A nanocomposite approach to develop biodegradable thermogels exhibiting excellent cell-compatibility for injectable cell delivery†
Naho
Oyama
a,
Hiromasa
Minami
a,
Daichi
Kawano
a,
Makoto
Miyazaki
b,
Tomoki
Maeda
b,
Kazunori
Toma
c,
Atsushi
Hotta
b and
Koji
Nagahama
*a
aDepartment of Nanobiochemistry, Frontiers of Innovative Research in Science and Technology, Konan University, 7-1-20 Minatojima-minamimachi, Kobe 650-0047, Japan. E-mail: nagahama@center.konan-u.ac.jp; Tel: +81-78-303-1328
bDepartment of Mechanical Engineering, Keio University, 3-14-1 Hiyoshi, Yokohama 223-8522, Japan
cAsahi Kasei Corporation, 2-1, Samejima, Fuji 416-0934, Japan
First published on 23rd June 2014
Abstract
A new class of injectable nanocomposite thermogels having excellent cell-compatibility were developed through cooperative self-assembly of biodegradable poly(lactide-co-glycolide)-b-poly(ethylene glycol)-b-poly(lactide-co-glycolide) copolymer micelles and clay nanosheets for effective cell delivery. This study will be valuable for the establishment of injectable cell delivery technology.
Hydrogels are widely studied as the biomaterials for a wide range of biomedical applications because of their tissue-like mechanical properties, highly hydrophilic character, and high permeability for water-soluble molecules such as bioactive agents, nutrients, and metabolites.1–4 Especially, in situ gel forming biodegradable polymer systems have recently received much attention as cell-delivery carriers and scaffolds,5–10 because of their potential applications in the development of an injectable cell therapy as the next generation of regenerative medicine. Injectable cell therapy is implemented by injection of precursor solutions containing cells directly into the damaged site in the body, and then the solutions are gelated and cells are encapsulated in the fixed hydrogel in situ. Injectable hydrogels as cell delivery carriers for cell therapy should preferably have an excellent cell-compatibility and non-animal origin and be chemically defined and easily prepared. To date, various types of injectable gels have been studied and are currently being investigated based on covalently cross-linked networks of synthetic polymers or biopolymers by utilizing click chemistry and enzymatic reactions, such as chitosan,11 hyaluronic acid,12 gelatin,13 collagen,14 alginate,15 poly(ethylene glycol) (PEG),16–18 and poly(methacrylic acid).19 However, these hydrogels require considerable manipulation (e.g., complex chemistry) to prepare hydrogel precursors and to use harmful regents/catalysts/enzyme for the gel formations. Moreover, controllability of the reaction rate for gel forming is quite poor, and the safety profile from the long-term use of these gels is still unclear. Thus, there is a need to have alternative injectable gels useful as cell delivery carriers.
A biodegradable thermo-gelling copolymer consisting of PEG and poly(lactide-co-glycolide) (PLGA), PLGA-PEG-PLGA, can be a base material to develop new injectable gels, owing to its advantages for easy and reproducible synthesis on a large scale and gelation in response to the body temperature at a sufficiently rapid rate without using harmful regents. Moreover, their safety for long-term use has been widely recognized through their investigation as drug delivery carriers for various kinds of low molecular weight drugs.20 However, aqueous PLGA-PEG-PLGA solutions indeed exhibit strong cytotoxicity during the cell loading process (thermo-responsive sol–gel transition process), because PLGA segments are released from the destabilized micelle core by thermal stimulus and directly disrupt the structures of cytomembranes of loaded cells through hydrophobic interactions (Fig. S1†), leading to necrosis.20 As PLGA-PEG-PLGA has a high critical gelation concentration (CGC, at above 15 wt%) to form hydrogels in situ, the high CGC facilitates the cytotoxicity. Additionally, high CGC causes poor space within the hydrogel to create spatial freedom for efficient cell loading, cell growth, and three-dimensional cell assembly. Both the high cytotoxicity against loading cells and poor space derived from the high CGC of PLGA-PEG-PLGA are obstacles to apply it for injectable cell therapy.
Here we show a new method to develop excellent cell-compatible, injectable gels having ultralow gelation concentration (minimum conc.: 0.05 wt%) by simple mixing of PLGA-PEG-PLGA and clay nanosheets. Interestingly, our nanocomposite strategy using clay nanosheets not only completely quenches the cytotoxicity of PLGA-PEG-PLGA but also lowers the CGC drastically. In the present paper, we report a novel strategy to create PLGA-PEG-PLGA based cell-compatible thermogels and to lower the CGC effectively, results of our studies on the thermo-gelation, and discussion of crucial parameters for achieving excellent cell-compatibility.
