
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceRational design of a “dual lock-and-key” supramolecular photosensitizer based on aromatic nucleophilic substitution for specific and enhanced photodynamic therapy†
Kun-Xu
Teng
,
Li-Ya
Niu
 *,
Yan-Fei
Kang
and
Qing-Zheng
Yang
*
*,
Yan-Fei
Kang
and
Qing-Zheng
Yang
*
Key Laboratory of Radiopharmaceuticals, Ministry of Education, College of Chemistry, Beijing Normal University, Beijing 100875, P. R. China. E-mail: niuly@bnu.edu.cn; qzyang@bnu.edu.cn
First published on 25th August 2020
Abstract
Photosensitizing agents are essential for precise and efficient photodynamic therapy (PDT). However, most of the conventional photosensitizers still suffer from limitations such as aggregation-caused quenching (ACQ) in physiological environments and toxic side-effects on normal tissues during treatment, leading to reduced therapeutic efficacy. Thus, integrating excellent photophysical properties and accurate carcinoma selectivity in a photosensitizer system remains highly desired. Herein, a “dual lock-and-key” supramolecular photosensitizer BIBCl–PAE NPs for specific and enhanced cancer therapy is reported. BIBCl–PAE NPs are constructed by encapsulating a rationally designed glutathione (GSH)-activatable photosensitizer BIBCl in a pH-responsive diblock copolymer. In normal tissues, BIBCl is “locked” in the hydrophobic core of the polymeric micelles due to ACQ. Under the “dual key” activation of low pH and high levels of GSH in a tumor microenvironment, the disassembly of micelles facilitates the reaction of BIBCl with GSH to release water-soluble BIBSG with ideal biocompatibility, enabling the highly efficient PDT. Moreover, benefiting from the Förster resonance energy transfer effect of BIBSG, improved light harvesting ability and 1O2 production are achieved. In vitro and vivo experiments have demonstrated that BIBCl–PAE NPs are effective in targeting and inhibiting carcinoma. BIBCl–PAE NPs show superior anticancer efficiency relative to non-activatable controls.
Introduction
Photodynamic therapy (PDT) is a promising local tumor treatment method combining photosensitizers (PSs), oxygen molecules and light.1 In comparison with conventional cancer therapy methods, it possesses several unique advantages including minimal invasion, fewer side effects, low toxicity, negligible drug resistance, and great spatiotemporal selectivity.2–4 Undoubtedly, PSs are essential for valid PDT,5 and researchers have been devoted to developing PSs with high performance in recent years, such as porphyrins,6–8 cyanines9,10 and BODIPY derivatives.11–14 Nonetheless, most traditional PSs still suffer from intrinsic drawbacks,15 especially common organic PSs have a strong tendency to aggregate by stacking in water, resulting in quenching of the electronic excited state and significantly reducing the quantum yield of 1O2.16 Thus, aggregation-caused quenching (ACQ) of PSs is an urgent problem to be solved. To suppress ACQ, several strategies have been proposed to design novel PSs. For example, Chen et al. fabricated a supramolecular PS from porphyrin derivatives with cucurbit[7]uril based on host–guest interactions, which enhanced the efficiency of the supramolecular PS to generate 1O2 attributed to the suppressed self-quenching of the excited state of porphyrins.17 Jin and co-workers designed an alternating copolymer, inhibiting the aggregation of porphyrins to enhance the efficacy of PDT.18 PSs with aggregation-induced emission (AIE) characteristics have received much attention in recent years.19,20 Tang and Liu have developed a series of PSs with AIE properties for PDT.21,22 Although these strategies turned “always-off” PSs into “always-on” ones, they still suffer from nonspecific phototoxicity and side effects on normal tissues.23,24In order to realize precise PDT, instead of eradicating self-quenching, ACQ can be taken advantage of as a “lock” to switch off the photoactivity in noncancerous tissues, turning waste into treasure.25,26 By the “key” stimulation of cancer-specific microenvironments or biomarkers, for example, low pH,27,28 hypoxia,29,30 or high levels of glutathione (GSH),31,32 ROS,33 and proteases,34,35 PSs are activated to generate 1O2 under irradiation, and thereby to realize tumor-specific PDT.36 However, single-factor activatable PSs are still unsatisfactory in terms of precise control of 1O2 release, because they may potentially pose the issue of nonspecific activation and even cause “false positive” results in the complex environment in vivo.37–40 Therefore, to further improve the accuracy of phototherapy, smart PSs that can be activated by the coexistence of multiple stimuli are highly desired to provide robust and precise specificity for therapeutic treatments.
Herein, we report a dual-stimuli cooperatively activatable supramolecular photosensitizer BIBCl–PAE NPs using the “dual lock-and-key” strategy for high-specificity and enhanced photodynamic therapy (Scheme 1). We encapsulate a rationally designed GSH-activatable photosensitizer BIBCl (first lock-and-key) in a pH-responsive diblock copolymer poly(ethylene glycol)–poly(β-amino ester) (PEG–PAE) to form BIBCl–PAE NPs (second lock-and-key).41,42 In blood and normal tissues with neutral pH and low GSH concentrations, the hydrophobic nature of BIBCl and amphiphilic polymer PEG–PAE act as “double locks” to lock the PS in a tightly aggregated state, and cannot sensitize oxygen to 1O2 due to ACQ. In a tumor microenvironment, the low pH (pH = 5.0–6.8) and high level of GSH (1–15 mM) function as “dual keys” to unlock the ACQ state of PSs. Protonation of PEG–PAE promotes the reaction of BIBCl with GSH to afford water-soluble BIBSG with high PDT performance. Moreover, benefiting from the Förster resonance energy transfer (FRET) effect, BIBSG showed improved light-harvesting ability and 1O2 production.43 These merits endow this intelligent supramolecular photosensitizer with a significantly specific and enhanced phototherapeutic response both in vitro and in vivo.
