
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceBinary and ternary Pt-based clusters grown in a plasma multimagnetron-based gas aggregation source: electrocatalytic evaluation towards glycerol oxidation
W.
Chamorro-Coral
 *a,
A.
Caillard
a,
P.
Brault
a,
S.
Baranton
b and
C.
Coutanceau
b
*a,
A.
Caillard
a,
P.
Brault
a,
S.
Baranton
b and
C.
Coutanceau
b
aGroupe de Recherches sur l’Energétique des Milieux Ionisés (GREMI), Université d’Orléans, CNRS, 14 rue d'Issoudun, BP6744, 45067 Orléans cedex 2, France. E-mail: wachamorroc@unal.edu.co
bInstitut de Chimie des Milieux et Matériaux de Poitiers (IC2MP), Université de Poitiers, CNRS, 4 rue Michel Brunet, TSA 51106, 86073 Poitiers cedex 9, France
First published on 19th January 2021
Abstract
Platinum (Pt), platinum–bismuth (PtBi), platinum–copper (PtCu) and platinum–bismuth–copper (PtCuBi) clusters were grown in a gas aggregation source (GAS) equipped with three in-plane plasma magnetrons located in a single region of the gas aggregation zone. The X-ray diffraction results have shown that PtCu clusters form alloys as the decrease of the lattice parameter occurs when the Cu atomic content increases. PtBi clusters do not form alloys, but the presence of secondary Bi oxide phases was detected. Scanning transmission electron microscope mapping images revealed that simultaneously adding Bi and Cu to Pt leads to PtCu alloyed clusters decorated with Bi or CuBi species on the surface. The electrochemical results indicated that the shell might be composed of a metastable CuBi phase. Electrochemical measurements have shown that the addition of Bi or Cu to the Pt clusters enhances the catalytic activity for glycerol oxidation by decreasing the oxidation onset potential.
Introduction
The biofuel industry is an interesting alternative to the traditional fossil fuel-based one to satisfy the growing energetic demand and mitigate the depletion of fossil reserves as well as the release of greenhouse gases.1,2 Biofuel is produced from vegetable oils from different varieties of palm or from colza, and glycerol is one of the main by-products generated during biodiesel production (ca. 10 wt%). Raw glycerol can be used in different industrial applications. But the high production of biodiesel has led to an excess of glycerol decreasing its sale price. Therefore, a conversion process of the glycerol molecule into more value-added compounds is needed to reduce the biofuel production costs.3 The conversion of glycerol may lead to monoglycerides, glycerol carbonates (useful for colors, varnishes, glues, cosmetics, and pharmaceuticals), ethers (useful in cosmetics, food-additives, and lubricants) or acetals and ketals (flavoring agents).4,5 Glycerol oxidation is mainly carried out using strong oxidants that are not selective, leading to the production of undesirable molecules or to total oxidation (CO2 formation).6,7 Heterogeneous catalysts with noble metals have already been explored but two main challenges raised have to be overcome: the cost and, to avoid a separation step of complex mixtures, the selectivity. Electrocatalytic processes are surface reactions; therefore, the use of metallic clusters (diameter < 5 nm) dispersed on a high surface carbon support can increase the specific surface area of electrocatalytic materials and help to drastically decrease the noble metal amount used in electrodes. Among these metals, Pt has been proved to be a good candidate due to its high activity and efficiency; however, some of its flaws are its high price, low availability, and high strategic character. To overcome these drawbacks, the use of multimetallic clusters that will decrease the Pt content while maintaining or even increasing the Pt performance is proposed. It has been shown that adding bismuth to Pt clusters decreases the glycerol oxidation onset potential and therefore the electric energy consumption.8,9 The negative shift of the onset potential occurs because Bi atoms promote the formation of intermediate species that modify the glycerol oxidation mechanism. The modification of Pt with Bi also allows improving the selectivity by avoiding the dissociative adsorption of glycerol with C–C bond breaking at low electrode potentials. Copper is also known to be a good co-catalyst. The modification of Pt with Cu enhances the catalytic activity for alcohol electro-oxidation and also allows decreasing the oxidation onset potential.10–12The aim of the present work is to grow multimetallic catalysts based on clusters with an admixture of Pt, Bi and Cu. The clusters were produced in a plasma-based gas aggregation source (GAS) using a multimagnetron approach, where each magnetron is powered independently. The multimetallic clusters supported on carbon form the electrocatalytic powders that will be tested for the electrochemical conversion of glycerol. Production of Pt and Cu clusters by means of a GAS based on magnetron sputtering has already been reported,13–17 while the growth of Bi clusters with less than 100 atoms using a GAS has only been described for pressures between 100 Pa and 2000 Pa and using thermal evaporation as the source for the metallic vapour.18,19 Recent reports have shown that the multimagnetron approach is a straightforward method to modify the composition of clusters during the synthesis of trimetallic nanoclusters.20–24 However, to our knowledge, this is the first time that multimetallic Pt-based clusters modified with Cu and/or Bi have been synthesized by means of a GAS assisted by a multimagnetron sputtering source. The electrochemical responses of Pt-cluster based electrodes, i.e. oxidation current density and/or onset potential, depend on the clusters properties that could be influenced by the experimental parameters of synthesis. Therefore, an objective of this work is to provide an insight about the relationship between the composition, nanostructure, and catalytic activity of the Pt-based clusters.
Experimental setup
Pt-based catalysts are synthesized using a GAS based on a multimagnetron source, allowing the production of clusters in a gas phase with optimal size control.25 The GAS setup is based on a commercial Nanogen 50 from Mantis Deposition Ltd and has been modified to include three magnetrons (ONYX, 1-inch in diameter, from Angstrom Sciences) with an independent positioning system that allows adjusting the position of each magnetron inside the aggregation source. Each of the Pt, Cu and Bi targets (99.99% purity) is mounted on one of the three magnetrons. The GAS chamber has dimensions of 30 cm length and 13 cm diameter: the distance between the three magnetrons and the GAS outlet orifice, 0.5 cm in diameter, is fixed to 7 cm (magnetrons are in an in-plane configuration). The argon flow rate is fixed to 80 sccm (standard cubic centimeter) giving a total pressure in the aggregation chamber of 42 Pa, which allows the gas-phase growth of clusters. By using such a device, alloyed, core–shell and Janus nanoclusters could be obtained depending on the experimental conditions and on the nature of the materials.20,24 The GAS is placed inside a high vacuum (HV) deposition chamber with a lower total pressure of about 0.4 Pa for an 80 sccm Ar flow rate (residual pressure of 10−4 Pa without argon introduction). An MKS type 1739A controller coupled to an MKS Type 247 readout was used to control the argon flow rate. The constant argon flow and the pressure difference between the GAS and the HV chamber generate a cluster beam from the GAS outlet orifice. The cluster beam passes through a quadrupole mass spectrometer (QMS, MesoQ from Mantis Deposition Ltd) collecting only the negatively charged clusters, which allows following in situ the cluster mass distribution. Using this procedure at least three curves were recorded for each deposition condition.A mobile substrate-holder is placed in the HV deposition chamber between the GAS and the QMS, with a distance of 9 cm between the GAS orifice and the substrate. Fig. 1 gives an overview of the experimental setup based on the plasma multimagnetron. The deposition rate is estimated for each pure material using a quartz crystal microbalance (QCM) attached to a mobile holder and placed at the same distance from the GAS orifice as that for the substrate holder. When the monometallic Pt target is sputtered, the deposition rates vary between 0.7 and 1.1 mg h−1 cm−2 in the power range from 20 W to 30 W, whereas for Cu the deposition rates vary from 0.1 to 0.6 mg h−1 cm−2 over the same power range as that for Pt. The deposition rate is not measurable for Bi owing to its low melting point (271.4 °C) that limits the condensation process in the GAS18 and avoids the formation of Bi clusters. In the case of co-sputtering of multiple targets, the composition of clusters being a priori unknown, it is not possible to measure the cluster deposition rate with the quartz crystal microbalance.
