
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceInvestigation of Au/Co3O4 nanocomposites in glycol oxidation by tailoring Co3O4 morphology†
Xuejiao
Wei
ab,
Sami
Barkaoui
b,
Jingwen
Chen
a,
Guiping
Cao
a,
Zeying
Wu
a,
Fei
Wang
*c and
Gao
Li
 *b
*b
aSchool of Chemical Engineering and Materials, Changzhou Institute of Technology, Changzhou 213032, China
bState Key Laboratory of Catalysis, Dalian Institute of Chemical Physics, Chinese Academy of Sciences, Dalian 116023, China. E-mail: gaoli@dicp.ac.cn
cAdvanced Catalysis and Green Manufacturing Collaborative Innovation Center, School of Petrochemical Engineering, Changzhou University, Changzhou 213164, China. E-mail: wangfei@cczu.edu.cn
First published on 5th February 2021
Abstract
The interfacial perimeter of nanogold and supports is often deemed as the catalytically active site for multiple reactions while the geometrical configuration of the interfacial perimeter at atomic scale is less studied. Herein, gold nanoparticles (NPs) of ca. 2.0 nm are dispersed on Co3O4 support in the shape of nanocubes (dominant Co3O4(001) facet) and nanoplates (Co3O4(111)), which forms different Au–Co3O4 interfaces with respect to the specific facet of the oxide support. A comparison is made on the basis of the interfacial structures and catalytic behavior of ethylene glycol oxidation. STEM analysis identifies that these metallic Au NPs interact with Co3O4 with an orientation relationship of Au/Co3O4(001) and Au/Co3O4(111). XPS and Raman spectroscopy investigations reveal the important variations in the reactivity of surface oxygen, surface Oads/OL ratio, and evolution of surface oxygen vacancies upon variation of the Co3O4 shape. Au/Co3O4-P exhibits much better catalytic activity than the Au/Co3O4-C counterpart in the aerobic oxidation of ethylene glycol, which is promoted by surface oxygen vacancies and intrinsic defects. It has been revealed that the surface oxygen vacancies participate in activating O2, thus making Co3O4-P a superior support for Au NPs in the catalysis of ethylene glycol oxidation.
Introduction
Methyl glycolate (MG) is an industrially important chemical, which can be used as solvents, extractants or as food additives. In addition, they are applied in the synthesis of macromolecule polymer material as platform molecules.1–3 To date, many strategies have been developed to prepare MG from the different conventional substrates.4,5 Moreover, since the heterogeneous Au catalysts were first reported to be active in the oxidation of ethylene glycol to methyl glycolate by Hayashi et al.,6 a series of studies have shown that Au catalysts supported on various supports are effective in the oxidation of various alcohols.7–12For analyzing the heterogeneous catalyst, the type of metal (especially Au), particle size and morphology have been extensively explored on the side of anchored metals.13 For example, Bianchi et al. have substantiated that the decrease in the Au particle diameter of supported catalysts could lead to the increase in catalytic activity in the selective liquid phase oxidation of glycols.14 However, Comotti et al. revealed that the catalytic activity was inversely proportional to the diameter of Au particles (3–6 nm) in the oxidation of glucose.15 These findings notably indicate that an Au size effect is apparent in different reactions. Whereas on the side of support, the morphology of oxide supports can mediate the catalytic activity, which is mainly due to their atomic arrangements and electronic structures (e.g., surface defects or surface oxygen vacancies, reconstructions and surface orientations).16–18 For instance, the Co3O4 morphology of nanotubes,19 nanosheets,20 nanowires21 and nanocubes22 showed distinct shape effect in CO oxidation. Note that the different properties of these Co3O4 nanomaterials are strongly associated with exposed crystal planes. Similar to Co3O4 nanowires, enclosed by the Co3O4(111) facets, show a high CO conversion rate of 161.75 μmol CO g−1 s−1 at 248 °C.21 Moreover, the Co3O4 nanorods (40% of the Co3O4(110) facets) exhibited excellent CO oxidation activity.23
Although the oxidation of ethylene glycol has been intensively investigated in the field of heterogeneous Au catalysts, identification of active sites and clarification of catalytic mechanism on the supported gold nanoparticles have not yet been fully investigated. Recently, additional studies favour that the reaction should occur at the Au-oxide interfacial perimeter over which the reactants are adsorbed and activated on the gold nanoparticle and the molecular oxygen is active by oxide surfaces (e.g. defects).24–26 With this respect, both the size of gold particles and the chemical properties of oxide surfaces played critical roles in determining the Au-oxide interfacial structures and consequently catalytic activity, which involves electronic and geometric interactions between Au particles and oxide surfaces at their interfaces.
Herein, we have initially prepared the gold NPs of ∼2 nm supported on Co3O4 with different morphology (nanocubes, mainly exposing Co3O4{111} and nanoplate, exposing Co3O4{001}) using gold colloids (∼2 nm Au![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :PVA particles) as a precursor. Moreover, the size and morphology of Au nanoparticles on the surface of Co3O4-C and Co3O4-P are similar based on the mathematical statistics, which could simplify the protocols of relationship between Au and oxide support and shed light on the mechanism of oxidation reaction over supported Au NPs. Furthermore, Au/Co3O4-C and Au/Co3O4-P catalysts show a remarkable improvement on the activity than the corresponding plain Co3O4 in the glycol oxidation reaction. Furthermore, XPS and Raman spectroscopy results confirmed that the larger number of defects in Co3O4-P than in Co3O4-C could participate in activating O2 and in the activation of oxygen species, which demonstrates that Co3O4-P is a superior support for Au NPs for catalyzing ethylene glycol oxidation.
