
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceAcetalization strategy in biomass valorization: a review
Jian
He
 acd,
Qian
Qiang
be,
Li
Bai
ac,
Wentao
Su
be,
Huazhong
Yu
acd,
Shima
Liu
acd and
Changzhi
Li
*be
acd,
Qian
Qiang
be,
Li
Bai
ac,
Wentao
Su
be,
Huazhong
Yu
acd,
Shima
Liu
acd and
Changzhi
Li
*be
aKey Laboratory of Hunan Forest Products and Chemical Industry Engineering, Jishou University, Zhangjiajie, 427000, China
bCAS Key Laboratory of Science and Technology on Applied Catalysis, Dalian Institute of Chemical Physics, Chinese Academy of Sciences, Dalian 116023, China. E-mail: licz@dicp.ac.cn
cCollege of Chemistry and Chemical Engineering, Jishou University, Jishou, 416000, China
dNational and Local United Engineering Laboratory of Integrative Utilization Technology of Eucommia Ulmoides, Jishou University, Jishou, 416000, China
eUniversity of Chinese Academy of Sciences, Beijing, 100049, China
First published on 28th June 2023
Abstract
Acetalization represents an appealing approach for the valorization of biobased platform molecules into valuable chemicals and fuels. Typically, it serves as both a synthesis tool for renewable cyclic acetals and a protection strategy to improve selectivity in biomass conversion. This contribution provides an overview on the application of the acetalization strategy in biomass valorization including synthesis of cyclic acetal fuel additives from the acetalization of biobased furanic compounds with biogenic ethylene glycol/glycerol and acetalization as a protection approach to improve product selectivity in biomass valorization. The latest progresses in the development of catalytic systems for the acetalization of biobased furanic compounds and biogenic ethylene glycol/glycerol are systematically summarized and discussed, with an emphasis on the reaction pathway, relationship between catalyst structures and their performance, and relevant catalytic mechanism. Moreover, the application of the acetalization strategy for protecting carbonyl groups/diol structure functionalities to improve the target products' selectivity in lignin depolymerization, 5-hydroxymethylfurfural oxidation, sorbitol dehydration, and xylose hydrogenation is also highlighted. Eventually, the prospects and challenges in the synthesis of cyclic acetal fuel additives as well as applying acetalization as a protection strategy in biomass valorization are outlined.
Keywords: Oxygenated fuel additives; Furanic compounds; Bioalcohols; Acetalization; Chemocatalysis.
 Jian He | Jian He is a lecturer at Key Laboratory of Hunan Forest Products and Chemical Industry Engineering, National and Local United Engineering Laboratory of Integrative Utilization Technology of Eucommia Ulmoides, and College of Chemistry and Chemical Engineering, Jishou University. He obtained his Ph.D. from Guizhou University in 2019 and studied at Technical University of Denmark as a guest PhD student for one year (2016.09–2017.09). His research focuses on the synthesis of biofuels and value-added chemicals from renewable biomass-based molecules with heterogeneous catalytic materials. |
 Changzhi Li | Changzhi Li is currently a professor at Dalian Institute of Chemical Physics (DICP), from where he received his Ph.D. degree in 2009 under the supervision of Prof. Zongbao (kent) Zhao. Then, he joined Prof. Tao Zhang's group, where he was promoted to full professor in 2019. The main theme of his research program is biomass catalytic conversion. He has published more than 70 papers in international journals and has over 40 patents. His current research focuses on the catalytic valorization of lignin into value-added chemicals and high-density fuels. |
1 Introduction
The synthesis of fuels and fine chemicals from renewable biomass is acclaimed as a viable strategy to reduce reliance on nonrenewable petroleum resources and mitigate CO2 emissions as well as achieve carbon neutrality.1–7 Lignocellulose and vegetable oil, the main solid and liquid components of biomass, represent sustainable carbon sources to produce transport fuels and valuable chemicals.8–15 The catalytic conversion of lignocellulose/vegetable oil and their related derivatives has attracted great attention in biomass valorization.16–25 Many elegant methods including hydrolysis, dehydration, hydrogenation, hydrogenolysis, oxidation, etherification, esterification, amination, aldol condensation, Diels–Alder, Knoevenagel condensation, and acetalization have been developed for the valorization of lignocellulose/vegetable oil derivatives toward value-added chemicals and biofuels.26–40In particular, acetalization is advocated as an appealing method in biomass valorization because it serves as both a synthesis tool for renewable acetal products and a protection strategy to improve product selectivity.41–43 Acetalization is a well-known acid-catalyzed reversible reaction between carbonyl compounds and alcohols, accompanied by water as the sole byproduct.44,45 Thanks to the high stability and low reactivity of the acetal compounds in basic medium, acetalization is the most frequently used and powerful tool to protect carbonyl functionalities in organic synthesis.46,47 Interestingly, the acetalization of biobased furanic compounds and diols or triols can lead to the formation of cyclic acetals, which serve as interesting oxygenated additives in fuels since cyclic acetals can enhance the octane number and cold flow properties and can synchronously reduce particulate emissions and gum formation (Fig. 1).48–50 In addition, the synthesis of a single product with high selectivity during the valorization of biomass derivatives is very challenging due to the presence of multiple functional groups (e.g., C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O, CC, and C–O) in biomass molecules.26,27 A protection strategy based on reversible acetalization reaction has been recently established to temporarily passivate the reactive groups (e.g., carbonyl groups or diol structure functionalities) with the aim of improving the selectivity toward target products in biomass valorization such as, e.g., lignin depolymerization, oxidation of 5-hydroxymethylfurfural into 2,5-furandicarboxylic acid, dehydration of sorbitol into isosorbide, and synthesis of xylitol from xylose (Fig. 1).
O, CC, and C–O) in biomass molecules.26,27 A protection strategy based on reversible acetalization reaction has been recently established to temporarily passivate the reactive groups (e.g., carbonyl groups or diol structure functionalities) with the aim of improving the selectivity toward target products in biomass valorization such as, e.g., lignin depolymerization, oxidation of 5-hydroxymethylfurfural into 2,5-furandicarboxylic acid, dehydration of sorbitol into isosorbide, and synthesis of xylitol from xylose (Fig. 1).
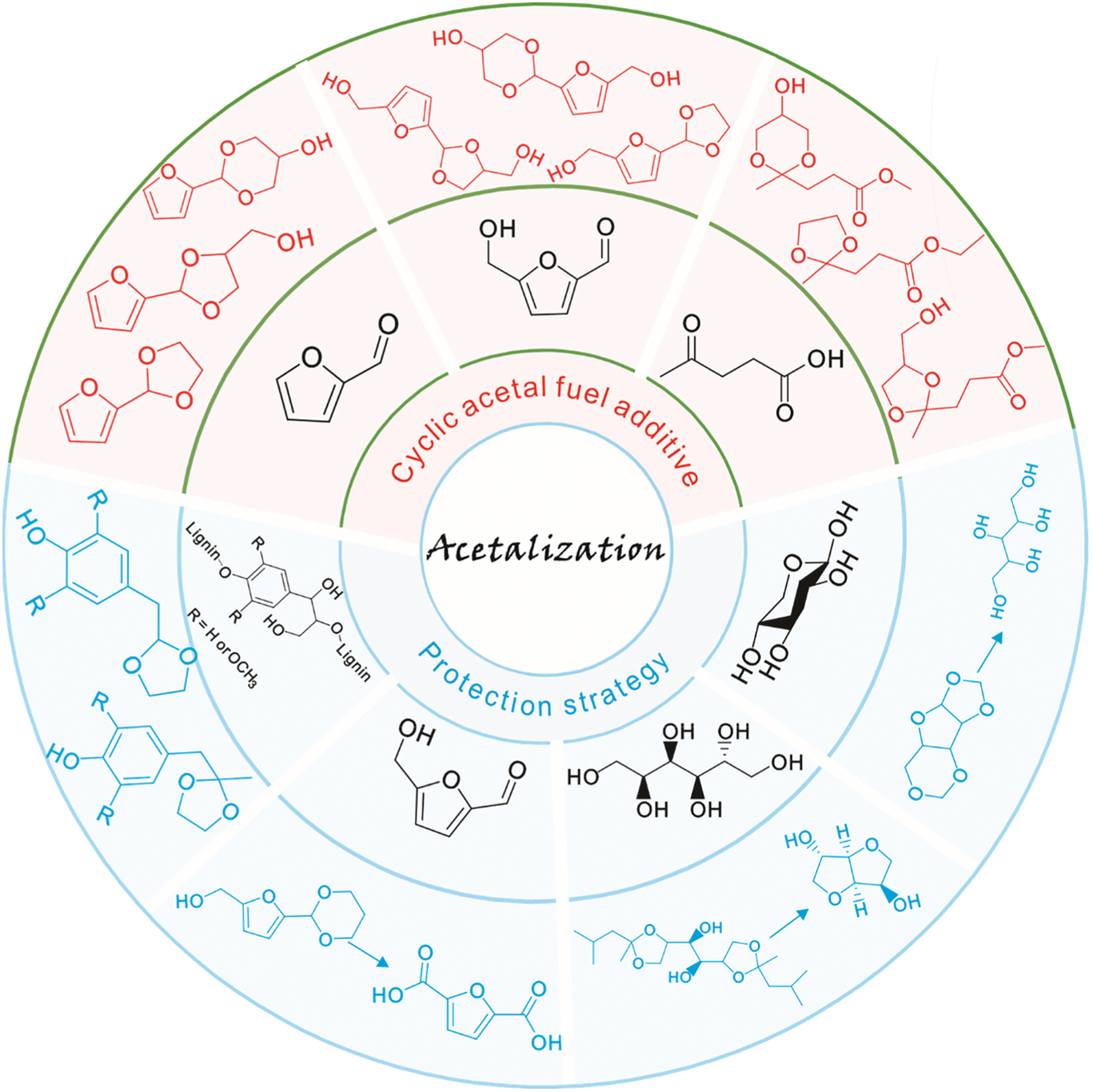 | ||
| Fig. 1 Representative examples of application of the acetalization strategy in biomass valorization. | ||
The process for the acetalization of biobased furanic compounds and diols/triols can be considered as a sustainable process if all the substrates are derived from renewable biomass.51,52 As a typical diol, ethylene glycol is a critical chemical with widespread industrial applications,53 which can be directly obtained from cellulose over tungsten-based catalyst via cascade hydrolysis, retro-aldol condensation, and subsequent hydrogenation.54–56 Another important biogenic polyol is glycerol. As the main byproduct of biodiesel production process, glycerol has gained much attention from both the academic and industrial sectors owing to an excessive amount of glycerol yielded with the expansion of biodiesel.57–60 Therefore, both ethylene glycol and glycerol are accessible from biomass with rich production capacity, which has provoked the development of efficient approaches for downstream valorization.61–63 An interesting destination for the valorization of biobased ethylene glycol/glycerol is to combine the simultaneous production of cyclic acetals fuel additives via the acetalization strategy (Fig. 2).51,52 Specially, the synthesized cyclic acetals with octane-improving, antidetonant, and antifreezing properties can not only serve as pour point, cold filter plugging point, and cloud point improvers in biodiesel/gasoline blends but can also be involved the improvement in vapor pressure (in gasoline blends) or flash point (in diesel/biodiesel blends).48–52 Moreover, the oxygenated additives based on cyclic acetals can be synthesized via the acetalization method under mild conditions, avoiding high reaction temperature and gaseous hydrogen, which are generally used in most other fuel additives/biofuels synthesis, thus endowing the merits of low cost and environmental friendliness.
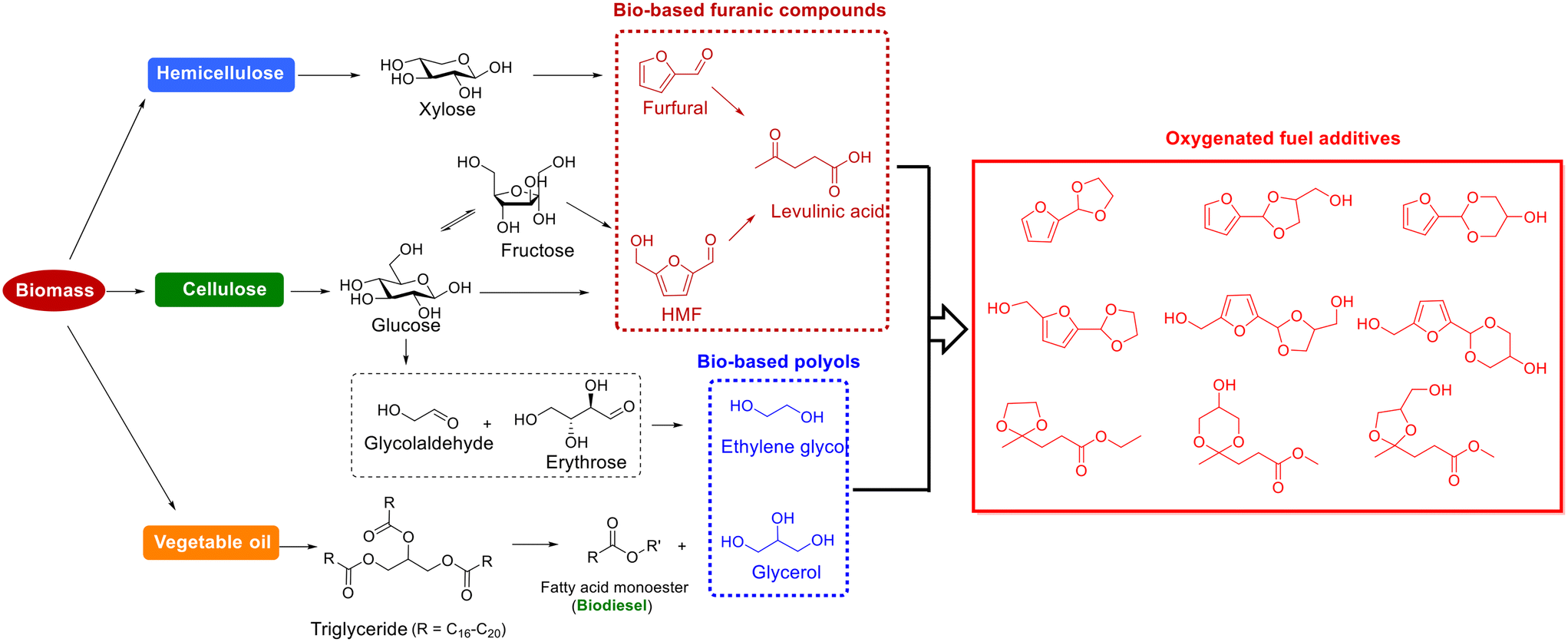 | ||
| Fig. 2 Representative oxygenated fuel additives from the acetalization of biobased furanic compounds and polyols. | ||
Much effort has been devoted to applying the acetalization strategy in biomass valorization owing to the synthesized cyclic acetals as fuel additives with excellent properties and acetalization endowing the temporary passivation of reactive groups as a protection protocol to improve the target product selectivity.48–52,64 Several reviews on the acetalization strategy have primarily focused on solketal fuel additives synthesized from the acetalization of glycerol with acetone.65–67 To the best of our knowledge, no systematic review paper is dedicated to the acetalization strategy in biomass valorization.
To further push the process based on acetal synthesis forward to industrial implementation and get more insights into the selectivity valorization of biomass derivatives via acetalization, in this contribution, a comprehensive overview of the acetalization strategy in biomass valorization has been presented. Concretely, the synthesis of cyclic acetal fuel additives from the acetalization of biobased furanic compounds and biogenic diol/triol as well as applying acetalization as a protection approach to improve product selectivity in biomass valorization are systematically summarized and discussed. Firstly, the latest advances in the development of catalytic systems for the acetalization of biobased furanic compounds and diol/triol are considered, with an emphasis on the corresponding reaction pathway, relationship between the catalyst structures and their performance, and relevant catalytic mechanism. Next, the application of the acetalization strategy for protecting carbonyl groups/diol structure functionalities to improve the target products' selectivity in biomass conversion is also summarized and discussed in this review, and focus is laid on discussing the application of the acetalization strategy in lignin depolymerization, 5-hydroxymethylfurfural oxidation, sorbitol dehydration, and xylose hydrogenation.
2 Synthesis of cyclic acetals from furfural
2.1 Acetalization of furfural with ethylene glycol
The acetalization between furfural and ethylene glycol typically in the presence of acid catalyst, as shown in Scheme 1, results in the production of 2-(furan-2-yl)-1,3-dioxolane (FD) along with an equal equivalent of water. Early reports on the acetalization of furfural with ethylene glycol are focused on using homogeneous acid catalysts. For example, the use of p-toluenesulfonic acid, as described by Lu and collaborators, enabled the acetalization of furfural with ethylene glycol in refluxing benzene to proceed successfully with 87% yield of FD (entry 1, Table 1).68 The authors discovered that the addition of anhydrous magnesium sulfate as a dehydrating agent into the reaction mixture was crucial for acquiring high FD yield because acetalization is a reversible reaction where the removal of formed water is conducive to driving the reaction to completion. Moreover, the FD yield could be further improved to 98% with tartaric acid as a catalyst in refluxing benzene (entry 2, Table 1). As compared to tartaric acid, the inferior catalytic performance in p-toluenesulfonic acid is attributed to its higher acidity, which induces other side reactions.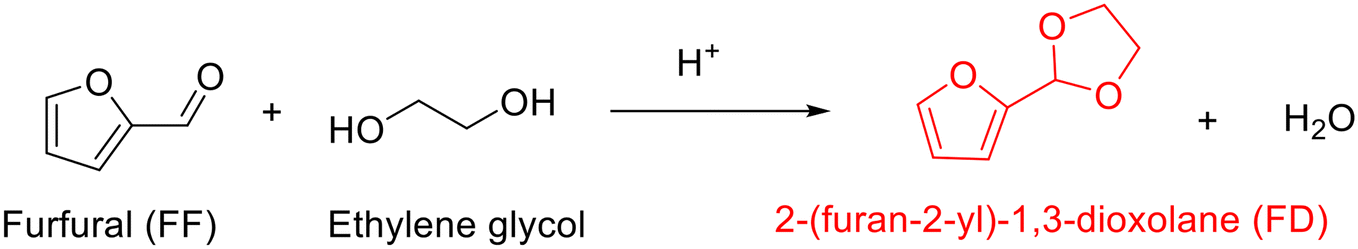 | ||
| Scheme 1 Acetalization of furfural with ethylene glycol to yield cyclic acetal. | ||
| Entry | Catalyst | Solvent | Reaction conditions | FF conv.a (%) | FD yield (%) | Ref. |
|---|---|---|---|---|---|---|
| a FF is the abbreviation of furfural. b A catalytic membrane based on polymeric acid generated from the complexation of poly(4-styrenesulfonic acid) with poly(4-vinylpyridine). c The name of the photocatalyst is eosin Y. | ||||||
| 1 | p-Toluenesulfonic acid | Benzene | FF (50 mmol), ethylene glycol (100 mmol), MgSO4, reflux, 14 h | — | 87 | 68 |
| 2 | Tartaric acid | Benzene | FF (50 mmol), ethylene glycol (100 mmol), MgSO4, reflux, 12 h | — | 98 | 68 |
| 3 | Kaolinitic clay | Benzene | FF (10 mmol), ethylene glycol (11 mmol), reflux, 2 h | — | 93 | 70 |
| 4 | SG-[(CH2)3SO3H-HIM]HSO4 | Cyclohexane | FF (70 mmol), ethylene glycol (126 mmol), 110 °C, 3 h | 85.0 | 85.0 | 71 |
| 5 | AlPO-5(1) | 1,4-Dioxane | FF (1 mmol), ethylene glycol (2 mL), 150 °C, 24 h | 97.2 | 95.7 | 72 |
| 6b | Catalytic membrane based on polymeric acid | Toluene | FF (0.1 mol L−1, 0.1 μL min−1), ethylene glycol (0.1 μL min−1), 80 °C, 38 s | — | 86 | 74 |
| 7c |
|
Acetonitrile | FF (0.5 mmol), ethylene glycol (0.5 mL), green LEDs, 12 h | — | 95 | 80 |
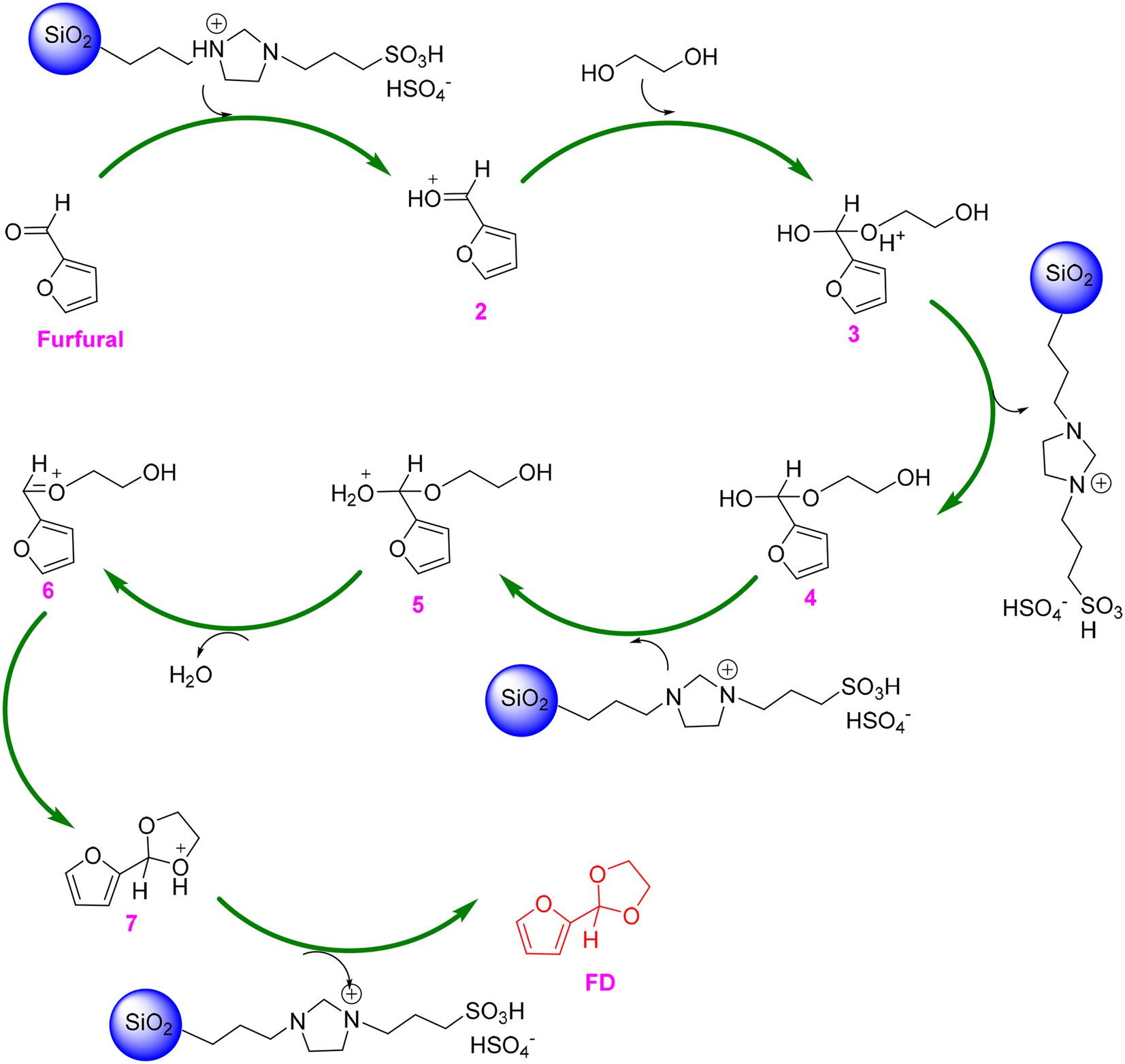
From the perspective of green and sustainable development, the substitution of homogeneous catalysts with heterogeneous catalysts is highly desirable as heterogeneous catalysts are known as their merits of noncorrosiveness, separation efficiency, and recyclability.69 For instance, kaolinitic clay, after three-step treatment (purification via separating coarser mineral impurities, calcination at 550 °C, and acidification with 2 mol L−1 HCl aqueous solution), was found to be an effective catalyst for the acetalization of furfural with ethylene glycol, giving 93% yield of FD in refluxing benzene (entry 3, Table 1).70 The dramatic improvement of the catalytic performance of the acid-activated clay, compared to natural kaolinitic clay, stemmed from the significantly higher amount of both Lewis acidity (due to Al remaining in the edges of the platelets) and Brønsted acidity (due to the coordinated hydroxyl groups of Al3+, Fe3+, and Ti4+ ions relocated in the interlamellar space of the clay). Moreover, the established catalytic system based on kaolinitic clay catalyst allowed the production of a broad range of acetal products from different carbonyl compounds and alcohols. Later, acidic ionic liquid 1-(propyl-3-sulfonate)imidazolium hydrosulfate (i.e., [(CH2)3SO3H-HIM]HSO4) immobilized on silica, named SG-[(CH2)3SO3H-HIM]HSO4, was proved to effectively catalyze the acetalization of furfural with ethylene glycol, affording 85% yield of FD in cyclohexane at 110 °C within 3 h (entry 4, Table 1).71 Excitingly, the SG-[(CH2)3SO3H-HIM]HSO4 catalyst exhibited no significant deactivation in ten consecutive reuse tests, highlighting the excellent recyclability of SG-[(CH2)3SO3H-HIM]HSO4. In addition, a plausible mechanism for acetalization over the SG-[(CH2)3SO3H-HIM]HSO4 catalyst was proposed by the authors, as shown in Scheme 2.71 The reaction starts with the carbonyl group of furfural taking a proton from SG-[(CH2)3SO3H-HIM]HSO4, followed by the nucleophilic addition of intermediate 2 and ethylene glycol and subsequent deprotonation to form hemiacetal 4, which then undergoes a cascade protonation–dehydration-nucleophilic addition–deprotonation pathway to finally yield the FD product.
