
Carboxy-functionalized pH responsive capsule polymer particles fabricated by particulate interfacial photocrosslinking†
Yukiya
Kitayama
 * and
Atsushi
Harada
*
* and
Atsushi
Harada
*
Department of Applied Chemistry, Graduate School of Engineering, Osaka Prefecture University, 1-1, Gakuen-cho, Naka-ku, Sakai, Osaka 599-8531, Japan. E-mail: kitayama@chem.osakafu-u.ac.jp; harada@chem.osakafu-u.ac.jp
First published on 1st March 2022
Abstract
pH-responsive capsule particles show promise for various applications, such as self-healing materials, micro/nanoreactors, and drug delivery systems. Herein, carboxy-functionalized capsule polymer particles possessing neutral–alkaline pH responsive controlled release capability were newly fabricated by interfacial photocrosslinking of spherical photoreactive polymer [poly(2-carboxyethyl acrylate-co-2-cinnamoylethyl methacrylate): P(CEA-CEMA)] particles and a subsequent encapsulation process. Using P(CEA-CEMA) particles, the shell-crosslinked hollow polymer particles were fabricated by the particulate interfacial photocrosslinking procedure. Furthermore, the encapsulation of sulforhodamine B as a model dye into the hollow particles was also performed. Under acidic pH conditions, encapsulated molecules were stably retained in the P(CEA-CEMA) capsules with negligible release of sulforhodamine B. However, the encapsulated sulforhodamine B was gradually or drastically released from the capsule particles under neutral or basic conditions, respectively, indicating that the neutral–alkaline pH responsive controlled release from the capsules was successfully achieved by regulating the release kinetics. These results demonstrate that the fabrication routes of hollow and capsule particles based on particulate interfacial photocrosslinking can be successfully applied to carboxy-functionalized photoreactive polymer particles, and the capsule polymer particles possessing pH-responsive release properties under neutral–basic conditions were successfully fabricated.
 Yukiya Kitayama | Yukiya Kitayama is assistant professor of the Graduate School of Engineering at Osaka Prefecture University. He received his PhD under the supervision of Prof. Dr Masayoshi Okubo from Kobe University. From April to October in 2012, he was a postdoctoral fellow in Prof. Dr Craig J. Hawker group in the University of California at Santa Barbara. From November 2012 to January in 2020, he got academic position as an assistant professor in Prof. Dr Toshifumi Takeuchi group at Kobe University, Japan. In February 2020, he won Leading Initiative for Excellent Young Researchers (LEADER) program from Japan Society for the Promotion of Science, and he moved to the current position in Osaka Prefecture University. His research interests are in polymer chemistry, colloid chemistry, molecular recognition, and drug delivery. |
1. Introduction
Polymer capsules with controlled-release properties triggered by pH changes are of great importance in a wide range of applications, such as self-healing materials,1–3 micro/nanoreactors,4–8 and drug delivery systems.9–11 Hollow polymer particles have great potential for the above applications because they can stably encapsulate various functional molecules. Several fabrication routes of hollow polymer particles have been widely developed to date. The sacrificial template-assisted approach has been widely developed utilizing various approaches for shell-fabrication such as layer-by-layer,12,13 seeded polymerization,14,15 surface-initiated living radical polymerization,16 and phenol–metal coordination.17,18 Self-assembly of block copolymer or colloidal particles (i.e., by formation of colloidsomes19–21 and polymer vesicles using block copolymer self-assembly,22–24 or polymerization-induced self-assembly which was pioneered by Armes and coworkers22,25,26) is also a powerful approach for hollow/capsule fabrication. Inverse miniemulsion periphery polymerization has been developed as another fabrication route for hollow/capsules by Zetterlund and coworkers.27,28 The self-assembled phase-separated polymer method has been developed by Okubo, Minami and coworkers, which is categorized as one of the polymerization-induced self-assembly approaches in the (micro)suspension polymerization system.29–31Recently, we developed a new fabrication route for shell-crosslinked hollow polymer particles based on the interfacial photocrosslinking of polymer particles.32,33 In this approach, [2π + 2π] photoinduced dimerization between alkene groups of different polymer chains occurs only at the interface of the polymer particles (Scheme 1). After the photoirradiation, the non-crosslinked polymers are removed from the particle core, resulting in the shell-crosslinked hollow polymer particles being obtained. Furthermore, capsule polymer particles were successfully fabricated by a posteriori encapsulation of various functional molecules in the shell-crosslinked hollow polymer particles utilizing the solvent exchange process (Scheme 2). Furthermore, functionalization of the capsules, in particular, the controlled release of encapsulated molecules and materials, is of great importance in various applications. Various stimuli responsive capsules have also been successfully developed through interfacial photocrosslinking by designing photoreactive monomers. For example, redox-responsive capsules were developed by using the designed photoreactive monomer possessing a disulfide linkage between the vinyl group and the cinnamoyl group.34 In the capsules, the polymer main chain and crosslinking points (cinnamoyl dimer) were cleaved by the addition of reducing agents, leading to the degradation of polymer capsules. The use of a coumarin-based photoreactive monomer for the interfacial photocrosslinking approach led to the fabrication of photodegradable capsule particles.35 As another approach, stimuli-responsive capsules were fabricated using a simple copolymer of stimuli-responsive comonomers with photoreactive monomers. Recently, we have successfully fabricated acidic pH-responsive polymer capsules by the interfacial photocrosslinking of photoreactive spherical polymer particles possessing tertiary amine groups as pH-responsive groups.36,37 An advantage of the fabrication route using a copolymer of pH responsive and photoreactive monomers is the high simplicity of procedures and starting materials. However, only tertiary amine-functionalized capsules possessing acidic pH-responsiveness have been reported to date, but the versatility of this fabrication route of pH responsive capsules is unclear.