A series of PLGA-PEG-PLGA copolymers using different molecular weights of PEG (Mw: 1.5k and 3.0k) were synthesized by ring-opening polymerization of DL-lactide and glycolide in the presence of PEG (Scheme S1†), and these copolymers are called P1.5k and P3k, respectively. The molecular composition, the molecular weight and its distribution of the obtained copolymers were determined by 1H-NMR spectroscopy in CDCl3 (Fig. S2†) and gel permeation chromatography (GPC), respectively. P1.5k and P3k copolymers showed a unimodal GPC curve with a polydispersity of less than 1.2. These data are summarized in Table S1.† Both the P1.5k and P3k amphiphilic copolymers were found to form micelles with PEG shells and the PLGA core in aqueous solution by the hydrophobic dye (pyrene) method. For the preparation of nanocomposite gels, we used laponite (LP) as fully synthetic biocompatible clay nanosheets. LP is a disk-shaped nanosheet of approximately 25 nm diameter and 1 nm thickness, possessing a negative face charge and a weak positive rim charge. LP is known for its ability to adsorb PEG chains due to the large and highly charged surface area (>350 m2 g−1).21
We examined how LP influences the micelle forming ability of P3k copolymers in water by comparing their critical micelle concentration (CMC) in the presence of LP with different concentrations (Fig. 1a). It was found that P3k copolymers form micelles in the presence of LP and the CMC values were almost the same, suggesting that the micelle forming ability of P3k copolymers is not affected by the presence of LP. The 1H-NMR technique was used to monitor the changes in molecular environment of PEG and PLGA segments in copolymer micelles when the LP concentration and temperature were individually changed. 1H-NMR spectra of P3k (0.1 wt%) in the absence or presence of LP (0.1 wt%) in D2O at different temperatures are displayed in Fig. S3 and S4,† respectively. Since chloroform is a good solvent for PEG and PLGA segments, both the signals of methylene groups in PEG (3.7 ppm) and methyl groups attributed to the lactic acid unit in PLGA segments (1.6 ppm) were observed as sharp peaks in CDCl3 (Fig. S2†). In contrast, the methyl proton signal of PLGA segments were broadened and drastically weaken in D2O, whereas the methylene proton signal of PEG segments was detected as a sharp peak from 20 to 50 °C (Fig. S3a–d†), indicating that both PLGA end segments basically form micelle cores, whereas a portion of PLGA end segments was released from the core and the major part of PEG middle segments stretched out to the aqueous environment to form micelle shells. As the temperature increases, no significant changes in the shape and the intensity of the PEG peak were observed, whereas the relative intensity of the PLGA peak gradually increased (Fig. 1b), suggesting that the micelle structure was retained even at high temperature, but the stability decreased and the amounts of released PLGA segments increased. On the other hand, the PEG peak obviously collapsed at 20 °C in the presence of 0.1 wt% LP (Fig. S4†), and the PLGA peak also weakens and broadens as compared with the PLGA peak shown in Fig. S3,† suggesting that LPs interacted with PEG segments forming micelle shells at the micelle surface and the amount of released PLGA segments decreased. The collapsed PEG peak was observed even at 50 °C, indicating that nanostructures composed of micelles and LPs were formed and their molecular environments were almost unchanged even at high temperature. Note that changes in the relative intensity of the PLGA peak were hardly observed even at high temperature in the presence of 0.1 wt% of LP (Fig. 1b), suggesting that the stability of the micelle structure was enhanced and the amounts of released PLGA segments significantly decreased because the micelle surfaces were moderately capped with LPs. Moreover, it is suggested that the micelle stability would be controllable by varying the molecular compositions of PLGA-PEG-PLGA copolymers and LPs.