 | ||
| Scheme 2 Plausible reaction mechanism for the acetalization of furfural with ethylene glycol over the SG-[(CH2)3SO3H-HIM]HSO4 catalyst. Adapted with permission from ref. 71. Copyright 2011, Elsevier. | ||
Recently, Riisager and colleagues prepared aluminum phosphate with Al/P molar ratio of 1 (named AlPO-5(1)) for the acetalization of furfural with ethylene glycol in 1,4-dioxane.72 A high FD yield of 95.7% at furfural conversion of 97.2% was obtained over AlPO-5(1) under 150 °C temperature for 24 h (entry 5, Table 1). The Brønsted acidic sites (0.0012 mmol g−1) together with adsorptive action were verified to be determinate factors in the catalytic behavior of AlPO-5(1) material, as evidenced from the poisoning experiments and FT-IR tests of furfural.72 Due to their advantages of enhancing the productivity and reducing the time consumption, continuous flow reactors have become more attractive in practical application.73 Uozumi and coworkers performed pioneering work using continuous flow reactor for the acetalization of furfural with ethylene glycol, where a catalytic membrane based on polymeric acid generated from the complexation of poly(4-styrenesulfonic acid) with poly(4-vinylpyridine) was fabricated in a microflow reactor (Fig. 3).74 The catalytic membrane formed at the interface between two parallel laminar flows and furnished a permeable barrier between the water-containing layer and organic layer in the reaction, which allowed water discharge to the outlet of the microreactor to drive the reaction to completion. The length, thickness, and width of the catalytic membrane, as evidenced from optical micrographs, were 20–40 mm, 20 μm, and 40 μm (Fig. 3, right top), respectively. Moreover, the catalytic membrane possessed a mesoporous structure, as reflected by a high-resolution scanning electron micrograph (Fig. 3, right bottom), which was conducive for providing more available active sites. Interestingly, the acetalization of furfural with ethylene glycol over the catalytic membrane in the microflow device proceeded successfully, giving 86% yield of FD at 80 °C with a residence time of 38 s (entry 6, Table 1).74 More importantly, the established continuous flow system allowed the spontaneous separation of acetal product and water, in which the acetal product existed in the toluene layer while water was present in the diol layer.
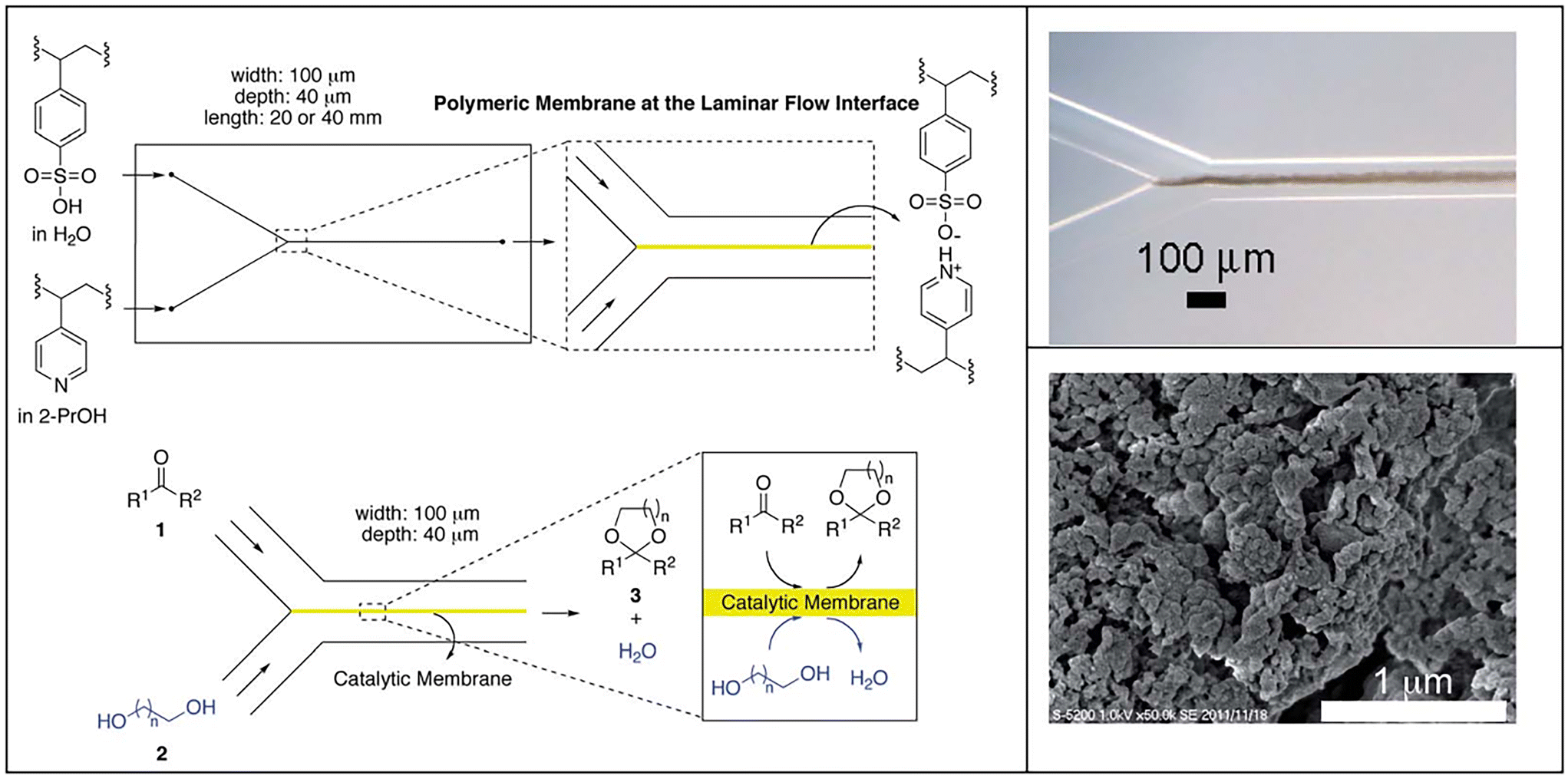 | ||
| Fig. 3 Schematic showing the preparation of microchannel reactor based on the polymeric acid catalyst membrane and its application in acetalization by a flow reaction. Reprinted with permission from ref. 74. Copyright 2014, Royal Society of Chemistry. | ||
Photocatalysis appears to be an attractive approach in biorefinery research because it can achieve the efficient utilization of solar energy and provides an ecofriendly alternative to the conventional thermocatalysis.75–79 A pioneering attempt in the photocatalytic acetalization of furfural with ethylene glycol was achieved by Lei and his collaborators, where eosin Y was used as the photocatalyst. With the catalysis of eosin Y and the irradiation of visible light (3 W green LEDs), the yield of FD reached up to 95% with a reaction time of 12 h (entry 7, Table 1).80 The authors speculated that the in situ photogenerated acidic species was probably responsible for the excellent behavior of the catalytic system.80 Encouragingly, a wide variety of aldehydes and alcohols are viable in this photocatalytic system, highlighting the universality of the eosin Y photocatalyst in acetalization.
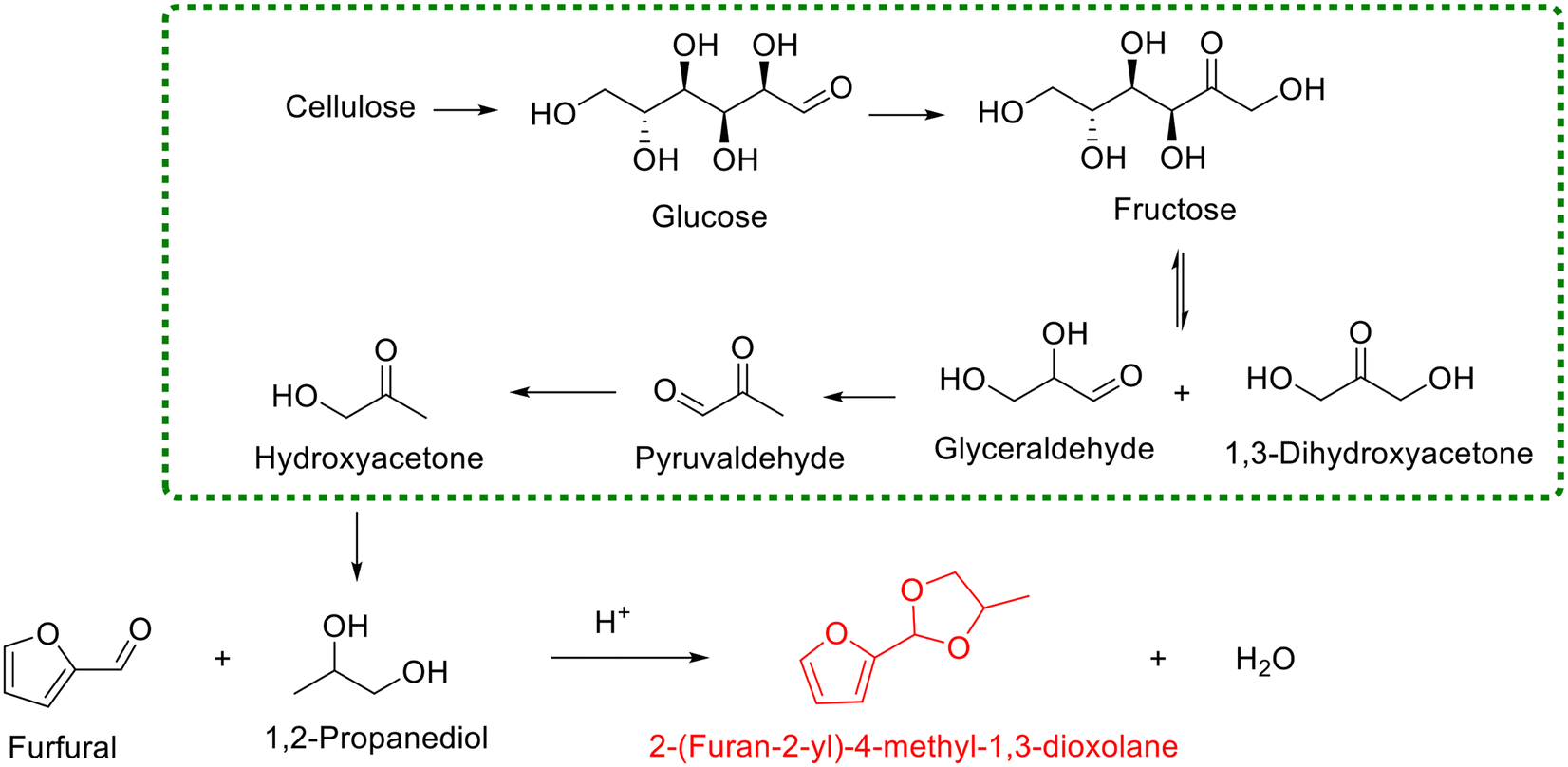
Analogous to ethylene glycol, other biogenic diols can also serve as starting materials for the synthesis of cyclic acetals via reaction with furfural. For example, 1,2-propanediol represents a renowned biobased diol with broad interest in food, pharmaceuticals, and polymer science, which has received exceptional attention in the cellulosic biorefinery system (Scheme 3).81,82 Currently, several heterogeneous catalysts are available for the acetalization of furfural with 1,2-propanediol, including SAPO-34 zeolite83 and sulfonic acid-functionalized metal–organic framework MIL-101 (ref. 84) and MIL-100(Fe).85 Song and coworkers used several commercial zeolites (i.e., SAPO, USY, HY, and H-ZSM-5) as catalysts for the acetalization of furfural with 1,2-propanediol under solvent-free conditions, aiming at revealing the effect of the structure of zeolites on the product yield.83 Interestingly, SAPO-34 displayed the best catalytic performance among the tested zeolites, giving 88.2% yield of 2-(furan-2-yl)-4-methyl-1,3-dioxolane when the reaction was performed with a 1,2-propanediol/furfural molar ratio of 10 at 80 °C for 6 h (Scheme 3). The authors pointed out that the superiority of SAPO-34 in the catalytic behavior, after the careful scrutiny of the used zeolites via various characterization techniques, was primarily associated with its higher microporosity, which was conducive to the diffusion of substrates and products.83 In addition, sulfonic acid-functionalized MIL-101 (ref. 84) and MIL-100(Fe),85 as described by Zhang and his collaborators, enabled the acetalization of aldehydes with diols to proceed smoothly in cyclohexane at 80 °C. Also, the universality tests of sulfonic acid-functionalized MIL-101 and MIL-100(Fe) exhibited that they could effectively facilitate the acetalization of furfural with 1,2-propanediol to produce 2-(furan-2-yl)-4-methyl-1,3-dioxolane in yields of 87.8% and 90.8%, respectively. Specially, the high catalytic performance of MIL-100(Fe) was rationalized by its Lewis acidity of the available unsaturated Fe metal sites together with abundant mesoporous cages within the MIL-100(Fe) catalyst.85 The synthesis of cyclic acetal from 1,2-propanediol highlights that various biogenic diols are viable in this protocol, enabling broad access to diverse cyclic acetals.
 | ||
| Scheme 3 Acetalization of furfural with 1,2-propanediol to yield cyclic acetal. | ||
In conclusion, both Brønsted and Lewis acidic sites are active for the acetalization of furfural with ethylene glycol. In addition, for zeolites and porous materials, higher microporosity has a significant positive effect of enhancing the catalytic activity, which improves the diffusion of substrates and products during the reaction.
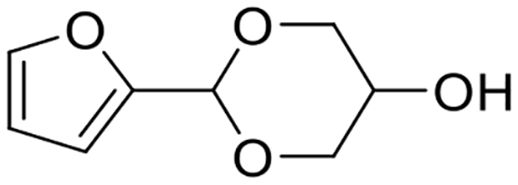
2.2 Acetalization of furfural with glycerol
Glycerol, the main byproduct in the biodiesel industry, is gradually surplus with the increase in biodiesel production. The attention toward the acetalization of furfural with glycerol to give fuel additives has been rising rapidly (Table 2).86–101 The acetalization of furfural with glycerol, nevertheless, is more challenging in comparison with the acetalization of furfural with ethylene glycol owing to the high viscosity of glycerol. Moreover, the acetal products originated from the acetalization of furfural with glycerol are more complicated because glycerol embraces two kinds of hydroxyl groups, which results in acetalization between furfural and 1,2-diol (affording dioxolane product) or 1,3-diol (giving dioxane product) in glycerol. Generally, two acetal products, namely, (2-(furan-2-yl)-1,3-dioxolan-4-yl)methanol (i.e., FDM) and 2-(furan-2-yl)-1,3-dioxan-5-ol (i.e., FDO), are achieved from the acetalization of furfural with glycerol, wherein each of the acetal products includes two stereoisomers, as shown in Scheme 4. Early research using homogeneous acid catalysts (e.g., p-toluenesulfonic acid,86 4-methylbenzenesulfonic acid,87 oxorhenium(V) oxazoline complex,88 and FeCl3·6H2O (ref. 89)) contributed to affording high yield of acetals from the acetalization of furfural with glycerol (entries 1–3, Table 2). Despite the promising achievement over homogeneous acid catalysts, extra neutralization steps, recycling difficulty, and corrosive issue plague their large-scale application in practical production. Recent emphasis has shifted toward developing effective and stable heterogeneous acid catalysts for the acetalization of furfural with glycerol.| Entry | Catalyst | Solvent | Reaction conditions | FF conv.a (%) | Yield (%) | Ref. | |
|---|---|---|---|---|---|---|---|
|
|
|
||||||
| a FF is the abbreviation of furfural. b The name of the catalyst is [2-(2′-hydroxyphenyl)-2-oxazolinato(−2)]oxorhenium(V). c The name of [BPy]HSO4 is N-butyl-pyridinium bisulfate. d The catalyst is 12-tungstosilicic acid anchored to ordered nanoporous MCM-48. | |||||||
| 1 | 4-Methylbenzenesulfonic acid | Benzene | 80 °C | — | 39.52 | 36.48 | 87 |
| 2b | Oxorhenium(V) oxazoline complex | Solvent-free | n(FF)/n(glycerol) = 5/1, 100 °C, 4 h | — | 53.9 | 23.1 | 88 |
| 3 | FeCl3·6H2O | Tetrahydrofuran | FF (1 mmol), glycerol (1 mmol), 60 °C, 16 h | 80 | 36 | 44 | 89 |
| 4 | Acidified bentonite | Solvent-free | n(FF)/n(glycerol) = 1/2, rt, 15 h | 72 | 41.04 | 30.96 | 90 |
| 5 | Amberlyst-15 | Cyclohexane | FF (0.75 mol), glycerol (0.67 mol), reflux, 3–4 h | — | 80 | 91 | |
| 6c | [BPy]HSO4 | Solvent-free | n(FF)/n(glycerol) = 2/1, 25 °C, 2 h | — | 28.2 | 64.3 | 92 |
| 7d | TSA/nMCM-48 | Solvent-free | n(FF)/n(glycerol) = 1/1, 30 °C, 40 min | 89 | 27.59 | 61.41 | 93 |
| 8 | SAPO 34 | Solvent-free | n(FF)/n(glycerol) = 1/1, 150 °C, 45 min | — | 23.76 | 41.58 | 94 |
| 9 | ZnCl2 | Solvent-free | n(FF)/n(glycerol) = 5/1, 100 °C, 2 h | — | 90 | 95 | |
| 10 | Aluminosilicate MCM-41 | Solvent-free | n(FF)/n(glycerol) = 5/1, 100 °C, 2 h | — | 80 | 95 | |
| 11 | Zr-Mont | Solvent-free | n(FF)/n(glycerol) = 1/1, rt, 4 h | 84 | 52.12 | 25.89 | 96 |
| 12 | ZrO2-350 | Solvent-free | FF (3.75 mmol), glycerol (48 mmol), 80 °C, 4 h | 87 | 29.58 | 45.24 | 97 |
| 13 | Al-SBA-15 | Solvent-free | FF (1 mmol), glycerol (1 mmol), 100 °C, 12 h | 74 | 50.32 | 23.68 | 98 |
| 14 | Fe/Al-SBA-15 | Solvent-free | FF (1 mmol), glycerol (1 mmol), 100 °C, 12 h | 56 | 29.68 | 26.32 | 99 |
| 15 | MCM-41-16Alanine | Solvent-free | FF (30 mmol), glycerol (30 mmol), 80 °C, 0.5 h | 90 | 70.2 | 19.8 | 100 |
| 16 | Graphene | Solvent-free | FF (10 mmol), glycerol (1 mmol), 100 °C, 2 h | — | 57.8 | 27.2 | 101 |
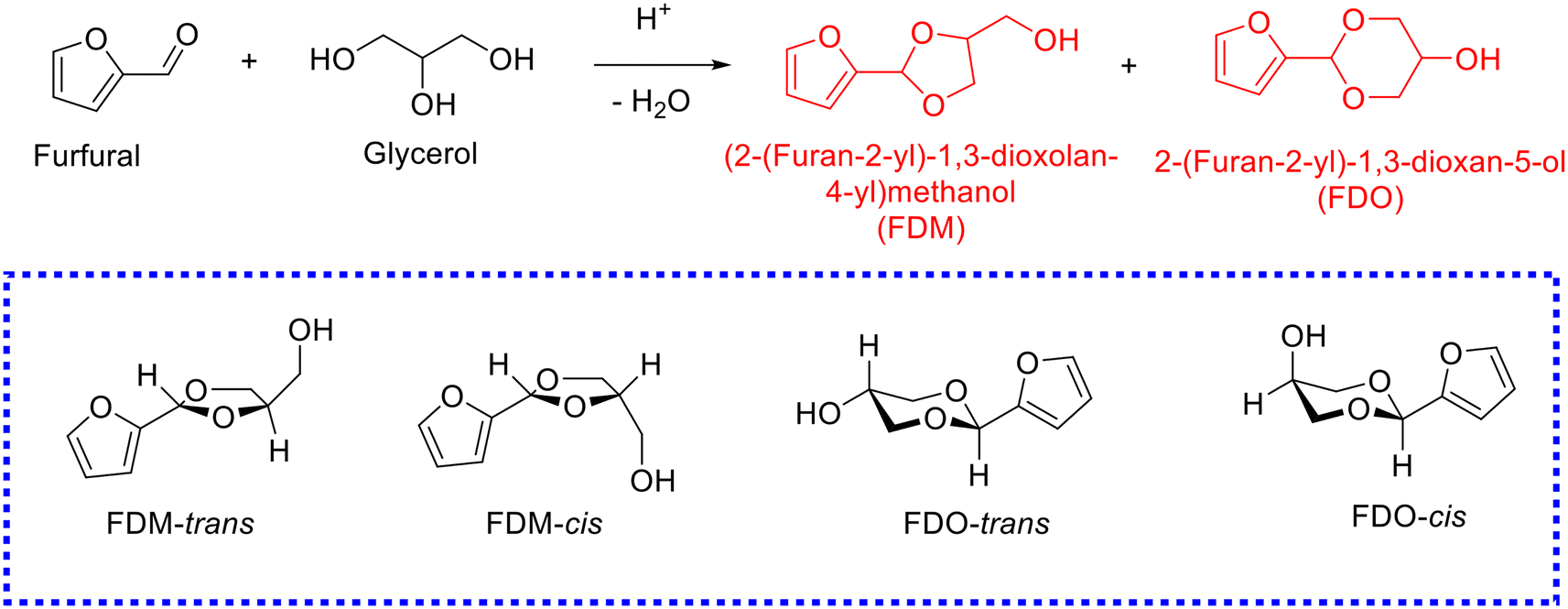 | ||
| Scheme 4 Acetalization of furfural with glycerol to produce cyclic acetals. | ||
Various heterogeneous Brønsted acid catalysts have been exploited for the acetalization of furfural with glycerol.90–94 For instance, acidified bentonite with sulphuric acid, as described by Bajaj and coworkers, was capable of catalyzing the acetalization of furfural with glycerol at room temperature, affording 41.04% yield of FDM and 30.96% yield of FDO at 72% furfural conversion in 15 h (entry 4, Table 2).90 Catalyst characterization revealed that the amount of acid sites, specific surface area, and pore volume of bentonite were increased after activation with sulphuric acid, which was proposed to be responsible for its profound catalytic performance. In addition, the established catalytic system based on the use of acidified bentonite as the catalyst is successful for the acetalization of different carbonyl compounds with glycerol. Analogously, commercial Amberlyst-15 was also active for the acetalization of furfural with glycerol, giving an 80% combined yield of FDM and FDO in refluxing cyclohexane (entry 5, Table 2).91
Subsequently, Wang and his collaborators originally reported the acetalization of furfural with glycerol over [BPy]HSO4 acidic ionic liquid (i.e., N-butyl-pyridinium bisulfate), giving 28.2% FDM yield and 64.3% FDO yield under mild conditions (25 °C and 2 h) (entry 6, Table 2).92 Interestingly, [BPy]HSO4 could be recycled and appeared as a heterogeneous catalyst during the reaction owing to acetal products and ionic liquid separated automatically after reaction. Concretely, a micro water-removing reactor was constructed in the reaction mixture under vigorous stirring caused by the fact that glycerol is soluble in [BPy]HSO4 but immiscible in furfural. The acetalization reaction occurred at the interface between polar phase (glycerol) and organic phase (furfural), in which cyclic acetals were produced and transported to organic phase while water was reverse transported to polar phase (Fig. 4). The micro water-removing reactor not only permitted the timely removal of the byproduct water to drive the reaction completion but also achieved the spontaneous separation of the acetal products and ionic liquid catalyst (Fig. 4).