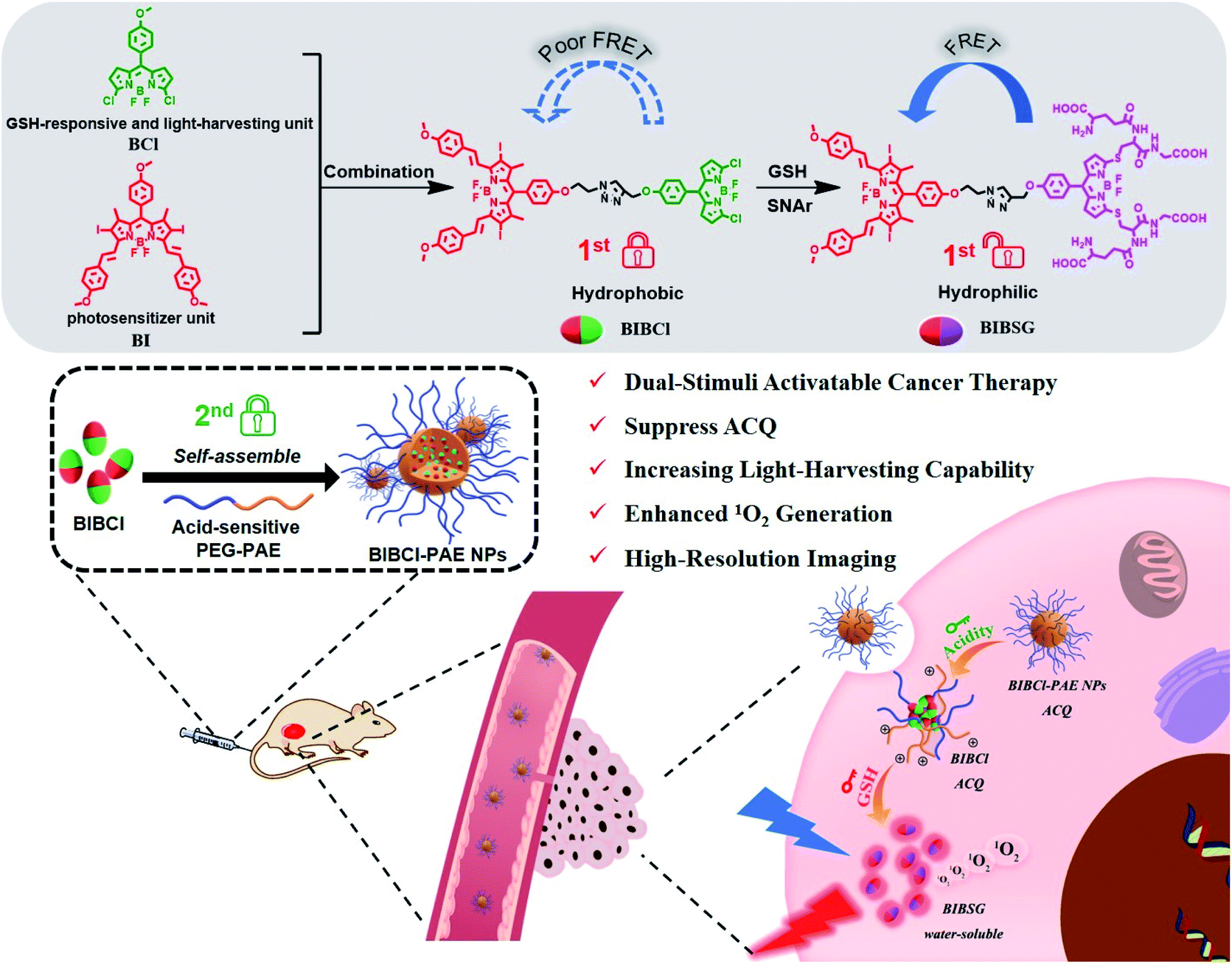 | ||
| Scheme 1 Schematic illustration for the fabrication of BIBCl–PAE NPs, and the processes of the “dual lock-and-key” strategy in a tumor microenvironment to achieve activated and enhanced generation of 1O2. | ||
Results and discussion
Design principles of the PS molecule and the supramolecular PS
We designed BIBCl as a new PS that consists of two moieties, an iodine-substituted BODIPY (BI) unit and chlorine-substituted BODIPY (BCl) unit. Specifically, the BI unit contains heavy atomic iodine, which can generate 1O2 through intersystem crossing (ISC) due to the heavy atom effects and energy transfer with oxygen.44 On the other hand, the BCl unit is designed as both a GSH-responsive moiety and a light-harvesting unit. According to previous reports of our group, the chlorine of the BCl moiety can be substituted with GSH to form a hydrophilic molecule.45,46 The resulting hydrophilic BIBSG is expected to avoid the ACQ problem of conventional PSs. In addition, the GSH substitution is accompanied by a 70 nm bathochromic-shift in the fluorescence spectrum, which results in a good overlap with the absorption spectrum of BI to enable the efficient FRET (Fig. S2 and S3†). Therefore, BIBSG will exhibit a broad absorption peak with strong light-harvesting ability and intensified 1O2 production. It is worth mentioning that it is the first example of a GSH-activated PS based on aromatic nucleophilic substitution. This new strategy not only markedly improves the water solubility, but also modulates the photophysical properties of the PS, showing distinguished advantages compared to the most commonly used cleavage of S–S bonds.47–49In order to further improve the precise capability of cancer treatments, PSs that can be activated by the coexistence of multiple cancer-associated stimulations are highly desired. The diblock copolymer PEG–PAE is a pH-responsive surfactant. And it forms self-assembling polymeric micelles in neutral or basic aqueous solutions (pH ≥ 7.0) and disassembles in an acidic environment (PH < 6.8).50 At pH 7.4, the critical micelle concentration (CMC) of PEG5000–PAE10000 is determined to be 80.2 μg mL−1 (Fig. S4†). BIBCl is encapsulated in the hydrophobic core of PEG–PAE polymeric micelles to form nanoparticles by hydrophobic interaction between the BIBCl and PAE segment in a neutral aqueous solution. The aggregated BIBCl results in quenching of the excited state with no PDT effect. Upon accumulation due to the enhanced permeability and retention (EPR) effect in a tumor microenvironment with weakly acidic pH and high levels of GSH, PEG–PAE will translate into hydrophilic molecules and disassemble, and the released BIBCl will react with GSH to produce water-soluble BIBSG with the PDT effect. Such nanoparticles represent a dual-activatable PDT with high specificity to tumors. By contrast, the nanoparticles remain in a tightly aggregated state in blood and normal tissues on account of their neutral pH and low GSH concentrations, and cannot sensitize oxygen to 1O2 due to ACQ.
BIBCl was synthesized (ESI, Schemes S1–S3†) and fully characterized by 1H NMR, 13C NMR and HRMS. BIBCl was encapsulated within biodegradable copolymer PEG5000–PAE10000 by a single-step sonication method (Scheme S5†) to form water-dispersible nanoparticles (BIBCl–PAE NPs). We also synthesized an analogue BIBH without GSH-activation capability as a control photosensitizer and prepared control nanoparticles BIBCl–PCL NPs encapsulated using pH-insensitive PCL10000–PEG5000, BIBH–PAE NPs and BIBH–PCL NPs (Schemes S4, S5 and Fig. S1†).
