Additionally, a comparative study encompassing conventional, microwave, and ultrasound techniques was undertaken. The results revealed a 10% reduction in yield when utilizing the conventional method over 4–7 h compared with a mere 2 min with microwave irradiation. Furthermore, the ultrasound technique also exhibited a decline in yield (10–15%) in comparison with the microwave-assisted approach. These findings clearly indicate that microwave irradiation is the preferred choice for conducting such experiments.
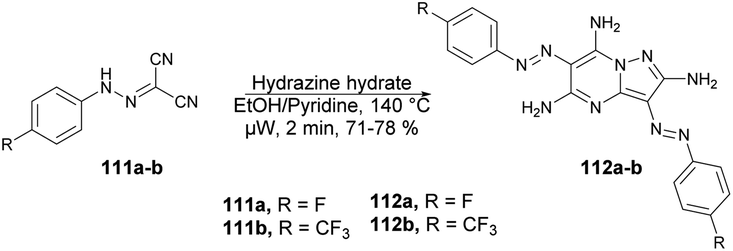 | ||
| Scheme 39 Synthesis of diarylazo-2,5,7-triaminopyrazolo[1,5-a]pyrimidines 112a–b. | ||
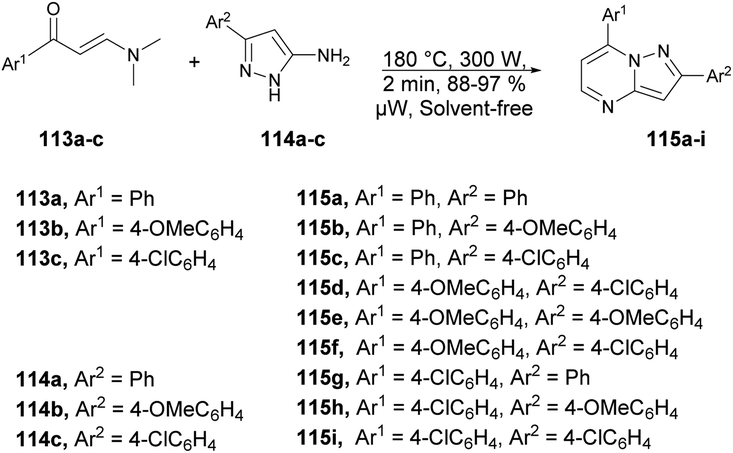
A microwave-assisted development of a series of 2,7-diarylpyrazolo[1,5-a]pyrimidine derivatives and investigation of their antitumor activity against a colorectal carcinoma cell line was reported by Miscione and co-workers.104 They performed the reaction between various β-enaminones 113a–c and NH-5-aminopyrazole 114a–c under microwave conditions at 180 °C and 300 W for 2 min to afford good isolated yields (88–97%) of pyrazolo[1,5-a]pyrimidines 115a–i (Scheme 40). The reaction proceeds through condensation of β-enaminones and NH-5-aminopyrazole followed by regioselective electrophilic substitution.
 | ||
| Scheme 40 Synthesis of diverse substituted pyrazolo[1,5-a]pyrimidines 115a–i. | ||
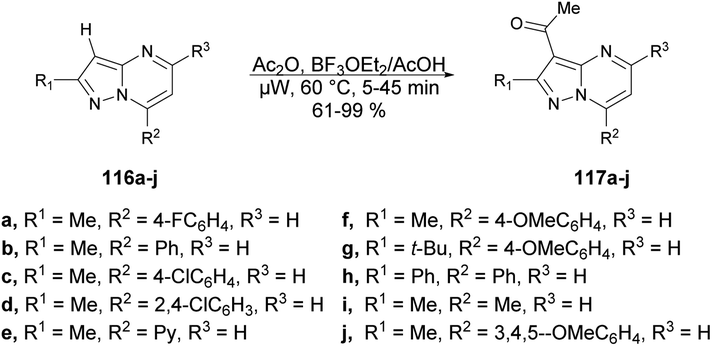
Portilla and co-workers described a straightforward microwave-assisted BF3-mediated acetylation reaction of pyrazolo[1,5-a]pyrimidines.105 The synthesis of this fundamental building block was reported in good yield, even with substrates containing electron-deficient or strongly hindered groups under mild reaction conditions. Importantly, several N-hetero aromatic substrates were effectively used for acetylation using a newly found technique. They performed the Lewis acid (BF3OEt2)-promoted acetylation reaction with pyrazolo[1,5-a]pyrimidines 116a–j using acetic acid as a solvent under microwave conditions at 60 °C for 5–45 min, which furnished good to excellent isolated yields (61–99%) of acetylated products 117a–j (Scheme 41).
 | ||
| Scheme 41 Synthesis of diverse acetylated pyrazolo[1,5-a]pyrimidines 117a–j. | ||
2.3. Coumarin and its derivatives
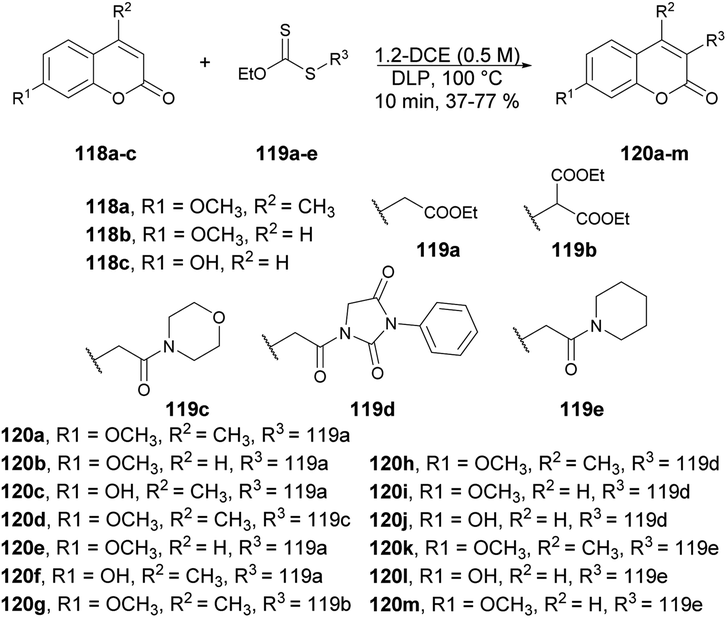
Coumarin derivatives have gained recognition for their diverse range of biological activities, such as antimicrobial,106,107 antioxidant and anti-inflammatory properties.108 Coumarin derivatives, with their unique and versatile oxygen-containing heterocyclic structure, are widely distributed in nature and can be found in numerous naturally occurring and synthetic bioactive molecules.109 This structure makes them a privileged scaffold in medicinal chemistry. Consequently, many coumarin derivatives have been extracted from natural sources, designed, synthesized, and assessed for various pharmacological targets.110 Furthermore, coumarin-based ion receptors,111 fluorescent probes,112–114 and biological stains are experiencing rapid growth, with extensive applications in monitoring enzyme activity, complex biological events, and precise pharmacological and pharmacokinetic properties within living cells.115 As a result, the extraction, synthesis, and biological evaluation of coumarins have become highly attractive and rapidly evolving areas of research.116–118 Thus, we have highlighted the latest synthetic pathways that use microwave-irradiated techniques and theoretical studies. This special issue places a strong emphasis on exploring the diverse facets of coumarin derivatives.Miranda and co-workers developed an efficient, microwave-assisted xanthate-based direct alkylation of the coumarin ring system.119 They carried out the C-3 radical alkylation of various derivatives of coumarin 118a–c and xanthate of ethyl acetate 119a–e using dilauril peroxide in toluene at 100 °C for 10 min under microwave irradiation to afford the desired products 120a–m in low-to-moderate isolated yields (Scheme 42). Furthermore, a comparative study between conventional and microwave methods was carried out, revealing a 5% decrease in yield when using conventional heating as opposed to microwave heating.
 | ||
| Scheme 42 C-3 radical alkylation of coumarins and formation of alkylated products 120a–m. | ||
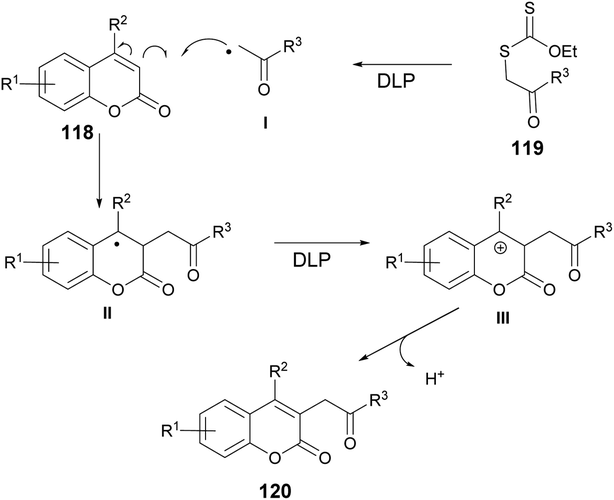
According to the suggested mechanism depicted in Scheme 43, the addition of radical I (formed from xanthate 119 through the action of dilauroyl peroxide (DLP)) to the coumarin system 118 could result in the formation of stabilized benzyl radical II. In the presence of an appropriate amount of DLP in the reaction medium, radical intermediate II might undergo oxidation, leading to the formation of benzyl carbocation III. The deprotonation of carbocation III would subsequently restore the double bond, ultimately yielding the alkylated product 120.
 | ||
| Scheme 43 Proposed mechanism for the oxidative radical addition products 120a–m. | ||
A copper-catalyzed four-component synthesis of 3-N-sulfonylamidine coumarins using microwaves was reported by Punniyamurthy and co-workers.120 They performed the coupling reaction with salicylaldehydes 121a–p, propiolate 122, sulfonyl azides 123, and secondary amines 124. Using a copper source along with a basic solvent under microwave conditions at 130 °C for 1 h afforded poor-to-good isolated yields of desired products 125a–p (Scheme 44). They found microwaves a more effective tool for these chemical reactions compared with traditional heating processes. This was due to the ability to produce higher reactivity, operate under milder conditions, and yield greater selectivity, all of which offer significant practical benefits. The results of that study suggest that use of microwave irradiation may lead to new opportunities for advancing the synthesis of highly functionalized coumarin derivatives through multiple components. Moreover, microwave-assisted organic synthesis offers the benefits of increased reactivity, gentle reaction conditions, and enhanced selectivity. The catalytic cycle depicted in Scheme 45 implies that the reaction could proceed through intermediates III and IV to generate the desired heterocycle 125a–p. It means that the combination of 122 and 123 in a cycloaddition reaction may lead to the formation of ketenimine II through I. The nucleophilic addition of amine to intermediate II could give rise to intermediate III, which, in turn, may react with the aldehyde to form intermediate IV. Further intermediate IV may subsequently undergo cyclization through transesterification, ultimately resulting in the synthesis of the target compound 125a–p as illustrated in Scheme 45.
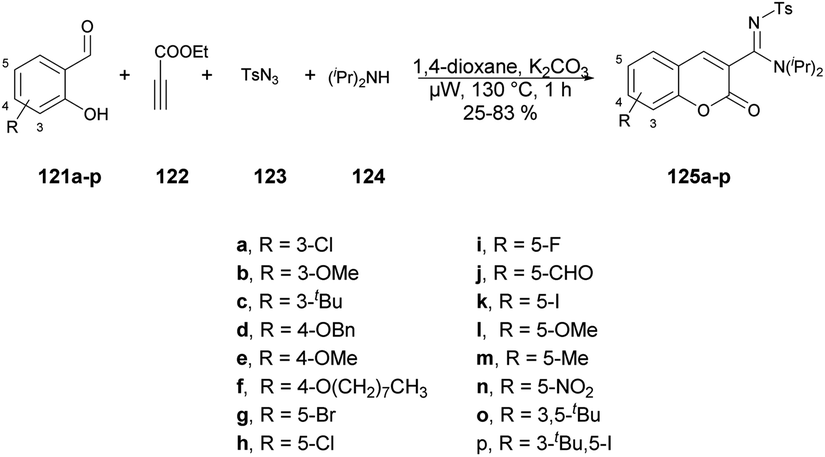 | ||
| Scheme 44 Synthesis of 3-N-sulfonylamidine coumarins 125a–p. | ||
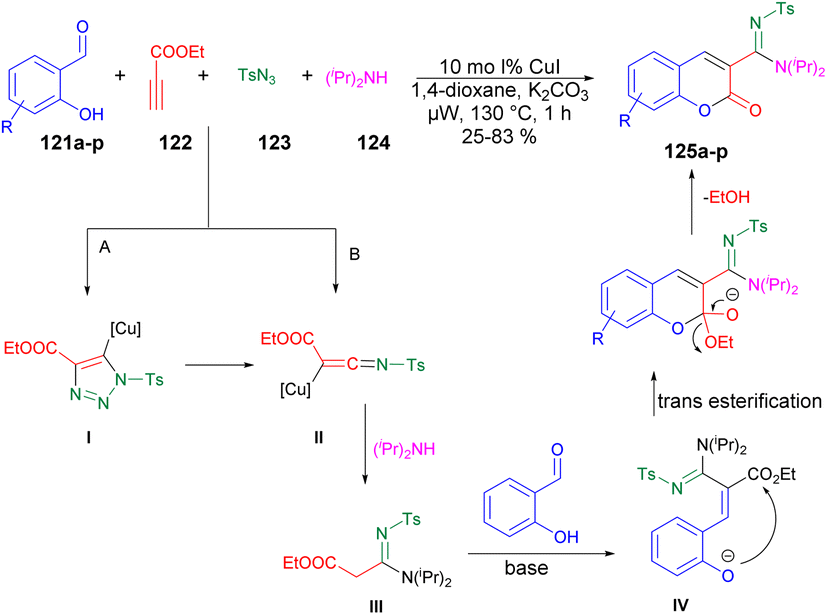 | ||
| Scheme 45 Catalytic cycle for the synthesis of 3-N-sulfonylamidine coumarins 125a–p. | ||
A facile and catalyst-free protocol was reported by Chen and co-workers to synthesize pentacyclic compounds containing coumarin, pyrrole and indene.121 They carried out the reaction with N-substituted 4-aminocoumarins 126a–p and ninhydrin 127 using toluene as a solvent under microwave heating at 110 °C. All reactions were carried out in a catalyst-free and regioselective manner, which afforded good-to-excellent yields (70–92%) of designed products 128a–p (Scheme 46). Microwave heating offers several advantages over conventional heating, including accelerated reaction rates, reduced energy consumption, and the production of cleaner products with fewer impurities. As a result of these benefits, microwave-assisted organic synthesis has garnered significant attention in the scientific community.
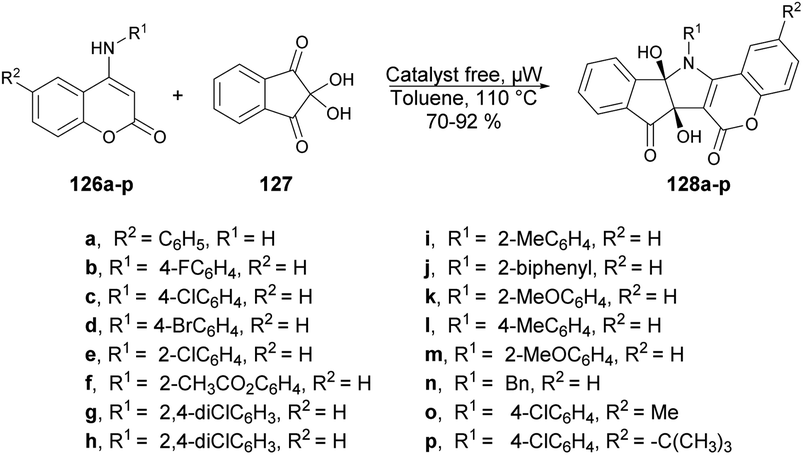 | ||
| Scheme 46 Catalyst free synthesis of coumarin derivatives 128a–p. | ||
Zhang and co-workers reported a microwave-assisted synthesis of coumarin[8,7-e][1,3]oxazine derivatives and evaluation of their antifungal activity.122 They accomplished a one-pot three-component Mannich reaction in this synthesis. Thus, they initially synthesized the various coumarin derivatives via a Pechmann condensation reaction. Then, using their designed 7-hydroxycoumarins 129a–j, they carried out the reaction with various amines in the presence of a catalytic amount of 4-N,N-dimethylaminopyridine (DMAP) using 1,4-dioxane as a solvent under microwave irradiation at 100 °C to afford the coumarin[8,7-e][1,3]oxazine derivatives 130a–j in good isolated yields (57–80%) (Scheme 47). The antifungal activities of derived compounds were evaluated against Alternaria mali, Rhizoctonia solani, Alternaria solani, Gibberella zeae, Colletotrichum capsica and Botrytis cinerea. It was found that the most active compound was 130i with an EC50 value of 0.0021 μM against Botrytis cinerea. In their initial study, they employed microwave irradiation for the synthesis of the designed coumarin–oxazines instead of conventional heating. The results indicated that the reaction times could be significantly reduced, from approximately 6 h to just 30 min.
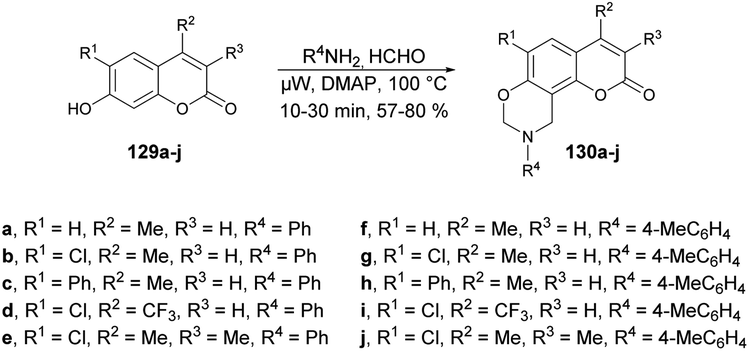 | ||
| Scheme 47 Synthesis of coumarin[8,7-e][1,3]oxazine derivatives 130a–j. | ||
Genovese and co-workers reported a microwave-assisted development of coumarin derivatives using Yb(OTf)3 as a catalyst.123 They performed the reaction with various substituted phenols 131a–j and propiolic acid 132 using Yb(OTf)3 as a catalyst under microwave irradiation at 80 °C and 200 W for 2 min to afford excellent yields (91–98%) of designed coumarin derivatives 133a–j (Scheme 48). Instead of microwave-assisted synthesis they carried out the reactions via a traditional heating method but, due to the poor yields of designed compounds, they opted for the microwave-irradiated technique.
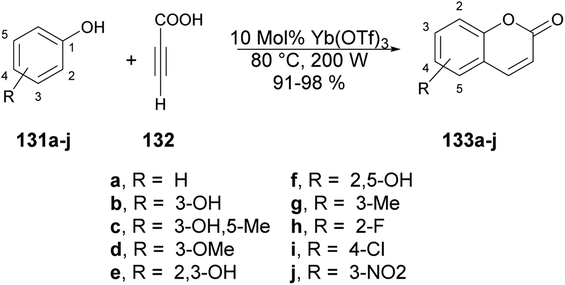 | ||
| Scheme 48 Yb(OTf)3-catalyzed synthesis of various coumarins 133a–j. | ||
A solvent-free microwave-assisted methodology for the synthesis of 7-hydroxy-4-methylcoumarin was reported by Bouasla and co-workers.124 They selected phenol, resorcinol and ethyl acetoacetate as reactants for a Pechmann condensation reaction. The catalytic activity of various heterogenous acid catalysts, including Amberlyst-15, zeolite, and hybrid silica functionalized with sulfonic acid, was examined in the solvent-free microwave-assisted synthesis of corresponding coumarin derivatives. They found that Amberlyst-15 to be the most active catalyst in microwave conditions and yielded 7-Hydroxy-4-methylcoumarin 136 in 97% yield (Scheme 49). The Pechmann reaction was also conducted using conventional thermal heating. However, even after 20 min, the phenol conversion did not exceed 18%. This outcome underscores the potential of microwave irradiation as a viable alternative to conventional heating methods for this type of reaction.
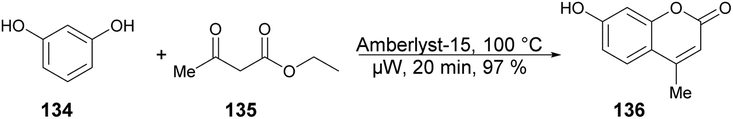 | ||
| Scheme 49 Synthesis of 7-hydroxy-4-methylcoumarin 136. | ||
A facile and efficient protocol for the synthesis of hetero[5]helicene-like molecules and coumarin derivatives was reported by Lin and co-workers. They used silica sulfuric acid (SSA) as a catalyst with various solvents and reaction temperatures for reaction selectivity to afford coumarin derivatives or hetero[5]helicene-like molecules.125 They found the best results when they carried out the reaction with coumarin and 3-aminopyrazole using SSA as a catalyst in ethylene glycol (EG) under microwave conditions at 130 °C (Scheme 50). After optimising the reaction conditions, various substrates with substituted functional groups were investigated. The results indicated that incorporating an electron-withdrawing group as the substituent R3 led to higher yields and a shorter reaction time. Conversely, when an electron-donating group was utilized as the R3 substituent, lower yields were observed, and the reaction time was prolonged.
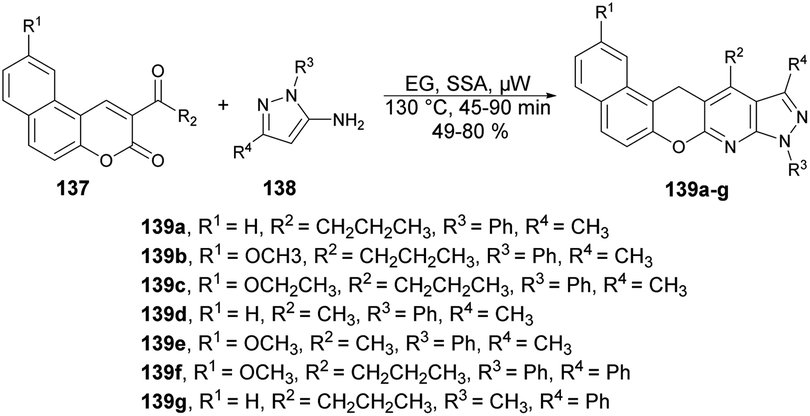 | ||
| Scheme 50 Synthesis of coumarin derivatives 139a–g. | ||
Inamdar and co-workers synthesized 5-methyl-4-[(2Z)-4-(2-oxo-2H-chromen-3-yl)-2-(4-arylimino)thiazol-3(2H)-yl]-2-(4-aryl)-2H-1,2,4-triazol-3(4H)-ones using a microwave-assisted technique.126 They performed the one-pot multi-component reaction with 3-(2-Bromoacetyl)-2H-chromen-2-one, various substituted triazoles 141a–e and diverse isothiocyanatobenzenes 142a–e in DMF under microwave irradiation at 120 °C for 15 min to afford good-to-excellent isolated yields (78–84%) of designed products 143a–n (Scheme 51). This microwave-assisted approach was solvent-free and catalyst-free, which provides environmentally friendly conditions to develop various derivatives of coumarin. Thus, this is one of the best green approaches for a complex synthesis.