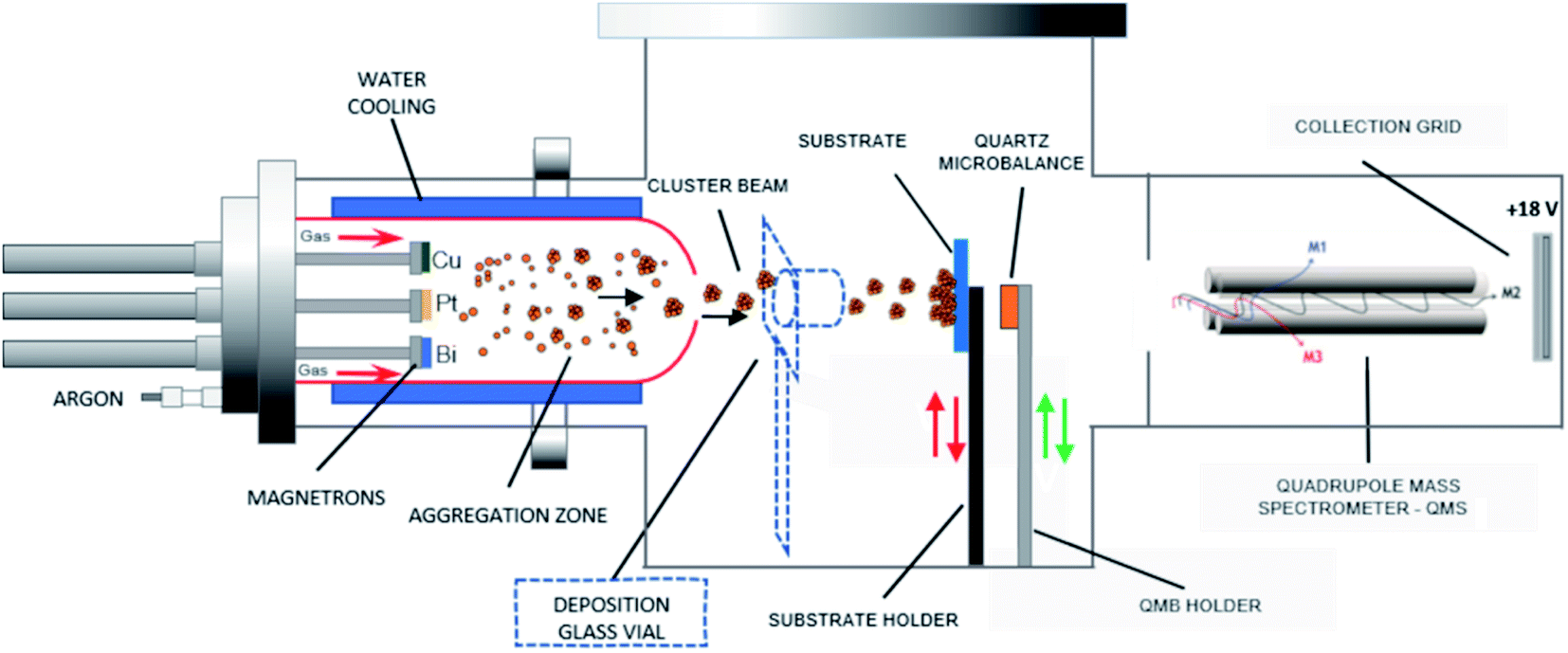 | ||
| Fig. 1 Schematic of the adjustable multimagnetron based GAS used for the multimetallic cluster synthesis, adapted from a Mantis Nanogen 50 system. | ||
Four types of clusters are produced: pure Pt clusters, bimetallic PtCu and PtBi clusters and trimetallic PtCuBi clusters. Magnetron sputtering was performed in direct current (DC) mode using a Pinnacle Plus generator (Advanced Energy) for Pt and Cu targets and an ion generator (Materials Science, Inc.) for the Bi target. To vary the cluster composition, the power applied on each magnetron (PtX, CuY and BiZ) is varied between 20 W and 30 W for Pt and Cu, and between 8 and 13 W for Bi. X, Y and Z correspond to the power applied to the respective Pt, Cu and Bi targets. Before each set of experiments, the targets are cleaned by sputtering during at least 5 min to obtain a stable voltage before moving the substrate holder in the beam. The influence of sputtering power on the cluster growth was evaluated by recording the in situ mass distribution curves measured by QMS. The curves follow a lognormal behaviour and after fitting, the average cluster mass (Cavg-mass) is obtained. The grid current intensity is directly related to the number of negatively charged clusters and the area under the curve corresponds to the sum of all the clusters of different masses that come from the cluster beam, which can be interpreted as the total mass deposited (mtotal).26
For physical characterization, clusters are deposited for 3 min onto N-type doped Si (100, p-doped) and for 3 seconds on carbon-coated Ni or Cu TEM grids (300 mesh, SPI) depending on the presence or the absence of Cu in the clusters. Physical characterization includes grazing incidence X-ray diffraction (GIXRD) performed on a Bruker diffractometer fitted with a Cu X-ray source (λK = 1.5418 Å), energy-dispersive X-ray spectroscopy (EDX) using a Bruker QUANTAX X-ray detector mounted in a Zeiss Supra 40 scanning electron microscope and transmission electron microscopy (TEM) investigations using a JEM-ARM 200F Cold FEG TEM/STEM operating at 200 kV, equipped with a spherical aberration (Cs) probe and image correctors (point resolution 0.12 nm in TEM mode and 0.078 nm in STEM mode). The experimental diffraction signals from GIXRD and TEM are compared with calculated XRD patterns using VESTA software.27 Crystal structures are obtained from the Materials Project (MP) database where each structure has a unique identification number in the form mp-xxxx.28
The clusters used to prepare the electrodes for the electrochemical measurements are collected for 10 minutes in a glass vial placed 3 cm from the cluster beam exit (Fig. 1). The resulting clusters are then dispersed in ethanol using an ultrasonic bath. The mass of collected clusters is obtained from the mass difference between the vial before and after deposition (instrumental error of ±0.0001 g). A calculated amount of carbon (Vulcan XC72 thermally treated at 400 °C under a nitrogen atmosphere) is added to obtain a metal loading of 40 wt%. Carbon powder and metal clusters are mixed and dispersed in ethanol. The suspension is sonicated for at least 20 minutes to ensure a uniform repartition of clusters on the carbon support. The suspension mixture is then filtered on a commercial alumina oxide membrane purchased from Sigma Aldrich (pore size of 0.2 μm, thickness of 0.1 mm, diameter of 13 mm) and dried. An ink of clusters/carbon powder in suspension in isopropanol is prepared and a 3 μL aliquot is deposited onto a 3 mm diameter glassy carbon electrode, as described elsewhere.3 The Pt/C, PtCu/C, PtBi/C and PtCuBi/C electrodes are loaded with 0.08 ± 0.01 mgPt cm−2, 0.09 ± 0.01 mgPtCu cm−2, 0.09 ± 0.01 mgPtBi cm−2 and 0.10 ± 0.01 mgPtCuBi cm−2, respectively. The electrochemical measurements are performed in a classical three-electrode cell with a reversible hydrogen electrode (RHE) as the reference, Pt-based catalytic powder deposited on a glassy carbon disk as the working electrode, and a glassy carbon plate as the counter electrode. Cyclic voltammetry (CV) measurements performed in a N2-purged 0.10 mol L−1 NaOH electrolyte are recorded using a VersaSTAT 4 potentiostat between +0.050 and +1.200 V vs. RHE with a scan rate of 0.050 V s−1. The electrocatalytic activity is evaluated at a scan rate of 0.005 V s−1 in the presence of 0.10 mol L−1 glycerol in a N2-purged 0.10 mol L−1 NaOH electrolyte at 20 °C after correction of the metal loading to assess mass activity. Because the accurate determination of the onset potential is very difficult,29 it is arbitrarily and reasonably defined in this work as the potential at which the anodic (oxidative) current reaches 1% of the maximum current peak, which is in the range of generally considered current values for the onset potential determination.8,30
Results
