
Carbon-based materials as heterogeneous antioxidants for biodiesel: efficiency and synergy with soluble antioxidants†
Monique R.
Jesus
ab,
Tais da C.
Soares
a,
Paulo R. M.
Silva
a,
Gilberto A.
Romeiro
c,
Mauricio G.
Fonseca
a and
Luciano N.
Batista
 *a
*a
aLaboratory of Engines and Fuels, National Institute of Metrology, Quality and Technology, Nossa senhora das graças avenue, 50, 25250020 Rio de Janeiro, Brazil. E-mail: lnbatista@inmetro.gov.br
bNational Institute of Technology, Venezuela Avenue, 82, 20081-312 Rio de Janeiro, Brazil
cFluminense Federal University, Institute of Chemistry, Niterói, RJ 24210-150, Brazil
First published on 6th January 2017
Abstract
Antioxidants are compounds widely used in several areas as fuels, foods, and cosmetics among others. In this work, we analyze the use of biochar produced from Jatropha curcas seeds as an antioxidant. The biochar presented low antioxidant activity, although the antioxidant capacity of traditional antioxidants and modified biochar has a particular synergy. The oxidation stability of biodiesel can be improved more than fivefold in comparison with that using only a traditional antioxidant. This interaction presents an excellent synergy that allows oxidation stability to be increased to relatively high levels without requiring an increase in the antioxidant concentration. At the end of the experiment the biochar is easily separated by simple filtration and can be reused. We expect that these results would allow a better use of this toxic biomass improving the sustainability of biodiesel storage.
Introduction
In the near future, the circular economy is likely to influence the prospects of the world economy. Materials and products within this new type of economy should be designed from the outset to be reused, renewed or recycled; these characteristics allow the reinsertion of previously used materials into the production chain, and thus the circular economy can reduce the use of primary feedstock, reuse the residues and increase the recovery of final products.1Lignocellulosic materials are extensively used worldwide, and this has become a serious environmental problem in some countries and regions due to the high potential methane emissions associated with these materials.2 These materials therefore require urgent commercial or industrial applications to avoid expensive waste treatments.
Some of the most widespread applications of biomass residues in chemistry are:
• The production of liquid fuels from biomass through pyrolysis,3,4 fermentation5,6 or chemical conversion;7–9
• Direct burning of biomass for heating;10,11
• The conversion of lignocellulosic materials to activated carbon with high adsorption capacity, to be applied as a pollutant control and remediation;12,13
• The conversion of biomass to biochar for use as a soil additive;14,15
• The conversion of biomass to a biochar structure through a heterogeneous catalyst by the addition of acid sites.16–18
Traditionally, the synthesis of biochar catalysts has been performed by high-temperature pyrolysis. The high temperatures are required to remove the oxygenated groups and to promote the cyclization process. During pyrolysis, ordering of the carbon sheets takes place, which tends to increase the surface area. Following the pyrolysis process, a solution of sulphuric acid is used in order to initiate sulphonation of the aromatic rings. The insertion of hydrogen sulphonic groups increases the number of acid sites and the strength of the material.
Biodiesel is one of the primary liquid biofuels in the world; 27 billion litres were produced in 2015, and the production is expected to reach more than 38 billion litres by 2024.19 Although it is widely used, the majority of the biodiesel in the world requires the addition of antioxidants to retard the oxidation process which, in some cases, could cause the fuel to be unusable or even toxic to human health.20 The main reason for the ready oxidation process of biodiesel is the presence of unsaturated esters which have allylic hydrogen (hydrogen linked to carbon neighbours which have a double bond) or bis-allylic hydrogen (hydrogen linked to carbon atoms with two double bonds). The bond dissociation energy of these specific C–H bonds is lower than that of the carbon–hydrogen bonds in methylenic hydrogen (C–H bond in a saturated chain) and methylic hydrogen (C–H bond of the terminal carbon chain CH3). The cleavage of the C–H bonds of allylic or bis-allylic carbon generates the alkyl radical, starting the process of fatty-acid oxidation. At this point, the antioxidant compound donates an hydrogen radical to the alkyl radical, returning the methyl ester to the original form. If antioxidants are not present or have insufficient concentration in the biofuel, the alkyl radical reacts with O2, forming lipid hydroperoxides and propagating the oxidation process. The final products of the lipid oxidation include aldehydes, acids, ketones21 and small oligomers derived from vinyl polymerization or the Diels–Alder cyclization.22
Results and discussion
In this communication, we report the production of biochar from Jatropha curcas cake, using a low-temperature pyrolysis process (<380 °C),23 and an evaluation of the antioxidant capacity of the biochar after treatment with sulfuric acid. We also analyse the efficiency of biochar as an antioxidant, in comparison with more traditional antioxidants.Jatropha curcas cake is used here due to the toxicity of its seeds, which means it cannot be used as animal feed24 or for any other application25 without treatment. The use of low-temperature pyrolysis aims to produce biochar with a larger quantity of phenol groups, which are required for antioxidant capacity. The sulphuric acid treatment produces biochar with acid catalyst properties and, at the same time, promotes the cleaning of the biochar surface.
The antioxidant capacity was determined by the induction period using the Rancimat method. This approach was chosen since it is an ASTM method, accepted worldwide for the analysis of the oxidative stability of biodiesel. Samples using biochar and traditional antioxidants were analysed together.