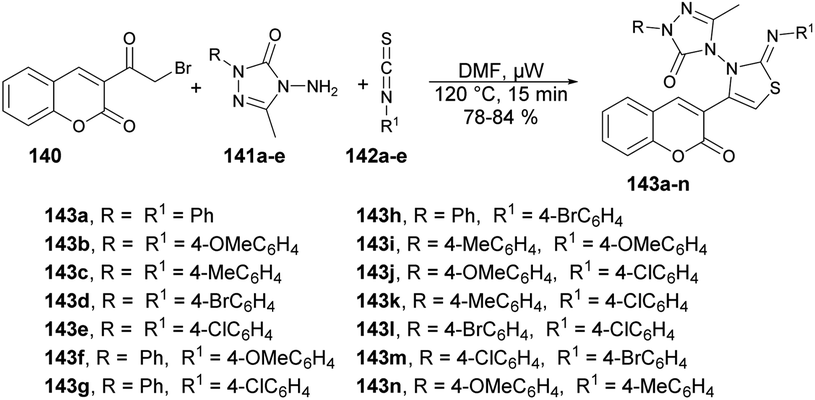 | ||
| Scheme 51 Synthesis of 5-methyl-4-[(2Z)-4-(2-oxo-2H-chromen-3-yl)-2-(4-arylimino)thiazol-3(2H)-yl]-2-(4-aryl)-2H-1,2,4-triazol-3(4H)-ones 143a–n. | ||
Seferoğlu and co-workers reported that a range of fluorescent compounds were produced by synthesizing a novel series of 2-coumarin-3-yl-imidazo[1,2-a]pyrimidines 146a–g using functionalized coumarin and pyrimidine units with different substitutions (Scheme 52).127 Another set of compounds, which included unsubstituted coumarin and 7-dialkylaminocoumarin 149a–f as a fluorophore, were obtained through palladium-catalyzed cross-coupling reactions. This was achieved by coupling 6-bromo-2-coumarin-3-yl-imidazopyrimidine derivatives 147a–b with various arylboronic acids 148a–c (Scheme 53). Microwave irradiation was used in all the reaction steps and compared with the conventional method. It was observed that microwave synthesis was facile, productive and occurred in a short time (30 min) as compared with the conventional method (24 h).
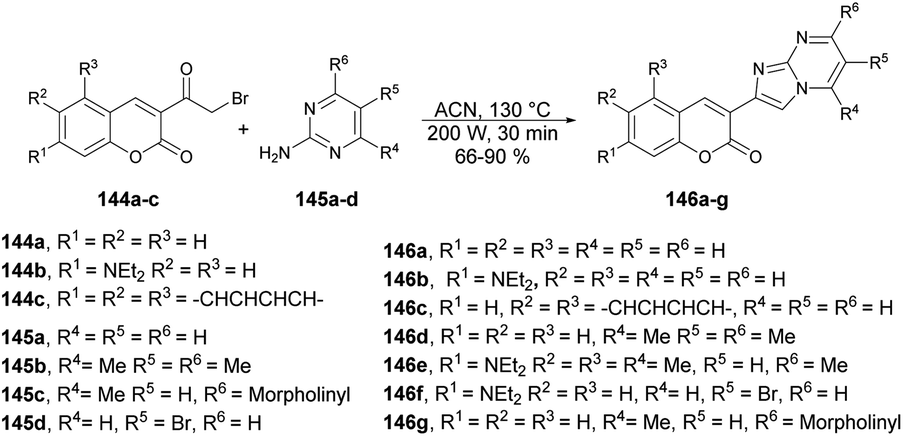 | ||
| Scheme 52 Synthesis of 2-coumarin-3-yl-imidazo[1,2-a]pyrimidines 146a–g. | ||
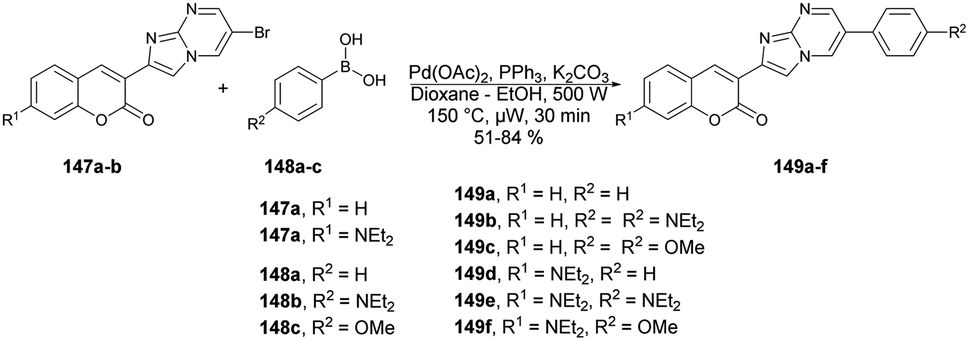 | ||
| Scheme 53 Synthesis of 6-aryl-2-coumarin-3-yl-imidazopyrimidines 149a–f. | ||
Bhuyan and co-workers reported a microwave-assisted solvent-free synthesis of diverse functionalized pyrido[3,2-c]coumarins 152a–q in excellent yields.128 Initially, they started to optimize the reaction conditions with 4-chloro-3-formylcoumarin 150, acetophenone 151 and ammonium acetate using various solvents at different temperatures under microwave conditions. The best condition obtained for completion of the reaction with a higher yield (84%) was a solvent-free condition at 130 °C and 3 min. Once the reaction conditions had been optimized, then various substrates were investigated, which afforded the diverse functionalized pyrido[3,2-c]coumarins 152a–q in good-to-excellent yields (71–95%) (Scheme 54).
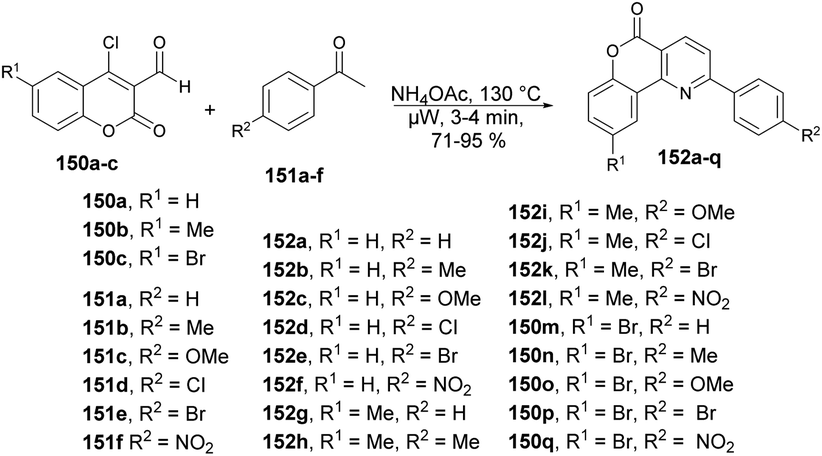 | ||
| Scheme 54 Synthesis of diverse pyrido[3,2-c]coumarins 152a–q. | ||
A microwave-irradiated multi-component synthesis of 6-(pyrrolyl) coumarin derivatives was reported by Ansary and co-workers.129 They performed the initial model reaction with 6-amino coumarin 153a, β-nitrostyrene 154b and diethyl acetylenedicarboxylate 155a to form 6-(pyrrolyl) coumarin 156a. They obtained best result (75% yield) when InCl3 was used as a catalyst in ethanol under microwave conditions at 100 °C for 1 h. After investigation of reaction conditions, they synthesized a series of 6-(pyrrolyl) coumarin derivatives 156a–j in 53–79% isolated yields using 6-amino coumarins 153a–b, β-nitrostyrenes 154a–d, and dialkyl acetylenedicarboxylates 155a–d (Scheme 55). It was found that nitrostyrenes with an electron-donating substituent at the aryl ring afforded better yields as compared with an electron-withdrawing substituent.
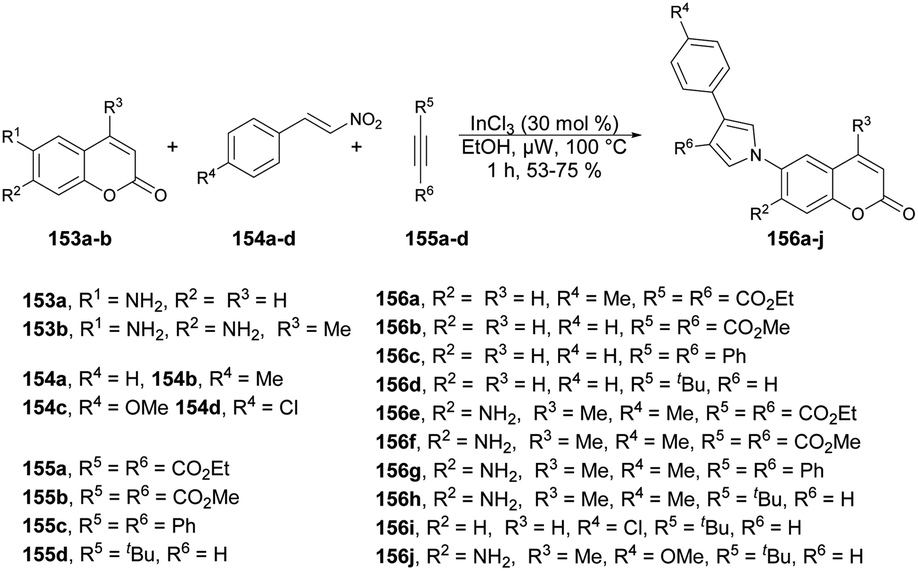 | ||
| Scheme 55 Synthesis of 6-(pyrrolyl) coumarin derivatives 156a–j. | ||
Schmidt and co-workers reported microwave-assisted development of new methodology for one-pot synthesis of 8-prenylcoumarins 159a–h.130 They performed the reaction with 1,1-dimethylallylated salicylaldehydes 157a–h, stabilized ylide [(ethoxycarbonyl)methylene]triphenylphosphorane 158 and N,N-diethylaniline under microwave conditions at 250 °C to afford poor-to-good isolated yields (22–91%) of designed products 159a–h (Scheme 56).
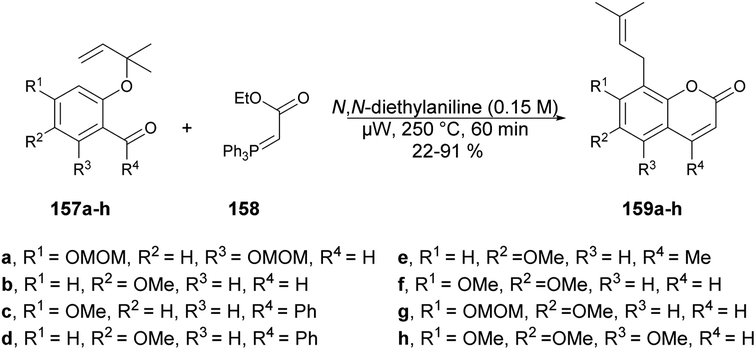 | ||
| Scheme 56 Synthesis of 8-prenylcoumarins 159a–h. | ||
Samanta and co-workers described a new class of diverse coumarins, dimedone, and naphthoquinone-fused pyrroles, which can be quickly synthesized through a one-pot, solvent-free annulation reaction.131 The reaction was catalyzed by a superior Lewis acid and assisted by microwave energy, and it involved various carbo- and heterocyclic enaminones with α-aroyl/heteroaryl/acetylidene malonates and 3-aroylidene-2-oxindoles. Initially, they started the model reaction between 4-aminocoumarin 160a as a C/N-binucleophile and benzoylidene malonate 161 as 1,2-bielectrophile. They investigated the reaction conditions using various solvent and neat conditions along with various acid catalysts under conventional and microwave conditions. Among all the tested conditions, the neat condition under microwave irradiation at 90 °C within 30 min provided a better yield (85%) of designed compound. After optimizing the reaction condition, they prepared a library of designed compounds 162a–m using similar microwave and catalytic conditions (Scheme 57). In addition, they conducted the experiment using the conventional method and noted that the reaction remained incomplete with 60% yield even after 12 h. However, when employing microwave irradiation, the reaction was completed within just 30 min, achieving higher yields. This microwave-based methodology offers several advantages, such as its environmentally friendly nature, superior synthetic efficiency, easy handling, high reactivity, and accelerated procedures, resulting in good yields with high purity.
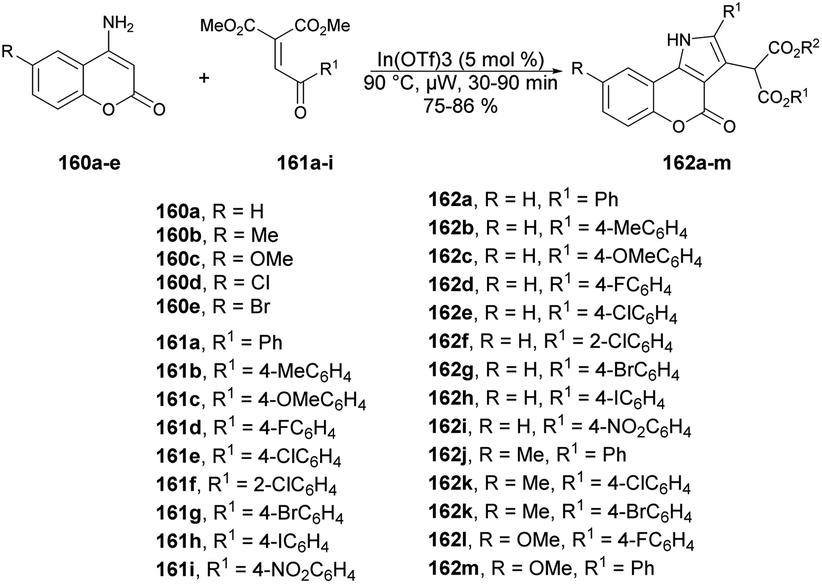 | ||
| Scheme 57 Synthesis of various coumarin derivatives 162a–m. | ||
Xu and co-workers developed a new method for synthesizing a wide variety of indolo[2,3-c]coumarins and indolo[2,3-c]quinolinones with high yields (up to 93%).132 They used a microwave-assisted base-free intramolecular cross dehydrogenative coupling (CDC) of 3-aniline substituted coumarins 163a–p under palladium catalysis using microwave irradiation at 140 °C to afford indolo[2,3-c]coumarins 164a–p (Scheme 58). This approach was considered to be one of the most convenient and uncomplicated ways to produce these fused polyheterocycles.
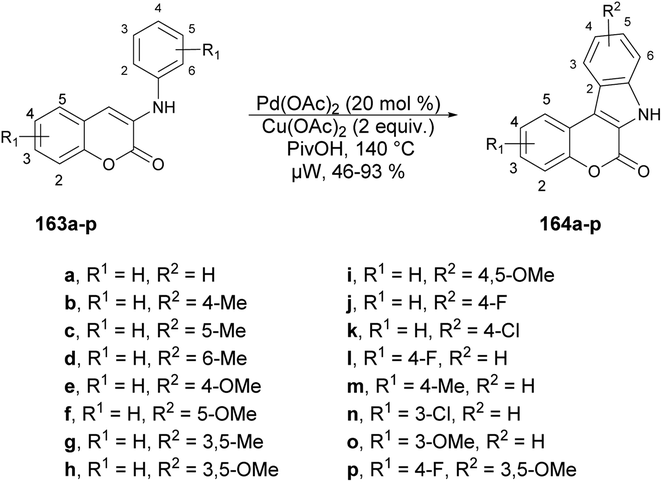 | ||
| Scheme 58 Synthesis of indolo[2,3-c]coumarins 164a–p. | ||
A reaction mechanism was proposed and is depicted in Scheme 59. It is hypothesized that the initial palladation occurs preferably at the coumarin nucleus and generates the intermediate I. The crucial intermediate III is likely formed through CeH activation in a concerted metalation–deprotonation process from the Pd(II) species I. Subsequently, reductive elimination leads to the formation of the desired indolo[2,3-c]coumarin product 164 and Pd(0). Finally, the oxidant Cu(II) salt serves to regenerate the active Pd(II) catalyst.
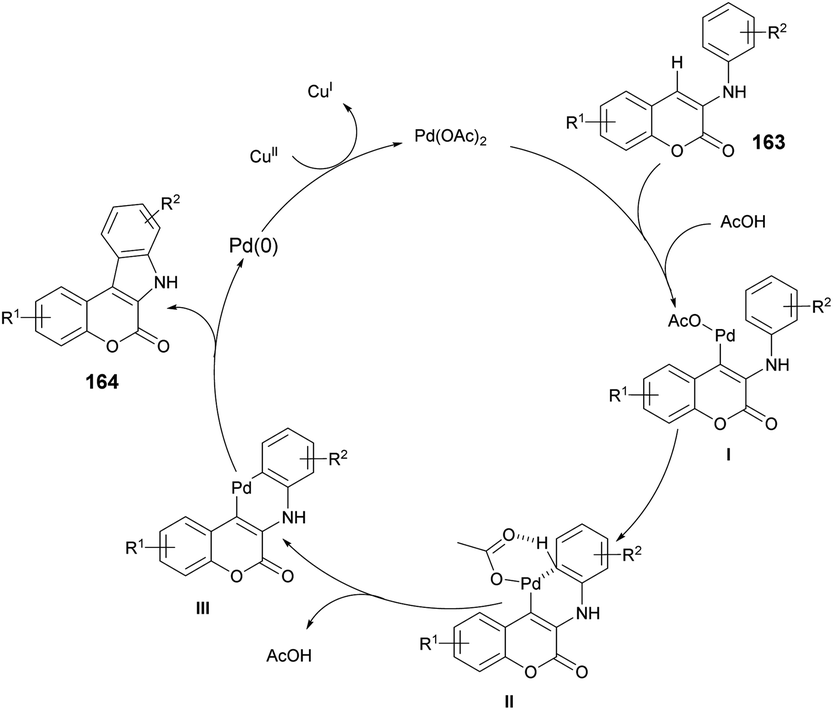 | ||
| Scheme 59 A plausible mechanism for the synthesis of indolo[2,3-c]coumarins 164. | ||
A new method for synthesizing 2-methylfuro[3,2-c]coumarins was developed by Cui and co-workers using a copper-catalyzed microwave-promoted propargylation and oxacyclization.133 The starting materials for this reaction were readily available 4-hydroxycoumarins and terminal propargyl acetates. They obtained moderate-to-good yields (up to 82%) of furo[3,2-c]coumarins 167a–l produced by this reaction. Interestingly, when they changed the solvent from dimethyl sulfoxide to 1,2-dichloroethane while retaining the conditions, the isomeric series of 2-methylene-2,3-dihydrofuro[3,2-c]coumarins 168a–d was obtained in low-to-acceptable yields (up to 85%) (Scheme 60).
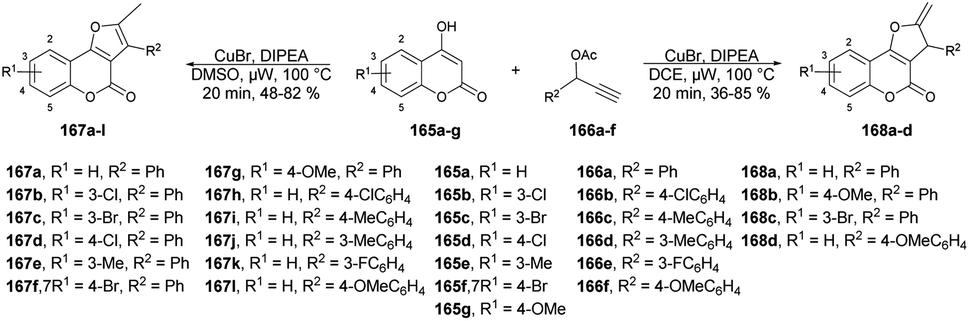 | ||
| Scheme 60 Synthesis of furo[3,2-c]coumarins 167a–l and isomeric series of 2-methylene-2,3-dihydrofuro[3,2-c]coumarins 168a–d. | ||
Litinas and co-workers developed a Pd-catalyzed oxidative coupling protocol for the synthesis of azacoumestans using microwave irradiation.134 The Pd-catalyzed oxidative coupling of 4-(arylamino)coumarins was carried out using Cu(OAc)2 in acetic acid under microwave irradiation. They synthesized azacoumestans, including those substituted with electron-withdrawing groups, with almost quantitative yield. The arylamines 170a–k were reacted with 4-bromocoumarin 169 in water or through Pd-catalyzed C–N coupling under microwave heating to achieve intermediate product 171a–k, which was again catalyzed by copper and Pd catalysts to afford the designed products 172a–k in good-to-excellent yields (85–99%) (Scheme 61).
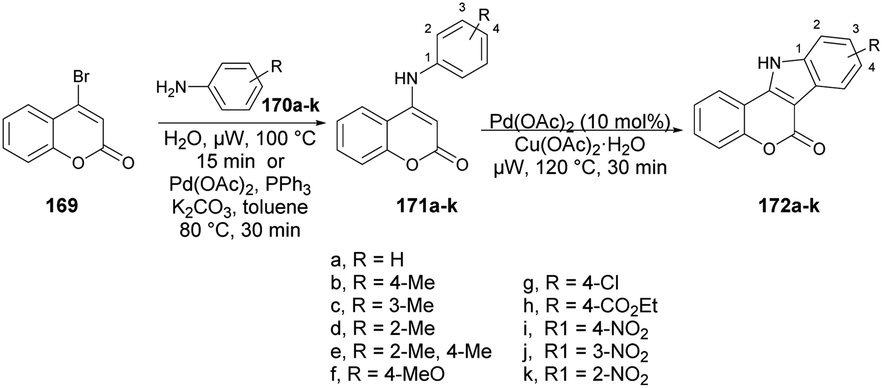 | ||
| Scheme 61 Synthesis of azacoumestans 172a–k. | ||
A microwave-assisted environmentally benign protocol to synthesize coumarin derivatives was developed by Mentese and co-workers.135 In order to synthesize designed molecules N-[(1,2,4-triazol-1-yl)acetyl]coumarin-3-carbohydrazides 175a–l, they carried out the reaction between 2-(3-alkyl/aryl-4-amino-5-oxo-4,5-dihydro-1H-1,2,4-triazol-1-yl)acetohydrazides 173a–f and 3-(1H-benzotriazol-1-ylcarbonyl)-2H-chromen-2-ones 174a–b in ethanol under microwave conditions at 130–145 °C for 20 ± 35 min to afford good isolated yields (58–72%) of designed products 175a–l (Scheme 62).