:PVA particles) as a precursor. Moreover, the size and morphology of Au nanoparticles on the surface of Co3O4-C and Co3O4-P are similar based on the mathematical statistics, which could simplify the protocols of relationship between Au and oxide support and shed light on the mechanism of oxidation reaction over supported Au NPs. Furthermore, Au/Co3O4-C and Au/Co3O4-P catalysts show a remarkable improvement on the activity than the corresponding plain Co3O4 in the glycol oxidation reaction. Furthermore, XPS and Raman spectroscopy results confirmed that the larger number of defects in Co3O4-P than in Co3O4-C could participate in activating O2 and in the activation of oxygen species, which demonstrates that Co3O4-P is a superior support for Au NPs for catalyzing ethylene glycol oxidation.
Experimental
Synthesis of Co3O4 oxides
Co3O4 was synthesized by typical procedures reported in the literature.27,28 The cubic-like Co3O4 (Co3O4-C for short) was synthesized by dissolving 10 mmol Co(NO3)2·6H2O in 45 mL of ethanol. Then, 10 mL triethylamine (TEA) was dissolved in 25 mL ethanol, which forms a transparent solution. The two solutions were mixed under vigorous stirring for 35 min. After stirring for 30 min, the suspension was transferred into a Teflon-lined stainless steel autoclave and aged at 160 °C for 24 h. The solution was filtered and washed by deionized water and ethanol dried at 80 °C overnight and then calcined at 350 °C for 3 h in air.For Co3O4 nanoplates (abbreviated as Co3O4-P), 0.9525 g CoCl2·6H2O was dissolved in 80 mL of deionized water. Moreover, 2 mL of TEA was added under magnetic stirring. After stirring for 10 min, the solution was transferred into a 100 mL autoclave and reacted at 180 °C for 20 h. The precipitate was filtered, washed several times with deionized water and ethanol until free of chloride ions (AgNO3 test). The solids were dried overnight at 80 °C and then calcined at 350 °C for 3 h in air. The surfactants in Co3O4-P and Co3O4-C were completely removed by the calcination and was confirmed by the temperature-programmed oxidation (TPO) measurements (Fig. S1 and S2 in the ESI†).
Preparation of Au/Co3O4 catalysts
Au:PVA colloids were prepared using a previously reported method.29 Typically, 120 mL of Au sols were deposited on Co3O4 support at room temperature and stirred for 3 h at 800 rpm. After stirring, the samples were washed with water and ethanol before being dried overnight at 50 °C. The final products were calcined at 300 °C for 2 h in the presence of a flow of air to remove all the capping surfactant, which was evidenced by TPO tests (Fig. S3 and S4 in the ESI†). The loading of Au was designed as 1.0 wt%. Moreover, inductively coupled plasma-atomic emission spectrometer (ICPS-8100, Shimadzu) analysis confirmed that the actual loading of Au was 0.62 wt% for both Au/Co3O4-C and Au/Co3O4-P.