First, the influence of co-sputtering Pt and Cu or Bi at different powers on the cluster mass distribution curves was analyzed. Fig. 2a and b show the mass distribution curves of Pt and Cu monometallic clusters determined for different sputtering powers. The grid current intensity obtained for the Pt clusters (Fig. 2a) is higher than that obtained for the monometallic Cu ones (Fig. 2b). Although the sputtering yield Y (thus the flux of sputtered atoms leaving the target) is higher for the Cu target than for the Pt one for the same sputtering power (at 30 W, YCu = 1.88, YPt = 1.53, calculated using SRIM software), more Pt clusters than Cu clusters are formed, showing that the condensation process is more effective for Pt atoms. Also, it is worth noting that a shift towards lower cluster average masses occurs in both Pt and Cu distribution curves when the sputtering power increases. This could be explained by a modification in the condensation process: high sputtering powers increase the number of atoms in the vapor phase, therefore increasing the probability of collisions, leading to the formation of more clusters but with lower average masses. Another aspect to consider is the area under the curves (Fig. 2, inset) which reveals an increase of the deposited mass (mtotal) with the sputtering power for both metals. This shows that an increase of the total number of clusters leads to an overall increase of mtotal. It is worth noting that the Pt cluster mass increases approximately by only 15%, while that of Cu increases by ca. 80% when the power is increased by 50% from 20 to 30 W. The mean size of clusters can be obtained from the lognormal fitting of the cluster mass curve by assuming that clusters are spherical in shape; the sizes range from 4.0 to 4.9 nm for Pt clusters and from 6.0 to 6.5 nm for Cu clusters.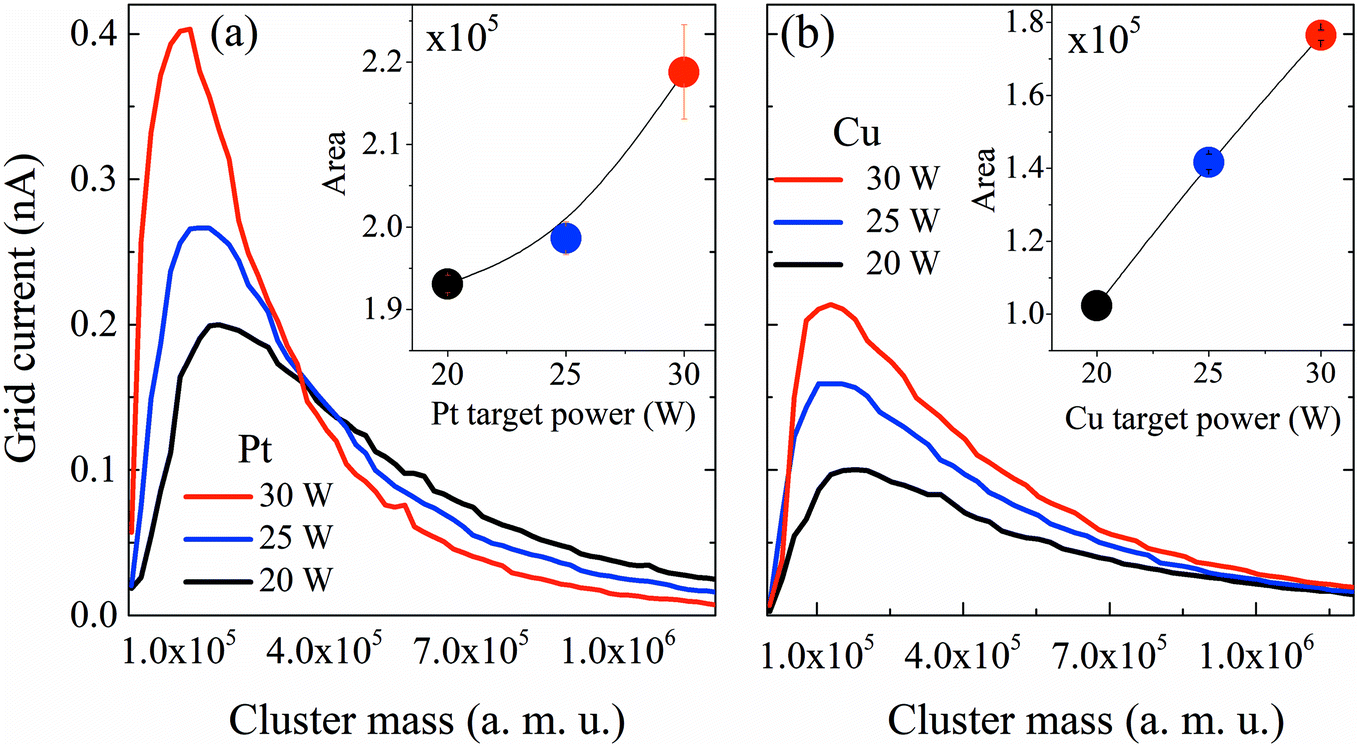 | ||
| Fig. 2 Mass distribution curves obtained using QMS for Pt and Cu monometallic clusters grown at different sputtering powers. (a) Pt and (b) Cu clusters. Inset: area under the curve representing the total mass deposited (mtotal). For sputtering powers between 20 W and 30 W, the mean cluster diameters, obtained from the lognormal fitting of the cluster mass distribution and assuming spherical clusters, range between 4.0 and 4.9 nm for Pt and between 6.0 and 6.5 nm for Cu. | ||
After reviewing the influence of the sputtering power on the average masses and mean cluster sizes of monometallic Pt and Cu clusters, the influence of the power applied to the Pt, Cu and Bi targets for the formation of Pt–Cu and Pt–Cu–Bi clusters by co-sputtering is also studied (Fig. 3). For Pt and Cu co-sputtering (Fig. 3a), the Pt sputtering power is fixed at 25 W, while the Cu sputtering power is increased from 0 W to 30 W. A positive jump of current occurs between 0 W and 15 W, but a further increase of the Cu sputtering power leads to a slight decrease of the mass distribution intensity as well as to a weak shift towards bigger cluster size (peak maximum at 1.2 × 105 amu for 20 W and 1.8 × 105 amu for 30 W). Considering the total mass deposited (Fig. 3a, inset), a jump between 0 and 15 W is also observed (increase of the area by more than 20%). For the formation of trimetallic clusters (Fig. 3b), the Pt and Cu sputtering powers are kept constant at 25 W and 30 W, respectively, and the Bi target sputtering power is increased from 0 W to 12 W. From 0 W to 10 W, the mass distribution curves remain almost unchanged except for a small but constant decrease of the mass distribution intensity. For a sputtering power of 12 W applied at the Bi target, the number of clusters decreases remarkably. Regarding the mass deposited, mtotal (inset of Fig. 3b), it increases until a sputtering power of 8 W is applied, then it starts to decrease especially for the sputtering power of 12 W. Finally, the results presented in Fig. 3 highlight the synergy between Pt and Cu that leads to the increase of the cluster size and of the deposited mass, while the addition of Bi seems to limit the condensation and growing processes for sputtering powers higher than 8 W.
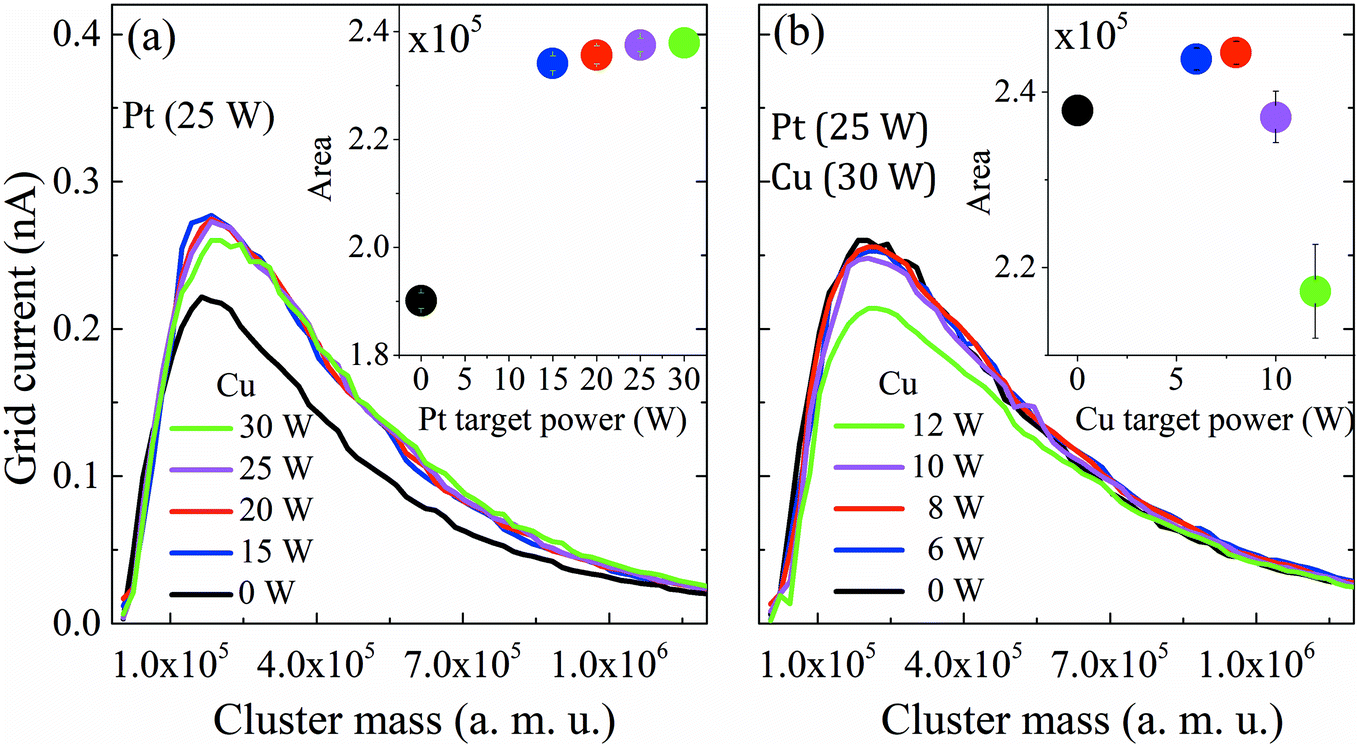 | ||
| Fig. 3 Mass distribution curves obtained from QMS for multimetallic clusters grown under different sputtering powers of Pt, Cu and Bi. (a) PtCu and (b) PtCuBi clusters. Inset: area under the curve representing the total deposited mass (mtotal). | ||
As stated previously, the cluster composition can be varied by modifying each magnetron sputtering power. However, factors such as the target erosion profile and surface target composition are difficult to control under given sputtering conditions, and continuous changes in these parameters during the synthesis process introduce uncertainties in atomic compositions. The composition of catalysts used for electrochemical characterization was estimated by the semiquantitative EDX method:31 (76 ± 8) at% and (23 ± 8) at% for Pt and Cu, respectively, in the PtCu catalyst, (92 ± 8) at% and (8 ± 6) at% for Pt and Bi, respectively, in the PtBi catalyst, and (79 ± 2) at%, (18 ± 11) at% and (4 ± 2 at%) for Pt, Cu and Bi, respectively, in the PtCuBi catalyst. It is also worth saying that the overlapping of the Si peak (from the substrate) with the Pt peak and that of Pt peaks with Bi peaks could result in an underestimation of Pt and/or Bi atomic compositions. The measurements were performed on clusters deposited onto silicon substrates. Oxygen is detected in all metallic clusters, but with higher contents in those containing Cu and/or Bi, showing the high reactivity of these metals towards the residual oxygen present in the GAS. This can be explained by the increasing Gibbs oxidation energy in the Pt, Bi and Cu series.32 Oxygen content was obtained by Rutherford Backscattering Spectroscopy (not shown here) using 2 MeV alpha particles. The RBS results give a ratio of ca. 30 at% of O but this value could be overestimated because the deposit of clusters on the Si substrate is inhomogeneous, which makes the RBS profile ambiguous and difficult to construe; therefore the fitting of the spectra could lead to inaccurate results.