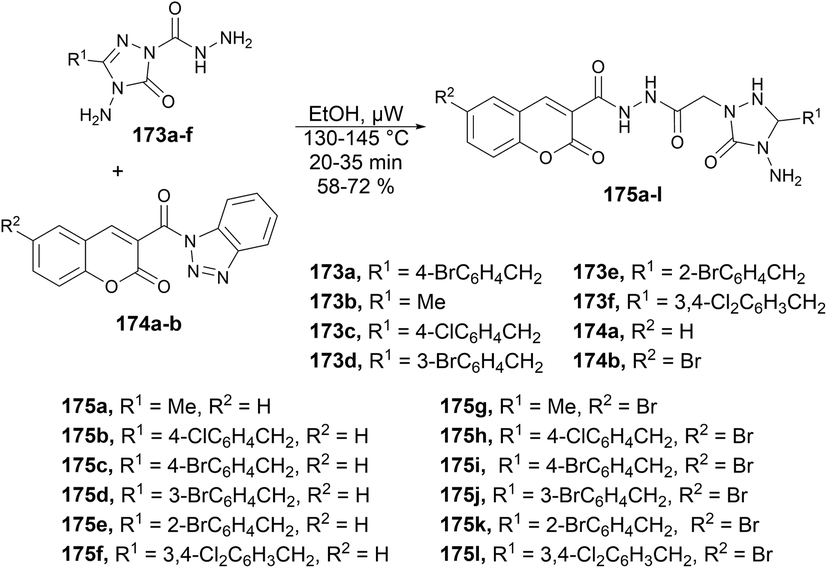 | ||
| Scheme 62 Synthesis of N-[(1,2,4-triazol-1-yl)acetyl]coumarin-3-carbohydrazides 119a–l. | ||
Hosamani and co-workers reported a microwave-irradiated rapid development of coumarin–thiazoline hybrids and their antitubercular activity.136 They performed the reaction between various substituted coumarins 176a–i and 4,5-dihydrothiazole-2-thiol 177 using K2CO3 as a base in ethanol under microwave conditions at 55 °C and 100 W for 5–9 min to afford good isolated yields (81–91%) of coumarin–thiazoline hybrids 178a–i (Scheme 63). After the synthesis, these compounds were investigated for their antitubercular activity. Compound 178b was found to be most potent antitubercular compound with an IC50 value of 280 μM. In order to compare the conventional method and microwave irradiation, various reactions were conducted. The conventional method produced 20% lower yields as compare with microwave irradiation. Thus, microwave irradiation was a better choice to perform this type of reaction.
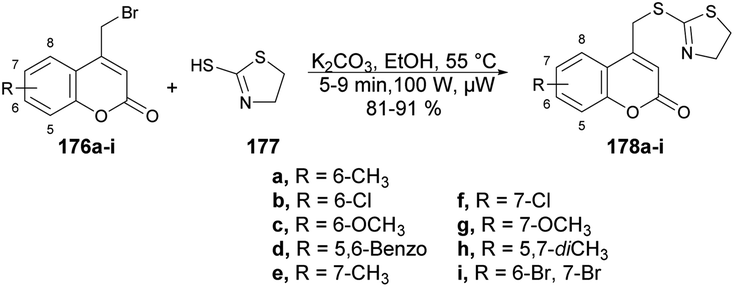 | ||
| Scheme 63 Synthesis of coumarin–thiazoline hybrids 178a–i. | ||
A new microwave-irradiated development of coumarin-3-carboxylic acids was reported by Genovese and co-workers.137 They performed the reaction between 2,2-dimethyl-1,3-dioxane-4,6-dione 179 and various carbonyl compounds 180a–k using Yb(OTf)3 as a catalyst with no solvent and microwave irradiation at 80 °C and 200 W for 5 min to afford a good isolated yield (92–98%) of coumarin-3-carboxylic acids 181a–k (Scheme 64). It is important to emphasize that when the same reaction was conducted under conventional conditions, it resulted in a complex mixture of products, as evidenced by TLC. This mixture partly originated from the decomposition of Meldrum's acid and the air-oxidation of benzaldehyde. Under these experimental conditions, the estimated yield of coumarin-3-carboxylic acid, as determined by GC, was ∼15%. Furthermore, attempts to use other β-dicarbonyl compounds (e.g., diethyl malonate) also yielded unsatisfactory results.
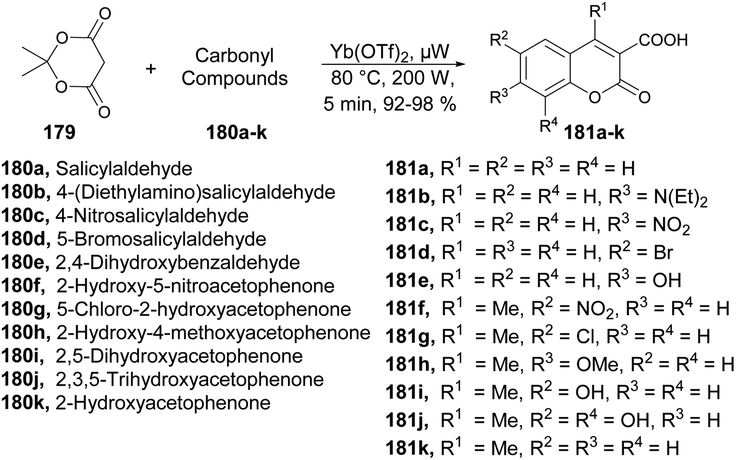 | ||
| Scheme 64 Synthesis of coumarin-3-carboxylic acids 181a–k. | ||
Coumarin derivatives have gained recognition for their diverse range of biological activities, such as antimicrobial, antioxidant and antitumor properties. In light of this, Rao and co-workers introduced a new method for developing hybrid chromene–coumarin derivatives 184a–i through an environmentally friendly and catalyst-free synthesis.138 Formation of the key C–C bond occurred in an aqueous medium using microwave irradiation at 105 °C, and resulted in a rapid reaction time, high conversion and exceptional regioselectivity (Scheme 65). Various solvents were tested but “in water” reactions proved to be the most efficient in terms of time and yields, achieving yields of 78–90% at 105 °C.
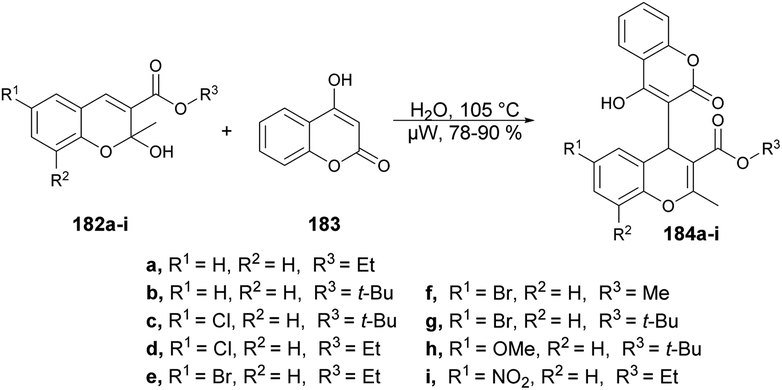 | ||
| Scheme 65 Synthesis of chromene–coumarin derivatives 184a–i. | ||
Zhang and co-workers developed a novel protocol for the synthesis of a group of pyrano [3,2-c]chromene-2,5-diones through microwave irradiation and also investigated their antifungal activity against Botrytis cinerea, Colletotrichum capsica, Alternaria solani, Gibberella zeae and Rhizoctonia solani.139 In order to synthesize pyrano[3,2-c]chromene-2,5-diones 187a–I, a reaction was carried out between various 4-hydroxycoumarin 185a–e and ethyl 2-methylacetoacetate 186 in the presence of DMAP as a catalyst in toluene under microwave irradiation at room temperature and 640 W for 15 min to afford 13–75% yield of desired products 187a–i (Scheme 66). Among all the derived compounds of pyrano [3,2-c]chromene-2,5-diones, compound 187g was found to be the most potent for antifungal activity with an EC50 value of 0.082 μM. Additionally, when compared with conventional heating, it was observed that the reaction times for target compounds could be significantly reduced from ∼6 h to just 15 min. Moreover, the isolated yields could be enhanced, reaching a maximum of 76% (compound 187a) as opposed to the previous 30%.
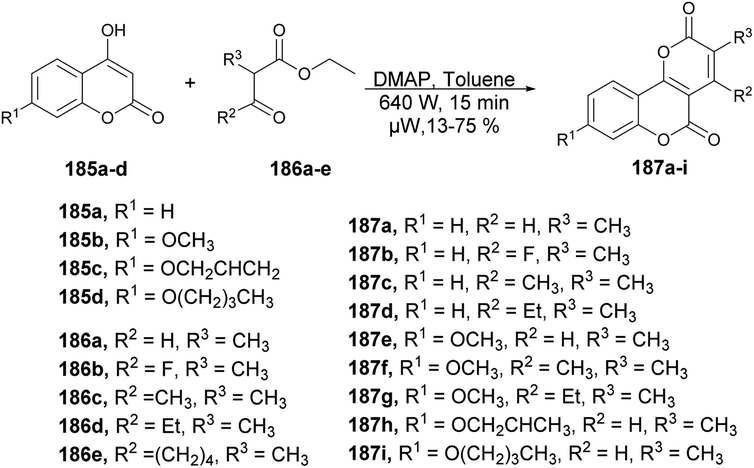 | ||
| Scheme 66 Synthesis of pyrano [3,2-c]chromene-2,5-diones 187a–i. | ||
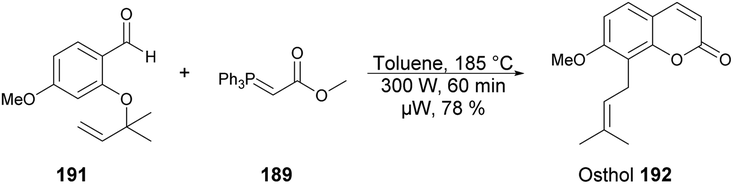
A microwave-assisted regioselective method for the synthesis of coumarin derivatives was developed by Pospisil and co-workers.140 They discovered an efficient and protecting group-free synthesis of coumarins. The reaction was carried out between various unprotected aldehydes 188a–e and stabilized Wittig ylide 189 in the presence of toluene in microwave conditions at 220 °C and 300 W for 60 min to afford a good isolated yield (74–89%) of coumarin derivatives 190a–e (Scheme 67). For the synthetic applications of the developed method, they also performed the reaction using a similar approach to develop natural product osthol 192 (Scheme 68).
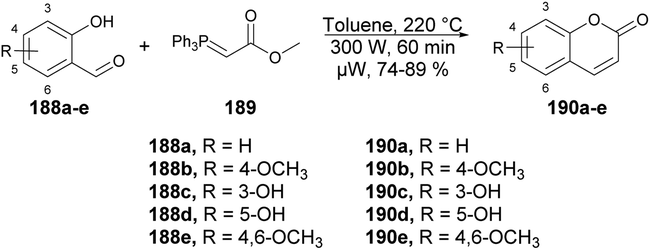 | ||
| Scheme 67 Synthesis of coumarin derivatives 190a–e. | ||
 | ||
| Scheme 68 Synthesis of natural product osthol 192. | ||
Desai and co-workers reported a microwave-assisted rapid and efficient method for the synthesis of novel coumarin derivatives and evaluation of activity against various bacteria (Escherichia coli, Pseudomonas aeruginosa, Staphylococcus aureus, Streptococcus pyogenes) and fungi (Candida albicans, Aspergillus niger, Aspergillus clavatus).141 To synthesize coumarin derivatives, the reaction was carried out between 3-acetyl-2H-chromen-2one 193, aromatic aldehydes 194a–m, malononitrile and ammonium acetate in microwave conditions using ethanol as a solvent to yield products 195a–m, which were again microwave-irradiated with another aldehyde 196 at 300 W and 100 °C for 8–10 min to afford a good isolated yield of desired products 197a–m (Scheme 69).
 | ||
| Scheme 69 Synthesis of coumarin derivatives 197a–m. | ||
Chavan and co-workers reported an expedient and high-yielding method for synthesizing coumarin, pyrazolone hybrids containing Schiff-base structures using microwave irradiation.142 For the synthesis of Schiff base analogues 200, the reaction was carried out between 2-hydroxy-1-naphthaldehyde 198 and 4-aminoantipyrene 199 in the presence of acetic acid in ethanol. On a condensation reaction with substituted 4-bromomethyl coumarins 201a–h in microwave conditions at 35 °C and 200 W for 8–12 min, a good isolated yield (82–95%) of various substituted coumarin, pyrazolone hybrids 202a–h were obtained (Scheme 70). The synthesized compounds were evaluated for their antibacterial and anti-inflammatory properties using the agar well-diffusion and egg albumin-denaturation methods, respectively. It was found that compound 202d and 202h exhibited antibacterial effects with a minimum inhibitory concentration (MIC) of 1.420 μM and MIC of 2.33 μM, respectively, against the Gram-positive Staphylococcus aureus strain, which was better than the standard drug (ciprofloxacin).
 | ||
| Scheme 70 Synthesis of various substituted coumarin pyrazolone hybrids 202a–h. | ||
The pyrano[3,2-c]pyranone structure is a crucial structure found in various natural products with different biological activities. Sagar and co-workers reported the efficient and rapid synthesis of two sets of pyrano[3,2-c]pyranone derivatives (205a–i and 207a–i) fused with carbohydrates utilizing 2 C-formyl galactal 203 and 2-C-formyl glucal 206. They were reacted with diverse 4-hydroxycoumarins 204a–i in a short time (10 min) using microwave-assisted conditions (Schemes 71 and 72).143 They also evaluated the anticancer potential of the newly derived pyrano[3,2-c]pyranones against MCF-7 (breast), MDA-MB-231 (breast), and HepG2 (liver) cancer cell lines via cellular assays and examined their ability to inhibit the growth and viability of MCF-7, MDA-MB-231, and HepG2 cell lines. It was found that compound 205d showed considerable growth-inhibitory effects with a MIC50 value of 10.9 μM.
 | ||
| Scheme 71 Synthesis of pyrano[3,2-c]pyranone derivatives 205a–i fused with 2-C-formyl galactal. | ||
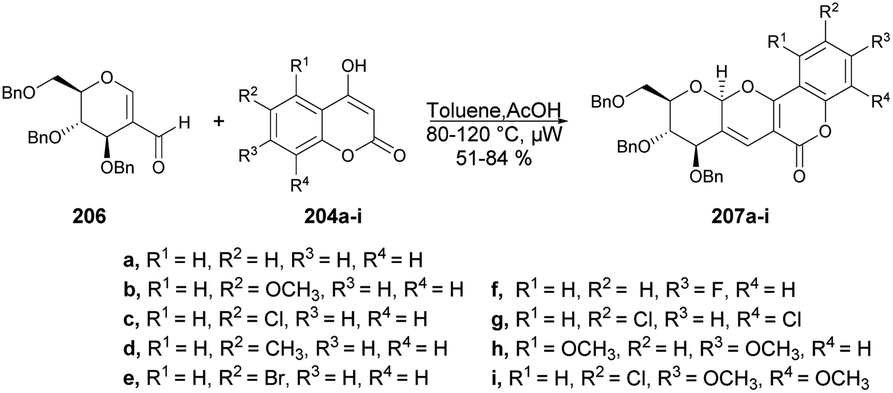 | ||
| Scheme 72 Synthesis of pyrano[3,2-c]pyranone hybrids 207a–i fused with 2-C-formyl glucal. | ||
Sagar and co-workers also reported the synthesis of two series of triazole-linked N-glycosides of coumarins and quinolones using 1-azido-2,3,4,6-tetra-O-acetyl-β-D-glucose 208 and 1-azido-2,3,4,6-tetra-O-acetyl-β-D-galactose 211 as starting materials.144 The reactions were performed under microwave-assisted conditions via click chemistry and resulted in a good isolated yield of triazole-linked N-glycosides of coumarins and quinolones (Schemes 73 and 74). They also evaluated the biological activity of diverse synthesized compounds against several cancer cell lines, and one of the library members 212d was found to have low micromolar IC50 (10.97 μM) and selective toxicity against the breast cancer cell line (MCF-7).
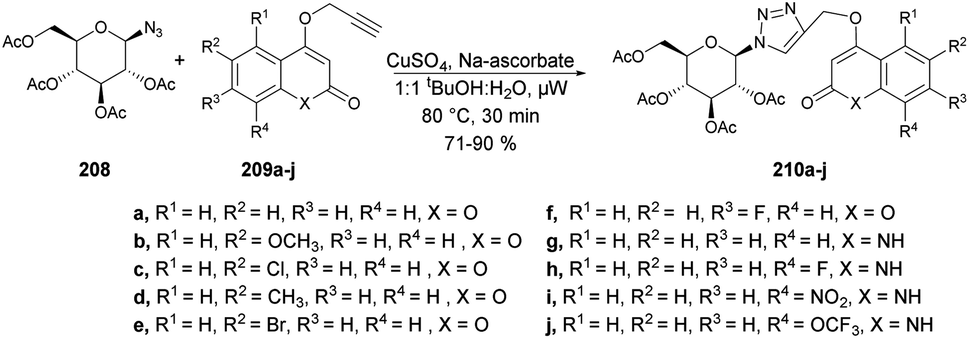 | ||
| Scheme 73 Synthesis of triazole-linked N-glucopyranosides 210a–j. | ||
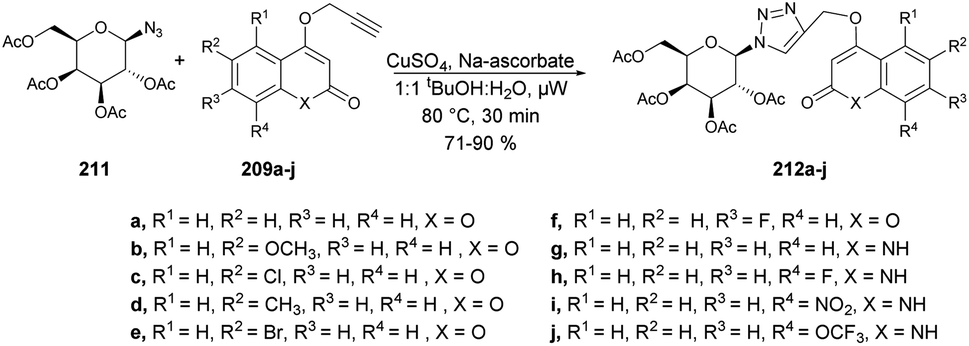 | ||
| Scheme 74 Synthesis of triazole linked N-galactopyranosides 212a–j. | ||
A highly efficient and environmentally friendly method was reported by Ashok and co-workers to synthesize 1,4-disubstituted 1,2,3-triazole derivatives 215a–l based on coumarin scaffolds.145 The synthesis involved a copper(I)-catalyzed click reaction between various substituted arylazides 214a–c and terminal alkynes 213a–d. It was carried out under microwave irradiation at 200 W and 40–100 °C for 5–8 min to afford the designed products 215a–l in excellent yields (Scheme 75). They also evaluated the synthesized compounds for their antimicrobial, antioxidant, and anti-inflammatory activities. Compounds 215g and 215l exhibited better anti-inflammatory activity with an IC50 value of 13.16 μM and 15.35 μM, respectively, than the standard drug diclofenac. In order to compare the synthesis of alkyne-substituted 3-aryl coumarin intermediates and coumarin-based 1,2,3-triazole the reaction was performed using conventional and microwave irradiation methods. In comparison, the microwave irradiation method yielded better results, with good-to-excellent yields ranging from 80% to 90%, while the conventional method resulted in yields of 68% to 79%.
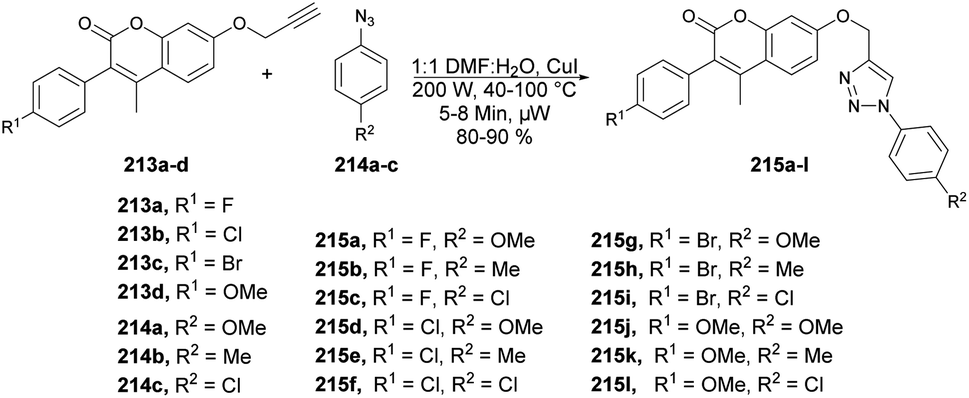 | ||
| Scheme 75 Synthesis of 1,4-disubstituted 1,2,3-triazole derivatives of coumarin 215a–l. | ||
A synthesis and evaluation of anticancer activity of α,β-unsaturated carbonyl-linked coumarin-triazole hybrids 218a–j using microwave assistance have been reported by Kumar and co-workers.146 The base-catalyzed reaction was carried out between 4-chloro-2-oxo-2H-chromene-3-carbaldehyde 216 and diverse aryl azides 217 in microwave conditions at 80 °C for 15–30 min afforded the α,β-unsaturated carbonyl-linked coumarin-triazole hybrids 218a–j (Scheme 76). Out of the series, compound 218d demonstrated greater potential for fighting cancer against PC-3 and DU-145 cell lines, exhibiting an IC50 value of 10.538 ± 0.3 μM and 9.845 ± 0.6 μM, respectively. Moreover, it had very low toxicity (10 μM) against normal cells.
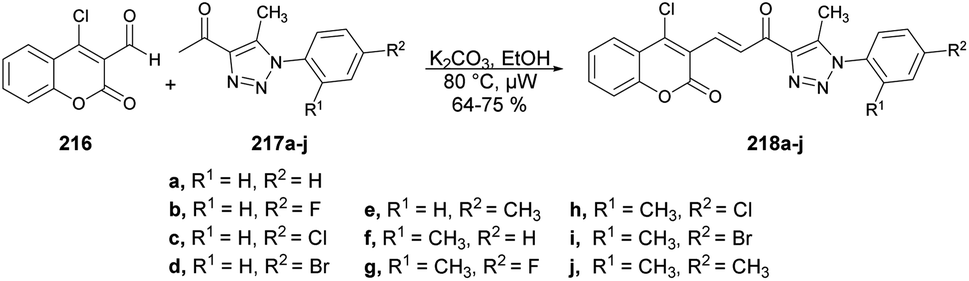 | ||
| Scheme 76 Synthesis of chalcone-linked coumarin-triazole hybrids 218a–j. | ||
Microwave-induced synthesis frequently leads to lower reaction temperatures and shorter reaction times. Target compounds were synthesized using conventional reflux and microwave irradiation methods. The optimization of reaction conditions demonstrated that microwave-assisted synthesis required significantly less reaction time compared with conventional heating methods.
