
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceA direct route to six and seven membered lactones via γ-C(sp3)–H activation: a simple protocol to build molecular complexity†
Jayabrata
Das‡
 a,
Pravas
Dolui‡
a,
Wajid
Ali
a,
Jyoti Prasad
Biswas
a,
Hediyala B.
Chandrashekar
a,
Gaurav
Prakash
a and
Debabrata
Maiti
*ab
a,
Pravas
Dolui‡
a,
Wajid
Ali
a,
Jyoti Prasad
Biswas
a,
Hediyala B.
Chandrashekar
a,
Gaurav
Prakash
a and
Debabrata
Maiti
*ab
aDepartment of Chemistry, IIT Bombay, Powai, Mumbai 400076, India. E-mail: dmaiti@chem.iitb.ac.in
bTokyo Tech World Research Hub Initiative(WRHI), Laboratory for Chemistry and Life Science, Tokyo Institute of Technology, Tokyo 152-8550, Japan
First published on 19th August 2020
Abstract
Lactones comprise a class of valuable compounds having biological as well as industrial importance. Development of a methodology to synthesize such molecules directly from readily available materials such as aliphatic carboxylic acid is highly desirable. Herein, we have reported synthesis of δ-lactones and ε-lactones via selective γ-C(sp3)–H activation. The γ-C–H bond containing aliphatic carboxylic acids provide six or seven membered lactones depending on the olefin partner in the presence of a palladium catalyst. A mechanistic investigation suggests that C–H activation is the rate-determining step. Further transformations of the lactones have been carried out to showcase the applicability of the present strategy.
Introduction
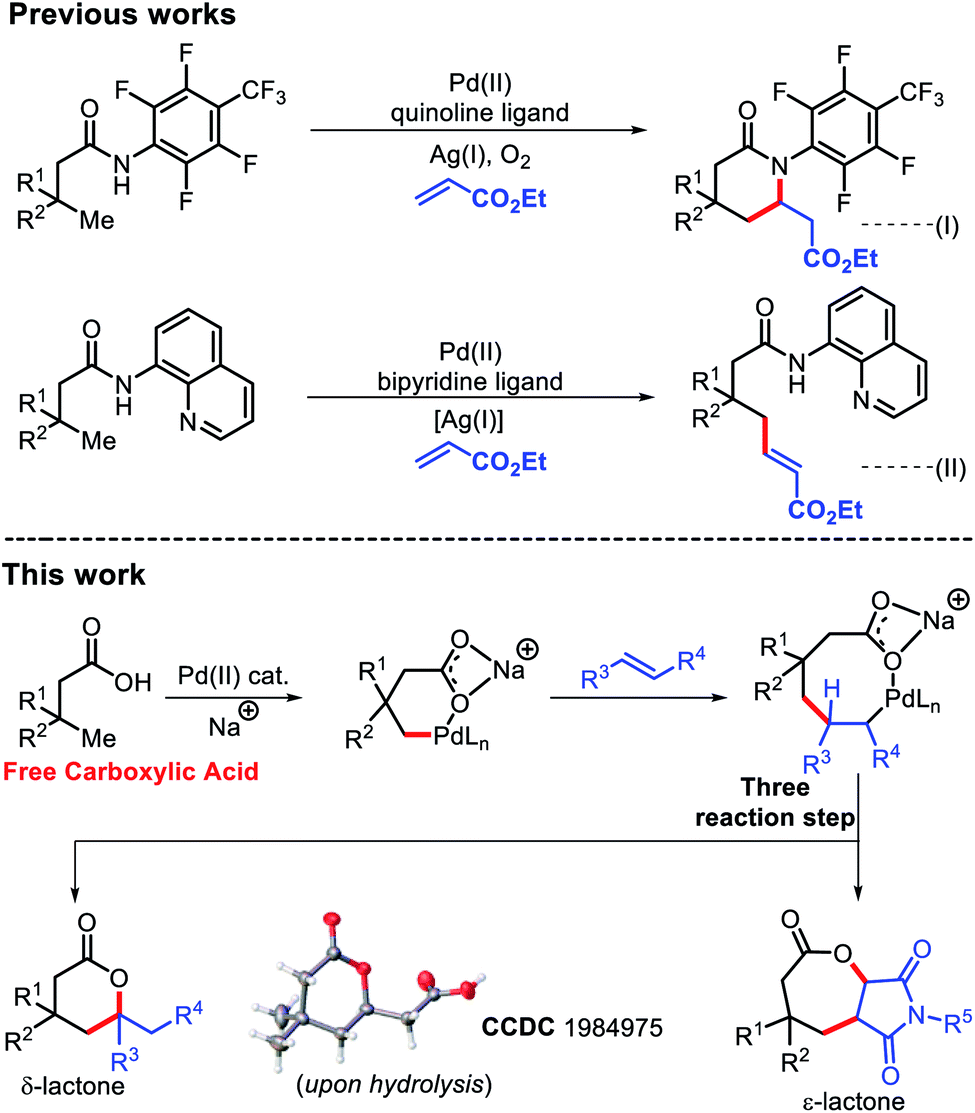
Lactones are an integral part of biological systems as well as industrially important moieties.1 Lactone motifs are abundantly present as small molecule signaling agents in different organisms.2 In particular, six and seven membered lactones are flavor and aroma constituents in many natural products.3 The wide availability and functional group interconversion ability of carboxylic acids makes them an excellent contender to synthesize lactones.4Transition metal catalyzed C–H bond activation has evolved as one of the most efficient tools to transform readily available materials into value added commodity chemicals.5 The last few decades have witnessed tremendous growth in the use of aromatic carboxylic acids as a substrate in C–H functionalization reactions.6 A similar success story has also been witnessed for aliphatic carboxylic acids with assistance from an exogenous template.7 But use of external templates decreases the efficiency of these protocols in terms of step as well as atom economy. Although, a few reports of directing group free reactions of aliphatic carboxylic acids are known, most are restricted to the proximal β position.8 This is due to formation of a favorable five membered metallacycle during β-C(sp3)–H activation. Moving towards further remote positions would require formation of less preferred metallacycles with the assistance from weakly coordinating carboxylates, which presents a significant challenge. Very recently, we have demonstrated the arylation reaction on such a free aliphatic carboxylic acid at the remote γ-position.9 Shi and coworkers have also contributed immensely in this area of aliphatic C–H functionalization.10
The Yu group reported β-olefination of aliphatic acids8d and γ-olefination using a highly acidic N-aryl amide auxiliary.7b However, this γ-olefination requires preinstallation of an expensive N-aryl amine template and removal of the auxiliary post functionalization, which limits its synthetic utility. In 2017, we faced the same problem while using an 8-amino quinoline template for γ-C(sp3)–H olefination.7f
We, therefore, strived to develop a methodology which can promote olefination without using any exogenous auxiliary. A γ-C(sp3)–H olefination of free aliphatic acids enabled by an acetyl protected amino acid ligand has been developed here (Fig. 1). δ-Lactones were formed as the products of γ-C(sp3)–H olefination via subsequent C–H activation/C–O cyclization. Considering the challenges in developing Heck coupling reactions with alkyl halides,11 direct olefination with readily available carboxylic acids would be highly useful. While we were preparing this manuscript, the Gemmeren12 and Yu13 groups reported similar approaches.