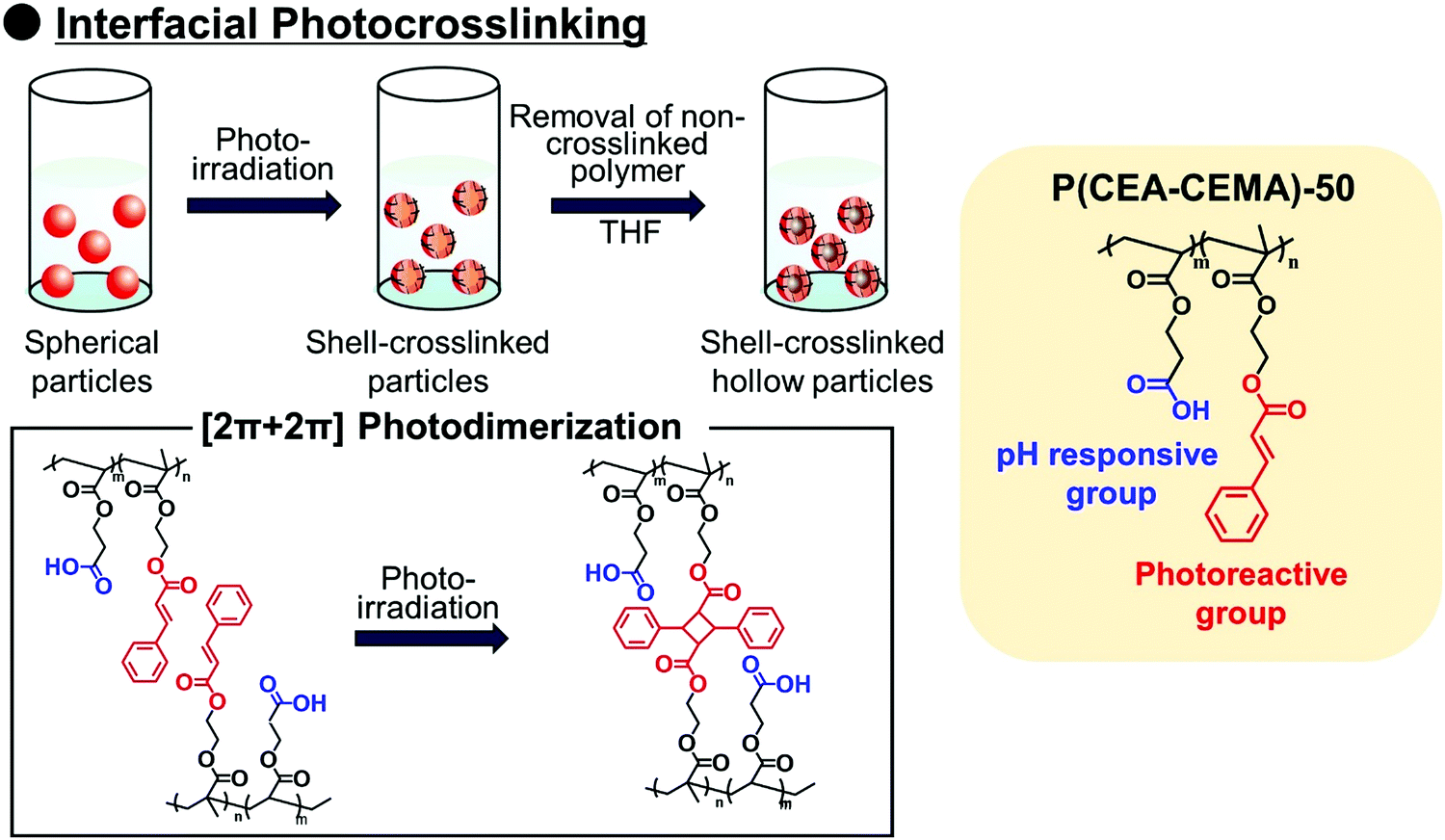 | ||
| Scheme 1 Schematic illustration of interfacial photocrosslinking of P(CEA-CEMA)-50 particles for the preparation of hollow particles. pH-responsive release of P(CEA-CEMA)-50 capsule particles. P(CEA-CEMA): poly(2-carboxyethylacrylate-co-2-cinnamoylethyl methacrylate). | ||
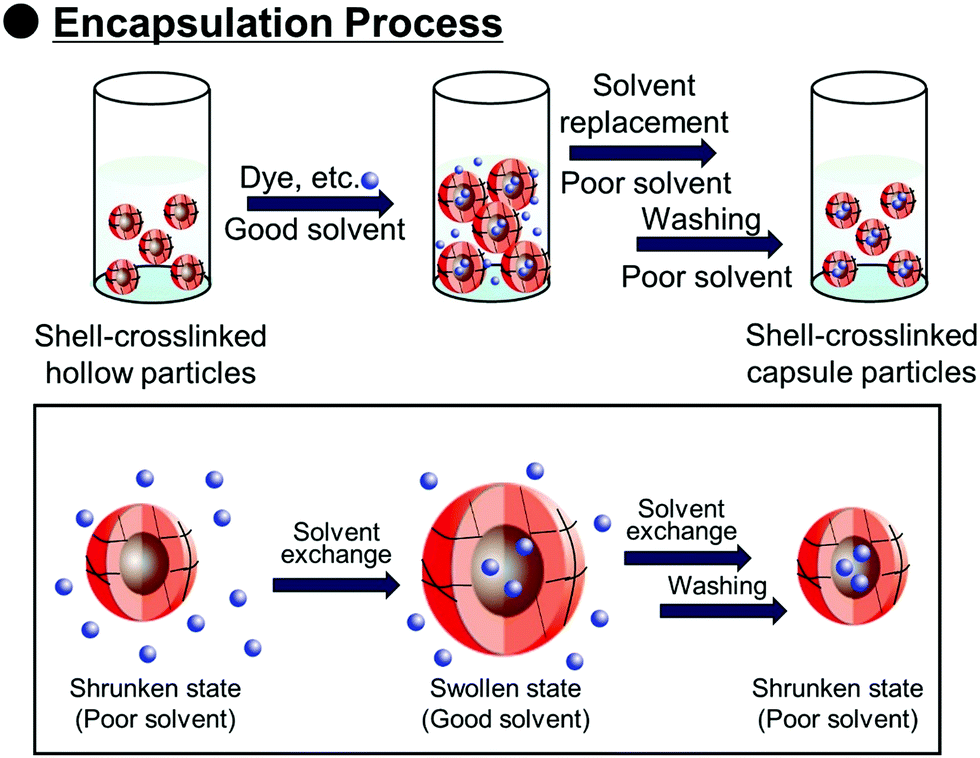 | ||
| Scheme 2 Schematic illustration of solvent exchange procedures applied to P(CEA-CEMA)-50 particles for the preparation of capsule particles. P(CEA-CEMA): poly(2-carboxyethylacrylate-co-2-cinnamoylethyl methacrylate). | ||
In this study, we fabricated carboxy-functionalized pH-responsive polymer capsules possessing controlled release capability in neutral–basic conditions to expand the variety of pH-responsive polymer capsules that can be prepared by the interfacial photocrosslinking of photoreactive random copolymer particles. Using the carboxy-functionalized capsules, the controlled release of the encapsulated molecules should be achieved at neutral–alkaline conditions. Through this research, we demonstrate the high versatility of the interfacial photocrosslinking approach using a simple random copolymer for creating a wide range of pH-responsive polymer capsules.
2. Results and discussion
For creation of neutral–alkaline responsive capsule polymer particles by interfacial photocrosslinking, functional polymers possessing photoreactivity and carboxy groups as pH responsive groups were prepared. In this study, 2-cinnamoylethyl methacrylate (CEMA), which was selected as a photoreactive monomer, was synthesized by the conjugation of cinnamoyl chloride and 2-hydroxyethyl methacrylate in the presence of triethylamine. CEMA possesses UV-absorbing properties with a maximum absorbance at λ = 280 nm, and the cinnamoyl group undergoes a [2π + 2π] photoinduced dimerization reaction by photoirradiation.38,39 2-Carboxyethyl acrylate (CEA), which was selected as an alkali-pH responsive monomer, has a pH-responsive carboxylic acid group, which means that the carboxylic acid is deprotonated under neutral–alkaline pH conditions, leading to increased water solubility.Conventional radical polymerization of CEA and CEMA was performed using 2,2′-azobis(isobutyronitrile) (AIBN) in a mixture of tetrahydrofuran (THF) and dimethylformamide (DMF). The number-average molecular weight of P(CEA-CEMA)-50 was estimated to be approximately 5800 Da (polydispersity index: 2.2) by gel permeation chromatography (GPC) after methylation of the carboxy groups of P(CEA-CEMA)-50. The CEMA molar content of the obtained polymer was estimated to be approximately 53 mol% by 1H-NMR spectroscopy, indicating that CEA and CEMA were copolymerized by conventional radical polymerization in a solution polymerization system (Fig. S1, ESI†). Furthermore, the cinnamoyl group was clearly observed in the 1H-NMR spectrum, indicating that side reactions with the cinnamoyl group did not occur during the polymerization. This conclusion is also supported by the UV-vis spectrum of the P(CEA-CEMA)-50 solution, which indicates that the obtained P(CEA-CEMA)-50 absorbs UV radiation at a maximum of λ = 280 nm (Fig. S2, ESI†). A polymer thin film was prepared on a quartz substrate to investigate the photoreactivity of P(CEA-CEMA)-50 in the polymer film state. According to the UV-vis spectra, the absorbance of the cinnamoyl groups in P(CEA-CEMA)-50 gradually decreased with increasing photoirradiation periods (Fig. 1). This phenomenon is a result of the [2π + 2π] photodimerization reaction in the polymer thin film state. It is worth noting that the absorbance at λ = 280 nm derived from free cinnamoyl groups did not completely disappear even after sufficient photoirradiation time. This result is reasonable because the cinnamoyl groups can dimerize only when two different cinnamoyl groups are placed close together in the polymer thin film. The degree of photodimerization of cinnamoyl groups in the film is roughly according to the A/A0 values shown in Fig. 1b because the maximum absorbance wavelength of the cinnamoyl group is shifted to a shorter wavelength after dimerization. Furthermore, the pH-responsive properties of P(CEA-CEMA)-50 in aqueous media were investigated by acid–base titration. The apparent pKa derived from the carboxy groups in P(CEA-CEMA)-50 was evaluated using an acid–base titration experiment, and the value was determined to be approximately 6.8, indicating that P(CEA-CEMA)-50 is pH-responsive (Fig. 1).
