Gelatin hydrogels formed by orthogonal thiol–norbornene photochemistry for cell encapsulation
Zachary
Mũnoz†
a,
Han
Shih†
ab and
Chien-Chi
Lin
*ab
aDepartment of Biomedical Engineering, Purdue School of Engineering & Technology, Indiana University-Purdue University Indianapolis, Indianapolis, IN 46202, USA. E-mail: lincc@iupui.edu
bWeldon School of Biomedical Engineering, Purdue University, West Lafayette, IN 47907, USA
First published on 23rd April 2014
Abstract
Covalently cross-linked gelatin hydrogels have received considerable attention in biomedical fields due to the inherent bioactivity of gelatin and the stability of covalent bonds linking the gelatin chains. Derivatives of gelatin, such as gelatin-methacrylamide (GelMA), can be cross-linked into covalent hydrogels through radical-mediated chain-growth photopolymerization. However, accumulating evidence suggests that chain-growth polymerized hydrogels may not be ideal for the encapsulation of cells and proteins prone to radical-mediated damage. The formation of heterogeneous kinetic chains following chain-growth polymerization of (meth)acrylates or (meth)acrylamides may also hinder molecular transport or alter cell–cell/cell–material interactions. This study presents a new synthesis route for preparing norbornene-functionalized gelatin (GelNB) that could be used to form orthogonally cross-linked gelatin-based hydrogels via a thiol–ene photo-click reaction. GelNB was synthesized through reacting gelatin with carbic anhydride in aqueous buffered solution, and the degree of norbornene substitution was controlled by adjusting the reaction time and the solution pH value. GelNB hydrogels were prepared by step-growth thiol–ene photopolymerization using multifunctional thiols as gel cross-linkers and the degree of GelNB hydrogel cross-linking was tuned by adjusting the thiol concentration, GelNB content, or cross-linker functionality. The cytocompatibility of orthogonally cross-linked GelNB hydrogels were demonstrated by in situ photo-encapsulation of human mesenchymal stem cells (hMSCs). When compared with the chain-growth GelMA hydrogels, the orthogonally cross-linked GelNB hydrogel promoted a faster and higher degree of cell spreading.
1. Introduction
Gelatin is a natural biomacromolecule derived from denatured collagen. Compared with its precursor, gelatin has higher water solubility and lower immunogenicity.1 Gelatin is inexpensive and contains peptide sequences critical for cell surface receptor recognition. For example, gelatin contains the Arg–Gly–Asp (RGD) sequence that can bind to cell surface integrins. Therefore, gelatin can be used to improve cell attachment, mostly through physical adsorption on the surface of a substrate that is otherwise non-adherent to cells. In addition to the cell affinity, gelatin can be cleaved by various proteases, such as matrix metalloproteinase 2 (MMP-2) and MMP-9. The protease sensitivity makes gelatin suited to the fabrication of hydrogels for three-dimensional (3D) cell studies. The most commonly used method to prepare gelatin hydrogel is through temperature-induced physical gelation. While this method requires no chemical modification on gelatin, it often uses temperature change beyond the physiologically acceptable range. Therefore, temperature-induced physical gelation is not applicable for in situ cell encapsulation. Physically gelled gelatin hydrogel also contains reversible crosslinks that lack stability for longer-term biomedical applications. Nonetheless, the inherent bioactivity of gelatin warrants its role as an important natural macromolecule for tissue engineering and regenerative medicine applications.2–5To increase the stability of gelation hydrogels while preserving the bioactivity offered by gelatin for 3D cell culture, chemical cross-linking methods are increasingly used for gelatin hydrogel fabrication. For example, Draye et al. prepared dextran–gelatin hybrid hydrogels through a Schiff base formation between oxidized dextran and gelatin.6 In this cross-linking scheme, primary amines on gelatin undergo nucleophilic addition with aldehydes on oxidized dextran to give stable imine bonds that cross-link gelatin chains into covalent hydrogels. Anseth and colleagues synthesized methacrylate modified gelatin (i.e., GelMA) that can be chain-polymerized into covalent gelatin hydrogels for in situ cell encapsulation and long-term 3D cell culture.7 Specifically, GelMA hydrogel-encapsulated valvular interstitial cells (VICs) exhibited higher viability and spreading when compared with the pure poly(ethylene glycol) (PEG) hydrogel system.7 Khademhosseini and colleagues have also utilized the GelMA hydrogel system for 3D cell culture and for micro-scale tissue engineering.8–13 GelMA could also be co-polymerized with PEG-dimethacrylate (PEGDMA) to yield hybrid hydrogels with highly tunable gel mechanical properties, cell-binding, and protease degradability for in situ cell encapsulation.14 In another example, thiolated gelatin was mixed with PEG-diacrylate (PEGDA) and gelation was achieved through a mixed-mode thiol–acrylate photopolymerization.15
Although photopolymerized GelMA hydrogels have proven powerful and versatile in 3D cell culture, the cross-linking of these gelatin hydrogels was a result of random chain-growth photopolymerization that has been shown to yield high initial radical concentrations and to produce heterogeneous hydrophobic kinetic chains following gelation.16,17 While convenient, chain-growth gelation may not be ideal for some cell types that are prone to radical-mediated damage.18 This disadvantage can be addressed by forming hydrogels with orthogonal cross-links or through ‘click chemistry’.18 Light-mediated thiol–norbornene (or thiol–ene) chemistry is one such example suitable for preparing cell-laden hydrogels with orthogonal cross-links.19 Thiol–ene hydrogels based on multifunctional PEG-norbornene macromer and cysteine-bearing peptide cross-linkers have been used to encapsulate a host of cells, such as human mesenchymal stem cells (hMSCs),20,21 valvular interstitial cells (VICs),22,23 pancreatic beta cells,18,21 and pancreatic epithelial cells.24 The thiol–ene photo-click reaction is not oxygen-inhibited and requires a lower radical concentration for initiation.19 Therefore, the cross-linking of thiol–ene hydrogels is extremely fast (with gel point of the order of a few seconds18,19,25) and produces a hydrogel network with an idealized and orthogonal structure.25 Since PEG-based thiol–ene hydrogels do not possess the necessary bioactivity for the encapsulated cells, cysteine-containing integrin-binding motifs (e.g., CRGDS) are often introduced within these hydrogels to provide the critical cell–matrix interactions. In addition, bis-cysteine-bearing MMP-sensitive peptides (e.g., CGPQG↓IWGQC, the arrow indicates a protease cleavage site) can be used as a gel cross-linker that permits cell-mediated local matrix cleavage.19,20
Recently, norbornene-functionalized hyaluronic acid (NorHA) was developed by the Burdick group for preparing photo-patternable thiol–ene hydrogels.26 To synthesize NorHA, hyaluronic acid was first converted to its tetrabutylammonium salt (HA-TBA). HA-TBA and 5-norbornene-2-carboxylic acid were then reacted in anhydrous DMSO in the presence of 4-(dimethylamino)pyridine (DMAP) and di-tert-butyl dicarbonate (Boc2O). The crude NorHA was obtained after 20 h of reaction at 45 °C. The synthesized NorHA could be cross-linked by dithiothreitol (DTT) at various thiol/ene stoichiometric ratios via a step-growth radical-mediated photopolymerization.26 The resulting NorHA-DTT hydrogels were cytocompatible and photo-patternable, two important characteristics shared with PEG-based thiol–ene hydrogels. The benefits of rapid and orthogonal thiol–ene reaction and bioactivity and biocompatibility of hyaluronic acid were retained in this new class of thiol–ene hydrogels. Although immobilized cell-adhesive ligands were still required in this system to support cell adhesion, this work has demonstrated the feasibility of synthesizing thiol–ene hydrogels using non-PEG based biomacromolecules.
In this contribution, we describe a new synthesis route for preparing norbornene-functionalized gelatin (GelNB) that can be stably cross-linked into 3D hydrogels for in situ cell encapsulation. Briefly, GelNB was prepared via reacting gelatin with carbic anhydride in aqueous buffer solutions at 50 °C. The reaction time and the buffer pH were adjusted to obtain GelNB with a sufficient degree of functionalization suitable for gel cross-linking. GelNB was reacted with bi- or tetra-functional thiols via a radical mediated step-growth thiol–ene reaction to form gelatin-based thiol–ene hydrogels. The properties of GelNB hydrogels were tuned by using different weight concentrations of GelNB in the precursor solution or thiol-containing linkers with different functionality (DTT or PEG-tetra-thiol). The cytocompatibility of GelNB hydrogels was evaluated by in situ encapsulation of human mesenchymal stem cells (hMSCs).