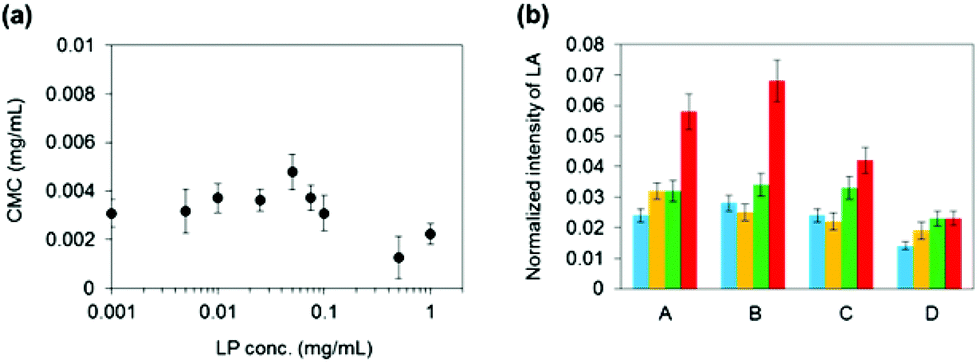 | ||
| Fig. 1 (a) Plots of CMC values of P3k in water as a function of the LP concentration. CMC values were measured at 20 °C. (b) The ratios of the peak intensity of lactic acid (LA) units in PLGA segments to PEG (LA/PEG) in 1H-NMR spectra measured in D2O at different temperatures. A: P3k, B: P3k0.1L0.001, C: P3k0.1L0.01, D: P3k0.1L0.1. Blue: 20 °C, yellow: 30 °C, green: 40 °C, red: 50 °C. | ||
We investigated the influence of the hierarchical capped micelle structures on their thermo-responsive sol–gel transition behaviour. Reference LP solutions did not exhibit thermo-responsive sol–gel transition at any concentration (Fig. S5a†), and P1.5k copolymer solutions exhibited a thermo-gelation above its CGC of 10 wt% in a reversible manner, as previously reported (Fig. S5b†). Fig. 2a shows the phase diagrams of P1.5k/LP nanocomposite aqueous solutions at a fixed concentration of copolymers (2 and 3 wt%) with varied LP concentrations (0.5–1.0 wt%). P1.5k/LP with certain composite ratios underwent thermo-responsive sol–gel transition in an irreversible manner despite the stabilized micelle structures through the capping with LP. Unexpectedly, the CGC value (2 wt%) of P1.5k/LP nanocomposites was five times lower than that of P1.5k copolymer. Moreover, the critical gelation temperature (CGT) of P1.5k copolymer solutions significantly decreased in the presence of LP with certain concentrations. These results mean that LP possesses an ability to promote the thermo-responsive formation of gel networks for PLGA-PEG-PLGA micelles. On the other hand, the gelation temperature of resulting P1.5k/LP composites was lower than room temperature and consequently their injectable property was lost.
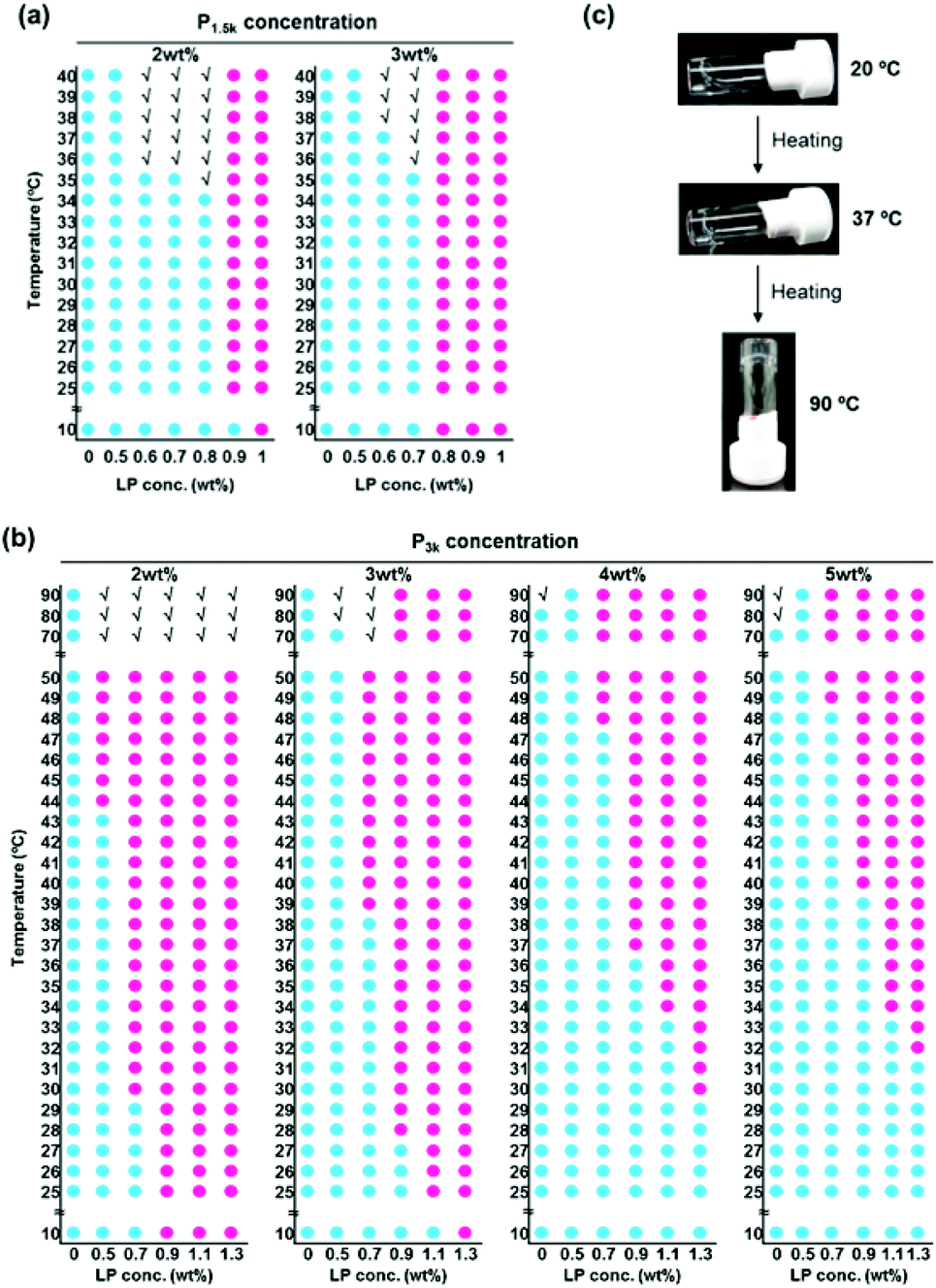 | ||
Fig. 2 Thermo-responsive sol–gel transition behavior. (a) Phase diagrams of P1.5k/LP aqueous solutions with different concentrations. (b) Phase diagrams of P3k/LP aqueous solutions with different concentrations.  : sol, : sol,  : gel, √: precipitation. (c) Photographs of temperature-responsive sol–gel transition of the P3k0.05L1.0 hybrid. : gel, √: precipitation. (c) Photographs of temperature-responsive sol–gel transition of the P3k0.05L1.0 hybrid. | ||
It is well known that the phase transition temperature of PEG-polyester copolymers increases with the increase in the molecular weight of PEG. Indeed, aqueous solutions of P3k copolymer showed relatively high phase transition temperature at around 80 °C (Fig. S5c†), but gelation did not occur at any concentration and temperature. In contrast, P3k/LP nanocomposites with certain mixing ratios underwent thermo-gelation between room temperature and the body temperature (Fig. 2b). Indeed, when an aqueous solution of P3k/LP composites (e.g. composite of 2 wt% of P3k and 0.7 wt% LP; P3k2L0.7) was heated to 37 °C, the transparent suspensions instantaneously became slightly translucent hydrogels, and the thermo-responsive phase transition was completely irreversible. Aqueous P3k solution never exhibited thermo-gelation at any concentration, suggesting cooperative formation of P3k bridged micelle network with LP network in the nanocomposite thermogels. These gels exhibited a very wide range of gel windows from 10 to 90 °C due to the stabilized micelle structure. Surprisingly, even at very low polymer concentrations, the thermo-gelation occurred between