Vedula and co-workers reported microwave irradiation of thiocarbohydrazide 221, aldehydes 220a–b, and 3-(2-bromoacetyl) coumarins 219a–j to synthesize a sequence of novel coumarin-based thiazoles 222a–n.147 The reaction was carried out under microwave conditions at 70 °C and 210 W for 5–10 min in ethanol/acetic acid to afford a good isolated yield of coumarin-based thiazoles 222a–n (89–93%) (Scheme 77). They evaluated the activity of synthesized compounds 222a–n against the Gram-positive bacterium Staphylococcus aureus (ATCC-12600). Compound 222e was discovered to exhibit good antibacterial activity with an IC50 value of 5.09 μM. They also performed the reactions using a conventional method and observed a lower yield of desired compounds as compared with use of microwave irradiation.
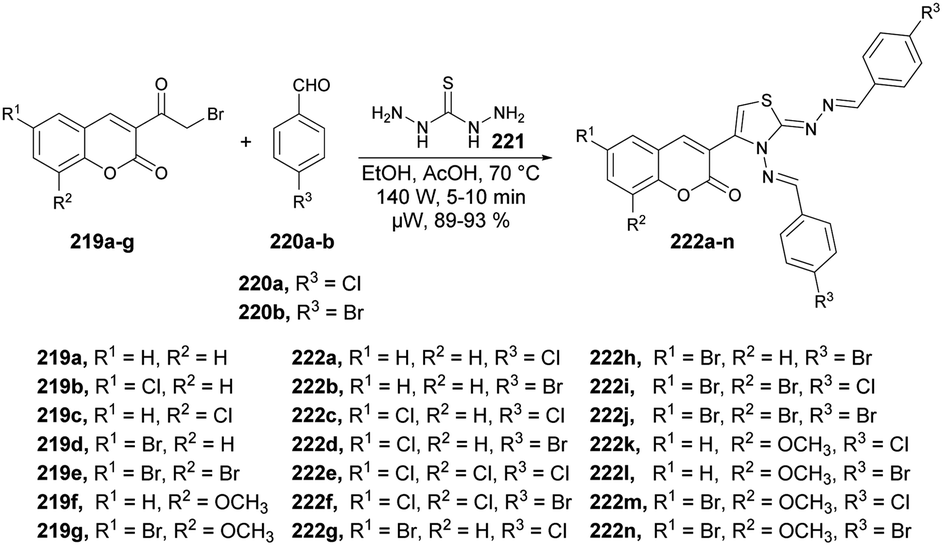 | ||
| Scheme 77 Synthesis of coumarin-based thiazoles 222a–n. | ||
In Scheme 78, a plausible mechanism for the formation of the target compound 222a–n is illustrated. Initially, two equivalents of aldehydes react with one equivalent of thiocarbohydrazide in the presence of acetic acid, resulting in the formation of bis-thiocarbohydrazone A. Subsequently, a SN2-type attack occurs, with the sulfur from bis-thiocarbohydrazone attacking the C–Br of 3-(2-bromoacetyl) to form intermediate II. This intermediate then undergoes acid-catalyzed dehydration, leading to the formation of the titled coumarin-based thiazole 222a.
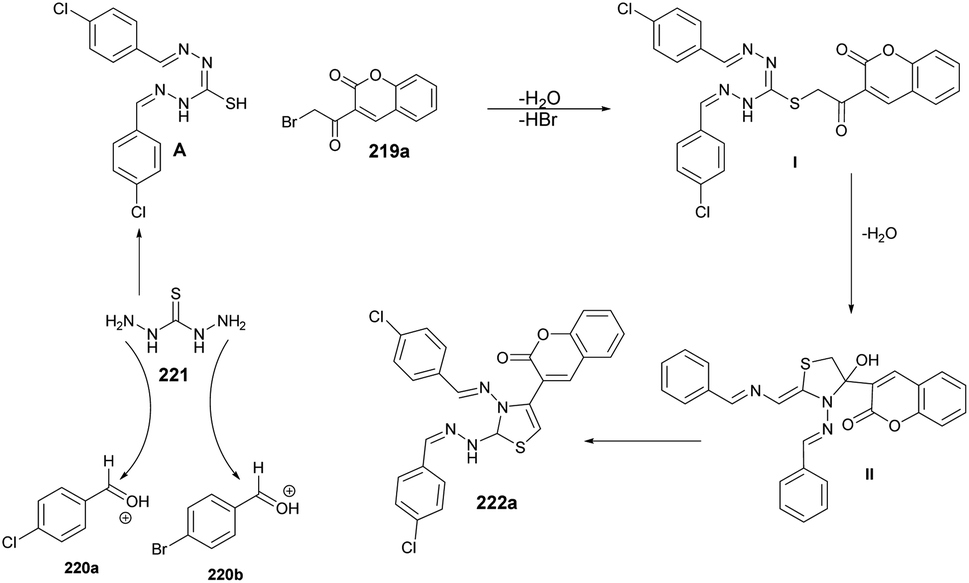 | ||
| Scheme 78 A plausible mechanism for the synthesis of coumarin-based thiazoles 222a–n. | ||
2.4. Isatins and their derivatives
Isatin is a heterocyclic compound used extensively as a foundational component in the synthesis and advancement of novel drug candidates.148–150 The remarkable versatility of isatins, which can serve as electrophiles and nucleophiles, coupled with their ready availability, positions them as highly prized building blocks in organic synthesis.151–153 They participate in a range of transformations, including electrophilic aromatic substitution at the C-5 and C-6 positions of the phenyl ring,154 N-substitutions,154 nucleophilic additions to the C-3 carbonyl group,154 selective oxidations, reductions, ring expansions, spiro-annulations, and more reactions.155 Using isatins as starting materials, various heterocyclic frameworks of significant biological importance, such as pyrrolidines, indoles, quinolines, β-lactams, and 2-oxindoles, have been synthesized.156–158Recent literature reflects a renewed interest in the chemistry and bioactivity of isatin derivatives, leading to advancements in established reaction procedures and the development of selective methods for producing isatin derivatives through microwave irradiation techniques.
Zakeri and co-workers have discovered microwave-assisted eco-friendly methods for producing spiro-4H-pyrans 226a–f through a one-pot, three-component condensation reaction using a domino Knoevenagel/Michael/cyclization sequence.159 These approaches were tested using various organocatalysts in water and solvent-free conditions under microwave irradiation and ball-milling conditions. The research team conducted numerous experiments to optimize the reaction parameters, such as catalyst type/amount, reaction temperature, and time. The most effective procedure was observed to be microwave irradiation in the presence of 4-dimethylaminopyridine (DMAP), resulting in good yields (78–90%) of final products 226a–f, in a short reaction time, environmentally sustainable and ease-of-operation manner (Scheme 79). Microwave-induced synthesis frequently leads to lower reaction temperatures and shorter reaction times. Target compounds were synthesized using conventional reflux and microwave irradiation methods. The optimization of reaction conditions demonstrated that microwave-assisted synthesis required significantly less reaction time compared with conventional heating methods.
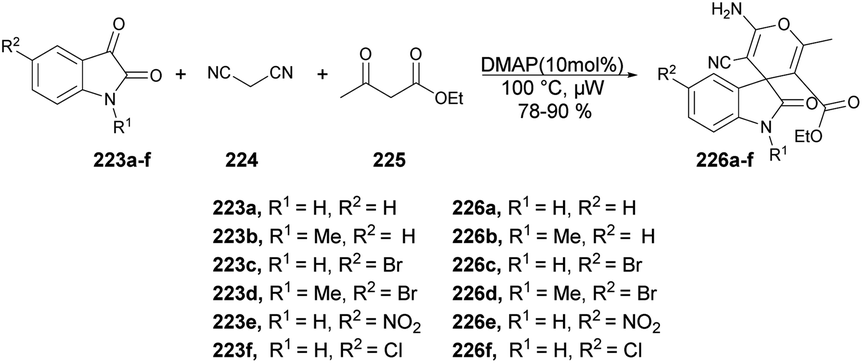 | ||
| Scheme 79 Synthesis of spiro-4H-pyrans 226a–f. | ||
Spiro-4H-pyrans can be formed through a series of chemical reactions, as depicted in Scheme 80. This scheme illustrates a plausible mechanism for the reaction between isatin and malononitrile with ethyl acetoacetate to yield 226 derivatives. The process involves a typical cascade reaction. First, isatin 223a undergoes a Knoevenagel condensation with malononitrile in the presence of DMAP, leading to the formation of an isatylidenemalononitrile derivative (intermediate I). Subsequently, the treatment of a β-keto ester with DMAP generates the major thermodynamic enolate product. Next, intermediate I is subjected to Michael addition, wherein the thermodynamic ethyl acetoacetate enolate attacks intermediate I, giving rise to the formation of intermediate II. Finally, the hydroxyl group participates in a cycloaddition reaction with the cyano moiety, resulting in the exclusive formation of the desired 226a product. Notably, the kinetic product was not observed in this process.
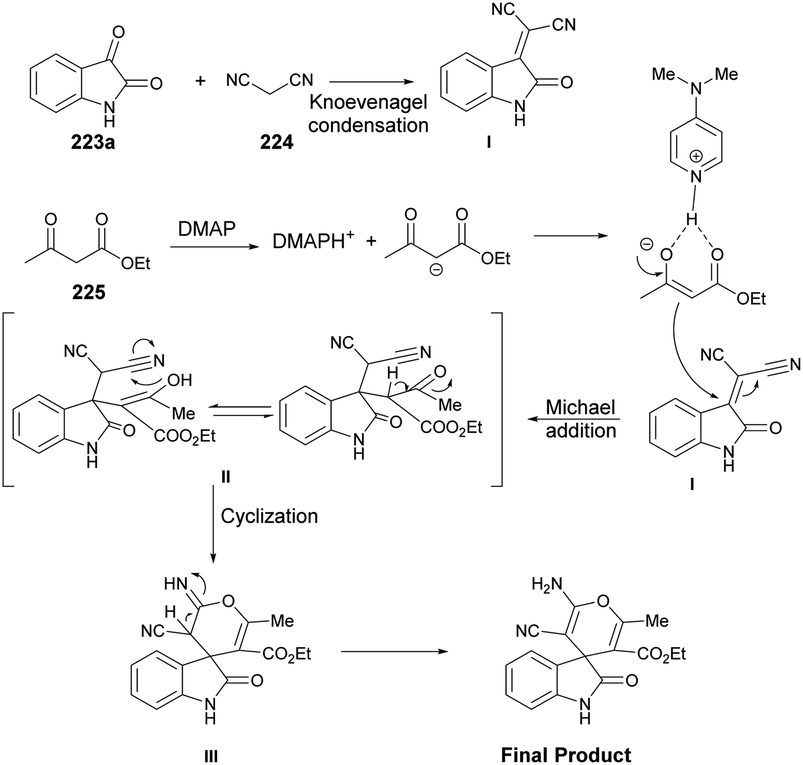 | ||
| Scheme 80 Plausible mechanism for the synthesis of spiro-4H-pyrans. | ||
Pardasani and co-workers developed a new catalyst-free method for synthesizing spiro-benzimidazoquinazolinones using multiple components under microwave irradiation.160 The method involves a one-pot three-component reaction of acenaphthoquinone or isatin 227a–h, 1,3-diketone 228, and 2-aminobenzimidazole 229 in ethanol at 160 °C and 180 W afforded good isolated yields (75–80%) of spiro-benzimidazoquinazolinones 230a–h (Scheme 81). One of the most attractive features of this method is the mild reaction conditions it employs, along with operational simplicity.
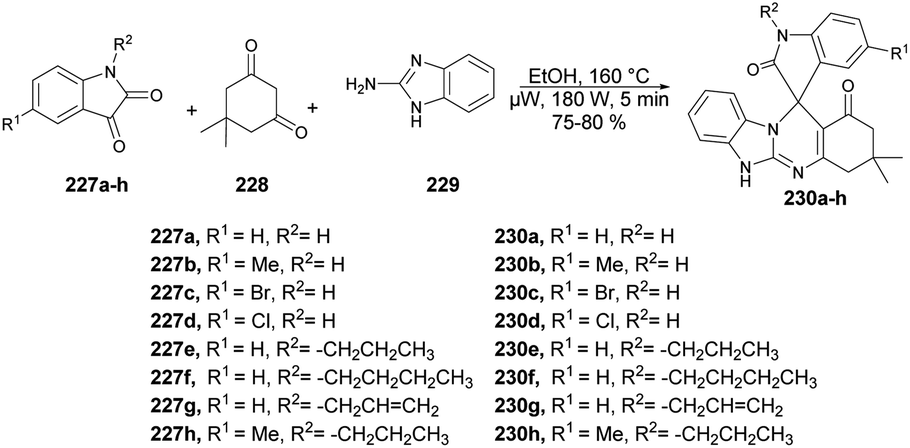 | ||
| Scheme 81 Synthesis of spiro-benzimidazoquinazolinones 230a–h. | ||
A microwave-assisted multicomponent reaction was developed by Meshram and co-workers to synthesize a series of isatin derivatives.161 The three-component reaction was performed between isatins 231a–g, amino acid 233 and but-2-ynedioates 232 in an aqueous medium, under microwave irradiated conditions at 80 °C for 10 min afforded a good isolated yield (70–93%) of the target compounds 234a–h (Scheme 82). The synthesis of other diverse functionalized spirooxindole derivatives can be achieved by utilizing this synthetic protocol. This reaction offers numerous benefits, including: (i) utilizing water as the reaction solvent; (ii) achieving a high atom economy; (iii) eliminating the need for a catalyst; (iv) accommodating a wide range of substrates.
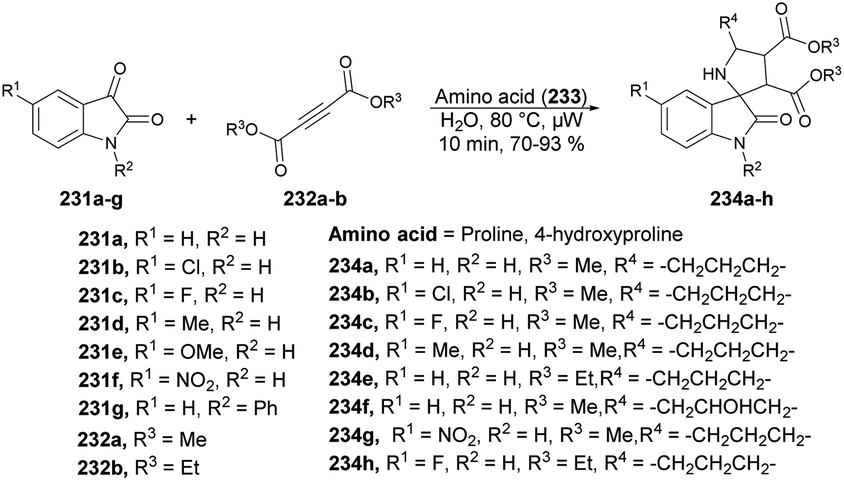 | ||
| Scheme 82 Synthesis of isatin based spiro compounds 234a–h. | ||
A plausible mechanism for the three-component reaction is proposed and depicted in Scheme 83. The reaction begins with the condensation of α-amino acid 233 and isatin 231, leading to the formation of imine intermediates II through the elimination of CO2. Subsequently, a 1,3-dipolar cycloaddition reaction (III) occurs between the imine intermediates and but-2-ynedioates 232, then undergoes cyclization, yielding the spiro compound 234. In the case of the four-component reaction, the spiro compound 234 undergoes a further reaction with phenacyl bromide, leading to the formation of a N-protected product.
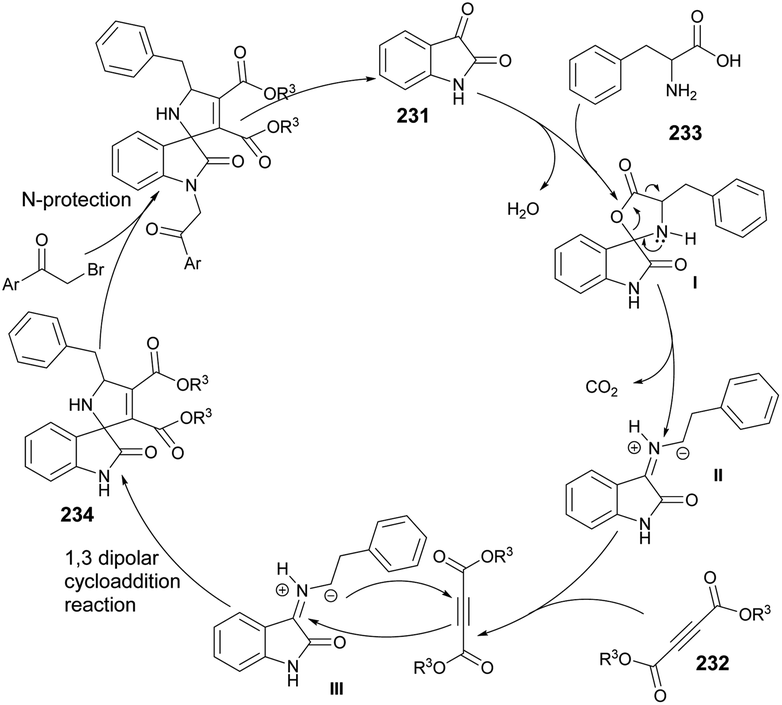 | ||
| Scheme 83 Plausible mechanism for the formation of pyrrolizine-1,2-dicarboxylate. | ||
Gohary and co-workers developed a microwave-assisted new range of isatin-β-thiocarbohydrazones by utilizing the key features required for metal chelation found in triapine.162 Their approach involved modifying the basic structure of triapine by substituting the pyridinyl group with isatin, which retained the three-pronged feature necessary for metal chelation. The reaction carried out between thiocarbohydrazide 235 and isatin derivatives 236a–b in ethanol under microwave irradiation afforded good isolated yields (73–91%) of 237a–b which, on further treatment with aryl aldehydes 238a–h, afforded the products 239a–j (Scheme 84). The newly developed compounds were also evaluated for their effectiveness in fighting cervical cancer (HeLa) and kidney fibroblast cancer (COS-7) cell lines utilizing doxorubicin as a standard drug. Compound 239c exhibited remarkable efficacy with an IC50 value of 1.51 μM against the HeLa cell line and 2.19 μM against the COS-7 cell line, among all the derived compounds.
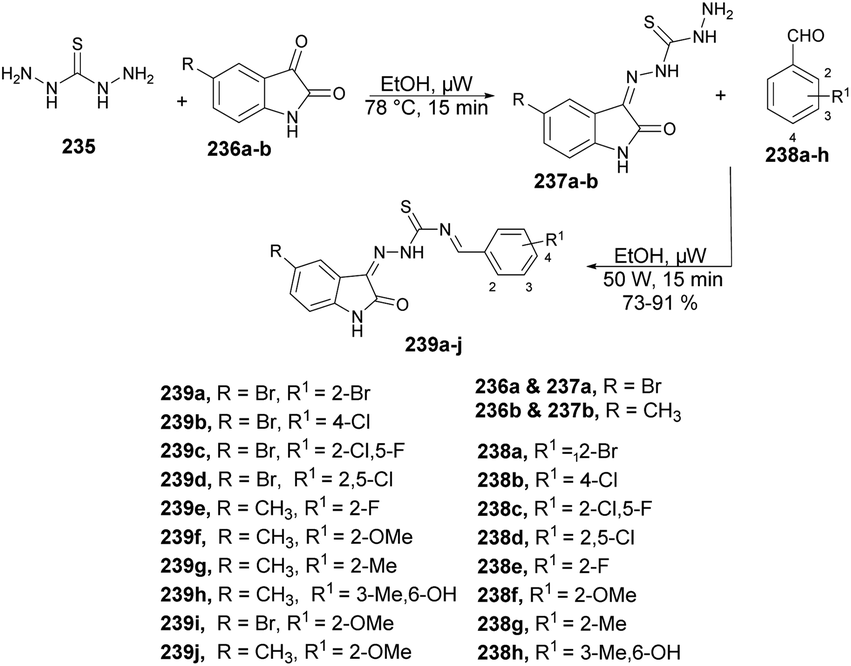 | ||
| Scheme 84 Synthesis of isatin-β-thiocarbohydrazones 239a–j. | ||
Greco and co-workers introduced a microwave-assisted Knoevenagel/Michael/cyclization multicomponent domino approach for producing isatin-based spiro compounds 241a–h, with ethanol serving as the solvent and 1-methylimidazolium chloride as a catalyst.163 They undertook a thorough methodological investigation involving alterations to solvent, catalyst, amount of catalyst, temperature, and heating mode to identify optimal reaction conditions. Additionally, the scope of the reaction was examined by varying the starting materials (isatin, malononitrile, and barbituric acid), which resulted in the synthesis of 12 spiro compounds with good yields ranging from 43% to 98% (Scheme 85).
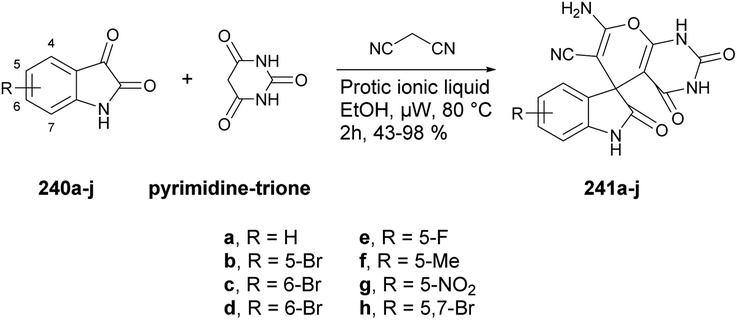 | ||
| Scheme 85 Synthesis of isatin derivatives 241a–h. | ||
Choudhury and co-workers reported a microwave-irradiated three-component reaction that could be switched depending on the reaction solvent and temperature, leading to the synthesis of two types of fused spirooxindoles.164 The reaction involves isatins 242a–e, 4-hydroxycoumarins 243a–b, and either aminopyrazole 244a–c under microwave irradiation. When the reaction is performed in acetonitrile at 85 °C, the resulting spirooxindoles 245a–g are fused with pyrazolo-tetrahydropyridinones due to the ring-opening of the hydroxycoumarin moiety (Scheme 86). On the other hand, when the reaction is performed in acetic acid at 130 °C, the resulting spirooxindoles 246a–g are fused with a tetracyclic coumarin-dihydropyridine-pyrazole/isoxazole moiety spiro fused with oxindoles (Scheme 87). This switchable multicomponent reaction has been used to synthesize a series of medicinally important spiroxindole-fused pyrazolo-tetrahydropyridinones and spirooxindole-fused coumarin-dihydropyridine-pyrazole/isoxazole tetracycles under metal-free conditions.