 | ||
| Fig. 1 γ-C(sp3)–H olefination of a carboxylic acid derivative. | ||
Results and discussion
We started to inspect the γ-C(sp3)–H olefination reaction with 3,3-dimethyl butanoic acid as the model substrate and benzyl acrylate as the olefin partner. An inorganic counter cation such as an alkali metal (Na+, K+, Cs+etc.) plays a pivotal role in transformation of carboxylic acid containing substrates. Such counter cations facilitate C–H activation by binding with a carboxylate in a κ2 coordination mode and thus Pd gets the opportunity to activate the accessible C–H bonds. A combination of Pd(OAc)2 (10 mol%), 2-pyridone (20 mol%), Ag2CO3 (2 equiv.), and Na2CO3 (1 equiv.) in the presence of 3,3-dimethyl butanoic acid and benzyl acrylate provided 28% isolated yield of 1a (Fig. 2).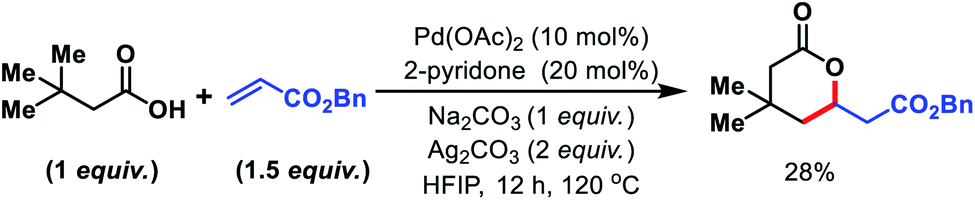 | ||
| Fig. 2 Initial reaction conditions. | ||
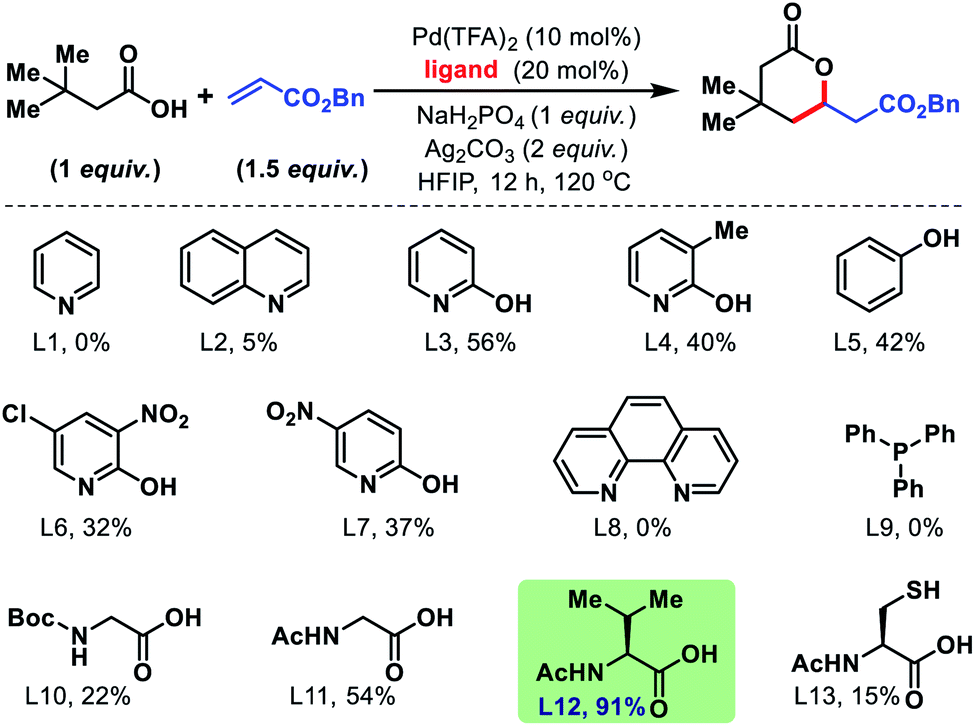
Encouraged by this outcome, we started optimization by varying different Pd salts. Among them Pd(TFA)2 was found to be the best catalyst (see the ESI†). To increase the efficiency of the reaction, we turned our attention towards finding a suitable ligand. We initially investigated pyridine and quinoline based ligands, but found them to be ineffective. Although pyridone ligands were superior, yields were not synthetically useful (Fig. 3).