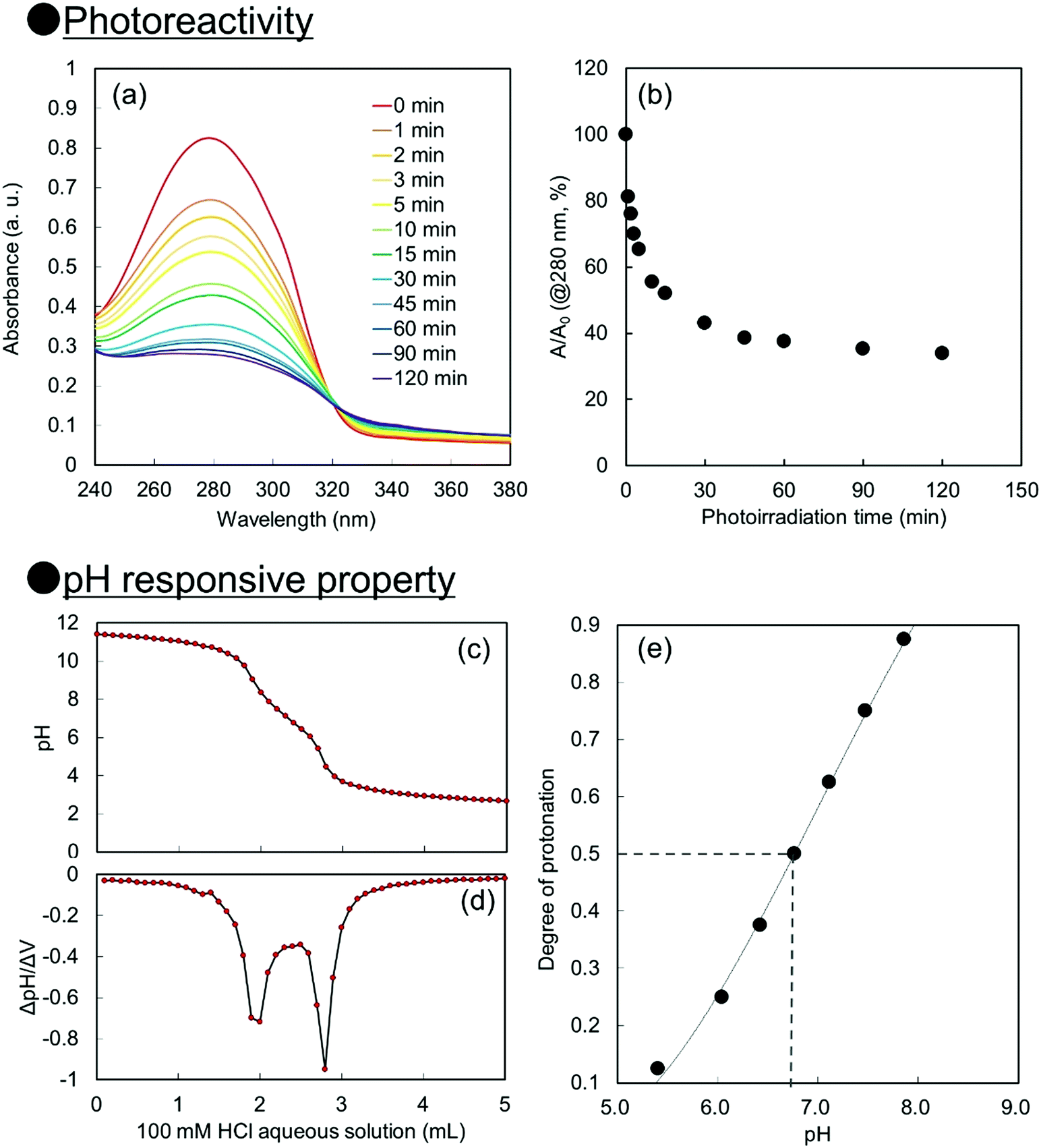 | ||
| Fig. 1 (a) UV-Vis spectra and (b) absorbance changes at λ = 280 nm of P(CEA-CEMA)-50 thin film prepared on a quartz substrate after various photoirradiation periods. (c) pH titration curve, (d) differential of the pH curve, and (e) degree of protonation under various pH conditions of P(CEA-CEMA)-50 in water. P(CEA-CEMA): poly(2-carboxyethylacrylate-co-2-cinnamoylethyl methacrylate). | ||
Solvent evaporation was carried out to prepare parent P(CEA-CEMA)-50 particles.40,41 In the solvent evaporation process, the organic solvent for dissolving P(CEA-CEMA)-50 must not be dissolved in aqueous media, and thus the organic solvent for the solvent evaporation method for P(CEA-CEMA)-50 was first investigated (Table S1, ESI†). P(CEA-CEMA)-50 was not dissolved in hexane, ethyl acetate, methanol, ethanol, and toluene. The poor solubility in these solvents might be caused by the high hydrogen bonding capabilities of the polymer chains. Acetone, THF, DMF, dimethylacetamide, and dimethyl sulfoxide (DMSO) can dissolve P(CEA-CEMA)-50, indicating that these solvents can work as a good solvent; however these solvents are not suitable for the solvent evaporation process due to their high miscibility with water. The polymer was dissolved in chloroform and methyl ethyl ketone. Due to the limited water miscibility of these solvents, these solvents are suitable for the solvent evaporation method for P(CEA-CEMA)-50. In this study, chloroform was selected as a solvent because it has lower boiling point and water solubility than methyl ethyl ketone (water solubility at 25 °C: 7.95 mg mL−1 for chloroform,42 223 mg mL−1 for methyl ethyl ketone;43 boiling point: 61.12 °C for chloroform,44 79.59 °C for methyl ethyl ketone44). P(CEA-CEMA)-50 was first dissolved in chloroform, and the polymer solution was homogenized in an acidic aqueous medium (pH 4.0) dissolving poly(vinyl alcohol) (PVA) using a homogenizer, where acidic water is a poor solvent of P(CEA-CEMA)-50. After homogenization, chloroform was slowly evaporated from the dispersion, leading to the formation of spherical P(CEA-CEMA)-50 particles (Fig. 2). Then, the interfacial photocrosslinking reaction of P(CEA-CEMA)-50 particles was carried out by photoirradiation (λ = 254 nm) to prepare the hollow polymer particles. The spherical particle morphology was maintained even after photoirradiation (Fig. 2). Non-photoirradiated P(CEA-CEMA)-50 particles dissolved in THF, whereas a part of photoirradiated P(CEA-CEMA)-50 particles remained even after THF washing. The removal ratio of P(CEA-CEMA)-50 from the photoirradiated particles after 6 h of photoirradiation was estimated to be approximately 50% by UV-vis analysis (Fig. S3, ESI†). These results confirm that photocrosslinking proceeded in the particle state. After the washing process, hollow P(CEA-CEMA)-50 particles were successfully obtained while maintaining a particle size similar to that before polymer removal, indicating that photoirradiation led to the formation of a crosslinked shell region at the particle interface of dispersed P(CEA-CEMA)-50 particles (Fig. 2). The selective crosslinking of the polymers at the interfacial region of the particles was well according to our previous studies.32,36,37 We previously clarified that the non-reacted cinnamoyl groups in the polymer shell shield core region from the photoirradiation, where the cinnamoyl group has a high absorption coefficient. The photocrosslinking degree of the cinnamoyl group of P(CEA-CEMA)-50 in the crosslinked shell region was evaluated to be approximately 32% by 1H-NMR after hydrolysis of photoirradiated P(CEA-CEMA)-50 particles (after removal of non-crosslinked polymers) (Fig. S4, ESI†).