Morphological and spectroscopic properties
The supramolecular photosensitizer BIBCl–PAE NPs was characterized as well-dispersed nanoparticles with a size distribution of 85 ± 11.0 nm by dynamic light scattering (DLS) and transmission electron microscopy (TEM) (Fig. 1a). These size distributions are in the size range suitable for the EPR effect, facilitating enrichment within tumor tissue. The hydrodynamic size of BIBCl–PAE NPs in PBS remained almost constant for 15 days (Fig. S5a†). Moreover, the zeta potential value of BIBCl–PAE NPs was determined to be ∼−89.0 mV, which provided further evidence for the high stability of nanoparticles (Fig. S6†). The absorption of BIBCl–PAE NPs is constant for 48 h in fetal calf serum, indicating that BIBCl–PAE NPs are remarkably stable under physiological conditions (Fig. S5b†). In addition, BIBCl–PAE NPs and BIBSG retained high absorbance (>90%) after 60 minutes of irradiation (Fig. S5c and d†), indicating their high photostability. | ||
Fig. 1 (a) DLS profiles of BIBCl–PAE NPs and TEM images (inset). (b) Time-dependent absorption spectra of BIBCl–PAE NPs (20 μM) in the presence of 50 equiv of GSH at pH 6.5. (c) Normalized absorption spectra of BIBCl–PAE NPs after 6 h under different conditions. (d) Centrifugation of BIBCl–PAE NPs after 6 h under different conditions (centrifugation at 12![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 rpm for 30 minutes). (e) Emission spectra of BIBSG excited at 580 nm and 670 nm. (f) Fluorescence decay curves of BSG and BIBSG, and the detection wavelength is 600 nm in DMSO. 000 rpm for 30 minutes). (e) Emission spectra of BIBSG excited at 580 nm and 670 nm. (f) Fluorescence decay curves of BSG and BIBSG, and the detection wavelength is 600 nm in DMSO. | ||
The time-dependent absorption response of BIBCl–PAE NPs (equivalent concentration of: 20.0 μM) in the presence of GSH (1.0 mM) at pH 6.5 was measured at 37 °C to simulate the tumor microenvironment (Fig. 1b). As the reaction progressed, the intensity of the original absorption band of the BCl unit centered at 510 nm decreased, while the intensity of two new absorption peaks at 543 nm and 580 nm increased, which are assigned to the mono- and di-substituted products of the BSG unit. The absorption peaks at 665 nm of the BI moiety showed a slight hypsochromic shift, suggesting the formation of water-soluble BIBSG is prone to disaggregate into free molecules. The final product was characterized by HRMS (Fig. S7†). The HRMS manifested a peak at 1830.3155, which was assigned to [BIBSG + H]+. This disassembly process was further confirmed by centrifugation experiments (Fig. 1d). After the centrifugation of the reaction mixture of BIBCl–PAE NPs and GSH at pH 6.5, the color of the solution remains blue-black, and no precipitate was observed. The absorption spectrum was overlapped with that of the free BIBSG in DMSO, indicating that water-soluble BIBSG was formed in the solution (Fig. S8c†). Moreover, after the reaction, BIBSG shows a broader absorption peak that suggests a stronger light-harvesting ability compared to that of BI only. Upon 580 nm excitation, the fluorescence of BSG at 600 nm was completely quenched, and the fluorescence of BI at 720 nm was observed (Fig. 1e), proving that efficient FRET occurred in BIBSG. In addition, we further measured the fluorescence lifetime of BSG and BIBSG. As shown in Fig. 1f, BIBSG presented a shorter fluorescence lifetime (1.80 ns) than BSG (4.97 ns), which demonstrated an efficient energy transfer from the BSG moiety to the BI moiety in BIBSG.
The “dual-lock” strategy was further confirmed by the control experiments (Fig. 1c and d). BIBCl–PAE NPs were centrifuged after the treatment with either high GSH concentrations or acidic solutions individually, and the solution changed from green to colourless and transparent. In the absence of GSH or weakly acidic conditions, negligible change was observed in the absorption spectra of BIBCl–PAE NPs under the identical conditions. These results indicate that the “single-key” is not able to release the photosensitizer out and ensure that PSs don't show photoactivity in blood or normal cells.
1O2 generation ability