2. Materials & methods
2.1. Materials
Type A gelatin (Bloom 238–282) and 4-arm PEG (M.W.: 5 kDa) were purchased from Amresco and JenKem Technology USA, respectively. Carbic anhydride (endo-cis-5-norbornene-2,3-dicarboxylic anhydride), dithiothreitol (DTT), and fluoraldehyde were purchased from Fisher Scientific. DPBS, fetal bovine serum (FBS), 100× antibiotic–antimycotic, and a live/dead staining kit for mammalian cells were obtained from Life Technologies. DMEM was acquired from HyClone. All other chemicals were obtained from Sigma-Aldrich unless otherwise noted.2.2. Synthesis, purification, and characterization of GelMA and GelNB
The synthesis and purification of methacrylamide-functionalized gelatin (GelMA) were performed according to a published protocol.8,13 The synthesis of norbornene-functionalized gelatin (GelNB) was carried out under similar reaction conditions to those for the synthesis of GelMA, except that carbic anhydride was used for the reaction. Briefly, 10 wt% gelatin was dissolved in DPBS at 50 °C under constant stirring. Carbic anhydride (20 wt/vol%) was added to the gelatin solution and the pH value of the buffer solution was adjusted using sodium hydroxide solution. The reaction was quenched by adding 5× warm DPBS (37 °C). After centrifugation to remove any undissolved carbic anhydride, GelNB solution was dialyzed in ddH2O at 40 °C for 3 days (MWCO: 6–8 kDa) and lyophilized to obtain dry product. The degree of norbornene substitution was determined with the fluoraldehyde assay using unmodified gelatin with known concentrations as standards.2.3. Synthesis and purification of LAP & PEG4SH
The photoinitiator lithium arylphosphinate (LAP) was synthesized as described previously.27 PEG4SH was synthesized as described below: (1) 4-arm PEG was first dissolved in anhydrous toluene and dried through solvent evaporation under reduced pressure. The dried PEG was re-dissolved in anhydrous tetrahydrofuran (THF), followed by addition of sodium hydride (1.5-fold excess to hydroxyl group) slowly. After the cessation of hydrogen gas, allyl bromide (6-fold excess to hydroxyl group) was added drop-wise to the PEG solution, which was kept at 40 °C under nitrogen purging overnight in the dark. Next, sodium bromide salt precipitate was filtered off to obtain 4-arm PEG-allylether (PEG4AE), which was precipitated in cold ethyl ether, filtered, and dried under reduced pressure. The desired amount of dried PEG4AE was dissolved in a dichloromethane (DCM) solution containing PEG4AE and the photoinitiator Irgacure I-2959 (0.5 wt%). With stirring, thioacetic acid (2-fold excess to allylether group) was added slowly to the solution. Thiol–ene conjugation was initiated by UV-light exposure (Omnicure S1000, 365 nm and 10 mW cm−2) for 15 min, followed by the addition of another portion of I-2959 (0.5 wt%) and another 30 min of reaction. After the thiol–ene photo-conjugation, 4-arm PEG-thioacetate was precipitated in cold ethyl ether, filtered and dried under reduced pressure. Thioacetate was hydrolyzed in a solution of sodium hydroxide (2 N) for 5 minutes, followed by solution neutralization with an equal volume of hydrochloride acid (2 N) solution. The volume of PEG4SH solution was reduced by half through rotary evaporation and dialyzed against ddH2O for 2 days at room temperature. PEG4SH was obtained by freeze drying and the purity was characterized by 1H NMR (>90%, Bruker 500).2.4. Preparation of hydrogels and swelling ratio measurements
Chain-growth GelMA or step-growth thiol–ene GelNB hydrogels were prepared by photopolymerization in PBS. Multifunctional thiol (i.e., di-thiol DTT or tetra-thiol PEG4SH) was also added to the GelNB precursor solution as a cross-linker. 1 mM of LAP was used as the photoinitiator and an ultraviolet light source (365 nm, 10 mW cm−2, 5 min) was used to initiate the gelation. After photo-crosslinking, hydrogels (50 μL) were incubated in ddH2O at 37 °C on an orbital shaker for 24 h to remove un-crosslinked (sol fraction) components. The gels were dried and weighed to obtain dry weight (WDry). The dried gels were incubated in 5 mL of buffer solution (pH 7.4 PBS) at 37 °C on an orbital shaker for 2 days for reaching equilibrium swelling. Next, hydrogel swollen weights were measured (WSwollen) and used to calculate the mass swelling ratio: qeq, which was defined as WSwollen/WDry.2.5. Rheometry
To monitor gelation kinetics, in situ photorheometry was conducted using a digital rheometer (Bohlin CVO 100) operated in an oscillatory rheometry time-sweep mode with 5% strain, 1 Hz frequency, and a gap size of 90 μm. Gelation was conducted in a UV cure cell at room temperature. A macromer solution (100 μL) was placed on a quartz plate in the UV cure cell and irradiated with UV light (Omnicure S1000, 365 nm, 10 mW cm−2) through a liquid light guide. UV light was turned on 10 seconds after the onset of rheometrical measurements. Gel points (i.e., crossover time) were defined as the time when storage modulus (G′) surpassed loss modulus (G′′).For gel stiffness characterization, gelatin-based hydrogel slabs were first fabricated between two glass slides separated by 1 mm thick spacers. Using a biopsy punch, circular gel discs (8 mm in diameter) were punched out from the gel slabs and incubated in pH 7.4 PBS for 2 days. At equilibrium swelling, oscillatory rheometry in the strain-sweep mode (0.1% to 5%) was performed. Gel moduli were measured using a parallel plate geometry (8 mm) with a gap size of 800 μm and were reported using averaged G′ values obtained in the linear viscoelastic region (LVR). In the strain range (0.1% to 5%), LVR was identified as the region where the modulus values (G′) do not deviate by more than 10% from a plateau value.
2.6. Cell encapsulation
hMSCs (used between passage 2 and 4 and a final cell density of 5 × 106 cells mL−1 in hydrogel) were suspended in a sterile polymer precursor solution containing either GelNB/DTT or GelMA with the desired gelatin weight content (4 or 8 wt%). All precursor solutions (25 μL) also contained 1 mM of LAP as the photoinitiator. Gelation and cell encapsulation were achieved simultaneously through long-wave UV light (365 nm, 6 mW cm−2) exposure for 5 min at room temperature. Following cell encapsulation, cell-laden hydrogels were maintained in hMSC growth media (low-glucose DMEM supplemented with 10% FBS, 1 ng mL−1 basic fibroblast growth factor (bFGF, Peprotech), and 1× antibiotic–antimycotic) and incubated at 37 °C in 5% of CO2. Cell culture media were refreshed every 2–3 days.2.7. Cell viability, actin staining, and imaging
Cell viability was monitored through live/dead staining where calcein AM (0.25 μL mL−1) and ethidium homodimer-1 (EthD-1, 2 μL mL−1) were used to stain live and dead cells, respectively. A confocal microscope (Olympus Fluoview, FV1000) was used to image the stained gels (100 μm thick and 10 μm depth increments). Four images were taken per hydrogel (n = 3 per condition) and the number of live and dead cells was counted per image. Cell viability was determined by the percent of live cells over the total cell counts. Cell spreading was characterized by measuring the longest cell end-to-end distance.Actin filaments of hMSCs were stained to visualize cell spreading. Briefly, cells were fixed with 4% paraformaldehyde for 45 minutes at room temperature on an orbital shaker. After cell fixation, hydrogels were washed twice with HBSS for 10 minutes and permeabilized with 0.5% of Triton X-100 in HBSS for 45 minutes, followed by washing twice with HBSS (10 minutes per wash). Hydrogels were blocked with HBSS solution containing equal volumes of BSA, FBS, and polyvinylpyrrolidone (5 vol% each) at 4 °C overnight in the dark on an orbital shaker. Next, cell-laden hydrogels were incubated in rhodamine phalloidin (cytoskeleton) solution (in HBSS with 1 vol% of Tween 20). Following two washings with HBSS (1 hour per wash) at room temperature, hydrogels were stored in blocking solution (HBSS with 1 vol% Tween 20) overnight at 4 °C. On the day of imaging, cell nuclei were counter-stained with DAPI for an hour at room temperature and washed three times with HBSS (30 minutes per wash). Hydrogels were imaged with a confocal microscope as described above.