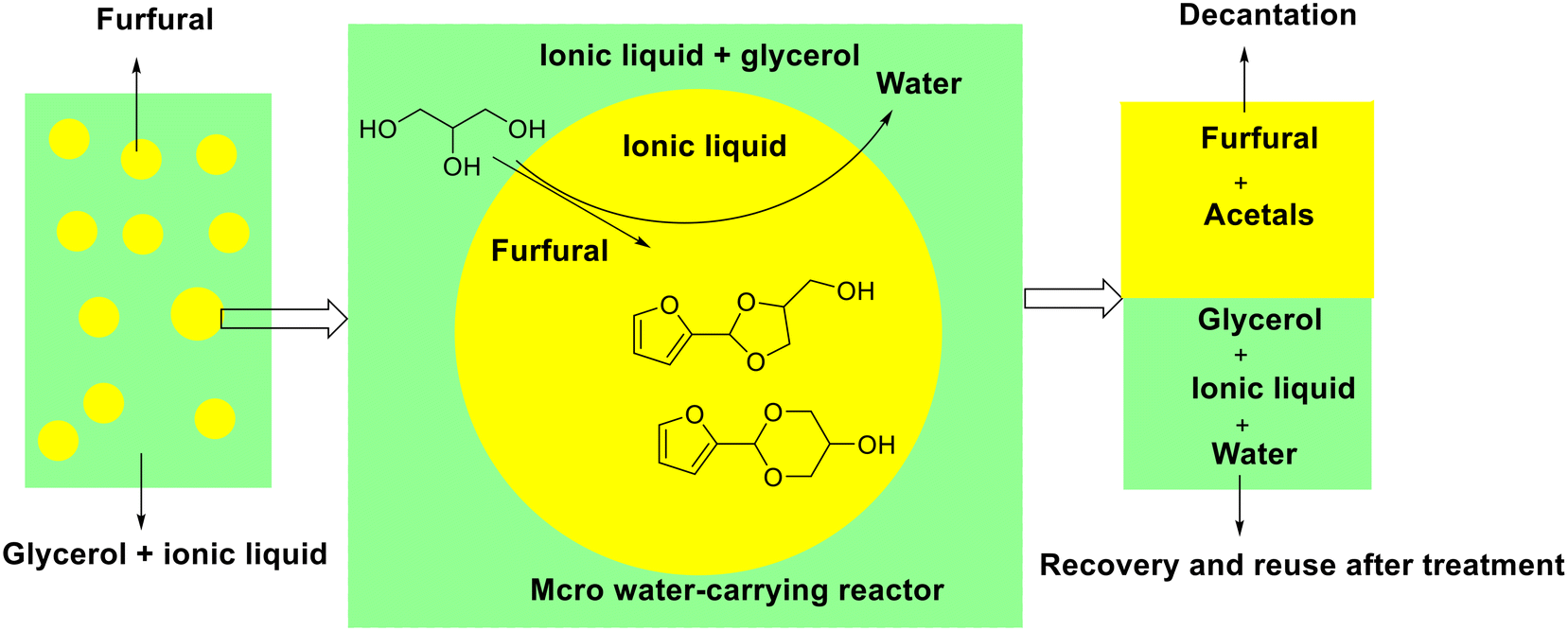 | ||
| Fig. 4 Schematic showing the acetalization of furfural with glycerol over [BPy]HSO4 ionic liquid in the micro water-removing reactor. Adapted with permission from ref. 92. Copyright 2014, Royal Society of Chemistry. | ||
Recently, 12-tungstosilicic acid (TSA) immobilized on ordered nanoporous MCM-48 (i.e., TSA/nMCM-48), as reported by Patel et al., exhibited a remarkable activity for the acetalization of furfural with glycerol, furnishing 27.59% and 61.41% yields of FDM and FDO (entry 7, Table 2),93 respectively. Also, the excellent catalytic performance of TSA/nMCM-48 was attributed to the strong acidity of TSA and 3D cubic geometry as well as the nanoporosity of MCM-48. Currently, most of the studies on the acetalization of furfural with glycerol, however, concentrated upon using commercial glycerol as the starting material. Optimally, the directly use of crude glycerol derived from the procedure of biodiesel production for acetalization is quite fascinating as it avoids expensive purification steps, thereby reducing the cost. Very recently, a successful attempt for the acetalization of furfural with crude glycerol, stemming from the transesterification of the coconut oil, was reported by Cesteros and coworkers.94 A silicoaluminophosphate acid catalyst (i.e., SAPO 34), prepared by microwave-assisted crystallization, was found to be active for the acetalization of furfural with crude glycerol at 150 °C with 45 min, resulting in 23.76% and 41.58% yield of FDM and FDO (entry 8, Table 2), respectively. Moreover, the catalytic performance of SAPO 34 prepared by microwave-heating was superior to the counterpart prepared by conventional heating, which was ascribed to the higher homogeneity of the microwave-heating contributing to the incorporation of higher amounts of silicon within the framework, thereby leading to a higher amount of protons.94
Besides Brønsted acidic sites, Lewis acidic sites also played an important role in promoting the acetalization of furfural with glycerol, which was exemplified by a 90% overall yield of FDM and FDO obtained over a simple Lewis acid salt (i.e., ZnCl2) under solvent-free condition (entry 9, Table 2), as described by Abu-Omar and coworkers.95 In light of the promising application prospect of heterogeneous catalysts, the authors utilized Lewis acidic aluminosilicate MCM-41 for the acetalization of furfural with glycerol, also affording an acceptable overall yield of FDM and FDO at 100 °C in 2 h (entry 10, Table 2).95 In addition, the aluminosilicate MCM-41 catalyst also worked well for crude glycerol (glycerol content of 87%), implying bright prospects in practical applications. Later, the activity of other Lewis acid catalysts such as metal ion-exchanged montmorillonite (e.g., Fe-Mont, Al-Mont, Sn-Mont, and Zr-Mont) in the acetalization of furfural with glycerol was explored in detail by Rode and coworkers.96 Zr-exchanged montmorillonite (i.e., Zr-Mont) was ultimately screened to be the optimal catalyst by providing 52.12% yield of FDM and 25.89% yield of FDO at 84% furfural conversion at room temperature in 4 h (entry 11, Table 2). Also, the excellent activity of Zr-Mont, as proposed by the authors, was related to its controlled acidity. Fortunately, the cyclic acetal products with high purity could be selectively separated from reaction mixture using cyclohexane as the extractant, which avoided the complicated separation process. Furthermore, the Zr-Mont catalyst exhibited good reusability with no significant deactivation through five consecutive tests.
Recently, a series of mesoporous ZrO2 with Lewis acidity were prepared via an inverse micelle method and developed for acetalization of furfural with glycerol.97 The results of catalytic evaluation pointed to the activity of ZrO2 decreased with the increase in the calcination temperature (from 350 to 600 °C), wherein ZrO2 calcined at 350 °C showed outstanding performance under the conditions of 80 °C and 4 h (29.58% FDM yield & 45.24% FDO yield) (entry 12, Table 2). The outstanding catalytic behavior of ZrO2-350 was associated with its high surface area and acidity, as evidenced from the results of catalyst characterization. In addition, a possible reaction mechanism of Lewis acid-catalyzed acetalization of furfural with glycerol was proposed, as shown in Scheme 5. The process involved the formation of a carbocation intermediate from the CO group of furfural activated by the Lewis acid sites on ZrO2, followed by the primary hydroxyl of glycerol attacking the carbocation to form the hemiacetal intermediate and subsequent cyclization to give two isomeric acetals.97
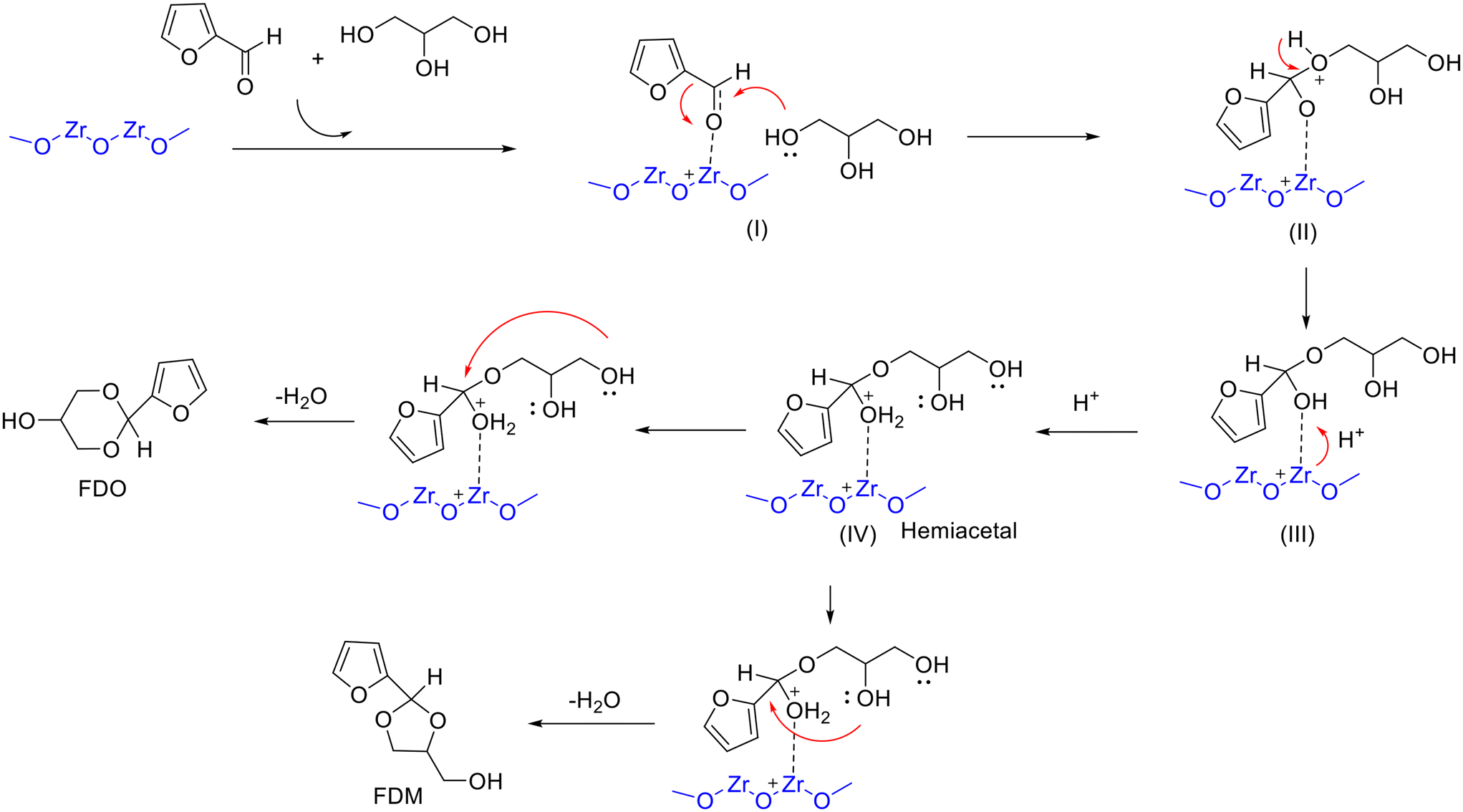 | ||
| Scheme 5 Possible reaction mechanism for the acetalization of furfural with glycerol over ZrO2 involving (I) to (IV) transition-state intermediates. Adapted with permission from ref. 97. Copyright 2021, Elsevier. | ||
Heterogeneous catalysts with both Brønsted and Lewis acid sites are also able to facilitate the acetalization of furfural with glycerol.98,99 Gonzalez-Arellano and colleagues employed Al-SBA-15, a Brønsted–Lewis dual acid sites material (75 μmol g−1 of Brønsted acid sites, 65 μmol g−1 of Lewis acid sites) for the acetalization of furfural with glycerol,98 which provided 50.32% yield of FDM and 23.68% yield of FDO at 100 °C within 12 h (entry 13, Table 2). The yield of FDM superior to that of FDO was proposed to be ascribed to the increasing torsional effect in 6-membered rings with cyclic and aromatic substrates, as described by the authors.98 However, another Brønsted–Lewis dual acidity catalyst with a large amount of Lewis acid sites (33 μmol g−1 of Brønsted acid sites, 200 μmol g−1 of Lewis acid sites), named Zr-SBA-16, showed less activity in acetalization. Based on the above observations, the acetalization reaction over Al-SBA-15, as referred by the authors, was largely facilitated by Brønsted acid sites, although both Brønsted and Lewis sites have the promotion effect on this reaction.98 In the subsequent research of the same group, Al-SBA-15 supported iron oxide nanoparticles (i.e., Fe/Al-SBA-15) were developed for the acetalization of furfural with glycerol, giving rise to appreciable results (entry 14, Table 2).99
In addition, alanine-functionalized MCM-41 catalyst (named MCM-41-16Alanine) with acid–base bifunctional properties has been recently discovered to be effective for the acetalization of furfural with glycerol, affording up to 70.2% yield of FDM and 19.8% yield of FDO at 80 °C within only 0.5 h (entry 15, Table 2).100 The preeminent catalytic performance of MCM-41-16Alanine, as inferred from the characterization analysis, was proposed to be a consequence of its larger amount of acidic sites (carboxylic acid), basic sites (amine group), and confinement effect from the mesopores structure of MCM-41. Moreover, Appaturi and coworkers firstly propose a possible reaction mechanism for the acid–base-catalyzed acetalization of furfural with glycerol. As described in Scheme 6, the hydroxyl hydrogen of glycerol was initially abstracted by the amine group (basic site) of MCM-41-16Alanine to give glycoxy nucleophile (step one). Simultaneously, the carboxylic acid group (acid site) of MCM-41-16Alanine activated the furfural molecule to form a stronger electrophile-carbocation (step one). Subsequently, the furfural carbocation was attacked by the glycoxy nucleophile to yield a furfural–glycerol intermediate (step two), followed by the carboxylate group of catalyst recapturing the proton from the intermediate to release the catalyst (steps three and four). The cyclic acetal products were eventually generated via the dehydration process with two different pathways, as shown in Scheme 6 (step five).
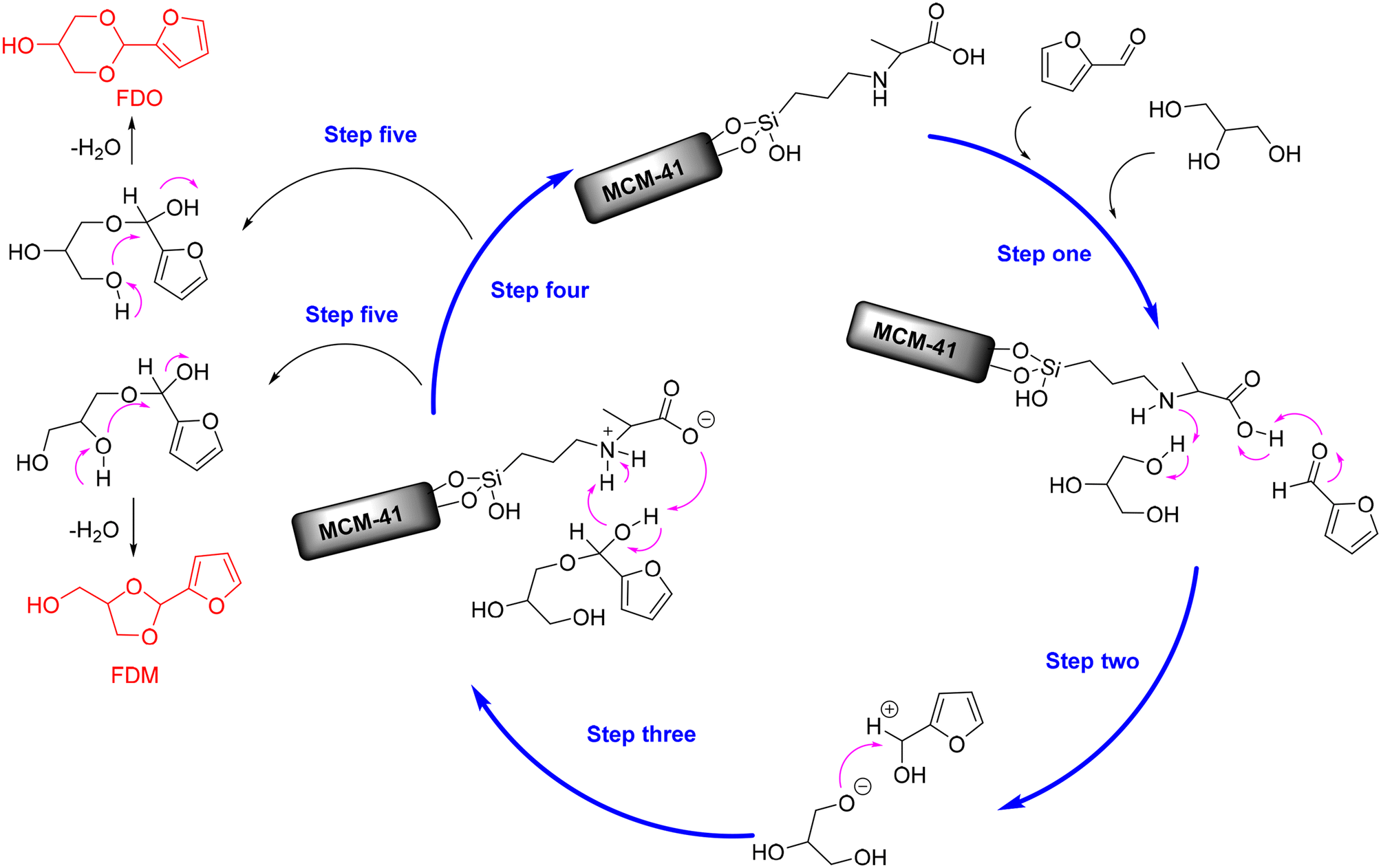 | ||
| Scheme 6 Possible reaction mechanism for the acetalization of furfural with glycerol over MCM-41-16Alanine catalyst. Adapted with permission from ref. 100. Copyright 2021, Elsevier. | ||
Amazingly, acid-free graphene also displayed a remarkable catalytic activity for the acetalization of furfural with glycerol at 100 °C, as serendipitously discovered by Felpin and coworkers, producing 57.8% yield of FDM and 27.2% yield of FDO within 2 h (entry 16, Table 2).101 This observation contradicted the proposed mechanism for the acetalization reaction over Brønsted and/or Lewis acid sites or acid–base sites. Considerable effort was therefore placed on investigating the acid-free mechanism for the acetalization reaction over grapheme.101 The results of XPS analysis and acid–base titration excluded the presence of acidic species (i.e., carboxylic acids and acidic protons) at the surface of graphene, which could preclude the previously proposed acid-catalyzed mechanism. Meanwhile, the absence of metal impurities (e.g., Mn) at the surface of graphene was corroborated by XPS analysis, which could rule out the Lewis acid- or acid–base-catalyzed mechanism. The excellent activity of graphene, as inferred by the authors, was probably rationalized by its peculiar electronic properties of the honeycomb-structured 2D material, notwithstanding the lack of direct evidence.101
Overall, the acetalization of furfural with glycerol proceeded smoothly on catalysis by Brønsted acid sites, Lewis acid sites, Brønsted–Lewis dual acid sites, or acid–base bifunctional sites. In terms of the observed catalytic results, the performance of different types of catalysts are almost comparable. However, for Brønsted–Lewis dual acidity catalyst, the synergistic effect of Brønsted and Lewis acid sites or which kind of acid plays a dominant role during the reaction needs further investigation. In addition, the ratio of FDM/FDO acetal products shows no obvious trend, which probably depends on the nature of the used catalyst and torsional effects in acetal rings.