The use of an insoluble solid antioxidant in biofuels means that, following its application as an antioxidant, it can be removed from the material using simple filtration. This recovery and reuse of the bio-based antioxidant offers ways to involve biochar in the circular economy, thus increasing the sustainability of biodiesel and the volume of biomass processing.
The pyrolysis process was applied to the Jatropha curcas cake at 380 °C for 1 hour (a detailed experimental description is given in the ESI†); the sample was then washed with hexane and then water to remove polar compounds. The sample was dried in a muffle. The sulphonation process was carried out by immersing 40 g of biochar in concentrated sulphuric acid at 130 °C for 13 hours, taking hourly samples. The samples were washed with deionized water until neutralization, and were then dried at 120 °C for 5 hours. The biochar was stored in a desiccator in an amber bottle.
Infrared spectrometry using Fourier transforms was then applied to detect changes in functional organic groups on the surface of the biochar. Fig. 1 presents the infrared spectrum of the biochar before and after the sulphonation process. Both spectra show absorption from the OH-stretching (3400 cm−1) with proportionally higher absorption after the sulphonation process. It appears that the sulphonation process oxidizes the biochar, converting some aldehyde groups into alcohol groups. The aliphatic C–H stretching appears between 2950 cm−1 and 2850 cm−1, indicating incomplete carbonization. Absorption from the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C stretching of aromatic groups appears between 1380 cm−1 and 1480 cm−1. Another indication of the aromatic presence is the absorption bands at 874, 792, and 723 cm−1 (not marked) arising from the plane vibrations in aromatic C–H. The absorption around 1260 cm−1 is attributed by the authors to the C–OH stretching in the aromatic phenolic group formed during the carbonization process.26 Absorption from the symmetric C–O–C stretching appears at 1097 cm−1 and is characteristic of the pyranose rings of cellulose and guaiacyl monomers.27 Following the sulphonation process, a relative decrease in the absorption of aliphatic components was observed (2950 cm−1 to 2850 cm−1). A symmetric SO stretching peak at 1035 cm−1 as well as the C–O–C asymmetric stretching at 1176 cm−1 is apparent. A broadened peak at 1720 cm−1 is attributable to SO3H groups.28 Both peaks confirm the incorporation of sulphonic groups into the biochar.
C stretching of aromatic groups appears between 1380 cm−1 and 1480 cm−1. Another indication of the aromatic presence is the absorption bands at 874, 792, and 723 cm−1 (not marked) arising from the plane vibrations in aromatic C–H. The absorption around 1260 cm−1 is attributed by the authors to the C–OH stretching in the aromatic phenolic group formed during the carbonization process.26 Absorption from the symmetric C–O–C stretching appears at 1097 cm−1 and is characteristic of the pyranose rings of cellulose and guaiacyl monomers.27 Following the sulphonation process, a relative decrease in the absorption of aliphatic components was observed (2950 cm−1 to 2850 cm−1). A symmetric SO stretching peak at 1035 cm−1 as well as the C–O–C asymmetric stretching at 1176 cm−1 is apparent. A broadened peak at 1720 cm−1 is attributable to SO3H groups.28 Both peaks confirm the incorporation of sulphonic groups into the biochar.