2.8. Data analysis & statistics
Data analysis and Student's t-test were performed on Prism 5 software. Unless otherwise noted, all experiments were conducted independently three times. All data presented are mean ± SEM.3. Results & discussion
3.1. Optimization of GelNB synthesis using carbic anhydride
To synthesize gelatin-norbornene (GelNB), we modified a protocol for gelatin-methacrylamide (GelMA) synthesis that was previously developed by the Anseth7 and the Khademhosseini groups.8,13 Here, the primary amines of gelatin served as nucleophiles for reaction with carbic anhydride to form amide-linked norbornene (Fig. 1A). The degree of norbornene substitution was characterized by a fluoraldehyde assay, which detects the concentration of primary amines on gelatin. Initially, the reaction was carried out by dissolving 20 wt/vol% of carbic anhydride in gelatin/DPBS (starting solution pH = 7.4), which was kept at 50 °C for 2 h. While these reaction conditions were optimized for the synthesis of GelMA with high degrees of substitution (>80%), we could only obtain a very low degree of norbornene substitution on gelatin (<20%, data not shown) using this protocol. When the reaction time was increased to 6 h and 70 h, however, the degree of norbornene substitution was increased to 27 ± 3% and 36 ± 2%, respectively (Fig. 1B). Further prolonging the reaction time did not yield an additional increase in the degree of substitution (data not shown). During the reaction, we noticed that the added carbic anhydride did not dissolve completely. When the pH value of the reaction buffer was increased to and maintained at 8, a clear reaction mixture was obtained. Adjusting pH values to slightly basic also increased the degree of substitution to 44 ± 2% (Fig. 1C). Further increasing the pH of the reaction to 9 did not enhance the degree of substitution (Fig. 1C). The lower degree of norbornene substitution regardless of the reaction conditions might be a result of the steric hindrance imposed by the strained norbornene. Although the reaction efficiency of carbic anhydride and gelatin was lower than that of methacrylic anhydride and gelatin, we were able to obtain step-growth gelatin hydrogels with various degrees of cross-linking using GelNB having at least 40% degree of substitution (see the section below). From the perspective of preserving gelatin bioactivity after chemical modification, a lower degree of substitution may actually be favorable since the majority of the bioactive primary amine groups remain available for cellular recognition.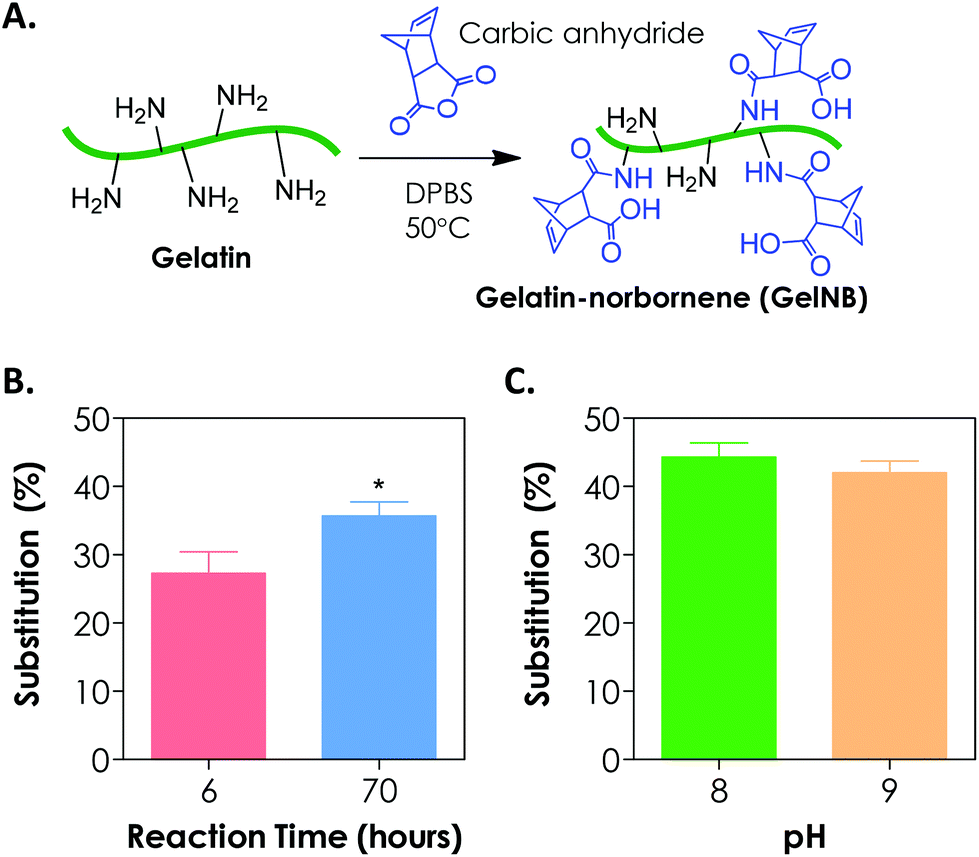 | ||
| Fig. 1 (A) Schematic of gelatin-norbornene (GelNB) synthesis. (B) Effect of reaction time (without pH adjustment) on the degree of norbornene substitution (*p < 0.05). (C) Effect of the solution pH value on the degree of norbornene substitution. In all reactions, carbic anhydride was added at 20 wt/vol%. | ||
3.2. Orthogonal gelation of GelNB hydrogels
To evaluate the cross-linking of gelatin hydrogels via an orthogonal step-growth thiol–ene photo-click reaction, we used a bi-functional cross-linker dithiothreitol (DTT) at various concentrations and the photoinitiator lithium arylphosphinate (LAP) at 1 mM (Fig. 2A). In situ photo-rheometry was used to monitor gelation kinetics under long-wave UV light exposure (365 nm, 10 mW cm−2). As shown in Fig. 2B, gelation took place rapidly after the UV light was turned on at 10 s. The gel point of this reaction was determined to be around 12 s. Although the onset of gel cross-linking was rapid, complete gelation (95% of the plateau G′ value, at 0.4 kPa) was roughly 300 seconds of light exposure, a rate significantly slower than that of the purely PEG-based thiol–ene gelation.18 We also compared the gelation kinetics of step-growth GelNB-DTT hydrogel to that of chain-growth GelMA hydrogel using gelatin-derivatives with similar degrees of functionalization (∼45%, Fig. 2C). Interestingly, the two systems did not exhibit substantial differences in gel points. At the end of the gelation experiment, however, chain-growth GelMA hydrogel yielded a higher shear modulus (∼0.9 kPa) than that in step-growth GelNB-DTT gelation (∼0.4 kPa). This result was unexpected since previous studies comparing photo-gelation of step-growth PEG-norbornene (PEGNB) and chain-growth PEG-diacrylate (PEGDA) have revealed that thiol–ene gelation was faster due to its non-oxygen inhibited nature.18 This was likely due to the complex amino acid sequences in gelatin that altered the kinetics of thiol–ene reaction, as studies have shown that the kinetics of thiol–ene or thiol–vinyl gelation, either light dependent or independent, could be affected by the amino acid sequences near the thiol-bearing linkers.25,28,29 We also performed controlled experiments to show that the gelation was through a light-initiated thiol–ene reaction. As shown in Fig. 2D (4 wt% GelNB) and 2E (8 wt% GelNB), the low G′ values (<0.5 Pa) and the lack of a cross-over point during the entire in situ rheometry test confirmed the light-mediated thiol–ene gelation mechanism. | ||
| Fig. 2 (A) Schematic of step-growth photopolymerization of GelNB hydrogels using bi-functional thiol as a cross-linker and LAP (1 mM) as a photoinitiator. (B) In situ photorheometry of GelNB (5 wt%) hydrogel gelation using DTT as a cross-linker ([SHDTT] = 15 mM). Light was turned on at 10 seconds (dashed line) after the start of the measurement. (C) Comparison of step-growth GelNB-DTT and chain-growth GelMA hydrogel gelation kinetics. Control experiments show that no gelation, physical or chemical, could be achieved using 4 wt% (D) and 8 wt% (E) of GelNB without light exposure. | ||
When compared with GelMA hydrogels, the lower moduli of GelNB-DTT hydrogels at equivalent gelatin content could be a result of the short DTT linker and/or the orthogonal crosslinks formed after the step-growth gelation. In the GelMA system, poly(methacrylamide) kinetic chains formed following chain-growth polymerization. The heterogeneous kinetic chains might cause a higher degree of chain entanglements in the GelMA system, which could increase the final gel modulus. Finally, we found that unmodified gelatin formed physical association and gelation within a few minutes at room temperature when its concentration was above 6 wt%. Chemical modification on gelatin (e.g., GelMA or GelNB) leads to decreased gelatin solubility at room temperature but increases the concentration threshold above which physical crosslinking forms. We did not observe physical gelation at GelNB concentrations below 8 wt% (Fig. 2E). Following chemical cross-linking of gelatin hydrogel, physical crosslinks could still form in the hydrogels, especially for gelatin gels formed from higher macromer concentrations. Future work may focus on characterizing the degree of physical cross-links contained in the highly chemically cross-linked gelatin hydrogel, as well as on exploiting the dual-mode crosslinking for manipulating cell fate processes.