Characterization
Scanning transmission electron microscopy (STEM) images were recorded on a F200 microscope operated at 300 kV. The specimen was prepared by ultrasonically dispersing the sample into ethanol, which deposits droplets of the suspensions onto a carbon-enhanced copper grid, and then dries them in air. Transmission electron microscopy (TEM) images were recorded on a FEI Tecnai G2 Spirit microscope operated at 120 kV. TPO analysis was measured with an AutoChem II 2920 instrument (Micromeritics) and analyzed using a thermal conductor detector (TCD). Before analysis, 50 mg of samples were pre-treated with a 5 vol% O2/He mixture (30 mL min−1) at 200 °C for 1 h to clean the sample surface. After cooling to room temperature, 5 vol% of O2/He was used as a feed gas to perform TPO from RT to 600 °C. X-ray photoelectron spectra (XPS) were then obtained using an ESCALAB MK-II spectrometer with an aluminium anode for Kα (hν = 1484.6 eV) radiation. The equipment base pressure was 1.7 × 10−10 mbar, and all samples were characterized at room temperature. Detailed spectra were recorded for the region of Co 2p, O 1s and Au 4f photoelectrons with a 0.1 eV step. Analysis was then performed using XPSPEAK41 software and charging effects were corrected by adjusting the binding energy (B.E.) of C 1s to 284.6 eV. The spectra were deconvoluted using the XPSPEAK program by curve fitting with a mixed Gaussian–Lorentzian function. Raman spectra were collected with Renishaw via a Raman microscope with a laser wavelength of 785 nm and a laser power of 3 mW and obtained at the same conditions after 60 s of exposure.Catalytic investigation
The oxidation reactions were conducted in a 10 mL continuous stirred-tank reactor. Typically, the reactions were performed at 90 °C under atmosphere of 4 MPa O2, stirring at 600 rpm, for 4 h using 2 mL methanol, 500 mg ethylene glycol and 100 mg catalysts. After the reaction, the reactor was cooled to less than 5 °C with an ice–water mixture and depressurized. An internal standard of n-amyl alcohol was added to liquid products. The conversion of ethylene glycol and selectivity toward the product of methyl glycolate (MG), dimethyl oxalate (DMO), and hydroxyethyl formate (HF) are analyzed by gas chromatography-mass spectrometry method on a GC/MS 7890B-5977A with a capillary column HP-5MS with He as carrier gas. The conversion of ethylene glycol (XEG) and selectivity toward desired product of methyl glycolate (SMG) were calculated using the following equations: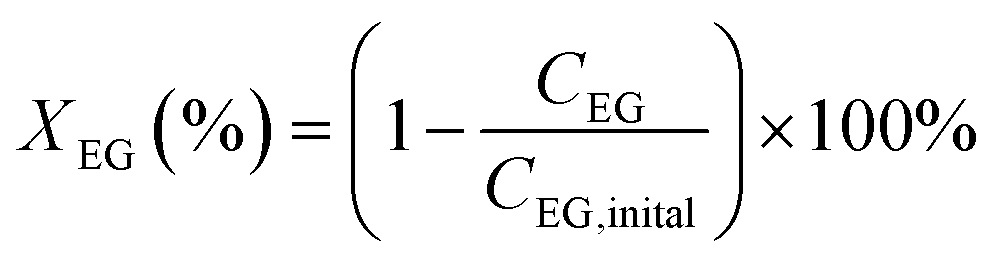 | (1) |
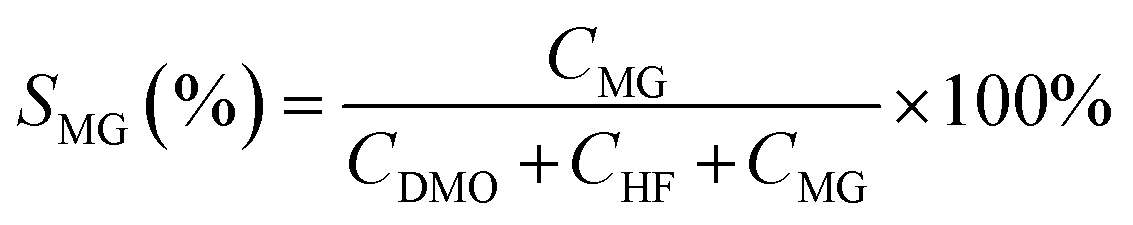 | (2) |
Results and discussion