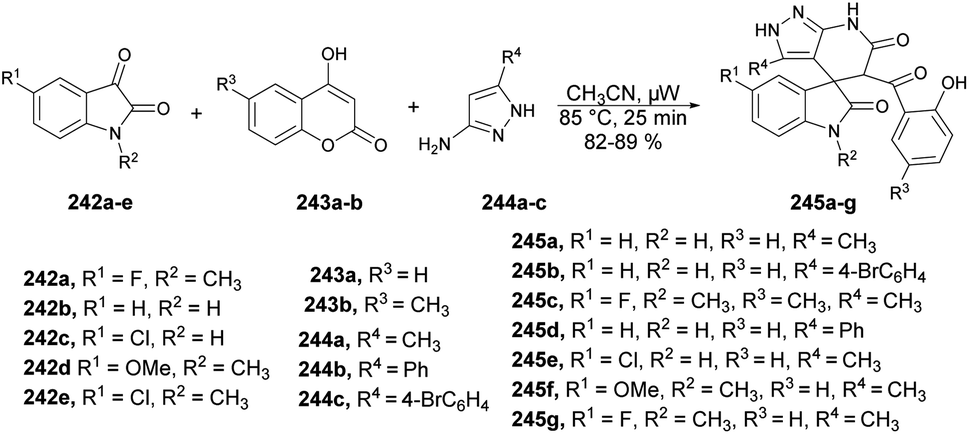 | ||
| Scheme 86 Synthesis of spirooxindoles 245a–g. | ||
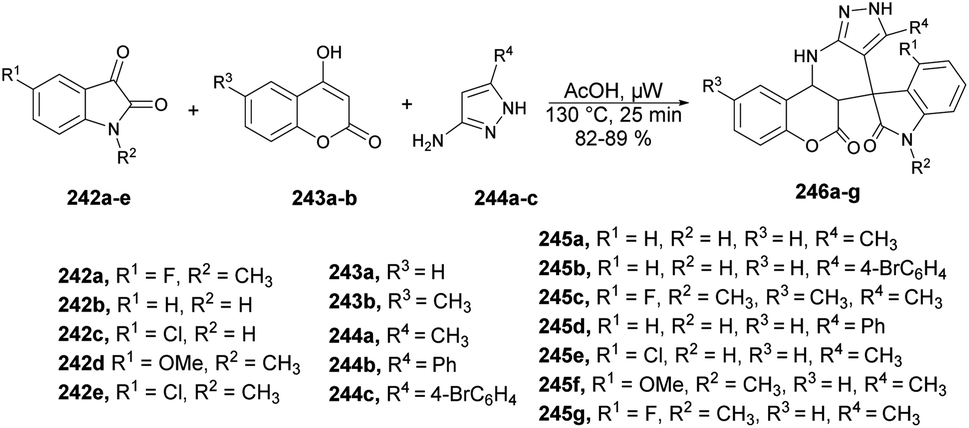 | ||
| Scheme 87 Synthesis of spirooxindoles 246a–g. | ||
Shankaraiah and co-workers devised an effective approach to synthesize pyrrolidine-fused bisspirooxindoles 250a–i using a one-pot method assisted by microwaves.165 The reaction was carried out under microwave-conditions at 100 °C. This method involves the generation of azomethine ylide through the decarboxylative condensation of isatins 247a–i and primary α-amino acids 248a–f, along with the [3 + 2] cycloaddition of 3-alkenyl oxindoles 249a–d (Scheme 88). The use of microwave irradiation in ethanol solvent without any additives or catalyst facilitates these reactions, and results in a broad substrate scope, good yields, eco-friendliness, and atom-economy.
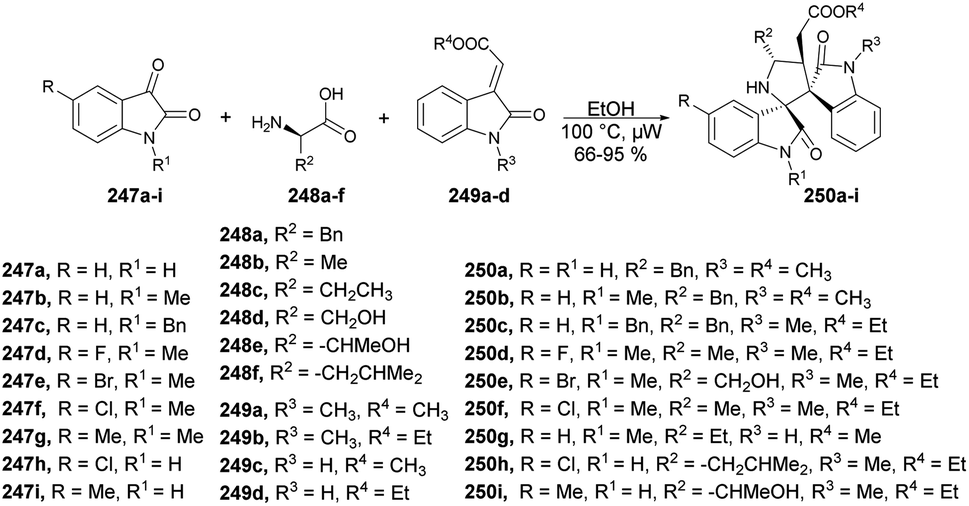 | ||
| Scheme 88 Synthesis of pyrrolidine fused bisspirooxindoles 250a–I. | ||
Microwave irradiation was used by Junior and co-workers to heat the reaction medium while synthesizing spiro 1,3,4-thiadiazolines from isatin-β-thiosemicarbazone acetylation.166 To produce thiosemicarbazones 253a–h, N-substituted isatin derivatives 251a–h were used as substrates, and thiosemicarbazide 252 was added to the isatin ketone carbonyl. The final step in the synthetic process involved the reaction of thiosemicarbazones with acetic anhydride under microwave irradiation at 120 °C to produce the spiro 1,3,4-thiadiazolines 254a–h within 6–18 min (Scheme 89). They also evaluated the antibacterial and antifungal activity of derived compounds and found that particularly spiro-thiadiazolines 254c derived from allylated isatins showed promising activity with an IC50 value of 360 μM in biological assays.
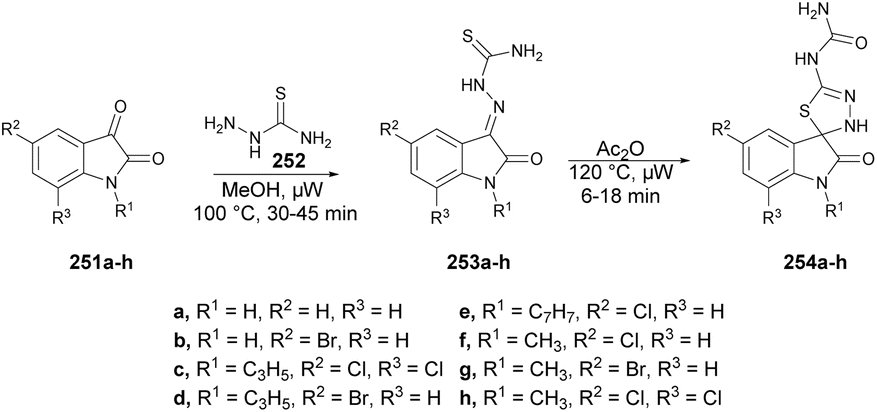 | ||
| Scheme 89 Synthesis of spiro 1,3,4-thiadiazolines from isatin-β-thiosemicarbazone 254a–h. | ||
In a one-pot process utilizing microwave irradiation and deep eutectic solvents (DES), Junior and co-workers discovered a simple and efficient alternative method for synthesizing 1,3,4-thiadialzolinic spiro compounds 257a–e.167 They explored a series of DES based on different acids as solvents in a three-component reaction which included isatin derivatives (monomeric and dimeric), thiosemicarbazide, and acetic anhydride. Of all the solvents screened, choline chloride (ChCl) and oxalic acid (in a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 ratio) were the best, producing yields of up to 79%. The yields for most of the monomeric compounds were either very close to or higher than those reported in the literature for a two-step synthesis (Scheme 90).
:1 ratio) were the best, producing yields of up to 79%. The yields for most of the monomeric compounds were either very close to or higher than those reported in the literature for a two-step synthesis (Scheme 90).
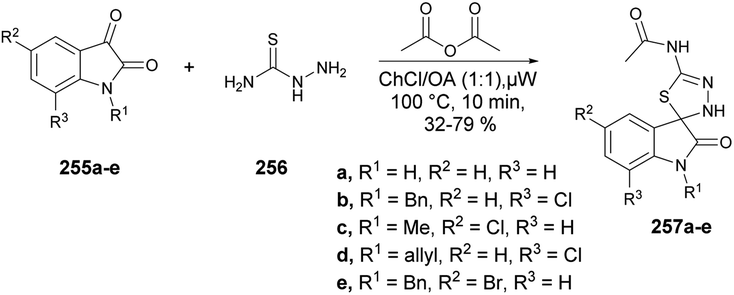 | ||
| Scheme 90 Synthesis of spiro 1,3,4-thiadiazolines from isatin 257a–e. | ||
Reddy and co-workers developed a series of oxazolo[5,4-b]quinoline-fused spirooxindoles using a microwave-assisted chemoselective synthesis.168 The three-component Knoevenagel/Michael addition reaction was performed between isatin 258a, β-diketone 259a and 5-amino-3-methylisoxazole 260 in the absence of a catalyst using solvent-free conditions under microwave irradiation at 120 °C at 700 W for 10 min to afford the oxazolo[5,4-b]quinoline-fused spirooxindole 261a in 95% yield. The optimized conditions were utilized to test the impact of altering the β-diketones and substituents in the isatin to determine the extent of this reaction. As a result, numerous derivatives of the intended compounds 261a–n were synthesized with outstanding yields (84–95%) (Scheme 91).
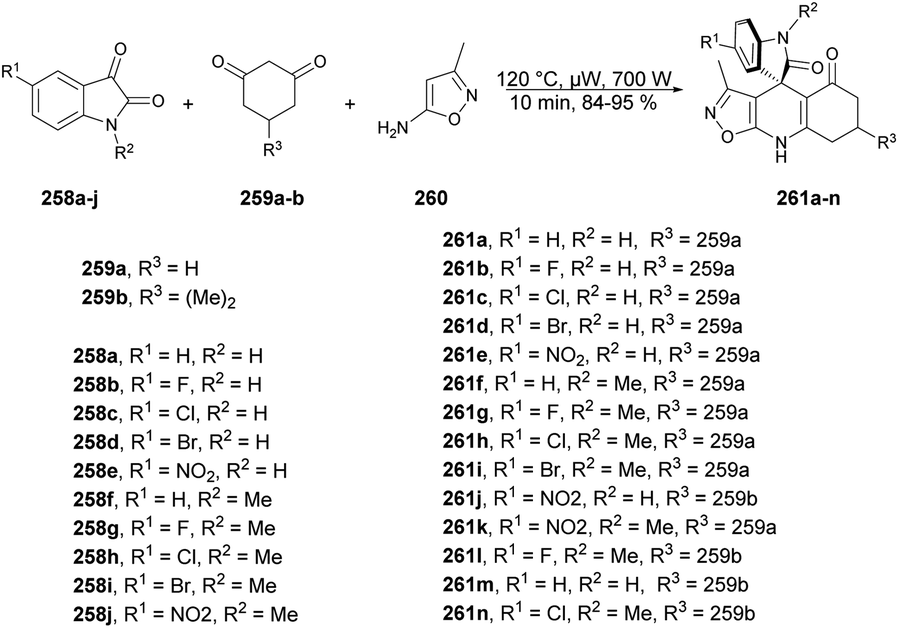 | ||
| Scheme 91 Synthesis of oxazolo[5,4-b]quinoline-fused spirooxindoles 261a–n. | ||
Praveen and co-workers reported a gold-catalyzed microwave-assisted synthesis of spirooxindoles using double condensation between isatin and 4-hydroxycoumarin.169 They performed the condensation reaction with various derivatives of isatin 262a–l and 4-hydroxy coumarin 263 using a gold catalyst (NaAuCl4·2H2O) under microwave irradiation at 40 °C for 15 min to afford good-to-excellent yields (86–96%) of designed products 264a–l (Scheme 92). They also observed that microwaves could lead to efficiently syntheses of these compounds as compared with a conventional heating method.
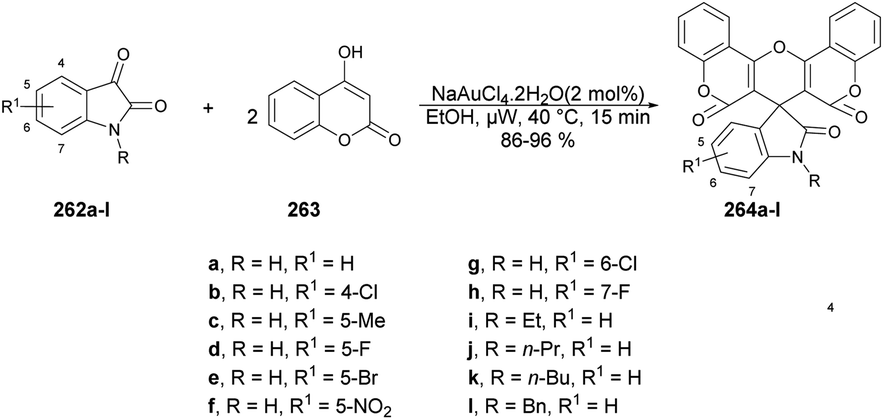 | ||
| Scheme 92 Gold-catalyzed synthesis of spirooxindoles 264a–l. | ||
Based on the observed results, a tentative mechanism (Scheme 93) was proposed to explain formation of the representative compound 264a. The process begins with 4-hydroxycoumarin undergoing nucleophilic addition at the carbonyl carbon of the [Au]-activated isatin, leading to the formation of intermediate I. Subsequently, dehydration of intermediate I takes place, resulting in the formation of intermediate II. The Lewis acidic [Au] species re-enters the catalytic cycle by interacting with the carbonyl oxygen of intermediate II. This interaction drives the addition of another molecule of 4-hydroxycoumarin, leading to the formation of intermediate III. Later, intramolecular cyclization occurs in intermediate III, resulting in the formation of intermediate IV. Through subsequent dehydration reactions, the final product 264a is obtained.
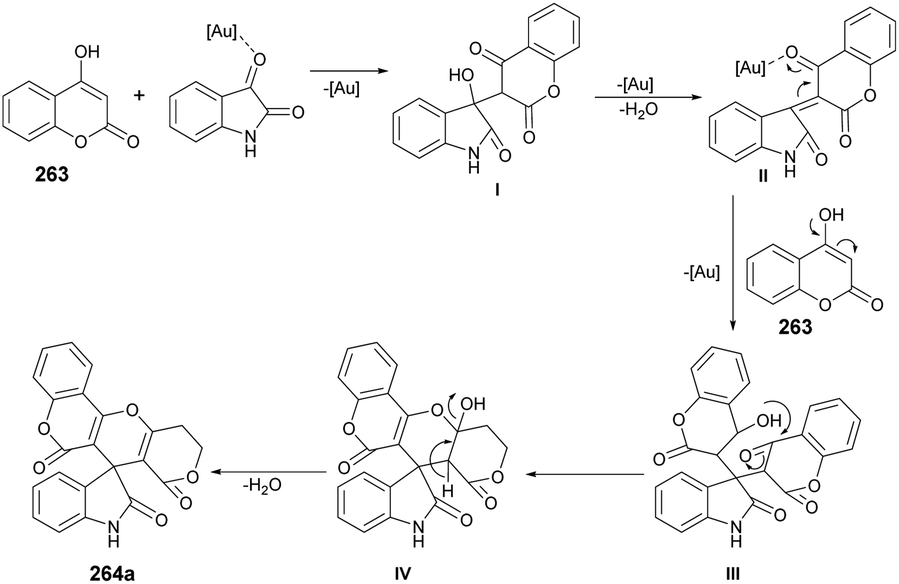 | ||
| Scheme 93 A plausible mechanism for the synthesis of spirooxindole. | ||
A new microwave-assisted development was reported by Thanh and co-workers for the synthesis of N-(2,3,4,6-tetra-O-acetyl-β-D-glucopyranosyl)thiosemicarbazones 268a–j with various isatin derivatives.170 Initially, they performed the reaction to convert N-(tetra-O-acetyl-β-D-glucopyranosyl)isothiocyanate 265 into the corresponding thiosemicarbazide 266 which, on further reaction with various isatin derivatives 267a–j under microwave irradiation at 100–200 °C, afforded excellent yields (74–96%) of designed products (268a–j) (Scheme 94). They also investigated the cytotoxic effects of the derived compounds. Compound 268f showed cytotoxic effects with an IC50 value of 0.78 μM against some cancer (LU-1, HepG2, MCF7, P338, SW480, KB) cell lines as well as a normal fibroblast cell line (NIH/3T3).
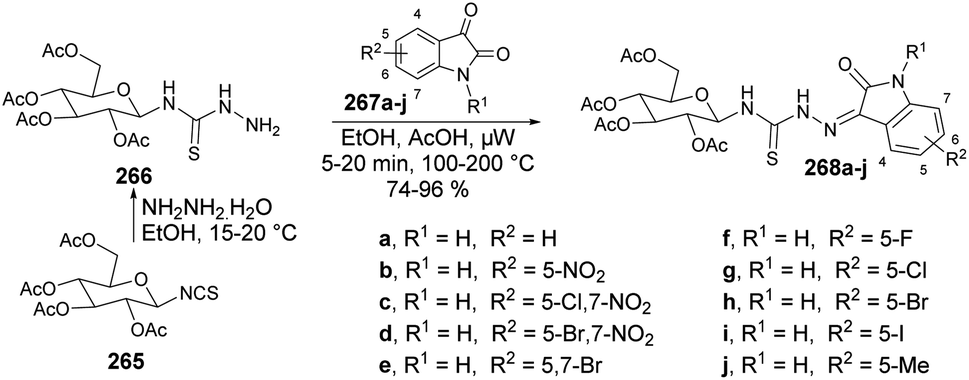 | ||
| Scheme 94 Synthesis of N-(2,3,4,6-tetra-O-acetyl-β-D-glucopyranosyl)thiosemicarbazones with isatin derivatives 268a–j. | ||
Meshram and co-workers developed a straightforward and environmentally friendly method that involves only one pot and three-components in an aqueous phase to synthesize functionalized spirooxindole derivatives.171 They performed the reaction with isatin 269a–d, β-nitrostyrene 270a–f, and aryl amines 271a–b in water under microwave irradiation. A library of spirooxindoles 272a–l was easily constructed with good diastereoselectivity and in moderate-to-good yields (64–92%) (Scheme 95). This approach is especially beneficial because it employs readily available starting materials, a green solvent and can be completed in short period of time.
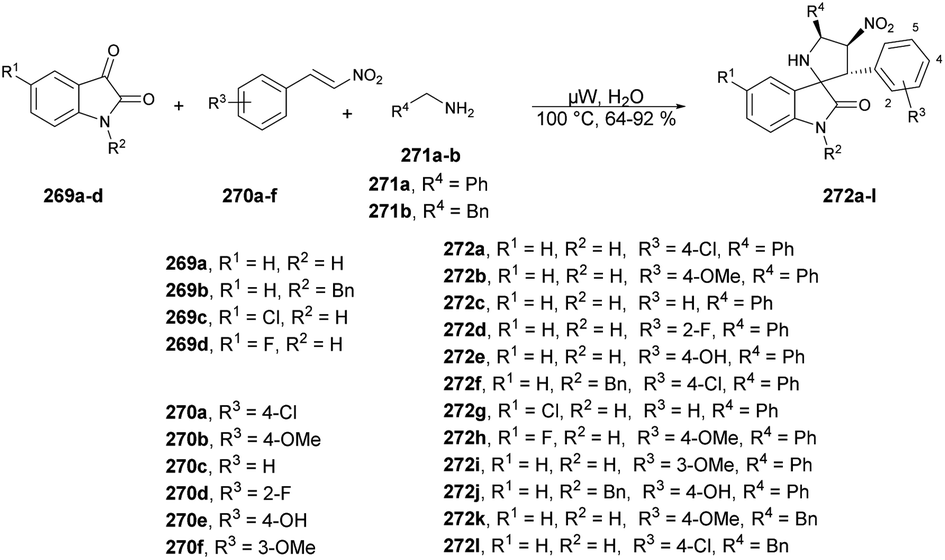 | ||
| Scheme 95 Synthesis of functionalized spirooxindole derivatives 272a–l. | ||
The α-amino acid or benzyl amine undergoes an initial reaction with isatin through a typical addition process, resulting in the formation of an imine. The formed imine then undergoes a reaction with β-nitrostyrenes, leading to a 1,3-cycloaddition reaction. During this cycloaddition, two nucleophilic carbons add to the electron-deficient carbon of the dipolarophile to form a transition state which, on transformation, is converted in compound 272a. This can be explained based on the transition state, with the observed product being more stable. The transition state is preferred if the spiro carbon is cis to the carbonyl of the oxindole moiety and trans to the nitro group (Scheme 96).
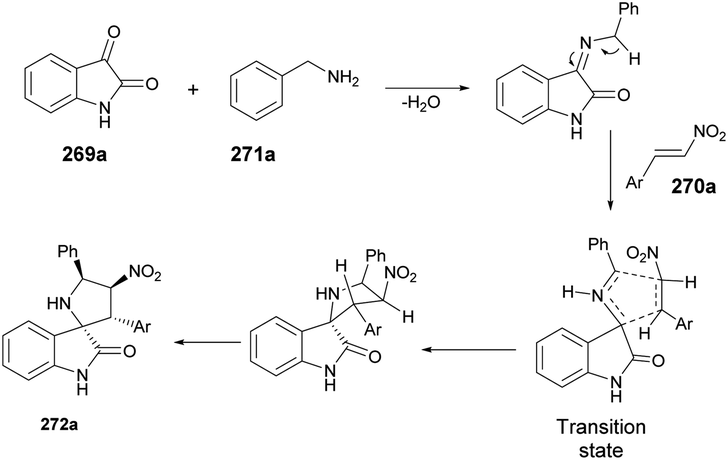 | ||
| Scheme 96 Plausible mechanism for the synthesis of representative molecule 272a. | ||
Dandia and co-workers developed a microwave-assisted, one-pot method for synthesizing spiropyrrolidine/thiapyrrolizidine oxindole derivatives with antimicrobial properties.172 The approach was simple, environmentally friendly, highly efficient, and involved two pharmacophoric groups (1,3-indanedione and pyrrolidine/thiapyrrolizidine) in a single molecule through a three-component 1,3-dipolar cycloaddition reaction. They carried out the reaction using substituted isatin 273a–c, thiazoles-4-carboxylic acid 275 and either 2-cyano-3-phenyl-acrylic acid ethyl ester or 2-benzylidene-malononitrile 274a–d reflux under microwave irradiation for 5–10 min to afford the desired products 276a–h (Scheme 97). The methodology exhibits good tolerance for functional groups and a wide range of substrates, and demonstrates high levels of chemo-, regio-, and stereoselectivity with acceptable yields (88–92%) of designed products.