3.3. Effect of macromer concentration on GelNB hydrogel crosslinking
Controlling the cross-linking density of a hydrogel is of paramount importance when designing matrices for tissue engineering and controlled release applications because cross-linking density not only determines molecular transport properties but also affects cell fate processes. One way of achieving a tunable gel cross-linking density in step-growth hydrogel is by adjusting the concentration of the cross-linker. In one example, we fixed [GelNB] at 5 wt% but varied the bi-functional thiol crosslinker (e.g., DTT) concentration to 10, 15, or 20 mM thiol (i.e., 5, 7.5, 10 mM DTT). Here, we defined the R ratio as “mM thiol per wt% GelNB” because even though we could characterize the degree of substitution on GelNB (based on a reduction in free amine group content), the exact molecular weights of gelatin and hence the molar concentrations of norbornene groups were difficult to define. The use of 10, 15, or 20 mM thiol would give rise to an R[SH]/[GelNB] value of 2, 3, or 4 (Fig. 3A). As shown in Fig. 3A, gelatin hydrogels prepared from these formulations exhibited the highest stiffness (G′ = 0.6 kPa) at 15 mM thiol (i.e., 7.5 mM DTT). Furthermore, the use of a lower thiol concentration (e.g., 10 mM) reduced slightly the gel modulus (not statistically significant) and the use of a higher thiol concentration (e.g., 20 mM) led to a significant reduction of gel cross-linking. This was likely caused by insufficient orthogonal gel cross-linking due to an excess amount of thiol groups. It is worth noting that the exact stoichiometric ratio of thiol to ene groups at R[SH]/[GelNB] = 3 was unknown. However, based on our gelation test (Fig. 3A), this R value gave rise to GelNB hydrogels with the highest moduli, indicating that the stoichiometric ratio of thiol-to-ene at this R value could be close to one. Since an R ratio (i.e., mM thiol per wt% GelNB) of 3 yielded the highest degree of gel cross-linking, this ratio was used in the subsequent studies.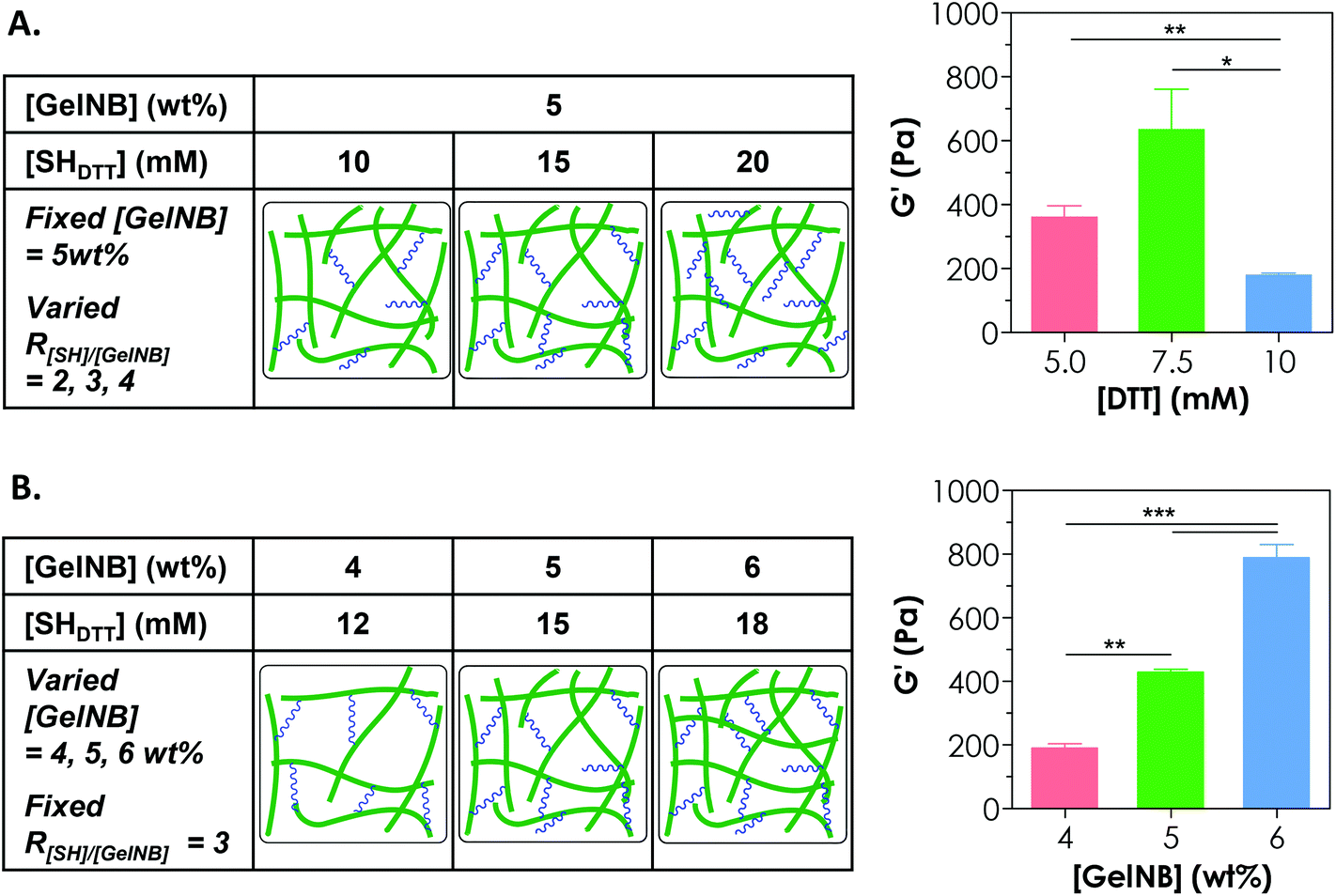 | ||
| Fig. 3 Controlling the cross-linking density of GelNB hydrogels via adjusting DTT and GelNB contents. (A) At a fixed GelNB content (5 wt%), gelatin hydrogel cross-linking density could be tuned by adjusting the DTT concentration. R is defined as mM thiol per wt% of GelNB. (B) At a fixed R, gelatin hydrogel cross-linking density could be tuned by adjusting the GelNB concentration. The experimental conditions were identical for 5 wt% GelNB and 15 mM SHDTT in both (A) and (B), but these experiments were conducted independently with at least three samples in each gel formulation. Data were presented as mean ± SEM. *, **, and *** represent p < 0.05, 0.01, and 0.001 respectively. | ||
Another approach for tuning the cross-linking density of a chemically cross-linked hydrogel is by adjusting the gelatin macromer concentration. For example, increasing the GelNB content in the precursor solution led to significant increases in the shear modulus (G′) of the hydrogels. Specifically, gelatin hydrogels formed from 4, 5, or 6 wt% GelNB had a shear modulus of approximately 0.2, 0.4, or 0.8 kPa, respectively (Fig. 3B). It is worth noting that the experiments in Fig. 3A and 3B were conducted independently. Although there was a mismatch between the data values in the 5 wt% GelNB + 15 mM SHDTT groups (middle bar in both graphs: G′ = 637 ± 124 Pa in Fig. 3A and 429 ± 10 Pa in Fig. 3B; mean ± SEM), the difference has no statistical significance. Similar to other chemically cross-linked hydrogels, step-growth GelNB-DTT hydrogels exhibited a lower equilibrium swelling ratio (qeq) at higher GelNB macromer concentration (data not shown).