 | ||
| Fig. 3 Ligand optimization. | ||
Employment of phenanthroline and phosphine-based ligands also remained ineffective. We then moved to employ mono-protected amino acids as ligands for the reaction. Although, Boc and Cbz protected amino acids did not show much promise, acetyl protected amino acids increased the yields significantly. Further studies with various acetyl protected amino acids revealed N-acetyl valine (L12) to be the best ligand with yield up to 91% (NMR yield, Fig. 3). We found an alkali metal base to be indispensable for the reaction. Na2HPO4 was found to be the suitable base and Ag2CO3 (2 equiv.) is the superior oxidant for the reaction. An extensive assessment of the reaction parameters revealed the optimum conditions, which are to treat 3,3-dimethyl butanoic acid (1 equiv.) with benzyl acrylate (1.5 equiv.) in the presence of Pd(TFA)2 (10 mol%), N-Ac-Val (20 mol%), Ag2CO3 (2 equiv.), and Na2HPO4 (1 equiv.) in HFIP solvent for 12 h at 120 °C to obtain 1a in 91% yield.
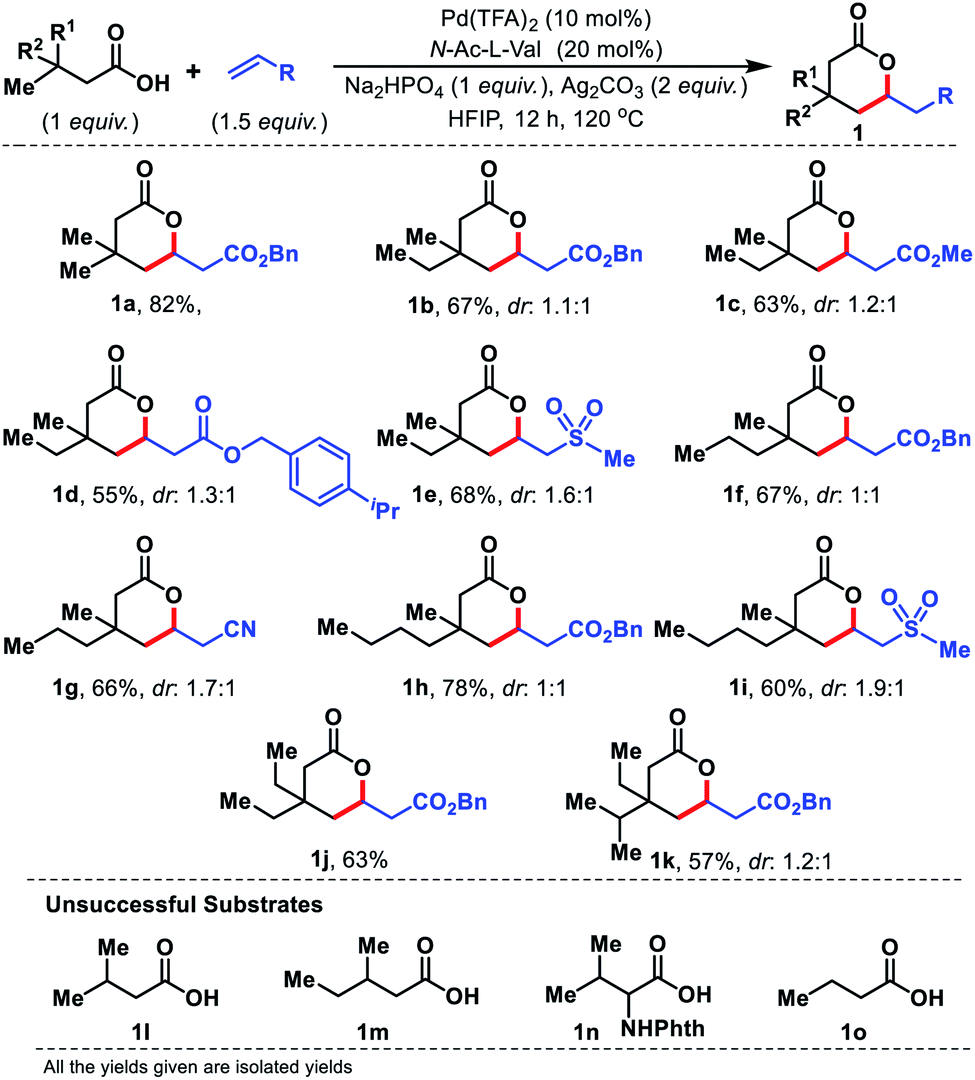
Having the optimal conditions for γ-C(sp3)–H olefination to form six membered lactones, we first explored the scope of aliphatic acids. To our delight, even when we performed the reaction in the presence of ethyl (1b–1e), propyl (1f and 1g), and butyl (1h and 1i) substituents at the β-position of the substrate, the reaction worked efficiently (Fig. 4). Carboxylic acids containing β-diethyl (1j) and ethyl-isopropyl (1k) substituents were also well compatible. Such substrates work well even when we changed the coupling partner from acrylates to sulphones or acrylonitrile. The β-quaternary center of the acid is necessary for this reaction as it provides a suitable geometrical alignment (via the Thorpe–Ingold effect) to form a thermodynamically less stable six membered palladacycle. Acids such as 1l–1o were unsuccessful under the present reaction conditions.