The 1O2 generation of the supramolecular PSs was measured with 9,10-anthracenediyl-bis(methylene)-dimalonic acid (ABDA) as the singlet oxygen scavenger. The 1O2 measurement mechanism of ABDA is illustrated in Fig. S9.† To determine whether the GSH facilitates enhancement of 1O2 generation of BIBCl–PAE NPs in acidic environments, we separately studied the 1O2 generation ability with or without GSH, and we also tested commercial PS Ce6 as a comparison. When BIBCl–PAE NPs were exposed to white LED light irradiation (400–800 nm, 10 mW cm−2), negligible change of the absorbance of ABDA was observed (Fig. 2b), indicating inefficient sensitization of oxygen due to self-quenching of the excited state of BIBCl caused by aggregation of the PS. By contrast, after reaction with GSH at pH 6.5 to yield BIBSG (Fig. 2a), the ABDA absorbance rapidly decreased (Fig. 2c), enabling the GSH/pH activated 1O2 generation. The 1O2 yield of BIBSG increased by a factor of 10-fold higher than that of BIBCl–PAE NPs without GSH (Fig. 2d). The 1O2 generation capacity of BIBSG is even better than that of commercial PS Ce6 (Fig. S10†). The 1O2 yield of BIBSG is ∼2-fold higher than that of Ce6 under identical conditions. We further employed the spin-trapping method to detect the generation of 1O2 by electron spin resonance (ESR) spectroscopy utilizing 2,2,6,6-tetramethylpiperidine (Temp) as the spin trappers to identify 1O2 (Fig. 2e). Only BIBCl–PAE NPs with GSH at pH 6.5 effectively generate 1O2 under irradiation, while no ESR signals were observed in other control groups. In addition, in order to demonstrate whether the light-harvesting ability of BIBSG would facilitate enhancement of 1O2 generation, we also obtained the decolorization curves of ABDA in the presence of BSG (energy donor), BI (energy acceptor), BIBCl (with poor FRET) and BIBSG respectively (Fig. S11†). As shown in Fig. 2f, the ability of BIBSG to generate 1O2 was about 1.5-fold higher than that of BIBCl and BI, and 16-fold higher than that of BSG. This revealed that BIBSG has an elevated photon utilization ability due to FRET, resulting in boosted 1O2 generation. The above results demonstrate the validity of our design. Upon the GSH/pH cooperative activation, the aggregated BIBCl in the self-quenched state is transformed into free molecule BIBSG with the FRET effect, eliminating ACQ and giving rise to an elevated photon utilization to boost 1O2 generation. | ||
| Fig. 2 (a) The absorption spectra of BIBCl–PAE NPs and GSH-treated BIBCl–PAE NPs. Absorption spectra of ABDA at different time points in the presence of (b) BIBCl–PAE NPs without GSH and (c) BIBCl–PAE NPs with GSH. (d) The absorbance changes of ABDA at 378 nm in the presence of different samples. (e) The ESR spectra to detect 1O2 generated by BIBCl–PAE NPs under panchromatic light illumination, using Temp as the spin trappers. (f) The absorbance changes of ABDA at 378 nm in the presence of different samples. | ||
Cellular uptake and the discrimination of cancer cells from normal cells
The cellular uptake of the supramolecular PS was tested in cancer cells and normal cells. Confocal laser scanning microscopy (CLSM) images were obtained after incubating HepG2 cells (liver cancer cells) with BIBCl–PAE NPs (equivalent concentration of BIBCl: 2.5 μg mL−1). As shown in Fig. 3a, as the incubation time was prolonged, fluorescence increase in the green and red channels was observed, indicating that BIBCl–PAE NPs were internalized into the HepG2 cells. After 6 hours, the fluorescence intensity in the green and red channels gradually reduced, whereas the fluorescence intensity in the near-infrared (NIR) channel kept increasing (Fig. 3b). This result suggests the reaction of BIBCl with intracellular GSH to generate BIBSG in which energy transfer from the BSG unit to the BI unit enhances the NIR emission of the BI moiety. Moreover, the NIR signal of the photosensitizer nicely coincided with the green fluorescence of lysosome tracking dye (correlation coefficient of 0.89) (Fig. S12†), which was ascribed to internalization of the BIBCl–PAE NPs by endocytosis and the subsequent release of BIBCl from the nanoparticles. In sharp contrast, when the same incubation procedure was applied in L02 cells (normal liver cells), the fluorescence intensity in the green and red channels was only about 1/15 of that in HepG2 cells after 6 hours of incubation (Fig. 3a and b). And when the incubation time was prolonged, fluorescence in the green and red channels did not decrease, indicating that no FRET occurred. We speculate that the BIBCl–PAE NPs remained in the tightly aggregated state due to the low concentration of GSH and neutral pH in normal cells. This result indicates that BIBCl–PAE NPs can be activated by GSH in the weakly acidic tumor microenvironment but remain aggregated without activity in normal cells. | ||
| Fig. 3 (a) CLSM images of HepG2 cells and L02 cells incubated with the BIBCl–PAE NPs at different time points (green channel: 500–550 nm, excited at 487 nm; red channel: 570–620 nm, excited at 562 nm; NIR channel: 663–738 nm, excited at 638 nm). The scale bar represents 20 μm. (b) Intracellular average fluorescence intensity at different time points after incubation with BIBCl–PAE NPs in HepG2 cells and L02 cells. The viability of cancer cell lines after treatment with various doses of BIBH–PCL NPs, BIBCl–PCL NPs, BIBH–PAE NPs and BIBCl–PAE NPs with irradiation for 10 min, as measured by CCK-8 assay: (c) HepG2 cells; (d) A549 cells; (e) HeLa cells. (f) Cell viability of L02 cells treated with BIBCl–PAE NPs at various doses under the same conditions. *P < 0.05 (one-way ANOVA). | ||