Adjusting the macromer concentration to tune the gel cross-linking density is a valid approach not only for chain-growth hydrogels bearing homo-polymerizable vinyl groups (e.g., methacrylamide on GelMA, acrylates on poly(ethylene glycol)-diacrylate or PEGDA), but also for step-growth hydrogels with mutually reactive functional groups (e.g., norbornene and thiol). Increasing the gelatin concentration in the pre-polymer solution directly increases the concentration of the cross-linkable moiety (e.g., methacrylamide in GelMA or norbornene in GelNB). In chain-growth photopolymerization, a higher methacrylamide concentration leads to accelerated gelation and increased cross-linking density. In step-growth polymerization, higher macromer and cross-linker concentrations improve the gelation efficiency and network ideality,25,30 which also results in the formation of a network with higher stiffness and lower swelling ratio. Although simple, adjusting the gel stiffness by increasing the GelNB (or GelMA) concentration in the precursor solution also increases the concentration of bioactive motifs. As described earlier, gelatin contains sequences for both integrin recognition and protease cleavage. The coupling of increased mechanical stimulation (due to increased crosslinking density) and cell–material interactions (due to higher concentrations of bioactive motifs) might confound the interpretation of experimental results relevant to cellular processes.
3.4. Effect of cross-linker functionality on GelNB hydrogel crosslinking
Adjusting the functionality of the cross-linker to affect the degree of hydrogel cross-linking in step-growth hydrogels has been exploited by us and other groups for the purpose of controlling the network ideality and permeability.25,30,31 Prior studies have mostly focused on synthetic hydrogels, including PEG-acrylate or PEG-vinylsulfone hydrogels formed by a conjugation addition reaction (i.e., Michael-type addition)30,31 and PEG-norbornene hydrogels formed by radical-mediated thiol–ene photopolymerization.18,25 These studies have shown that increasing cross-linker functionality enhances the cross-linking efficiency of step-growth hydrogels. Here, we used GelNB (at 5 wt% and ∼41% degree of norbornene substitution) together with a bi-functional or tetra-functional thiol linker (DTT or PEG4SH) to yield step-growth hydrogels with different degrees of network cross-linking (Fig. 4A). Fig. 4B shows that when 4-arm PEG-thiol (PEG4SH5 kDa, fB = 4) was used as the gel cross-linker, the equilibrium shear modulus of the GelNB-PEG4SH hydrogel was increased to roughly 5 kPa from 0.4 kPa when a bi-functional DTT (fB = 2) was used. The equilibrium swelling ratios (Fig. 4C) and shear modulus of the GelNB hydrogels also show an inverse correlation. In this example, the stoichiometric ratio between the thiol and ene groups was maintained in both GelNB-DTT and GelNB-PEG4SH hydrogel systems because both [GelNB] (5 wt%) and [SH] (15 mM) were kept constant and the only difference was the functionality of the cross-linkers (i.e. fB = 2 for DTT and fB = 4 for PEG4SH). To increase the degree of gelatin hydrogel swelling while maintaining the high cross-linking efficiency, one could use multi-arm (e.g., PEG4SH or PEG8SH) cross-linkers with higher molecular weights. This approach should increase hydrogel swelling due to the extended cross-linker chain length in between the gelatin chains.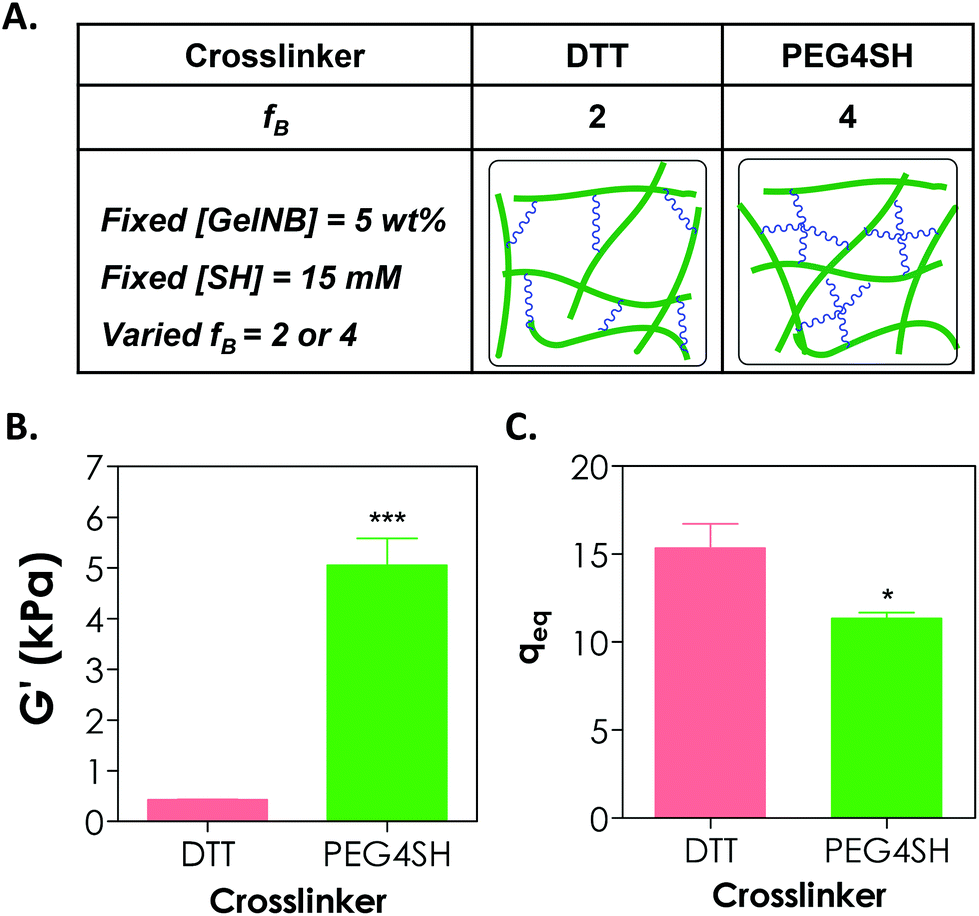 | ||
| Fig. 4 (A) Controlling the cross-linking density of GelNB hydrogels via adjusting the functionality of the cross-linker (fB). The hydrogel polymerized from higher cross-linker functionality has higher cross-linking density but a similar bioactive motif concentration. (B) Influence of cross-linker functionality (DTT, fB = 2; or PEG4SH, fB = 4) on gelatin hydrogel shear modulus (G′). (C) Influence of cross-linker functionality on equilibrium mass swelling ratio (qeq) of the hydrogel. [GelNB] = 5 wt%. [SH] = 15 mM (i.e., 7.5 mM DTT or 3.75 mM PEG4SH. R = 3). * and *** represent p < 0.05 and 0.001 respectively. | ||
It is worth noting that in chain-growth GelMA hydrogels, increasing the hydrogel stiffness by using higher concentration of GelMA inevitably increases the concentration of bioactive motifs. In the current GelNB hydrogel system, the gelatin used was kept at 5 wt% for both systems and the stiffness was tuned by using cross-linkers with different functionality (2 or 4). This indicates that the concentrations of bioactive motifs contributed by gelatin were similar for both systems (Fig. 4A). Although not explored in this contribution, gelatin hydrogels with independently tuned biophysical and biochemical properties may be used to understand the influence of individual ECM cues on cell fate processes.
