
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceProgress and perspectives of Pd-based catalysts for direct synthesis of hydrogen peroxide
Jiamei
Wei
,
Shen
Wang
,
Jianguo
Wu
,
Dong
Cao
* and
Daojian
Cheng
 *
*
State Key Laboratory of Organic-Inorganic Composites and Beijing Advanced Innovation Center for Soft Matter Science and Engineering, Beijing University of Chemical Technology, Beijing 100029, People's Republic of China. E-mail: caod@mail.buct.edu.cn; chengdj@mail.buct.edu.cn
First published on 20th July 2023
Abstract
Hydrogen peroxide (H2O2) is a green oxidant that has been widely used. The direct synthesis of hydrogen peroxide (DSHP) offers significant advantages in terms of high atomic economy and environmentally friendly effects. However, due to the inevitable side reactions and severe mass transfer limitations, it is still challenging to balance the selectivity and activity for the DSHP. Combining theoretical understanding with the controllable synthesis of nanocatalysts may significantly facilitate the design of “dream catalysts” for the DSHP. In this work, the main factors affecting the reaction performance of catalysts and the active sites of catalysts have been reviewed and discussed in detail. The development and design of catalysts with high efficiency were introduced from three aspects: the catalyst support, active component and atomic impurity. In addition, the coupling of DSHP and other oxidation reactions to realize one-pot in situ oxidation reactions was comprehensively emphasized, which showed essential guiding significance for the future development of H2O2.
Keywords: Direct synthesis of H2O2; Pd-based catalyst; Selectivity and activity; Catalytic mechanism; In situ oxidation reactions.
 Jiamei Wei | Jiamei Wei is a doctoral candidate under the supervision of Professor Daojian Cheng at the State Key Laboratory of Organic Inorganic Composites, Beijing University of Chemical Technology. The research field is based on the experimental synthesis of nanoclusters and atomically dispersed catalysts and their applications in DSHP, olefin hydroformylation and other fields. |
 Shen Wang | Shen Wang is a doctoral candidate under the supervision of Professor Cheng Daojian at the State Key Laboratory of Organic Inorganic Composites, Beijing University of Chemical Technology. He obtained his Master of Engineering Degree from Tiangong University in 2021, majoring in Chemical Engineering. Currently, his research field is the development of green oxidation technology of TS-1/H2O2. |
 Jianguo Wu | Jianguo Wu is a doctoral candidate under the supervision of Professor Cheng Daojian at the College of Chemical Engineering, Beijing University of Chemical Technology. His research directions are the preparation of atomically dispersed catalysts for hydrogenation and other fields. |
 Dong Cao | Dong Cao, Ph.D, is a lecturer at the College of Chemical Engineering, Beijing University of Chemical Technology. The research field is the design and controllable preparation of metal catalysts in the fields of electrocatalysis and fine chemicals. Currently, more than 20 SCI papers have been published, including Nat. Commun., Angew. Chem., Adv. Energy Mater., Appl. Catal. B Environ., Small, Chem. Eng. J., and so on. |
 Daojian Cheng | Daojian Cheng is a Professor of Beijing University of Chemical Technology, supervisor of doctoral candidates, National Excellent Youth Fund winner, and Fellow of the Royal Society of Chemistry. He was listed as one of the Highly Cited Chinese Researchers of Elsevier (2020–2022). He is interested in the design, preparation and application of metal nanocatalysts in the chemical industry. In recent years, more than 180 SCI papers have been published in international mainstream journals such as Nat. Catal., Nat. Energy, Nat. Commun., Proc. Natl. Acad. Sci., Angew Chem., ACS Catal., Adv. Energy Mater., and so on. |
1 Introduction