 | ||
| Fig. 4 Diversification of free carboxylic acids. | ||
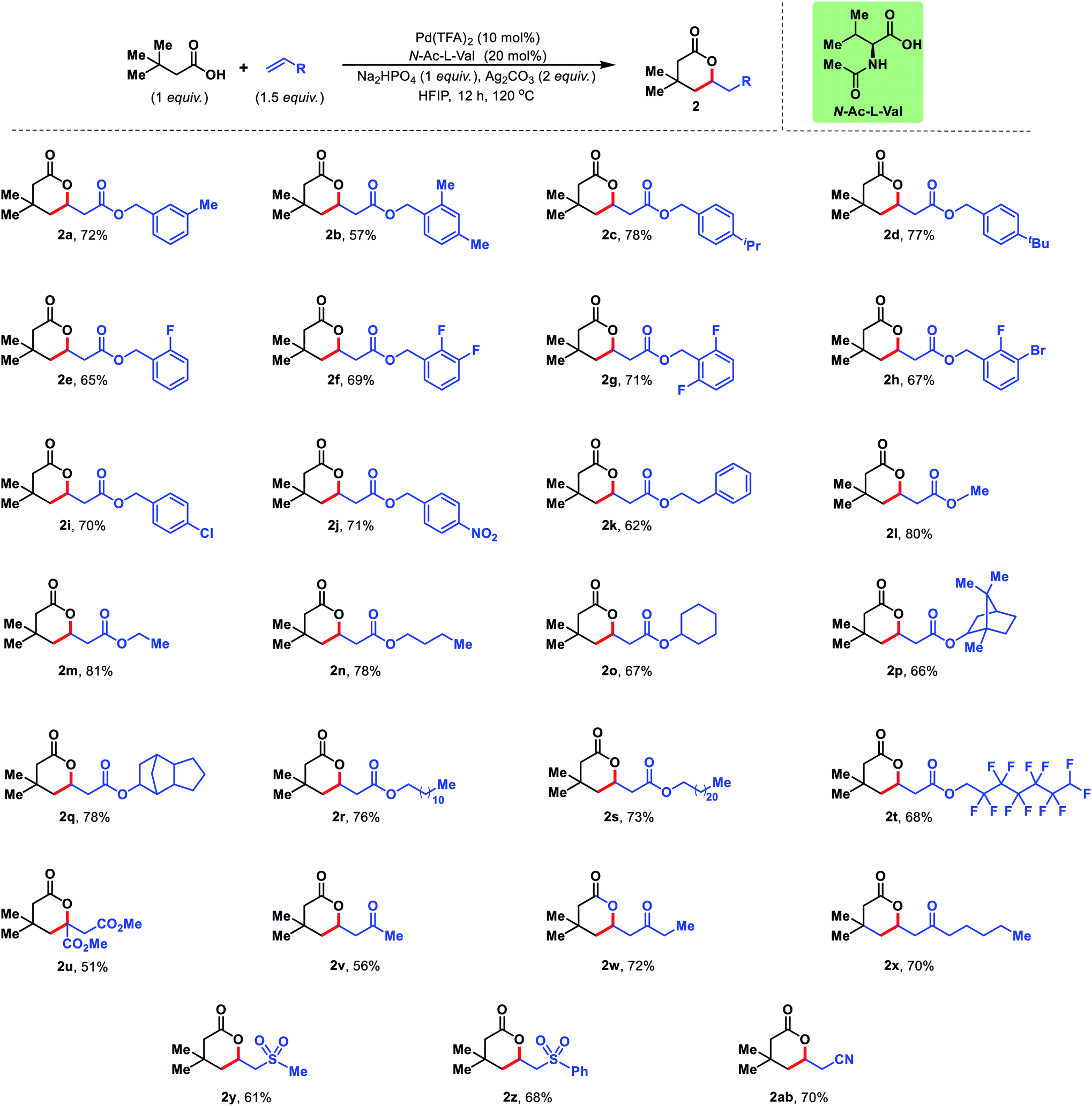
Next, we diversified the scope of olefin coupling partners. Various substituted benzyl acrylates were explored. Diverse electronic substitutions on the phenyl ring of benzyl acrylate did not affect the efficiency of the protocol (2a–2j). A phenethyl acrylate has also been utilized as the olefin coupling partner for γ-C(sp3)–H olefination (2k). Other aliphatic acrylates starting from simple methyl acrylate to bicyclic acrylate provided excellent yields under the reaction conditions (2l–2q). Long chain acrylates like dodecyl, dodecafluoro acrylate and docosyl acrylate were also found to be suitable coupling partners for this reaction (2r–2t) (Fig. 5).
 | ||
| Fig. 5 Scope of various olefins. | ||
The present strategy is not limited to esters of acrylic acid but various activated olefins can also be utilized in the reaction. Olefins containing sulphone, nitrile, ketone and diester groups were well compatible with the reaction conditions and provided corresponding δ-lactones in good to excellent yields (2u–2ab) (Fig. 5).