room temperature and the body temperature and the resulting gels were able to support their own weight during the vial inversion test; a sample of P3k passed the test at concentrations as low as 0.05 wt% in the presence of LP (Fig. 2c), which is the lowest CGC value for biodegradable thermo-gelling polymers previously reported and is about three orders of magnitude lower in CGC than those of many of the well-studied polymeric hydrogelators.22 Moreover, the resulting thermogels prepared from ultralow concentration of P3k exhibited very high thermal stability (keep transparency at above 90 °C), though the thermogel is formed only by weak non-covalent forces. Thus, hierarchical capped structure of P3k micelles with LP allows for the formation of new nano-colloidal gel networks and provides unique properties to the nanocomposite gels. The LP concentration necessary for thermo-gelation was prone to increase with the increase in P3k concentration. At a fixed P3k concentration, the CGT decreased with the increase in LP concentration. These results suggest that the thermo-gelation properties including CGC and CGT were governed by the structure of composite micelles. In other words, systematic variation of P3k and LP concentrations for preparing nanocomposites allows for diversity in the gelation concentration, gelation temperature and fine-tuning, which have never been achieved for previously reported biodegradable thermo-gelling polymers.
In order to clear the gel network structures of P3k/LP nanocomposite hydrogels and the formation process, FTIR measurement at gel state, DLS measurement at sol state, cryo-TEM measurements at both sol and gel states, and rheological analyses during thermo-gelation were carried out. Fig. S6† shows FTIR spectra of LP, P3k copolymer, and P3k3L0.9 composite gel. The peaks at 980 and 3440 cm−1 in FTIR spectra of LP can be attributed to the –Si–O– stretching vibration and –OH bending vibration, respectively.23 The peaks located at 2880, 1750 and 1100 cm−1 in FTIR spectra of P3k can be attributed to the –C–H– stretching vibration of PEG segments, C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O stretching vibration of PLGA segments, and –C–O– stretching vibration of PEG segments, respectively. The –OH bending vibration of LP was significantly weak in the presence of P3k (in the spectra of P3k3Lx composite gels), whereas the peak attributed to –Si–O– stretching vibration was detected as a sharp peak. The –C–H– stretching vibration and –C–O– stretching vibration of PEG segments in P3k copolymer were broadened in the presence of LP (in the spectra of P3k3Lx composite gels), whereas the peak attributed to CO stretching vibration of PLGA segments was detected as a sharp peak. These results indicate interactions between –OH groups of LP and PEG segments of P3k copolymers in the P3k3Lx composite gels. These IR results are well consistent with the results obtained from 1H-NMR data (Fig. S4†) measured in D2O. Especially, the –C–O– stretching vibration of PEG segments was significantly weak in the presence of LP, indicating that the main interactions between PEG segments and LP would be –OH groups of LP and ether oxygen molecules of PEG segments in the P3k3Lx composite gels. Thus, PEG segments in PLGA-PEG-PLGA copolymers bind on the disk-shaped LP nanosheet and these interactions result in the formation of the capped micelle structure.