As an important chemical raw material, hydrogen peroxide (H2O2) is mainly used in pulp bleaching, textile printing and dyeing, wastewater treatment, chemical synthesis and other fields.1–6 The by-product of the H2O2 oxidation reaction is only water and it has a lower active oxygen content than molecular oxygen. As an oxidizer, it has a high selectivity that fully reflects its environmental protection and efficiency.7–10 The global chemical industry and the government's demand for H2O2 is increasing year by year.11,12 At present, the traditional anthraquinone auto-oxidation manufacturing processes (the AO process) can achieve a high efficiency in H2O2, but the step is complicated, resulting in many harmful organic compounds.13,14 The AO process is also accompanied by costs and safety issues incurred in the transportation and storage of the product (Fig. 1a). The equipment for the electrochemical synthesis of H2O2 is relatively complex (Fig. 1b).15,16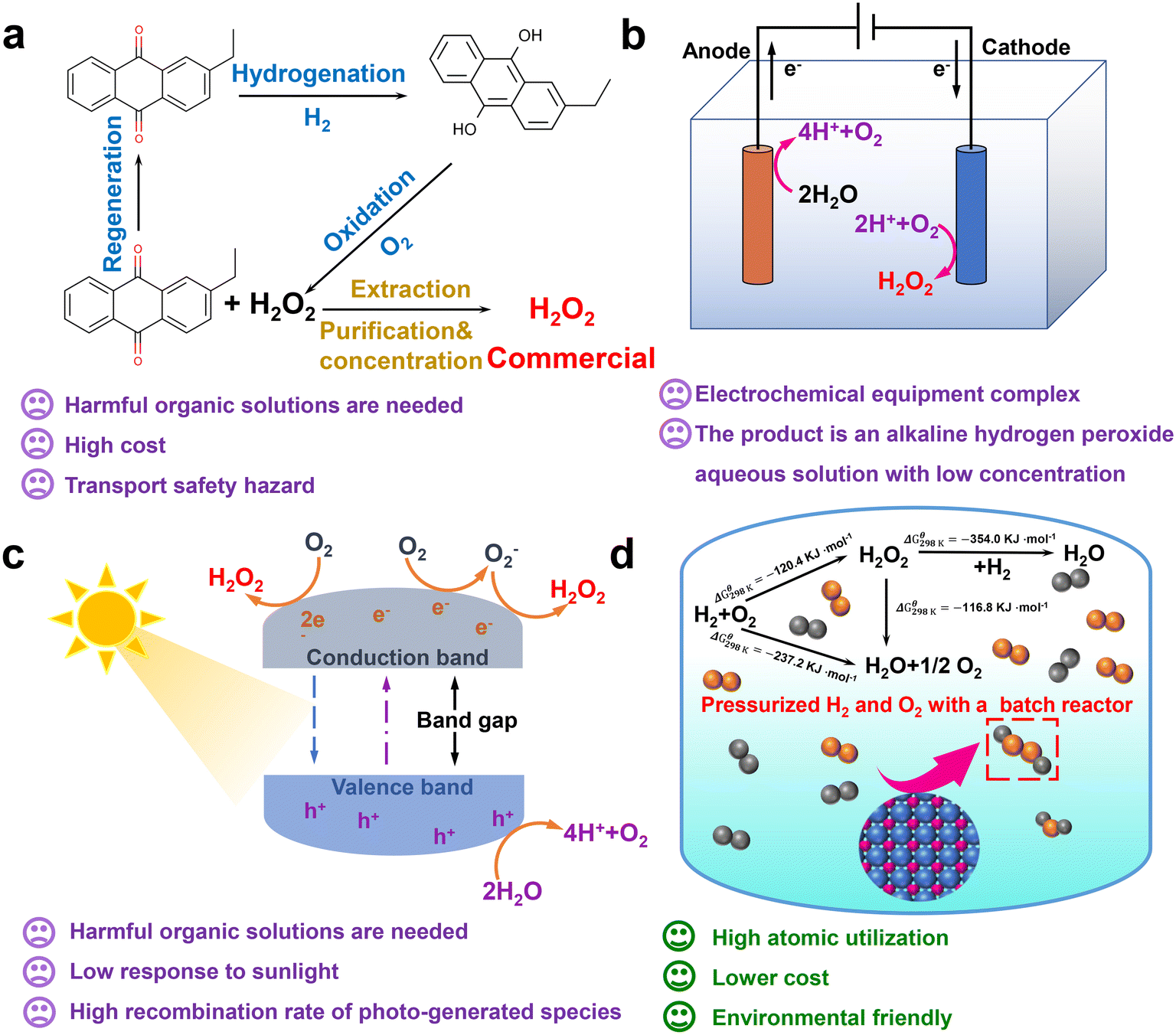 | ||
| Fig. 1 Introduction to the (a) anthraquinone method, reprinted with permission from ref. 12. Copyright 2022, Elsevier. (b) Electrochemical synthesis method, reprinted with permission from ref. 12. Copyright 2022, Elsevier. (c) Photocatalytic synthesis method, reprinted with permission from ref. 12. Copyright 2022, Elsevier. And (d) direct synthesis method, as well as graphical explanation of their related drawbacks and advantages. Reprinted with permission from ref. 11. Copyright 2020, Elsevier. | ||
The photocatalytic synthesis process of H2O2 requires organic solvents and has poor light reactivity (Fig. 1c).15,16 However, the direct synthesis of hydrogen peroxide (DSHP) is a more atomically efficient synthesis route than the current industrial process (Fig. 1d).17 The synthesis process of the DSHP can be achieved by a reactor system, which can perform small-scale on-site synthesis.18 Furthermore, the DSHP also has significant benefits such as high atom economics, lower relative costs, and environmental friendliness. The reaction mechanism for the DSHP is shown in Fig. 1d. Both the side reaction (H2 + O2 → H2O2, H2O2 → H2O, H2O2 → H2O + O2) and the main reaction are thermodynamically spontaneous, which is one of the reasons for its low selectivity.19 At the same time, this is a solid–liquid–gas three-phase reaction system with mass transfer limitations, which will result in a low yield of H2O2.19 Therefore, the development of high efficiency catalysts is key to solving the problems of poor selectivity, low activity, slow reaction rate and low product concentration of the DSHP.
The application of Pd-based catalysts has attracted significant attention in the DSHP because of their superior performance to other catalysts and has been a “hot area” of research. In recent years, the research on Au catalysts has gradually started, particularly Au–Pd bimetallic catalysts. Metal Pd has shown a better catalytic effect in the DSHP, but it has been more active in catalyzing the decomposition and hydrogenation of H2O2, which limits its application. At present, Pd-based catalysts have been well developed. Some catalysts showed a selectivity to H2O2 close to 100%, and some demonstrated high activity.17 In short, while advances have been made in Pd-based catalysts, it is still difficult to balance the selectivity and activity of the DSHP. Much more needs to be done in catalytic design. Factors that affect the performance of the catalyst to synthesize H2O2 include many aspects. Firstly, in terms of the mechanism, studies have shown that protons and organic solvents can significantly affect the catalytic mechanism of Pd-based catalysts. In recent years, researchers have also carried out extensive research into catalytic sites and have drawn many conclusions. Moreover, it will help us extend the mechanism to the experimental design of effective catalysts. Effective synthesis of H2O2 is achieved by optimization of support properties, optimization of active components and introduction of heteroatoms.
Here, we summarized the research progress of the DSHP and systematically analyzed the catalytic mechanism and catalytically active sites. The macro-control of the catalytic activity of DSHP was discussed from three aspects: the catalyst support, metal species and heteroatoms (Fig. 2). Additionally, there were a large number of species of hydroperoxides and reactive oxygen in the DSHP process. Many studies have shown that when the DSHP is combined with other oxidation reactions, it is possible to achieve in situ oxidation reactions. This significantly reduces manufacturing costs while fully utilizing the product to meet multiple process requirements. This work provided a comprehensive discussion of catalyst design and the in situ application of H2O2, which has some guiding significance for the future direction of the DSHP.