3 Synthesis of cyclic acetals from 5-hydroxymethylfurfural
3.1 Acetalization of 5-hydroxymethylfurfural with ethylene glycol
Similar to furfural, 5-hydroxymethylfurfural (HMF) derived from cellulosic hexoses, also represents a versatile platform molecule for the synthesis of cyclic acetal fuel additives. The acetalization of HMF with ethylene glycol, nevertheless, seems a little complicated relative to the acetalization of furfural due to the presence of hydroxyl group in HMF, which can induce side reaction (i.e., etherification). Usually, the acetalization of HMF with ethylene glycol results in the formation of (5-(1,3-dioxolan-2-yl)furan-2-yl)methanol (i.e., DFM), as delineated in Scheme 7, and concomitantly yielding etherification products such as 5-((2-hydroxyethoxy)methyl)furan-2-carbaldehyde and 2-((5-(1,3-dioxolan-2-yl)furan-2-yl)methoxy)ethan-1-ol. Current emphasis in the acetalization of HMF mainly focuses on the production of cyclic acetal products, albeit the etherized derivatives of HMF are also potential biofuels.102–105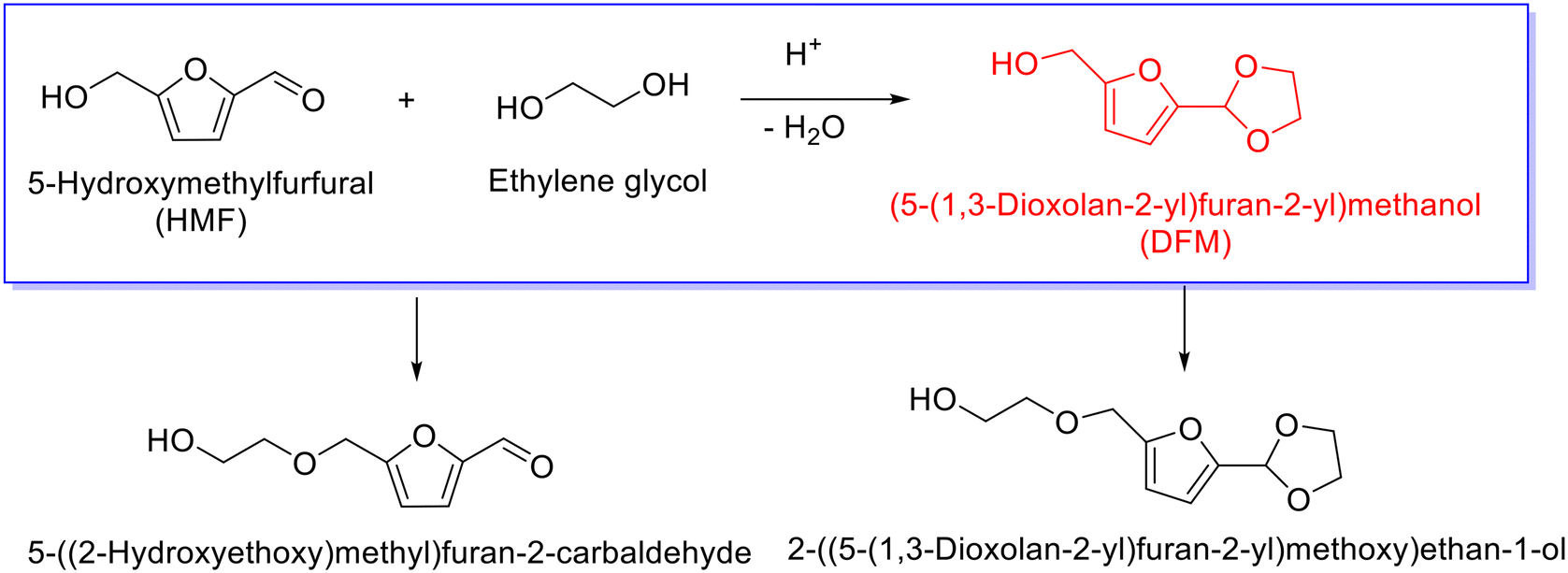 | ||
| Scheme 7 Acetalization of HMF with ethylene glycol to yield cyclic acetal. | ||
Analogously, acid sites play a vital role in promoting the acetalization of HMF with ethylene glycol. For instance, acidic resin Amberlyst-15, as reported by Dumesic and coworkers, succeeded in facilitating the acetalization of HMF with ethylene glycol at 35 °C, giving rise to a 55.6% yield of DFM at 2.2 molar ratio of ethylene glycol to HMF with a reaction time of 18.5 h.106 Moreover, the as-synthesized DFM, as highlighted by the authors, could serve as a starting feedstock for the manufacture of norcantharimide derivatives, which are therapeutic molecules, via Diels–Alder reaction with maleimides. Interestingly, the obtained norcantharimide derivatives were capable of releasing reactants (i.e., HMF, maleimides and ethylene glycol) through retro Diels–Alder reaction at ambient temperatures under acidic conditions, which was triggered by the reverse-conversion of the acetal group to an aldehyde leading to mismatches of the molecular orbitals in norcantharimides.106 The innovative strategy illustrates compelling opportunities for cyclic acetals with wide application in the synthesis of value-added chemicals and synchronously opens up the possibility of recycling biobased molecules.
Similar to the acetalization of furfural with ethylene glycol, the acetalization of HMF with ethylene glycol also occurs smoothly over synergistic effect of acid–base sites. For example, the CePO4 catalyst with acidity and basicity, prepared by a hydrothermal method, has demonstrated great promise in the acetalization of HMF with ethylene glycol, affording 80% yield of DFM at 80 °C with 1 h.107 Based on the excellent catalytic performance, IR measurements of CePO4 samples with adsorbed acetone and methanol were performed to provide a basis for inferring the reaction mechanism. Also, the observed results demonstrated that the synergistic effect of the acid–base sites within CePO4 was crucial for the acetalization of HMF with ethylene glycol, wherein the Lewis acid sites and weak base sites activated the HMF and ethylene glycol molecules,107 respectively. Furthermore, CePO4 worked in a heterogeneous catalysis form, as confirmed by hot-filtration tests and ICP-AES measurements for the filtrate. In addition, experiments with various carbonyl compounds and alcohols demonstrated CePO4 has a good level of generality for acetalization. However, the current reported systems able to catalyze the acetalization of HMF with ethylene glycol are limited. Therefore, tremendous effort to exploit effective heterogeneous catalysts for the acetalization of HMF are required.
3.2 Acetalization of 5-hydroxymethylfurfural with glycerol
As presented above, current researches concerning the acetalization of HMF with ethylene glycol over acid catalysts are limited, which is attributable to the HMF molecule bearing diverse functional groups (i.e., –OH, CO, and furan ring) that can bring about etherification, acetalization, and hydrolysis/alcoholysis to give levulinic acid or its esters.108–110 The selective synthesis of cyclic acetals from HMF and glycerol is thus not an easy task, and the acetalization of HMF with glycerol is also scarce in the current open literature.
In general, a mixture of five-membered-ring acetal and six-membered-ring acetal, namely, (5-(4-(hydroxymethyl)-1,3-dioxolan-2-yl)furan-2-yl)methanol (i.e., FDFM) and 2-(5-(hydroxymethyl)furan-2-yl)-1,3-dioxan-5-ol (i.e., FFDO), were obtained from the acetalization of HMF with glycerol, and each with trans and cis isomers (Scheme 8), respectively. To date, several successful examples on the acetalization of HMF with glycerol have been reported, as compiled in Table 3.111,112,116
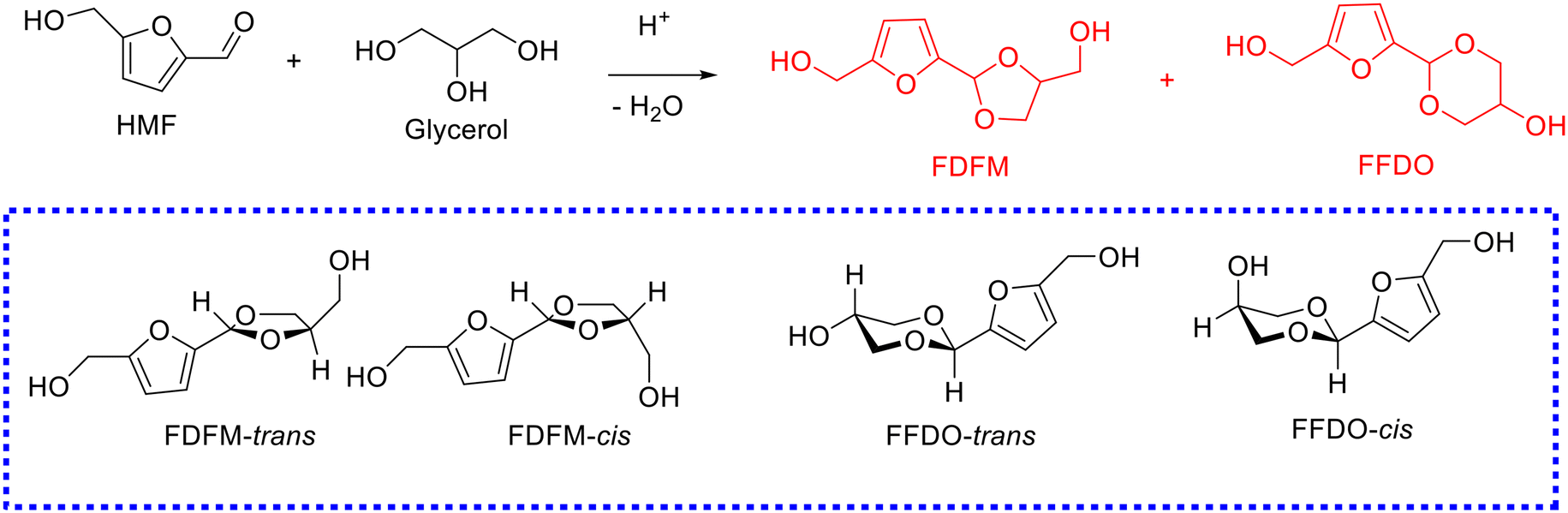 | ||
| Scheme 8 Acetalization of HMF with glycerol to yield cyclic acetals. | ||
| Entry | Catalyst | Solvent | Reaction conditions | HMF conv. (%) | Yield (%) | Ref. | |
|---|---|---|---|---|---|---|---|
|
|
|
||||||
| a The substrate is 5-methylfurfural (5-MF). b ITQ-2 was derived from the delamination of a layered precursor of MCM-22. c The number in parentheses denotes the mole ratio of silicon to aluminum within the catalyst. | |||||||
| 1 | MoO3/SnO2 | Solvent-free | n(HMF)/n(glycerol) = 1/1, rt, 0.5 h | — | 40.95 | 22.05 | 111 |
| 2a | MoO3/SnO2 | Solvent-free | n(5-MF)/n(glycerol) = 1/1, rt, 0.5 h | — | 40.8 | 19.2 | 111 |
| 3 | H-Beta(12)c | Acetonitrile–trifluorotoluene (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1) :1) |
n(HMF)/n(glycerol) = 1/2, 83 °C, 24 h | 82 | 44.06 | 16.94 | 112 |
| 4 | USY(12)c | Acetonitrile–trifluorotoluene (1:1) |
n(HMF)/n(glycerol) = 1/2, 83 °C, 24 h | 75 | 57.95 | 17.05 | 112 |
| 5b | ITQ-2(15)c | Acetonitrile–trifluorotoluene (1:1) |
n(HMF)/n(glycerol) = 1/2, 83 °C, 3 h | 98 | 72.21 | 25.79 | 112 |
| 6 | MCM-41(15)c | Acetonitrile–trifluorotoluene (1:1) |
n(HMF)/n(glycerol) = 1/2, 83 °C, 8 h | 94 | 74.82 | 19.18 | 112 |
| 7 | BC-SO3H | Solvent-free | n(HMF)/n(glycerol) = 2/1, 30 °C, 10 h | — | 51.6 | — | 116 |
Novel molybdenum and tungsten promoted stannic oxide solid acids (i.e., MoO3/SnO2 and WO3/SnO2), as described by Reddy and coworkers,111 were synthesized and applied for the acetalization of HMF with glycerol to produce cyclic acetals under solvent-free conditions. WO3/SnO2 turned out to be a superior catalyst owing to increased acid sites density (81.45 μmol g−1), enhanced surface area (56 m2 g−1), notable redox properties, and superior lattice defects after the doping of Mo into SnO2, affording 40.95% yield of FDFM and 22.05% yield of FFDO at room temperature in 0.5 h (entry 1, Table 3). The analysis by FTIR studies of adsorbed pyridine demonstrated that the WO3/SnO2 catalyst primarily possessed Brønsted acid sites (ratio of Brønsted acid sites to Lewis acid sites over 95). In addition, the WO3/SnO2 catalyst featured good catalytic performance toward the acetalization of 5-methylfurfural with glycerol under solvent-free and room temperature conditions, furnishing 40.8% yield of the five-membered-ring acetal and 19.2% yield of the six-membered-ring acetal (entry 2, Table 3).111 Furthermore, the WO3/SnO2 catalyst was found to be reusable for three consecutive runs without significant deactivation. In 2018, Iborra and colleagues originally reported that several zeolites played an important catalytic role in the acetalization of HMF with glycerol in the mixture of acetonitrile–trifluorotoluene (1/1 of volume ratio).112 Concretely, tridirectional large pore zeolites, such as H-Beta(12) and USY(12), were able to promote the acetalization of HMF with glycerol and secured good yields (44.06% FDFM yield & 16.94% FFDO yield over H-Beta(12), 57.95% FDFM yield & 17.05% FFDO yield over USY(12)) under mild reaction conditions (83 °C, 24 h) (entries 3–4, Table 3).112 Unfortunately, H-Beta(12) and USY(12) catalysts were susceptible to deactivation due to the strong adsorption of organic material, contributing to micropore blockage. Subsequently, a laminar zeolite (ITQ-2, derived from the delamination of a layered precursor of MCM-22) and a mesoporous aluminosilicate (MCM-41) were tested for the acetalization of HMF with glycerol, both of which gave excellent catalytic results at 83 °C (entries 5–6, Table 3).112 Relatively, ITQ-2 and MCM-41 were stable catalysts, as demonstrated by the fact that their deactivation rates were lower than that of H-Beta(12) and USY(12), owing to their smaller confinement effects, easy diffusion, and adequate surface polarity. In particular, pure silica MCM-41 material was inactive for the acetalization of HMF with glycerol (no products detected), demonstrating that the silanol groups were not acidic enough to facilitate the reaction. Thus, as the authors referred, the active sites for zeolites and ITQ-2 catalysts were possibly attributed to tetrahedrally-coordinated Al, which shows Brønsted acidity.112
Recently, various heterogeneous acid catalysts have been successfully fabricated using biomass as the starting materials,113–115 and it is predictable that biomass-derived heterogeneous acid catalysts will become more attractive options for biomass valorization thanks to both the reaction substrate and catalysts precursor stemming from biomass. The pioneering attempt on developing biomass-derived heterogeneous acid catalysts for the acetalization of HMF with glycerol was done by Biradar and coworkers,116 wherein SO3H-functionalized carbon catalyst (BC-SO3H) was prepared from waste bagasse by a one-step hydrothermal method using concentrated sulfuric acid. The as-prepared BC-SO3H exhibited remarkable catalytic performance for the acetalization of HMF with glycerol and procured good yield (51.6% yield of FDFM) at 30 °C in 10 h under solvent-free condition (entry 7, Table 3).116 Moreover, the universality test of the BC-SO3H catalyst displayed that it was capable of facilitating the acetalization of various aldehydes with glycerol to give diverse cyclic acetals with good yields (52–81% yields). This work provides an elegant strategy for the development of biomass-derived catalysts for biomass catalytic conversion, which helps to improve the utilization efficiency of biomass and thus shows great potential for industrial applications. In addition, the acetalization of HMF derivative (e.g., 5-(octyloxymethyl)furfural) with glycerol to yield biomass-derived nonionic surfactant (i.e., 5-(octyloxymethyl)furfural glyceryl acetal) over the acid catalyst was also reported by Corma and coworkers.117
To sum up, Brønsted acid sites are the main prevailing active sites for the acetalization of HMF with glycerol in literature. Also, for porous catalytic materials (e.g., zeolites and mesoporous aluminosilicate), the catalytic performance and reusability are greatly affected by their structures. Regrettably, information on the hydration of HMF to give levulinic acid in these catalytic systems is absent. Actually, acetalization leads to the formation of water, which allows the possibility of HMF hydration over Brønsted acid sites. Therefore, the effect of Brønsted acid density and acid strength on acetalization and/or its competing reaction (HMF hydration) deserve further study.