Determination of the compositions of the multimetallic clusters does not give any information on whether they form mixtures of metals or alloys. Hence, crystal structures of the Pt-based clusters have been explored. GIXRD is performed on the clusters deposited on a Si wafer. To avoid artefacts due to difference in mass deposited on the wafer, all diffractograms are normalized with respect to the total area under the corresponding GIXRD patterns. Fig. 4 presents the diffraction pattern of Pt clusters with signals peaking at 39.75° and 46.23° that correspond to the (111) and (200) diffraction planes of the face-centered cubic (fcc) phase of Pt (mp-126 (ref. 28)), respectively. The diffraction pattern of PtBi clusters displays the main diffraction peaks corresponding to the Pt fcc phase, but broader in shape and with lower intensities. Additionally, a broad signal centered at 29° is assigned to overlapped diffraction signals from phases such as Bi (mp-23152 (ref. 28)), PtBi (mp-1066078 (ref. 28)) and Bi oxide (mp-23262 (ref. 28)), which therefore prevents correct indexation. The peak broadening can be due to crystallographic domains of short coherence lengths that exist in clusters of small sizes (diameter < 1 nm) or of poor crystallinity. PtCu clusters lead to the same diffraction peaks as those for the Pt fcc phase but slightly shifted towards higher values. For the trimetallic clusters, the GIXRD pattern displays slight shifts of the diffraction peaks (compared to the pure Pt phase) towards higher 2θ values owing to the presence of the Cu atoms and a weak signal close to 29° that is assigned to the presence of Bi oxide phases. The inset in Fig. 4 compares the position of the (111) diffraction peak maxima recorded for the different mono-, bi- and tri-metallic clusters.
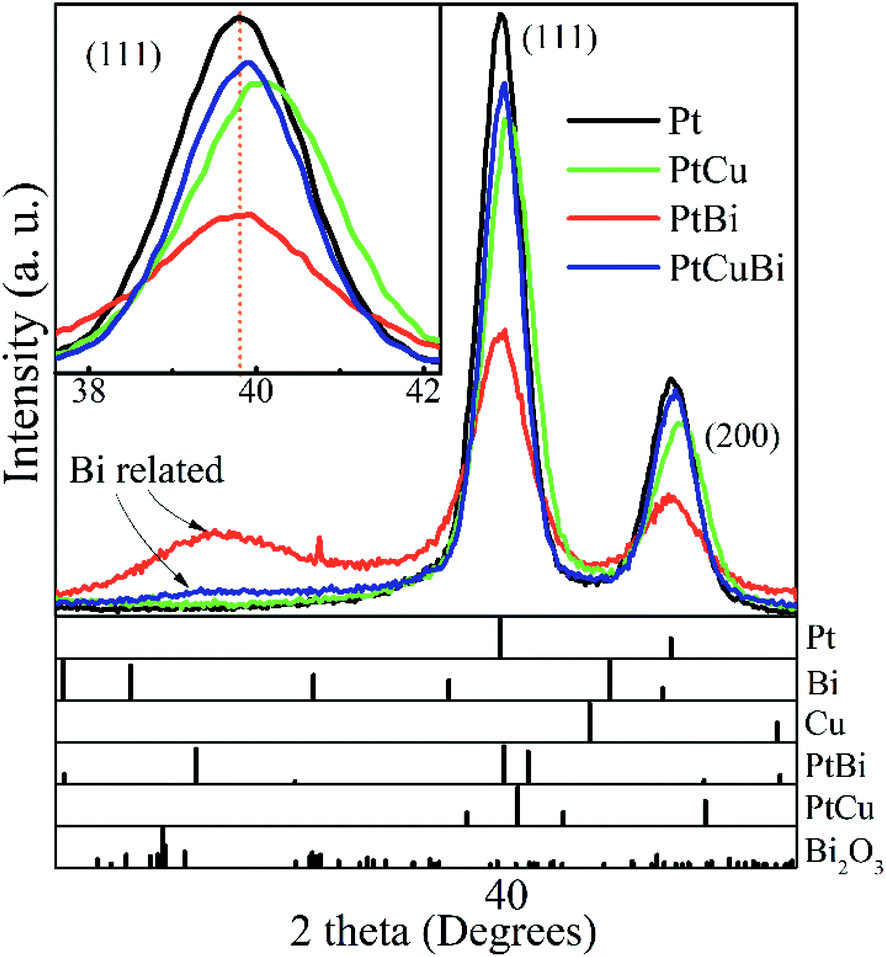 | ||
| Fig. 4 Grazing incident X-ray diffractograms of Pt, PtBi, PtCu and PtCuBi clusters and (bottom) calculated XRD patterns of Pt (mp-126 (ref. 28)), Bi (mp-23151 (ref. 28)), Cu (mp-30 (ref. 28)), PtBi (mp-1066078 (ref. 28)), PtCu (mp-644311 (ref. 28)) and Bi2O3 (mp-23262 (ref. 28)). Inset: zoomed-in image of the (111) diffraction peak of the Pt-based clusters; the dashed line (orange) is a visual reference. | ||
Changes in the crystal structure of bi- and tri-metallic clusters with respect to monometallic Pt/C were followed as a function of the sputtering power by focusing on the position of the (111) diffraction peak. Fig. 5 reveals the shift in position of the (111) diffraction peak and the induced change of the value of the lattice parameter, a, as a function of the ratio of the sputtering powers PBi/PPt (Fig. 5a) and PCu/PPt (Fig. 5b). The (111) peak of Pt clusters is located at 39.760°, close to that of bulk Pt (39.755°). When increasing the Bi content in clusters, the position of the (111) peak does not change, remaining in the measurement error bars. For the PtCu and the PtCuBi clusters, the position of the (111) peak shifts to higher 2θ values, and therefore to lower Pt lattice parameters.
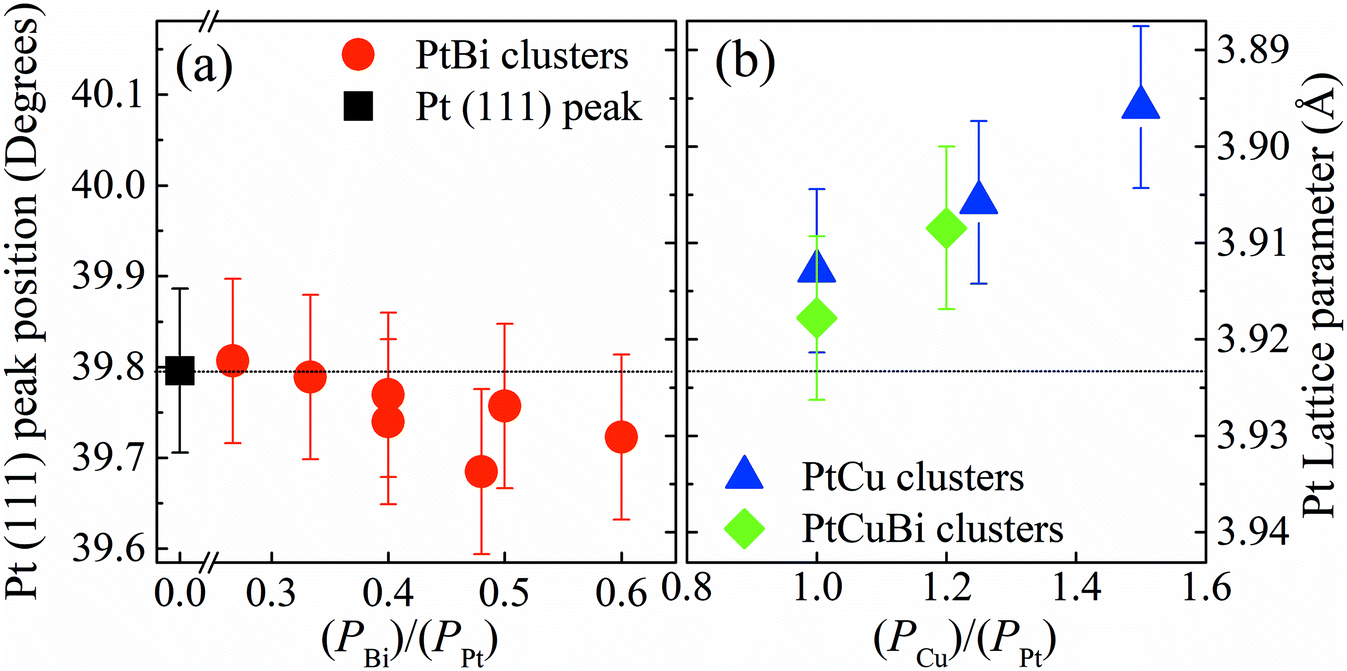 | ||
| Fig. 5 Left axis: X-ray diffraction peak position of the (111) plane of the Pt fcc crystal lattice. Right axis: lattice parameter for (a) Pt clusters (black square), PtBi clusters (red circles), (b) PtCu clusters (blue triangles) and PtCuBi clusters (green diamonds). The X axis corresponds to the ratio of the powers applied on Bi or Cu magnetrons compared to the power on the Pt magnetron (Px/PPt) with x = Bi, Cu. | ||
Cu and Pt both crystallize in the fcc structure and the decrease of the interatomic distances in the cell (shrinkage of the crystal lattice) is due to the formation of a PtCu alloy as already reported.33,34 If the Pt–Cu system followed Vegard's law, the Cu content in PtCu clusters would be approximately 5 at% while for the PtCuBi it would be less than 5%. These results are below the percentage expected from EDX and RBS measurements (∼20 at%); these under-estimations of the Cu content by Vegard's law are due to the fact that at the nanoscale, Vegard's law can lead to inaccurate results.35–37 A non-uniform distribution of the atoms within the clusters leads to deviations from Vegard's law; however, GIXRD data also fail to verify the presence of Janus-type, core–shell type or intermixed type structures.35,36 Such deviations are driven by compressive strain forces due to differences in atom sizes and thus different unit cell volumes (11.8 Å3 for Cu and 15.8 Å3 for Pt).