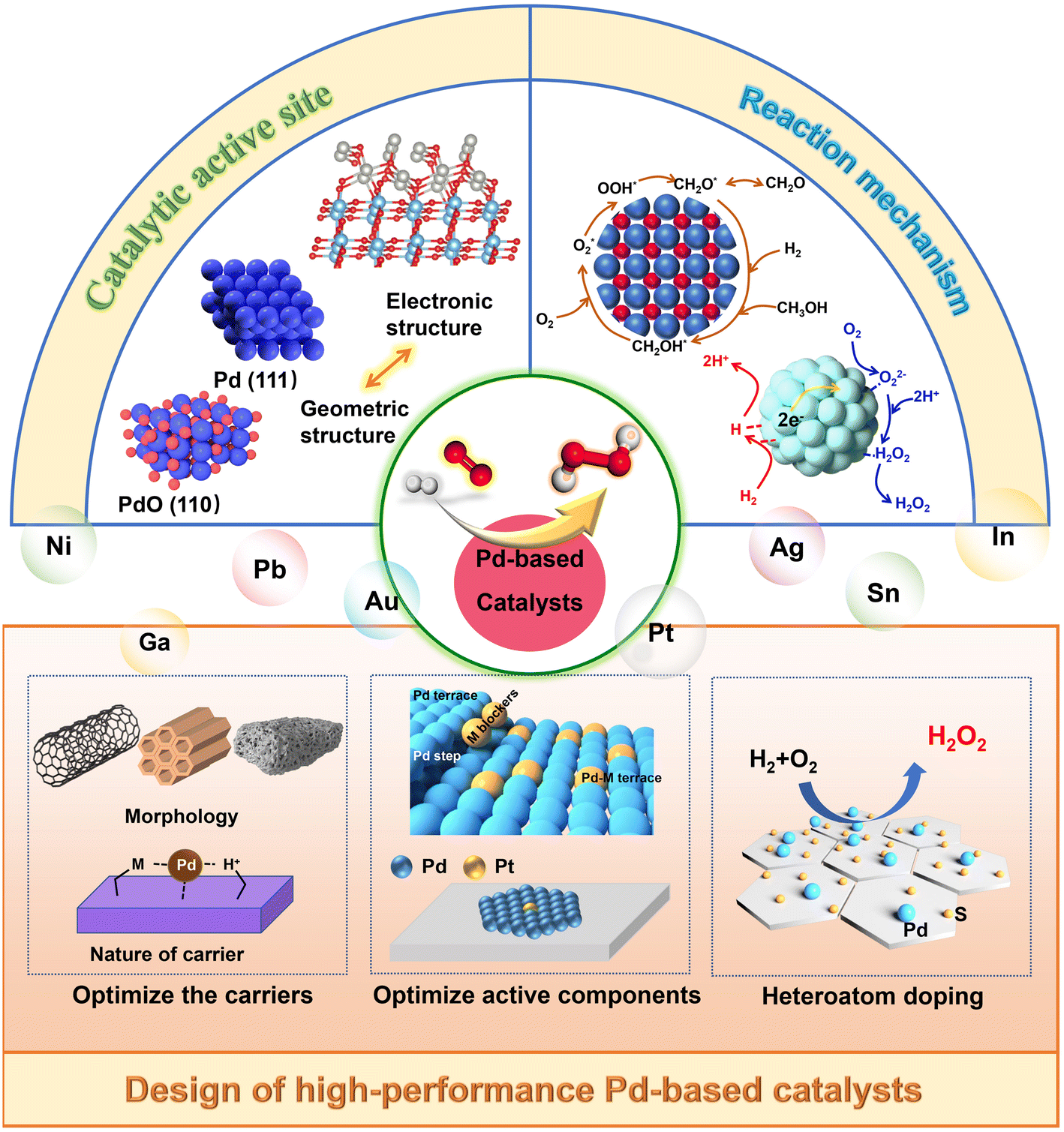 | ||
| Fig. 2 The catalytic mechanism, active site and design strategy of Pd-based catalysts for direct synthesis of hydrogen peroxide. | ||
2 Catalytic mechanism of Pd-based catalysts
2.1 Reaction mechanism
The reaction process for the DSHP, as shown in Fig. 1d, consists of four chemical reactions. From the thermodynamic point of view, it can be seen that both the main reaction for the production of H2O2 and the side reactions for the decomposition and hydrogenation of H2O2 are spontaneous reactions.19 And the reaction process of the side reactions is thermodynamically more favorable than the main reaction. This leads to a lower selectivity of the DSHP. Therefore, the key to current research is to design catalysts that balance selectivity and activity to inhibit the side reactions of H2O2 formation.The exact nature of the active sites responsible for the formation of H2O2 and its side reactions remains uncertain, although in combination with the results of recent studies.22 The generally accepted reaction pathway for the production of H2O2 is shown in Fig. 3a, where H2 is activated with O2 to 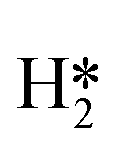 and
and 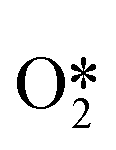 and further dissociates to form the intermediates H* and OOH* and finally H2O2.60 Hydrogenation and decomposition side reactions of H2O2 occur mainly because the O–O bonds in the intermediates
and further dissociates to form the intermediates H* and OOH* and finally H2O2.60 Hydrogenation and decomposition side reactions of H2O2 occur mainly because the O–O bonds in the intermediates 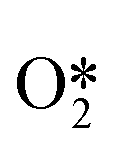 , OOH*, and
, OOH*, and 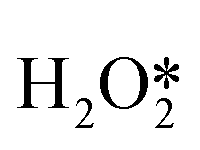 break to form O*, which reacts in H* to produce water (Fig. 3b).60 Therefore, perhaps the greatest challenge for the future design of efficient catalysts is to inhibit the cleavage of O–O bonds of reaction intermediates and increase the presence of superoxide radicals.
break to form O*, which reacts in H* to produce water (Fig. 3b).60 Therefore, perhaps the greatest challenge for the future design of efficient catalysts is to inhibit the cleavage of O–O bonds of reaction intermediates and increase the presence of superoxide radicals.
 | ||
| Fig. 3 Schematic diagram of the elementary reaction step network of the (a) main reaction and (b) side reaction for direct synthesis of H2O2 from H2 and O2.60 Copyright 2017, American Chemical Society. | ||
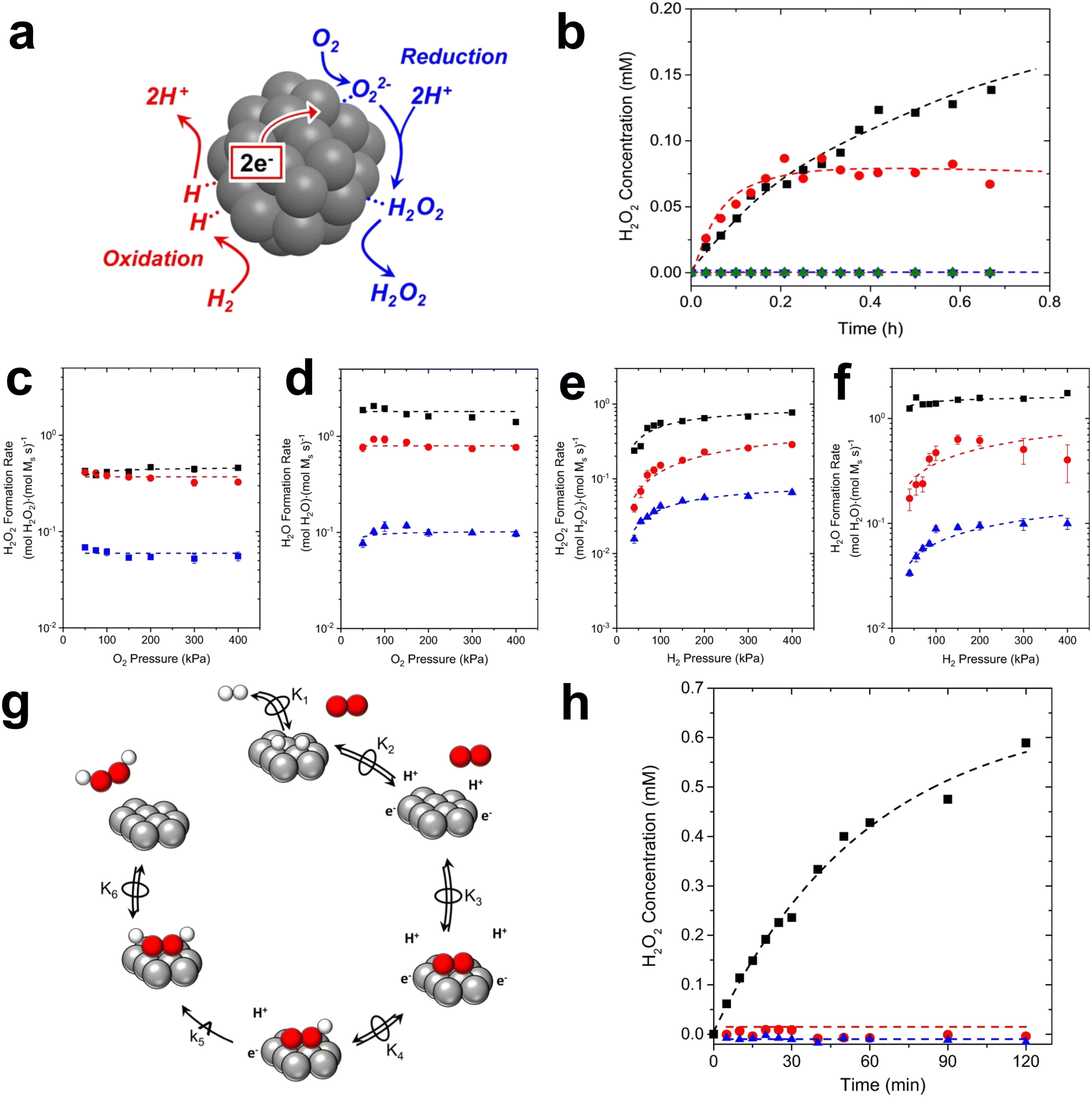
First of all, Wilson et al. conducted a detailed study on the DSHP from Pd nanoclusters.20 They have proposed that the DSHP does not proceed by a Langmuir–Hinshelwood mechanism, but by sequential proton–electron transfer to O2 and hydroperoxide intermediates to form H2O2. This was similar to a two-electron oxygen reduction reaction (ORR). A schematic diagram of this mechanism is shown in Fig. 4a. They have also concluded that protons are a critical factor in the synthesis of H2O2 and that synthesis in protonic solutions is superior to that in non-protonic solutions. The addition of proton donors increased the rate of H2O2 formation, and simultaneously increased the rate of H2O formation, increasing the selectivity to H2O2 (Fig. 4b). Secondly, Deguchi et al. carried out a kinetic analysis of DSHP using Pd-based catalysts in aqueous solution and discussed the role of protons in the DSHP.21 According to the kinetic study, H+ accelerated the elementary reaction steps OOH* + H* → 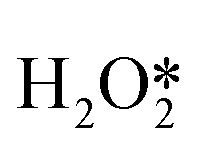 and