In vitro cellular toxicity evaluation
The quantitative evaluations of the PDT effect of BIBCl–PAE NPs and controls BIBH–PAE NPs, BIBCl–PCL NPs and BIBH–PCL NPs were carried out by cell counting kit-8 (CCK-8) assays on HepG2 cells (Fig. 3c). In the absence of light irradiation, both BIBCl–PAE NPs and controls did not show toxicity in HepG2 cells (Fig. S13†). Under irradiation with white LED light (50 mW cm−2, 400–800 nm) for 10 min, BIBCl–PAE NPs showed obvious cytotoxicity in HepG2 cells, with a half-maximal inhibitory concentration (IC50) of 4.5 μg mL−1, which was much lower than that of BIBH–PAE NPs (24.8 μg mL−1). The results indicated the PDT potency of BIBCl–PAE NPs was enhanced by about 5.5-fold compared to that of BIBH–PAE NPs without GSH-activation capability. There was almost no cytotoxicity with BIBH–PCL NPs and BIBCl–PCL NPs due to efficient excited state quenching caused by tight aggregation. Similar results were also obtained in other tumor cell lines, such as A549 cells (lung cancer cells) and HeLa cells (cervical cancer cells) (Fig. 3d, e and S13†), revealing the robust PDT ability of BIBCl–PAE NPs in cancer cells. Importantly, BIBCl–PAE NPs hardly caused normal cell (L02 cells) death under irradiation (Fig. 3f). Such encouraging results also demonstrated that, in the presence of illumination, BIBCl–PAE NPs only show phototoxicity in tumor cells, but have no toxic side-effects on normal cells.In vitro therapeutic mechanism
Reactive oxygen species (ROS) play an important role in PDT and are considered to be the main cause of cell death. The ROS probe, 2,7-dichlorodihydrofluorescein diacetate (DCFH-DA), was used to evaluate the cellular ROS during PDT. As shown in Fig. 4a, in the presence of white LED light illumination, the HepG2 cells treated with BIBCl–PAE NPs displayed bright fluorescence in the green channel compared to the control group without illumination, indicating effective ROS generation under light irradiation. Moreover, when NaN3 was added, the fluorescence in the green channel disappeared, meaning that the ROS induced by the PS was completely scavenged by NaN3, further implying the generation of ROS. Then, we employed the Singlet Oxygen Sensor Green reagent (SOSG, a commercial 1O2 probe) to further examine whether the generated ROS is 1O2 (Fig. 4a). Under light illumination, the PS-loaded cells incubated with SOSG showed obvious fluorescence in the green channel, whereas no fluorescence was observed under the identical conditions in the dark. Similarly, the addition of NaN3 effectively scavenged 1O2 and significantly reduced the intracellular fluorescence. In addition, DCFH-DA was also used to evaluate the ROS generation in normal cells. The results showed that the L02 cells treated with BIBCl–PAE NPs displayed negligible fluorescence in the green channel compared to HepG2 cells (Fig. S14†), indicating that there was almost no effective ROS generation in normal cells. These results indicate that our photosensitizer system can selectively generate 1O2 in tumor cells.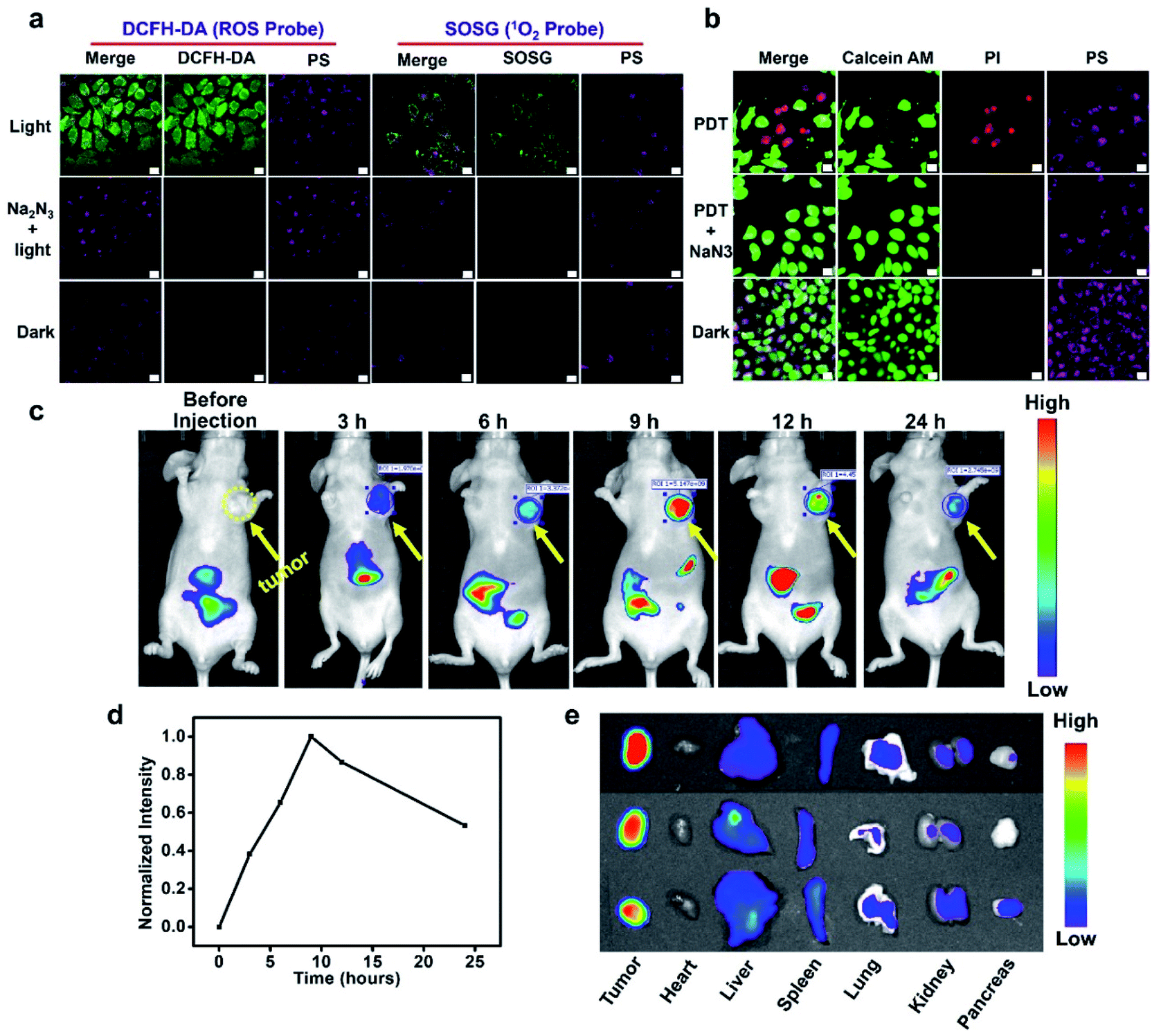 | ||
| Fig. 4 (a) Evaluation of 1O2 generation in HepG2 cells with DCFH-DA and SOSG. The scale bar represents 20 μm. (b) CLSM images of calcein AM/PI-stained HepG2 cells. The scale bar represents 20 μm. (c) In vivo fluorescence imaging of HepG2 tumor-bearing BALB/c mice after intravenous injection of BIBCl–PAE NPs (injected at a dose of 2 mg kg−1 BIBCl). (d) Normalized fluorescence intensity of the photosensitizer in tumor sites at different time points. (e) Fluorescence images of ex vivo organs harvested at 24 h post injection. | ||
To further observe cancer cell inhibition with BIBCl–PAE NPs, a calcein–AM and propidium iodide (PI) assay that respectively stains viable cells (emit fluorescence in the green channel) and apoptotic cells (emit fluorescence in the red channel) was performed. It can be seen in Fig. 4b that HepG2 cells treated with BIBCl–PAE NPs showed obvious red and green fluorescence after illumination, indicating partial apoptosis. By contrast, in the presence of NaN3 or without illumination, only fluorescence in the green channel was observed, which meant no apoptosis. Moreover, an Annexin V-FITC/PI apoptosis detection kit was used to further investigate the cell apoptosis due to BIBCl–PAE NPs by flow cytometry (Fig. S15†). These results proved that BIBCl–PAE NPs generate 1O2 to induce tumor cell apoptosis.