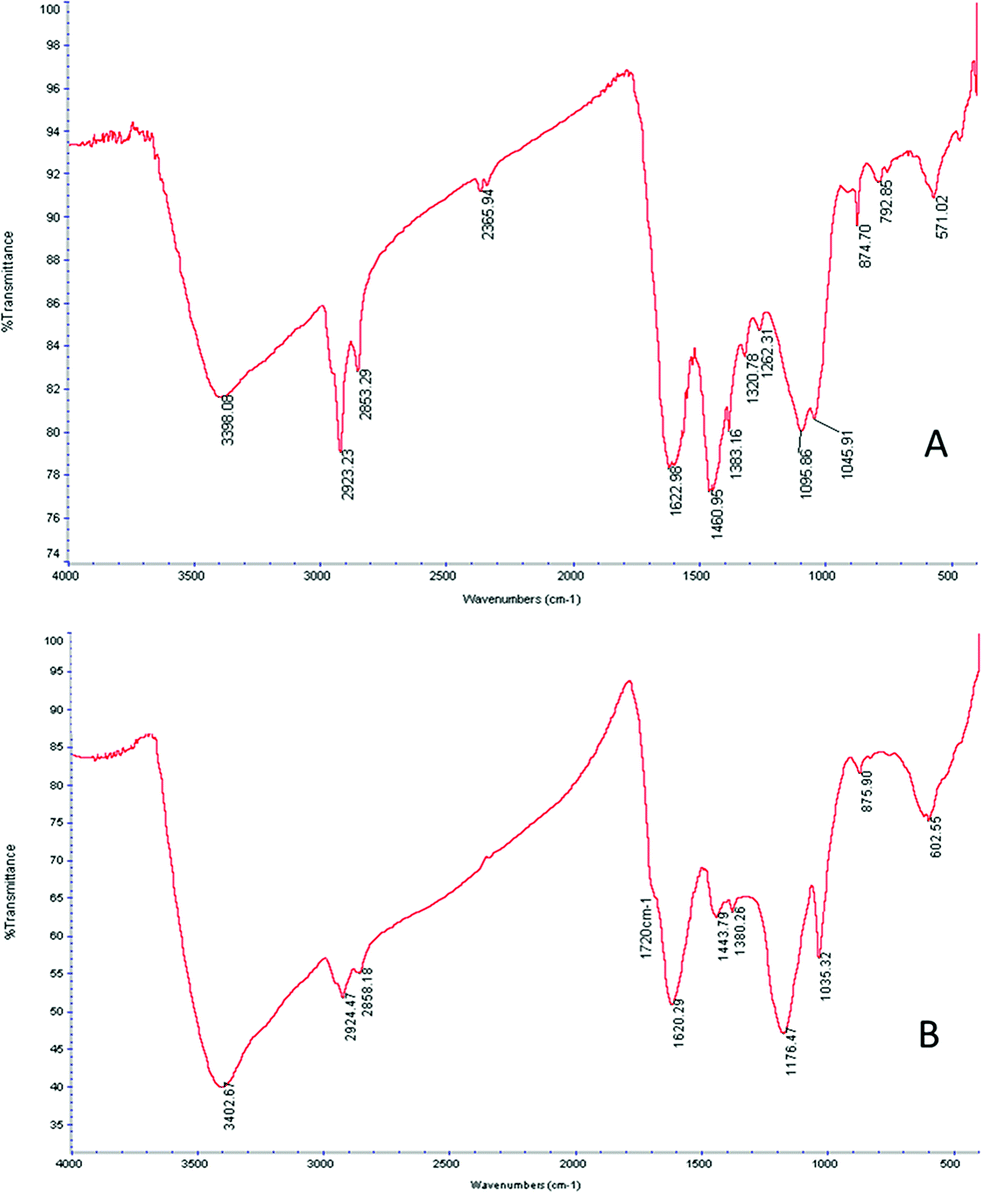 | ||
| Fig. 1 Infrared spectra of biochar (A) before and (B) after the sulphonation process. The presence of sulphonic groups is confirmed by the absorption bands at 1720 cm−1 and 1035 cm−1. The process of sulphonation does not remove the phenol groups, retaining the antioxidant potential, as confirmed by the –OH stretching absorption band close to 3400 cm−1 and the small absorption bands between 1260 and 1480 cm−1 which are associated with the CC from aromatic rings. | ||
The presence of phenol groups on the biochar surface indicates some antioxidant capacity; however, the number of groups present on the surface needs to be determined. The phenol and organic acid groups are the main groups on the biochar surface with acid properties, although the low acid strength of these organic groups is unable to promote most esterification or epoxidation reactions. The insertion of a hydrogen sulfonic group, a stronger acid group, enhances the acid strength. The difference in the acid strengths of the organic groups allows the determination of the number of sulphonic acid groups and the number of phenol groups per gram of the sample. If the phenol groups react during sulphonation, the concentration of weak acid groups is expected to decrease. The determination of the number of acid groups was made using Boehm's titration29,30 and the results are shown in Fig. 2.
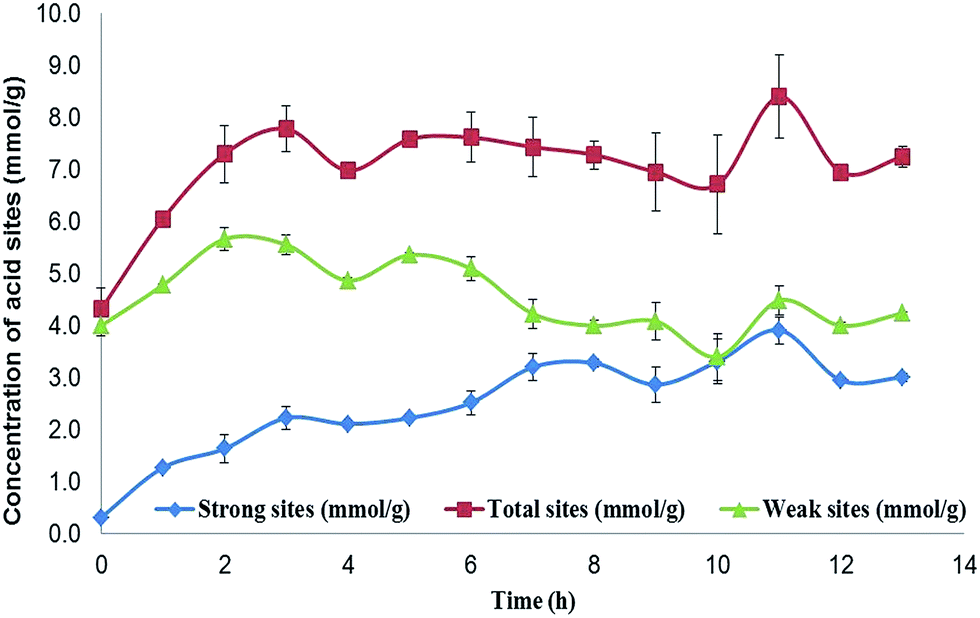 | ||
| Fig. 2 Concentration of acid sites on the biochar surface. The total number of acid sites reaches about 7.5 mmol g−1 in just 3 hours and remains close to this value over time. The number of strong acid sites increases constantly up to about 3.0 mmol g−1 over 7 hours, and these are attributed to the presence of HSO3− groups. The concentration of weak acid sites reaches 5.6 ± 0.3 mmol g−1 in 2–3 hours, and decays over time to about 4.0 mmol g−1. The main weak acid groups on the biochar are the phenol and carboxyl groups. Weak acid sites are present during the entire sulphonation process, although the concentration decays to close to zero. The origin of the weak acid sites on the final product may be different from those at the beginning of the process; however, there are sufficient phenol groups to retain the antioxidant capacity of the biochar (n = 3, the error bar is the standard deviation). | ||
The results of the Boehm titration showed that the number of total acid sites on the initial material was 4.0 × 10−3 mol per gram, exclusively formed by weak acid groups in the form of phenol. After 13 hours of immersion in sulphuric acid, the biochar showed 7.0 × 10−3 mol of acid sites per gram of biochar, while the number of weak acid sites remained at 4.0 × 10−3 mol. This result indicates that the sulphonation process has not affected the phenol groups; however, this cannot reflect the full extent of the process, since after the concentration of the weak acid sites reaches 5.5 × 10−3 mol g−1 during the first 2 hours, it then returns to 4.0 × 10−3 mol g−1 after 7 hours, and remains close to this value until the end of the process. Meanwhile, the number of HSO3 groups (strong acid sites) increases to 2 × 10−3 mol g−1 during the first 2 hours, and remains at this value for 3 hours. After 5 hours, the concentration of –HSO3 increases again to 3.0 × 10−3 mol g−1.