and 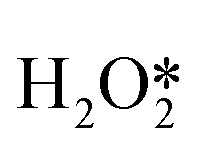 → H2O2; it also accelerated OOH* + H* → H2O + O*. The two aspects were in competition, with the former favoring the direct synthesis of H2O2 and the latter leading to the formation of water as a by-product. The H+ promoted the former more strongly, increasing the selectivity to H2O2.
→ H2O2; it also accelerated OOH* + H* → H2O + O*. The two aspects were in competition, with the former favoring the direct synthesis of H2O2 and the latter leading to the formation of water as a by-product. The H+ promoted the former more strongly, increasing the selectivity to H2O2.
 | ||
| Fig. 4 (a) Pd clusters catalyze both heterolytic H2 oxidation and O2 reduction steps in order to form H2O2. (b) H2O2 concentrations as a function of time during direct synthesis in a well-mixed semi-batch reactor using protic (methanol (black ■) and water (red ●)) or aprotic (dimethyl sulfoxide (green ▲), acetonitrile (blue ▼), and propylene carbonate (magenta ✦)) solvents.20 Copyright 2016, American Chemical Society. Turnover rates for the formation of (c) H2O2 and (d) H2O as a function of H pressure on silica-supported Pd (black), PdZn6 (red), and PdZn30 (blue). Their (e) H2O2 and (f) H2O conversion rate as a function of O2 pressure. (g) The basic step of directly synthesizing the formation of hydrogen peroxide by the reaction on the particles of Pd and PdZnx. The red ball represents O2 and the white ball represents H2. (h) H2O2 concentrations as a function of time during catalysis in a semi-batch reactor on PdZn30 in methanol (black), acetonitrile (red), and dimethyl sulfoxide (blue).23 Copyright 2018, Academic Press Inc. | ||
It has been generally accepted that protons modify the electronic structure of Pd. However, there are observable differences in the rate dependence and kinetics of H2O2 formation in alcoholic and aprotic solutions. The protons seem to participate directly in the catalytic cycle as a co-reactant or as a co-catalyst of the reaction.22 Wilson et al. proved the proton–electron transfer theory in a silicon-supported PdZn bimetallic catalyst by measuring the dependence of the steady-state H2O2 and H2O formation rate on the pressure of H2 and O2 and the involvement of the proton solution.23 As shown in Fig. 4c–f, the rate of H2O2 formation increased in an approximately linear fashion when the H2 pressure was less than 100 kPa H2. Above this value, the H2O2 rate was sub-linearly dependent on the H2 pressure. The rate of water formation was also reliant on the H2 pressure. The rate of formation of H2O2 and H2O remained essentially unchanged with the change of O2 pressure. The concentration of H2O2 in the proton solution gradually increased with time, while the concentration of H2O2 in the non-proton solution remained basically unchanged and zero (Fig. 4h). These have shown that the proton–electron transfer plays an important role in the formation of H2O2 (Fig. 4e).