4 Synthesis of cyclic ketals from levulinic acid
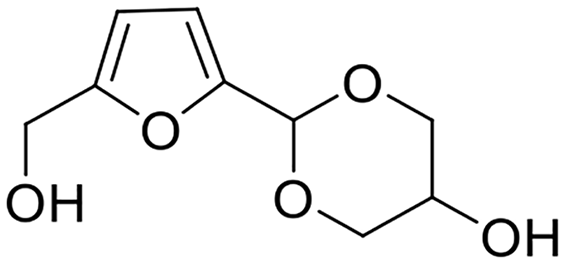
Levulinic acid and its esters, as shared derivatives from cellulose and hemicellulose, are obtained from HMF via rehydration/alcoholysis or accessible from furfural through reduction and consecutive ring-opening reaction (Scheme 9).118–121 The synthesis of cyclic ketals from levulinic acid or its esters and polyols is perceived as one of the promising applications of levulinic acid or its esters owing to cyclic ketals with a wide range of industrial applications from fuel additives to fine chemicals (e.g., surfactants, binders, plasticizers, and polymers).122–125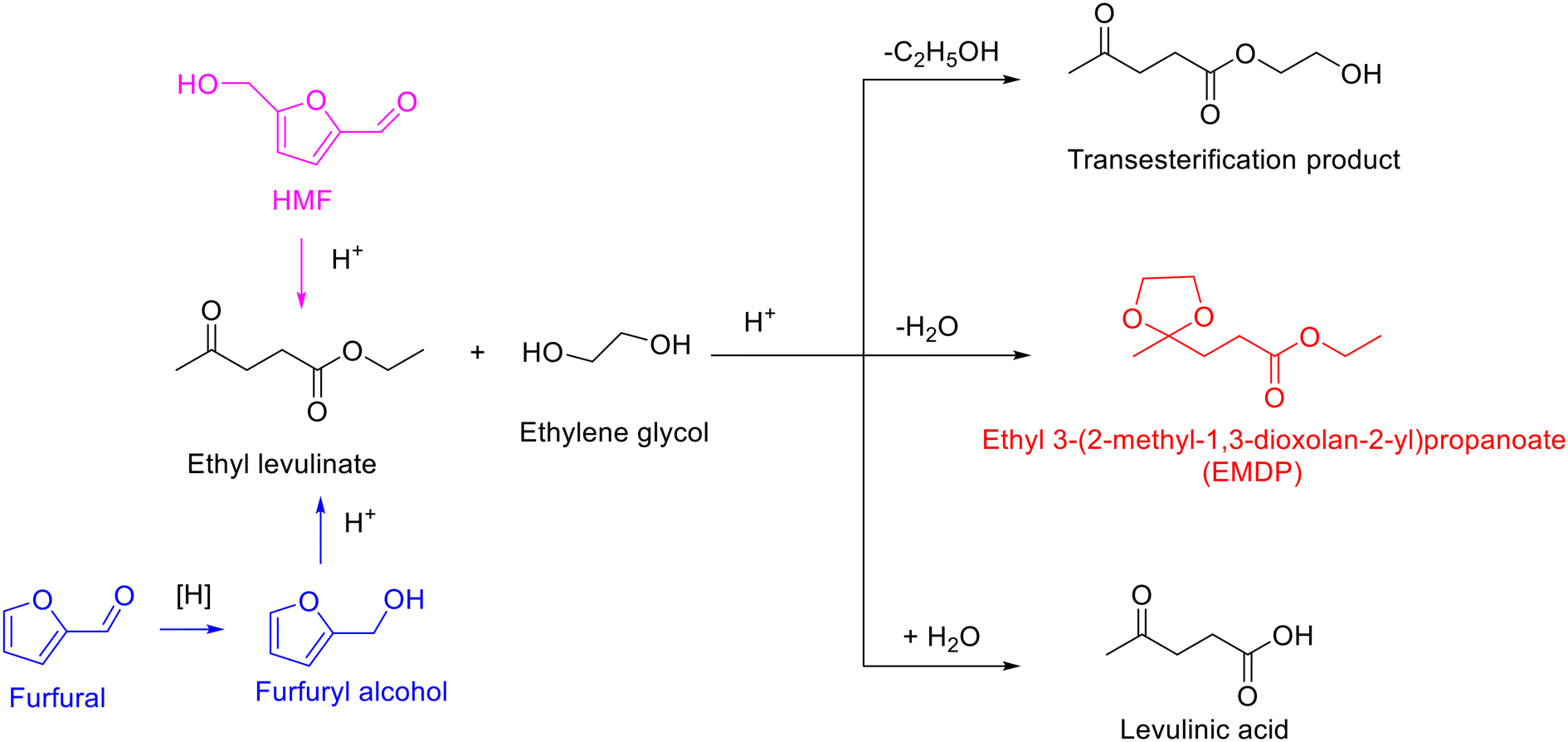 | ||
| Scheme 9 Acetalization of ethyl levulinate with ethylene glycol to form cyclic ketal and byproducts. | ||
As expected, ethylene glycol and glycerol as the biobased diol and triol are frequently used as starting materials for reacting with levulinic acid or its esters to produce cyclic ketal products.126–129 Likewise, the acetalization of levulinic acid or its esters with ethylene glycol requires acid sites and usually results in the formation of ethyl-3-(2-methyl-1,3-dioxolan-2-yl)propanoate (EMDP), along with the transesterification product and hydrolysis product (i.e., levulinic acid), as exhibited in Scheme 9. An early example on the acetalization of ethyl levulinate with ethylene glycol was reported by Lachter and coworkers applying heterogeneous acid catalysts such as Amberlyst-70, H-ZSM-5, and niobium phosphate.126 The three tested heterogeneous acid catalysts turned out to be capable of promoting acetalization of ethyl levulinate with ethylene glycol at 110 °C within 4 h (ethyl levulinate/ethylene glycol molar ratio = 0.5), affording 87%, 68%, and 54% ethyl levulinate conversion, respectively. Notably, the selectivity toward EMDP was 96% over Amberlyst-70 and H-ZSM-5 catalysts, while 100% EMDP selectivity was obtained in the presence of niobium phosphate.126 Moreover, both Amberlyst-70 and H-ZSM-5 exhibited apparent deactivation in reusability experiments while niobium phosphate was found to be reusable for five consecutive runs without any deactivation, strongly implying that niobium phosphate was a promising durable catalyst candidate for the acetalization of ethyl levulinate with ethylene glycol. Furthermore, the surface area of used niobium phosphate was close to that of fresh one (120 m2 g−1versus 138 m2 g−1), suggesting that the adsorption/deposition of organic matters is relatively less.
A further attempt on improving the EMDP yield from the acetalization of ethyl levulinate with ethylene glycol was done by Srinivasan and his collaborators using azeotropic distillation with the help of Dean–Stark apparatus in cyclohexane to remove in situ generated water.127 Various heterogeneous acid catalysts, including Na-beta, Na-Y, Na-ZSM-5, Amberlyst-15, and montmorillonite, were subjected to the acetalization of ethyl levulinate with ethylene glycol. Na-Beta zeolite, among the tested catalysts, exhibited the best catalytic performance owing to the presence of a sufficient amount of Brønsted and Lewis acidic sites, giving 99% ethyl levulinate conversion and up to 99% yield of EMDP at 120 °C within 1 h.127 The inferior catalytic activity of other tested catalysts (e.g., Na-Y, Na-ZSM-5, Amberlyst-15, and montmorillonite) was possible associated to the lack of strong Brønsted or Lewis acidic sites. In addition, acetalization is preferential over the transesterification side reaction in the established catalytic system because the carbonyl oxygen in ethyl levulinate has more electron density in comparison to carboxylic oxygen, which permits the acidic sites in the Na-beta zeolite to preferentially interact with the carbonyl oxygen lone pair instead of carboxylic oxygen.127
Analogously, several publications have demonstrated the synthesis of cyclic ketals from the acetalization of levulinic acid or its esters with glycerol over acid catalysts.128,129 Taking methyl levulinate as an example, a mixture of five-membered-ring ketal (i.e., methyl 3-(5-hydroxy-2-methyl-1,3-dioxan-2-yl)propanoate, MHMDP) and six-membered-ring ketal (i.e., methyl 3-(4-(hydroxymethyl)-2-methyl-1,3-dioxolan-2-yl)propanoate, MHMDE), as illustrated in Scheme 10, are generally accessible from the acetalization of methyl levulinate with glycerol. A successful attempt on the acetalization of methyl levulinate with glycerol was achieved by Zhao and his collaborators using low concentration of sulfuric acid.128 Under the assistance of continuous removal of water with Dean–Stark apparatus, a significantly high cyclic ketals (i.e., MHMDP and MHMDE) with yield of up to 97.2% was achieved over sulfuric acid at 123 °C with 0.5 h (a molar ratio of methyl levulinate to glycerol of 4.4, a vacuum degree of 90 kPa). Even though significant progress achieved, the conversion process is characterized by the issue of corrosion and difficulty of catalyst separation and recyclability arising from the homogeneous sulfuric acid.
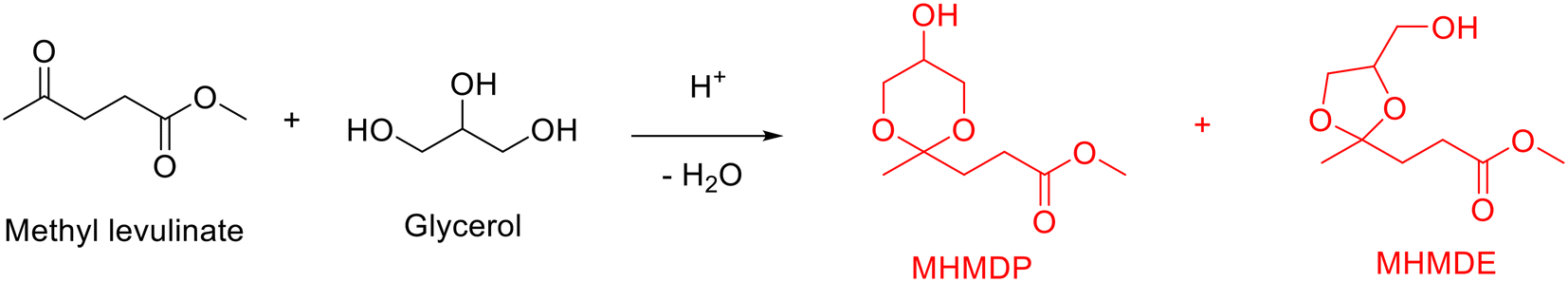 | ||
| Scheme 10 Acetalization of methyl levulinate with glycerol to form cyclic ketals. | ||
Recent interest in the acetalization of methyl levulinate with glycerol has been shifted from homogeneous acid catalysts to heterogeneous acid catalysts to avert the issues faced by homogeneous catalysis. For example, a macro/mesoporous solid acid, derived from sodium lignosulphonate via freeze drying, pyrolysis (350–450 °C) and H+ exchanging method, was found to be effective for the acetalization of methyl levulinate with glycerol.129 53% yield of MHMDP and 47% yield of MHMDE were obtained under the conditions of a 4 molar ratio of methyl levulinate to glycerol, 100 °C, 1 h, and 50 mL min−1 N2 flow. The outstanding catalytic performance of the as-prepared macro/mesoporous solid acid was linked with its surface endowing large amounts of –OH, –COOH, and –SO3H groups and its macro/mesoporous structure, which contributes to the good accessibility of the active sites.
In brief, recent studies on the acetalization of levulinic acid with ethylene glycol/glycerol primarily focus on the application of ketal products. Therefore, developing environment friendly, cost-effective, and effective heterogeneous catalysts as well as exploring the reaction mechanism via in situ characterization remains highly rewarding.
5 Applications of the acetalization protection strategy in biomass valorization
Apart from the production of fuel additives, the strategy of acetalization has been successfully applied in other biomass valorizations. As is well known, biomass-derived molecules are generally very reactive due to the presence of diverse chemical bonds including C–C, C–O, O–H, CC, and CO, thus resulting in various byproducts in the course of transformation.130–132 From the atomic economy and green chemistry viewpoints, the selective conversion of biobased molecules with high selectivity is highly desirable. Recently, the protection of reactive functional groups, for biobased oxygenated compounds, has been identified as an elegant way to improve the selectivity of target product during the procedure of catalytic upgradation.133 In particular, acetalization is a renowned reversible reaction and represents a powerful method to protect carbonyl compounds (e.g., aldehydes and ketones) due to the stability and low reactivity of acetals/ketals in basic media and easily returning to carbonyl compounds via deprotection.134–136 Currently, the strategy of acetalization has already been successfully applied in some important transformations in the field of biomass conversion. Several typical examples herein, on using acetalization strategy for protecting functional groups in biomass conversion to improve the target products selectivity, are summarized.
5.1 Depolymerization of lignin
Lignin, one of the main components of lignocellulose, is the most abundant aromatic polymer in nature, which represents a sustainable feedstock for the production of aromatic chemicals and fine chemicals.137,138 At present, lignin is still vastly underutilized due to the absence of effective method for lignin depolymerization, although several elegant approaches including thermal, acid/base catalyzed, reductive, and oxidative depolymerization are available for lignin depolymerization.139–144 Amongst acid-catalyzed depolymerization (i.e., acidolysis) method has received unique attention in lignin depolymerization because acidolysis is the most widely used approach in the depolymerization of lignocellulose.145 Various valuable functionalized phenolic monomers (e.g., aldehydes and ketones) are accessible from lignin acidolysis, as shown in Scheme 14; the yields of monomers nevertheless are generally low due to the recondensation of the formed fragments to give highly condensed C–C bonded products via aldol condensation and nucleophilic aromatic substitution reactions.To overcome the aforementioned issue encountered by lignin acidolysis, a pioneering attempt to improve phenolic monomers yields on introducing ethylene glycol to stabilize the formed phenolic monomers via acetalization was done by Deuss and coworkers (Scheme 11).145 Trapping the reactive phenolic monomers via acetalization to afford acetals/ketals could inhibit the repolymerization of functionalized phenolic monomers and consequently enhance the yield of monomers.146,147 Thus, the strategy of acetalization shows great potential in trapping/stabilizing reactive intermediates in biomass valorization.
 | ||
| Scheme 11 Representative products or acetal stabilized products from lignin β-O-4 linkage acidolysis. | ||
Likewise, the introduction of aldehydes/ketones can also stabilize lignin via acetalization between the introduced aldehydes/ketones and hydroxyl groups of lignin (e.g., the α,γ-diol on the side chain).148 The direct hydrogenolysis of native lignin in raw biomass represents a prevalent strategy for lignin depolymerization, which generally involves mixing solid biomass feedstock with a heterogeneous metal catalyst in the presence of hydrogen source such as high-pressure hydrogen gas or hydrogen-donating solvent.149–151 Despite the excellent results obtained, the difficult recovery of the heterogeneous catalysts from the solid residue and mass transfer limitations may hamper the further practical application of these systems. The use of extracted lignin for hydrogenolysis could avert these shortcomings.152 However, monomer yields produced from the hydrogenolysis of extracted lignin were far lower than that obtained from native lignin, mainly caused by the presence of lignin condensation reactions during its extraction.152 Exhilaratingly, Luterbacher and coworkers discovered the addition of formaldehyde during lignin extraction could result in near theoretical yields of lignin monomers from the hydrogenolysis of the extracted lignin,153 which was attributed to the formation of 1,3-dioxane structures from the reaction of formaldehyde with lignin side-chain hydroxyl groups, as evidenced from 2D HSQC NMR spectra, to prevent lignin condensation (Scheme 12). Along with the formation of a stable six-membered 1,3-dioxane structure (acetal), the electron-rich positions of ortho or para to methoxyl groups on the aromatic ring were electrophilically substituted by (protonated) formaldehyde to form hydroxymethyl groups during the lignin extraction process, blocking these reactive positions and subsequently preventing lignin condensation (Scheme 12). Concretely, one gram of air-dried biomass particles (i.e., beech wood or high-syringyl transgenic poplar) were mixed with 1,4-dioxane (9 mL), HCl solution (37%, 420 μL), and formaldehyde solution (36.5%, 1 mL) at 80 °C for 5 h to produce a soluble lignin fraction (i.e., extracted lignin), followed by hydrogenolysis over 5 wt% Ru/C at 250 °C for 15 h to give lignin monomers.153 Finally, 47% or 78% yield of monomers were achieved from the hydrogenolysis of formaldehyde-stabilized lignin (beech wood or high-syringyl transgenic poplar), far surpassing the monomers yield (7% yield for beech wood or 26% yield for high-syringyl transgenic poplar) from the hydrogenolysis of extracted lignin without formaldehyde addition. Ten major phenolic monomers were formed from formaldehyde-stabilized lignin while only five major lignin monomers were generated from extracted lignin without formaldehyde addition, as shown in Scheme 12.153 Furthermore, as described by the authors, the process for extracting soluble-stabilized lignin fractions and subsequent hydrogenolysis was compatible with current polysaccharide depolymerization approaches, bringing great potential for real application.
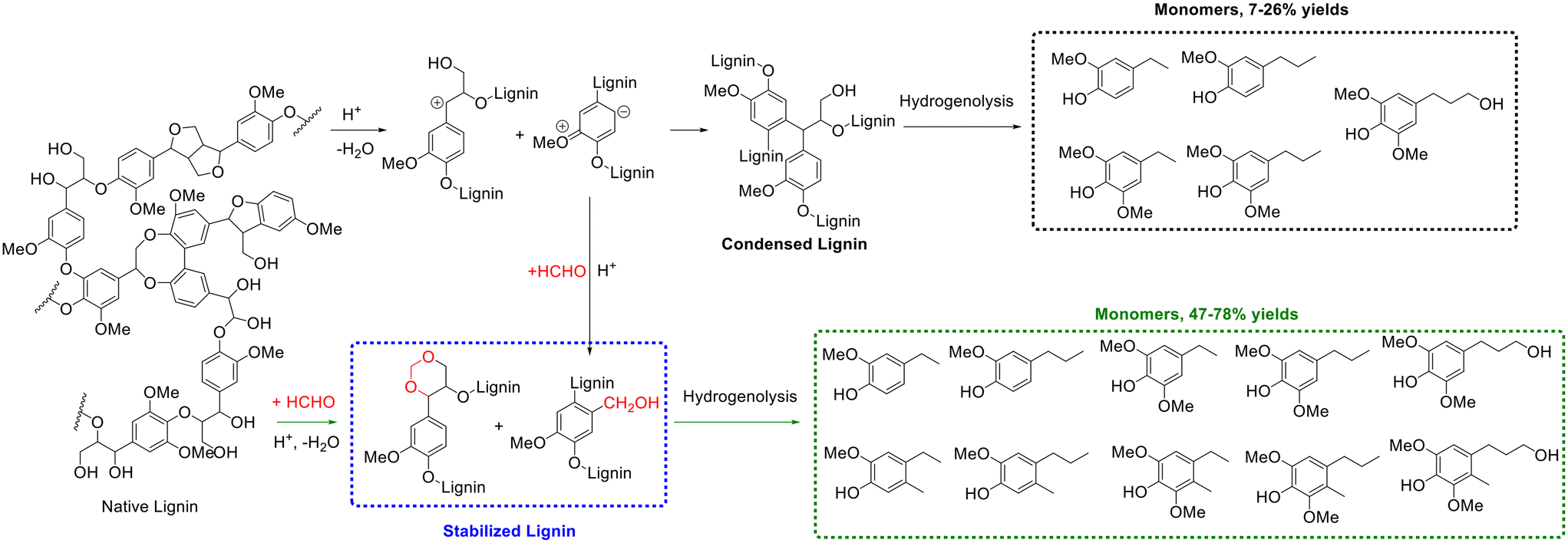 | ||
| Scheme 12 Monomers production from extracted lignin (stabilization with formaldehyde or condensed lignin), followed by hydrogenolysis. Adapted with permission from ref. 153. Copyright 2016, American Association for the Advancement of Science. | ||
In the subsequent research, different protection reagents, such as acetaldehyde, propionaldehyde, acetone, 2-butanone, 2-methoxypropene, 2-dimethoxypropane, dimethyl carbon, and phenylboronic acid, were added during biomass (i.e., birch, spruce, and high-syringyl transgenic poplar) pretreatment to stabilize lignin via the acetalization of protection reagents and α,γ-diol group in lignin, and the stabilized lignin were used for the production of monomers through hydrogenolysis with a Pd/C catalyst.154 Among all the tested protection reagents, aldehydes including formaldehyde, acetaldehyde, and propionaldehyde turned out to be the most effective reagents with superior yields of lignin monomers obtained from hydrogenolysis. Interestingly, the selectivity toward final lignin monomers was greatly increased together with the number of products being markedly reduced when acetaldehyde or propionaldehyde was used as the protection reagent in comparison with formaldehyde,154 which was ascribed to the absence of hydroxyethylation or hydroxypropylation during the reaction, as revealed from HSQC and homonuclear correlation spectra. For example, as high as 70% yield of monomers with an 80% selectivity toward 4-n-propanolsyringol was achieved from propionaldehyde-stabilized lignin derived from high-syringyl transgenic poplar.154
Beyond hydrogenolysis, oxidation has been proposed by the group of Luterbacher as an effective method for the selective synthesis of oxygenated aromatic molecules from propionaldehyde-stabilized lignin.155 The authors established a pioneering two-step process for the transformation of propionaldehyde stabilized lignin into oxygenated aromatic molecules that involves the selectivity deprotection of the acetal and oxidation of the α-OH into a ketone intermediate (i.e., oxidized lignin) using 2,3-dichloro-5,6-dicyano-1,4-benzoquinone (DDQ) as the oxidant/catalyst in acetic acid/water (95/5) medium, followed by the depolymerization of oxidized lignin to aromatic monomers using a formic acid/sodium formate system (Scheme 13).155 Eventually, the yield of syringyl propane dione reached up to 80% (based on the original Klason lignin), accompanied by 10–13% yield of guaiacyl propanedione.155 The unprecedented selectivity to a single lignin monomer, as inferred by the authors, was probably credited to the preservation of the lignin structure via the acetalization of propionaldehyde and α,γ-diols on the side chain of lignin.155
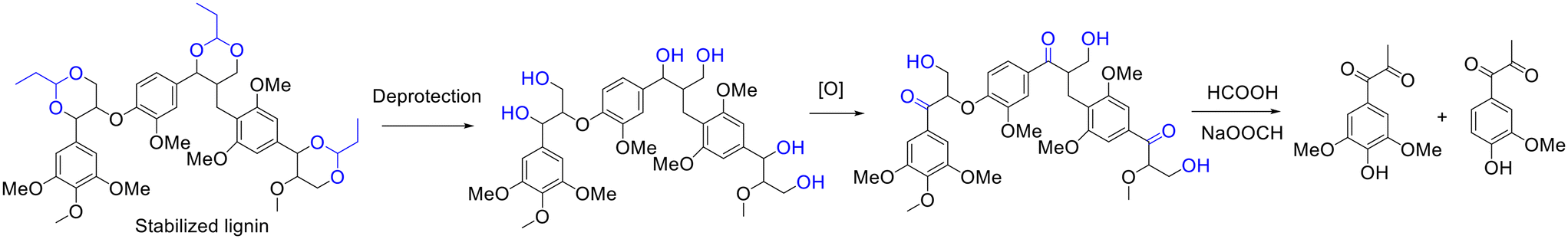 | ||
| Scheme 13 Synthesis of syringyl and guaiacyl propane diones from the oxidation of stabilized lignin, followed by depolymerization. Adapted with permission from ref. 155. Copyright 2019, John Wiley and Sons. | ||
In summary, the protection protocol for valorizing lignin or its derivatives based on the concept of in situ acetalization strategy to prevent their repolymerization can improve the selectivity toward monomer products.