In summary, the sulphonation process affects the biochar surface first by promoting new weak acid sites, and at the same time inserting hydrogen sulphonic acid groups (–HSO3), and secondly by promoting the destruction of some weak acid sites after several hours. The sulphonation process does not increase the surface area, since all samples of biochar have a surface area of less than 10 m2 g−1, and no meaningful changes were observed in X-ray diffraction, indicating that the biochar has not changed in terms of crystallinity. However, the cleaning process of the sample promotes the opening of large cavities (of μm order) on the biochar surface, as shown by scanning electron microscopy. All characterizations are given in the ESI.†
Evaluation of the antioxidant capacity of the material was carried out using the Rancimat method (see ESI†). Samples of sunflower and Jatropha biodiesel using several concentrations of the sulphonated biochar were tested both with and without the presence of a commercial antioxidant. Sunflower biodiesel was chosen due to its low oxidative resistance, while the Jatropha curcas biodiesel is an example of biofuel with high oxidative resistance. Jatropha curcas oil has been reported with several oxidative stability values, although the biodiesel from a Brazilian cultivar of Jatropha curcas has been reported with high oxidative stability, mainly after degumming, reaching 13 h of oxidative stability.31 Wang et al.,32 report 8 h for Jatropha curcas, a biodiesel oxidative stability value similar to the value obtained by us. The difference in the oxidative resistance of these biofuels can be explained by the content of unsaturated fatty esters (see ESI†).
Fig. 3 shows the induction period (IP) of biodiesel samples with different biochar contents. The first sample of sunflower biodiesel has an IP of about 0.89 ± 0.11 h; the addition of 0.17 g g−1 of biochar enhances the oxidative stability to 1.89 ± 0.10 h. In other words, the presence of 17% biochar in the sample was enough to increase the oxidation stability by 100%.
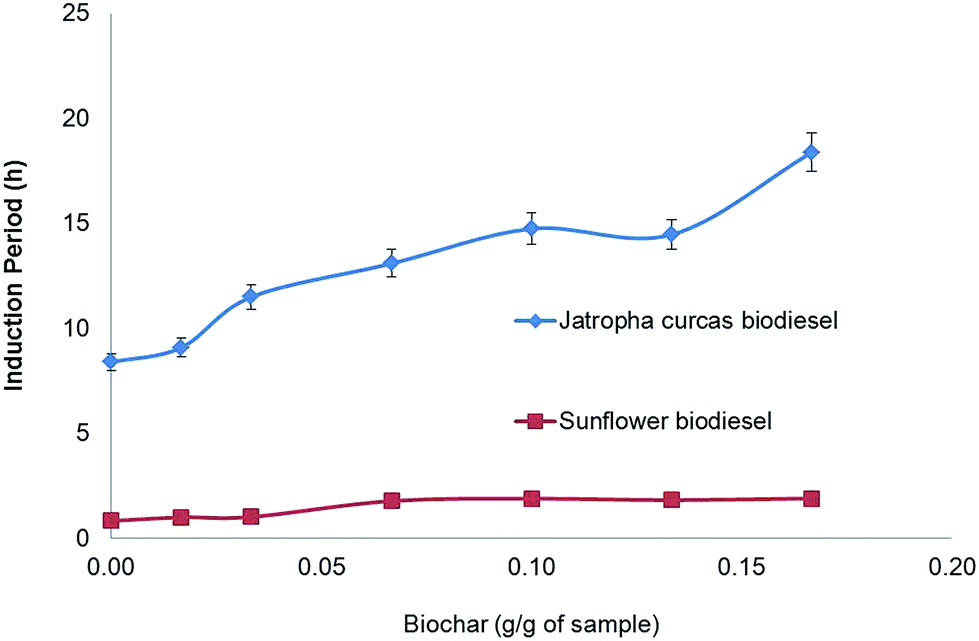 | ||
| Fig. 3 Induction period of methyl biodiesel from Jatropha curcas oil and sunflower oil, for several concentrations of biochar. Both showed an increase in the induction period. With the addition of 10% biochar, sunflower oil shows twice the induction period (1.89 h) of the initial sample. The same effect is shown by Jatropha curcas biodiesel; the induction period reaches more than 18 h with the addition of 17% biochar. These results show that biochar retains a significant antioxidant capacity after treatment with sulphuric acid, although this requires a relatively large quantity of biochar (n = 5, the error bar is the standard deviation). | ||
Fig. 4 shows a summary of the results for the IP of methylic sunflower biodiesel antioxidants with biochar. The first antioxidant is 2,2′-methylene-bis-(4-methyl-6-tert-butyl-phenol) (TBP) from Eastman Co. and N-(1,3-dimethylbutyl)-N′-phenyl-p-phenylenediamine (PPD) from Lanxess Co. Silicate (60 μm) from Sigma-Aldrich Co. is used to evaluate whether the sole addition of a solid material into the sample can affect O2 solubility and, consequently, the induction period.