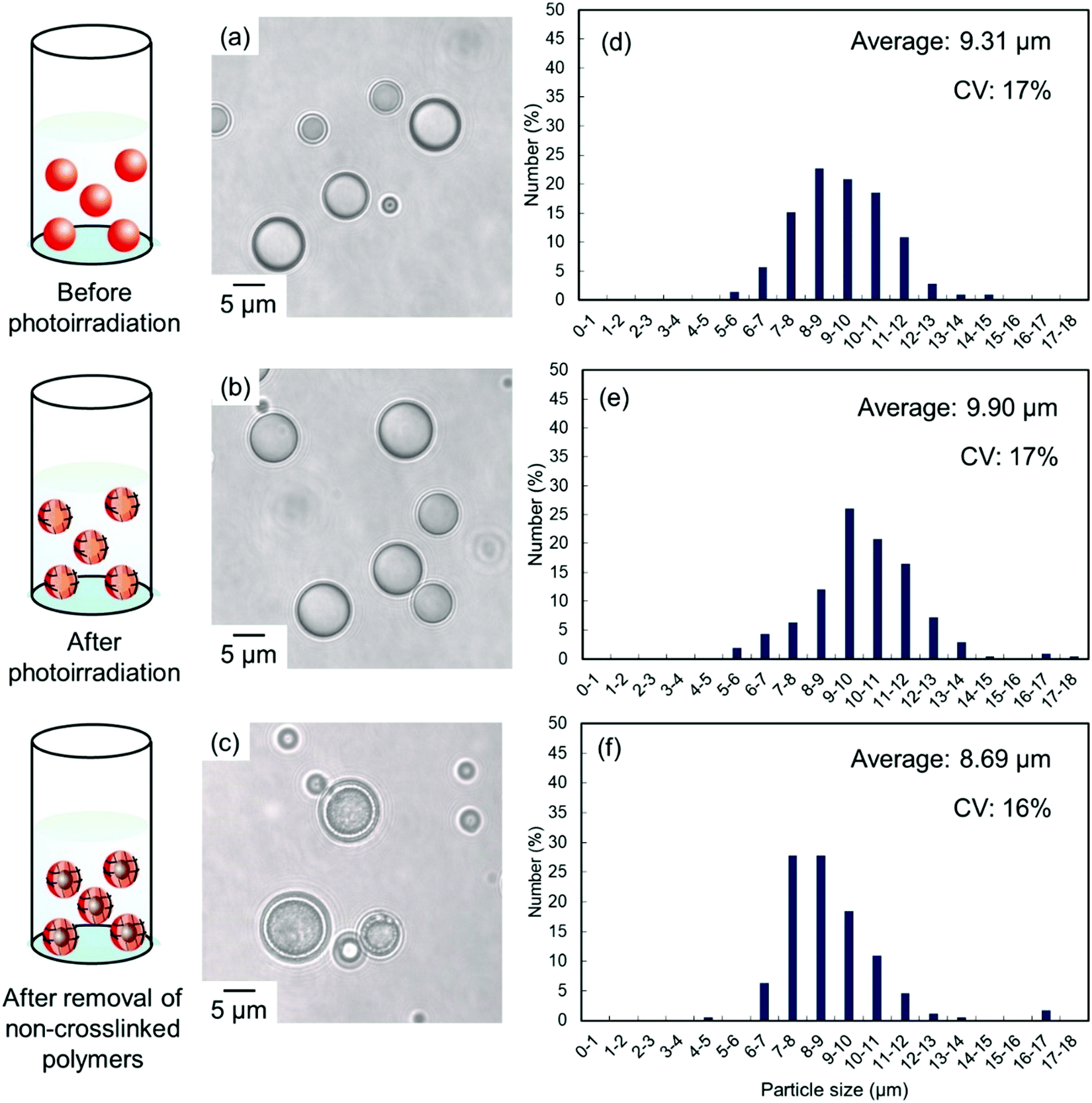 | ||
| Fig. 2 Optical micrographs (a–c) and particle size distributions (d–f) of P(CEA-CEMA)-50 particles (a) before and (b) after interfacial photocrosslinking, and (c) after subsequent removal of the non-crosslinked polymer. P(CEA-CEMA): poly(2-carboxyethylacrylate-co-2-cinnamoylethyl methacrylate). | ||
To evaluate the effect of the photoirradiation process on the particle crosslinking reaction, in situ pH-switching experiments were carried out (Fig. 3). Before photoirradiation, P(CEA-CEMA)-50 particles dispersed in acidic solution (pH 4.0) disappeared after addition of a small quantity of NaOH solid, when the aqueous medium became alkaline (good solvent for the polymer) from acidic conditions (poor solvent for the polymer) whereas, after photoirradiation, the P(CEA-CEMA)-50 particles were not dissolved and the swollen polymer particles remained even after addition of a small quantity of solid NaOH. To further clarify the successful preparation of hollow polymer particles by interfacial photocrosslinking of parent polymer particles, fluorescein-labeled P(CEA-CEMA)-50 was prepared by copolymerization with a small portion of fluorescein acrylamide. The fluorescence derived from the fluorescein-labeled P(CEA-CEMA)-50 was confirmed by fluorescence spectra (Fig. S4, ESI†). From the z-stack images obtained using confocal laser scanning microscopy (CLSM), the fluorescence derived from fluorescein-labeled P(CEA-CEMA)-50 was observed in the internal region (Fig. 4a). From the line analysis of the fluorescence image, polymer particles prepared using fluorescein-labeled P(CEA-CEMA)-50 had homogeneous internal fluorescence intensity (Fig. 4d). After the photoirradiation (without washing), the homogeneous internal morphology was maintained for P(CEA-CEMA)-50 particles; i.e. the fluorescence intensity derived from the polymers was observed from the internal region of the particles (Fig. 4b and e). However, the fluorescence derived from fluorescein-labeled polymers was only observed at the particle shell region with negligible fluorescence intensity in the internal core region of the particles after the washing process of the photoirradiated polymer particles to remove the non-crosslinked polymers (Fig. 4c and f). Furthermore, the internal hollow morphology can be directly observed in the washed P(CEA-CEMA)-50 by scanning electron microscopy (Fig. 4g). These results strongly suggest that the interfacial region of the P(CEA-CEMA)-50 particles was selectively crosslinked and non-crosslinked polymers were successfully removed from the photoirradiated P(CEA-CEMA)-50 particles, resulting in hollow polymer particles being prepared by the interfacial photocrosslinking of spherical polymer particles.