5.2 Oxidation of 5-hydroxymethylfurfural into 2,5-furandicarboxylic acid
2,5-Furandicarboxylic acid (FDCA), an oxidative derivative of 5-hydroxymethylfurfural (HMF), stands out as an ideal suitable replacement for petroleum-derived terephthalic acid in plastics production due to the molecular structure of FDCA similar to terephthalic acid.156 HMF oxidation is seen to be the most straightforward approach for producing FDCA, and thus the attention of HMF-to-FDCA has been boosted rapidly in recent years.157,158The majority of researches on the oxidation of HMF into FDCA, however, are limited to dilute HMF (0.5–2.1 wt%) solutions due to the intrinsic instability of HMF stemming from the highly reactive hydroxymethyl and formyl groups.159 In fact, the oxidation of HMF into FDCA generally goes through several intermediates with high reactivity such as 5-(hydroxymethyl)furan-2-carboxylic acid and 5-formylfuran-2-carboxylic acid, which induce the generation of byproducts including humins via condensation and polymerization (Scheme 14a) and subsequently result in low selectivity toward FDCA.159 Excitingly, a seminal work aiming at enhancing the FDCA selectivity in relatively concentrated HMF solutions (i.e., 10 wt%) was reported by Nakajima and his collaborators, wherein HMF initially converted into FDCA-acetal with high thermal stability through acetalization with 1,3-propanediol and subsequently underwent successive oxidation to produce FDCA.159 As shown in Scheme 14b, the oxidation of HMF-acetal into FDCA with DFF-acetal and FFCA-acetal served as intermediates, which are stable in the course of oxidation and thereby significantly reduced the side reactions and then substantially improved the FDCA selectivity. For example, under the catalysis of Au/CeO2, FDCA selectivity reached up to 94% using HMF-acetal as the substrate while FDCA selectivity was only 28% in HMF solutions under identical reaction conditions (140 °C, 0.5 MPa O2, and 15 h).159 Importantly, approximately 80% 1,3-propanediol could be recovered after the reaction, which highlighted the promising potential of the innovative protocol possesses for the large-scale production of FDCA from HMF-acetal. In the subsequent work of the same group, the effective production of bis(2-hydroxyethyl)furan-2,5-dicarboxylate from HMF-acetal, a six-membered ring acetal derived from the acetalization of HMF with 1,3-propanediol, was also reported.160
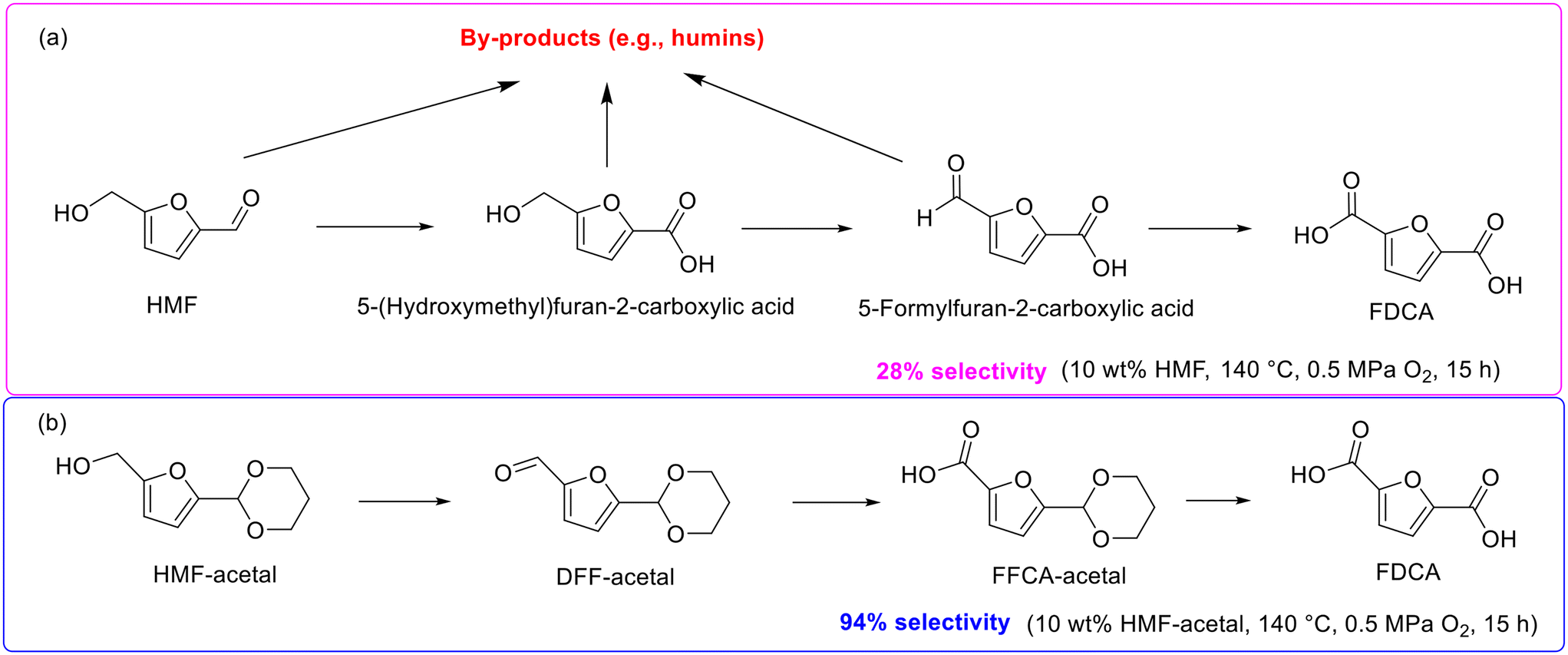 | ||
| Scheme 14 Reaction pathways for the synthesis of FDCA from HMF (a) and HMF-acetal (b). | ||
5.3 Dehydration of sorbitol into isosorbide
Sorbitol, produced from the catalytic hydrogenation of sugars such as glucose and cellulose, is a vital intermediate in the cellulosic biorefinery system.161,162 Due to the established commercial applications of its various derivatives, sorbitol has recently received exceptional attention. Isosorbide, accessible from double dehydration of sorbitol (namely double intramolecular etherification), represents a privileged molecule widely used in cosmetics, medicine, polymers, and plastics science.163,164 In particular, a prominent application is that isosorbide can serve as an alternative to bisphenol A for the manufacture of harmless plastic bottles (e.g., baby bottles and drinking bottles). The dehydration of sorbitol into isosorbide, therefore, has sparked growing interest in recent years.163,164However, high-selectivity conversion of sorbitol into isosorbide is still challenging owing to the presence of multiple etherification sites during the dehydration process arising from the similar chemical environment of six vicinal hydroxyl groups in the sorbitol molecule.165 As described in Scheme 15a, the transformation of sorbitol into isosorbide undergoes two sequential dehydration steps (i.e., two intramolecular etherification) involving 1,4-sorbitan as an intermediate, and the process is accompanied with the formation of 2,5-sorbitan and 1,5-sorbitan as dead-end byproducts. Numerous studies have demonstrated that the selectivity toward isosorbide from the direct dehydration of sorbitol is located in the range of 60–80% and generally needs high temperature and vacuum conditions (e.g., 5 kPa) to continually remove water from the reaction mixture.
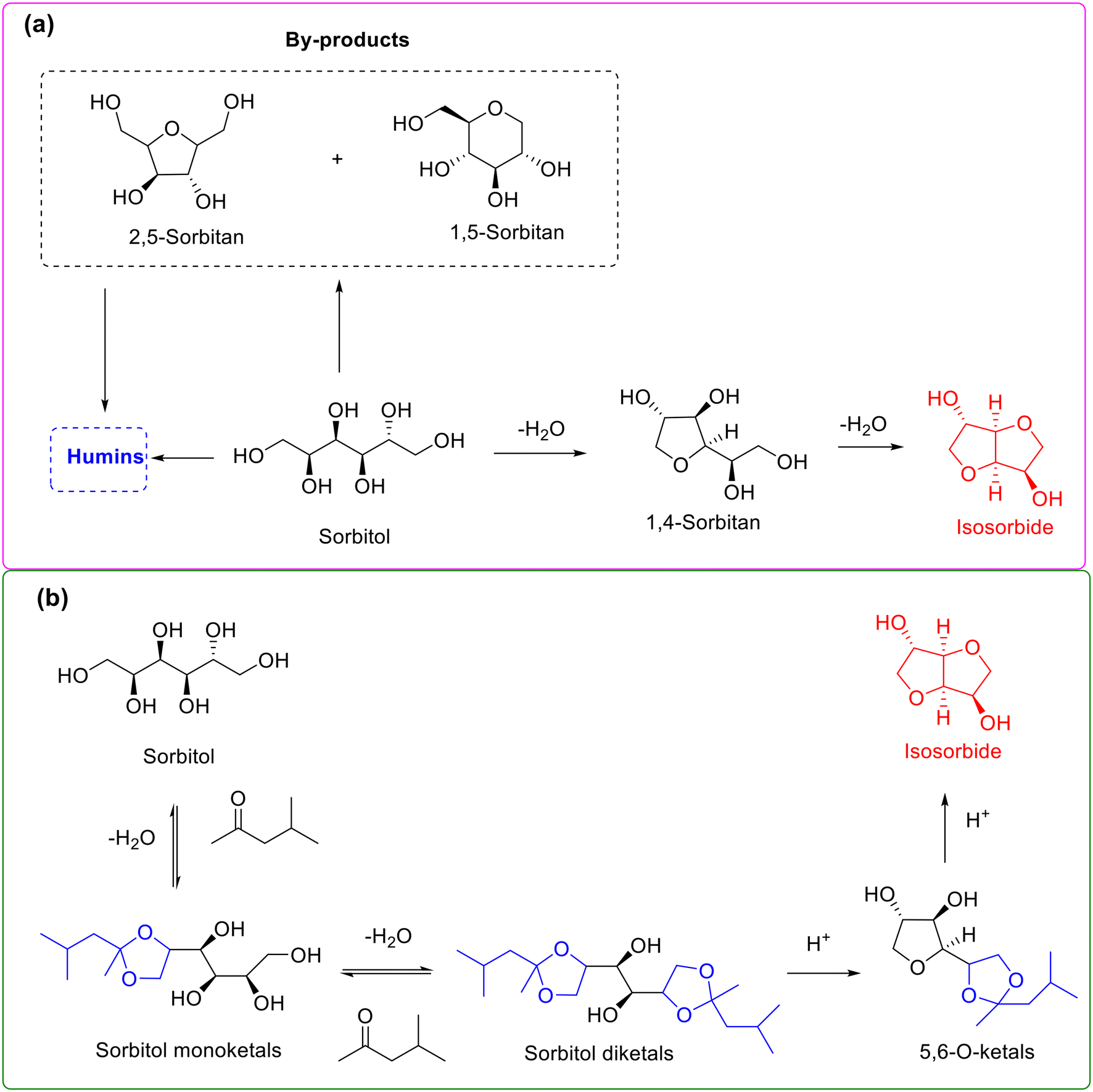 | ||
| Scheme 15 Reaction pathway for the synthesis of isosorbide from the dehydration of sorbitol without (a) or with (b) acetalization strategy. Adapted with permission from ref. 165. Copyright 2018, Royal Society of Chemistry. | ||
In attempts to obtain high-selectivity produced isosorbide, Xu and colleagues serendipitously discovered that the dehydration of sorbitol over H-beta in methyl isobutyl ketone medium afforded as high as 93% isosorbide selectivity, while the selectivity toward isosorbide was only 68.8% in a solvent-free system.165 The interesting results clearly indicated that the presence of methyl isobutyl ketone is capable of improving the isosorbide selectivity substantially. Hence, the authors tried to get insights into the role of methyl isobutyl ketone using 13C NMR spectroscopy to monitor the reaction.165 Also, the results of 13C NMR spectroscopy gave clear evidence to the formation of terminal 1,3-dioxolane ketal intermediates. Consequently, on the basis of the obtained results, the authors speculated that methyl isobutyl ketone initially reacted with the terminal vicinal-diol groups of sorbitol via ketalization to give the terminal five-membered 1,3-dioxolane intermediates, followed by two sequential intramolecular etherification to yield isosorbide (Scheme 15b). The unprecedented high selectivity toward isorobide was rationalized by the ketalization of the terminal vicinal-diol group in sorbitol, leading to reduction in the number of etherification active sites, thus subsequently effectively inhibiting the generation of byproducts and thereby substantially improving the isosorbide selectivity.165 Very recently, the protocol of using methyl isobutyl ketone to generate the terminal five-membered 1,3-dioxolane intermediates via ketalization was further successfully applied to the dehydration–esterification of sorbitol to isosorbide esters.166
5.4 Synthesis of xylitol from xylose
Xylitol, an attractive platform molecule obtained from the hydrolysis–hydrogenation of hemicellulose, has wide application in food and pharmaceutical industry (e.g., sweetener) and health products (e.g., toothpaste and mouthwash) as well as the chemical industry.167,168 The hydrogenation of xylose represents the most straightforward and frequently used approach to synthesize xylitol, which strongly depends on using a highly purified reactant. The separation and purification of xylose from the hydrolysate of hemicellulose, nevertheless, is intractable because xylose is nonvolatile and usually mixed with other nonvolatile sugars. Currently, xylose is generally separated by chromatography, which is costly in organic solvents and energy/time and thus constitutes a great challenge in xylitol production cost. Besides, the xylose in the hydrolysate of hemicellulose can also undergo dehydration to form furfural over acid sites, which inevitably leads to lower xylose yield. Interestingly, Luterbacher and coworkers demonstrated that the addition of an aldehyde (e.g., formaldehyde) during the organosolv pretreatment of biomass could result in the formation of diformylxylose via acetalization.169 Surprisingly, diformylxylose (i.e., acetal stabilized xylose), unlike xylose, is volatile and can be easily purified by distillation, which encouraged the same group to explore the synthesis of xylitol from the hydrogenation of diformylxylose.Diformylxylose, as reported by Luterbacher and coworkers, can be obtained from beech wood catalyzed by sulfuric acid in a mixed solution of water, γ-valerolactone, and formaldehyde.170 Importantly, highly purified (98%) diformylxylose can be obtained from the reaction mixture after filtration to remove cellulose and liquid CO2 extraction to remove stabilized lignin and subsequently undergoes distillation.170 Afterward, diformylxylose was subjected to the catalytic system based on using Pt/C as the catalyst to produce xylitol under the conditions of WHSV = 0.19 h−1, H2 flow = 50 mL min−1, T = 185 °C. Excitedly, the yield of xylitol was maintained at about 70% in long tests (over 100 h).170 The transformation of diformylxylose-to-xylitol, as shown in Scheme 16, started with the hydrolysis of diformylxylose into hydroxymethylxylofuranose and subsequently further hydrolysis to xylose (open chain), followed by hydrogenation to yield xylitol.170 The observed results unequivocally demonstrated that acetal-stabilized xylose (i.e., diformylxylose) represents an ideal alternative to xylose as the starting material for the production of xylitol.
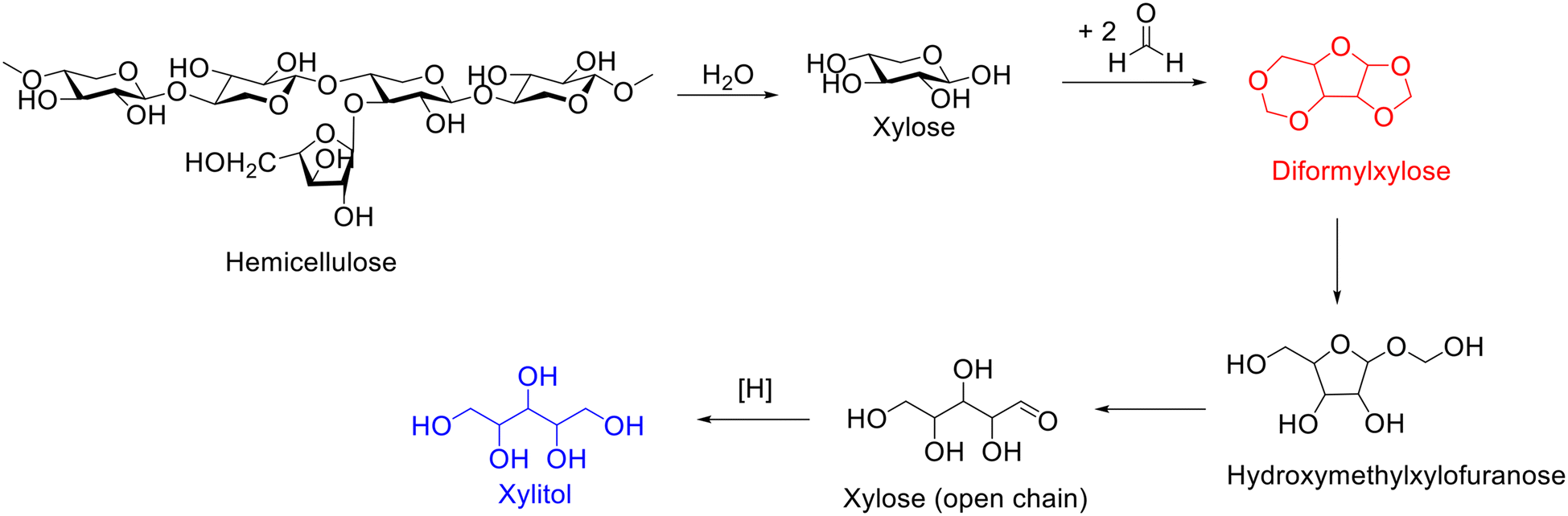 | ||
| Scheme 16 Reaction pathway for the synthesis of xylitol from diformylxylose. | ||
6 Conclusion and perspectives
Acetalization can be considered as a sustainable protocol for biomass valorization because not only can the substrates be renewable biomass derivatives with rich production capacity but the yielded cyclic acetals/ketals are also attractive fuel additives; moreover, the major byproduct is water. Recent advances in the acetalization of biogenic furanic compounds (i.e., furfural, 5-hydroxymethylfurfural and levulinic acid or its esters) and biobased diol or triol (i.e., ethylene glycol and glycerol) to access cyclic acetals/ketals fuel additives have been systematically discussed in this review, with emphasis on catalysts, structure–activity correlation, reaction pathway, and catalytic mechanism. In addition, acetalization as a protection strategy for improving the yields of target products in biomass valorization has also been discussed.Although remarkable progress has been achieved in the acetalization of furanic compounds and biobased diol or triol, there is still a long road to go to achieve the ultimate goal for industrial applications. Several aspects should be reinforced and improved in follow-up studies.
(1) Numerous investigations have concentrated upon obtaining excellent cyclic acetals/ketals yields from the acetalization of furanic compounds and ethylene glycol/glycerol and thus usually need excess of one of reactant to compel reversible acetalization completion. Nevertheless, studies on the recovery of excess reactant after the reaction are scarce. Moreover, the separation and purification of cyclic acetals/ketals deserve much attention. Expectedly, designing a suitable biphasic reaction system for this transformation may probably realize the recovery of the surplus substrate and/or separation of the acetals product.
(2) A five-membered-ring acetal (i.e., 1,3-dioxolane) and a six-membered-ring acetal (i.e., 1,3-dioxane) are available from the acetalization of furanic compounds and glycerol. However, it is still a significant challenge to achieve the selective synthesis of 1,3-dioxolane or 1,3-dioxane. Designing structurally adjustable catalysts or choosing suitable solvents may provide an opportunity for achieving the selective synthesis of 1,3-dioxolane or 1,3-dioxane. In addition, the use of crude glycerol, stemming from biodiesel production, for acetalization is more economically viable.
(3) Currently, the acetalization of furanic compounds and ethylene glycol/glycerol is strongly associated with the employed catalysts. Despite numerous catalysts that have been exploited in acetalization, these catalysts more or less suffer from some issues such as low activity, complicated preparation operation, or preparation process involving toxic/corrosive reagents. Therefore, the development of catalysts (e.g., biomass-derived catalysts) with economic benefits for acetalization is very attractive.
(4) Green catalysis technologies (e.g., photocatalysis and electrocatalysis) have the merits of energy efficiency and environmental friendliness, and they can emerge as alternative and promising low-carbon strategies to synthesize fuel additives. Thus, photocatalysis and electrocatalysis can be further developed for the acetalization of furanic compounds and ethylene glycol/glycerol. In addition, using continuous flow strategy featuring continuous manufacturing and high efficiency, the acetalization of furanic compounds and ethylene glycol/glycerol in fixed-bed continuous reactors should be emphasized.
(5) At present, the available researches for getting insights into detailed catalytic mechanism for the acetalization of furanic compounds and ethylene glycol/glycerol are limited. More effort should therefore be devoted to the fundamental understanding of the catalytic mechanism via in situ spectroscopic measurements and density functional theory calculation.
(6) Taking advantages of the fact that acetalization is a reversible reaction and the formed cyclic acetals/ketals are stable/low reactivity in basic media, acetalization as a protection strategy of the carbonyl group is worth further promotion in biomass valorization such as exclusive hydrogenation of CC in biobased multifunctional compounds while leaving the CO group unreduced.
Conflicts of interest
There are no conflicts of interest to declare.Acknowledgements
The authors gratefully acknowledge financial support from National Key R&D Program of China (2019YFC1905300), National Natural Science Foundation of China (No. 22268019) and Hunan Provincial Natural Science Foundation of China (No. 2021JJ40436).References
- Y. Ding, T. Guo, Z. Li, B. Zhang, F. E. Kühn, C. Liu, J. Zhang, D. Xu, M. Lei, T. Zhang and C. Li, Transition-metal-free synthesis of functionalized quinolines by direct conversion of β-O-4 model compounds, Angew. Chem., Int. Ed., 2022, 61, e202206284 CrossRef CAS
.