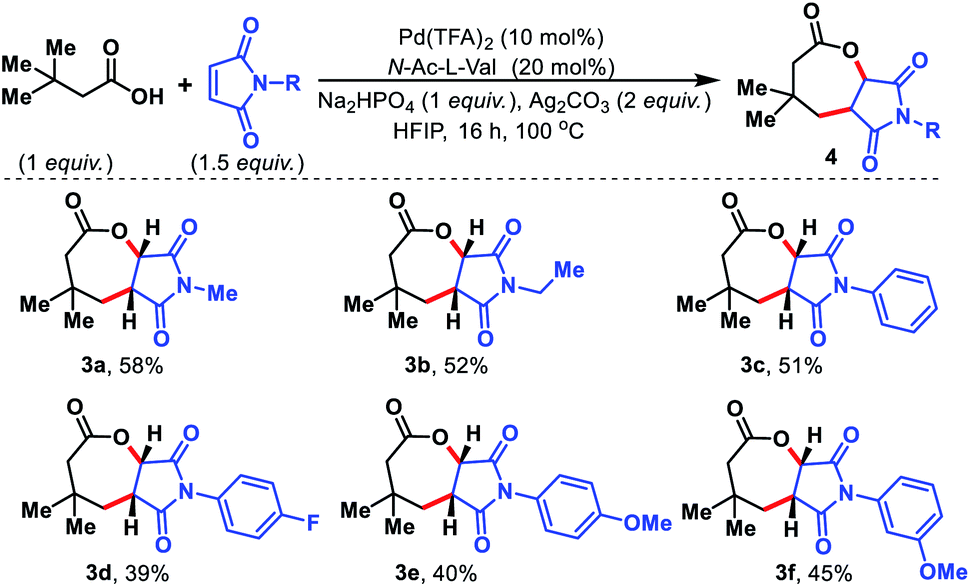
Maleimides are known in the literature as an excellent olefin coupling partner.14 Various N-substituted maleimides possess biological activities, especially antifungal properties15 which make them a valuable coupling partner in the reaction. We sought to utilize maleimides as the olefin partner in our protocol. To our surprise, such maleimides provided fused seven membered lactones under the present reaction conditions. Initially, we started to explore the seven membered lactone with N-alkyl maleimides (Fig. 6, 3a and 3b). To evaluate further, various N-aryl substituted maleimides were tested and all of them provided seven membered lactones (3c–3f). The fused seven membered lactones obtained are prevalent structural motifs in bioactive molecules. Moreover, synthesis of such seven membered lactones via γ-C(sp3)–H activation provides a straightforward and effective route compared to traditional methods of synthesizing similar molecules.
 | ||
| Fig. 6 Scope of maleimides. | ||
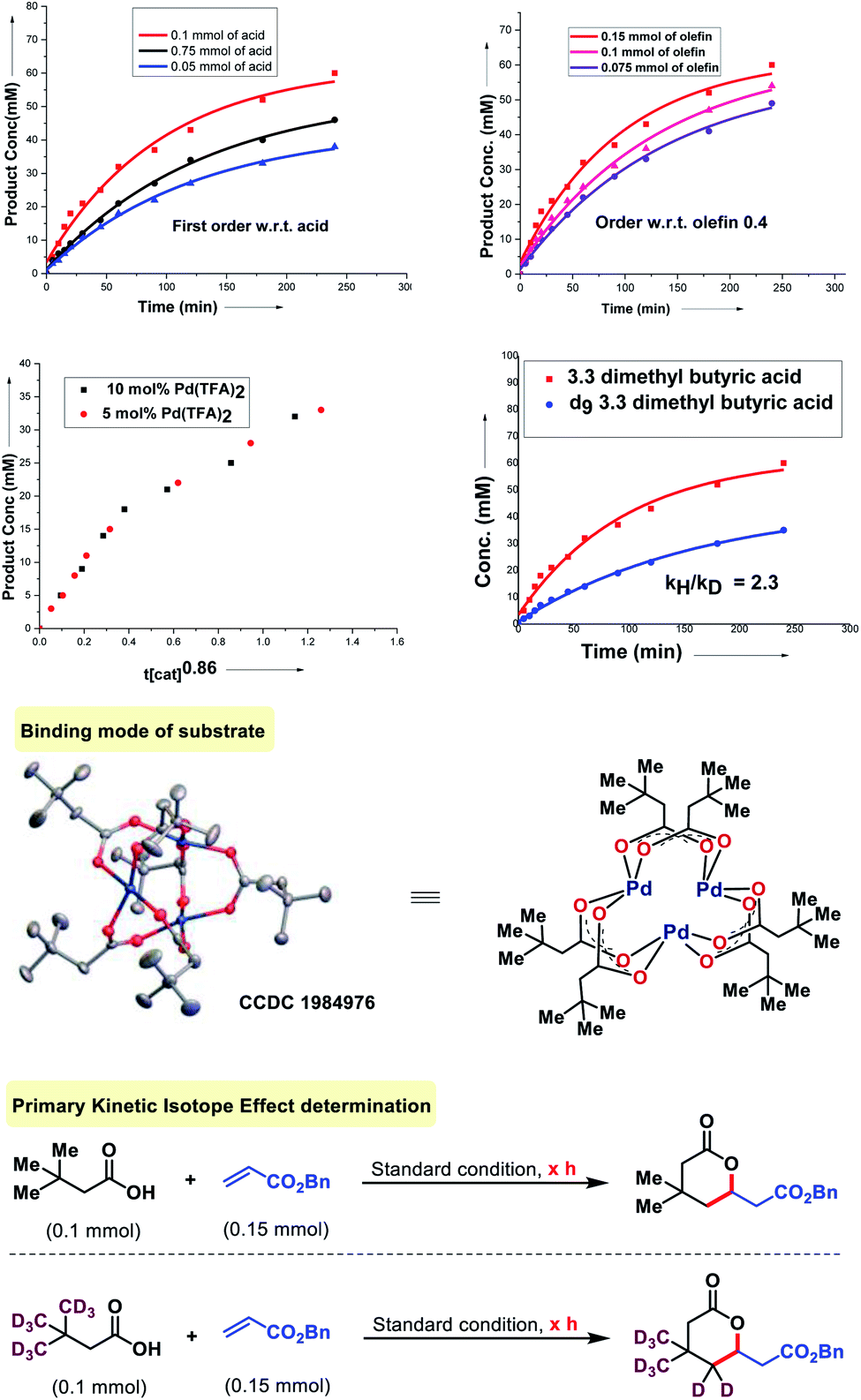
To probe the mechanism of the olefination with aliphatic acids, we first tested the relevance of the substrate-palladium species A (Fig. 7). We found that A is a catalytically and kinetically competent intermediate. A reversibility experiment of the substrate (3,3-dimethyl butanoic acid) in the presence of D2-HFIP showed no deuterium scrambling in the recovered carboxylic acid. To confirm further, a reversibility experiment was carried out with the deuterated substrate. No deuterium depletion was observed in the recovered substrate. This indicated the irreversible nature of the C–H activation step. The order with respect to the acid and olefin was studied. A first order dependency with respect to the acid and a 0.4 order dependency with respect to the olefin was determined which indicates that only the acid is likely to be involved in the rate determining step of the reaction. The order with respect to the catalyst was found to be nearly unit order (0.86, Fig. 7) which again indicates a similar interpretation.16 Further information was obtained by the measurement of the kinetic isotope effect (KIE). An apparent KIE value of 2.3 suggests that cleavage of the C–H bond is involved in the rate-determining step of the reaction.