In vivo tumor imaging and anticancer effect
Motivated by the excellent in vitro experimental results, we further evaluated the tumor enrichment effect and tumor growth inhibition effect of BIBCl–PAE NPs on immunodeficient mouse models by using a subcutaneous tumor model of human liver cancer HepG2 cells in BALB/c mice. To determine the enrichment time of supramolecular PSs in the tumor tissue, the tumor-bearing mice were intravenously injected at a dose of 2 mg kg−1 BIBCl, followed by in vivo fluorescence imaging at different time points. As shown in Fig. 4c, 3 hours after intravenous injection, significant fluorescence was observed in the tumor portion and had a high contrast ratio. With the increase in time, the fluorescence intensity of tumor sites increased further, reached maximum at 9 hours after injection, and then began to weaken (Fig. 4d). Notably, the tumor site exhibited strong fluorescence compared to surrounding sites with a high S/N ratio of 7.9, which is attributed to the EPR effect in tumor tissues and ACQ in normal tissues. Next, the ex vivo biodistribution of the photosensitizer was evaluated at 24 hours postinjection (Fig. 4e). Obviously, the fluorescence in the tumor was significantly stronger than that in other organs. Moreover, except for the stomach and intestines, other organs showed almost no fluorescence signal (Fig. S16†).After determining the enrichment time, we further proceeded to investigate the antitumor efficiency of BIBCl–PAE NPs in vivo by using immunocompetent BALB/c mice. BIBCl–PAE NPs (250 μg mL−1, 100 μL) were injected into the mice bearing HepG2 tumor cells (primary tumor volume: ∼100 mm3) by tail vein injection, followed by irradiation (white LED light, 400–800 nm, 100 mW cm−2) on the tumors at 9 hours and 24 hours postinjection. Then, the body weights (Fig. 5a) and tumor volumes (Fig. 5b) were measured during the subsequent 14 days. The weight of the mice increased slightly, suggesting the negligible systemic cytotoxicity of BIBCl–PAE NPs during PDT. We were pleased to find that the tumors in the mice were significantly reduced and the tumors of the two mice completely disappeared, indicating that BIBCl–PAE NPs could effectively suppress tumors. PBS with light irradiation as a control showed a 20-fold increase of tumor volumes, indicating that the light irradiation has no distinct influence on the tumor growth. BIBCl–PAE NPs without irradiation also exhibited similar tumor growth rates to PBS, suggesting that BIBCl–PAE NPs were non-toxic in the absence of irradiation. In contrast, unobvious inhibition of tumor growth was observed when mice were injected with BIBH–PAE NPs or BIBH–PCL NPs, indicating the strategy in which PSs react with GSH in a weakly acidic tumor microenvironment to form a water-soluble molecule to suppress ACQ and active PDT is valid. When the mice were sacrificed on the 15th day and all the tumor tissues were peeled, a considerable difference in the therapeutic efficacy was further intuitively reflected by the images of tumor tissues (Fig. 5c) and average tumor weight (Fig. 5d and S17†). Obviously, the group of BIBCl–PAE NPs light has the best anti-tumor effect (Fig. 5b–d). In addition, hematoxylin & eosin (H&E) staining was applied to examine the ability of different treatment groups to cause tumor damage under irradiation (Fig. 5f). Samples were taken 2 days after treatment and it was found that BIBCl–PAE NPs caused destructive cell necrosis in the tumor, indicating a desirable ability to destruct the tumor cells as compared to others. The results of H&E staining were consistent with the antitumor data observed in vivo. The H&E staining images of the paracancerous tissue and main tissues were also obtained (Fig. S18†). None of the tissues was significantly damaged compared with the non-administered group. More importantly, the above results demonstrated that the activatable BIBCl–PAE NPs could suppress ACQ of the PS in the tumor while achieving effective anti-tumor effects.
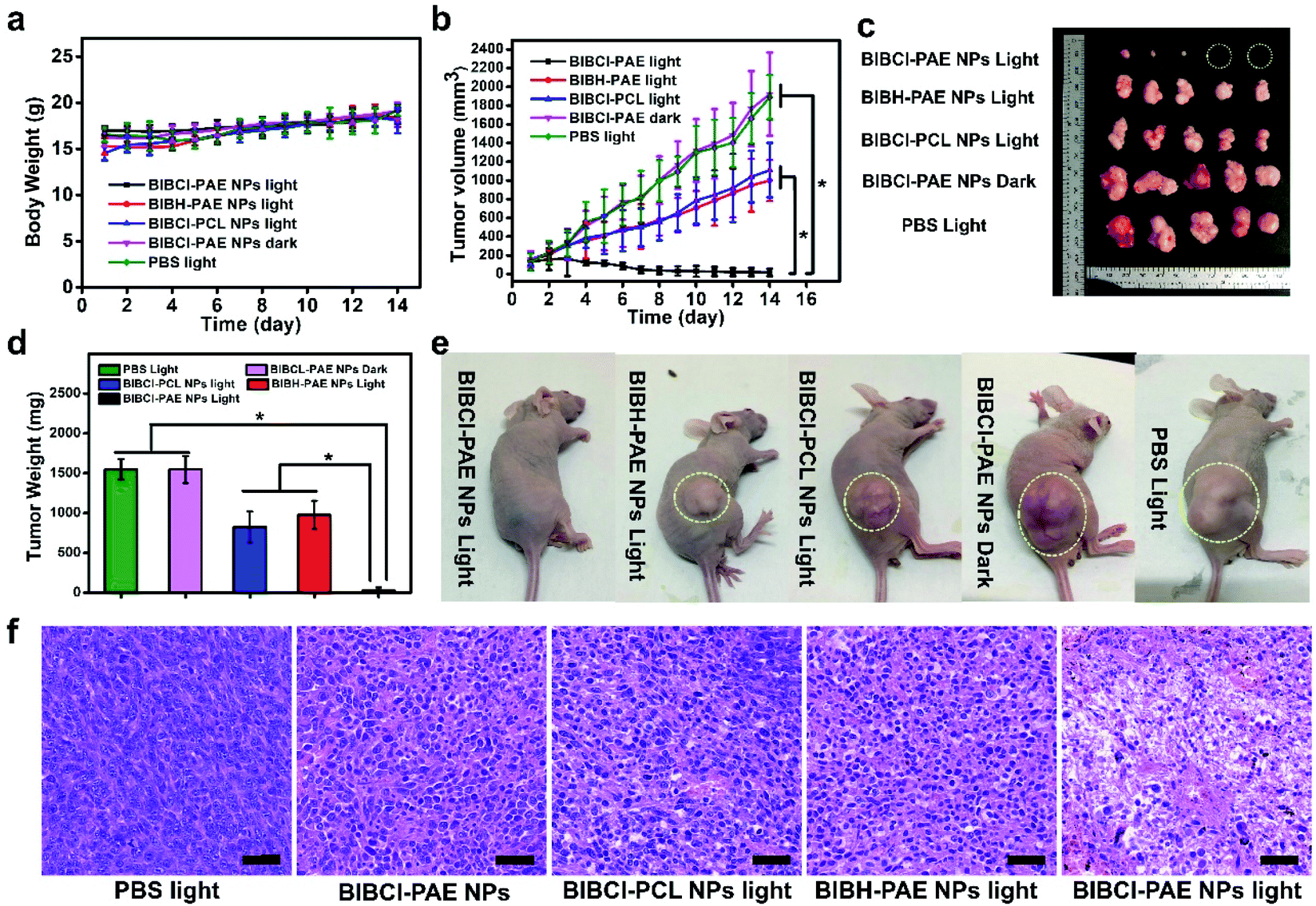 | ||
| Fig. 5 PDT was performed 9 hours after PS injection (250 μg mL−1, 100 μL). Values are mean ± s.e.m. (n = 5 mice per group) (a) body weights of the mice during the observation. (b) Tumor growth profiles during the observation. (c) Images of tumor tissues from different groups of tumor-bearing mice. (d) Average tumor weight of different groups of tumor-bearing mice. (e) Representative photos of mice from different groups on the 14th day. (f) H&E staining of tumor tissue sections from different treatment groups after 2 days of treatment, and the scale bar represents 40 μm. *P < 0.05 (one-way ANOVA). | ||