3.5. Cytocompatibility of GelNB-DTT hydrogels using in situ encapsulation of hMSCs
One attractive feature of gelatin-based hydrogels is the inherent bioactivity offered by the peptide sequences in gelatin. Gelatin contains numerous bioactive sites, notably the integrin binding sequence RGD and the substrate for protease cleavage (collagenase, gelatinase, etc.). Combining the aforementioned bioactivity with the network stability offered by the chemical cross-links (polymethacrylate in GelMA or thioether in GelNB), these modified gelatin hydrogels are an interesting class of biomaterials for cell-based regeneration applications. For example, the presence of cell-binding motifs in the gelatin sequence renders this class of hydrogels attractive because no additional cell-binding ligand is needed during network gelation. The cytocompatibility of chain-growth GelMA hydrogels has been extensively evaluated previously in cell lines and primary cells, including valvular interstitial cells,7 fibroblasts, and human vascular endothelial cells.8,12,32,33 Here, we evaluated the cytocompatibility of step-growth GelNB hydrogels using in situ encapsulation of human mesenchymal stem cells (hMSCs). Chain-growth GelMA hydrogels were used as the control. Both GelNB and GelMA used in this study had a similar degree of functional group substitution (∼40–45%). Fig. 5A shows representative confocal z-stack images of encapsulated hMSCs stained with calcium AM (green: live cells) and ethidium bromide homodimer (red: dead cells) one day post-encapsulation. Although most of the hMSCs remained viable in both hydrogel systems, slightly more dead cells could be seen in chain-growth GelMA hydrogels than in step-growth GelNB-DTT hydrogels (for both 4 and 8 wt% gelatin gels). We also analyzed semi-quantitatively the encapsulated cell viability by counting percentages of live cells and found that cell viability in step-growth GelNB-DTT gels was significantly higher than in chain-growth GelMA hydrogels (Fig. 5B). Specifically, ∼97% and ∼91% of the counted cells were stained green in 4 and 8 wt% of step-growth GelNB-DTT hydrogels, respectively. On the other hand, only ∼85% and 80% of the counted cells were alive in 4 and 8 wt% chain-growth GelMA hydrogels, respectively. Live/dead staining also revealed that, after 1-day in vitro culture, hMSCs encapsulated in these two gelatin hydrogel systems still retained the rounded morphology without visible cellular protrusion or spreading (Fig. 5A).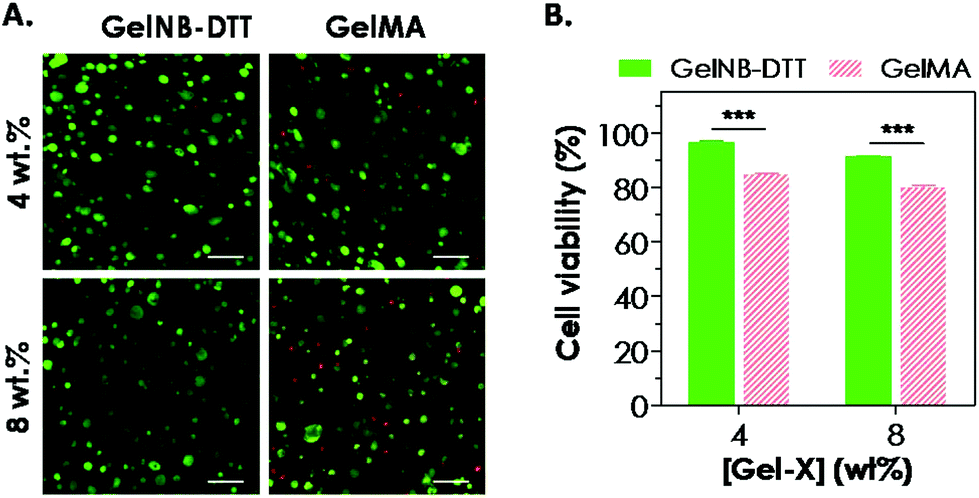 | ||
| Fig. 5 Cytocompatibility of step-growth GelNB-DTT and chain-growth GelMA hydrogels for in situ encapsulated hMSCs. (A) Representative confocal z-stack images of live/dead stained hMSCs encapsulated in step-growth GelNB-DTT or chain-growth GelMA hydrogels with two macromer concentrations (live cells stained green and dead cells stained red; scales: 100 μm). (B) Percentages of live cell count in gelatin hydrogels with different weight contents (Gel-X: GelNB-DTT or GelMA). The degree of substitution for GelNB and GelMA was both at ∼40–45%. | ||
Maintaining the viability of hMSCs in chemically cross-linked hydrogels is of critical importance. Results shown in Fig. 5 have demonstrated that the orthogonal thiol–ene reaction was highly cytocompatible for in situ encapsulation of hMSCs. We and other groups have previously reported the differential influences of chain-growth and step-growth gelation on the viability of in situ encapsulated proteins and cells.17,18,34 However, previous comparisons were all based on synthetic PEG-based hydrogels. McCall and Anseth have shown that higher initial radical concentration in chain-growth photopolymerization caused a significant reduction in encapsulated protein bioactivity.17 In another example, we showed that step-growth thiol–norbornene photo-click gelation was more cytocompatible than chain-growth polymerization for in situ encapsulation of pancreatic β-cells.18 We have also shown that PEG-based hydrogels formed by radical-mediated thiol–norbornene photochemistry supported the viability of hMSCs.21 In this study, the decrease in hMSC viability in GelMA hydrogels might also be the result of a higher initial radical concentration presented in the chain-growth polymerization systems. Finally, even though the initial viability of hMSCs encapsulated in step-growth GelNB hydrogels was only 11–12% higher than that in the chain-growth GelMA hydrogels, this difference might affect long-term cell proliferation, spreading, cell–cell interactions, matrix deposition, and differentiation.