To look in more detail at the nanostructure of the clusters, Fig. 6 presents TEM and high resolution TEM micrographs (HRTEM) of Pt-based clusters: Pt (Fig. 6a), PtCu (Fig. 6b) and PtCuBi (Fig. 6c). Fig. 6a – left shows mainly faceted clusters with an average size of 5 nm which is close to that obtained from the distribution curves of Fig. 2. The fast Fourier transformation (FFT) pattern in Fig. 6a – right confirms the presence of (111), (100) and (110) families of diffraction planes with distances of 0.228 ± 0.003 nm, 0.198 ± 0.002 nm and 0.140 ± 0.001 nm, respectively, corresponding to the values for a bulk Pt material. For clusters that are smaller than 4 nm the shape of the projection of faceted clusters resembles that obtained for truncated octahedra (Fig. 6a – center), with a higher proportion of low surface energy (111) facets (1.49 J m−2) and lower proportions of (110) facets (1.68 J m−2 for the reconstructed facet) and (200) facets (1.86 J m−2).38 For such small clusters, interplanar distances of 0.224 ± 0.003 nm, 0.194 ± 0.002 nm and 0.135 ± 0.002 nm for the (111), (200) and (110) diffraction planes, respectively, are lower than those determined for bigger clusters, which could indicate a shrinkage of the crystal lattice due to internal stress as observed for fcc clusters at the nanoscopic scale due to their high surface stress.39,40
 | ||
| Fig. 6 High resolution TEM micrographs (left), zoomed-in image of single clusters (center) and corresponding fast Fourier transformation patterns (right) for (a) Pt, (b) PtCu and (c) PtCuBi clusters. | ||
Regarding PtCu bimetallic clusters, the TEM micrograph (Fig. 6b – left) shows highly aggregated clusters of 4.5 nm average size, and regions with shorter coherence domains compared to pure Pt clusters. As observed in Fig. 3, when Pt and Cu are co-sputtered, the number of clusters formed is higher than that for Pt sputtering alone, and this phenomenon could be responsible for a cluster aggregation process. In agreement with GIXRD results (Fig. 4), the crystallographic structure of clusters does not change when their aggregation occurs as confirmed by the FFT pattern on Fig. 6b – right showing that the crystallographic planes correspond to those of the Pt fcc phase. A zoomed-in image of a highly ordered region (Fig. 6b – center) confirms the presence of the Pt crystallographic planes. The TEM micrograph of ternary clusters PtCuBi (Fig. 6c – left) shows highly aggregated clusters with a lower crystal quality. Also, only round-shaped clusters are observed. Isolated clusters can have a size as low as 3.5 nm while aggregated clusters can reach 5.5 nm. The FFT pattern (Fig. 6c – right) shows the presence of Pt diffraction planes, but also that of an additional region of relatively high intensity that could be related to diffraction planes of Bi phases (metallic, oxide or alloyed with Pt) as observed by GIXRD in Fig. 4. A zoomed-in image of the micrograph (Fig. 6c – center) reveals mostly diffraction planes that have short coherence distances, such as (110) Pt planes, confirming the poor crystallinity of tri-metallic clusters.
To understand the poor crystallinity of ternary clusters, detailed nano-structural analysis is needed. EDX-STEM maps of ternary PtCuBi clusters are obtained. Fig. 7 shows, separately, the HAADF-STEM image of the PtBiCu clusters (Fig. 7a), the distribution of the three elements in the clusters (Fig. 7b) and the distribution of each element, Pt, Cu and Bi, in the clusters (Fig. 7c–e, respectively). On the one hand, Pt and Cu are homogeneously distributed along each cluster, which is in accordance with the formation of a PtCu alloy. On the other hand, Bi is mainly distributed on the borders of the clusters and on the grain-boundaries. This could give us the insight that ternary PtCuBi clusters present the structure of an alloyed PtCu core surrounded by a Bi shell. Molecular dynamic simulations have already confirmed such a kind of core–shell structure for the Pt–Bi system grown using a GAS, having its origins in stronger Pt–Bi bonding energy than for Pt–Pt.8 This result agrees with other studies where Bi atoms always decorate the Pt surface and bond to two or three Pt atoms depending on the Bi content.8,41–43 The MP database28 indicates that three stable alloys can be found for the Pt–Cu system, while two stable alloys are present for the Pt–Bi system. There are no data for binary alloys between Cu and Bi or ternary alloys between Pt, Cu and Bi but a metastable Cu–Bi alloy could be formed.44,45 Considering only the stable phases, Pt will preferentially alloy with Cu (and not with Bi) and Cu will be segregated from Bi; however the database compounds are calculated at 0 K and 0 atm and sputtering is a synthesis technique far-from-equilibrium. Therefore, the presence of metastable phases cannot be discarded.
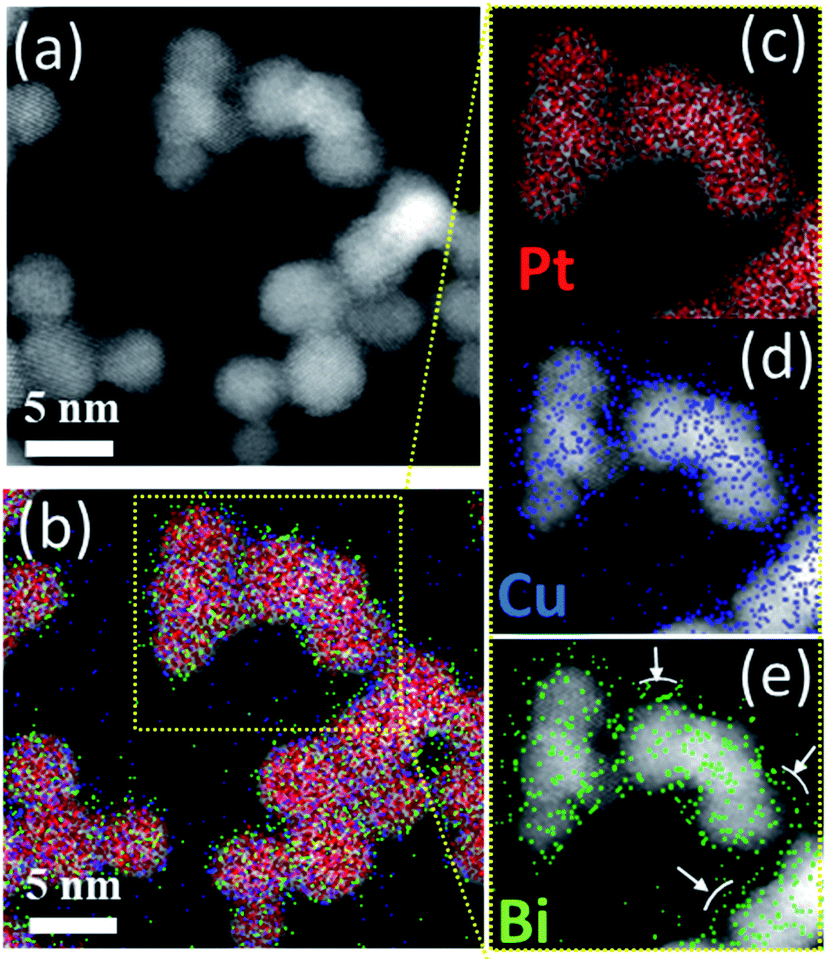 | ||
| Fig. 7 (a) HAADF-STEM and (b) STEM/EDX elemental mapping images of Pt, Cu and Bi atom distribution in PtCuBi clusters. Enlarged STEM/EDX elemental mapping images for (c) Pt, (d) Cu and (e) Bi atom distribution in PtCuBi clusters. | ||
Due to differences in the growing process, properties such as crystallinity, lattice parameters and the nanostructure of the Pt-based clusters slightly change depending on the formation of mono-, bi- and tri-metallic clusters and these differences could affect their performances as electrocatalysts. For example, atom size differences between Pt and Cu in binary PtCu and ternary PtCuBi materials lead to differences in electronegativity that are responsible for d-band edge downshift,36 with a possible change in the oxidative catalytic activity. Therefore, their electroactivity was evaluated by cyclic voltammetry. This electrochemical technique allows obtaining detailed information on the cluster surfaces because only electroactive surface atoms are probed. Fig. 8 displays the fifth stable cyclic voltammogram (CV) of Pt-based electrodes recorded in a N2-purged 0.10 mol L−1 NaOH electrolyte between +0.050 and +1.200 V vs. RHE at 0.050 V s−1.