Conclusions
We developed a smart GSH/pH cooperatively activatable supramolecular photosensitizer BIBCl–PAE NPs based on a “dual lock-and-key” strategy for specific and enhanced PDT. The hydrophobic characteristic of BIBCl and diblock copolymer PEG–PAE acted as “dual locks” to keep the PS in the aggregation state to realize the initial ACQ in blood and normal tissues. In the tumor microenvironment, the low pH and high concentrations of GSH served as “dual keys”that promote the reaction of BIBCl with GSH to form water-soluble BIBSG, facilitating the activation of the photosensitizer. Moreover, the FRET effect of BIBSG enabled the increased light-harvesting ability and enhanced 1O2 production. The in vitro study demonstrated that our photosensitizer not only discriminated cancer cells from normal cells in live cell imaging, but also exhibited robust PDT ability in cancer cells without toxicity and side effects to normal cells. The in vivo results revealed that BIBCl–PAE NPs were rapidly enriched in tumor sites to selectively “light up” the tumor area and showed irreversible cytotoxicity to the tumor tissue without affecting other normal tissues. Both in vitro and in vivo results indicated that the supramolecular PSs serve as a potentially new class of PDT agents for use in future cancer theranostics based on their favorable features such as the cooperative activation, high-specificity cancer therapy and light-harvesting ability to overcome limitations associated with conventional PSs. We speculate that our design may inspire others to develop new effective photosensitizers for cancer treatment.Ethical statement
Procedures related to animal experiments were implemented in compliance with the China Animal Management Regulations (2017 Edition), and were approved by the animal care committee of Beijing Normal University.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was financially supported by the National Natural Science Foundation of China (21525206 and 21971023).Notes and references
- D. E. J. G. J. Dolmans, D. Fukumura and R. K. Jain, Nat. Rev. Cancer, 2003, 3, 380–387 CrossRef CAS.
- A. P. Castano, P. Mroz and M. R. Hamblin, Nat. Rev. Cancer, 2006, 6, 535–545 CrossRef CAS.
- Z. Zhou, J. Song, L. Nie and X. Chen, Chem. Soc. Rev., 2016, 45, 6597–6626 RSC.
- X. Li, S. Lee and J. Yoon, Chem. Soc. Rev., 2018, 47, 1174–1188 RSC.
- R. Bonnett, Chem. Soc. Rev., 1995, 24, 19–33 RSC.
- E. D. Sternberg, D. Dolphin and C. Bruckner, Tetrahedron, 1998, 54, 4151–4202 CrossRef CAS.
- M. Ethirajan, Y. Chen, P. Joshi and R. K. Pandey, Chem. Soc. Rev., 2011, 40, 340–362 RSC.
- W.-S. Li and T. Aida, Chem. Rev., 2009, 109, 6047–6076 CrossRef CAS.
- M. H. Lee, E.-J. Kim, H. Lee, H. M. Kim, M. J. Chang, S. Y. Park, K. S. Hong, J. S. Kim and J. L. Sessler, J. Am. Chem. Soc., 2016, 138, 16380–16387 CrossRef CAS.
- E. Delaey, F. van Laar, D. De Vos, A. Kamuhabwa, P. Jacobs and P. de Witte, J. Photochem. Photobiol. B Biol., 2000, 55, 27–36 CrossRef CAS.
- A. Kamkaew, S. H. Lim, H. B. Lee, L. V. Kiew, L. Y. Chung and K. Burgess, Chem. Soc. Rev., 2013, 42, 77–88 RSC.
- J. Zhao, K. Xu, W. Yang, Z. Wang and F. Zhong, Chem. Soc. Rev., 2015, 44, 8904–8939 RSC.
- R. C. Wang, K. K. Dong, G. Xu, B. Shi, T. L. Zhu, P. Shi, Z. Q. Guo, W. H. Zhu and C. C. Zhao, Chem. Sci., 2019, 10, 2785–2790 RSC.
- J. Zhou, Y. Zhang, G. Yu, M. R. Crawley, C. R. P. Fulong, A. E. Friedman, S. Sengupta, J. Sun, Q. Li, F. Huang and T. R. Cook, J. Am. Chem. Soc., 2018, 140, 7730–7736 CrossRef CAS.
- W. Fan, P. Huang and X. Chen, Chem. Soc. Rev., 2016, 45, 6488–6519 RSC.
- F. Helmich, C. C. Lee, M. M. L. Nieuwenhuizen, J. C. Gielen, P. C. M. Christianen, A. Larsen, G. Fytas, P. E. L. G. Leclere, A. P. H. J. Schenning and E. W. Meijer, Angew. Chem., Int. Ed., 2010, 49, 3939–3942 CrossRef CAS.
- L. Chen, H. Bai, J.-F. Xu, S. Wang and X. Zhang, ACS Appl. Mater. Interfaces, 2017, 9, 13950–13957 CrossRef CAS.
- J. G. Jin, Y. C. Zhu, Z. H. Zhang and W. A. Zhang, Angew. Chem., Int. Ed., 2018, 57, 16354–16358 CrossRef CAS.
- J. Mei, N. L. C. Leung, R. T. K. Kwok, J. W. Y. Lam and B. Z. Tang, Chem. Rev., 2015, 115, 11718–11940 CrossRef CAS.
- G. Feng and B. Liu, Acc. Chem. Res., 2018, 51, 1404–1414 CrossRef CAS.
- Y. Y. Yuan, C. J. Zhang, M. Gao, R. Y. Zhang, B. Z. Tang and B. Liu, Angew. Chem., Int. Ed., 2015, 54, 1780–1786 CrossRef CAS.