3.6. Spreading of encapsulated hMSCs in GelNB-DTT or GelMA hydrogels
Previous work has shown that chain-growth GelMA hydrogels supported the spreading of fibroblasts and vascular endothelial cells. Here, we examined the ability of step-growth GelNB hydrogels to support hMSCs spreading in 3D. While Fig. 5A shows that hMSCs encapsulated in both gelatin hydrogel systems remained rounded one day post-encapsulation, we found that these cells started to extend long processes 2-day post encapsulation (Fig. 6A) when encapsulated in 4 wt% GelNB-DTT hydrogels. Interestingly, cells encapsulated in chain-growth GelMA hydrogels (4 or 8 wt%) did not show spreading at day-2 (Fig. 6A). At day-7 post-encapsulation, encapsulated hMSCs showed a higher degree of spreading for both gelatin hydrogel systems and in both weight contents (Fig. 6B). Cells in step-growth GelNB-DTT hydrogels, however, extended longer processes than all other formulations. By two-week post encapsulation, all cells exhibited long processes but the degree of cell spreading was more pronounced in step-growth GelNB hydrogels than in chain-growth GelMA hydrogels (Fig. 6C). The higher degree of cell spreading in step-growth gelatin hydrogels could also be easily observed from the staining of F-actin (Fig. 7). While encapsulated hMSCs formed an interconnected network in 4 wt% step-growth GelNB-DTT hydrogels, cells were more closely packed in the chain-growth GelMA gels.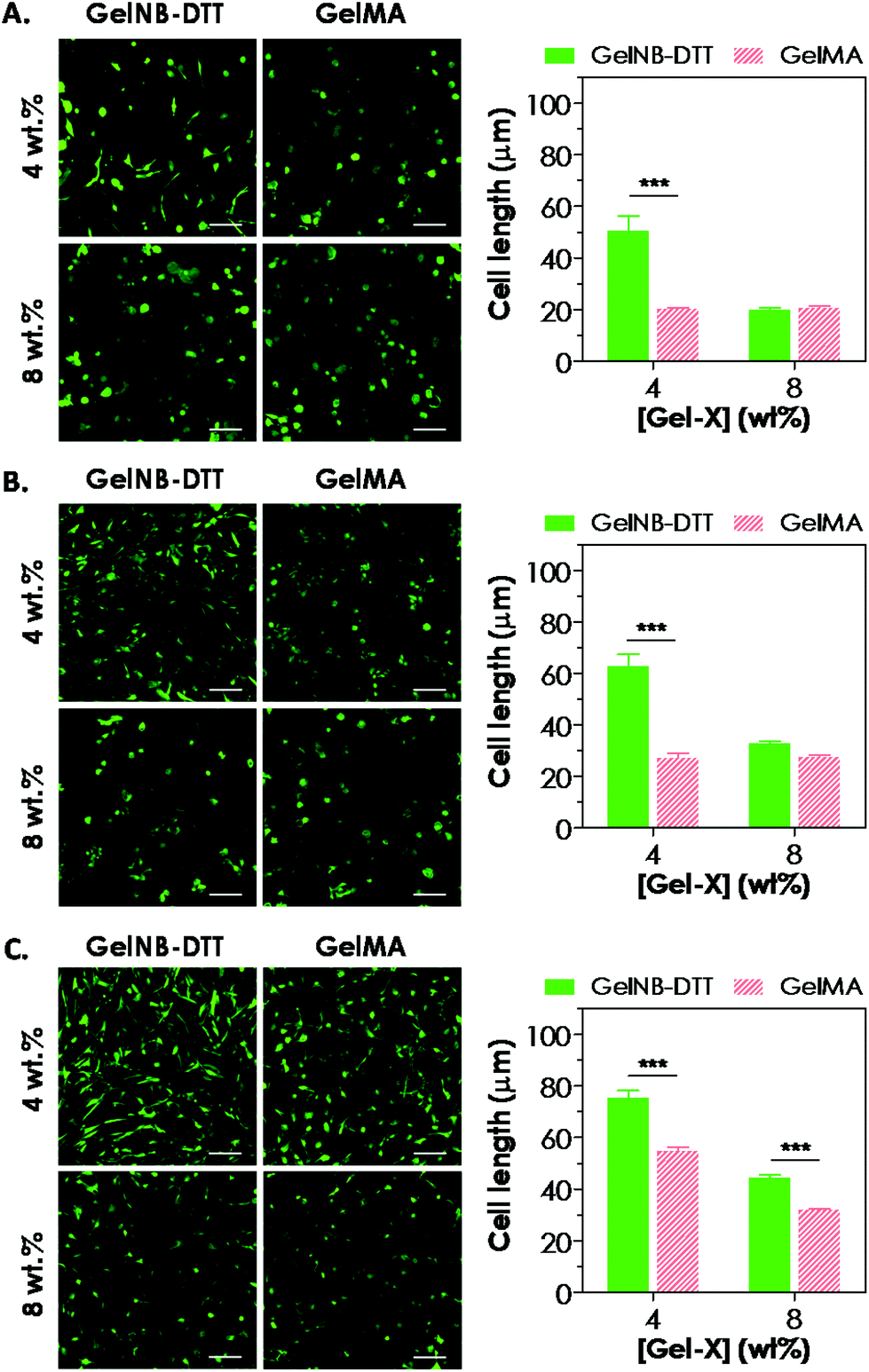 | ||
| Fig. 6 Spreading of hMSCs encapsulated in step-growth GelNB-DTT or chain-growth GelMA hydrogels (4 or 8 wt% gelatin). Cell-laden hydrogels were stained with a live/dead staining kit at day-2 (A), day-7 (B), and day-13 (C) post-encapsulation, followed by imaging with confocal microscopy (scales: 100 μm). Accompanying each set of live/dead staining images are the average cell lengths quantified by measuring the longest end-to-end distance on a cell using ImageJ software. Results were reported as mean ± SEM. | ||
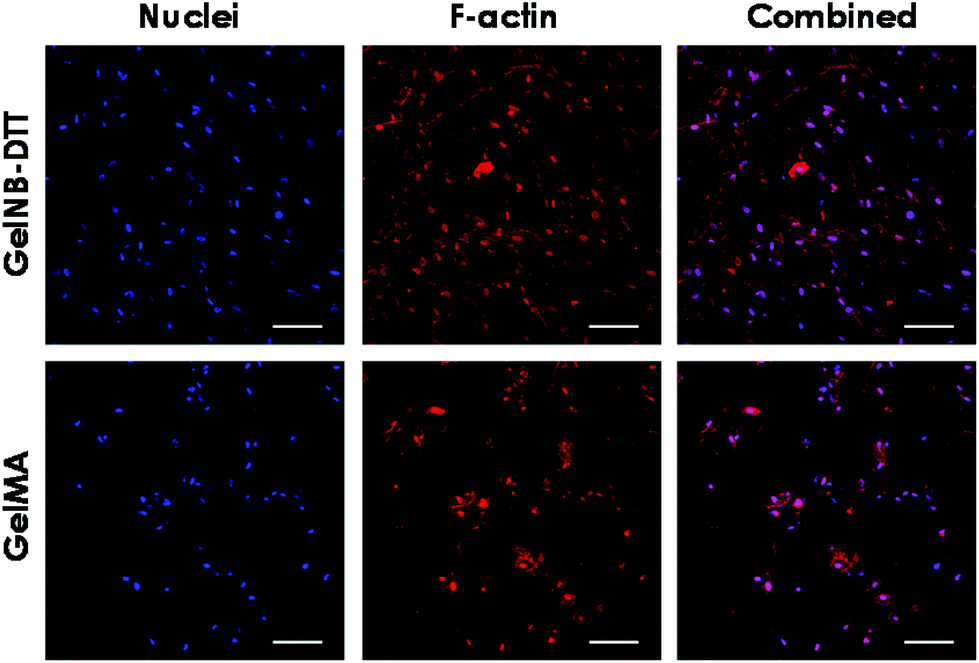 | ||
| Fig. 7 F-actin (red) staining in hMSCs encapsulated in step-growth GelNB-DTT or GelMA hydrogels (scales: 100 μm). The gelatin concentration was 4 wt% and cells were stained at day-13 post-encapsulation. Cell nuclei were counter-stained with DAPI (blue). | ||
When using hydrogels as a carrier to encapsulate hMSCs, there are two requirements that need to be fulfilled if one desires to see cell spreading in 3D: (1) protease-sensitivity and (2) cell-adhesiveness. When hMSCs were encapsulated in PEG-based hydrogels without the presence of protease cleavage sites, cells might be viable but they could not spread or extend cellular processes even in the presence of a cell-adhesive motif (e.g., RGDS).20,21 On the other hand, when protease-sensitive sites (e.g., peptide sequences such as GPQG↓IWGQ) were incorporated into the otherwise non-degradable hydrogels, hMSCs were able to locally degrade a hydrogel mesh and extend protrusions only if cell-adhesive ligands were also present in the hydrogel. Much work has been done to engineer synthetic hydrogel matrices bearing these two important features for supporting cell viability, function, and morphogenesis. In gelatin-based hydrogels, however, these two criteria are simultaneously fulfilled due to the inherent bioactivity of peptide sequences in gelatin. Gelatin not only contains integrin binding motifs, but also has protease-sensitive sequences that can be degraded enzymatically. Interestingly, hMSCs encapsulated in gelatin hydrogels formed from different chemistries showed different levels of spreading (Fig. 6 and 7). Gel cross-linking chemistry (step-growth or chain-growth) likely affected the nanoscopic structure of the gelatin hydrogel network (i.e., orthogonal cross-links in GelNB-DTT hydrogels and heterogeneous polymethacrylamide kinetic chains in GelMA hydrogels). The presence of the heterogeneous kinetic chains likely influenced cell–material interaction and the degree of cell spreading in the encapsulated hMSCs. This study also revealed the profound influence of hydrogel network cross-linking on cell spreading in three dimensions. While hMSCs spread readily in 4 wt% GelNB hydrogels, cell spreading in 8 wt% GelNB hydrogels was comparable to that in GelMA hydrogels in the first week post-encapsulation. This result suggests that a high matrix cross-linking density could restrict or delay cell spreading even in hydrogels with orthogonal cross-links and bioactive motifs.