Characterization of Au/Co3O4
TEM images showed that the Co3O4-C oxides are monodisperse with a uniform size of ∼22 nm (Fig. S5 in the ESI†). Co3O4-P with a hexagonal shape was prepared with an average edge length of ∼160 nm and thickness of ∼25 nm (Fig. S6 in the ESI†). Further, the Au:PVP colloids of 2 nm were adsorbed and deposited onto the surface of the defined Co3O4 oxides (Fig. S7 in the ESI†). Au/Co3O4 samples primarily kept their initial Co3O4 morphologies after the introduction of Au NPs and annealing at 300 °C. HAADF-STEM images showed that the gold particles were dispersed uniformly in space on the oxide-supports (Fig. 1a–f). The gold particles exhibited a mean size of 2.0 ± 0.5 nm and a quasi-truncated octahedron enclosed by Au{111} and {100} facets in the Au/Co3O4-C samples. Moreover, Au NPs uniformly anchored on the Co3O4(001) facet as the measured inter-planar distance of 0.29 nm corresponds to the (220) crystal plane of cubic Co3O4 oxides (Fig. 1b and c).30
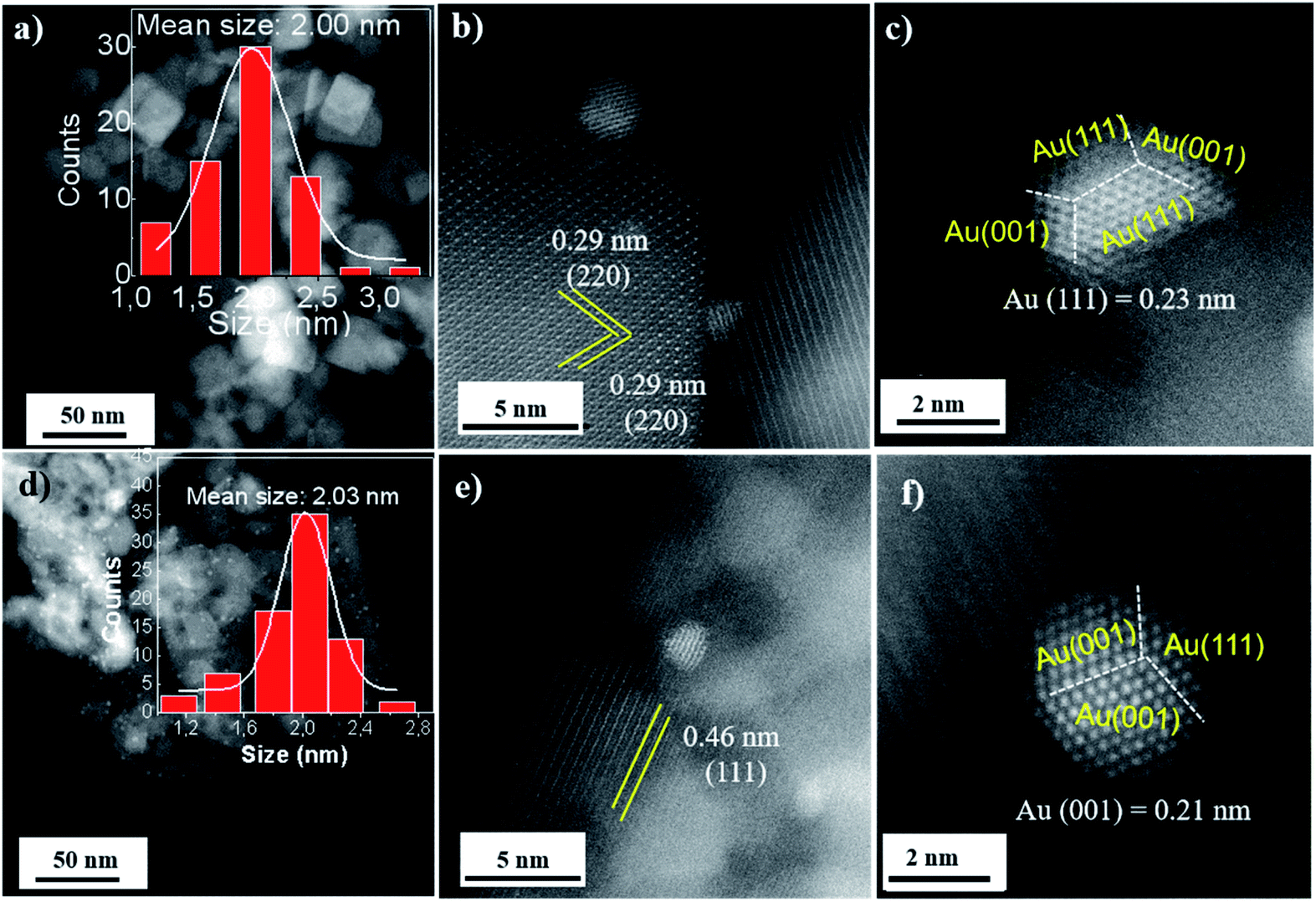 | ||
| Fig. 1 STEM images of Au particles on the Co3O4(001) in the Au/Co3O4-C samples (a–c) and Co3O4(111) in the Au/Co3O4-P (d–f). Insets, in (a) and (c), are the size distribution of the gold nanoparticles. | ||
For Au/Co3O4-P catalysts, lattice fringes with an inter-planar spacing of 0.46 nm can be attributed to the Co3O4(111) facets of Co3O4-P oxides.27,31 Gold particles uniformly loaded onto the Co3O4(111) facet and exhibited a quasi-truncated octahedron enclosed by Au{111} and {100} facets, as seen in Fig. 1d and e. Au NPs showed a particle size of 2.0 ± 0.6 nm (Fig. 1d), which is similar with the particle size in the Au/Co3O4-C and the parent Au colloids. It demonstrates that the small gold particles exhibited strong interaction with both Co3O4(001) and Co3O4(111) facets, and the Au NPs should have stronger interaction with Co3O4(111) facets with a short lattice spacing of 0.21 nm than the Au NPs on the Co3O4(001) plane.
Further, XPS analysis was performed to explore the chemical state of Au/Co3O4 catalysts. The spectra of Au 4f, Co 2p and O 1s are compiled in Fig. 2. Au 4f XPS spectra showed two same sets of the Au 4f7/2 and Au 4f5/2 signals at bonding energies (BEs) of 83.9 and 87.6 eV (Fig. 2a and d), which indicates that the nanogold chemical states are metallic (Au 4f7/2 BE of gold foil is ∼83.8 eV) in both Au/Co3O4-C and Au/Co3O4-P samples.32 TEM and XPS results suggest that the chemical property of gold nanoparticles (e.g. Au loading, Au mean size, and oxidation state) are similar, which can relieve the factors (e.g. size-dependence, charge effects, etc.) and focus on the Au–Co3O4 interfaces in the aerobic oxidation of ethylene glycol to methyl glycolate (vide infra).