- Y. Y. Yuan, C. J. Zhang, S. D. Xu and B. Liu, Chem. Sci., 2016, 7, 1862–1866 RSC.
- S. Ozlem and E. U. Akkaya, J. Am. Chem. Soc., 2009, 131, 48–49 CrossRef CAS.
- W. A. Velema, W. Szymanski and B. L. Feringa, J. Am. Chem. Soc., 2014, 136, 2178–2191 CrossRef CAS.
- B. M. Luby, C. D. Walsh and G. Zheng, Angew. Chem., Int. Ed., 2019, 58, 2558–2569 CrossRef CAS.
- X. Li, C. y. Kim, S. Lee, D. Lee, H.-M. Chung, G. Kim, S.-H. Heo, C. Kim, K.-S. Hong and J. Yoon, J. Am. Chem. Soc., 2017, 139, 10880–10886 CrossRef CAS.
- J. W. Tian, L. Ding, H. J. Xu, Z. Shen, H. X. Ju, L. Jia, L. Bao and J. S. Yu, J. Am. Chem. Soc., 2013, 135, 18850–18858 CrossRef CAS.
- J. Zou, P. Wang, Y. Wang, G. Liu, Y. Zhang, Q. Zhang, J. Shao, W. Si, W. Huang and X. Dong, Chem. Sci., 2019, 10, 268–276 RSC.
- W. Piao, K. Hanaoka, T. Fujisawa, S. Takeuchi, T. Komatsu, T. Ueno, T. Terai, T. Tahara, T. Nagano and Y. Urano, J. Am. Chem. Soc., 2017, 139, 13713–13719 CrossRef CAS.
- H. Chen, J. Tian, W. He and Z. Guo, J. Am. Chem. Soc., 2015, 137, 1539–1547 CrossRef CAS.
- H. H. Fan, G. B. Yan, Z. L. Zhao, X. X. Hu, W. H. Zhang, H. Liu, X. Y. Fu, T. Fu, X. B. Zhang and W. H. Tan, Angew. Chem., Int. Ed., 2016, 55, 5477–5482 CrossRef CAS.
- L.-Y. Niu, Y.-Z. Chen, H.-R. Zheng, L.-Z. Wu, C.-H. Tung and Q.-Z. Yang, Chem. Soc. Rev., 2015, 44, 6143–6160 RSC.
- M. Wang, Y. Zhai, H. Ye, Q. Lv, B. Sun, C. Luo, Q. Jiang, H. Zhang, Y. Xu, Y. Jing, L. Huang, J. Sun and Z. He, ACS Nano, 2019, 13, 7010–7023 CrossRef CAS.
- H. S. Jung, J. Han, H. Shi, S. Koo, H. Singh, H.-J. Kim, J. L. Sessler, J. Y. Lee, J.-H. Kim and J. S. Kim, J. Am. Chem. Soc., 2017, 139, 7595–7602 CrossRef CAS.
- X. Li, S. Yu, Y. Lee, T. Guo, N. Kwon, D. Lee, S. C. Yeom, Y. Cho, G. Kim, J.-D. Huang, S. Choi, K. T. Nam and J. Yoon, J. Am. Chem. Soc., 2019, 141, 1366–1372 CrossRef CAS.
- X. Li, S. Kolemen, J. Yoon and E. U. Akkaya, Adv. Funct. Mater., 2017, 27, 1604053 CrossRef.
- S. Li, Q. Zou, Y. Li, C. Yuan, R. Xing and X. Yan, J. Am. Chem. Soc., 2018, 140, 10794–10802 CrossRef CAS.
- Y. Tang, Y. Li, X. Hu, H. Zhao, Y. Ji, L. Chen, W. Hu, W. Zhang, X. Li, X. Lu, W. Huang and Q. Fan, Adv. Mater., 2018, 30, 11801140 Search PubMed.
- X. Zhao, C. X. Yang, L. G. Chen and X. P. Yan, Nat. Commun., 2017, 8, 14998 CrossRef CAS.
- L. Teng, G. Song, Y. Liu, X. Han, Z. Li, Y. Wang, S. Huan, X.-B. Zhang and W. Tan, J. Am. Chem. Soc., 2019, 141, 13572–13581 CrossRef CAS.
- G. H. Gao, M. J. Park, Y. Li, G. H. Im, J.-H. Kim, H. N. Kim, J. W. Lee, P. Jeon, O. Y. Bang, J. H. Lee and D. S. Lee, Biomaterials, 2012, 33, 9157–9164 CrossRef CAS.
- Y.-F. Kang, L.-Y. Niu and Q.-Z. Yang, Chin. Chem. Lett., 2019, 30, 1791–1798 CrossRef CAS.
- M. Li, S. Long, Y. Kang, L. Guo, J. Wang, J. Fan, J. Du and X. Peng, J. Am. Chem. Soc., 2018, 140, 15820–15826 CrossRef CAS.
- J. Zhao, W. Wu, J. Sun and S. Guo, Chem. Soc. Rev., 2013, 42, 5323–5351 RSC.
- L. Y. Niu, Y. S. Guan, Y. Z. Chen, L. Z. Wu, C. H. Tung and Q. Z. Yang, J. Am. Chem. Soc., 2012, 134, 18928–18931 CrossRef CAS.
- L. Y. Niu, Y. S. Guan, Y. Z. Chen, L. Z. Wu, C. H. Tung and Q. Z. Yang, Chem. Commun., 2013, 49, 1294–1296 RSC.
- F. Zhang, Q. Ni, O. Jacobson, S. Cheng, A. Liao, Z. Wang, Z. He, G. Yu, J. Song, Y. Ma, G. Niu, L. Zhang, G. Zhu and X. Chen, Angew. Chem., Int. Ed., 2018, 57, 7066–7070 CrossRef CAS.
- F. Kong, Z. Liang, D. Luan, X. Liu, K. Xu and B. Tang, Anal. Chem., 2016, 88, 6450–6456 CrossRef CAS.
- Z. Sheng, D. Hu, M. Zheng, P. Zhao, H. Liu, D. Gao, P. Gong, G. Gao, P. Zhang, Y. Ma and L. Cai, ACS Nano, 2014, 8, 12310–12322 CrossRef CAS.
- G. H. Gao, G. H. Im, M. S. Kim, J. W. Lee, J. Yang, H. Jeon, J. H. Lee and D. S. Lee, Small, 2010, 6, 1201–1204 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Procedures for synthesis, characterization data, and supplementary figures. See DOI: 10.1039/d0sc01122c |
| This journal is © The Royal Society of Chemistry 2020 |