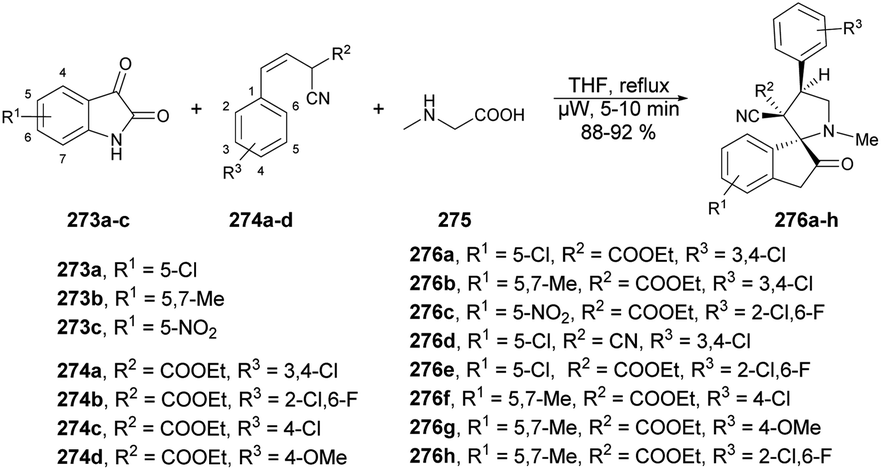 | ||
| Scheme 97 Synthesis of spiropyrrolidine oxindole derivatives 276a–h. | ||
Besbes and co-workers synthesized a series of isatin-aminorhodanine hybrids 279a–l using a microwave-irradiated technique.173 They performed the condensation reaction of isatin derivatives 278a–l and 3-aminorhodanine 277 in a microwave condition at 100 °C for 5 min to afford moderate-to-acceptable yields (55–92%) of hybrid products 279a–l (Scheme 98). This microwave approach is facile, effective for the development of various biologically important hybrid molecules, ecological, fast and energy efficient. Compared with the microwave method, the conventional method is slow, time-consuming and provides a low yielding product. When a similar reaction was carried out using the conventional heating method, it was completed in 24 h, compared with 5 min using microwaves.
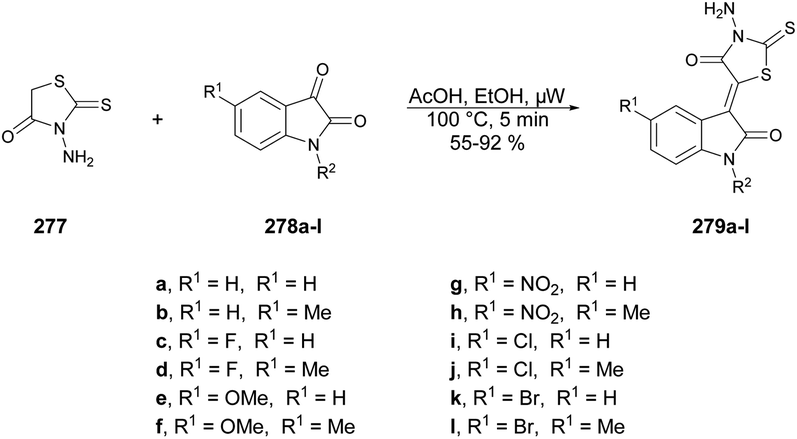 | ||
| Scheme 98 Synthesis of isatin-aminorhodanine hybrids 279a–l. | ||
The antiviral properties of spiro-oxindoles have led to their broad application. Initially, Nino and co-workers utilized a rapid and efficient synthetic method to produce ketonitrones using a solvent-free microwave-assisted reaction between various hydroxylamines and isatin or indanone derivatives.174 This synthetic technique was simple, efficient, fast, clean, and stereoselective. Subsequently, they investigated the feasibility of generating spiro-isoxazolidines 284a–g containing nucleobases with isatin and indanone nuclei through solvent-free microwave-assisted 1,3-dipolar cycloaddition, which yielded good outcomes in terms of yields (74–85%) of regio- and diastereoselective products (Schemes 99 and 100).
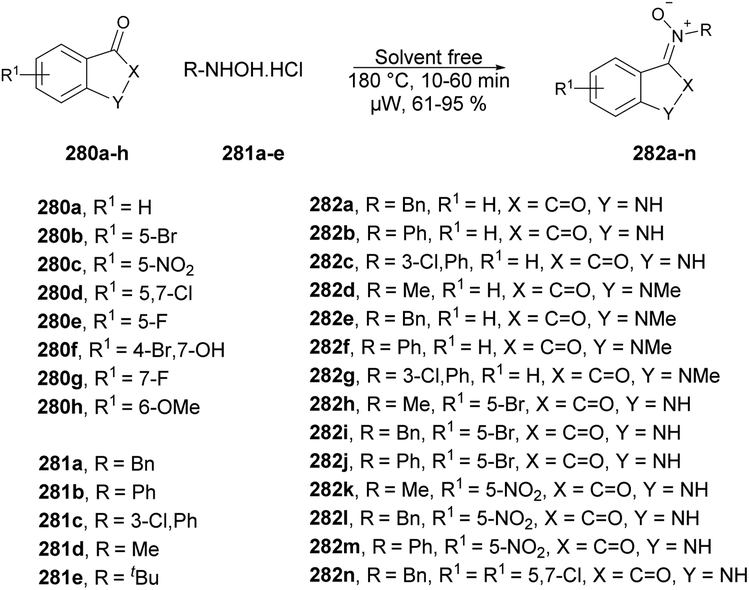 | ||
| Scheme 99 Synthesis of ketonitrones 282a–n. | ||
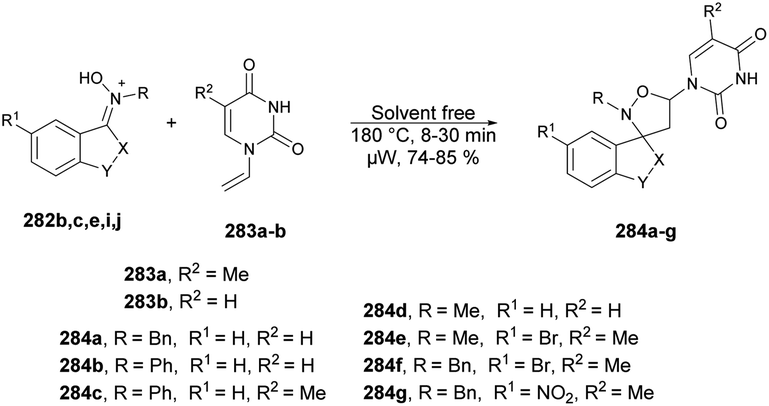 | ||
| Scheme 100 Synthesis of spiro-isoxazolidines containing nucleobases with isatin 284a–g. | ||
Ashok and co-workers developed a group of 2-indolinone-based bis-1,2,3-triazole derivatives via microwave irradiation.175 They performed the copper-catalyzed azide–alkyne click reaction between various isatin derivatives containing terminal alkyne and azides under microwave irradiation at 80 °C for 3–5 min to afford excellent yields (85–93%) of designed products 287a–f. The microwave-assisted method that was involved in this reaction completed the reaction in short period of time with higher yields (85–93%) as compared with yields using conventional heating (43–54%) (Scheme 101).
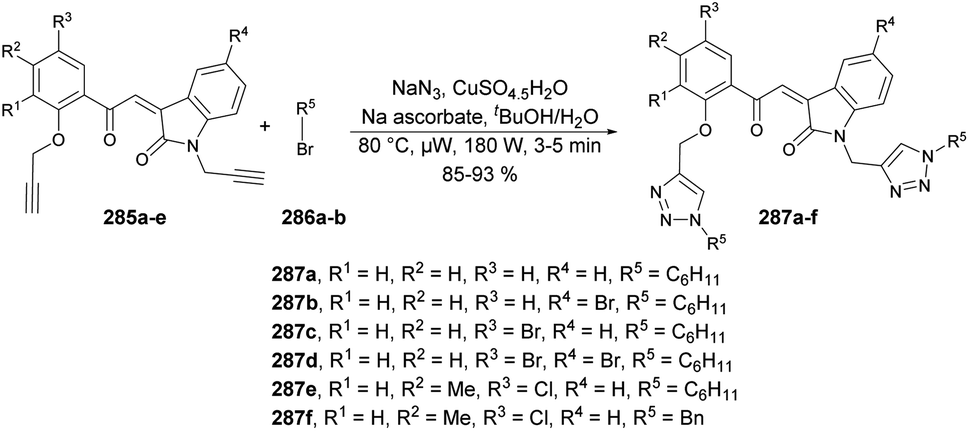 | ||
| Scheme 101 Synthesis of 2-indolinone-based triazole derivatives 287a–f. | ||
To save the lives of patients infected with multidrug resistant-tuberculosis (MDR-TB), there is an urgent need for new chemotherapeutic agents. Karpoormath and co-workers addressed this issue by synthesizing and characterizing novel isatin hydrazones 290a–o and their thiomorpholine-tethered analogues 291a–o.176 They started with isatins 288a–b which, on reaction with hydrazine hydrate, afforded compounds 289a–b. These compounds were again treated with various aromatic aldehydes under microwave irradiation at 80 °C to afford the designed compounds 290a–o. After that, isatin hydrazones 290a–o reacted with thiomorpholine and formaldehyde at room temperature to afford the designed products 291a–o in good isolated yields (68–85%) (Scheme 102). They investigated the anti-mycobacterial activity of derived compounds against the H37Rv strain of Mycobacterium tuberculosis. All of the synthesized compounds were initially screened for their anti-mycobacterial activity. Notably, five compounds (290f, 290h, 290n, 291f, and 291m) demonstrated high activity, with 290f (IC50 = 1.9 μM) exhibiting the highest inhibition of H37Rv, indicating potential as effective treatment options.
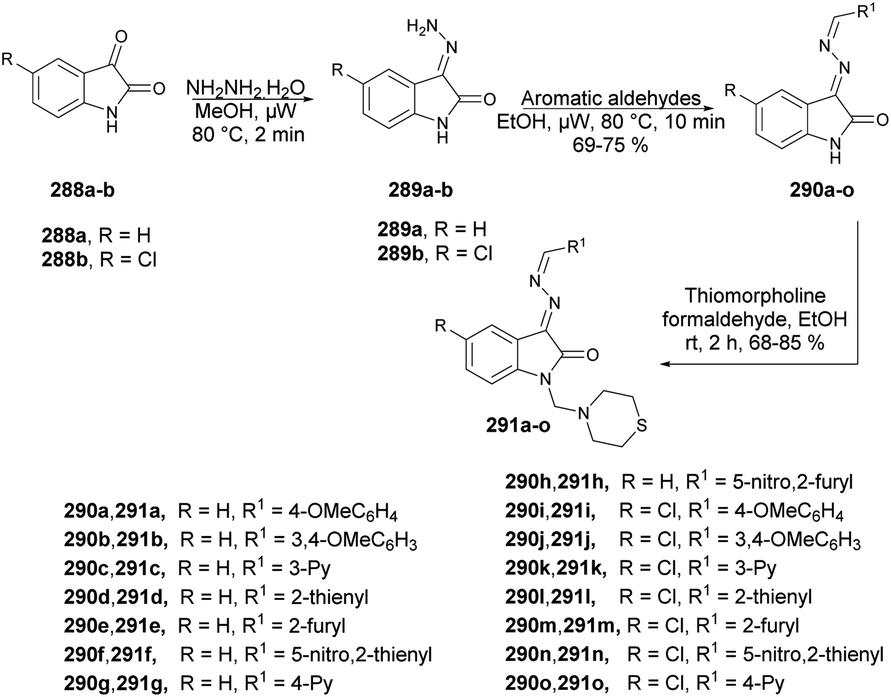 | ||
| Scheme 102 Synthesis of isatin hydrazones 290a–290o and their thiomorpholine-tethered analogues 291a–291o. | ||
Maniam and co-workers reported a microwave-assisted, one-pot, three-component, 1,3-dipolar cycloaddition approach to generate azomethine ylides from L-proline and isatin, followed by their reaction with various β-nitrostyrenes to rapidly synthesize a library of Sox-pyrrolizidines.177 These compounds exhibited remarkable binding affinities to the intensely amyloidogenic regions of HEWL, leading to the prevention of fibril aggregation and prompting fibrillation to adopt a less structured form than β-sheets. Notably, the Sox molecules showed negligible cell toxicity, and compound 295b was capable of safeguarding HEK cells against α,β-induced toxicity (Scheme 103).
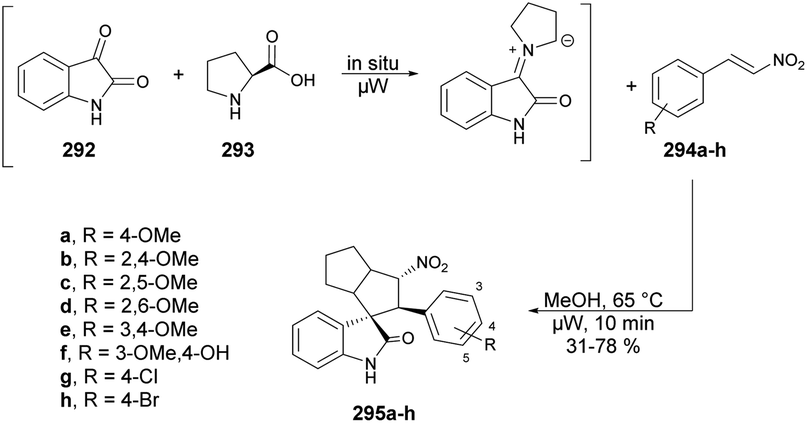 | ||
| Scheme 103 Synthesis of Sox-pyrrolizidines 295a–h. | ||
The author of that study proposed a reaction mechanism involving two main steps. First, a condensation reaction takes place to form an intermediate (Scheme 104). Following this, a cycloaddition reaction occurs between the two nucleophilic carbons and electrophilic β-carbon of the β-nitrostyrene. This addition results in the formation of a spirocyclic structure involving three new C–C bonds and generating four contiguous stereocenters, including a quaternary carbon. Consequently, two potential regioisomers can be formed due to the different transition states, namely TS1 and TS2.
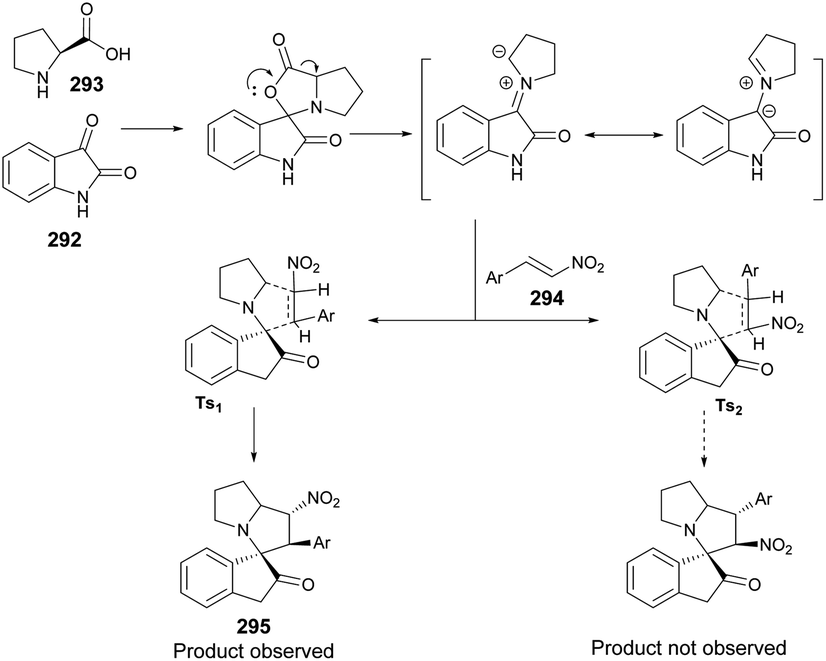 | ||
| Scheme 104 Proposed mechanism for the synthesis of nitro-Sox compounds. | ||
However, the sterically hindered nature of TS2 compared with TS1 leads to an unfavorable and less stable product, which is not observed experimentally. This observation was consistent with the exclusive formation of only one regioisomer from TS1. Despite the presence of four stereocenters, the reaction tends to favor the aromatic group attached to the pyrrolizidine moiety being cis to the carbonyl group of the oxindole and trans to the nitro group. This specific arrangement minimizes steric hindrance between the aryl group attached to the pyrrolizidine moiety and the benzo group of the oxindole moiety.
Junior and co-workers developed a novel approach to synthesize N-(2-oxoindolin-3-ylidene)benzohydrazide derivatives, which are extensively explored in pharmacological research.178 Conventionally, their production relies on organic solvents, leading to increased costs, the generation of residues and is a time-consuming process. To address these challenges, researchers developed a more environmentally friendly and cost-effective method utilizing bentonite clay as a catalyst and microwave irradiation. The study involved optimizing various parameters, such as the type and amount of catalyst, solvent, and heating method. Ultimately, the most favorable conditions were found to be a solvent-free reaction in the presence of bentonite as the catalyst, resulting in yields of up to 96% for isolated products and reaction times of 15–60 min. Remarkably, these outcomes were comparable to or even surpassed those reported in the literature, yet with the added advantage of a greener approach through heterogeneous catalysis using a microwave technique (Scheme 105).
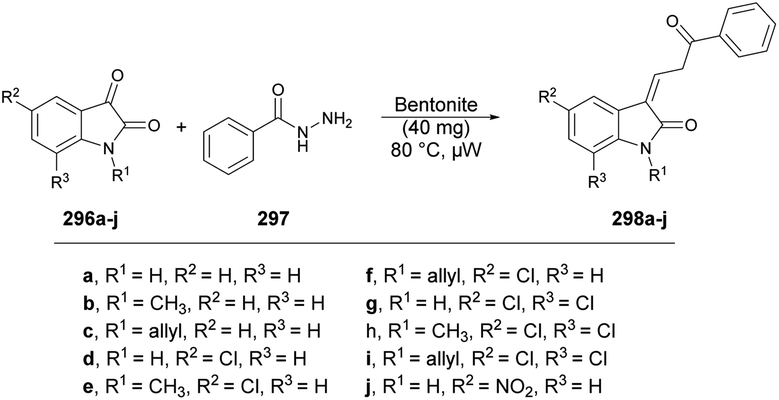 | ||
| Scheme 105 Synthesis of N-(2-oxoindolin-3-ylidene)benzohydrazide derivatives 298a–j. | ||
Furthermore, researchers demonstrated that the bentonite catalyst could be recycled at least three times without significant loss in yields or reaction time, showcasing its practicality as a low-cost and sustainable alternative. This innovative methodology opens up new possibilities for the efficient and environmentally compatible synthesis of isatin-derived Schiff bases with potential implications in pharmacological applications.
Chaudhary and co-workers developed a novel series of functionalized spiro[indoline-3,20-pyrrolidin]-2-one/spiro[indoline-3,30-pyrrolizin]-2-one compounds (denoted as 302a–d, 303a–e, and 304a–d).179 These compounds were derived from pharmaceutically privileged bioactive sub-structures, namely isatins 299a–f, various substituted chalcones 300a–f, and 301a–c amino acids, using 1,3-dipolar cycloaddition reactions. Microwave-assisted 1,3-dipolar cycloaddition reactions were carried out in MeOH at 80 °C for 5 min involving substituted isatins 299a–f, substituted chalcones 300a–f, and various amino acids 301a–c. These reactions yielded the desired chalcone-isatin-amino acid-based spirooxindole compounds 302a–d, 303a–e, and 304a–d in excellent yields of up to 98%, and the process displayed high diastereoselectivity. The reaction involved the [3 + 2] cycloaddition of substituted chalcones with in situ-generated azomethine ylides, which resulted from the microwave-assisted decarboxylative condensation of substituted isatins and various secondary amino acids. Compared with traditional methods, the microwave-assisted approach yielded higher quantities of compounds with improved quality, all while requiring less time for synthesis.
After the successful synthesis of derived molecules, researchers evaluated the in vitro antileishmanial activity against Leishmania donovani and performed structure–activity relationship (SAR) studies. Among the compounds tested, 302a, 303e, and 304d demonstrated the most potent activity within the series, exhibiting IC50 values of 2.43 μM, 0.96 μM, and 3.55 μM, respectively, when compared with the standard reference drug amphotericin B (IC50 = 0.60 μM). Those findings suggest that the newly synthesized compounds hold promise as potential candidates with antileishmanial properties (Scheme 106).