- K. Lee, Y. Jing, Y. Wang and N. Yan, A unified view on catalytic conversion of biomass and waste plastics, Nat. Rev. Chem., 2022, 6, 635–652 CrossRef
- E. Lizundia, F. Luzi and D. Puglia, Organic waste valorisation towards circular and sustainable biocomposites, Green Chem., 2022, 24, 5429–5459 RSC
- P. Sun, C. Liu, H. Wang, Y. Liao, X. Li, Q. Liu, B. F. Sels and C. Wang, Rational positioning of metal ions to stabilize open tin sites in beta zeolite for catalytic conversion of sugars, Angew. Chem., Int. Ed., 2022, 62, e202215737 CrossRef PubMed
- S. Yang, C. Shi, Z. Shen, L. Pan, Z. Huang, X. Zhang and J. Zou, Conversion of lignin oil and hemicellulose derivative into high-density jet fuel, J. Energy Chem., 2023, 77, 452–460 CrossRef CAS
- C. Yang, H. Chen, T. Peng, B. Liang, Y. Zhang and W. Zhao, Lignin valorization toward value-added chemicals and fuels via electrocatalysis: A perspective, Chin. J. Catal., 2021, 42, 1831–1842 CrossRef CAS
- H. J. Cho, M. J. Kuo, M. Ye, Y. Kurz, Y. Yuan and R. F. Lobo, Selective synthesis of 4,4′-dimethylbiphenyl from 2-methylfuran, ACS Sustainable Chem. Eng., 2021, 9, 3316–3323 CrossRef CAS
- N. K. Gupta, P. Reif, P. Palenicek and M. Rose, Toward renewable amines: recent advances in the catalytic amination of biomass-derived oxygenates, ACS Catal., 2022, 12, 10400–10440 CrossRef CAS
- F. Ebrahimian, J. F. M. Denayer and K. Karimi, Potato peel waste biorefinery for the sustainable production of biofuels, bioplastics, and biosorbents, Bioresour. Technol., 2022, 360, 127609 CrossRef CAS PubMed
- X. Liu, S. Xing, L. Yang, J. Fu, P. Lv, X. Zhang, M. Li and Z. Wang, Highly active and durable Ca-based solid base catalyst for biodiesel production, Fuel, 2021, 302, 121094 CrossRef CAS
- J. He, Z. Yu, H. Wu, H. Li and S. Yang, Mesoporous tin phosphate as an effective catalyst for fast cyclodehydration of bio-based citral into p-cymene, Mol. Catal., 2021, 515, 111887 CrossRef CAS
- O. Y. Abdelaziz, I. Clemmensen, S. Meier, C. A. E. Costa, A. E. Rodrigues, C. P. Hulteberg and A. Riisager, On the oxidative valorization of lignin to high-value chemicals: A critical review of opportunities and challenges, ChemSusChem, 2022, 15, e202201232 CrossRef CAS PubMed
- N. Li and M. Zong, (Chemo)biocatalytic upgrading of biobased furanic platforms to chemicals, fuels, and materials: A comprehensive review, ACS Catal., 2022, 12, 10080–10114 CrossRef CAS
- S. Xiang, L. Dong, Z. Wang, X. Han, Y. Guo, X. Liu, X. Gong and Y. Wang, Co@CoO: An efficient catalyst for the depolymerization and upgrading of lignocellulose to alkylcyclohexanols with cellulose intact, J. Energy Chem., 2023, 77, 191–199 CrossRef CAS
- D. Wang, W. Gong, J. Zhang, M. Han, C. Chen, Y. Zhang, G. Wang, H. Zhang and H. Zhao, Encapsulated Ni-Co alloy nanoparticles as efficient catalyst for hydrodeoxygenation of biomass derivatives in water, Chin. J. Catal., 2021, 42, 2027–2037 CrossRef CAS
- X. Du, J. Liu, D. Li, H. Xin, X. Lei, R. Zhang, L. Zhou, H. Yang, Y. Zeng, H. Zhang, W. Zheng, X. Wen and C. Hu, Structural and electronic effects boosting Ni-doped Mo2C catalyst toward high-efficiency C-O/C-C bonds cleavage, J. Energy Chem., 2022, 75, 109–116 CrossRef CAS
- D. Chu, Z. Luo and C. Zhao, Selective production of acetol or methyl lactate from cellulose over RuSn catalysts, J. Energy Chem., 2022, 73, 607–614 CrossRef CAS
- Z. Xu, J. Luo and Y. Huang, Recent advances in the chemical valorization of cellulose and its derivatives into ester compounds, Green Chem., 2022, 24, 3895–3921 RSC
- L. Zhou, Y. Li, Y. Lu, S. Wang and Y. Zou, Ph-Induced selective electrocatalytic hydrogenation of furfural on Cu electrodes, Chin. J. Catal., 2022, 43, 3142–3153 CrossRef CAS
- A. Jaswal, P. P. Singh and T. Mondal, Furfural--a versatile, biomass-derived platform chemical for the production for renewable chemicals, Green Chem., 2022, 24, 510–551 RSC
- H. Guo, T. Dowaki, F. Shen, X. Qi and R. L. Smith, Jr., Critical assessment of reaction pathways for next-generation biofuels from renewable resources: 5-ethoxymethylfurfural, ACS Sustainable Chem. Eng., 2022, 10, 9002–9021 CrossRef CAS
- J. Wu, L. Xu, Y. Li, C. Dong, Y. Lu, T. T. T. Nga, Z. Kong, S. Li, Y. Zou and S. Wang, Anodic cross-coupling of biomass platform chemicals to sustainable biojet fuel precursors, J. Am. Chem. Soc., 2022, 144, 23649–23656 CrossRef CAS
- Y. Zhang, S. Niu, Y. Hao, S. Liu, J. Liu, K. Han, Y. Wang and C. Lu, Preparation of SrZrAl multiple oxide catalyst for produce biodiesel from acidified palm oil, Renewable Energy, 2023, 207, 116–127 CrossRef CAS
- W. Fang, S. Liu, L. Schill, M. Kubus, T. Bligaard and A. Riisager, On the role of Zr to facilitate the synthesis of diesel and jet fuel range intermediates from biomass-derived carbonyl compounds over aluminum phosphate, Appl. Catal., B, 2023, 320, 121936 CrossRef CAS
- J. Zhang, Z. Wang, M. Chen, Y. Zhu, Y. Liu, H. He, Y. Cao and X. Bao, N-doped carbon layer-coated Au nanocatalyst for H2-free conversion of 5-hydroxymethylfurfural to 5-methylfurfural, Chin. J. Catal., 2022, 43, 2212–2222 CrossRef CAS
- L. Ricciardi, W. Verboom, J. Lange and J. Huskens, Production of furans from C5 and C6 sugars in the presence of polar organic solvents, Sustain. Energy Fuels, 2022, 6, 11–28 RSC
- C. Xu, E. Paone, D. Rodríguez-Padrón, R. Luque and F. Mauriello, Recent catalytic routes for the preparation and the upgrading of biomass derived furfural and 5-hydroxymethylfurfural, Chem. Soc. Rev., 2020, 49, 4273–4306 RSC
- J. He, L. Chen, S. Liu, K. Song, S. Yang and A. Riisager, Sustainable access to renewable N-containing chemicals from reductive amination of biomass-derived platform compounds, Green Chem., 2020, 22, 6714–6747 RSC
- B. Liu, Z. Zheng, Y. Liu, M. Zhang, Y. Wang, Y. Wan and K. Yan, Efficient electrooxidation of biomass-derived aldehydes over ultrathin NiV-layered double hydroxides films, J. Energy Chem., 2023, 78, 412–421 CrossRef CAS
- M. T. Bender, X. Yuan, M. K. Goetz and K. Choi, Electrochemical hydrogenation, hydrogenolysis, and dehydrogenation for reductive and oxidative biomass upgrading using 5-hydroxymethylfurfural as a model system, ACS Catal., 2022, 12, 12349–12368 CrossRef CAS
- Z. An and J. Li, Recent advances in catalytic transfer hydrogenation of furfural to furfuryl alcohol over heterogeneous catalysts, Green Chem., 2022, 24, 1780–1808 RSC
- Y. Pang, Z. Zhang, W. Qiu, N. Chen, J. Wei, X. Li, L. Lin and H. Huang, Chemo-catalytic synthesis of biomass-derived furanyl diethers: Green and renewable bio-diesel components, Sustain. Energy Fuels, 2022, 6, 4845–4859 RSC
- J. He, Q. Qiang, S. Liu, K. Song, X. Zhou, J. Guo, B. Zhang and C. Li, Upgrading of biomass-derived furanic compounds into high-quality fuels involving aldol condensation strategy, Fuel, 2021, 306, 121765 CrossRef CAS
- L. Chen, S. Liu, H. Pan, K. Song, X. Zhou, J. Guo, O. Zhuo and J. He, Naturally biodegradable polymer as an effective heterogeneous catalyst for synthesis of biofuels via Knoevenagel condensation strategy, Biomass Convers. Biorefin., 2022 DOI:10.1007/s13399-021-02253-8
- M. J. Climent, A. Corma and S. Iborra, Conversion of biomass platform molecules into fuel additives and liquid hydrocarbon fuels, Green Chem., 2014, 16, 516–547 RSC
- J. He, X. Liu, L. Bai, S. Liu, K. Song, X. Zhou, J. Guo, X. Meng and C. Li, Effective one-pot tandem synthesis of γ-valerolactone from biogenic furfural over zirconium phosphate catalyst, Biofuels, Bioprod. Biorefin., 2022, 16, 1613–1626 CrossRef CAS
- X. Liu, L. Chen, H. Xu, S. Jiang, Y. Zhou and J. Wang, Straightforward synthesis of beta zeolite encapsulated Pt nanoparticles for the transformation of 5-hydroxymethyl furfural into 2,5-furandicarboxylic acid, Chin. J. Catal., 2021, 42, 994–1003 CrossRef CAS
- W. Zhang, Y. Shi, Y. Yang, J. Tan and Q. Gao, Facet dependence of electrocatalytic furfural hydrogenation on palladium nanocrystals, Chin. J. Catal., 2022, 43, 3116–3125 CrossRef CAS
- A. Kumar, A. S. Chauhan, R. Bains and P. Das, Catalytic transformations for agro-waste conversion to 5-hydroxymethylfurfural and furfural: Chemistry and scale-up development, Green Chem., 2023, 25, 849–870 RSC
- F. Xu, Z. Li, L. Zhang, S. Liu, H. Li, Y. Liao and S. Yang, Synthesis of renewable isoindolines from bio-based furfurals, Green Chem., 2023, 25, 3297–3305 RSC
- M. G. Teixeira, R. Natalino and M. J. da Silva, A kinetic study of heteropolyacid-catalyzed furfural acetalization with methanol at room temperature via ultraviolet spectroscopy, Catal. Today, 2020, 344, 143–149 CrossRef CAS
- X. Yue and Y. Queneau, 5-Hydroxymethylfurfural and furfural chemistry toward biobased surfactants, ChemSusChem, 2022, 15, e202102660 CrossRef CAS PubMed
- P. Sudarsanam, B. Mallesham, A. N. Prasad, P. S. Reddy and B. M. Reddy, Synthesis of bio-additive fuels from acetalization of glycerol with benzaldehyde over molybdenum promoted green solid acid catalysts, Fuel Process. Technol., 2013, 106, 539–545 CrossRef CAS
- J. N. Hall and P. Bollini, Metal-organic framework MIL-100 catalyzed acetalization of benzaldehyde with methanol: Lewis or Brønsted acid catalysis?, ACS Catal., 2020, 10, 3750–3763 CrossRef CAS
- N. Narkhede and A. Patel, Room temperature acetalization of glycerol to cyclic acetals over anchored silicotungstates under solvent free conditions, RSC Adv., 2014, 4, 19294–19301 RSC
- X. Han, J. Cai, X. Mao, X. Yang, L. Qiu, F. Li, X. Tang, Y. Wang and S. Liu, Highly active solid oxide acid catalyst for the synthesis of benzaldehyde glycol acetal, Appl. Catal., A, 2021, 618, 118136 CrossRef CAS
- J. Kim, C. Lim, B. M. Reddy and S. Park, Hierarchical porous organic polymer
as an efficient metal-free catalyst for acetalization of carbonyl compounds with alcohols, Mol. Catal., 2018, 451, 43–50 CrossRef CAS
- A. L. G. Pinheiro, J. V. C. do Carmo, D. C. Carvalho, A. C. Oliveira, E. Rodríguez-Castellón, S. Tehuacanero-Cuapa, L. Otubo and R. Lang, Bio-additive fuels from glycerol acetalization over metals-containing vanadium oxide nanotubes (MeVOx-NT in which, Me = Ni, Co, or Pt), Fuel Process. Technol., 2019, 184, 45–56 CrossRef CAS
- S. Kirchhecker, A. Dell'Acqua, A. Angenvoort, A. Spannenberg, K. Ito, S. Tin, A. Taden and J. G. de Vries, HMF-glycerol acetals as additives for the debonding of polyurethane adhesives, Green Chem., 2021, 23, 957–965 RSC
- M. Kapkowski, J. Popiel, T. Siudyga, M. Dzida, E. Zorębski, M. Musiał, R. Sitko, J. Szade, K. Balin, J. Klimontko, M. Zubko and J. Polanski, Mono- and bimetallic nano-Re systems doped Os, Mo, Ru, Ir as nanocatalytic platforms for the acetalization of polyalcohols into cyclic acetals and their applications as fuel additives, Appl. Catal., B, 2018, 239, 154–167 CrossRef CAS
- R. R. Pawar, S. V. Jadhav and H. C. Bajaj, Microwave-assisted rapid valorization of glycerol towards acetals and ketals, Chem. Eng. J., 2014, 235, 61–66 CrossRef CAS
- L. A. Bailey, T. Bere, T. E. Davies, S. H. Taylor and A. E. Graham, Preparation of biomass-derived furfuryl acetals by transacetalization reactions catalyzed by nanoporous aluminosilicates, ACS Sustainable Chem. Eng., 2022, 10(41), 13759–13764 CrossRef CAS
- A. Wang and T. Zhang, One-pot conversion of cellulose to ethylene glycol with multifunctional tungsten-based catalysts, Acc. Chem. Res., 2013, 46, 1377–1386 CrossRef CAS
- M. Zheng, J. Pang, R. Sun, A. Wang and T. Zhang, Selectivity control for cellulose to diols: Dancing on eggs, ACS Catal., 2017, 7, 1939–1954 CrossRef CAS
- N. Ji, T. Zhang, M. Zheng, A. Wang, H. Wang, X. Wang and J. G. Chen, Direct catalytic conversion of cellulose into ethylene glycol using nickel-promoted tungsten carbide catalysts, Angew. Chem., Int. Ed., 2008, 47, 8510–8513 CrossRef CAS
- Y. Liu, C. Luo and H. Liu, Tungsten trioxide promoted selective conversion of cellulose into propylene glycol and ethylene glycol on a ruthenium catalyst, Angew. Chem., Int. Ed., 2012, 51, 3249–3253 CrossRef CAS PubMed
- S. G. Khan, M. Hassan, M. Anwar, Zeshan, U. M. Khan and C. Zhao, Mussel shell based CaO nano-catalyst doped with praseodymium to enhance biodiesel production from castor oil, Fuel, 2022, 330, 125480 CrossRef
- J. Sahar, M. Farooq, A. Ramli, A. Naeem and N. S. Khattak, Biodiesel production from Mazari palm (Nannorrhops ritchiana) seeds oil using tungstophosphoric acid decorated SnO2@Mn-ZIF bifunctional heterogeneous catalyst, Appl. Catal., A, 2022, 643, 118740 CrossRef CAS
- M. Liu, H. Liu, N. Li, C. Zhang, J. Zhang and F. Wang, Selective oxidation of glycerol into formic acid by photogenerated holes and superoxide radicals, ChemSusChem, 2022, 15, e202201068 CrossRef CAS PubMed
- S. Xu, Q. Tian, Y. Xiao, W. Zhang, S. Liao, J. Li and C. Hu, Regulating the competitive reaction pathway in glycerol conversion to lactic acid/glycolic acid selectively, J. Catal., 2022, 413, 407–416 CrossRef CAS
- H. Yue, Y. Zhao, X. Ma and J. Gong, Ethylene glycol: Properties, synthesis, and applications, Chem. Soc. Rev., 2012, 41, 4218–4244 RSC
- S. He, T. S. Kramer, D. S. Santosa, A. Heeres and H. J. Heeres, Catalytic conversion of glycerol and co-feeds (fatty acids, alcohols, and alkanes) to bio-based aromatics: Remarkable and unprecedented
synergetic effects on catalyst performance, Green Chem., 2022, 24, 941–949 RSC
- J. Wu, X. Yang and M. Gong, Recent advances in glycerol valorization via electrooxidation: Catalyst, mechanism and device, Chin. J. Catal., 2022, 43, 2966–2986 CrossRef CAS
- J. Cho, Y. Kim, B. D. Etz, G. M. Fioroni, N. Naser, J. Zhu, Z. Xiang, C. Hays, J. V. Alegre-Requena, P. C. S. John, B. T. Zigler, C. S. McEnally, L. D. Pfefferle, R. L. McCormick and S. Kim, Bioderived ether design for low soot emission and high reactivity transport fuels, Sustainable Energy Fuels, 2022, 6, 3975–3988 RSC
- A. R. Trifoi, P. S. Trifoi and T. Pap, Glycerol acetals and ketals as possible diesel additives. A review of their synthesis protocols, Renewable Sustainable Energy Rev., 2016, 62, 804–814 CrossRef CAS
- I. Zahid, M. Ayoub, B. B. Abdullah, M. H. Nazir, M. Ameen, Zulqarnain, M. H. M. Yusoff, A. Inayat and M. Danish, Production of fuel additive solketal via catalytic conversion of biodiesel-derived glycerol, Ind. Eng. Chem. Res., 2020, 59, 20961–20978 CrossRef CAS
- A. Cornejo, I. Barrio, M. Campoy, J. Lázaro and B. Navarrete, Oxygenated fuel additives from glycerol valorization. Main production pathways and effects on fuel properties and engine performance: A critical review, Renewable Sustainable Energy Rev., 2017, 79, 1400–1413 CrossRef
- T. Lu, J. Yang and L. Sheu, An efficient method for the acetalization of α,β-unsaturated aldehydes, J. Org. Chem., 1995, 60, 2931–2934 CrossRef CAS
- F. Poovan, V. G. Chandrashekhar, K. Natte and R. V. Jagadeesh, Synergy between homogeneous and heterogeneous catalysis, Catal. Sci. Technol., 2022, 12, 6623–6649 RSC
- D. E. Ponde, V. H. Deshpande, V. J. Bulbule, A. Sudalai and A. S. Gajare, Selective catalytic transesterification, transthiolesterification, and protection of carbonyl compounds over natural kaolinitic clay, J. Org. Chem., 1998, 63, 1058–1063 CrossRef
- J. Miao, H. Miao, Y. Shao, G. Guan and B. Xu, Acetalization of carbonyl compounds catalyzed by acidic ionic liquid immobilized on silica gel, J. Mol. Catal. A: Chem., 2011, 348, 77–82 CrossRef CAS
- W. Fang and A. Riisager, Efficient valorization of biomass-derived furfural to fuel bio-additive over aluminum phosphate, Appl. Catal., B, 2021, 298, 120575 CrossRef CAS
- M. Ventura, J. Mazarío and M. E. Domine, Isomerization of glucose-to-fructose in water over a continuous flow reactor using Ca-Al mixed oxide as heterogeneous catalyst, ChemCatChem, 2022, 14, e202101229 CrossRef CAS
- M. Minakawa, Y. M. A. Yamada and Y. Uozumi, Driving an equilibrium acetalization to completion in the presence of water, RSC Adv., 2014, 4, 36864–36867 RSC
- X. Yang, K. Liu, J. Ma and R. Sun, Carbon quantum dots anchored on 1,2,3,5-tetrakis (carbazole-9-yl)-4,6-dicyanobenzene for efficient selective photo splitting of biomass-derived sugars into lactic acid, Green Chem., 2022, 24, 5894–5903 RSC
- Z. Xue, S. Wu, Y. Fu, L. Luo, M. Li, Z. Li, M. Shao, L. Zheng, M. Xu and H. Duan, Efficient light-driven reductive amination of furfural to furfurylamine over ruthenium-cluster catalyst, J. Energy Chem., 2023, 76, 239–248 CrossRef CAS
- S. Lv, H. Liu, J. Zhang, Q. Wu and F. Wang, Water promoted photocatalytic transfer hydrogenation of furfural to furfural alcohol over ultralow loading metal supported on TiO2, J. Energy Chem., 2022, 73, 259–267 CrossRef CAS
- J. Wang, H. Zhao, P. Liu, N. Yasri, N. Zhong, M. G. Kibria and J. Hu, Selective superoxide radical generation for glucose photoreforming into arabinose, J. Energy Chem., 2022, 74, 324–331 CrossRef CAS
- H. Wang, X. Li, X. Zhao, C. Li, X. Song, P. Zhang, P. Huo and X. Li, A review on heterogeneous photocatalysis for environmental remediation: From semiconductors to modification strategies, Chin. J. Catal., 2022, 43, 178–214 CrossRef CAS
- H. Yi, L. Niu, S. Wang, T. Liu, A. K. Singh and A. Lei, Visible-light-induced acetalization of aldehydes with alcohols, Org. Lett., 2017, 19, 122–125 CrossRef CAS
- A. Aho, S. Engblom, K. Eränen, V. Russo, P. Russo, N. Kumar, J. Wärnå, T. Salmi and D. Y. Murzin, Glucose transformations over a mechanical mixture of ZnO and Ru/C catalysts: Product distribution, thermodynamics and kinetics, Chem. Eng. J., 2021, 405, 126945 CrossRef CAS
- X. Liu, X. Liu, H. Wang, T. Xiao, Y. Zhang and L. Ma, A mechanism study on efficient conversion of cellulose to acetol over Sn-Co catalysts with low Sn content, Green Chem., 2020, 22, 6579–6587 RSC
- H. Song, F. Jin, Q. Liu and H. Liu, Zeolite-catalyzed acetalization reaction of furfural with alcohol under solvent-free conditions, Mol. Catal., 2021, 513, 111752 CrossRef CAS
- Y. Jin, J. Shi, F. Zhang, Y. Zhong and W. Zhu, Synthesis of sulfonic acid-functionalized MIL-101 for acetalization of aldehydes with diols, J. Mol. Catal. A: Chem., 2014, 383–384, 167–171 CrossRef CAS
- F. Zhang, J. Shi, Y. Jin, Y. Fu, Y. Zhong and W. Zhu, Facile synthesis of MIL-100(Fe) under HF-free conditions and its application in the acetalization of aldehydes with diols, Chem. Eng. J., 2015, 259, 183–190 CrossRef CAS
- E. V. Gromachevskaya, F. V. Kvitkovsky, E. B. Usova and V. G. Kulnevich, Investigation in the area of furan acetal compounds. 13*. synthesis and structure of 1,3-dioxacyclanes based on furfural and glycerol, Chem. Heterocycl. Compd., 2004, 40, 979–985 CrossRef CAS
- L. A. Oparina, O. A. Vysotskaya, A. V. Stepanov, N. K. Gusarova, K. A. Chernyshev, L. B. Krivdin and B. A. Trofimov, Nucleophilic addition to acetylenes in superbasic catalytic systems: XVII. Vinyl ethers with furyl and cycloacetal fragments: Synthesis and structure, Russ. J. Org. Chem., 2010, 46, 1383–1387 CrossRef CAS