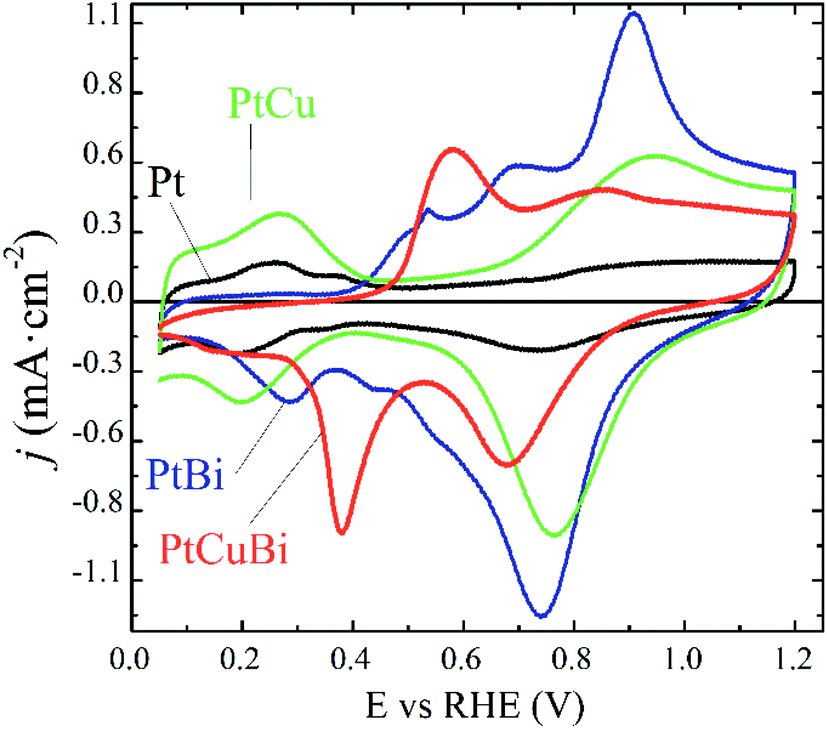 | ||
| Fig. 8 Fifth stable cyclic voltammograms of Pt/C (black line), PtCu/C (green line), PtBi/C (blue line) and PtCuBi/C (red line) materials recorded between +0.050 and +1.200 V vs. RHE (v = 0.050 V s−1, N2-saturated 0.10 mol L−1 NaOH electrolyte, T = 20 °C). | ||
The CVs can be differentiated between those recorded on materials without Bi, Pt/C and PtCu/C catalysts, and those recorded on materials containing Bi, PtBi/C and PtCuBi/C catalysts. The Pt/C catalyst (black line) displays the typical CV signature of polycrystalline platinum nanoparticles in alkaline solution with the appearance of redox features related to the underpotential H adsorption (HUPD) and desorption of adsorbed HUPD in the +0.050 to +0.400 V vs. RHE range, and the features are related to platinum surface oxidation from ca. 0.600 V vs. RHE for the positive potential variation. In the negative potential variation, the Pt surface oxides are reduced giving rise to the reduction peak centered at ca. +0.750 V vs. RHE.
The PtCu/C catalyst (green line) also shows H and O interactions with the Pt surface. It is worth noting that no redox peak related to Cu oxidation/reduction is detected, revealing the stability of the PtCu surface alloy.46 Higher oxidation and reduction currents in the +0.600 V vs. RHE to +1.200 V vs. RHE range, i.e. in the potential region of Pt surface oxidation and reduction, are recorded, which seems to indicate that Pt surface atoms and Cu surface atoms are in strong interactions leading to the simultaneous formation/reduction of Pt and Cu surface oxides.
The addition of Bi to Pt and PtCu leads to two main changes in the CVs. First, the HUPD adsorption/desorption peaks are suppressed, as observed elsewhere,9 revealing changes in the cluster surface. These changes come from physical alterations such as the Pt surface covering of bismuth atoms or from electronic perturbations at the Pt surface due to Bi–Pt interactions. The second feature is the presence of additional peaks centered at +0.530, +0.680 and +0.880 V vs. RHE for the PtBi catalyst and at +0.580 and +0.850 V vs. RHE for the PtCuBi catalyst, for the positive potential variation.
In the case of PtBi/C, the shape of the CV greatly resembles that obtained on a Pt0.8Bi0.2/C catalyst prepared by a wet chemistry method (water in oil microemulsion),8 which contradicts the EDX analysis. The oxidation peaks located at ca. +0.500 V, +0.700 V and +0.900 V vs. RHE in the CV of a Pt0.8Bi0.2/C material prepared with the water in oil microemulsion8 have been assigned to the oxidation reaction of bismuth according to eqn (1). The existence of different redox features is related to the level of interaction between Bi species and Pt surface atoms: the lower the interaction between both metals, the lower the oxidation potential of Bi.8
| Bi + 6OH− ⇌ Bi2O3 + 3H2O + 6e− | (1) |
The fact that plasma sputtering and wet chemistry methods lead to materials exhibiting the same electrochemical behavior indicates that the surfaces of both catalysts have the same structure. Therefore, the oxidation peak above +0.800 V vs. RHE can be related to the simultaneous formation of Pt and Bi oxides, as in the cases of PtBi materials from wet chemistry synthesis, and the signals between +0.500 and +0.800 V vs. RHE are related to the formation of Bi oxides.9 These oxidation peaks give rise to a main reduction peak centered at ca. +0.730 V vs. RHE, shoulders at ca. +0.550 and ca. +0.450 V vs. RHE and a second peak at ca. +0.280 V vs. RHE for the negative potential variation.
In the case of the PtCuBi catalyst, only two oxidation peaks (at +0.580 and +0.850 V vs. RHE) are observed for the positive potential variation (instead of three for the PtBi catalyst), giving rise to only two reduction peaks for the negative variation of potential. The oxidation peak at higher potential is already visible for PtCu and PtBi and could reasonably by assigned to the simultaneous oxidation of Pt, Bi and Cu surface atoms. The one at lower potential is certainly related to the oxidation of species containing Bi or/and Cu atoms, according to eqn (1) and (2).32
| Cu + 2OH− ⇌ Cu2O + H2O + 2e− | (2) |
The existence of a unique redox feature in the low potential region (oxidation peak at ca. +0.600 V vs. RHE and reduction peak at ca. +0.400 V vs. RHE) for the PtCuBi/C catalyst could be translated to a weaker interaction between the Pt and the oxide species from Cu and Bi that could be present at the surface47 compared to that observed in the higher potential range (oxidation peak at ca. +0.900 V vs. RHE and reduction peak at ca. +0.700 V vs. RHE).
The GIXRD and electrochemical results confirm that in PtCu clusters, Pt and Cu form a uniform alloy with a strong interaction between Pt and Cu atoms at the surface. The electrochemical response of the PtBi catalyst is aligned with the assumption that Bi atoms are on the cluster surface as observed by STEM in Fig. 7 and decorate Pt clusters. In the case of the PtCuBi/C catalyst, the results for both PtCu/C and PtBi/C could indicate the possible electrochemical response of an alloyed PtCu cluster decorated with Bi species. However, the CVs show that a portion of the Cu atoms is on the cluster surface, and the unique oxidation peak at 0.580 V vs. RHE represents the formation of a structure where Bi and Cu surface atoms are also in strong interaction. As mentioned before, the presence of a Cu–Bi metastable phase cannot be discarded and a structure consisting in a PtCu core decorated with amorphous Cu–Bi could also be proposed.
The activity of the Pt-based catalysts towards the electrooxidation of 0.10 mol L−1 glycerol in a N2-purged 0.10 mol L−1 NaOH electrolyte is compared in Fig. 9. The metal loadings on electrodes are in a very narrow range from 0.08 ± 0.01 mgmetal cm−2 to 0.10 mgmetal cm−2, increasing with addition of Cu, Bi and CuBi to Pt; therefore, the influence of the metal loading on the electrode will have a negligible effect compared with that of the cluster composition. The Pt/C catalyst (black curve) leads to an oxidation onset potential of ca. +0.400 V vs. RHE, which is consistent with other previous studies realized on Pt/C and PtxBi1−x materials prepared by wet chemistry.3,9,47 The addition of Bi to Pt leads to a slight shift of the onset potential towards a lower value and slightly higher activity between ca. +0.380 V vs. RHE and ca. +0.600 V vs. RHE, as higher currents (mass activities) are recorded over this potential range. For potentials higher than 0.700 V vs. RHE, Pt/C displays higher activity than PtBi/C. The low increase of mass activity at low potentials and the lower mass activity achieved at higher potentials demonstrate that the Bi atomic ratio is relatively high, higher than 10 at%,8 in agreement with cyclic voltammetry measurements, hindering the active platinum surface. This definitively confirms that the estimation of the Bi atomic ratio in the clusters is underestimated by EDX. In contrast, the addition of Cu to Pt leads to a significant shift of the onset potential towards a lower value and to a huge increase of the mass activities in the potential range from +0.280 V to +0.800 V vs. RHE. PtCuBi/C leads to a lower oxidation onset potential of ca. +0.250 V vs. RHE and to slightly higher activity in the +0.250 V to +0.600 V vs. RHE potential range. Between +0.600 and +0.800 V vs. RHE, PtCu becomes the best catalyst. For example, the mass activity achieved at +0.600 V vs. RHE on the PtCu and PtCuBi/C catalysts (ca. 55 A gmetal−1) is twice higher than that recorded on the Pt/C catalyst (ca. 30 A gmetal−1). These activity values are very close to those reported under the same experimental conditions (0.10 mol L−1 glycerol, N2-saturated 0.10 mol L−1 NaOH electrolyte, scan rate of 0.005 V s−1 and 20 °C) by Kouamé et al.8 on Pt/C and PtBi/C catalysts prepared by wet chemistry (“water in oil” microemulsion), i.e. between 40 and 70 A gmetal−1. At 0.800 V vs. RHE, the Pt/C catalyst prepared using the GAS leads to higher activity (mass activity of 120 A gmetal−1) than that prepared by wet chemistry (mass activity of 100 A gmetal−1).8 These results seem to indicate that the Pt/C catalyst prepared using the GAS is more prone to poisoning at potentials lower than 0.700 V vs. RHE, but more active at potentials higher than 0.800 V vs. RHE than the one prepared by wet chemistry. Both catalysts are composed of Pt NPs with a mean size of ca. 5 nm dispersed on the same carbon support (Vulcan XC 72), with the same metal weight loading (40 wt%) and almost the same surface concentrations (0.100 mg cm−2 for the wet chemistry Pt/C catalyst and 0.080 for the GAS Pt/C catalyst). Therefore, the difference in activity is not due to the size effect or agglomeration effect but rather seems to be due to the Pt surface structure effect. Indeed, the TEM image in Fig. 5a displays faceted Pt NPs, whereas Pt nanoparticles obtained by wet chemistry were round-shaped,8 and it is well known that the surface structure of Pt NPs has a huge effect on poisoning and electro-activity.48–50