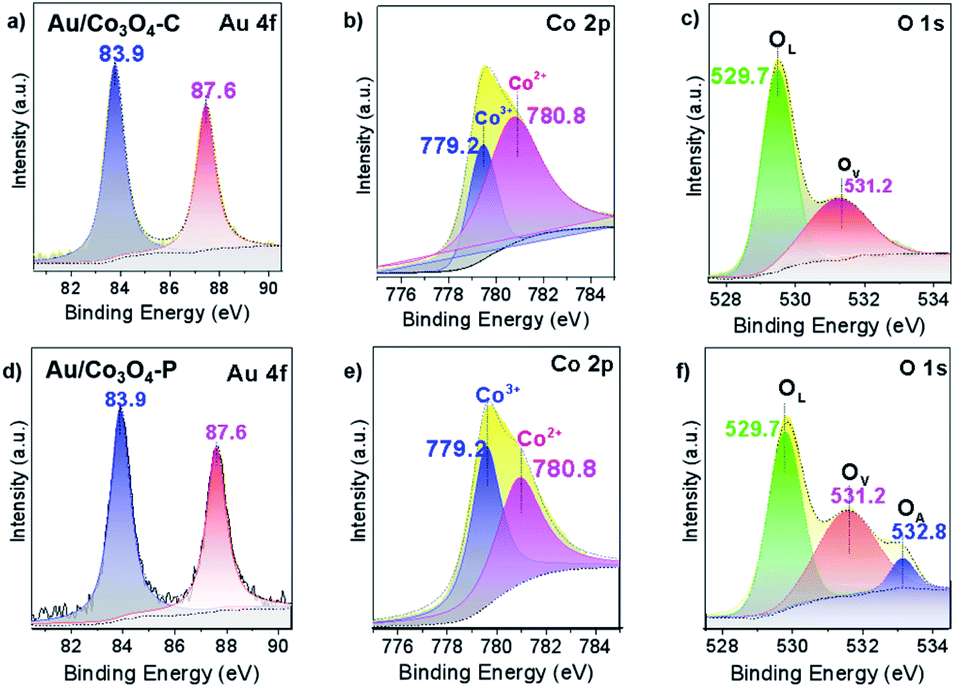 | ||
| Fig. 2 Au 4f, Co 2p and O1s XPS survey spectra of (a–c) Au/Co3O4-C and (d–f) Au/Co3O4-P. Note that the Co3+, Co2+, Ov, OA and Olatt species are fixed at 779.2, 780.8, 531.2, 532.8 and 529.7 eV, respectively. | ||
In addition, two XPS sets were found at 795.1 and 780.0 eV with an energy difference of ∼15.1 eV, which corresponds to Co 2p1/2 and Co 2p3/2 spin–orbit-split doublet peak of Co3O4 spinel.33–37 The Co 2p3/2 peak of 780.0 eV is fitted and deconvoluted into two peaks of 780.8 and 779.2 eV (Fig. 2b and e), which is attributed to Co2+ and Co3+ species, respectively. In addition, the surface ratio of Co3+/Co2+ is similar in the Au/Co3O4-P and Au/Co3O4-C catalysts (1.07 vs. 1.09, Table 1).
| Catalysts | Au loading (wt%) | Au mean size (nm) | Co3+/Co2+ ratio | OV/OL ratio |
|---|---|---|---|---|
| Au/Co3O4-C | 0.62 | 2.00 | 1.07 | 0.60 |
| Au/Co3O4-P | 0.62 | 2.03 | 1.09 | 0.79 |
Moreover, the O 1s electronic levels were examined. The asymmetric O 1s peaks could be deconvoluted to two components at 531.2 and 529.7 eV for Au/Co3O4-C (Fig. 2c). However, the O 1s peaks can be best fitted by three components centred at ∼529.7, ∼531.2, and ∼532.8 eV, which can be ascribed to lattice oxygen atoms (OL), oxygen vacancies (OV), and surface oxygen species (OA), respectively.38 Furthermore, the existence of peak at 532.8 eV (Fig. 2f) for the Au/Co3O4-P is assigned to surface chemisorbed or dissociated oxygen.39 The OV/OL ratio of Au/Co3O4-P (0.79) is much higher than that of Au/Co3O4-C (0.60, Table 1). Above all, it is obvious that the XPS strength of surface oxygen species increases for Au/Co3O4-P, which indicates the existence of more oxygen vacancy sites on the Au/Co3O4-P than that of Au/Co3O4-C.
To clearly identify the surface oxygen vacancies of Au/Co3O4–C and Au/Co3O4–P, Raman analysis was applied. As shown in Fig. 3, the Au/Co3O4 catalysts displayed five vibration peaks (A1g + Eg + 3 F2g) in the range of 150–800 cm−1. The peaks at 484, 523 and 622 cm−1 are assigned to the Eg, F2g(2) and F2g(1) symmetry, respectively. The Raman peak at 196 cm−1 is attributed to the F2g(3) mode of tetrahedral sites (CoO4) while the peak at 691 cm−1 with A1g symmetry is attributed to the characteristics of octahedral CoO6 sites, which correspond to the unique characteristics of spinel-type cubic Co3O4 phase, which is in great agreement with these reported for Co3O4.40 The full width at half-maximum (FWHM's) of all peaks in the Au/Co3O4-P are higher than that of Au/Co3O4-C catalysts, which could account for the high concentration of surface oxygen vacancies and intrinsic defects for Au/Co3O4-P.40 To summarize, Au/Co3O4-P has more defective structures than Au/Co3O4-C which can promote the oxidation reactions as these surface defective structures can facilitate adsorption and activation of oxygen species.