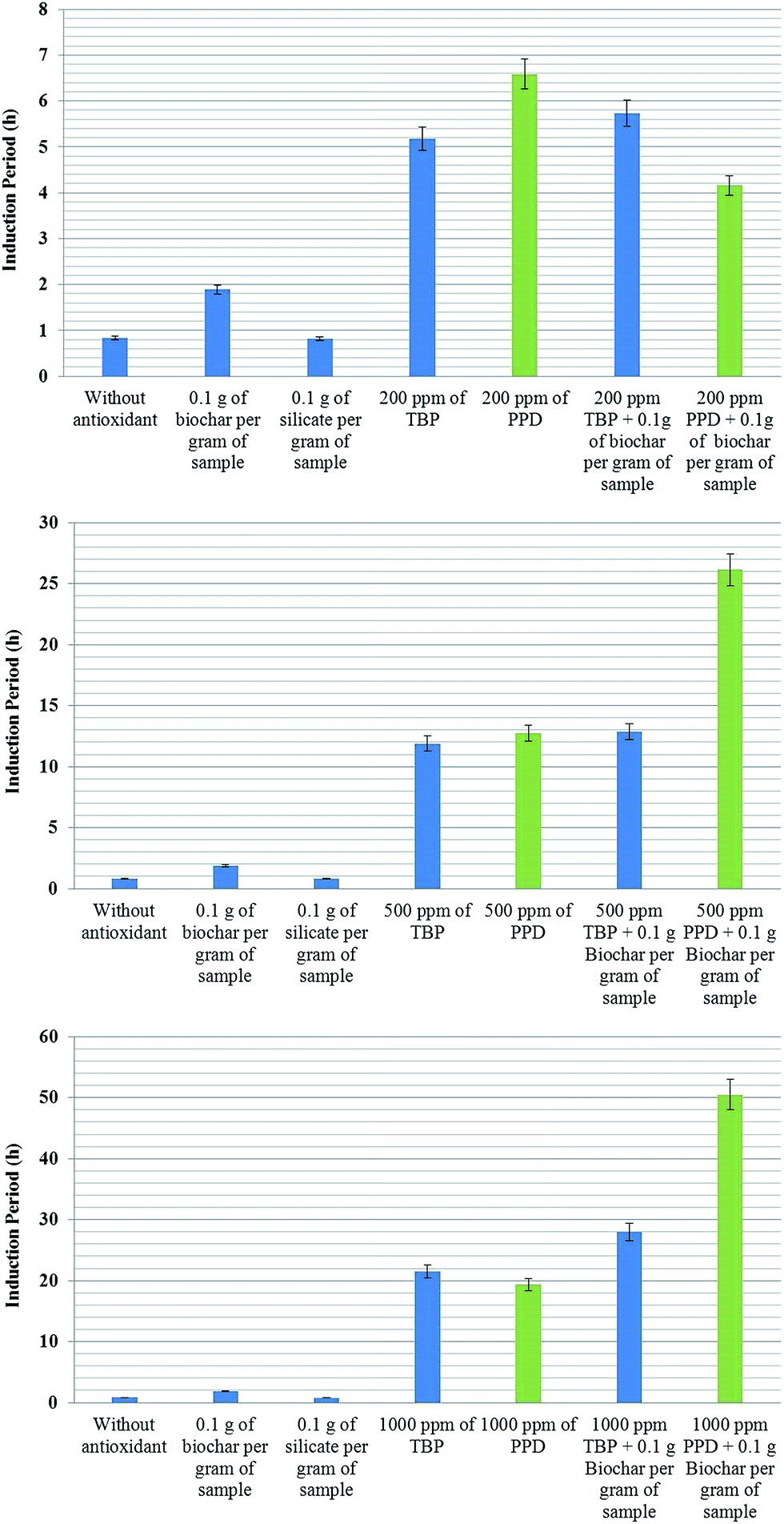 | ||
| Fig. 4 Induction period (IP) of methyl biodiesel from sunflower oil with antioxidants TBP (2,2′-methylene-bis-(4-methyl-6-tert-butyl-phenol)) and PPD (N-(1,3-dimethylbutyl)-N′-phenyl-p-phenylenediamine) with three differing concentrations. The use of 10% of the silicate gave rise to the same IP as that of the biofuel without antioxidants. This indicates that the presence of the solid does not affect the IP. The samples with TBP and biochar show only a small increase compared to the samples with only TBP; these results are very similar to the sum of the induction periods of the TBP samples and biochar samples separately. The sample with PPD shows the synergistic behavior in the presence of biochar. In the sample with 500 ppm of PPD plus biochar, the IP reaches 26 h, twice that of the sample with only PPD. The combination of biochar and 1000 ppm of PPD improves the IP to more than 50 h, two and a half times higher than the sample with only PPD (n = 5, the error bar is the standard deviation). | ||
Sunflower biodiesel was used to demonstrate the effect of synergy because it requires less time for analysis allowing a higher number of replicates.