 | ||
| Fig. 7 Mechanistic investigation. | ||
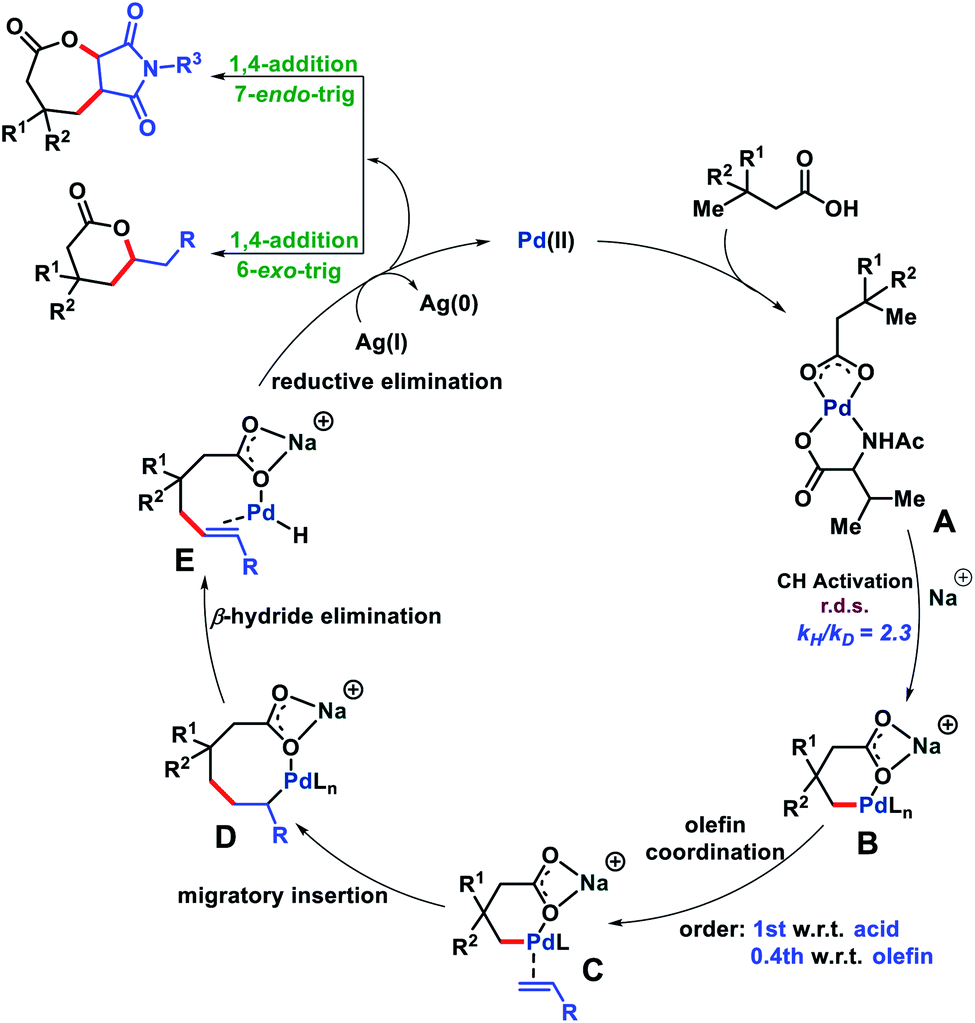
Based on the evidence we obtained from the aforementioned experiments and related literature reports,17 we propose a reaction mechanism in Fig. 8. The carboxylate group of the substrate first binds to the metal catalyst to form a metal coordinated complex A which upon treatment with an alkali metal base generates a γ-C–H activated six membered intermediate B. Next, olefin binding to B and 1,2-migratory insertion lead to an eight membered cyclo-palladated intermediate D. From D, β-hydride elimination affords E which upon reductive elimination followed by Michael addition forms a six or seven membered lactone (Baldwin's rules). The formed Pd(0) oxidized to Pd(II) aided by Ag(I) and completed the catalytic cycle. The reason for the formation of seven membered lactones we believe lies in the cyclization step after reductive elimination. The more substituted position of maleimide disfavors the cyclization and therefore, cyclization occurred at the less substituted side of maleimide to form the seven membered lactone.18
 | ||
| Fig. 8 Mechanistic cycle. | ||
The practicality of the reaction has been demonstrated by performing the reaction up to the gram scale (see the ESI†). The decrease in yield on a large scale is perhaps due to improper heat and mass transfer in a bigger flask. Homogeneity could be an additional issue here. To further harness the utility of the six membered lactones, it has been transformed into structurally different synthons. Reduction with LiAlH4 provided 1,5,7-triol (4e). Selective hydrolysis of the ester group with HCl generated another δ-lactone with a carboxylic group (4a). Upon hydrolysis with NaOH an unsaturated pimelic acid derivative was formed (4b, 4c), which was further reduced to its saturated analogue (4f) that is widely used for industrial application. The dicarboxylic acid (4f) has been cyclized to generate an eight membered anhydride upon treatment with SOCl2 (4g). In a similar fashion, a sulphone containing δ-lactone was hydrolysed to form an unsaturated carboxylic acid (4d) (Fig. 9).