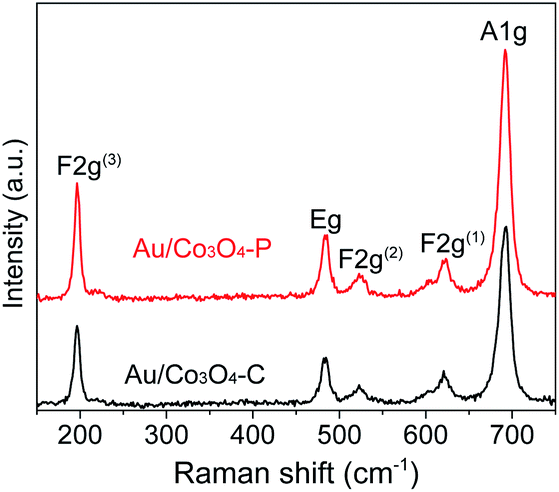 | ||
| Fig. 3 Raman spectra of the as-prepared Au/Co3O4-C and Au/Co3O4-P catalysts. | ||
Catalytic performance
The aerobic oxidation of ethylene glycol is chosen as a probe reaction to evaluate the catalytic efficiency of Au/Co3O4 catalysts and Co3O4 catalysts. The aerobic oxidations were carried out in a liquid phase: a 4 MPa O2 gas at 90 °C for 4 h (details see Experimental section), and the catalytic results are compiled in Fig. 4. Note that both Co3O4-C and Co3O4-P supports were inactive during the aerobic oxidation of ethylene glycol under such conditions. Clearly, the presence of Au NPs has notably improved the catalytic activity in the EG oxidation reactions. There major products of methyl glycolate (MG), dimethyl oxalate (DMO), and hydroxyethyl formate (HF) were observed by a GC-MS method in the Au/Co3O4 catalyzed oxidation reaction of ethylene glycol (EG). Note that DMO can be considered as the further oxidative esterification of MG product and HF is other oxidative esterification of methanol with EG (Scheme S1†).41 Au/Co3O4-P catalysts gave a promising activity in the oxidation of ethylene glycol; a 67% conversion was achieved while a medium EG conversion of ∼41% was obtained over the Au/Co3O4-C catalysts. Moreover, Au/Co3O4-P catalysts gave a higher selectivity (∼87%) towards the desired MG product (an important chemical) than Au/Co3O4-C catalysts, as shown in Fig. 4. In all, the Au/Co3O4-C catalysts are considered inferior to that of the Au/Co3O4-P counterpart.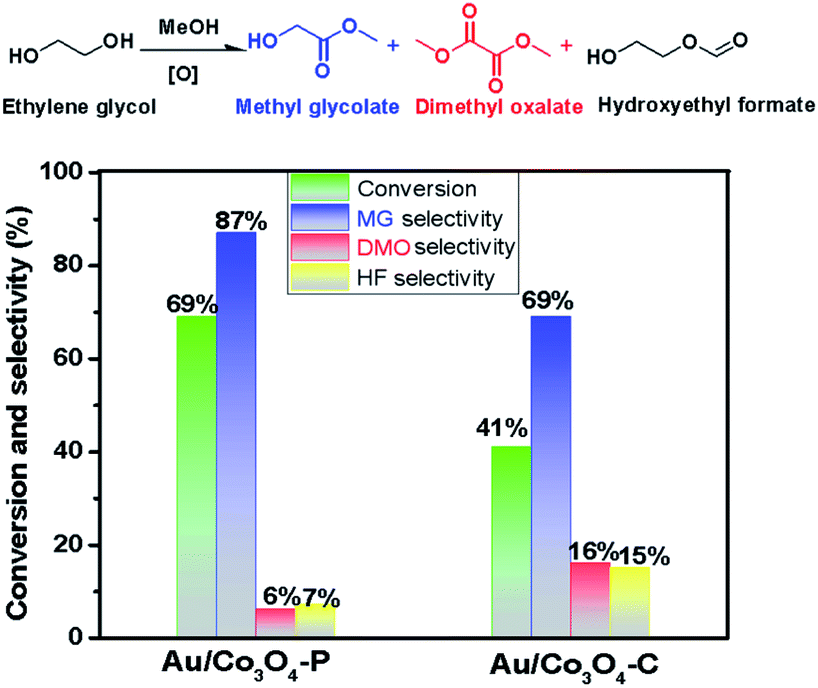 | ||
| Fig. 4 Catalytic performance of Au/Co3O4-P and Au/Co3O4-C over oxidation of ethylene glycol. Reaction conditions: 500 mg ethylene glycol and 100 mg catalysts in a 2 mL methanol, 4 MPa O2, 90 °C, for 4 h. | ||
Previously, Dong et al. observed that the correlation between the activity and diameter is consistent with the nature of active exposed atoms.42 However, when compared to previous studies, our results show that the interfacial environment between Au and Co3O4 support seems to impact the catalytic activity largely, while the Au particles on both Au/Co3O4-C and Au/Co3O4-P catalysts have similar structure based on our statistical calculation (Table S1 in the ESI†). Indeed, the difference between the activity can be mainly attributable to the change of interfacial environment due to the introduction of Au on the different crystal plane (Co3O4(111) for Co3O4-P and Co3O4(001) for Co3O4-C).