As seen previously in Fig. 3, the addition of 10% biochar increases the IP of sunflower biodiesel by more than 90%; however, the presence of a solid in the biofuel does not explain the increase in the IP, since the solid silicate did not modify the IP. The biodiesel with 200 ppm of TBP increases the IP to 5.1 ± 0.5 h, and the addition of 0.1 g of biochar together with 200 ppm of TBP enhances the IP by a further 15%, reaching 5.8 ± 0.2 h (topmost graph in Fig. 4). The improvement shown by the biochar with TBP shows the same value of IP as that of the biodiesel plus biochar as compared with the initial biodiesel. These results indicate that the final value of IP is simply the sum of the separate effects of the antioxidants.
The biodiesel with 200 ppm of PPD has an IP of 6.4 ± 0.5 h. In the presence of biochar, the PPD appears to lose some of its antioxidant capacity, giving an IP of only 4.1 ± 0.3 h. The lower graph in Fig. 4 shows the results of the same experiment, but with an increase in the concentration of antioxidants to 500 and 1000 ppm. The results show a significant difference. Biodiesel with 1000 ppm of TBP shows an IP of about 21 h, whilst the sample with both biochar and TBP has an IP of about 28 h, more than 30% higher. The IP of biodiesel with 1000 ppm PPD was about 20 h; however, in the presence of both biochar and PPD the IP reaches more than 50 ± 5 h, an entirely unexpected result. This experiment was repeated five times with similar results. At higher concentrations of PPD, there is a notable synergy with biochar, amplifying the original antioxidant capacity, although at lower concentrations, these effects are not apparent. A possible mechanism which may explain these results is presented below (Fig. 5).
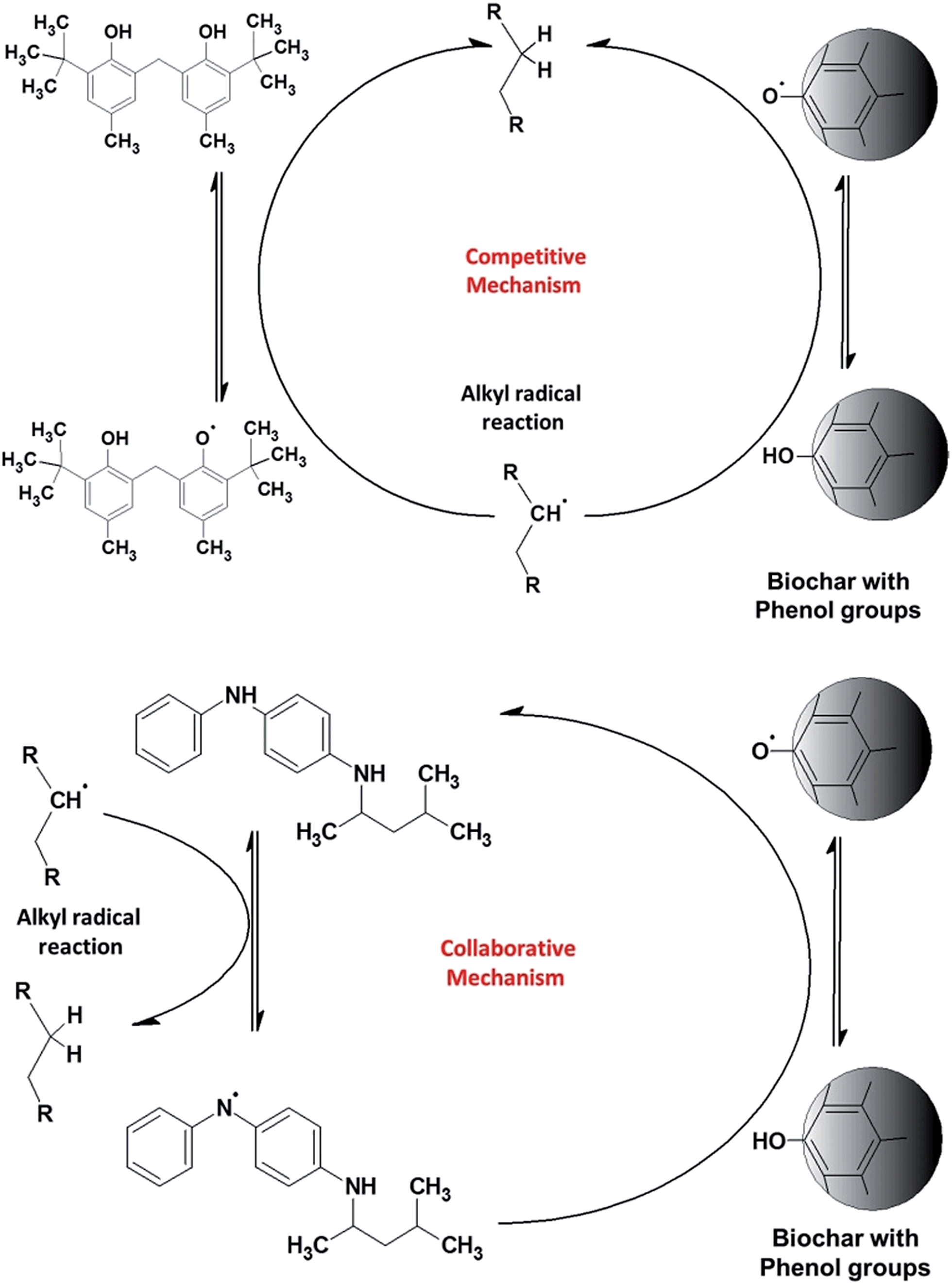 | ||
| Fig. 5 The competitive mechanism proposed to explain the phenol-type antioxidant. The presence of biochar increases the number of phenol groups competing to transfer the hydrogen radical to the alkenyl radical formed (R = –CH2CH2–). Since the phenol is located on the solid surface, it has low mobility and consequently its low contribution to IP increases. The energies evolved by both phenol radicals are similar, this similarity makes the donation of the hydrogen radical from biochar to the antioxidant on the mixture difficult. The amine-type antioxidants act in a collaborative way. The amine antioxidants donate the hydrogen radical to the alkyl radical quickly, and over time the amine antioxidants are regenerated by the phenol groups located on the biochar surface. The bond dissociation energy is lower for the phenol group than that for the amine group, thus favouring the amine. | ||
TBP is a phenol-type antioxidant, as the phenol group on the biochar surface and the scavenging activity of antioxidant compounds depend on hydrogen atom transfer (HAT). The HAT tendency is associated with the lower bond dissociation energies (BDE) of the O–H bond, which tend to be similar for phenol groups. The BDE of amines is higher than that of phenol, which tends to have lower scavenging activity. Thus, when TBP and biochar are added to the biofuel, there is a competition in the reaction with alkyl radicals (topmost diagram in Fig. 5). It is therefore possible that TBP and biochar may have a simple additive effect, since their actions have few or no interactions and solely compete to regenerate the radicals in the original molecule. Since the phenol groups are on the solid material with low mobility, the most antioxidant capacity therefore arises from TBP.