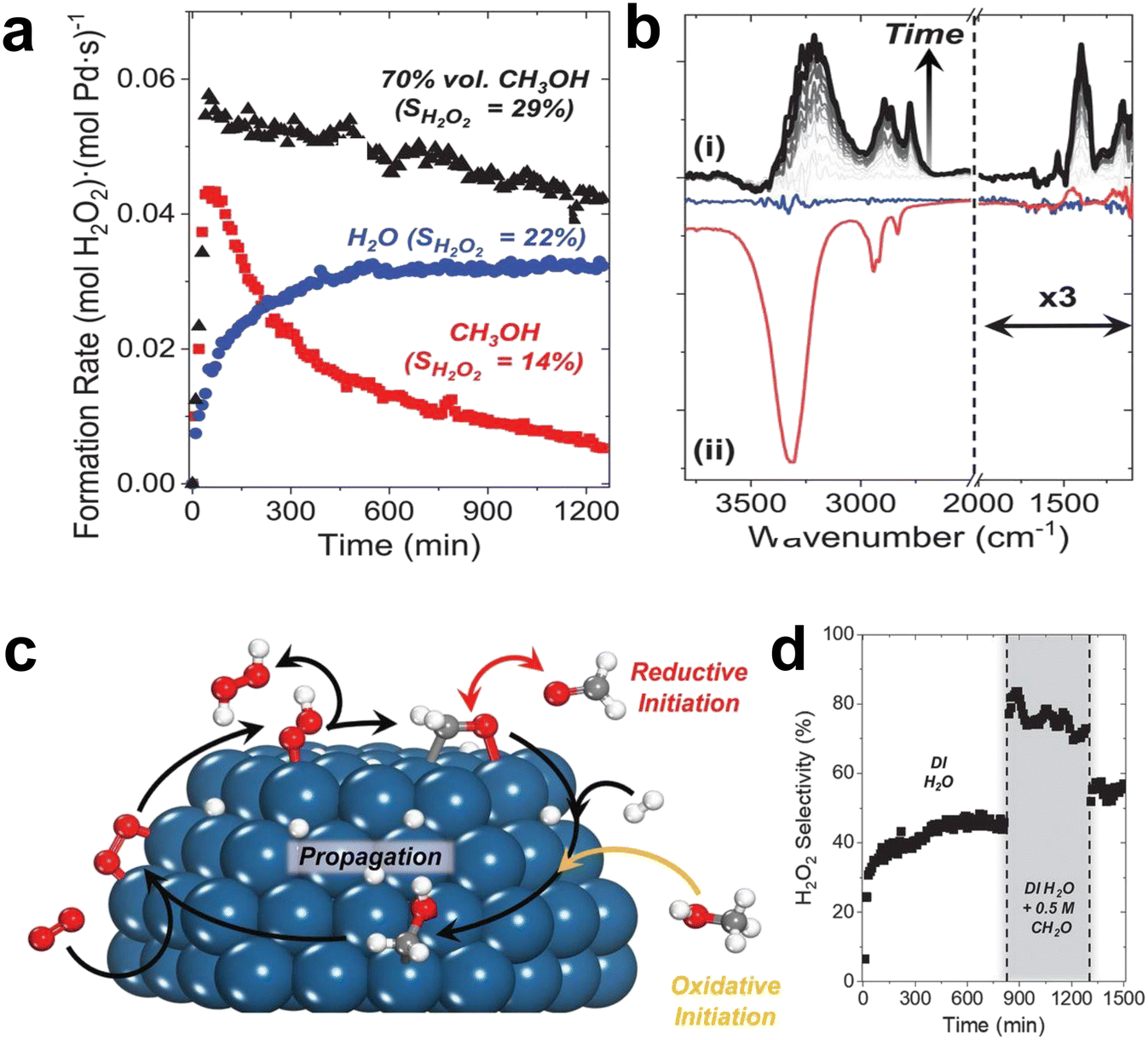 | ||
| Fig. 5 (a) The rate of hydrogen peroxide formation as a function of time in an aqueous solution of methanol, water and 70 vol% methanol in a fixed bed reactor. (b) The difference between the in situ infrared spectrum of the methanol-derived intermediate accumulated on Pd/SiO2 within 8 hours (i) and the steady-state spectrum of the species on Pd–SiO2 (blue line) and SiO2 (red line) under oxygen-rich and reaction conditions (ii). Catalytic O2 reduction with surface redox mediators. (c) Schematic diagram of the formation of hydroxymethyl (CH2OH*) and its subsequent role as a redox mediator. (d) Steady-state H2O2 selectivity is increased when formaldehyde (0.5 M CH2O) is added to deionized (DI) H2O.24 Copyright 2021, American Association for the Advancement of Science. | ||
Methanol and water co-catalyze the reduction of oxygen by promoting the proton–electron transfer reaction (Fig. 5c). Methanol generates hydroxymethyl intermediates on the surface of Pd, which effectively transfer protons and electrons to oxygen to form H2O2 and formaldehyde. The formaldehyde then oxidizes hydrogen to regenerate hydroxymethyl. The reaction of solvent molecules at the solid–liquid interface could generate redox media in situ, offering the possibility of greatly improving the rate and selectivity of the catalytic reaction.
In addition, Akram et al. recently studied the DSHP using a hydrophobic alcohol (decan-1-ol) and water as a solvent.25 The catalytic effect was significantly improved as the catalyst tended to be retained in hydrophobic organic components and the contact of the formed H2O2 with the catalyst was limited. The degradation of H2O2 was significantly reduced by optimized combination of the solvent and catalyst to achieve industrially acceptable concentrations of H2O2 under mild conditions.
This work has provided a promising basis for further exploration of a wider range of solvents and catalysts for the DSHP.
2.2 Catalytically active sites
Choudhary and colleagues have believed that Pd2+ is the active center for the catalytic synthesis of H2O2, because Pd2+ can adsorb peroxides, and Pd0 is beneficial for the decomposition reaction.26 Song et al. combined DFT calculations and microkinetic studies and found that co-adsorbed O plays a key role in the catalytic activity and selectivity of the DSHP on the surface of Pd (111) and Pd (100).27 The selectivity of the Pd (111) surface for the formation of H2O2 was nearly 99% when some Pd–O was present on the catalyst surface. Wang et al. used the density functional theory to study the active state of surface Pd and obtained completely different reaction modes on the surface of Pd (111) and PdO (101).28 The binding energy of H atoms on the Pd (111) surface was significantly higher than on the PdO (101) surface, and the reaction was endothermic on the PdO (101) surface. The reaction of OOH* with a surface-bound H atom would then require the cleavage of a very strong Pd–H bond on the Pd (111) surface to form H2O2. The report showed that the main product on the surface of PdO (101) is H2O2, while that on the surface of Pd (111) is H2O. However, a number of other studies have also shown that Pd0 is also an important active site. Kanungo et al. used a microreactor and in situ XAS characterization to ensure that Pd was completely reduced during the reaction and only existed in the metallic state.29 They believed that Pd0 was the reaction phase in the synthesis of H2O2. Liu et al. added acid and halide promoters to a reacted ethanol solution and found that Pd0/SiO2 was the most active, followed by partially reduced Pd0/SiO2, so they also believed that Pd0 was more conducive to the production of H2O2.30
When Pd nanoparticles are exposed to H2 and O2, PdO and PdH phases are formed and coexist during the reaction. If there are additives such as halides, the intermediates (for example, O*, OH*) or halide-covered surface of Pd nanoparticles, PdO, PdH phase surfaces, or soluble Pd composite surfaces may generate H2O2. Dissanayake et al. were the first to show that the rate of H2O2 production in the presence of H+ was independent of the amount of Pd in the catalyst.31 Pd colloid was considered to be the main active substance for the synthesis of H2O2, but the possibility that soluble Pd2+ complexes were also active sites cannot be ruled out. However, Priyadarshini et al. showed that the rate and selectivity of H2O2 production are independent of the number of detectable Pd2+ complexes, suggesting that the active substance is Pd0 rather than Pd2+.32 Chen et al. compared the potential energy of directly forming H2O2 on Pd (111), β-PdH (111), and β-PdH (211). They believed that Pd exists as a hydride phase, and the stepped PdH (211) surface could meet the conditions of O2 adsorption and the non-dissociation of the surface intermediates 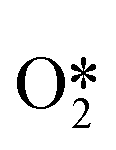 , OOH* and
, OOH* and 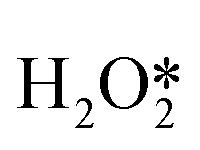 .33 In addition, Flaherty et al. observed a certain induction time (about half an hour) for the catalytic production of H2O2 by PdO, in contrast to the immediate production of H2O2 by Pd without induction time.22 The DSHP requires the presence of a certain reduced state of Pd nanoparticles. In summary, there is no precise statement about the active site of Pd, but it can be seen that there seem to be different conclusions depending on the reaction environment, the type of support and the preparation method. In the future, the design of catalysts with more suitable Pd states for different reaction systems will lead to better results.