O stretching vibration of PLGA segments, and –C–O– stretching vibration of PEG segments, respectively. The –OH bending vibration of LP was significantly weak in the presence of P3k (in the spectra of P3k3Lx composite gels), whereas the peak attributed to –Si–O– stretching vibration was detected as a sharp peak. The –C–H– stretching vibration and –C–O– stretching vibration of PEG segments in P3k copolymer were broadened in the presence of LP (in the spectra of P3k3Lx composite gels), whereas the peak attributed to CO stretching vibration of PLGA segments was detected as a sharp peak. These results indicate interactions between –OH groups of LP and PEG segments of P3k copolymers in the P3k3Lx composite gels. These IR results are well consistent with the results obtained from 1H-NMR data (Fig. S4†) measured in D2O. Especially, the –C–O– stretching vibration of PEG segments was significantly weak in the presence of LP, indicating that the main interactions between PEG segments and LP would be –OH groups of LP and ether oxygen molecules of PEG segments in the P3k3Lx composite gels. Thus, PEG segments in PLGA-PEG-PLGA copolymers bind on the disk-shaped LP nanosheet and these interactions result in the formation of the capped micelle structure.
Micelle aggregation in response to temperature increase (20–50 °C) was not observed for DLS data of P3k solutions (Fig. S7†). In contrast, peak broadening and the rise in average diameter were observed for P3k/LP composite solutions at a fixed temperature (20 °C), indicating disordered micelle aggregation. These phenomena were clearly observed in the presence of high mixing ratios of LP, suggesting that LP located at the micelle surface lead to the formation of aggregated micelles. It is noteworthy that dissociation of the aggregated micelles was started by heating, and then nanostructures with very narrow size distribution were formed at 50 °C. The diameter of the resulting nanostructures was slightly large as compared with bare P3k micelles and the diameter increased with the increase in LP concentrations. The above evidence together with the formation and the maintenance of capped micelle structure even at 50 °C via CMC and 1H-NMR data (Fig. 1) implies that the nanostructures possess hierarchical structures composed of P3k and LP with a high uniformity in size and shape. In contrast, PLGA-PEG-PLGA micelles form aggregates with disordered structures through hydrophobic interaction in response to the temperature increase and form gel networks composed of micelle aggregates above certain concentrations.20 The disordered micelle aggregation is the main cause for the high CGC values of PLGA-PEG-PLGA. In order to analyze visually the nanostructures of P3k/LP composites in sol and gel states, cryo-TEM measurements were carried out at 10 and 37 °C, respectively. Cryo-TEM images of the P3k3L0.9 solution sample prepared at 10 °C are shown in Fig. 3a. Micelle aggregates with a large variety in size and shape were in solution, and the result is well consistent with the DLS data. In contrast, nanostructures with uniform size and shape were observed in the gel sample prepared at 37 °C, and the result is also well consistent with the DLS data. Moreover, the nanostructures were well dispersed with almost the same distance in the composite hydrogels, suggesting that the drastic structural changes of P3k/LP nanostructures in response to the temperature increase are one of the crucial factors for thermo-gelation at very low concentrations.