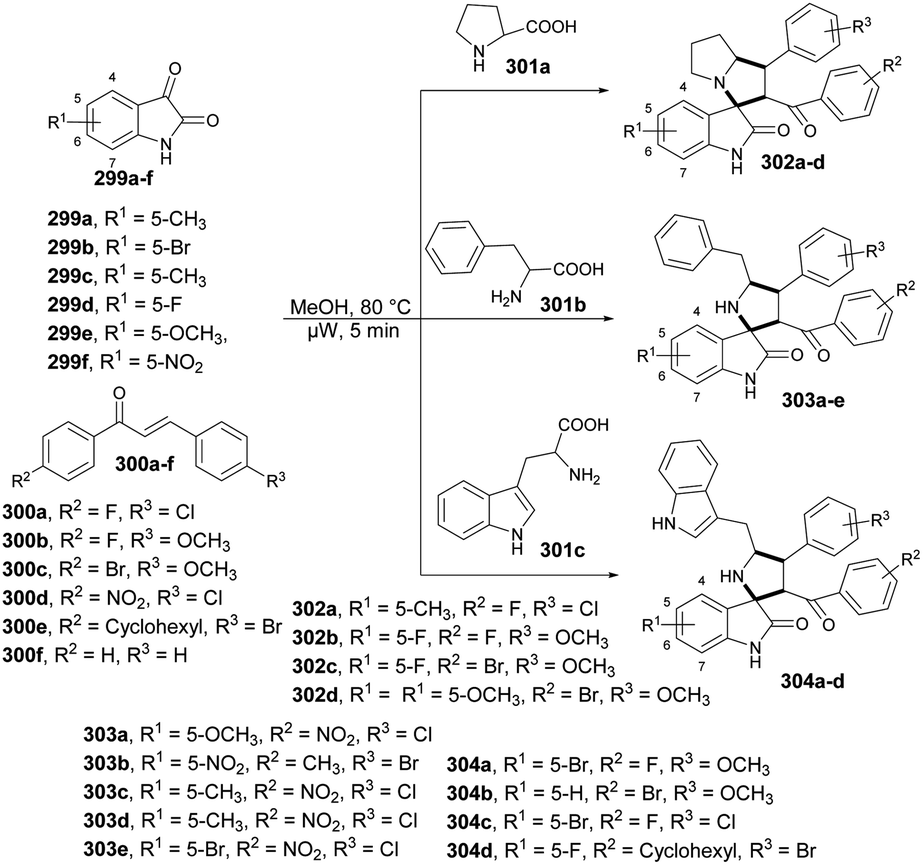 | ||
| Scheme 106 Synthesis of spiro[indoline-3,20-pyrrolidin]-2-one/spiro[indoline-3,30-pyrrolizin]-2-one 302a–d, 303a–e, and 304a–d. | ||
Zhao and co-workers developed an environmentally friendly and efficient microwave-promoted multicomponent reaction involving isatins, α-amino acids, and 1,4-dihydro-1,4-epoxynaphthalene.180 Within a remarkably short time of 15 min, the reaction produced oxygen-bridged spirooxindoles in good-to-excellent yields. Notably, the 1,3-dipolar cycloaddition process exhibited compatibility with various primary amino acids and showcased high efficiency due to its rapid reaction time (Scheme 107). Furthermore, they scaled up the reaction and performed synthetic transformations of spiropyrrolidine oxindole, showcasing its practical synthetic utility. The introduction of this new method expands the structural diversity of spirooxindoles, making them a promising scaffold for novel-drug discovery. With these attractive features and potential applications, that work presents a powerful approach to enhance the repertoire of spirooxindoles as a platform for exploring new drug candidates.
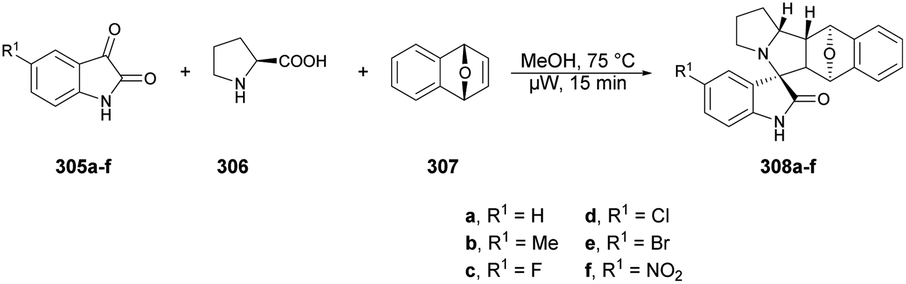 | ||
| Scheme 107 Synthesis of oxygen-bridged spirooxindoles 308a–f. | ||
Initially, the reaction involves the condensation of isatin with proline, resulting in the formation of the ylide intermediate A through subsequent decarboxylation. This ylide intermediate A serves as a 1,3-dipole and reacts with 3a through two potential pathways, labeled as A and B. Path A leads to the production of the thermodynamically more stable endo-cycloadduct 4a, while the formation of the exo-cycloadduct 4a′ through path B is likely less favored.
3. Conclusions and perspectives
Based on the discussions stated above, it is evident that microwave-assisted organic synthesis is a highly effective method for conducting a diverse range of chemical reactions in a shorter reaction of time with high yield and selectivity. Ultimately, the use of microwave irradiation has greatly enhanced the process of drug discovery and contributed towards efficient synthesis of complex natural products and natural product-like molecules. This review presented the microwave-assisted organic synthesis of N- and O-containing heterocyclic compounds. Various quinoline derivatives with structural diversity could be prepared in shorter reaction time and high yields. Similarly, pyrazolopyrimidine derivatives have been synthesized under microwave irradiation which were otherwise difficult to prepare. Comarins, isatins and their diverse derivatives with structural complexity were prepared in efficient manners recently adopting microwave assisted-organic synthesis. Thus, this technique has scope to prepare various natural-product scaffolds, which are complex and filling 3D chemical space.In conclusion, microwave-assisted synthesis has proven to be a game-changer in the field of organic synthesis and materials science. Its ability to accelerate reactions, improve yields, and reduce energy consumption has made it an invaluable tool for researchers. As we delve deeper into its mechanisms and explore new applications, microwave-assisted synthesis is poised to play an increasingly significant part in shaping the future of chemistry and materials research. The data provided showcase several successful applications of microwave-assisted synthesis in various chemical reactions, leading to enhanced yields, shortened reaction times, and improved selectivity. The examples presented highlight the effectiveness of microwave irradiation in different synthetic transformations.
Suzuki–Miyaura cross-coupling reactions: Ababri and co-workers demonstrated an optimized microwave-assisted Suzuki–Miyaura cross-coupling reaction protocol. By utilizing microwave irradiation at 135 °C, the reaction time was significantly reduced from 12 h to just 40 min, and the yield of the coupled product was impressive (67–89%). Moreover, the unwanted debromination reaction was effectively avoided, resulting in high product purity. Friedlander synthesis of 8-hydroxyquinolines: Microwave-assisted Friedlander synthesis provided a single-step conversion of various derivatives of 8-hydroxyquinolines with a higher yield (72%) compared with conventional heating (34% yield). This demonstrates the efficiency and effectiveness of microwave-assisted synthesis in complex transformations. Copper-catalyzed four-component synthesis of 3-N-sulfonylamidine coumarins: Punniyamurthy and co-workers reported a copper-catalyzed four-component synthesis of coumarins using microwave irradiation. The use of microwave conditions resulted in higher reactivity, milder reaction conditions, and greater selectivity, leading to good isolated yields of the desired products. Microwave-assisted synthesis of isatin-β-thiocarbohydrazones: Gohary and co-workers developed a new range of isatin-β-thiocarbohydrazones using microwave irradiation. The approach utilized the key features required for metal chelation found in triapine and resulted in good isolated yields of the desired products. Moreover, some of the newly developed compounds showed promising efficacy against cancer cell lines. Microwave-assisted synthesis of quinoline fused 1,4-benzodiazepine: Kamble and co-workers reported an efficient microwave-assisted protocol for the synthesis of quinoline fused 1,4-benzodiazepines. The use of microwave irradiation at 80 °C afforded excellent isolated yields of the desired products, but the conventional method did not afford a good yield in a limited time. The Pechmann reaction was conducted using conventional thermal heating. However, even after 20 min, the phenol conversion did not exceed 18%. This outcome underscores the potential of microwave irradiation as a viable alternative to conventional heating methods for this type of reaction.
Continuous efforts are being made by different research groups across the globe using microwave irradiation techniques for the successful synthesis of a variety of biologically active compounds. This strategy shows great promises for its use in the near future as a powerful tool for constructing efficiently natural product-like scaffolds. Microwave-assisted synthesis has already demonstrated its potential as a powerful and efficient tool for chemical transformations. Looking ahead, this innovative technology holds promising future perspectives that could further revolutionize synthetic chemistry and materials research.
Despite the successful applications of microwave-assisted synthesis, further research is needed to gain deeper understanding of the underlying mechanisms responsible for its enhanced reactivity and selectivity. A deeper understanding of these mechanisms can guide the optimization of reactions and the development of new synthetic strategies. The success of microwave-assisted synthesis in various reactions encourages the exploration of new transformations that can benefit from this technique. Researchers can investigate unexplored reactions that can be accelerated and improved through microwave irradiation. Green and sustainable chemistry: the energy efficiency and reduced reaction times associated with microwave-assisted synthesis align well with the principles of green and sustainable chemistry. Microwave-assisted synthesis has shown its potential for promoting green chemistry principles. Future research should continue to explore environmentally friendly reaction conditions, such as solvent-free and catalytic microwave reactions, to reduce waste and energy consumption.
Researchers need to address scale-up challenges to make microwave-assisted synthesis more applicable to industrial processes. The development of scalable and economically viable methodologies is essential. Bridging the gap between laboratory-scale experiments and industrial applications is a critical aspect of future perspectives. Researchers should focus on developing scalable and economically viable microwave-assisted synthetic methodologies to facilitate the adoption of this technology in industrial processes. Researchers can explore the combination of microwave-assisted synthesis with other innovative techniques, such as flow chemistry, photochemistry, and catalysis, to achieve synergistic effects and expand the scope of synthetic possibilities. The development of advanced microwave reactors with precise control over power, frequency, and temperature will allow researchers to optimize and scale-up reactions effectively. Integration of automation and artificial-intelligence technologies in reactor design could lead to more efficient and autonomous synthesis processes. Multicomponent reactions: microwave irradiation has proven to be advantageous in multicomponent reactions, offering rapid access to complex molecules. Future efforts should focus on developing novel multicomponent reactions that take advantage of the enhanced reactivity and selectivity offered by microwave technology.
The pharmaceutical industry can benefit significantly from microwave-assisted synthesis for drug discovery and development. Integrating microwave technology in medicinal chemistry research can expedite the synthesis of potential drug candidates and facilitate the production of complex molecules. Microwave technology can be applied to the synthesis of advanced materials with “tailored” properties. Researchers should explore the use of microwave irradiation in the preparation of nanomaterials, catalysts, and functional materials for various applications. safety and standardization:
As microwave-assisted synthesis gains wider adoption, it is crucial to establish standardized safety protocols and guidelines for its use in laboratories and industrial settings. Safety training and awareness will ensure responsible and efficient operation.
These forward-looking perspectives provide valuable guidance to researchers interested in advancing this field. The review emphasizes the practical benefits of microwave-assisted synthesis, such as shorter reaction times, improved yields, and enhanced reactivity. It also highlights the applicability of microwave technology to complex reactions, which can lead to the synthesis of highly functionalized compounds. The review includes examples of microwave-assisted synthesis applied to drug discovery, showcasing its relevance and potential impact in the pharmaceutical industry.
The future perspectives of microwave-assisted synthesis hold great promise for advancing synthetic chemistry and materials science. With a focus on understanding mechanisms, developing green and sustainable methodologies, and exploring new applications, microwave-assisted synthesis is poised to become an indispensable tool in modern chemistry, paving the way for faster, cleaner, and more efficient chemical transformations.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
Authors are thankful to the Banaras Hindu University (BHU) and Jawaharlal Nehru University (JNU) for providing facilities to compile this review. GT & AK are grateful to BHU for the research fellowship. VKM is thankful to UGC New Delhi for the JRF/SRF Fellowship.References
- Q. Cheng, L. Xiang, M. Izumikawa, D. Meluzzi and B. S. Moore, Nat. Chem. Biol., 2007, 3, 557–558 CrossRef CAS PubMed.
- G. Castro-Falcón, D. Hahn, D. Reimer and C. C. Hughes, ACS Chem. Biol., 2016, 11, 2328–2336 CrossRef PubMed.
- T. Grkovic, R. K. Akee, C. C. Thornburg, S. K. Trinh, J. R. Britt, M. J. Harris, J. R. Evans, U. Kang, S. Ensel, C. J. Henrich, K. R. Gustafson, J. P. Schneider and B. R. O'Keefe, ACS Chem. Biol., 2020, 15, 1104–1114 CrossRef CAS PubMed.
- J. H. Miller, J. J. Field, A. Kanakkanthara, J. G. Owen, A. J. Singh and P. T. Northcote, J. Nat. Prod., 2018, 81, 691–702 CrossRef CAS PubMed.
- M. Feher and J. M. Schmidt, J. Chem. Inf. Comput. Sci., 2003, 43, 218–227 CrossRef CAS PubMed.
- D. J. Newman, G. M. Cragg and D. G. I. Kingston, in The Practice of Medicinal Chemistry, ed. C. G. Wermuth, Academic Press, New York, 3rd edn, 2008, pp. 159–186 Search PubMed.
- C. G. Wermuth and J. de la Fontaine, The practice of medicinal chemistry, 1996, vol. 2, pp. 203–237 Search PubMed.
- A. L. Harvey, R. Edrada-Ebel and R. J. Quinn, Nat. Rev. Drug Discovery, 2015, 14, 111–129 CrossRef CAS PubMed.
- T. Rodrigues, D. Reker, P. Schneider and G. Schneider, Nat. Chem., 2016, 8, 531–541 CrossRef CAS PubMed.
- B. Shen, Cell, 2015, 163, 1297–1300 CrossRef CAS PubMed.
- E. K. Davison and J. Sperry, J. Nat. Prod., 2017, 80, 3060–3079 CrossRef CAS PubMed.
- T. Ma, L. Liu, H. Xue, L. Li, C. Han, L. Wang, Z. Chen and G. Liu, J. Med. Chem., 2008, 51, 1432–1446 CrossRef CAS PubMed.
- S. Anzabi Zadeh, W. N. Street and B. W. Thomas, J. Biomed. Inf., 2023, 137, 104267 CrossRef PubMed.
- A. O. Dada, A. A. Inyinbor, O. S. Bello and B. E. Tokula, Biomass Convers. Biorefin., 2023, 13, 9181–9193 CrossRef CAS.
- P. Ashrafi, D. Nematollahi, A. Shabanloo, A. Ansari, M. Eslamipanah and B. Jaleh, Electrochim. Acta, 2023, 458, 142555 CrossRef CAS.
- W. M. Eldehna, R. I. Al-Wabli, M. S. Almutairi, A. B. Keeton, G. A. Piazza, H. A. Abdel-Aziz and M. I. Attia, J. Enzyme Inhib. Med. Chem., 2018, 33, 867–878 CrossRef CAS PubMed.
- J. Ren, S. Ding, X. Li, R. Bi and Q. Zhao, J. Org. Chem., 2021, 86, 12762–12771 CrossRef CAS PubMed.
- J.-J. Chen, Y.-L. Sun, A. Tiwari, Z.-J. Xiao, K. Sodani, D.-H. Yang, S. Vispute, W.-Q. Jiang, S.-D. Chen and Z.-S. Chen, Cancer Sci., 2012, 103, 1531–1537 CrossRef CAS PubMed.
- D. Dolles and M. Decker, in Design of Hybrid Molecules for Drug Development, ed. M. Decker, Elsevier, 2017, pp. 137–165 Search PubMed.
- B. Nişancı, in Nontraditional Activation Methods in Green and Sustainable Applications, ed. B. Török and C. Schäfer, Elsevier, 2021, pp. 329–347 Search PubMed.
- J. Yoon and J. S. Ryu, Bioorg. Med. Chem. Lett., 2010, 20, 3930–3935 CrossRef CAS PubMed.
- E. Bálint and G. Keglevich, in Milestones in Microwave Chemistry, ed. G. Keglevich, Springer International Publishing, Cham, 2016, pp. 1–10 Search PubMed.
- C. O. Kappe and D. Dallinger, Nat. Rev. Drug Discovery, 2006, 5, 51–63 CrossRef CAS PubMed.
- S. Banerjee, Curr. Org. Chem., 2020, 24, 2525–2526 CrossRef CAS.
- N. Kaur, Synth. Commun., 2014, 44, 3509–3537 CrossRef CAS.
- P. Khanna, L. Khanna, S. J. Thomas, A. M. Asiri and S. S. Panda, Curr. Org. Chem., 2018, 22, 67–84 CrossRef CAS.
- J. D. Moseley and C. O. Kappe, Green Chem., 2011, 13, 794–806 RSC.
- X.-L. Wei, Q.-L. Shi, L. Jiang and Y. Qin, New J. Chem., 2023, 47, 15525–15533 RSC.
- C. Leonelli, P. Veronesi and G. Cravotto, in Microwave-assisted Extraction for Bioactive Compounds: Theory and Practice, ed. F. Chemat and G. Cravotto, Springer US, Boston, MA, 2013, pp. 1–14 Search PubMed.
- J. Gising, L. R. Odell and M. Larhed, Org. Biomol. Chem., 2012, 10, 2713–2729 RSC.
- K. Pei, D. Li, W. Qi and D. Wu, Molecules, 2023, 28, 1892 CrossRef CAS PubMed.
- A. Adhikari, S. Bhakta and T. Ghosh, Tetrahedron, 2022, 126, 133085 CrossRef CAS.
- F. Zhu, H. Lu and Y. Lu, J. Mater. Sci., 2023, 58, 4399–4415 CrossRef CAS.
- P. Lidström, J. Tierney, B. Wathey and J. Westman, Tetrahedron, 2001, 57, 9225–9283 CrossRef.
- V. Martinez, T. Stolar, B. Karadeniz, I. Brekalo and K. Užarević, Nat. Rev. Chem, 2023, 7, 51–65 CrossRef CAS PubMed.
- M. Baghbanzadeh, L. Carbone, D. Cozzoli and C. O. Kappe, Angew. Chem., Int. Ed. Engl., 2012, 50, 11312–11359 CrossRef PubMed.
- C. O. Kappe, Angew. Chem., Int. Ed., 2004, 43, 6250–6284 CrossRef CAS PubMed.
- A. Lew, P. Krutzik, M. Hart and A. Chamberlin, J. Comb. Chem., 2002, 4, 95–105 CrossRef CAS PubMed.
- E. Grant and B. J. Halstead, Chem. Soc. Rev., 1998, 27, 213–224 RSC.
- Y. Mendoza Tolentino, G. Y. Romero-Zúñiga, M. A. Ceniceros Reyes, P. C. Flores Silva, Á. V. Ramírez, R. Yáñez-Macías, E. Hernández Hernández and P. González Morones, J. Appl. Polym. Sci., 2023, 140, e53793 CrossRef CAS.
- D. Dallinger and C. O. Kappe, Chem. Rev., 2007, 107, 2563–2591 CrossRef CAS PubMed.
- A. Rodriguez Garcia, P. Prieto, A. Hoz, A. Díaz-Ortiz, R. Ml and J. García, ChemistryOpen, 2015, 2015, 308–317 CrossRef PubMed.
- R. Javahershenas and S. Nikzat, RSC Adv., 2023, 2023, 16619–16629 RSC.
- J. R. Vanhoof, R. Dirix and D. E. De Vos, Green Chem., 2023, 25, 978–985 RSC.
- V. Flores-Morales, A. P. Villasana-Ruíz, I. Garza-Veloz, S. González-Delgado and M. L. Martinez-Fierro, Molecules, 2023, 28, 2413 CrossRef CAS PubMed.
- A. Arif and M. Khan, RSC Adv., 2023, 13, 11652–11684 RSC.
- A. Mushtaq, U. Azam, S. Mehreen and M. M. Naseer, Eur. J. Med. Chem., 2023, 249, 115119 CrossRef CAS PubMed.
- A. Weyesa and E. Mulugeta, RSC Adv., 2020, 10, 20784–20793 RSC.
- C. Verma, M. A. Quraishi and E. E. Ebenso, Surf. Interfaces, 2020, 21, 100634 CrossRef CAS.
- F. W. Bergstrom, Chem. Rev., 1944, 35, 77–277 CrossRef CAS.
- B. S. Matada, R. Pattanashettar and N. G. Yernale, Bioorg. Med. Chem., 2021, 32, 115973 CrossRef CAS PubMed.
- R. R. Baker, J. R. Pereira da Silva and G. Smith, Food Chem. Toxicol., 2004, 42, 3–37 CrossRef PubMed.
- K. Mezgebe, Y. Melaku and E. Mulugeta, ACS Omega, 2023, 8, 19194–19211 CrossRef CAS PubMed.
- J. R. Angupale, J. Tusiimire and N. C. Ngwuluka, Malar. J., 2023, 22, 1–19 CrossRef PubMed.
- X. Wang, Y. Zhuang, Y. Wang, M. Jiang and L. Yao, Eur. J. Med. Chem., 2023, 260, 115710 CrossRef CAS PubMed.
- M. S. S. Chauhan, T. Umar and M. K. Aulakh, ChemistrySelect, 2023, 8, e202204960 CrossRef CAS.
- J.-X. An, Y. Ma, W.-B. Zhao, Y.-M. Hu, Y.-R. Wang, Z.-J. Zhang, X.-F. Luo, B.-Q. Zhang, Y.-Y. Ding and Y.-Q. Liu, J. Antibiot., 2023, 76, 131–182 CrossRef CAS PubMed.
- S. Kumar, A. Patel and N. Ahmed, RSC Adv., 2015, 5, 93067–93080 RSC.
- Á. Baji, A. Gyovai, J. Wölfling, R. Minorics, I. Ocsovszki, I. Zupkó and É. Frank, RSC Adv., 2016, 6, 27501–27516 RSC.
- N. A. Liberto, J. B. Simões, S. de Paiva Silva, C. J. da Silva, L. V. Modolo, Â. de Fátima, L. M. Silva, M. Derita, S. Zacchino, O. M. P. Zuñiga, G. P. Romanelli and S. A. Fernandes, Bioorg. Med. Chem., 2017, 25, 1153–1162 CrossRef CAS PubMed.
- W. Ali, A. Dahiya, R. Pandey, T. Alam and B. K. Patel, J. Org. Chem., 2017, 82, 2089–2096 CrossRef CAS PubMed.
- A. T. Garrison, Y. Abouelhassan, H. Yang, H. H. Yousaf, T. J. Nguyen and R. W. Huigens III, MedChemComm, 2017, 8, 720–724 RSC.
- B. Asadi, A. Landarani-Isfahani, I. Mohammadpoor-Baltork, S. Tangestaninejad, M. Moghadam, V. Mirkhani and H. A. Rudbari, Tetrahedron Lett., 2017, 58, 71–74 CrossRef CAS.
- D. Chandra, A. K. Dhiman, R. Kumar and U. Sharma, Eur. J. Org Chem., 2019, 2019, 2753–2758 CrossRef CAS.
- S. B. Marganakop, R. Kamble, A. Nesaragi, P. Bayannavar, S. D. Joshi, P. P. Kattimani and B. S. Sudha, Asian J. Chem., 2021, 33, 1107–1114 CAS.
- R. Munir, M. Zia-ur-Rehman, S. Murtaza, S. Zaib, N. Javid, S. J. Awan, K. Iftikhar, M. M. Athar and I. Khan, Molecules, 2021, 26, 656 CrossRef CAS PubMed.
- M. Robert Khumalo, S. N. Maddila, S. Maddila and S. B. Jonnalagadda, RSC Adv., 2019, 9, 30768–30772 RSC.
- P. Chidurala, V. Jetti, R. Pagadala, J. S. Meshram and S. B. Jonnalagadda, J. Heterocycl. Chem., 2015, 52, 1302–1307 CrossRef CAS.
- D. Insuasty, R. Abonia, B. Insuasty, J. Quiroga, K. K. Laali, M. Nogueras and J. Cobo, ACS Comb. Sci., 2017, 19, 555–563 CrossRef CAS PubMed.
- P. Fedoseev and E. Van der Eycken, Chem. Commun., 2017, 53, 7732–7735 RSC.