Recently, it is widely accepted that the catalytic behavior of supported Au catalyst is strongly associated with Au particles and the nature of the support as well as the Au–support interaction.43,44 For liquid oxidation reactions, the situation is more complicated due to the catalyst's surface properties.45–47 Herein, the selective oxidation only occurs in the presence of Au, and the Au/Co3O4-P shows better activity than that of Au/Co3O4-C. Combined with similar Au dispersion in Au/Co3O4-P and Au/Co3O4-C samples, the oxygen vacancy site rooted in the Au–Co3O4 interfacial structure should play a vital role in this glycol oxidation. Nevertheless, we believe that it is justified that the Au/Co3O4-P exhibits better catalytic activity than Au/Co3O4-C in the oxidation of ethylene glycol, which is promoted by the surface oxygen vacancies and intrinsic defects confirmed by XPS and Raman results.48,49
Conclusions
In summary, gold NPs of ca. 2.0 nm immobilized on Co3O4 oxides have been synthesized through a simple impregnation of gold colloids and oxides. Au/Co3O4-C and Au/Co3O4-P catalysts show a remarkable improvement on the activity than the corresponding plain Co3O4 oxides in the glycol oxidation reaction. The sensitive factors, e.g. size-hierarchy and chemical state of the gold particles, are effectively excluded during the nanogold catalyzed reactions. STEM analyses identified that Au particles with Au{111} and Au{100} exposures interact with Co3O4{111} and Co3O4{001}. Therefore, Au/Co3O4{111} exhibited better catalytic activity and selectivity in the catalyzing glycol oxidation. XPS and Raman spectroscopy analyses confirmed that the surface oxygen vacancies and intrinsic defects on Co3O4{111} on the surface of gold NPs are responsible for the high catalytic behavior in catalyzing glycol oxidation.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We thank the financial support by the fund of National Natural Science Foundation of China (No. 21701168 and No. 21802008), Liaoning Natural Science Foundation (2020-MS-024), and Liaoning Revitalization Talents Program (XLYC1807121).Notes and references
- Y. Cui, B. Wang, C. Wen, X. Chen and W. L. Dai, ChemCatChem, 2016, 8, 527–531 CrossRef CAS.
- G. Dong, Y. Cao, S. Zheng, J. Zhou, W. Li, F. Zaera and X. Zhou, J. Catal., 2020, 391, 155–162 CrossRef CAS.
- H. Fan, J. Tan, Y. Zhu, H. Zheng and Y. Li, J. Mol. Catal. A: Chem., 2016, 425, 68–75 CrossRef CAS.
- B. Wang, Q. Xu, H. Song and G. Xu, J. Energy Chem., 2016, 16, 78–80 Search PubMed.
- A. Y. Yin, C. Wen, W. L. Dai and K. N. Fan, Appl. Catal., B, 2011, 108, 90–99 CrossRef.
- T. Hayashi, T. Inagaki, N. Itayama and H. Baba, Catal. Today, 2006, 117, 210–213 CrossRef CAS.
- C. Zhang, Y. Chen, H. Wang, Z. Li, K. Zheng, S. Li and G. Li, Nano Res., 2018, 11, 2139–2148 CrossRef CAS.
- C. Fan, R. Wang, P. Kong, X. Wang, J. Wang, X. Zhang and Z. Zheng, Catal. Commun., 2020, 140, 106002 CrossRef CAS.
- G. L. Brett, P. J. Miedziak, N. Dimitratos, J. A. LopezSanchez, N. F. Dummer, R. Tiruvalam, C. J. Kiely, D. W. Knight, S. H. Taylor, D. J. Morgan, A. F. Carley and G. J. Hutchings, Catal. Sci. Technol., 2012, 2, 97–104 RSC.
- R. K. P. Purushothaman, J. van Haveren, D. S. van Es, I. Melian-Cabrera and H. J. Heeeres, Green Chem., 2012, 14, 2031–2037 RSC.
- K. Suzuki, T. Yamaguchi, K. Matsushita, C. Iitsuka, J. Miura, T. Akaogi and H. Ishida, ACS Catal., 2013, 3, 1845–1849 CrossRef CAS.
- C. Liu, J. Zhang, J. Huang, C. Zhang, F. Hong, Y. Zhou, G. Li and M. Haruta, ChemSuSChem, 2017, 10, 1976–1980 CrossRef CAS.
- A. Taketoshi and M. Haruta, Chem. Lett., 2014, 43, 380–387 CrossRef CAS.
- C. Bianchi, F. Porta, L. Prati and M. Rossi, Top. Catal., 2000, 13, 231–236 CrossRef CAS.
- M. Comotti, C. D. Pina, R. Matarrese and M. Rossi, Angew. Chem., Int. Ed., 2004, 43, 5812–5815 CrossRef CAS.
- P. Bhojane, L. Sinha, R. S. Devan and P. M. Shirage, Nanoscale, 2018, 10, 1779–1787 RSC.
- X. Zhang, P. Yang, Y. Liu, J. Pan, D. Li, B. Wang and J. Feng, J. Catal., 2020, 385, 146–159 CrossRef CAS.
- J. Wang, R. Gao, D. Zhou, Z. Chen, Z. Wu, G. Schumacher, Z. Hu, X. Liu, J. Wang and R. Gao, ACS Catal., 2017, 7, 6533–6541 CrossRef CAS.
- Y. Lv, Y. Li and W. Shen, Catal. Commun., 2013, 42, 116–120 CrossRef CAS.
- C. Xu, Y. Liu, C. Zhou, L. Wang, H. Geng and Y. Ding, ChemCatChem, 2011, 3, 399–407 CrossRef CAS.
- Y. Sun, P. Lv, J. Y. Yang, L. He, J. C. Nie, X. Liu and Y. Li, Chem. Commun., 2011, 47, 11279–11281 RSC.
- Y. Teng, Y. Kusano, M. Azuma, M. Haruta and Y. Shimakawa, Catal. Sci. Technol., 2011, 1, 920–922 RSC.