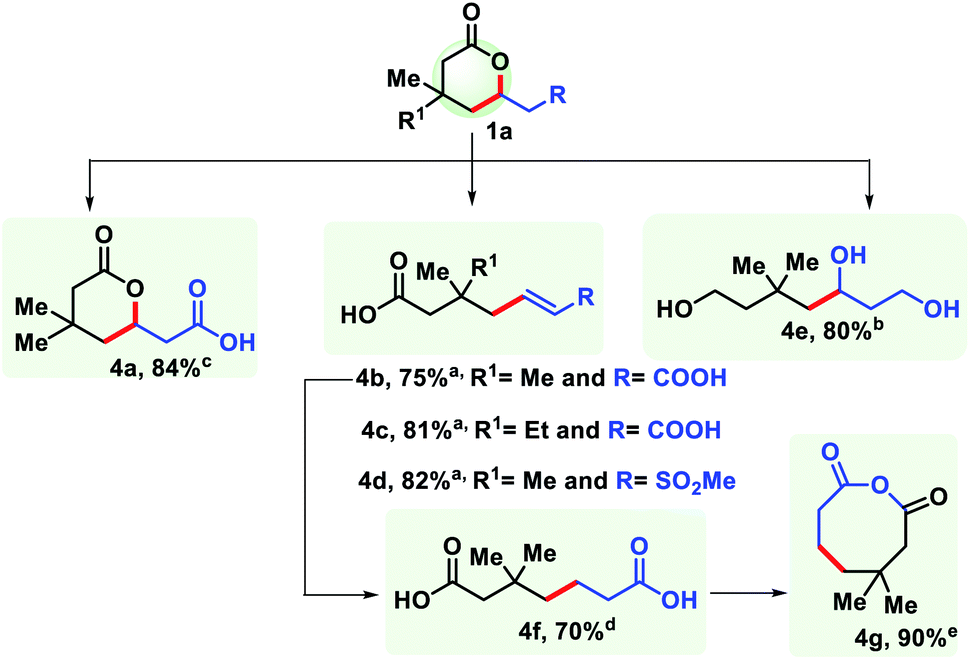 | ||
| Fig. 9 Side product formation and synthetic applications of δ-lactones. a Condition A: 1a (0.2 mmol), NaOH (4 equiv.), EtOH/H2O reflux, overnight. b Condition B: 1a (0.2 mmol), LiAlH4 (3 equiv.), THF, rt, 12 h. c Condition C: 1a (0.2 mmol), 6N HCl, 12 h, rt. d Condition D: 4b (0.1 mmol) Pd/C (10 mol%), H2, MeOH, rt, 12 h. e Condition E: 4f (0.1 mmol), SOCl2 (3 equiv.), 8 h, rt. | ||
Conclusion
In conclusion, a ligand enabled γ-C(sp3)–H olefination protocol has been developed for aliphatic free carboxylic acids. The potential of the methodology has been showcased via synthesis of δ-lactones that contain ester, ketone, sulphone, and nitrile groups. Various N-substituted maleimides have been utilized to form fused seven-membered lactones. A mechanistic investigation has been carried out to shed light on the reaction mechanism. Further, the application potential of the δ-lactones has been demonstrated through various transformations. Considering the severe challenges involved in developing analogous Heck coupling reactions, this transformation provides an exciting complementary pathway.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We thank SERB India (CRG/2018/003915) for financial support. Fellowships from CSIR India (JD, JP and GP) and UGC India (PD and CHB) are greatly acknowledged. We thank S. S. Anjana for her generous help in crystallography.Notes and references
- T. Janecki, Natural Lactones and Lactams: Synthesis, Occurrence and Biological Activity, John Wiley & Sons., 2013 Search PubMed.
- S. Schulz and S. Hötling, Nat. Prod. Rep., 2015, 32, 1042 RSC.
- M. Ebine, Y. Suga, H. Fuwa and M. Sasaki, Org. Biomol. Chem., 2010, 8, 39 RSC.
- L. J. Goossen, N. Rodriguez and K. Gooßen, Angew. Chem., Int. Ed., 2008, 47, 3100 CrossRef CAS PubMed.
- (a) Z. Chen, B. Wang, J. Zhang, W. Yu, Z. Liu and Y. Zhang, Org. Chem. Front., 2015, 2, 1107 RSC; (b) T. Gensch, M. Hopkinson, F. Glorius and J. Wencel-Delord, Chem. Soc. Rev., 2016, 45, 2900 RSC; (c) C. Sambiagio, D. Schönbauer, R. Blieck, T. Dao-Huy, G. Pototschnig, P. Schaaf, T. Wiesinger, M. F. Zia, J. Wencel-Delord and T. Besset, Chem. Soc. Rev., 2018, 47, 6603 RSC.
- (a) R. Giri and J.-Q. Yu, J. Am. Chem. Soc., 2008, 130, 14082 CrossRef CAS PubMed; (b) L. Ackermann, Chem. Rev., 2011, 111, 1315 CrossRef CAS PubMed; (c) K. M. Engle, T.-S. Mei, M. Wasa and J.-Q. Yu, Acc. Chem. Res., 2012, 45, 788 CrossRef CAS PubMed; (d) M. Pichette Drapeau and L. J. Gooßen, Chem.–Eur. J., 2016, 22, 18654 CrossRef CAS PubMed.