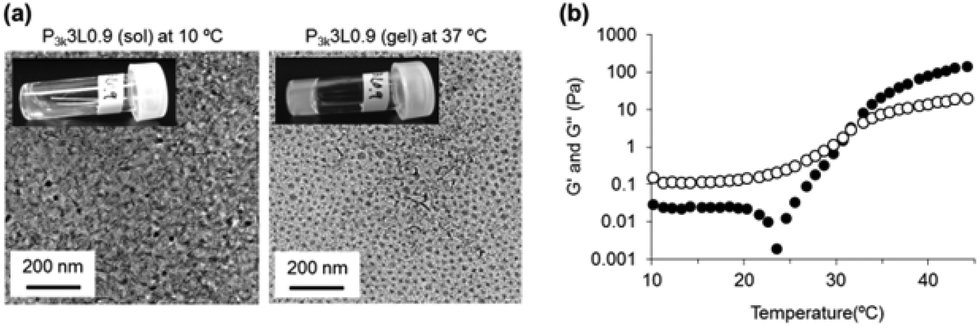 | ||
| Fig. 3 (a) Cryo-TEM images of P3k3L0.9 aqueous solution at 10 °C (sol state) and at 37 °C (gel state). (b) Plots of storage modulus (G′) (closed circle) and loss modulus (G′′) (open circle) of P3k/LP hybrid aqueous solutions vs. temperature. | ||
In order to investigate the characteristic structural changes of the P3k/LP composite solution during its thermo-gelation, dynamic rheological measurements of P3k3L0.9 solution were carried out. The temperature dependent changes in storage modulus (G′) and loss modulus (G′′) were not observed between 10 and 45 °C for both 3 wt% of P3k and 0.9 wt% Lp solutions (Fig. S8†). Typically, an abrupt increase of G′ from the plateau value was observed prior to thermo-gelation for the widely studied biodegradable thermo-gelling PEG-polyester copolymers including PLGA-PEG-PLGA. In contrast, P3k3L0.9 composite solutions exhibited remarkable changes in G′ value as follows: as the temperature increases, the G′ value drastically dropped from the plateau value just prior to an abrupt increase of G′ value (Fig. 3b), suggesting the dynamic dissociation of nanostructures and subsequent reorganization of new synergistic nanostructures to form gel networks during the thermo-responsive sol–gel transition. From the collated results of the micelle and gelation studies described in this paper, we can propose a temperature-responsive sol–gel transition mechanism for the P3k/LP composite system as follows: a restricted portion of PLGA segments are released from the micelle core by thermal stimulus, whereas the stability of the copolymer micelle is not decreased because PEG shells located at the micelle surface are capped with LP. Consequently, disordered aggregation of copolymer micelles through hydrophobic interaction is inhibited, whereas capped micelles aggregate through electrostatic face–edge interaction of LP located on the micelle surface. Further temperature increase leads to dissociation of the aggregates, and the hierarchical nanostructures with a high uniformity in size and shape are spontaneously organized. Released PLGA segments of the nanostructures anchor in the neighbouring micelle core, and then the middle PEG segment bridges between micelles and bridged micelle networks were uniformly formed. The high uniformity would lead to ultralow CGC of P3k/LP nanocomposites. From a practical standpoint, the P3k/LP composite solutions were flowing liquids with a low viscosity at room temperature, suggesting good injectability. In fact, P3k/LP composite solutions loading L929 cells (5 × 105 cells/200 μL) can be readily injected into the subcutaneous layer of mice by a 26-gauge needle and the solutions instantaneously form hydrogels in situ without any loss in cell numbers and gel volume (Fig. S9†).
As described in the Introduction section, PLGA-PEG-PLGA exhibits strong cytotoxicity during its thermo-gelation because PLGA segments released from the destabilized micelles core directly interact with the cytomembrane via hydrophobic interactions, leading to necrosis. We therefore investigated the hemolytic activity of P3k and P3k/LP dilute solutions. The hemolytic activity of the samples was monitored by measuring light absorption (at 570 nm) of the released hemoglobin from the lysed red blood cells (RBCs). Bare P3k copolymers revealed significant hemolytic activities in a dose dependent manner (Fig. S10†). The hemolytic activities obviously increased with the increase in treated temperature, proving that the released PLGA segments are the main cause of cytotoxicity. On the other hand, completely no hemolytic activity was observed for P3k micelles stabilized by LP capping (P3k/LP composites) even at high polymer concentration (10 mg mL−1) and temperature (37 °C), as shown in Fig. 4a. Also, the photograph of the P3k treated RBCs shows the hemoglobin (red color) released to the supernatant, whereas no red color was observed for P3k/LP composites treated RBCs, suggesting excellent cell compatibility of P3k/LP nanocomposite solutions. Next, we examined the cell compatibility of P1.5k, P3k, and P3k/LP during thermo-gelation using L929 fibroblast cells. Although P1.5k and P3k solutions showed strong cytotoxicity in a dose-dependent manner (Fig. S11a and b†), P3k/LP solutions showed 100% cell viability even at long time treatment (for 60 minutes), as shown in Fig. S10c.† P3k/LP composites (e.g. P3k5L1.1) can readily encapsulate large numbers of L929 cells (maximum cell number: 5 × 106 cells per 100 μL of gel) by using its thermo-gelling property, and as we expected, the encapsulated cells were completely alive, whereas P1.5k gels exhibit cytotoxicity in a dose-dependent manner and almost 100% of the cells encapsulated in 15 wt% and 20 wt% P1.5k gels were dead after 4 hours (Fig. 4b and c). Moreover, L929 cells cultured on P3k/LP composite gels were completely alive, indicating no cytotoxicity to the tissue around the gel formed in situ. These results evidence the excellent cell-compatibility of P3k/LP nanocomposite thermogels, and the nanocomposite strategy is very useful to develop injectable cell delivery carriers having rich space within the hydrogel in order to create spatial freedom for growth and three-dimensional assembly of encapsulated cells.