 | ||
| Fig. 3 Time lapse images of NaOH added aqueous dispersion of P(CEA-CEMA)-50 particles (a) before and (b) after interfacial photocrosslinking. P(CEA-CEMA): poly(2-carboxyethylacrylate-co-2-cinnamoylethyl methacrylate). | ||
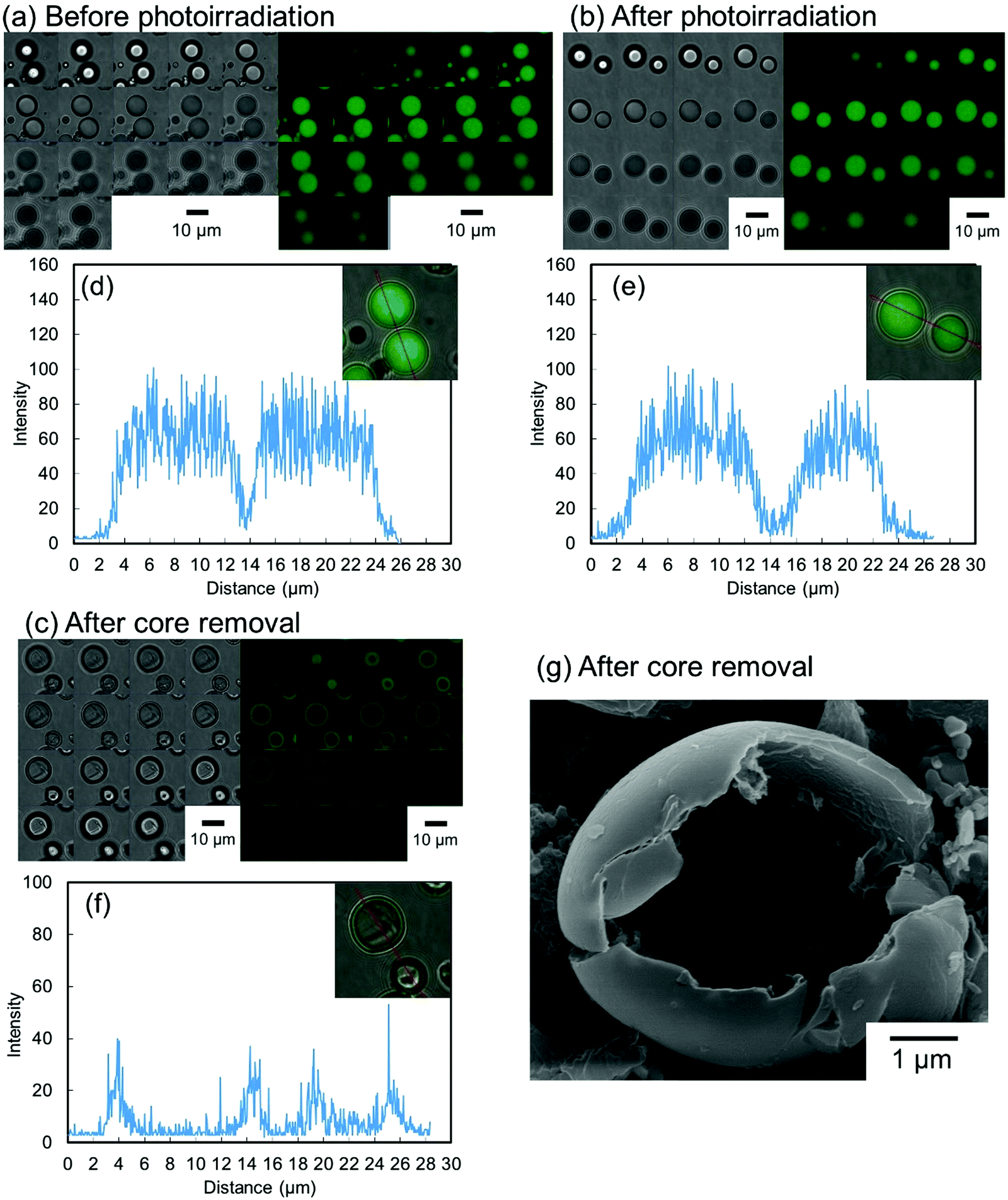 | ||
| Fig. 4 Confocal laser scanning micrographs (z-stack images) of fluorescein-labeled P(CEA-CEMA)-50 particles (a) before and (b) after interfacial photocrosslinking, and (c) after removal of the non-crosslinked polymer. Interval of Z-stack images is 1 μm. Left: Bright image. Right: Fluorescence image. Line analysis of the internal fluorescence intensity of fluorescein-labeled P(CEA-CEMA)-50 particles (d) before and (e) after interfacial photocrosslinking, and (f) after removal of the non-crosslinked polymer. (g) SEM image of photoirradiated P(CEA-CEMA)-50 particles after removal of the non-crosslinked polymer. P(CEA-CEMA): poly(2-carboxyethylacrylate-co-2-cinnamoylethyl methacrylate). | ||
Encapsulation by the P(CEA-CEMA)-50 hollow particles was carried out using a solvent exchange procedure.34 Shell-crosslinked P(CEA-CEMA)-50 hollow particles were allowed to swell through incubation in a THF/DMSO mixture containing sulforhodamine B, a model fluorescent molecule. The swelling allowed the diffusion of sulforhodamine B into the capsules. Subsequently, the solvent was exchanged for an acidic aqueous solution, a poor solvent, which shrunk the polymer shells, leading to the physical encapsulation of the molecules diffused into hollow particles. After the solvent exchange process, the z-stack image of the P(CEA-CEMA)-50 capsule particles was observed by CLSM. Fluorescence derived from sulforhodamine B was observed in the core region of the P(CEA-CEMA)-50 capsule particles, indicating that sulforhodamine B was successfully encapsulated in P(CEA-CEMA)-50 hollow particles by solvent exchange (Fig. 5).