- A. Rani, A. Viljoen, Sumanjit, L. Kremer and V. Kumar, ChemistrySelect, 2017, 2, 10782–10785 CrossRef CAS.
- S. Moloi, S. Maddila and S. B. Jonnalagadda, Res. Chem. Intermed., 2017, 43, 6233–6243 CrossRef CAS.
- X.-Y. Li, Y. Liu, X.-L. Chen, X.-Y. Lu, X.-X. Liang, S.-S. Zhu, C.-W. Wei, L.-B. Qu and B. Yu, Green Chem., 2020, 22, 4445–4449 RSC.
- D. B. Patel, D. P. Rajani, S. D. Rajani and H. D. Patel, J. Heterocycl. Chem., 2020, 57, 1524–1544 CrossRef CAS.
- C. K. Chan, C.-Y. Lai and C. C. Wang, Synthesis, 2020, 52, 1779–1794 CrossRef CAS.
- B. A. L. Sacchelli, B. C. Rocha and L. H. Andrade, Org. Lett., 2021, 23, 5071–5075 CrossRef CAS PubMed.
- B. Saroha, G. Kumar, M. Kumari, R. Kaur, N. Raghav, P. K. Sharma, N. Kumar and S. Kumar, Int. J. Biol. Macromol., 2022, 222, 2270–2308 CrossRef CAS PubMed.
- P. J. Choi, G.-L. Lu, H. S. Sutherland, A. C. Giddens, S. G. Franzblau, C. B. Cooper, W. A. Denny and B. D. Palmer, Tetrahedron Lett., 2022, 90, 153611 CrossRef CAS.
- J.-L. Yue, X.-W. Chang, J.-W. Zheng, L. Shi, Y.-J. Xiang, J.-Y. Que, K. Yuan, J.-H. Deng, T. Teng, Y.-Y. Li, W. Sun, H.-Q. Sun, M. V. Vitiello, X.-D. Tang, X.-Y. Zhou, Y.-P. Bao, J. Shi and L. Lu, Sleep Med. Rev., 2023, 68, 101746 CrossRef CAS PubMed.
- J. K. Walsh, A. Moscovitch, J. Burke, R. Farber and T. Roth, Sleep Med., 2007, 8, 753–759 CrossRef PubMed.
- R. Wang, H. Wang, G. Jiang, Y. Sun, T. Liu, L. Nie, A. Shavandi, K. E. Yunusov, U. E. Aharodnikau and S. O. Solomevich, Biomater. Sci., 2023, 11, 1704–1713 RSC.
- S. Abu-Melha, Arabian J. Sci. Eng., 2023, 1–21 Search PubMed.
- A. M. Naglah, A. A. Askar, A. S. Hassan, T. K. Khatab, M. A. Al-Omar and M. A. Bhat, Molecules, 2020, 25, 1431 CrossRef CAS PubMed.
- S. Sheikhi-Mohammareh, F. Oroojalian, H. Beyzaei, M. Moghaddam-Manesh, A. Salimi, F. Azizollahi and A. Shiri, Talanta, 2023, 262, 124723 CrossRef CAS PubMed.
- A. Kumar, K. K. Bhagat, A. K. Singh, H. Singh, T. Angre, A. Verma, H. Khalilullah, M. Jaremko, A.-H. Emwas and P. Kumar, RSC Adv., 2023, 13, 6872–6908 RSC.
- H. Galons, N. Oumata and L. Meijer, Expert Opin. Ther. Pat., 2010, 20, 377–404 CrossRef CAS PubMed.
- K. Ahmed, M. I. Choudhary and R. S. Z. Saleem, Eur. J. Med. Chem., 2023, 259, 115701 CrossRef CAS PubMed.
- M. A. Khedr, W. A. Zaghary, G. E. Elsherif, R. A. Azzam and G. H. Elgemeie, Nucleosides, Nucleotides Nucleic Acids, 2023, 42, 77–104 CrossRef CAS PubMed.
- X. Gao, F. Zhao, Y. Wang, X. Ma, H. Chai, J. Han and F. Fang, Bioorg. Med. Chem., 2023, 91, 117402 CrossRef CAS PubMed.
- M. Abirami, B. Karan Kumar, Faheem, S. Dey, S. Johri, R. M. Reguera, R. Balaña-Fouce, K. V. Gowri Chandra Sekhar and M. Sankaranarayanan, Eur. J. Med. Chem., 2023, 257, 115471 CrossRef CAS PubMed.
- Z. M. Alamshany, R. R. Khattab, N. A. Hassan, A. A. El-Sayed, M. A. Tantawy, A. Mostafa and A. A. Hassan, Molecules, 2023, 28, 739 CrossRef CAS PubMed.
- C.-C. Tseng, S.-E. Tsai, S.-M. Li and F. F. Wong, J. Org. Chem., 2019, 84, 16157–16170 CrossRef CAS PubMed.
- B. Jismy, G. Guillaumet, M. Akssira, A. Tikad and M. Abarbri, RSC Adv., 2021, 11, 1287–1302 RSC.
- J. D. Singleton, R. Dass, N. R. Neubert, R. M. Smith, Z. Webber, M. D. H. Hansen and M. A. Peterson, Bioorg. Med. Chem. Lett., 2020, 30, 126813 CrossRef CAS PubMed.
- J. H. Ng, E. R. T. Tiekink and A. V. Dolzhenko, RSC Adv., 2022, 12, 8323–8332 RSC.
- J.-C. Castillo, D. Estupiñan, M. Nogueras, J. Cobo and J. Portilla, J. Org. Chem., 2016, 81, 12364–12373 CrossRef CAS PubMed.
- I. Bassoude, Z. Tber, E. M. Essassi, G. Guillaumet and S. Berteina-Raboin, RSC Adv., 2016, 6, 3301–3306 RSC.
- J. Zhang, J.-F. Peng, Y.-B. Bai, P. Wang, T. Wang, J.-M. Gao and Z.-T. Zhang, Mol. Diversity, 2016, 20, 887–896 CrossRef CAS PubMed.
- J.-C. Castillo, H.-A. Rosero and J. Portilla, RSC Adv., 2017, 7, 28483–28488 RSC.
- J.-C. Castillo, A. Tigreros and J. Portilla, J. Org. Chem., 2018, 83, 10887–10897 CrossRef CAS PubMed.
- A. K. Pathak, S. Gupta, P. B. Punjabi, G. Ameta and C. Ameta, J. Heterocycl. Chem., 2019, 56, 2056–2062 CrossRef CAS.
- A. M. Alsaedi, T. A. Farghaly and M. R. Shaaban, Molecules, 2019, 24, 4009 CrossRef PubMed.
- M. Fouda, H.-A. S. Abbas, E. H. Ahmed, A. A. Shati, M. Y. Alfaifi and S. E. I. Elbehairi, Molecules, 2019, 24, 1080 CrossRef PubMed.
- A. Ballesteros-Casallas, M. Paulino, P. Vidossich, C. Melo, E. Jiménez, J.-C. Castillo, J. Portilla and G. P. Miscione, Eur. J. Med. Chem. Rep., 2022, 4, 100028 CAS.
- S.-L. Aranzazu, A. Tigreros, A. Arias-Gómez, J. Zapata-Rivera and J. Portilla, J. Org. Chem., 2022, 87, 9839–9850 CrossRef CAS PubMed.
- Y. Xu, H. Li, S. Xu, X. Liu, J. Lin, H. Chen and Z. Yuan, J. Med. Chem., 2022, 65, 424–435 CrossRef CAS PubMed.
- S. K. Konidala, V. Kotra, R. C. S. R. Danduga, P. K. Kola, R. R. Bhandare and A. B. Shaik, Arabian J. Chem., 2021, 14, 103154 CrossRef CAS.
- H. M. Alshibl, E. S. Al-Abdullah, M. E. Haiba, H. M. Alkahtani, G. E. Awad, A. H. Mahmoud, B. M. Ibrahim, A. Bari and A. Villinger, Molecules, 2020, 25, 3251 CrossRef CAS PubMed.
- L. K. A. M. Leal, A. A. G. Ferreira, G. A. Bezerra, F. J. A. Matos and G. S. B. Viana, J. Ethnopharmacol., 2000, 70, 151–159 CrossRef CAS PubMed.
- T. Ojala, S. Remes, P. Haansuu, H. Vuorela, R. Hiltunen, K. Haahtela and P. Vuorela, J. Ethnopharmacol., 2000, 73, 299–305 CrossRef CAS PubMed.
- S. Gond, P. Yadav, A. K. Singh and V. P. Singh, J. Mol. Struct., 2023, 1273, 134317 CrossRef CAS.
- Y. Cao, X. Liu, J. Zhang, Z. Liu, Y. Fu, D. Zhang, M. Zheng, H. Zhang and M.-H. Xu, ACS Chem. Neurosci., 2023, 14, 829–838 CrossRef CAS PubMed.
- Y. Huo, Y. Ji, D. Chen and S. Pu, Dyes Pigm., 2023, 209, 110940 CrossRef CAS.
- X.-L. Xue, Y. Wang, H. Zhang, S. Chen, S.-Y. Niu, L. Cui, K.-P. Wang and Z.-Q. Hu, Spectrochim. Acta, Part A, 2023, 292, 122410 CrossRef CAS PubMed.
- M. R. Antonijević, E. H. Avdović, D. M. Simijonović, Ž. B. Milanović, A. D. Amić and Z. S. Marković, Int. J. Mol. Sci., 2022, 23, 490 CrossRef PubMed.
- X.-T. Xu, X.-Y. Deng, J. Chen, Q.-M. Liang, K. Zhang, D.-L. Li, P.-P. Wu, X. Zheng, R.-P. Zhou, Z.-Y. Jiang, A.-J. Ma, W.-H. Chen and S.-H. Wang, Eur. J. Med. Chem., 2020, 189, 112013 CrossRef PubMed.
- V. Pardo-Jiménez, P. Navarrete-Encina and G. Díaz-Araya, Molecules, 2019, 24, 739 CrossRef PubMed.
- V. Kumar Pasala, G. Gudipudi, V. Sankeshi, M. Basude, R. Gundla, S. singh Jadav, B. Srinivas, E. Yadaiah Goud and D. Nareshkumar, Bioorg. Chem., 2021, 114, 104970 CrossRef CAS PubMed.
- L. D. Miranda, E. Icelo-Ávila, Á. Rentería-Gómez, M. Pila and J. G. Marrero, Eur. J. Org Chem., 2015, 2015, 4098–4101 CrossRef CAS.
- G. Murugavel and T. Punniyamurthy, J. Org. Chem., 2015, 80, 6291–6299 CrossRef CAS PubMed.
- Z.-W. Chen, J.-B. Hou, Z.-R. Dai and X.-F. Yang, Chin. Chem. Lett., 2016, 27, 1622–1625 CrossRef CAS.
- M.-Z. Zhang, R.-R. Zhang, W.-Z. Yin, X. Yu, Y.-L. Zhang, P. Liu, Y.-C. Gu and W.-H. Zhang, Mol. Diversity, 2016, 20, 611–618 CrossRef CAS PubMed.
- S. Fiorito, F. Epifano, V. A. Taddeo and S. Genovese, Tetrahedron Lett., 2016, 57, 2939–2942 CrossRef CAS.
- S. Bouasla, J. Amaro-Gahete, D. Esquivel, M. I. López, C. Jiménez-Sanchidrián, M. Teguiche and F. J. Romero-Salguero, Molecules, 2017, 22, 2072 CrossRef PubMed.
- W. Lin, X. Hu, S. Song, Q. Cai, Y. Wang and D. Shi, Org. Biomol. Chem., 2017, 15, 7909–7916 RSC.
- S. K. Shaikh, M. S. Sannaikar, M. Kumbar, P. Bayannavar, R. Kamble, S. Inamdar and S. Joshi, ChemistrySelect, 2018, 3, 4448–4462 CrossRef CAS.
- B. Aydıner and Z. Seferoğlu, Eur. J. Org. Chem., 2018, 2018, 5921–5934 CrossRef.
- S. Kolita and P. J. Bhuyan, ChemistrySelect, 2018, 3, 1411–1414 CrossRef CAS.
- A. Das, H. Roy and I. Ansary, ChemistrySelect, 2018, 3, 9592–9595 CrossRef CAS.
- C. Schultze and B. Schmidt, J. Org. Chem., 2018, 83, 5210–5224 CrossRef CAS PubMed.
- A. Yadav, S. K. Gudimella and S. Samanta, ChemistrySelect, 2019, 4, 12768–12773 CrossRef CAS.
- C.-X. Gu, W.-W. Chen, B. Xu and M.-H. Xu, Tetrahedron, 2019, 75, 1605–1611 CrossRef CAS.
- X.-Y. Zhang, L.-L. Hu, Z. Shen, Z.-Z. Chen, Z.-G. Xu, S.-Q. Li, J.-W. Xie and H.-L. Cui, Synlett, 2015, 26, 2821–2825 CrossRef CAS.
- T. Balalas, A. Abdul-Sada, D. J. Hadjipavlou-Litina and K. E. Litinas, Synthesis, 2017, 49, 2575–2583 CrossRef CAS.
- B. Kahveci, F. Yılmaz, E. Menteşe and S. Ülker, Chem. Heterocycl. Compd., 2015, 51, 447–456 CrossRef CAS.
- D. S. Reddy, K. M. Hosamani, H. C. Devarajegowda and M. M. Kurjogi, RSC Adv., 2015, 5, 64566–64581 RSC.
- S. Fiorito, S. Genovese, V. A. Taddeo and F. Epifano, Tetrahedron Lett., 2015, 56, 2434–2436 CrossRef CAS.
- L. C. Rao, N. S. Kumar, V. Dileepkumar, U. S. N. Murthy and H. M. Meshram, RSC Adv., 2015, 5, 28958–28964 RSC.
- R.-R. Zhang, J. Liu, Y. Zhang, M.-Q. Hou, M.-Z. Zhang, F. Zhou and W.-H. Zhang, Eur. J. Med. Chem., 2016, 116, 76–83 CrossRef CAS PubMed.
- D. Konrádová, H. Kozubíková, K. Doležal and J. Pospíšil, Eur. J. Org Chem., 2017, 2017, 5204–5213 CrossRef.
- N. C. Desai, H. M. Satodiya, K. M. Rajpara, V. V. Joshi and H. V. Vaghani, J. Saudi Chem. Soc., 2017, 21, S153–S162 CrossRef CAS.
- R. R. Chavan and K. M. Hosamani, R. Soc. Open Sci., 2018, 5, 172435 CrossRef PubMed.
- P. Kumari, S. Gupta, C. Narayana, S. Ahmad, N. Vishnoi, S. Singh and R. Sagar, New J. Chem., 2018, 42, 13985–13997 RSC.
- P. Kumari, S. Dubey, S. Venkatachalapathy, C. Narayana, A. Gupta and R. Sagar, New J. Chem., 2019, 43, 18590–18600 RSC.
- R. Dharavath, N. Nagaraju, M. R. Reddy, D. Ashok, M. Sarasija, M. Vijjulatha, T. Vani, K. Jyothi and G. Prashanthi, RSC Adv., 2020, 10, 11615–11623 RSC.
- C. B. Vagish, K. Kumara, H. K. Vivek, S. Bharath, N. K. Lokanath and K. Ajay Kumar, J. Mol. Struct., 2021, 1230, 129899 CrossRef CAS.
- S. Mamidala, S. R. Peddi, R. K. Aravilli, P. C. Jilloju, V. Manga and R. R. Vedula, J. Mol. Struct., 2021, 1225, 129114 CrossRef CAS.
- S. Dhadda, S. Sharma, P. Jakhar and H. Sharma, RSC Adv., 2023, 13, 3723–3742 RSC.
- M. Daraie, M. Mirzaei, M. Bazargan, V. S. Amiri, B. A. Sanati and M. M. Heravi, Sci. Rep., 2022, 12, 12004 CrossRef CAS PubMed.
- P. Brandão, Ó. López, L. Leitzbach, H. Stark, J. G. Fernández-Bolaños, A. J. Burke and M. Pineiro, ACS Med. Chem. Lett., 2021, 12, 1718–1725 CrossRef PubMed.
- Y. Zhang, J. Ge, L. Luo, S.-Q. Yan, G.-W. Lai, Z.-Q. Mei, H.-Q. Luo and X.-L. Fan, Org. Lett., 2020, 22, 7952–7957 CrossRef CAS PubMed.
- D. V. Osipov, M. R. Demidov, V. A. Osyanin and Y. N. Klimochkin, Chem. Heterocycl. Compd., 2021, 57, 1045–1050 CrossRef CAS.
- H. Li, L. Shan, C. Liu, N. Liu, X. Wang and Y. Hu, Org. Lett., 2023, 25, 4656–4660 CrossRef CAS PubMed.
- S. Kumar, A. S. Nair, M. A. Abdelgawad and B. Mathew, ACS Omega, 2022, 7, 16244–16259 CrossRef CAS PubMed.
- T. Sarkar, B. K. Das, K. Talukdar, T. A. Shah and T. Punniyamurthy, ACS Omega, 2020, 5, 26316–26328 CrossRef CAS PubMed.
- G. S. Singh, in Green Synthetic Approaches for Biologically Relevant Heterocycles, ed. G. Brahmachari, Elsevier, 2nd edn, 2021, pp. 505–535 Search PubMed.
- M. A. Borad, M. N. Bhoi, N. P. Prajapati and H. D. Patel, Synth. Commun., 2014, 44, 897–922 CrossRef CAS.
- S. S. Panda, M. N. Aziz, J. Stawinski and A. S. Girgis, Molecules, 2023, 28, 668 CrossRef CAS PubMed.
- M. Zakeri, M. M. Nasef, E. Abouzari-Lotf, A. Moharami and M. M. Heravi, J. Ind. Eng. Chem., 2015, 29, 273–281 CrossRef CAS.
- P. Maloo, T. K. Roy, D. M. Sawant, R. T. Pardasani and M. M. Salunkhe, RSC Adv., 2016, 6, 41897–41906 RSC.
- P. R. Mali, P. K. Shirsat, N. Khomane, L. Nayak, J. B. Nanubolu and H. M. Meshram, ACS Comb. Sci., 2017, 19, 633–639 CrossRef CAS PubMed.
- M. T. Gabr, N. S. El-Gohary, E. R. El-Bendary, M. M. El-Kerdawy and N. Ni, Eur. J. Med. Chem., 2017, 128, 36–44 CrossRef CAS PubMed.
- R. Westphal, E. Venturini Filho, L. B. Loureiro, C. F. Tormena, C. Pessoa, C. D. Guimarães, M. P. Manso, R. G. Fiorot, V. R. Campos, J. A. Resende, F. Medici and S. J. Greco, Molecules, 2022, 27, 8051 CrossRef CAS PubMed.
- R. Mishra, A. Jana, A. K. Panday and L. H. Choudhury, New J. Chem., 2019, 43, 2920–2932 RSC.
- S. Bhandari, S. Sana, B. Sridhar and N. Shankaraiah, ChemistrySelect, 2019, 4, 1727–1730 CrossRef CAS.
- D. P. da Costa, A. C. de Castro, G. A. da Silva, C. G. Lima-Junior, F. P. de Andrade Júnior, E. de Oliveira Lima, B. G. Vaz and L. C. da Silva, J. Heterocycl. Chem., 2021, 58, 766–776 CrossRef CAS.
- A. Castro, I. M. G. Andrade, M. C. Coelho, D. P. da Costa, D. d. N. Moreira, R. A. Maia, G. d. S. Lima, G. F. dos Santos, B. G. Vaz, G. C. G. Militão, P. B. N. da Silva, M. L. A. d. A. Vasconcellos and C. G. Lima-Junior, J. Heterocycl. Chem., 2023, 60, 392–405 CrossRef CAS.
- P. Yuvaraj, K. Manivannan and B. S. R. Reddy, Tetrahedron Lett., 2015, 56, 78–81 CrossRef CAS.
- K. Parthasarathy, C. Praveen, J. C. Jeyaveeran and A. A. M. Prince, Bioorg. Med. Chem. Lett., 2016, 26, 4310–4317 CrossRef CAS PubMed.
- N. D. Thanh, N. T. K. Giang, T. H. Quyen, D. T. Huong and V. N. Toan, Eur. J. Med. Chem., 2016, 123, 532–543 CrossRef PubMed.
- P. R. Mali, L. Chandrasekhara Rao, V. M. Bangade, P. K. Shirsat, S. A. George, N. Jagadeesh babu and H. M. Meshram, New J. Chem., 2016, 40, 2225–2232 RSC.
- A. Dandia, S. Khan, P. Soni, A. Indora, D. K. Mahawar, P. Pandya and C. S. Chauhan, Bioorg. Med. Chem. Lett., 2017, 27, 2873–2880 CrossRef CAS PubMed.
- K. Khaldoun, A. Safer, N. Boukabcha, N. Dege, S. Ruchaud, M. Souab, S. Bach, A. Chouaih and S. Saidi-Besbes, J. Mol. Struct., 2019, 1192, 82–90 CrossRef CAS.
- L. Maiuolo, P. Merino, V. Algieri, M. Nardi, M. L. Di Gioia, B. Russo, I. Delso, M. A. Tallarida and A. De Nino, RSC Adv., 2017, 7, 48980–48988 RSC.
- D. Ashok, S. Gundu, V. K. Aamate and M. G. Devulapally, Mol. Diversity, 2018, 22, 57–70 CrossRef CAS PubMed.
- S. Karunanidhi, B. Chandrasekaran, R. Karpoormath, H. M. Patel, F. Kayamba, S. R. Merugu, V. Kumar, S. Dhawan, B. Kushwaha and M. C. Mahlalela, Bioorg. Chem., 2021, 115, 105133 CrossRef CAS PubMed.
- N. H. de Silva, S. Pyreddy, E. W. Blanch, H. M. Hügel and S. Maniam, Bioorg. Chem., 2021, 114, 105128 CrossRef CAS PubMed.
- G. A. da Silva, A. C. de Castro, R. K. S. Mendes, D. d. N. Moreira, G. R. d. S. Cavalcanti, M. G. da Fonseca, J. P. A. Gomes, E. B. de Alencar-Filho, B. G. Vaz, G. Franco dos Santos, G. d. S. Lima, F. F. da Silva and C. G. Lima-Junior, J. Mol. Struct., 2022, 1270, 133914 CrossRef CAS.
- N. K. Sahu, R. Sharma, K. P. Suhas, J. Joshi, K. Prakash, R. Sharma, R. Pratap, X. Hu, S. Kaur and M. Jain, Molecules, 2023, 28, 4817 CrossRef CAS PubMed.
- Y. Shi, H. Zhao and Y. Zhao, Molecules, 2023, 28, 3508 CrossRef CAS PubMed.
Footnote |
| † This review is dedicated to Arun K. Shaw on his 68th birthday. |
| This journal is © The Royal Society of Chemistry 2023 |