While the cell studies presented in this contribution show that the current step-growth gelatin hydrogel system could serve as an attractive alternative to the existing covalent gelatin hydrogel systems, challenges exist for the reported GelNB system and future improvement is required. For example, the current synthesis route for norbornene-functionalization on gelatin took more than 70 h to complete (Fig. 1B). Compared to GelNB synthesis, the synthesis of GelMA was not only much faster, but also yielded a higher degree of substitution when using the same ratio of reactant to gelatin (methacrylic anhydride for GelMA and carbic anhydride for GelNB synthesis). Furthermore, we found that the gelation kinetics of step-growth GelNB hydrogel was not significantly different from that of chain-growth GelMA hydrogels (Fig. 2C), suggesting that modification and refinement of the synthesis route are needed in the future.
4. Conclusions
In summary, we have developed a new synthesis route for preparing norbornene-functionalized gelatin (GelNB). GelNB could be photopolymerized into chemically cross-linked hydrogels by means of light-mediated thiol–ene photoclick chemistry and the degree of network cross-linking could be controlled by adjusting the concentrations of GelNB and the cross-linker, or the functionality of the multi-functional linker. The step-growth GelNB hydrogels were cytocompatible for in situ cell encapsulation, and the encapsulated hMSCs exhibited high viability. Furthermore, the presence of cell adhesive motifs and protease cleavage sequences permitted 3D adhesion and spreading of the encapsulated hMSCs to a higher degree when compared with chain-growth gelatin hydrogels. This new step-growth gelatin hydrogel system expands the utility of current gelatin hydrogel systems and should be useful in various tissue engineering and regenerative medicine applications.Acknowledgements
This project was supported in part by the Department of Biomedical Engineering at IUPUI (Faculty Start-up Fund), a pilot grant from IUPUI Biomechanics and Biomaterials Research Center (BBRC), and partial student support (to ZM) from the Undergraduate Research Opportunity Program (UROP) of IUPUI Center for Research & Learning (CRL). The authors thank Dr Chang Seok Ki for his technical assistance on gelatin functionalization and material characterization.References
- J. M. Zhu and R. E. Marchant, Expert Rev. Med. Devices, 2011, 8, 607–626 CrossRef CAS PubMed.
- K. P. Rao, J. Biomater. Sci., Polym. Ed., 1995, 7, 623–645 CAS.
- J. D. Kretlow and A. G. Mikos, AIChE J., 2008, 54, 3048–3067 CrossRef CAS PubMed.
- S. Huang and X. B. Fu, J. Controlled Release, 2010, 142, 149–159 CrossRef CAS PubMed.
- Y. L. Li, J. Rodrigues and H. Tomas, Chem. Soc. Rev., 2012, 41, 2193–2221 RSC.
- J. P. Draye, B. Delaey, A. Van de Voorde, A. Van Den Bulcke, B. De Reu and E. Schacht, Biomaterials, 1998, 19, 1677–1687 CrossRef CAS.
- J. A. Benton, C. A. DeForest, V. Vivekanandan and K. S. Anseth, Tissue Eng., Part A, 2009, 15, 3221–3230 CrossRef CAS PubMed.
- Y. C. Chen, R. Z. Lin, H. Qi, Y. Z. Yang, H. J. Bae, J. M. Melero-Martin and A. Khademhosseini, Adv. Funct. Mater., 2012, 22, 2027–2039 CrossRef CAS PubMed.
- R. Gauvin, Y. C. Chen, J. W. Lee, P. Soman, P. Zorlutuna, J. W. Nichol, H. Bae, S. C. Chen and A. Khademhosseini, Biomaterials, 2012, 33, 3824–3834 CrossRef CAS PubMed.
- M. Nikkhah, N. Eshak, P. Zorlutuna, N. Annabi, M. Castello, K. Kim, A. Dolatshahi-Pirouz, F. Edalat, H. Bae, Y. Z. Yang and A. Khademhosseini, Biomaterials, 2012, 33, 9009–9018 CrossRef CAS PubMed.
- J. Ramon-Azcon, S. Ahadian, R. Obregon, G. Camci-Unal, S. Ostrovidov, V. Hosseini, H. Kaji, K. Ino, H. Shiku, A. Khademhosseini and T. Matsue, Lab Chip, 2012, 12, 2959–2969 RSC.
- J. W. Nichol, S. T. Koshy, H. Bae, C. M. Hwang, S. Yamanlar and A. Khademhosseini, Biomaterials, 2010, 31, 5536–5544 CrossRef CAS PubMed.
- H. Aubin, J. W. Nichol, C. B. Hutson, H. Bae, A. L. Sieminski, D. M. Cropek, P. Akhyari and A. Khademhosseini, Biomaterials, 2010, 31, 6941–6951 CrossRef CAS PubMed.
- C. B. Hutson, J. W. Nichol, H. Aubin, H. Bae, S. Yamanlar, S. Al-Haque, S. T. Koshy and A. Khademhosseini, Tissue Eng., Part A, 2011, 17, 1713–1723 CrossRef CAS PubMed.
- Y. Fu, K. D. Xu, X. X. Zheng, A. J. Giacomin, A. W. Mix and W. Y. J. Kao, Biomaterials, 2012, 33, 48–58 CrossRef CAS PubMed.
- C. C. Lin, S. M. Sawicki and A. T. Metters, Biomacromolecules, 2008, 9, 75–83 CrossRef CAS PubMed.
- J. D. McCall and K. S. Anseth, Biomacromolecules, 2012, 13, 2410–2417 CrossRef CAS PubMed.
- C. C. Lin, A. Raza and H. Shih, Biomaterials, 2011, 32, 9685–9695 CrossRef CAS PubMed.
- B. D. Fairbanks, M. P. Schwartz, A. E. Halevi, C. R. Nuttelman, C. N. Bowman and K. S. Anseth, Adv. Mater., 2009, 21, 5005–5010 CrossRef CAS.
- S. B. Anderson, C. C. Lin, D. V. Kuntzler and K. S. Anseth, Biomaterials, 2011, 32, 3564–3574 CrossRef CAS PubMed.
- A. Raza and C. C. Lin, Macromol. Biosci., 2013, 13, 1048–1058 CrossRef CAS PubMed.
- J. A. Benton, B. D. Fairbanks and K. S. Anseth, Biomaterials, 2009, 30, 6593–6603 CrossRef CAS PubMed.
- S. T. Gould, N. J. Darling and K. S. Anseth, Acta Biomater., 2012, 8, 3201–3209 CrossRef CAS PubMed.
- A. Raza, C. S. Ki and C. C. Lin, Biomaterials, 2013, 34, 5117–5127 CrossRef CAS PubMed.
- H. Shih and C. C. Lin, Biomacromolecules, 2012, 13, 2003–2012 CrossRef CAS PubMed.
- W. M. Gramlich, I. L. Kim and J. A. Burdick, Biomaterials, 2013, 34, 9803–9811 CrossRef CAS PubMed.
- B. D. Fairbanks, M. P. Schwartz, C. N. Bowman and K. S. Anseth, Biomaterials, 2009, 30, 6702–6707 CrossRef CAS PubMed.
- M. P. Lutolf and J. A. Hubbell, Biomacromolecules, 2003, 4, 713–722 CrossRef CAS PubMed.
- C. N. Salinas and K. S. Anseth, Macromolecules, 2008, 41, 6019–6026 CrossRef CAS.
- A. Metters and J. Hubbell, Biomacromolecules, 2005, 6, 290–301 CrossRef CAS PubMed.
- A. Shikanov, R. M. Smith, M. Xu, T. K. Woodruff and L. D. Shea, Biomaterials, 2011, 32, 2524–2531 CrossRef CAS PubMed.
- R. Z. Lin, Y. C. Chen, R. Moreno-Luna, A. Khademhosseini and J. M. Melero-Martin, Biomaterials, 2013, 34, 6785–6796 CrossRef CAS PubMed.
- H. Shin, B. D. Olsen and A. Khademhosseini, Biomaterials, 2012, 33, 3143–3152 CrossRef CAS PubMed.
- J. J. Roberts and S. J. Bryant, Biomaterials, 2013, 34, 9969–9979 CrossRef CAS PubMed.
Footnote |
| † These authors contributed equally to the work. |
| This journal is © The Royal Society of Chemistry 2014 |