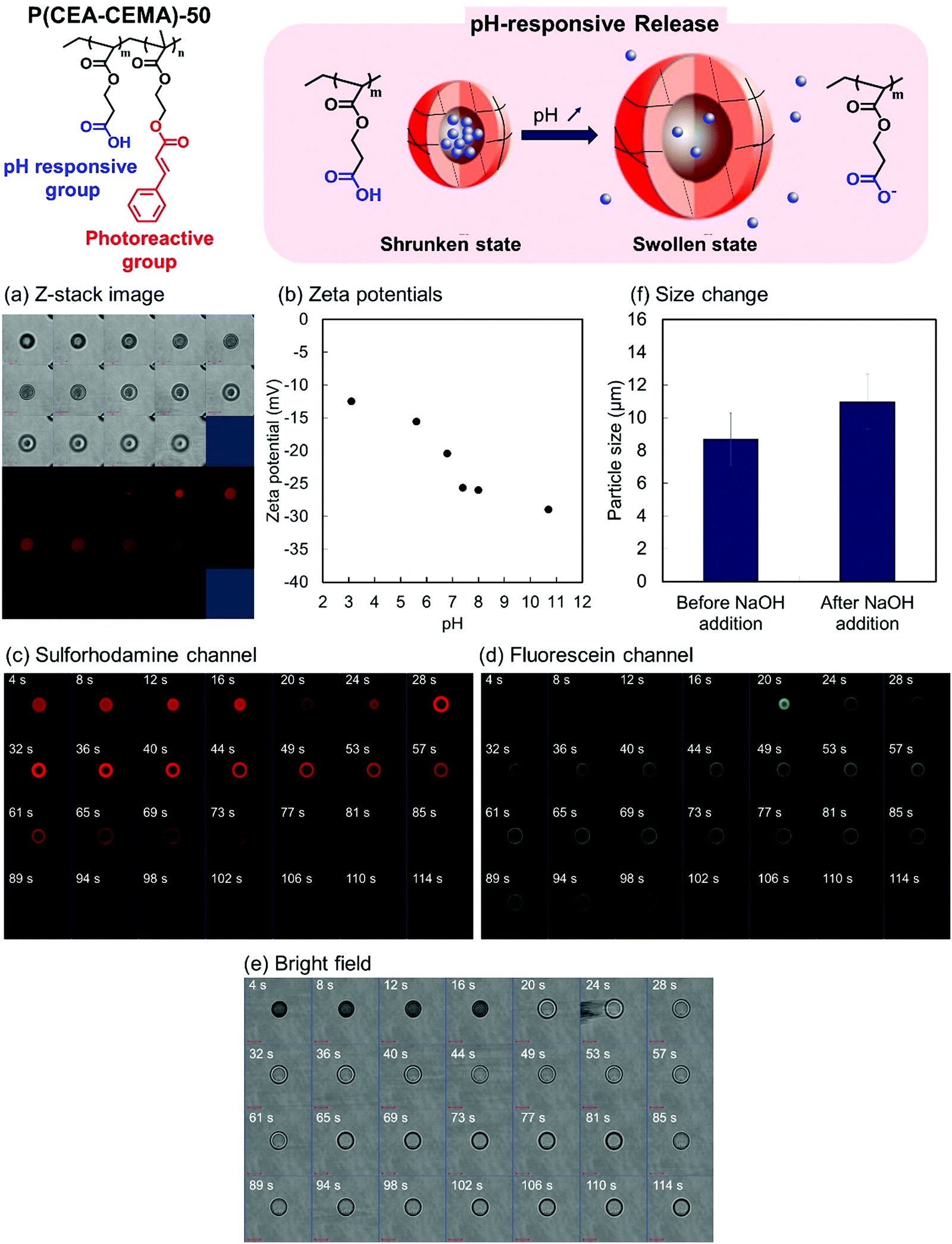 | ||
| Fig. 5 (a) CLSM images (z-stack images) of P(CEA-CEMA)-50 capsule particles containing sulforhodamine B. Interval of z-stack images is 1 μm. Top: Bright image. Bottom: Fluorescence image. (b) Zeta potential of P(CEA-CEMA)-50 hollow particles at various pH conditions. Time-lapse CLSM images of P(CEA-CEMA)-50 capsule particles containing sulforhodamine B after addition of NaOH granules: (c) sulforhodamine B channel, (d) fluorescein channel, and (e) bright field. (f) Average particle size of polymer capsules before (left) and after (right) NaOH addition. P(CEA-CEMA): poly(2-carboxyethylacrylate-co-2-cinnamoylethyl methacrylate). | ||
The pH-responsive properties of the P(CEA-CEMA)-50 hollow polymer particles were investigated from the change in the zeta potential of the hollow polymer particles with pHs. Under increasing pH conditions, the zeta potential of P(CEA-CEMA)-50 hollow particles gradually decreased (Fig. 5). As discussed above, P(CEA-CEMA)-50 has a pH-responsive carboxy group, and the apparent pKa of P(CEA-CEMA)-50 existed at pH 6.8. Notably, the zeta potential behavior was in good agreement with the water solubility of the non-photoirradiated P(CEA-CEMA)-50 particles. That is, non-photoirradiated P(CEA-CEMA)-50 particles were partially dissolved at pH 6.8, whereas they were almost completely dissolved at pH 7.4–11, according to the transmittance results (Fig. S5, ESI†). Therefore, the carboxy groups at the surface of the P(CEA-CEMA)-50 hollow particles were deprotonated under neutral and alkaline conditions in the aqueous medium.
pH-switching experiments were then carried out to investigate the pH-responsive release properties of the P(CEA-CEMA)-50 capsule particles. Here, a small quantity of solid NaOH was added to the aqueous dispersion of fluorescein-labeled P(CEA-CEMA)-50 polymer capsules containing sulforhodamine B. As fluorescein is a pH-dependent fluorescent molecule, the fluorescence intensity drastically increased when the pH changed from acidic to neutral/basic. Sulforhodamine B, in contrast, is not pH-dependent in the range of pH 3.0–10.0.45 Just after the addition of NaOH, the fluorescence intensity derived from fluorescein was negligible and the fluorescence derived from sulforhodamine B was observed from the particles. After several seconds, the fluorescence intensity derived from fluorescein increased and the fluorescence derived from sulforhodamine B rapidly disappeared from the particle core region (Fig. 5c–e and Fig. S6, ESI†). However, the fluorescence derived from sulforhodamine B slowly decreased at the particle shell layer, possibly due to non-specific π–π stacking or hydrophobic interactions between the polymer and dyes. Notably, the average particle size increased after the addition of NaOH, indicating that the P(CEA-CEMA)-50 capsule particles swelled in alkaline conditions (Fig. 5f).
The pH-responsive release capability of P(CEA-CEMA)-50 capsule particles containing sulforhodamine B was investigated. The P(CEA-CEMA)-50 capsule particles were incubated in various aqueous media at different pH conditions. After several incubation periods, the absorbance of the released sulforhodamine B in the aqueous media was measured by UV-vis spectroscopy. In acidic aqueous media (pH 4.0 and 5.5), the absorbance did not increase even after 24 h incubation, indicating that the P(CEA-CEMA)-50 capsule particles rigidly retained sulforhodamine B under acidic conditions (Fig. 6). In reality, fluorescence derived from sulforhodamine B was clearly observed from the capsule particles in the CLSM image. At neutral pH (6.8), the absorbance derived from sulforhodamine B gradually increased with incubation time. Furthermore, the absorbance drastically increased in the initial stage of incubation in alkaline aqueous media (pH 8.5 and 10). From the absorbance derived from sulforhodamine B, the loading efficiency and the loading amount into the capsules were approximately 0.15% and 34 μg/1 mg-particles. These results indicate that whereas neutral and alkaline conditions stimulated the continuous and burst release of encapsulated sulforhodamine B, respectively. From CLSM images, the fluorescence derived from sulforhodamine B was absent from the capsule particles incubated at pH 6.8, 8.5, and 10. the P(CEA-CEMA)-50 capsule particles were successfully endowed with pH-responsive controlled-release properties triggered by neutral–alkaline pH conditions.