- (a) J. He, M. Wasa, K. S. L. Chen, Q. Shao and J.-Q. Yu, Chem. Rev., 2017, 117, 8754 CrossRef CAS PubMed; (b) S. Li, G. Chen, C.-G. Feng, W. Gong and J.-Q. Yu, J. Am. Chem. Soc., 2014, 136, 5267 CrossRef CAS PubMed; (c) G. Liao, X.-S. Yin, K. Chen, Q. Zhang, S.-Q. Zhang and B.-F. Shi, Nat. Commun., 2016, 7, 12901 CrossRef PubMed; (d) R.-Y. Zhu, L.-Y. Liu and J.-Q. Yu, J. Am. Chem. Soc., 2017, 139, 12394 CrossRef CAS PubMed; (e) A. Dey, S. Pimparkar, A. Deb, S. Guin and D. Maiti, Adv. Synth. Catal., 2017, 359, 1301 CrossRef CAS; (f) N. Thrimurtulu, S. Khan, S. Maity, C. M. Volla and D. Maiti, Chem. Commun., 2017, 53, 12457 RSC; (g) A. Deb, S. Singh, K. Seth, S. Pimparkar, B. Bhaskararao, S. Guin, R. B. Sunoj and D. Maiti, ACS Catal., 2017, 7, 8171 CrossRef CAS; (h) S. Guin, A. Deb, P. Dolui, S. Chakraborty, V. K. Singh and D. Maiti, ACS Catal., 2018, 8, 2664 CrossRef CAS; (i) L. Jin, J. Wang and G. Dong, Angew. Chem., Int. Ed., 2018, 57, 12352 CrossRef CAS PubMed; (j) B. Li, B. Lawrence, G. Li and H. Ge, Angew. Chem., Int. Ed., 2019, 59, 3078 CrossRef PubMed.
- (a) R. Giri, N. Maugel, J.-J. Li, D.-H. Wang, S. P. Breazzano, L. B. Saunders and J.-Q. Yu, J. Am. Chem. Soc., 2007, 129, 3510 CrossRef CAS PubMed; (b) K. K. Ghosh and M. van Gemmeren, Chem.–Eur. J., 2017, 23, 17697 CrossRef CAS PubMed; (c) Y. Zhu, X. Chen, C. Yuan, G. Li, J. Zhang and Y. Zhao, Nat. Commun., 2017, 8, 14904 CrossRef CAS PubMed; (d) Z. Zhuang, C.-B. Yu, G. Chen, Q.-F. Wu, Y. Hsiao, C. L. Joe, J. X. Qiao, M. A. Poss and J.-Q. Yu, J. Am. Chem. Soc., 2018, 140, 10363 CrossRef CAS PubMed; (e) K. K. Ghosh, A. Uttry, A. Koldemir, M. Ong and M. van Gemmeren, Org. Lett., 2019, 21, 7154 CrossRef CAS PubMed; (f) Z.-T. He and J. F. Hartwig, J. Am. Chem. Soc., 2019, 141, 11749 CrossRef CAS PubMed; (g) Z. Zhuang and J.-Q. Yu, Nature, 2020, 577, 656 CrossRef CAS PubMed.
- P. Dolui, J. Das, H. B. Chandrashekar, S. Anjana and D. Maiti, Angew. Chem., Int. Ed., 2019, 58, 13773 CrossRef CAS PubMed.
- L. Liu, Y.-H. Liu and B.-F. Shi, Chem. Sci., 2020, 11, 290 RSC.
- (a) C. M. McMahon and E. J. Alexanian, Angew. Chem., Int. Ed., 2014, 53, 5974 CrossRef CAS PubMed; (b) D. Kurandina, M. Parasram and V. Gevorgyan, Angew. Chem., Int. Ed., 2017, 56, 14212 CrossRef CAS PubMed; (c) G.-Z. Wang, R. Shang, W.-M. Cheng and Y. Fu, J. Am. Chem. Soc., 2017, 139, 18307 CrossRef CAS PubMed.
- For related recent elegant approach of δ-lactone synthesis, see K. K. Ghosh, A. Uttry, A. Mondal, F. Ghiringhelli and M. van Gemmeren, Angew. Chem., Int. Ed., 2020, 59, 12848 CrossRef CAS PubMed . While our manuscript was under review, this seminal contribution on ligand-enabled γ-C(sp3)–H olefination of free carboxylic acids has been reported.
- H. S. Park, Z. Fan, R. Y. Zhu and J. Q. Yu, Angew. Chem., Int. Ed., 2020, 59, 12853 CrossRef CAS PubMed.
- (a) B. Ramesh, M. Tamizmani and M. Jeganmohan, J. Org. Chem., 2019, 84, 4058 CrossRef CAS PubMed; (b) S. Jambu, R. Sivasakthikumaran and M. Jeganmohan, Org. Lett., 2019, 21, 1320 CrossRef CAS PubMed.
- (a) X. L. Chen, L. J. Zhang, F. G. Li, Y. X. Fan, W. P. Wang, B. J. Li and Y. C. Shen, Pest Manage. Sci., 2015, 71, 433 CrossRef CAS PubMed; (b) M. Asif, Acta Sci. Pharm. Sci., 2019, 3, 51 Search PubMed.
- J. Burés, Angew. Chem., Int. Ed., 2016, 55, 2028 CrossRef PubMed.
- A. Deb, A. Hazra, Q. Peng, R. S. Paton and D. Maiti, J. Am. Chem. Soc., 2017, 139, 763 CrossRef CAS PubMed.
- A. L. J. Beckwith, C. J. Easton and A. K. Serelis, J. Chem. Soc., Chem. Commun., 1980, 482 RSC.
Footnotes |
| † Electronic supplementary information (ESI) available. CCDC 1984975 and 1984976. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/d0sc03144e |
| ‡ These authors contributed equally. |
| This journal is © The Royal Society of Chemistry 2020 |