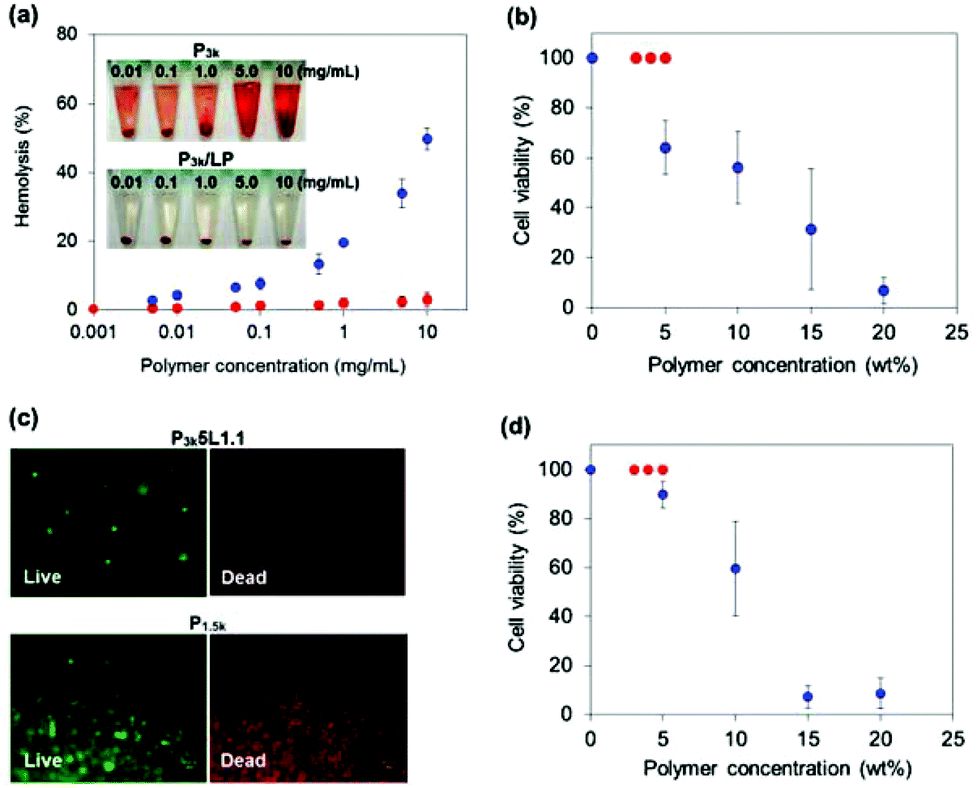 | ||
Fig. 4 (a) Hemolysis activity of P3k ( ) and P3k/LP ( ) and P3k/LP ( ) at 37 °C. The inset shows photographs of RBCs treated with these polymer samples at different concentrations. (b) Cell viability of L929 cells encapsulated in P3k/LP gels ( ) at 37 °C. The inset shows photographs of RBCs treated with these polymer samples at different concentrations. (b) Cell viability of L929 cells encapsulated in P3k/LP gels ( ) and P1.5k gels ( ) and P1.5k gels ( ) with different polymer concentrations after 4 hours incubation. (c) Live/dead staining of L929 cells encapsulated in P3k5L1.1 gels and P1.5k 15 wt% gel after 4 hours incubation. (d) Cell viability of L929 cells cultured on P3k/LP gels ( ) with different polymer concentrations after 4 hours incubation. (c) Live/dead staining of L929 cells encapsulated in P3k5L1.1 gels and P1.5k 15 wt% gel after 4 hours incubation. (d) Cell viability of L929 cells cultured on P3k/LP gels ( ) and P1.5k gels ( ) and P1.5k gels ( ) with different polymer concentrations after 4 hours incubation. ) with different polymer concentrations after 4 hours incubation. | ||
Conclusions
In conclusion, we demonstrated a facile method to develop PLGA-PEG-PLGA based cell-compatible injectable gels. The significantly low CGC of the composite gels should be advantageous not only for practical costs, but also for injectable cell scaffolds having sufficient space for cell migration, cell growth, and cell 3D organization inside the hydrogels. Since LP is known to adsorb various kinds of proteins including growth factors and cytokines,24 PLGA-PEG-PLGA/LP nanocomposite thermogels could be applied as injectable cell delivery carries for various types of cells.Acknowledgements
The authors thank Prof. Y. Ohya (Kansai Univ.) for rheometer measurements. LP was supplied by Wilbur-Ellis Co. This work was partly supported by JSPS KAKENHI grant number 24700487.Notes and references
- D. Seliktar, Science, 2012, 336, 1124–1128 CrossRef CAS PubMed.