.33 In addition, Flaherty et al. observed a certain induction time (about half an hour) for the catalytic production of H2O2 by PdO, in contrast to the immediate production of H2O2 by Pd without induction time.22 The DSHP requires the presence of a certain reduced state of Pd nanoparticles. In summary, there is no precise statement about the active site of Pd, but it can be seen that there seem to be different conclusions depending on the reaction environment, the type of support and the preparation method. In the future, the design of catalysts with more suitable Pd states for different reaction systems will lead to better results.
In addition, the interface effect between the metal Pd and other metals and the interaction between Pd and the support are also essential to improve the catalytic performance. Huynh et al. successfully synthesized an efficient catalyst with good selectivity for DSHP by exploiting the synergistic effect of a hybrid C and TiO2 support with an ordered alloy of PdNi.36 The TiO2–C support material could promote the strong interaction between the metal and the support. The ordered and evolved structure formed stronger electronic interactions and contributed to the synthesis of highly reactive H2O2, and the significant increase in selectivity was attributed to the mixed TiO2–C supports (Fig. 6). The heterometallic bond in the ordered alloy and the hybrid support significantly improves the performance of the DSHP reaction, which will greatly guide our future work. In the catalyst preparation and design process, more attention should be paid to the interaction between the metal supports. Li et al. recently successfully synthesized a series of Pd–Sn bimetallic nanocrystals with a hollow structure (Fig. 7a–i), which can completely inhibit the decomposition/hydrogenation side reaction.37 The structure of Pd–Sn bimetallic nanocrystals was closely related to the amount of tin chloride and the heat treatment temperature. The selectivity to H2O2 can be further improved by using water as the reaction solvent (Fig. 7j–l). More importantly, further analysis showed that the presence of PdO, the overall effect between Pd and Sn and the interfacial effect between Pd/SnOx and PdO/SnOx were responsible for the significant improvement in catalyst performance. From these research results, it can be seen that the interface effect of Pd metal particles is a very important aspect that can greatly improve the selectivity of the catalyst, and the use of metal bonding and electronic structure changes can fundamentally inhibit side reactions.
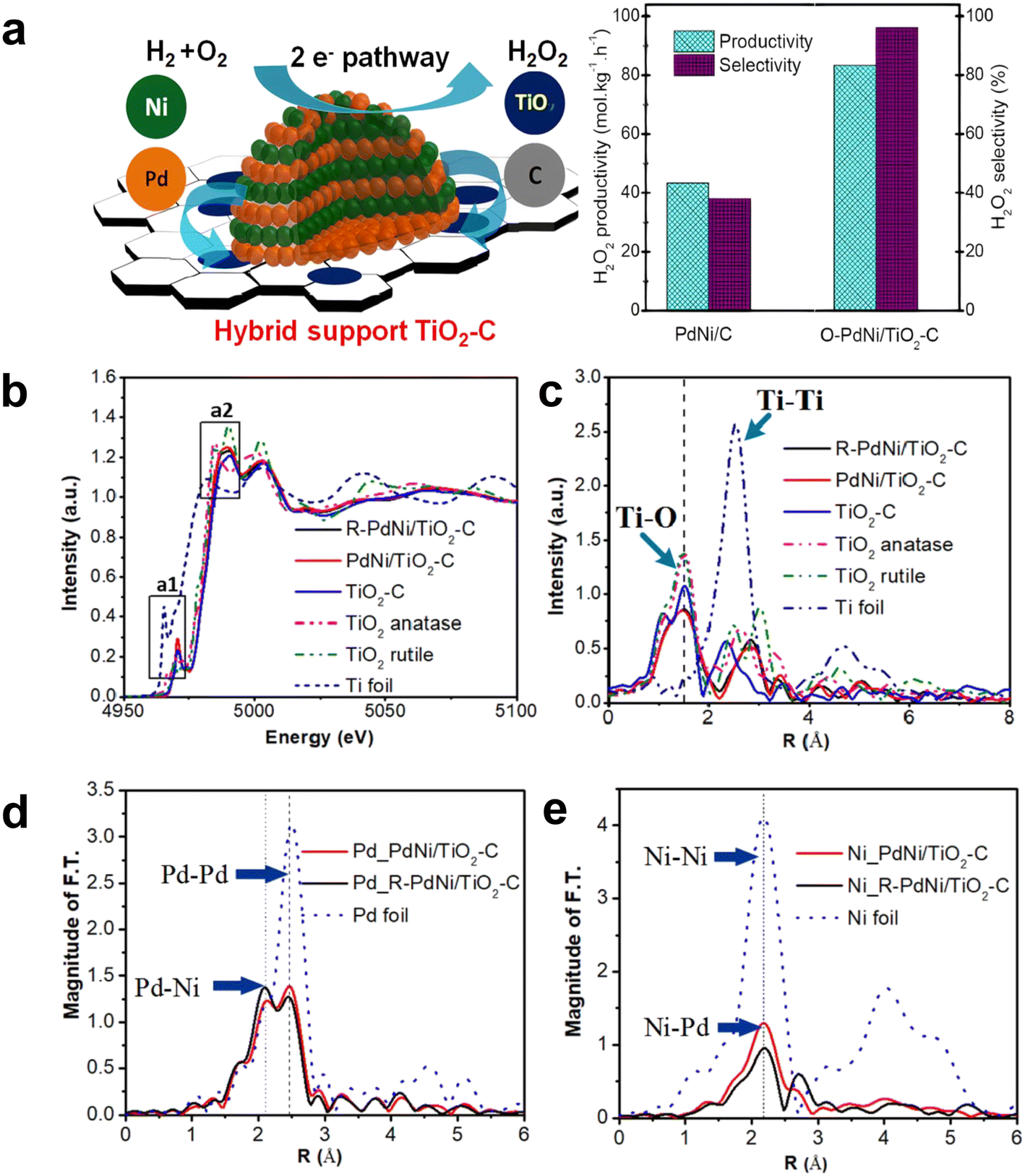 | ||
| Fig. 6 (a) Schematic diagram of the reaction mechanism and performance comparison of the PdNi bimetallic catalyst supported by the C and TiO2 mixed support. (b) XANES spectra of Ti K-edge and (c) EXAFS spectra of TiO2–C, PdNi/TiO2–C, and O–PdNi/TiO2–C. EXAFS spectra of (d) Pd and (e) Ni in PdNi/TiO2–C and R–PdNi/TiO2–C.36 Copyright 2021, American Chemical Society. | ||