- B. L. Wegenhart and M. M. Abu-Omar, A Solvent-free method for making dioxolane and dioxane from the biorenewables glycerol and furfural catalyzed by oxorhenium(V) oxazoline, Inorg. Chem., 2010, 49, 4741–4743 CrossRef CAS PubMed
- S. Zaher, L. Christ, M. A. E. Rahim, A. Kanj and I. Karamé, Green acetalization of glycerol and carbonyl catalyzed by FeCl3·6H2O, Mol. Catal., 2017, 438, 204–213 CrossRef CAS
- R. R. Pawar, K. A. Gosai, A. S. Bhatt, S. Kumaresan, S. M. Lee and H. C. Bajaj, Clay catalysed rapid valorization of glycerol towards cyclic acetals and ketals, RSC Adv., 2015, 5, 83985–83996 RSC
- J. R. Dodson, T. Avellar, J. Athayde and C. J. A. Mota, Glycerol acetals with antioxidant properties, Pure Appl. Chem., 2014, 86, 905–911 CAS
- B. Wang, Y. Shen, J. Sun, F. Xu and R. Sun, Conversion of platform chemical glycerol to cyclic acetals promoted by acidic ionic liquids, RSC Adv., 2014, 4, 18917–18923 RSC
- A. Patel and D. Pithadia, Low temperature synthesis of bio-fuel additives via valorisation of glycerol with benzaldehyde as well as furfural over a novel sustainable catalyst, 12-tungstosilicic acid anchored to ordered cubic nano-porous MCM-48, Appl. Catal., A, 2020, 602, 117729 CrossRef CAS
- F. Guerrero-Ruíz, E. Yara-Varón, M. D. González, M. Torres, P. Salagre, R. Canela-Garayoa and Y. Cesteros, Use of biobased crude glycerol, obtained biocatalytically, to obtain biofuel additives by catalytic acetalization of furfural using SAPO catalysts, Fuel, 2022, 319, 123803 CrossRef
- B. L. Wegenhart, S. Liu, M. Thom, D. Stanley and M. M. Abu-Omar, Solvent-free methods for making acetals derived from glycerol and furfural and their use as a biodiesel fuel component, ACS Catal., 2012, 2, 2524–2530 CrossRef CAS
- A. Patil, S. Shinde, S. Kamble and C. V. Rode, Two-step sequence of acetalization and hydrogenation for synthesis of diesel fuel additives from furfural and diols, Energy Fuels, 2019, 33, 7466–7472 CrossRef CAS
- C. A. Akinnawo, L. Mosia, O. A. Alimi, C. O. Oseghale, D. P. Fapojuwo, N. Bingwa and R. Meijboom, Eco-friendly synthesis of valuable fuel bio-additives from glycerol, Catal. Commun., 2021, 152, 106287 CrossRef CAS
- C. Gonzalez-Arellano, R. A. D. Arancon and R. Luque, Al-SBA-15 catalysed cross-esterification and acetalisation of biomass-derived platform chemicals, Green Chem., 2014, 16, 4985–4993 RSC
- C. Gonzalez-Arellano, S. De and R. Luque, Selective glycerol transformations to high value-added products catalysed by aluminosilicate-supported iron oxide nanoparticles, Catal. Sci. Technol., 2014, 4, 4242–4249 RSC
- J. N. Appaturi, R. J. Ramalingam, H. A. Al-Lohedan, F. Khoerunnisa, T. C. Ling and E. Ng, Selective synthesis of dioxolane biofuel additive via acetalization of glycerol and furfural enhanced by MCM-41-alanine bifunctional catalyst, Fuel, 2021, 288, 119573 CrossRef CAS
- N. Oger, Y. F. Lin, E. L. Grognec, F. Rataboul and F. Felpin, Graphene-promoted acetalisation of glycerol under acid-free conditions, Green Chem., 2016, 18, 1531–1537 RSC
- S. Fulignati, C. Antonetti, T. Tabanelli, F. Cavani and A. M. R. Galletti, Integrated cascade process for the catalytic conversion of 5-hydroxymethylfurfural (HMF) to furanic and tetrahydrofuranic diethers as potential bio-fuels, ChemSusChem, 2022, 15, e202200241 CrossRef CAS PubMed
- W. Wang, H. Wang, X. Jiang, Z. He, Y. Yang, K. Wang and Z. Liu, Biomass-modified zirconium-based catalyst for one-pot reductive etherification of bioderived aldehydes to furanic diether, ACS Sustainable Chem. Eng., 2022, 10, 4969–4979 CrossRef CAS
- H. Guo, T. Dowaki, F. Shen, X. Qi and R. L. Smith, Jr., Critical assessment of reaction pathways for next-generation biofuels from renewable resources: 5-ethoxymethylfurfural, ACS Sustainable Chem. Eng., 2022, 10, 9002–9021 CrossRef CAS
- J. E. Rorrer, A. T. Bell and F. D. Toste, Synthesis of biomass-derived ethers for use as fuels and lubricants, ChemSusChem, 2019, 12, 2835–2858 CrossRef CAS PubMed
- H. Chang, G. W. Huber and J. A. Dumesic, Chemical-switching strategy for synthesis and controlled release of norcantharimides from a biomass-derived chemical, ChemSusChem, 2020, 13, 5213–5219 CrossRef CAS PubMed
- S. Kanai, I. Nagahara, Y. Kita, K. Kamata and M. Hara, A bifunctional cerium phosphate catalyst for chemoselective acetalization, Chem. Sci., 2017, 8, 3146–3153 RSC
- O. M. Portilla-Zuñiga, J. J. Martínez, M. Casella, D. I. Lick, A. G. Sathicq, R. Luque and G. P. Romanelli, Etherification of 5-hydroxymethylfurfural using a heteropolyacid supported on a silica matrix, Mol. Catal., 2020, 494, 111125 CrossRef
- Y. Wang, Y. Dou, H. Zhang, B. Gu, C. Oldani, Q. Tang, F. Jing, Q. Cao and W. Fang, Direct conversion of fructose to levulinic acid in water medium catalyzed by a reusable perfluorosulfonic acid Aquivion® resin, Mol. Catal., 2022, 520, 112159 CrossRef CAS
- D. Song, J. Liu, C. Zhang and Y. Guo, Design of Brønsted acidic ionic liquid functionalized mesoporous organosilica nanospheres for efficient synthesis of ethyl levulinate and levulinic acid from 5-hydroxymethylfurfural, Catal. Sci. Technol., 2021, 11, 1827–1842 RSC
- B. Mallesham, P. Sudarsanam, G. Raju and B. M. Reddy, Design of highly efficient Mo and W-promoted SnO2 solid acids for heterogeneous catalysis: Acetalization of bio-glycerol, Green Chem., 2013, 15, 478–489 RSC
- K. S. Arias, A. Garcia-Ortiz, M. J. Climent, A. Corma and S. Iborra, Mutual valorization of 5-hydroxymethylfurfural and glycerol into valuable diol monomers with solid acid catalysts, ACS Sustainable Chem. Eng., 2018, 6, 4239–4245 CrossRef CAS
- X. Ge, H. Li, M. Liu, Z. Zhao, X. Jin, X. Fan and X. Gao, Microwave-assisted catalytic alcoholysis of fructose to ethoxymethylfurfural (EMF) over carbon-based microwave-responsive catalyst, Fuel Process. Technol., 2022, 233, 107305 CrossRef CAS
- S. Wang, T. L. Eberhardt, D. Guo, J. Feng and H. Pan, Efficient conversion of glucose into 5-HMF catalyzed by lignin-derived mesoporous carbon solid acid in a biphasic system, Renewable Energy, 2022, 190, 1–10 CrossRef CAS
- M. Niakan, M. Masteri-Farahani, S. Karimi and H. Shekaari, Sulfonic acid functionalized dendrimer-grafted cellulose as a solid acid catalyst for the high-yield and green production of 5-hydroxymethylfurfural, Sustainable Energy Fuels, 2022, 6, 2514–2522 RSC
- K. Ravi and A. V. Biradar, Highly active and scalable SO3H functionalized carbon catalyst synthesized from bagasse for transformation of bio-based platform chemicals into fuel precursors and its in-depth characterization studies, Fuel, 2022, 321, 124008 CrossRef CAS
- A. Garcia-Ortiz, K. S. Arias, M. J. Climent, A. Corma and S. Iborra, One-pot synthesis of biomass-derived surfactants by reacting hydroxymethylfurfural, glycerol, and fatty alcohols on solid acid catalysts, ChemSusChem, 2018, 11, 2870–2880 CrossRef CAS PubMed
- D. Song, J. Liu, C. Zhang and Y. Guo, Design of Brønsted acidic ionic liquid functionalized mesoporous organosilica nanospheres for efficient synthesis of ethyl levulinate and levulinic acid from 5-hydroxymethylfurfural, Catal. Sci. Technol., 2021, 11, 1827–1842 RSC
- Y. Wang, Y. Huang, L. Liu, L. He, T. Li, C. Len and W. Yang, Molecular oxygen promoted synthesis of methyl levulinate from 5-hydroxymethylfurfural, ACS Sustainable Chem. Eng., 2020, 8, 14576–14583 CrossRef CAS
- L. Peng, X. Gao, Y. Liu, J. Zhang and L. He, Coupled transfer hydrogenation and alcoholysis of furfural to yield alkyl levulinate over multifunctional zirconia-zeolite-supported heteropoly acid, Energy Fuels, 2021, 35, 4182–4190 CrossRef CAS
- K. Y. Nandiwale, M. Vishwakarma, S. Rathod, I. Simakova and V. V. Bokade, One-pot cascade conversion of renewable furfural to levulinic acid over a bifunctional H3PW12O40/SiO2 catalyst in the absence of external H2, Energy Fuels, 2021, 35, 539–545 CrossRef CAS
- W. Xuan, M. Hakkarainen and K. Odelius, Levulinic acid as a versatile building block for plasticizer design, ACS Sustainable Chem. Eng., 2019, 7, 12552–12562 CAS
- A. Sinisi, M. D. Esposti, M. Toselli, D. Morselli and P. Fabbri, Biobased ketal-diester additives derived from levulinic acid: Synthesis and effect on the thermal stability and thermomechanical properties of poly(vinyl chloride), ACS Sustainable Chem. Eng., 2019, 7, 13920–13931 CrossRef CAS
- B. D. Mullen, V. Badarinarayana, M. Santos-Martinez and S. Selifonov, Catalytic selectivity of ketalization versus transesterification, Top. Catal., 2010, 53, 1235–1240 CrossRef CAS
- A. S. Amarasekara and S. A. Hawkins, Synthesis of levulinic acid-glycerol ketal-ester oligomers and structural characterization using NMR spectroscopy, Eur. Polym. J., 2011, 47, 2451–2457 CrossRef CAS
- F. A. Freitas, D. Licursi, E. R. Lachter, A. M. R. Galletti, C. Antonetti, T. C. Brito and R. S. V. Nascimento, Heterogeneous catalysis for the ketalisation of ethyl levulinate with 1,2-dodecanediol: Opening the way to a new class of bio-degradable surfactants, Catal. Commun., 2016, 73, 84–87 CrossRef CAS
- S. Gundekari, M. Mani, J. Mitra and K. Srinivasan, Selective preparation of renewable ketals from biomass-based carbonyl compounds with polyols using β-zeolite catalyst, Mol. Catal., 2022, 524, 112269 CrossRef CAS
- P. Li, Z. Xiao, C. Chang, S. Zhao and G. Xu, Efficient synthesis of biobased glycerol levulinate ketal and its application for rigid polyurethane foam production, Ind. Eng. Chem. Res., 2020, 59, 17520–17528 CrossRef CAS
- L. J. Konwar, A. Samikannu, P. Mäki-Arvela, D. Boström and J. Mikkola, Lignosulfonate-based macro/mesoporous solid protonic acids for acetalization of glycerol to bio-additives, Appl. Catal., B, 2018, 220, 314–323 CrossRef CAS
- Y. Wang, H. Wang, X. Kong and Y. Zhu, Catalytic conversion of 5-hydroxymethylfurfural to some highvalued derivatives by selective activation of C-O, C=O and C=C bonds, ChemSusChem, 2022, 15, e202200421 CrossRef CAS PubMed
- X. Wang, M. Arai, Q. Wu, C. Zhang and F. Zhao, Hydrodeoxygenation of lignin-derived phenolics--a review on the active sites of supported metal catalysts, Green Chem., 2020, 22, 8140–8168 RSC
- K. Tomishige, M. Yabushita, J. Cao and Y. Nakagawa, Hydrodeoxygenation of potential platform chemicals derived from biomass to fuels and chemicals, Green Chem., 2022, 24, 5652–5690 RSC
- F. J. A. G. Coumans, Z. Overchenko, J. J. Wiesfeld, N. Kosinov, K. Nakajima and E. J. M. Hensen, Protection strategies for the conversion of biobased furanics to chemical building blocks, ACS Sustainable Chem. Eng., 2022, 10, 3116–3130 CrossRef CAS
- J. M. Rubio-Caballero, S. Saravanamurugan, P. Maireles-Torres and A. Riisager, Acetalization of furfural with zeolites under benign reaction conditions, Catal. Today, 2014, 234, 233–236 CrossRef CAS
- Y. Du, X. Liu, X. Wu, Q. Cheng, C. Ci and W. Huang, Tunable fabrication of NiAl-LDHs containing acid activity sites as green catalyst for acetalization of furfural to furfural diethyl acetal, ChemistrySelect, 2018, 3, 7996–8002 CrossRef CAS
- M. J. da Silva and M. G. Teixeira, Assessment on the double role of the transition metal salts on the acetalization of furfural: Lewis and Brønsted acid catalysts, Mol. Catal., 2018, 461, 40–47 CrossRef CAS
- B. Zhang, T. Guo, Z. Li, F. E. Kühn, M. Lei, Z. Zhao, J. Xiao, J. Zhang, D. Xu, T. Zhang and C. Li, Transition-metal-free synthesis of pyrimidines from lignin β-O-4 segments via a one-pot multicomponent reaction, Nat. Commun., 2022, 13, 3365 CrossRef CAS PubMed
- B. Zhang, T. Guo, Y. Liu, F. E. Kühn, C. Wang, Z. K. Zhao, J. Xiao, C. Li and T. Zhang, Sustainable production of benzylamines from lignin, Angew. Chem., Int. Ed., 2021, 60, 20666–20671 CrossRef CAS PubMed
- X. Shen, C. Zhang, B. Han and F. Wang, Catalytic self-transfer hydrogenolysis of lignin with endogenous hydrogen: Road to the carbon-neutral future, Chem. Soc. Rev., 2022, 51, 1608–1628 RSC
- Y. Jing, L. Dong, Y. Guo, X. Liu and Y. Wang, Chemicals from lignin: A review of the catalytic conversion involving hydrogen, ChemSusChem, 2020, 13, 4181–4198 CrossRef CAS PubMed
- K. Wu, W. Wang, H. Guo, Y. Yang, Y. Huang, W. Li and C. Li, Engineering Co nanoparticles supported on defect MoS2-x for mild deoxygenation of lignin-derived phenols to arenes, ACS Energy Lett., 2020, 5, 1330–1336 CrossRef CAS
- B. Zhang, Z. Qi, X. Li, J. Ji, W. Luo, C. Li, A. Wang and T. Zhang, ReOx/AC-catalyzed cleavage of C−O bonds in lignin model compounds and alkaline lignins, ACS Sustainable Chem. Eng., 2019, 7, 208–215 CrossRef CAS
- X. Liu, F. P. Bouxin, J. Fan, V. L. Budarin, C. Hu and J. H. Clark, Recent advances in the catalytic depolymerization of lignin towards phenolic chemicals: A review, ChemSusChem, 2020, 13, 4296–4317 CrossRef CAS PubMed
- J. Pang, B. Zhang, Y. Jiang, Y. Zhao, C. Li, M. Zheng and Z. Zhang, Complete conversion of lignocellulosic biomass to mixed organic acids and ethylene glycol via cascade steps, Green Chem., 2021, 23, 2427–2436 RSC
- P. J. Deuss, M. Scott, F. Tran, N. J. Westwood, J. G. de Vries and K. Barta, Aromatic monomers by in situ conversion of reactive intermediates in the acid-catalyzed depolymerization of lignin, J. Am. Chem. Soc., 2015, 137, 7456–7467 CrossRef CAS PubMed
- P. J. Deuss, C. S. Lancefield, A. Narani, J. G. de Vries, N. J. Westwood and K. Barta, Phenolic acetals from lignins of varying compositions via iron(III) triflate catalysed depolymerisation, Green Chem., 2017, 19, 2774–2782 RSC
- Z. Zhang, C. W. Lahive, J. G. M. Winkelman, K. Barta and P. J. Deuss, Chemicals from lignin by diol-stabilized acidolysis: Reaction pathways and kinetics, Green Chem., 2022, 24, 3193–3207 RSC
- X. Luo, Y. Li, N. K. Gupta, B. Sels, J. Ralph and L. Shuai, Protection strategies enable selective conversion of biomass, Angew. Chem., Int. Ed., 2020, 59, 11704–11716 CrossRef CAS PubMed
- J. Park, A. Riaz, D. Verma, H. J. Lee, H. M. Woo and J. Kim, Fractionation of lignocellulosic biomass over core-shell Nialumina catalysts with formic acid as a co-catalyst and hydrogen source, ChemSusChem, 2019, 12, 1743–1762 CrossRef CAS PubMed
- X. Diao, N. Ji, X. Li, Y. Rong, Y. Zhao, X. Lu, C. Song, C. Liu, G. Chen, L. Ma, S. Wang, Q. Liu and C. Li, Fabricating high temperature stable Mo-Co9S8/Al2O3 catalyst for selective hydrodeoxygenation of lignin to arenes, Appl. Catal., B, 2022, 305, 121067 CrossRef CAS
- Q. Wang, L. Xiao, Y. Lv, W. Yin, C. Hou and R. Sun, Metal-organic-framework-derived copper catalysts for the hydrogenolysis of lignin into monomeric phenols, ACS Catal., 2022, 12, 11899–11909 CrossRef CAS
- J. S. Luterbacher, A. Azarpira, A. H. Motagamwala, F. Lu, J. Ralph and J. A. Dumesic, Lignin monomer production integrated into the γ-valerolactone sugar platform, Energy Environ. Sci., 2015, 8, 2657–2663 RSC
- L. Shuai, M. T. Amiri, Y. M. Questell-Santiago, F. Héroguel, Y. Li, H. Kim, R. Meilan, C. Chapple, J. Ralph and J. S. Luterbacher, Formaldehyde stabilization facilitates lignin monomer production during biomass depolymerization, Science, 2016, 354, 329–333 CrossRef CAS PubMed
- W. Lan, M. T. Amiri, C. M. Hunston and J. S. Luterbacher, Protection group effects during α,γ-diol lignin stabilization promote high-selectivity monomer production, Angew. Chem., Int. Ed., 2018, 57, 1356–1360 CrossRef CAS PubMed
- W. Lan, J. B. de Bueren and J. S. Luterbacher, Highly selective oxidation and depolymerization of α,γ-diolprotected lignin, Angew. Chem., Int. Ed., 2019, 58, 2649–2654 CrossRef CAS PubMed
- D. Zhao, T. Su, C. Len, R. Luque and Z. Yang, Recent advances in oxidative esterification of 5-hydroxymethylfurfural to furan-2,5-dimethylcarboxylate, Green Chem., 2022, 24, 6782–6789 RSC
- G. Totaro, L. Sisti, P. Marchese, M. Colonna, A. Romano, C. Gioia, M. Vannini and A. Celli, Current advances in the sustainable conversion of HMF into FDCA, ChemSusChem, 2022, 15, e202200501 CrossRef CAS PubMed
- J. Cai, K. Li and S. Wu, Recent advances in catalytic conversion of biomass derived 5-hydroxymethylfurfural into 2,5-furandicarboxylic acid, Biomass Bioenergy, 2022, 158, 106358 CrossRef CAS
- M. Kim, Y. Su, A. Fukuoka, E. J. M. Hensen and K. Nakajima, Aerobic oxidation of HMF-cyclic acetal enables selective FDCA formation with CeO2-supported Au catalyst, Angew. Chem., Int. Ed., 2018, 57, 8235–8239 CrossRef CAS PubMed
- M. Kim, Y. Su, T. Aoshima, A. Fukuoka, E. J. M. Hensen and K. Nakajima, Effective strategy for high-yield furan dicarboxylate production for biobased polyester applications, ACS Catal., 2019, 9, 4277–4285 CrossRef CAS
- Y. Fu, F. Devred, P. Eloy, T. Haynes, M. L. Singleton and S. Hermans, The effect of Ni particle size and carbon support on catalytic activity for glucose hydrogenation reaction, Appl. Catal., A, 2022, 644, 118833 CrossRef CAS
- A. R. Mankar, A. Modak and K. K. Pant, High yield synthesis of hexitols and ethylene glycol through one-pot hydrolytic hydrogenation of cellulose, Fuel Process. Technol., 2021, 218, 106847 CrossRef CAS
- L. Wang, X. Liu, Y. Wang, W. Sun and L. Zhao, Thermodynamics and reaction kinetics of the sorbitol dehydration to isosorbide using NbOPO4 as the catalyst, Ind. Eng. Chem. Res., 2022, 61, 7833–7841 CrossRef CAS
- J. Guo, Y. Song, S. Liu, L. Huang, X. Wang, S. Liu and C. Li, Sequential dehydration of sorbitol to isosorbide over acidified niobium oxides, Catal. Sci. Technol., 2021, 11, 4226–4234 RSC
- P. Che, F. Lu, X. Si, H. Ma, X. Nie and J. Xu, A strategy of ketalization for catalytic selective dehydration of biomass-based polyols over H-beta zeolite, Green Chem., 2018, 20, 634–640 RSC
- P. Che, H. Ma, X. Nie, W. Yu and J. Xu, Methyl isobutyl ketone-enabled selective dehydrationesterification of sorbitol to isosorbide esters over H-beta catalyst, Green Chem., 2022, 24, 7545–7555 RSC
- M. Audemar, W. Ramdani, T. Junhui, A. R. Ifrim, A. Ungureanu, F. Jérôme, S. Royer and K. O. Vigier, Selective hydrogenation of xylose to xylitol over Co/SiO2 catalysts, ChemCatChem, 2020, 12, 1973–1978 CrossRef CAS
- N. Ullah, F. Jérôme and K. O. Vigier, Efficient nickel-iron bimetallic nanoparticles catalysts for the selective hydrogenation of biomass-derived sugars to sugar alcohols, Mol. Catal., 2022, 529, 112558 CrossRef CAS
- Y. M. Questell-Santiago, R. Zambrano-Varela, M. T. Amiri and J. S. Luterbacher, Carbohydrate stabilization extends the kinetic limits of chemical polysaccharide depolymerization, Nat. Chem., 2018, 10, 1222 CrossRef CAS PubMed
- Y. M. Questell-Santiago, J. H. Yeap, M. T. Amiri, B. P. Le Monnier and J. S. Luterbacher, Catalyst evolution enhances production of xylitol from acetalstabilized xylose, ACS Sustainable Chem. Eng., 2020, 8, 1709–1714 CrossRef CAS
| This journal is © Institute of Process Engineering of CAS 2024 |