- N. A. Peppas, J. Z. Hilt, A. Khademhosseini and R. Langer, Adv. Mater., 2006, 18, 1345–1360 CrossRef CAS.
- T. N. Vo, F. K. Kasper and A. G. Mikos, Adv. Drug Delivery Rev., 2012, 64, 1292–1309 CrossRef CAS PubMed.
- M. W. Tibbitt and K. S. Anseth, Biotechnol. Bioeng., 2009, 103, 655–663 CrossRef CAS PubMed.
- M. K. Nguyen and D. S. Lee, Macromol. Biosci., 2010, 10, 563–579 CrossRef CAS PubMed.
- L. Yu and J. Ding, Chem. Soc. Rev., 2008, 37, 1473–1481 RSC.
- M. P. Lutolf and J. A. Hubbell, Nat. Biotechnol., 2005, 23, 47–55 CrossRef CAS PubMed.
- C. Wang, R. R. Varshney and D. A. Wang, Adv. Drug Delivery Rev., 2010, 62, 699–710 CrossRef CAS PubMed.
- L. Haines-Butterick, K. Rajagopal, M. Branco, D. Salick, R. Rughani, M. Pilarz, M. S. Lamm, D. J. Pochan and J. P. Schneider, Proc. Natl. Acad. Sci. U. S. A., 2007, 104, 7791–7796 CrossRef CAS PubMed.
- S. J. Bidarra, C. C. Barrias, K. B. Fonseca, M. A. Barbosa, R. A. Soares and P. L. Granja, Biomaterials, 2011, 32, 7897–7904 CrossRef CAS PubMed.
- N. Q. Tran, Y. K. Joung, E. Lih and K. D. Park, Biomacromolecules, 2011, 12, 2872–2880 CrossRef CAS PubMed.
- X. Z. Shu, Y. Liu, Y. Luo, M. C. Roberts and G. D. Prestwich, Biomacromolecules, 2002, 3, 1304–1311 CrossRef CAS PubMed.
- Y. Fu, K. Xu, X. Zhang, A. J. Giacomin, A. W. Mix and W. J. Kao, Biomaterials, 2012, 33, 48–58 CrossRef CAS PubMed.
- C. Deng, F. Li, J. M. Hackett, S. H. Chaudhry, F. N. Toll, B. Toye, W. Hodge and M. Griffith, Acta Biomater., 2010, 6, 187–194 CrossRef CAS PubMed.
- O. Jeon, C. Powell, L. D. Solorio, M. D. Krebs and E. Alsberg, J. Controlled Release, 2011, 154, 258–266 CrossRef CAS PubMed.
- G. N. Grover, J. Lam, T. H. Nguyen, T. Segura and H. D. Maynard, Biomacromolecules, 2012, 13, 3013–3017 CrossRef CAS PubMed.
- D. J. Menzies, A. Cameron, T. Munro, E. Wolvetang, L. Grøndahl and J. J. Cooper-White, Biomacromolecules, 2013, 14, 413–423 CrossRef CAS PubMed.
- V. X. Truong, M. P. Ablett, H. T. J. Gilbert, J. Bowen, S. M. Richardson, J. A. Hoyland and A. P. Dove, Biomater. Sci., 2014, 2, 167–175 RSC.
- Y. F. Poon, Y. Cao, Y. Liu, V. Chan and M. B. Chan-Park, ACS Appl. Mater. Interfaces, 2012, 2, 2012–2025 Search PubMed.
- A. Alexander, J. Khan, S. Saraf and S. Saraf, J. Controlled Release, 2013, 172, 715–729 CrossRef CAS PubMed.
- J. I. Dawson, J. M. Kanczler, X. B. Yang, G. S. Attard and R. O. C. Oreffo, Adv. Mater., 2011, 23, 3304–3308 CrossRef CAS PubMed.
- T. Vermonden, R. Censi and W. E. Hennink, Chem. Rev., 2012, 112, 2853–2888 CrossRef CAS PubMed.
- S. Wang, F. Zheng, Y. Huang, M. Shen, M. Zhu and X. Shi, ACS Appl. Mater. Interfaces, 2012, 4, 6393–6401 CAS.
- C.-W. Chiu and J.-J. Lin, Prog. Polym. Sci., 2012, 37, 406–444 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental details and characterization data. See DOI: 10.1039/c4bm00074a |
| This journal is © The Royal Society of Chemistry 2014 |