 | ||
| Fig. 6 pH responsive release properties of P(CEA-CEMA)-50 capsule particles containing sulforhodamine B. (a) Release of sulforhodamine B from P(CEA-CEMA)-50 capsule particles after different incubation times under different pH conditions. (b–f) Bright field (left) and fluorescence field (right) images obtained from CLSM of P(CEA-CEMA)-50 particles after incubation in various aqueous media under different pH conditions. CLSM: confocal laser scanning microscopy. P(CEA-CEMA): poly(2-carboxyethylacrylate-co-2-cinnamoylethyl methacrylate). | ||
3. Conclusions
In this study, P(CEA-CEMA)-50 capsule particles capable of neutral–alkaline pH responsive controlled release were successfully prepared by the interfacial photocrosslinking of spherical polymer particles. P(CEA-CEMA)-50 prepared by conventional radical copolymerization of pH-responsive and photoreactive monomers exhibited [2π + 2π] photoreactive and pH-responsive properties. P(CEA-CEMA)-50 hollow particles were successfully prepared by interfacial photocrosslinking, and the subsequent encapsulation procedure allowed sulforhodamine B to be trapped inside the capsule. Under acidic conditions, the polymer capsules rigidly retained sulforhodamine B in the internal space, whereas neutral and alkaline conditions stimulated the continuous and burst release of encapsulated sulforhodamine B, respectively, from the capsule particles. The alkali-pH-responsive capsule particles can be applied for research in a variety of fields, such as mitochondria-targeted drug delivery systems, after minor adjustments to optimize the response to pH conditions. Through this research, we have successfully demonstrated the high versatility of the fabrication route of pH-responsive capsules utilizing interfacial photocrosslinking of simple random copolymer particles prepared from pH-responsive and photoreactive monomers. Utilizing the approach, various pH responsive capsules possessing different pH-responsive groups such as imidazole, primary amine, secondary amine, phosphate, and pyridine groups will be created by a simple procedure based on the interfacial photocrosslinking.4. Experimental section
4.1. Synthesis of carboxy-functionalized photoreactive polymers
Functional polymers possessing pH-responsive and photo-reactive groups were synthesized as follows. For the preparation of P(CEA-CEMA)-50, CEA (1.5 mmol), CEMA (1.5 mmol), and the initiator, AIBN (12.4 mg), were dissolved in a mixture of THF (10 mL) and DMSO (2 mL) in a 15 mL Schlenk tube. To prepare fluorescein-labeled P(CEA-CEMA)-50, a small quantity of fluorescein acrylamide (30 μmol) was added to the prepolymerization solution, where fluorescein acrylamide was synthesized as previously reported.46 After N2/vacuum cycles, conventional radical polymerization was performed at 60 °C for 16 h. The polymer was precipitated in water, and the precipitated polymer was further washed with methanol to remove the remaining monomers and initiators. The obtained polymer was dried in vacuo. Notably, the number after the polymer represents the initial molar ratio of CEMA in the monomers. For GPC analysis, carboxy groups of P(CEA-CEMA)-50 were methylated by trimethylsilyldiazomethane as the reported procedure.47–49 In brief, P(CEA-CEMA)-50 (20 mg) was dissolved in THF (5 mL), and a hexane solution of trimethylsilyldiazomethane (10%, 0.1 mL) was added to the polymer solution. After overnight reaction at room temperature, excess acetic acid was added to quench the trimethylsilyldiazomethane.4.2. Preparation of polymer particles
The solvent evaporation method36 was used to prepare polymer particles possessing pH-responsive and photoreactive polymers. P(CEA-CEMA)-50 (50 mg) was dissolved in chloroform (1 mL), and each solution was homogenized in phthalic acid buffer (pH 4.0) containing PVA (25 mL, 0.0067 wt%) using a homogenizer at 12![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 rpm for 1 min. The chloroform in the polymer droplets was slowly evaporated at room temperature with gentle stirring. The average sizes of more than 100 particles in the optical micrographs were estimated using ImageJ software.50
000 rpm for 1 min. The chloroform in the polymer droplets was slowly evaporated at room temperature with gentle stirring. The average sizes of more than 100 particles in the optical micrographs were estimated using ImageJ software.50
4.3. Interfacial photocrosslinking for hollow particle fabrication
Interfacial photocrosslinking was performed as follows: P(CEA-CEMA)-50 particles (solid content: 2 mg mL−1) dispersed in phthalic acid buffer (pH 4.0, 3 mL) containing PVA (0.0067 wt%) were separately placed in 6 mL vials. Each dispersion was photoirradiated (λ = 254 nm, 2 mW cm−2, GL 15, Toshiba, Tokyo, Japan) for different lengths of time, with gentle stirring at room temperature. Each dispersion was then washed five times with THF by centrifugation to remove the non-crosslinked polymers. After centrifugation, the solvent was replaced with fresh THF/DMSO (9/1, v/v, ∼1 mL), followed by 10 mM phthalic acid buffer (pH 4.0, 1 mL) containing PVA (0.0067 wt%). Before SEM observation, the P(CEA-CEMA)-50 hollow particles dispersed in the acidic aqueous solution was freeze-dried, and the dried particles were used for SEM observation after platinum coating.4.4. Encapsulation of sulforhodamine B
Hollow P(CEA-CEMA)-50 particles were prepared from the corresponding seed particles (2 mg mL−1, 3 mL) using the above procedure (photoirradiation time: 6 h). The hollow particles were then dispersed in THF (0.9 mL) and subsequently mixed with DMSO (0.1 mL) containing sulforhodamine B (final concentration: 1 mg mL−1). After 3 h of incubation, the particles were separated by centrifugation, and the solvent was replaced with phthalic acid buffer (pH 4.0, 1 mL). The particles were then washed with 10 mM phthalic acid buffer (pH 4.0, 1 mL) five times by centrifugation to remove the non-encapsulated sulforhodamine B. Finally, the washed capsule particles were dispersed in fresh 10 mM phthalic acid buffer (pH 4.0, 1 mL).4.5. Determination of pH-responsive release properties
Hollow P(CEA-CEMA)-50 particles were prepared by the above procedure using seed P(CEA-CEMA)-50 particles (solid content: 2 mg mL−1; 3 mL × 15). The hollow P(CEA-CEMA)-50 particles were then dispersed in THF (40.5 mL) before mixing with DMSO (4.5 mL) containing sulforhodamine B (final concentration: 1 mg mL−1). After 3 h of incubation, the particles were separated by centrifugation, and the solvent was replaced with 10 mM phthalic acid buffer (pH 4.0, 45 mL). The particles were then washed five times with 10 mM phthalic acid buffer (pH 4.0) to remove non-loaded free sulforhodamine B. The washed capsule particles were then dispersed in 10 mM phthalic acid buffer (pH 4.0, 45 mL). A solvent of capsule particle dispersion (1 mL) prepared by the above-described procedure was then changed to the aqueous solution with different pH conditions (final pH: 4.0, 5.5, 6.8, 8.5, and 10.0) by centrifugation. After various incubation periods, the supernatant of each capsule particle dispersion was corrected by centrifugation. The UV-vis spectra of the supernatants (λ = 565 nm) were recorded to estimate the pH-dependent controlled-release capabilities of the capsule polymers.Abbreviations
AIBN, 2,2′-azobis(isobutyronitrile); CEMA, 2-cinnamoylethyl methacrylate; CLSM, confocal laser scanning microscopy; DMF, dimethylformamide; GPC, gel permeation chromatography; P(CEA-CEMA), poly(2-carboxyethyl acrylate-co-2-cinnamoylethyl methacrylate); TEA, trimethylamine; THF, tetrahydrofuran.Conflicts of interest
The authors declare no competing financial interest.Acknowledgements
This work was supported by JSPS KAKENHI (Grant number 21H02004 for Y. K.) and partially supported by the KOSE Cosmetology Research Foundation, Takahashi Industrial and Economic Research Foundation, and the Leading Initiative for Excellent Young Researchers, MEXT, Japan (Y. K.).References
- S. R. White, N. R. Sottos, P. H. Geubelle, J. S. Moore, M. R. Kessler, S. R. Sriram, E. N. Brown and S. Viswanathan, Nature, 2001, 409, 794–797 CrossRef CAS PubMed.
- H. Jin, C. L. Mangun, A. S. Griffin, J. S. Moore, N. R. Sottos and S. R. White, Adv. Mater., 2014, 26, 282–287 CrossRef CAS PubMed.
- D. G. Shchukin, M. Zheludkevich, K. Yasakau, S. Lamaka, M. G. S. Ferreira and H. Mohwald, Adv. Mater., 2006, 18, 1672–1678 CrossRef CAS.
- J. Gaitzsch, X. Huang and B. Voit, Chem. Rev., 2016, 116, 1053–1093 CrossRef CAS PubMed.
- L. Klermund and K. Castiglione, Bioprocess Biosyst. Eng., 2018, 41, 1233–1246 CrossRef CAS PubMed.
- X. Liu, D. Appelhans and B. Voit, J. Am. Chem. Soc., 2018, 140, 16106–16114 CrossRef CAS PubMed.
- S. Varlas, J. C. Foster, P. G. Georgiou, R. Keogh, J. T. Husband, D. S. Williams and R. K. O’Reilly, Nanoscale, 2019, 11, 12643–12654 RSC.
- J. Su, H. Chen, Z. Xu, S. Wang, X. Liu, L. Wang and X. Huang, ACS Appl. Mater. Interfaces, 2020, 12, 41079–41087 CrossRef CAS PubMed.