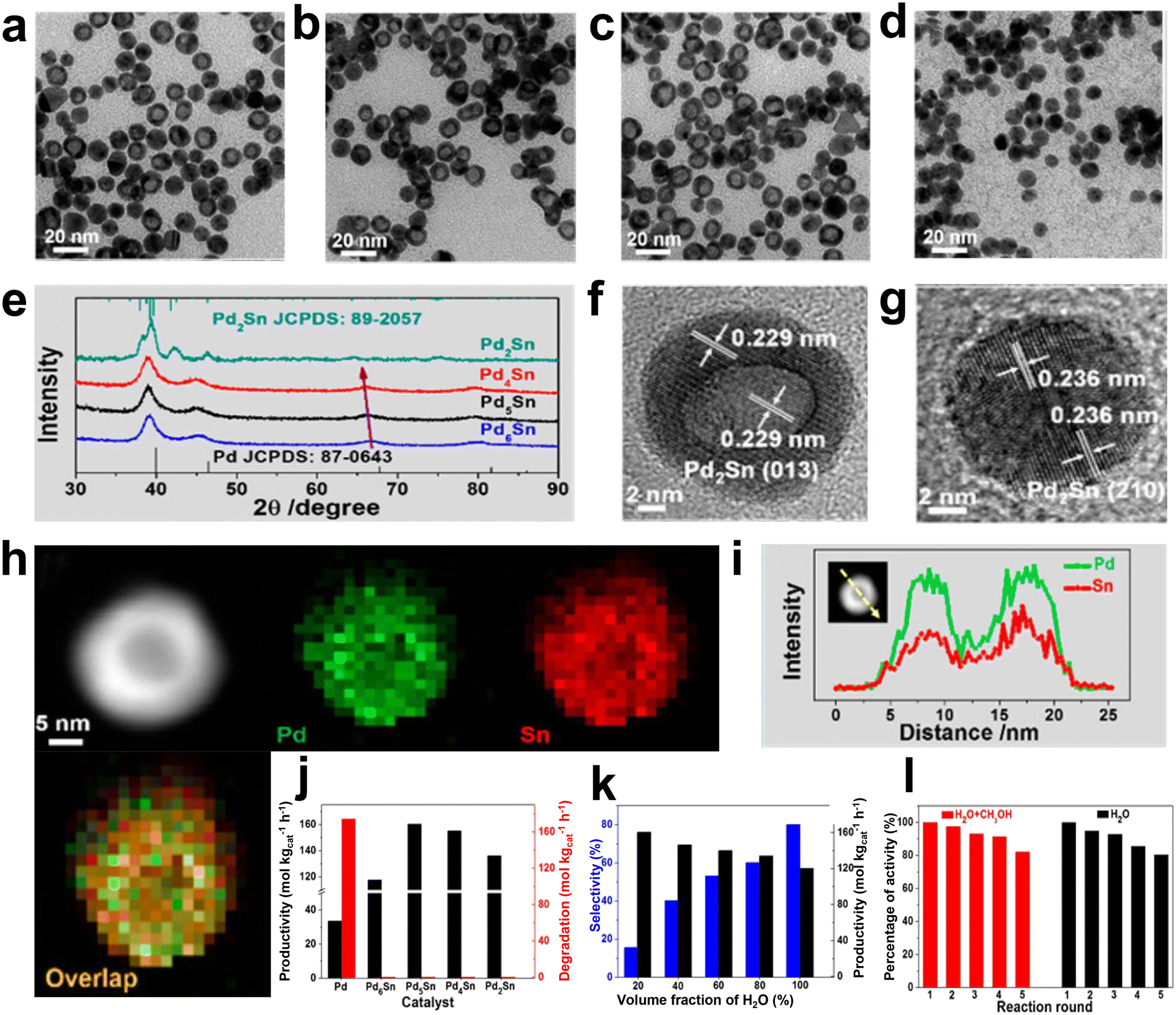 | ||
| Fig. 7 (a–d) TEM images of Pd2Sn, Pd4Sn, Pd5Sn, and Pd6Sn nanocrystals. (e) PXRD patterns of these four Pd–Sn nanocrystals. HRTEM images of the typical (f) hollow and (g) solid Pd2Sn nanocrystals. (h) HAADF-STEM image and EDX elemental mappings of Pd2Sn nanocrystals. Pd: green, Sn: red. (i) EDX line scanning of Pd2Sn nanocrystals. (j) H2O2 productivity and degradation of different catalysts after rapid thermal treatment at 350 °C for 6 min. (k) H2 selectivity and H2O2 productivity of Pd5Sn/TiO2 in different reaction media. (l) Recycling performance of Pd5Sn/TiO2 in a methanol/water mixture and pure water.37 Copyright 2018, American Chemical Society. | ||
Further, Yang et al. demonstrated for the first time that Pd–S NCs with atomically isolated Pd sites are highly robust and versatile catalysts that can be used for the direct, electrocatalytic and photocatalytic synthesis of H2O2.39 In the DSHP, the presence of the S element in Pd–S NCs contributes to the formation of isolated Pd atoms, which improves the selectivity towards H2O2, resulting in excellent activity and selectivity (133.6 mol kgcat−1 h−1, 89.5%). In addition, Ding et al. prepared a PdSb/TiO2 bimetallic catalyst with Sb uniformly distributed on the surface of the Pd–Sb catalyst, isolating a continuous Pd site, which is conducive to increasing the proportion of Pd monomer sites.40 XPS characterization showed that Sb inhibited the oxidation of Pd, forming a layer of Sb2O3 that partially encapsulated the surface of the Pd particles, thereby inhibiting H2 activation and subsequent H2O2 hydrogenation. The side reaction rate gradually decreased with increasing Sb/Pd ratio, indicating that the electronic structure and geometry of the PdSb/TiO2 catalyst were influenced by the Sb concentration. It was also demonstrated that the isolated Pd site is responsible for the non-dissociative activation of O2 for the selective synthesis of H2O2. And the continuous distribution of Pd atoms tends to lead to the dissociation of the O–O bond and the production of water as a by-product. Ouyang et al. also confirmed the advantages of isolated Pd sites in the selective synthesis of H2O2.41 Ricciardulli et al. proved that the separation of Pd atoms in the Au domain forms the active site for the preferential synthesis of H2O2, and that the selectivity increases with the distance between the surface Pd atoms.42 Both simulations and experiments have shown that both H2O2 and H2O are formed by the reduction of OOH* intermediates through the proton–electron transfer step of water molecules on Pd and Pd1Aux nanoparticles. The change in the geometric structure is a critical factor in improving selectivity, and the increase in H2O2 selectivity reflects the difference in active site requirements for the formation of the H2O2 and H2O transition states. The separation of monomer active sites is a catalyst design strategy to improve H2O2 selectivity.