PPD is an amine-type antioxidant that acts in a collaborative way. The PPD reacts with alkyl radicals (among other radicals) transferring hydrogen radicals and forming amine radicals. Thus, the PPD is regenerated by the phenol groups (BDE of phenol is lower than those of anilines) on the surface of the biochar, retaining the concentration of PPD in the biofuel for a significant length of time. The higher concentration of PPD allows sufficient time to regenerate the amine radicals by the phenol groups on the biochar surface. This mechanism explains why the synergy arises only with the higher concentration of PPD; a concentration of 200 ppm is not high enough to give an adequate equilibrium level between amine radical regeneration and amine radical formation.
We therefore suggest that the total antioxidant capacity of the association of PPD with biochar arises from a collaborative mechanism, enhancing the IP of the biofuel with high efficiency (Fig. 5, lower diagram). A similar mechanism was proposed by Rawat et al. to explain the synergy of some combination of two soluble phenol antioxidants.33
Regardless of which mechanism is used, however, the oxidative stability of biofuels shows a marked increase with a combination of PPD and biochar. This phenomenon can be used to produce new antioxidant systems for fuels, paints or foods and new antioxidant materials, to promote new uses for biomass, and to aid in a fuller comprehension of the effect of the combinations of antioxidants.
Conclusion
We have demonstrated the notable synergistic effect of biochar and aromatic diamines on the oxidative stability of biofuels and have proposed a mechanism to explain this. We have also shown that the typical sulphuric acid treatment for producing strong acid sites on biochar surfaces retains the phenol group on the biochar surface. Thus, the same product can be used as a catalyst and as a heterogeneous antioxidant material.The information in this article can be used in the development of new antioxidant systems for fuels, paints or foods, and to gain a fuller understanding of the effects of antioxidant combinations in biofuels.
Acknowledgements
The authors are grateful to Fundação de Amparo a Pesquisa do Estado do Rio de Janeiro-FAPERJ for funding support and S. R. Albinante from Instituto de Macromoleculas for the SEM images.References
- W. R. Stahel, Nature, 2016, 531, 435–438 CrossRef CAS PubMed.
- P. Bousquet, P. Ciais, J. B. Miller, E. J. Dlugokencky, D. A. Hauglustaine, C. Prigent, G. R. Van der Werf, P. Peylin, E.-G. Brunke, C. Carouge, R. L. Langenfelds, J. Lathiere, F. Papa, M. Ramonet, M. Schmidt, L. P. Steele, S. C. Tyler and J. White, Nature, 2006, 443, 439–443 CrossRef CAS PubMed.
- T. Kan, V. Strezov and T. J. Evans, Renewable Sustainable Energy Rev., 2016, 57, 1126–1140 CrossRef CAS.
- A. Raheem, W. A. K. G. Wan Azlina, Y. H. Taufiq Yap, M. K. Danquah and R. Harun, Renewable Sustainable Energy Rev., 2015, 49, 990–999 CrossRef CAS.
- A. Romaní, F. Pereira, B. Johansson and L. Domingues, Bioresour. Technol., 2015, 179, 150–158 CrossRef PubMed.
- D. Kennes, H. N. Abubackar, M. Diaz, M. C. Veiga and C. Kennes, J. Chem. Technol. Biotechnol., 2016, 91, 304–317 CrossRef CAS.
- C.-H. Zhou, X. Xia, C.-X. Lin, D.-S. Tong and J. Beltramini, Chem. Soc. Rev., 2011, 40, 5588–5617 RSC.