- X. W. Xie, Y. Li, Z. Q. Liu, M. Haruta and W. J. Shen, Nature, 2009, 458, 746 CrossRef CAS.
- H. Li, S. Wang, F. Hong, Y. Gao, B. Zeng, R. S. Haider, F. Fan, J. Huang and C. Li, J. Chem. Phys., 2020, 152, 194702 CrossRef CAS.
- I. X. Green, W. J. Tang, M. Neurock and J. T. Yates, Science, 2011, 333, 736–739 CrossRef CAS.
- C. Wang, H. Wang, Q. Yao, H. Yan, J. Li and J. Lu, J. Phys. Chem. C, 2016, 120, 478–486 CrossRef CAS.
- Z. Chen, S. Wang, W. Liu, X. Gao, D. Gao, M. Wang and S. Wang, Appl. Catal., A, 2016, 525, 94–102 CrossRef CAS.
- Y. Zhan, G. Du, S. Yang, Ch. Xu, M. Lu, Z. Liu and J. Y. Lee, ACS Appl. Mater. Interfaces, 2015, 7, 12930–12936 CrossRef CAS.
- B. Shao, J. Zhang, J. Huang, B. Qiao, Y. Su, Sh. Miao, Y. Zhou, D. Li, W. Huang and W. Shen, Small Methods, 2018, 2, 1800273 CrossRef.
- Y. J. Mergler, J. Hoebink and B. E. Nieuwenhuys, J. Catal., 1997, 167, 305–313 CrossRef CAS.
- H. Chen, M. Yang, S. Tao and G. Chen, Appl. Catal., B, 2017, 209, 648–656 CrossRef CAS.
- O. F. Odio, L. Lartundo-Rojas, P. Santiago-Jacinto, R. Martínez and E. Reguera, J. Phys. Chem. C, 2014, 118, 2776–2791 CrossRef CAS.
- S. Fan, Y. Zhang, X. Ma, E. Yan, X. Liu, S. Li, W. Liang and X. Zhai, Int. J. Electrochem. Sci., 2013, 8, 10498–10505 CAS.
- J. Wang, C. Zhang, S. Yang, H. Liang and Y. Men, Catal. Sci. Technol., 2019, 9, 6379–6390 RSC.
- J. Wang, S. Yang, H. Sun, J. Qiu and Y. Men, J. Colloid Interface Sci., 2020, 577, 355–367 CrossRef CAS.
- Z. S. Wu, W. Ren, L. Wen, L. Gao, J. Zhao, Z. Chen, G. Zhou, F. Li and H. M. Cheng, ACS Nano, 2010, 4, 3187–3194 CrossRef CAS.
- B. M. Abu-Zied, S. M. Bawaked, S. A. Kosa and W. Schwieger, Appl. Surf. Sci., 2015, 351, 600–609 CrossRef CAS.
- C. Zhang, F. Zheng, Z. Zhang, D. Xiang, C. Cheng, Z. Zhuang, P. Li, X. Li and W. Chen, J. Mater. Chem. A, 2019, 7, 9059–9067 RSC.
- K. Song, E. Cho and Y. M. Kang, ACS Catal., 2019, 9, 3773–3782 CrossRef.
- G. Zhai, J. Wang, Z. Chen, W. An and Y. Men, Chem. Eng. J., 2018, 337, 488–498 CrossRef CAS.
- Y. Shi, S. Tian, Q. Shi, A. Waheed, Y. Cao and G. Li, Nanoscale Adv., 2019, 1, 3654–3659 RSC.
- Y. Ke, X. Qin, C. L. Liu, R. Z. Yang and W. S. Dong, Catal. Sci. Technol., 2014, 4, 3141–3150 RSC.
- A. Waheed, Q. Shi, N. Maeda, D. M. Meier, Z. Qin, G. Li and A. Baiker, Catalysts, 2020, 10, 933 CrossRef CAS.
- Q. Fang, Z. Qin, Y. Shi, F. Liu, S. Barkaoui, H. Abroshan and G. Li, ACS Appl. Energy Mater., 2019, 2, 2654–2661 CrossRef CAS.
- T. Ishida, N. Kinoshita, H. Okatsu, T. Akita, T. Takei and M. Haruta, Angew. Chem., Int. Ed., 2008, 47, 9265–9268 CrossRef CAS.
- R. Jin, G. Li, S. Sharma, Y. Li and X. Du, Chem. Rev., 2021, 121(2), 567–648 CrossRef CAS.
- X. Du, Y. Huang, X. Pan, B. Han, Y. Su, Q. Jiang, M. Li, H. Tang, G. Li and B. Qiao, Nat. Commun., 2020, 11, 5811 CrossRef CAS.
- Q. Shi, Z. Qin, A. Waheed, Y. Gao, H. Xu, H. Abroshan and G. Li, Nano Res., 2020, 13, 939–946 CrossRef CAS.
- S. Guo, G. Zhang, Z.-K. Han, S. Zhang, D. Sarker, W. W. Xu, X. Pan, G. Li and A. Baiker, ACS Appl. Mater. Interfaces, 2021, 13, 622–630 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1na00053e |
| This journal is © The Royal Society of Chemistry 2021 |