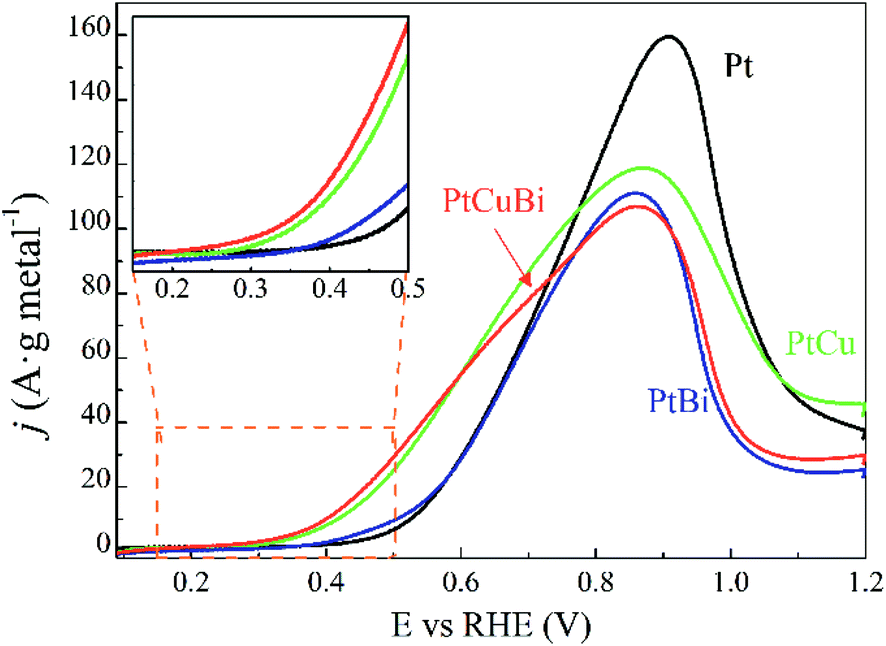 | ||
| Fig. 9 Polarization curves for the oxidation of 0.10 mol L−1 glycerol recorded for Pt/C (black line), PtBi/C (blue line), PtCu/C (green line) and PtCuBi/C (red line) materials (v = 0.005 V s−1, N2-saturated 0.10 M NaOH electrolyte, T = 20 °C). Inset: zoomed-in image of the oxidation potential onset region. | ||
Moreover, the potential range over which the PtCu/C and PtCuBi/C catalysts are active is wider than that for Pt/C, which can have an impact on the selectivity of the reaction with respect to the electrode potential. Indeed, at least two oxidation processes occur on PtCu/C and PtCuBi/C leading to the main peak with a shoulder at lower potentials, whereas Pt/C leads only to one oxidation peak. However, it was previously shown8,51 that the species formed during the glycerol electrooxidation on Pt-based catalysts also depend on the electrode potential. In alkaline media, the formations of products such as glyceraldehyde and dihydroxyacetone in a lower proportion are promoted below +0.600 V vs. RHE. Above +0.700 V vs. RHE, mostly carboxylate species such as glycerate or tartronate are observed.52,53
The role of bismuth in the activity enhancement at low potentials was proposed to come from the promotion of both a geometric effect (dilution of adjacent Pt atoms) that favors a particular adsorption mode of glycerol on Pt sites avoiding multi-bonding and/or dissociative adsorption leading to adsorbed CO poisoning the catalyst surface and from a strong electronic effect between Pt and Bi that favors the desorption of less oxidized species like glyceraldehyde and dihydroxyacetone (DHA). But for a Bi ratio higher than 10 at%, the activity was not significantly improved, likely due to too high Pt surface coverage.8
The presence of Cu in the catalyst can also modify the oxidation mechanism at low potentials.54 It has been shown that alloying Cu with Pt led to a downshift of the d-band edge.33,36,55–57 This could be the origin of changes in the electrochemical reactivity by favoring, at low electrode potentials, the adsorption of hydroxyl groups on copper atoms that can be used as extra-oxygen atoms to complete the electro-oxidation reaction towards carboxylate (bifunctional mechanism) according to eqn (3) (considering the only formation of glycerate):
| CH2OH–CHOH–CH2OH + 5OH− → CH2OH–CHOH–COO− + 4H2O + 4e− | (3) |
This mechanism will increase the desorption rate, liberating free Pt sites for further glycerol adsorption and increasing the overall reaction rate.52,53,58 But in this case, the selectivity will be towards carboxylate species even in the low potential region. Therefore, the use of Bi as well as of Cu can improve the activity and alter the selectivity and the oxidation rate that lead to molecules with high added-value.52,59,60
Conclusions
Modification of the sputtering power applied to the three metallic targets (Pt, Bi, and Cu) located in a gas aggregation source allows production of mono, bi and trimetallic clusters with average sizes below 6 nm. In situ (quadrupole mass spectrometry) and ex situ (EDX-STEM mapping and Grazing Incidence X-ray diffraction) techniques show the effect of the co-sputtering of Pt and Cu or Bi on the growth and on the characteristics of the resulting clusters. Increasing the sputtering power enhances the total deposited mass and the number of Pt and PtCu clusters. However, in the case of Pt and Bi co-sputtering, the total mass deposited and the number of produced clusters decrease for a Bi sputtering power exceeding the limit value of 8 W. Addition of Cu leads to the formation of a homogeneous PtCu alloy (as confirmed by GIXRD and EDX-STEM mapping images) that decreases the Pt lattice parameter as a function of the sputtering power. For ternary clusters, detailed information on the nanostructure obtained from EDX-STEM mapping images indicated the formation of a PtCu alloyed core surrounded by Bi atoms. However, electrochemical characterization of the PtCuBi/C catalysts could also indicate an additional structure such as an alloyed PtCu core decorated with amorphous Cu–Bi species.This structure has a deep impact on the electrochemical response of the Pt-based clusters during the electrooxidation of glycerol. Addition of Cu and Bi leads to a decrease of the oxidation onset potential. The presence of Cu increases the oxidation current in a broad range of potentials while the presence of Bi only increases the current in a narrower potential range. The PtCuBi/C catalyst reveals both Cu and Bi features: higher oxidation currents and a larger oxidation potential range that lead to the lowest onset potential in our series of Pt-based catalysts and to the higher activity below +0.600 V vs. RHE, with PtCu remaining the most active between +0.600 V and +0.800 V vs. RHE. Therefore, a plasma magnetron-based gas aggregation source can be used for tailoring Pt-based multimetallic clusters for applications in electrocatalysis.
Conflicts of interest
The authors declare no competing financial or commercial interest.Acknowledgements
This work was funded by the French “Agence Nationale de la Recherche” under grant ANR-16-CE29-0007 (ECO-PLAN project) and by the CNRS PEPS project Dust4Pem. All the authors acknowledge Olivier Weldling and Thierry Sauvage from CEMHTI laboratory (Orléans) for the RBS analysis. The authors thank Audrey Sauldubois (CME, Orléans) and Jaafar Ghanbaja (IJL, Nancy) for the HRTEM measurements and Thomas Lecas and Nicolas Dumuis for technical support on the gas aggregation source.References
- S. K. Duran, Mater. Today: Proc., 2020, 26, 261–265 CAS.
- K. Hanaki and J. Portugal-Pereira, in Biofuels and Sustainability: Holistic Perspectives for Policy-making, ed. K. Takeuchi, H. Shiroyama, O. Saito and M. Matsuura, Springer Japan, Tokyo, 2018, pp. 53–71 Search PubMed.
- M. Simões, S. Baranton and C. Coutanceau, ChemSusChem, 2012, 5, 2106–2124 CrossRef.
- H. W. Tan, A. R. Abdul Aziz and M. K. Aroua, Renewable Sustainable Energy Rev., 2013, 27, 118–127 CrossRef CAS.
- M. Pagliaro, R. Ciriminna, H. Kimura, M. Rossi and C. Della Pina, Angew. Chem., Int. Ed., 2007, 46, 4434–4440 CrossRef CAS.
- J. C. Beltrán-Prieto, K. Kolomazník and J. Pecha, Aust. J. Chem., 2013, 66, 511–521 CrossRef.
- G. Dodekatos, S. Schünemann and H. Tüysüz, ACS Catal., 2018, 8, 6301–6333 CrossRef CAS.
- B. S. R. Kouamé, S. Baranton, P. Brault, C. Canaff, W. Chamorro-Coral, A. Caillard, K. De Oliveira Vigier and C. Coutanceau, Electrochim. Acta, 2020, 329, 135161 CrossRef.
- M. Simões, S. Baranton and C. Coutanceau, Electrochim. Acta, 2010, 56, 580–591 CrossRef.
- A. A. Alekseenko, S. V. Belenov, V. S. Menshikov and V. E. Guterman, Russ. J. Electrochem., 2018, 54, 415–425 CrossRef CAS.