- T. Parsell, S. Yohe, J. Degenstein, T. Jarrell, I. Klein, E. Gencer, B. Hewetson, M. Hurt, J. I. Kim, H. Choudhari, B. Saha, R. Meilan, N. Mosier, F. Ribeiro, W. N. Delgass, C. Chapple, H. I. Kenttamaa, R. Agrawal and M. M. Abu-Omar, Green Chem., 2015, 17, 1492–1499 RSC.
- T. Ennaert, J. Van Aelst, J. Dijkmans, R. De Clercq, W. Schutyser, M. Dusselier, D. Verboekend and B. F. Sels, Chem. Soc. Rev., 2016, 45, 584–611 RSC.
- J. Shen, C. Igathinathane, M. Yu and A. K. Pothula, Bioresour. Technol., 2015, 185, 89–98 CrossRef CAS PubMed.
- Y. Chen, G. Tian, M. Zhou, Z. Huang, C. Lu, P. Hu, J. Gao, Z. Zhang and X. Tang, Environ. Sci. Technol., 2016, 50, 5825–5831 CrossRef CAS PubMed.
- G.-X. Yang and H. Jiang, Water Res., 2014, 48, 396–405 CrossRef CAS PubMed.
- X. Tan, Y. Liu, G. Zeng, X. Wang, X. Hu, Y. Gu and Z. Yang, Chemosphere, 2015, 125, 70–85 CrossRef CAS PubMed.
- G. Yang, Z. Wang, Q. Xian, F. Shen, C. Sun, Y. Zhang and J. Wu, RSC Adv., 2015, 5, 40117–40125 RSC.
- V. Hansen, D. Müller-Stöver, J. Ahrenfeldt, J. K. Holm, U. B. Henriksen and H. Hauggaard-Nielsen, Biomass Bioenergy, 2015, 72, 300–308 CrossRef CAS.
- L. J. Konwar, J. Boro and D. Deka, Renewable Sustainable Energy Rev., 2014, 29, 546–564 CrossRef CAS.
- L. J. Konwar, R. Das, A. J. Thakur, E. Salminen, P. Mäki-Arvela, N. Kumar, J.-P. Mikkola and D. Deka, J. Mol. Catal. A: Chem., 2014, 388–389, 167–176 CrossRef CAS.
- C. Poonjarernsilp, N. Sano and H. Tamon, Appl. Catal., B, 2014, 147, 726–732 CrossRef CAS.
- OECD, FAO, oecd-fao-agricultural-outlook-2015_agr_outlook-2015-en (2), Organisation for Economic Co-operation and Development, Paris, 2015 Search PubMed.
- L. d. N. Batista, V. F. Da Silva, É. C. G. Pissurno, T. da Conceição Soares, M. R. de Jesus, C. N. Kunigami, M. G. Brasil and M. G. da Fonseca, Environ. Chem. Lett., 2015, 13, 353–358 CrossRef CAS.
- E. Choe and D. B. Min, Compr. Rev. Food Sci. Food Saf., 2006, 5, 169–186 CrossRef CAS.
- J. Pullen and K. Saeed, Renewable Sustainable Energy Rev., 2012, 16, 5924–5950 CrossRef CAS.
- M. K.-K. Figueiredo, G. A. Romeiro and R. N. Damasceno, J. Anal. Appl. Pyrolysis, 2009, 86, 53–57 CrossRef CAS.
- C. Li, A. Ng, L. Xie, H. Mao, C. Qiu, R. Srinivasan, Z. Yin and Y. Hong, Plant Cell Rep., 2016, 35, 103–114 CrossRef CAS PubMed.
- Y. Li, L. Chen, Y. Lin, Z. F. Fang, L. Q. Che, S. Y. Xu and D. Wu, Anim. Feed Sci. Technol., 2015, 204, 18–27 CrossRef CAS.
- R. Xing, Y. Liu, Y. Wang, L. Chen, H. Wu, Y. Jiang, M. He and P. Wu, Microporous Mesoporous Mater., 2007, 105, 41–48 CrossRef CAS.
- J. W. Lee, M. Kidder, B. R. Evans, S. Paik, A. C. Buchanan III, C. T. Garten and R. C. Brown, Environ. Sci. Technol., 2010, 44, 7970–7974 CrossRef CAS PubMed.
- M.-H. Zong, Z.-Q. Duan, W.-Y. Lou, T. J. Smith and H. Wu, Green Chem., 2007, 9, 434–437 RSC.
- L. Zhu, H. Lei, L. Wang, G. Yadavalli, X. Zhang, Y. Wei, Y. Liu, D. Yan, S. Chen and B. Ahring, J. Anal. Appl. Pyrolysis, 2015, 115, 149–156 CrossRef CAS.
- R. Azargohar and A. K. Dalai, Microporous Mesoporous Mater., 2008, 110, 413–421 CrossRef CAS.
- F. Diana da Silva Araújo, I. C. Araújo, I. C. G. Costa, C. V. Rodarte de Moura, M. H. Chaves and E. C. E. Araújo, Renewable Energy, 2014, 71, 495–501 CrossRef.
- R. Wang, M. A. Hanna, W.-W. Zhou, P. S. Bhadury, Q. Chen, B.-A. Song and S. Yang, Bioresour. Technol., 2011, 102, 1194–1199 CrossRef CAS PubMed.
- D. S. Rawat, G. Joshi, B. Y. Lamba, A. K. Tiwari and P. Kumar, Energy, 2015, 84, 643–655 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c6se00017g |
| This journal is © The Royal Society of Chemistry 2017 |