- K. Kataoka, A. Harada and Y. Nagasaki, Adv. Drug Delivery Rev., 2001, 47, 113–131 CrossRef CAS PubMed.
- R. P. Brinkhuis, F. Rutjes and J. C. M. van Hest, Polym. Chem., 2011, 2, 1449–1462 RSC.
- L. J. De Cock, S. De Koker, B. G. De Geest, J. Grooten, C. Vervaet, J. P. Remon, G. B. Sukhorukov and M. N. Antipina, Angew. Chem., Int. Ed., 2010, 49, 6954–6973 CrossRef CAS PubMed.
- A. P. R. Johnston, C. Cortez, A. S. Angelatos and F. Caruso, Curr. Opin. Colloid Interface Sci., 2006, 11, 203–209 CrossRef CAS.
- J. J. Richardson, J. W. Cui, M. Bjornmalm, J. A. Braunger, H. Ejima and F. Caruso, Chem. Rev., 2016, 116, 14828–14867 CrossRef CAS PubMed.
- X. L. Xu and S. A. Asher, J. Am. Chem. Soc., 2004, 126, 7940–7945 CrossRef CAS PubMed.
- K. Oyama, M. Seike, K. Mitamura, S. Watase, T. Suzuki, T. Omura, H. Minami, T. Hirai, Y. Nakamura and S. Fujii, Langmuir, 2021, 37, 4599–4610 CrossRef CAS PubMed.
- T. Morinaga, M. Ohkura, K. Ohno, Y. Tsujii and T. Fukuda, Macromolecules, 2007, 40, 1159–1164 CrossRef CAS.
- J. Chen, S. Pan, J. Zhou, Q. Z. Zhong, Y. Qu, J. J. Richardson and F. Caruso, Chem. Mater., 2020, 32, 6975–6982 CrossRef CAS.
- J. Zhou, Z. Lin, Y. Ju, M. A. Rahim, J. J. Richardson and F. Caruso, Acc. Chem. Res., 2020, 53, 1269–1278 CrossRef CAS PubMed.
- S. Shilpi, A. Jain, Y. Gupta and S. K. Jain, Crit. Rev. Ther. Drug Carrier Syst., 2007, 24, 361–391 CrossRef CAS PubMed.
- R. W. Jaggers and S. A. F. Bon, Soft Matter, 2018, 14, 6949–6960 RSC.
- J. C. Cazotti, S. E. Smeltzer, N. M. B. Smeets, M. A. Dubé and M. F. Cunningham, Polym. Chem., 2020, 11, 2653–2665 RSC.
- N. J. Warren and S. P. Armes, J. Am. Chem. Soc., 2014, 136, 10174–10185 CrossRef CAS PubMed.
- Y. Anraku, A. Kishimura, M. Kamiya, S. Tanaka, T. Nomoto, K. Toh, Y. Matsumoto, S. Fukushima, D. Sueyoshi, M. R. Kano, Y. Urano, N. Nishiyama and K. Kataoka, Angew. Chem., Int. Ed., 2016, 55, 560–565 CrossRef CAS PubMed.
- S. Ohno, K. Ishihara and S. Yusa, Langmuir, 2016, 32, 3945–3953 CrossRef CAS PubMed.
- D. W. Zhou, S. M. Dong, R. P. Kuchel, S. Perrier and P. B. Zetterlund, Polym. Chem., 2017, 8, 3082–3089 RSC.
- X. F. Xu, C. Y. Pan, W. J. Zhang and C. Y. Hong, Macromolecules, 2019, 52, 1965–1975 CrossRef CAS.
- R. H. Utama, Y. Guo, P. B. Zetterlund and M. H. Stenzel, Chem. Commun., 2012, 48, 11103–11105 RSC.
- R. H. Utama, M. H. Stenzel and P. B. Zetterlund, Macromolecules, 2013, 46, 2118–2127 CrossRef CAS.
- H. Minami, H. Kanamori, Y. Hata and M. Okubo, Langmuir, 2008, 24, 9254–9259 CrossRef CAS PubMed.
- H. Minami, H. Kobayashi and M. Okubo, Langmuir, 2005, 21, 5655–5658 CrossRef CAS PubMed.
- P. Chaiyasat, S. Noppalit, M. Okubo and A. Chaiyasat, Phys. Chem. Chem. Phys., 2015, 17, 1053–1059 RSC.
- Y. Kitayama, K. Yoshikawa and T. Takeuchi, Langmuir, 2016, 32, 9245–9253 CrossRef CAS PubMed.
- Y. Kitayama, Polym. J., 2019, 51, 963–974 CrossRef CAS.
- Y. Kitayama and T. Takeuchi, Chem. – Eur. J., 2017, 23, 12870–12875 CrossRef CAS PubMed.
- Y. Kitayama and T. Takeuchi, ACS Appl. Polym. Mater., 2020, 2, 3813–3820 CrossRef CAS.
- Y. Kitayama and A. Harada, ACS Appl. Mater. Interfaces, 2021, 13, 10359–10375 CrossRef CAS PubMed.
- Y. Kitayama and A. Harada, ACS Appl. Mater. Interfaces, 2021, 13, 34973–34983 CrossRef CAS PubMed.
- Y. Ito, B. Borecka, J. Trotter and J. R. Scheffer, Tetrahedron Lett., 1995, 36, 6083–6086 CrossRef CAS.
- M. Khan, G. Brunklaus, V. Enkelmann and H. W. Spiess, J. Am. Chem. Soc., 2008, 130, 1741–1748 CrossRef CAS PubMed.
- T. Tanaka, R. Nakatsuru, Y. Kagari, N. Saito and M. Okubo, Langmuir, 2008, 24, 12267–12271 CrossRef CAS PubMed.
- T. Tanaka, M. Okayama, Y. Kitayama, Y. Kagawa and M. Okubo, Langmuir, 2010, 26, 7843–7847 CrossRef CAS PubMed.
- D. Mackay, A. Bobra, W. Y. Shiu and S. H. Yalkowsky, Chemosphere, 1980, 9, 701–711 CrossRef CAS.
- R. W. Taft, M. H. Abraham, R. M. Doherty and M. J. Kamlet, Nature, 1985, 313, 384–386 CrossRef CAS.
- W. M. Haynes, CRC Handbook of Chemistry and Physics. 95th Edition, CRC Press LLC, Boca Raton, 2014-2015 Search PubMed.
- J. Coppeta and C. Rogers, Exp. Fluids, 1998, 25, 1–15 CrossRef CAS.
- T. Takeuchi, Y. Kitayama, R. Sasao, T. Yamada, K. Toh, Y. Matsumoto and K. Kataoka, Angew. Chem., Int. Ed., 2017, 56, 7088–7092 CrossRef CAS PubMed.
- S. M. North and S. P. Armes, Polym. Chem., 2021, 12, 4846–4855 RSC.
- H. Moribe, Y. Kitayama, T. Suzuki and M. Okubo, Macromolecules, 2011, 44, 263–268 CrossRef CAS.
- H. Moribe, Y. Kitayama, T. Suzuki and M. Okubo, Polym. J., 2012, 44, 205–210 CrossRef CAS.
- C. A. Schneider, W. S. Rasband and K. W. Eliceiri, Nat. Methods, 2012, 9, 671–675 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Materials, apparatus, 1H-NMR and UV-vis spectra of P(CEA-CEMA)-50, fluorescence spectrum of P(CEA-CEMA)-50, photocrosslinking and pH-responsive properties of P(CEA-CEMA)-50 particles, and release kinetics of sulforhodamine B from the capsules. See DOI: 10.1039/d1tb02866a |
| This journal is © The Royal Society of Chemistry 2022 |