- H. Yang, L. Dai, D. Xu, J. Fang and S. Zou, Electrochim. Acta, 2010, 55, 8000–8004 CrossRef CAS.
- J. Maya-Cornejo, R. Carrera-Cerritos, D. Sebastián, J. Ledesma-García, L. G. Arriaga, A. S. Aricò and V. Baglio, Int. J. Hydrogen Energy, 2017, 42, 27919–27928 CrossRef CAS.
- O. Kylián, V. Valeš, O. Polonskyi, J. Pešička, J. Čechvala, P. Solař, A. Choukourov, D. Slavínská and H. Biederman, Mater. Lett., 2012, 79, 229–231 CrossRef.
- O. Kylián, J. Prokeš, O. Polonskyi, J. Čechvala, J. Kousal, J. Pešička, J. Hanuš and H. Biederman, Thin Solid Films, 2014, 571, 13–17 CrossRef.
- M. Vaidulych, J. Hanuš, J. Kousal, S. Kadlec, A. Marek, I. Khalakhan, A. Shelemin, P. Solař, A. Choukourov, O. Kylián and H. Biederman, Plasma Processes Polym., 2019, 16, 1900133 CrossRef CAS.
- O. Kylián, J. Kratochvíl, J. Hanuš, O. Polonskyi, P. Solař and H. Biederman, Thin Solid Films, 2014, 550, 46–52 CrossRef.
- D. Nelli, A. Krishnadas, R. Ferrando and C. Minnai, J. Phys. Chem. C, 2020, 124, 14338–14349 CrossRef CAS.
- A. Wurl, M. Hyslop, S. A. Brown, B. D. Hall and R. Monot, Eur. Phys. J. D, 2001, 16, 205–208 CrossRef CAS.
- M. Broyer, B. Cabaud, A. Hoareau, P. Melinon, D. Rayane and B. Tribollet, in Physics and Chemistry of Small Clusters, ed. P. Jena, B. K. Rao and S. N. Khanna, Springer US, Boston, MA, 1987, pp. 277–281 Search PubMed.
- L. Martínez, M. Díaz, E. Román, M. Ruano, D. Llamosa and P. Y. Huttel, Langmuir, 2012, 28, 11241–11249 CrossRef.
- G. E. Johnson, R. Colby, M. Engelhard, D. Moon and J. Laskin, Nanoscale, 2015, 7, 12379–12391 RSC.
- V. Singh, C. Cassidy, P. Grammatikopoulos, F. Djurabekova, K. Nordlund and M. Sowwan, J. Phys. Chem. C, 2014, 118, 13869–13875 CrossRef CAS.
- G. E. Johnson, R. Colby and J. Laskin, Nanoscale, 2015, 7, 3491–3503 RSC.
- D. Llamosa, M. Ruano, L. Martínez, A. Mayoral, E. Roman, M. García-Hernández and Y. Huttel, Nanoscale, 2014, 6, 13483–13486 RSC.
- A. Caillard, S. Cuynet, T. Lecas, P. Andreazza, M. Mikikian, A.-L. Thomann and P. Brault, J. Phys. D: Appl. Phys., 2015, 48, 475302 CrossRef.
- W. Chamorro-Coral, A. Caillard, P. Brault, P. Andreazza, C. Coutanceau and S. Baranton, Plasma Processes Polym., 2019, 16, e1900006 Search PubMed.
- K. Momma and F. Izumi, J. Appl. Crystallogr., 2011, 44, 1272–1276 CrossRef CAS.
- Materials Project, https://materialsproject.org/, accessed 10 September 2019 Search PubMed.
- A. J. R. Botz, M. Nebel, R. A. Rincón, E. Ventosa and W. Schuhmann, Electrochim. Acta, 2015, 179, 38–44 CrossRef CAS.
- J. Gonzalez-Cobos, S. Baranton and C. Coutanceau, ChemElectroChem, 2016, 3, 1694–1704 CrossRef CAS.
- D. E. Newbury and N. W. E. Ritchie, Scanning, 2013, 35, 141–168 CrossRef CAS.
- W. M. Haynes, CRC handbook of chemistry and physics, CRC Press, 97th edn, 2016 Search PubMed.
- X. Peng, Y. Zhao, D. Chen, Y. Fan, X. Wang, W. Wang and J. Tian, Electrochim. Acta, 2014, 136, 292–300 CrossRef CAS.
- M. Gao, W. Yang and Y. Yu, Int. J. Hydrogen Energy, 2018, 43, 14293–14300 CrossRef CAS.
- L. Leppert, R. Q. Albuquerque and S. Kümmel, Phys. Rev. B: Condens. Matter Mater. Phys., 2012, 86, 241403 CrossRef.
- V. Petkov, Y. Maswadeh, J. A. Vargas, S. Shan, H. Kareem, Z.-P. Wu, J. Luo, C.-J. Zhong, S. Shastri and P. Kenesei, Nanoscale, 2019, 11, 5512–5525 RSC.
- K. T. Jacob, S. Raj and L. Rannesh, Int. J. Mater. Res., 2007, 98, 776–779 CAS.
- R. Tran, Z. Xu, B. Radhakrishnan, D. Winston, W. Sun, K. A. Persson and S. P. Ong, Sci. Data, 2016, 3, 160080 CrossRef CAS.
- D. Nafday, S. Sarkar, P. Ayyub and T. Saha-Dasgupta, ACS Nano, 2018, 12, 7246–7252 CrossRef CAS.
- H. J. Wasserman and J. S. Vermaak, Surf. Sci., 1972, 32, 168–174 CrossRef CAS.
- J. Solla-Gullón, P. Rodríguez, E. Herrero, A. Aldaz and J. M. Feliu, Phys. Chem. Chem. Phys., 2008, 10, 1359–1373 RSC.
- P. Rodríguez, J. Solla-Gullón, F. J. Vidal-Iglesias, E. Herrero, A. Aldaz and J. M. Feliu, Anal. Chem., 2005, 77, 5317–5323 CrossRef.
- J. Z. Chen, Z. Wu, X. Zhang, S. Choi, Y. Xiao, A. Varma, W. Liu, G. Zhang and J. T. Miller, Catal. Sci. Technol., 2019, 9, 1349–1356 RSC.
- O. Teppo, J. Niemelä and P. Taskinen, Thermochim. Acta, 1990, 173, 137–150 CrossRef CAS.
- D. J. Chakrabarti and D. E. Laughlin, Bull. Alloy Phase Diagrams, 1984, 5, 148–155 CrossRef CAS.
- R. M. Castagna, J. M. Sieben, A. E. Alvarez and M. M. E. Duarte, Int. J. Hydrogen Energy, 2019, 44, 5970–5982 CrossRef CAS.
- C. Coutanceau, A. Zalineeva, S. Baranton and M. Simoes, Int. J. Hydrogen Energy, 2014, 39, 15877–15886 CrossRef CAS.
- J. F. Gomes, F. B. C. de Paula, L. H. S. Gasparotto and G. Tremiliosi-Filho, Electrochim. Acta, 2012, 76, 88–93 CrossRef CAS.
- A. Zalineeva, S. Baranton and C. Coutanceau, Electrochim. Acta, 2015, 176, 705–717 CrossRef CAS.
- R. M. L. M. Sandrini, J. R. Sempionatto, G. Tremiliosi-Filho, E. Herrero, J. M. Feliu, J. Souza-Garcia and C. A. Angelucci, ChemElectroChem, 2019, 6, 4238–4245 CrossRef CAS.
- C. Coutanceau, S. Baranton and R. S. B. Kouamé, Front. Chem., 2019, 7, 100 CrossRef CAS.
- Y. Kwon, Y. Birdja, I. Spanos, P. Rodriguez and M. T. M. Koper, ACS Catal., 2012, 2, 759–764 CrossRef CAS.
- N. Worz, A. Brandner and P. Claus, J. Phys. Chem. C, 2010, 114, 1164–1172 CrossRef.
- P. N. Amaniampong, Q. T. Trinh, J. J. Varghese, R. Behling, S. Valange, S. H. Mushrif and F. Jérôme, Green Chem., 2018, 20, 2730–2741 RSC.
- B. Chen, D. Cheng and J. Zhu, J. Power Sources, 2014, 267, 380–387 CrossRef CAS.
- Y. Kang, B. Jiang, Z. A. Alothman, A. Y. Badjah, M. Naushad, M. Habila, S. Wabaidur, J. Henzie, H. Li and Y. Yamauchi, Chem.–Eur. J., 2019, 25, 343–348 CrossRef CAS.
- O. B. Ayodele, R. Cai, J. Wang, Y. Ziouani, Z. Liang, M. C. Spadaro, K. Kovnir, J. Arbiol, J. Akola, R. E. Palmer and Y. V. Kolen’ko, ACS Catal., 2020, 10, 451–457 CrossRef CAS.
- A. C. Garcia, Y. Y. Birdja, G. Tremiliosi-Filho and M. T. M. Koper, J. Catal., 2017, 346, 117–124 CrossRef CAS.
- R. Ciriminna, A. Fidalgo, L. M. Ilharco and M. Pagliaro, ChemistryOpen, 2018, 7, 233–236 CrossRef CAS.
- D. Liu, J.-C. Liu, W. Cai, J. Ma, H. B. Yang, H. Xiao, J. Li, Y. Xiong, Y. Huang and B. Liu, Nat. Commun., 2019, 10, 1779 CrossRef.
| This journal is © The Royal Society of Chemistry 2021 |