3 Design of high-performance Pd-based catalysts
3.1 Optimization of the properties of the support
Edwards et al. studied the DSHP from Au, Pd and AuPd catalysts by using SiO2 as the support.43 The acid pretreatment of the SiO2 support would enhance the catalytic activity of the Pd catalyst. Moreover, when Au atoms were added, it displayed enhanced activity due to the synergistic effect. Notably, acid pretreatment could increase the concentration of hydroxyl groups on the surface of the support, which contributed to uniform dispersion of Pd metal on the support and effectively inhibited the hydrogenation activity of H2O2. In addition, Edwards and Solsona et al. found that acid pretreatment of carbon supports with Pd–Au catalysts would reduce the size of alloy nanoparticles.44 These smaller nanoparticles could reduce the activity of H2O2 decomposition or inhibit the decomposition of active sites. Liang et al. prepared a series of activated carbon-supported Pd catalysts pretreated with nitric acid and tested the performance of the DSHP at room temperature.45 The characterization results showed that nitric acid pretreatment mainly affected the surface acidity and structural properties of activated carbon. However, the pore properties of activated carbon had no significant effect on the catalytic performance. The acid functional groups increased the Pd2+ content, enhanced the dispersion of palladium and ultimately increased the selectivity to H2O2 and reduced its hydrogenation activity. Hang Thi Thuy Vu et al. systematically studied the effect of a Pd catalyst with a sulfonated carbon material as a solid acid support on the DSHP.46 The selectivity to H2O2 decreased with increasing Pd NP size. However, higher Pd2+/Pd0 ratios increased the selectivity to H2O2. From the above studies, it can be seen that acidity regulation inhibits the side reactions by increasing the acidity of the support, regulating the electronic properties and others, which improves the H2O2 yield and selectivity.
Furthermore, directly using heteropolyacid as the support is also a method to increase the acidity of the support. Alotaibi et al. studied the performance of a Pd-based catalyst with cesium-containing heteropolyacid as a support for the DSHP and subsequent degradation of H2O2.47 The experimental results showed that the 0.5 wt% Pd/Cs2.5H0.5PW12O40 catalyst showed good catalytic activity, and the heteropolyacid Pd catalyst was more effective than pure copper or bimetallic Pd–Cu prepared by the same preparation method. Ntainjua et al. found that Au–Pd bimetallic catalysts containing Cs heteropolyacid loadings achieved optimal catalysis at ambient temperature, in pure aqueous solvent, and in the absence of acids and halides.48 The presence of Au in the supported heteropolyacid catalyst was of vital importance. At the same time, the heteropolyacid inhibited the side reactions of hydrogenation and decomposition of H2O2.
During the reaction process, the acidity of the reaction system has a significant impact on the performance of direct synthesis of H2O2. The presence of protic acid can inhibit the hydrogenation and direct decomposition of H2O2, thereby improving the selectivity of the reaction.
Gudarzi et al. showed that the performance of ACC-supported Pd catalysts was related to the oxidation state of Pd in the metal phase and the surface chemistry of the support.49 The surface chemistry of the support is determined by the nature and number of surface functional groups. Oxidized ACC-supported catalysts exhibit higher selectivity than non-oxidized catalysts, and oxygen-containing surface functional groups can increase the selectivity of the catalyst. Platero et al. studied a series of Au–Pd/SBA-15 catalysts supported by mesoporous surface functionalized silica SBA-15. SBA-15 was functionalized with –SO3H, –NH2 and –SH organic groups, and then the metal was loaded by the ion exchange method.50 The experimental results showed that the catalyst prepared by functionalized SBA-15 had better catalytic performance than the non-functionalized SBA-15 catalyst prepared by the impregnation method. The XPS results showed that the non-functionalized catalyst Au–Pd/SBA-15 prepared by early wet impregnation had two pairs of peaks (Pd2+ and Pd0), while the functionalized catalyst only had Pd in the oxidized state. At the same time, the position of the binding energy of Pd was closely related to the particle size of the metal, and the high binding energy was attributed to PdO2 or extremely small PdO particles.51 These samples had very low Pd/Au ratios by XPS, which is indicative of abnormal aggregation of gold clusters. The metal dispersion could be controlled by functionalization with –SO3H and –SH groups, and the stable oxidation states of the metals were Au+ and Pd2+, respectively. The average metal Pd particle size of these catalyst samples was a good compromise between high metal dispersion and low energy sites. Oxygen was in a state of non-dissociation during chemical adsorption, and the catalytic activity was good. The decomposition side reaction of H2O2 could be inhibited by the formation of larger Au clusters. Although the NH2 group functionalization did not affect the particle size distribution, it could reduce the hydrogenation and decomposition activity of H2O2 and increase the H2O2 generation rate and selectivity.
Blanco-Brieva et al. prepared a Pd-supported catalyst supported on resin or silicon functionalized with acid and bromine groups.52 The direct synthesis of higher concentrations (>5 wt%) of H2O2 in the absence of dissolved acids and halides has been achieved. The bifunctional silica (Si–C6H4–SO3H) support had aryl sulfonate groups and aryl bromine groups, while resin supports introduced brominated groups into a sulphonate functionalized resin styrene–divinylbenzene copolymer. The catalytic performance of such catalysts did not depend on the concentration of bromine, but on the Br/Pd ratio. The catalysts performed better if a higher proportion of palladium species interacted with the sulphonate group.
Yook et al. systematically studied AuPd catalysts supported on different nanostructured carbon materials (activated carbon, carbon nanotubes, carbon black, and ordered mesoporous carbon) to understand the surface properties of the carbon support and the catalysis of the porous structure (Fig. 8a).53 The results showed that the high density of oxygen functional groups on the carbon surface was an important factor in obtaining an effective high dispersion bimetallic AuPd alloy catalyst (Fig. 8b and d). At the same time, single mesoporous supports were more effective than microporous supports, as the microporous structure leads to less efficient diffusion and slower conversion of H2 (Fig. 8c). Moreover, the H2O2 produced in the carbon micropores was more readily hydrogenated to produce water as a by-product, thus reducing the selectivity to H2O2. Ghedini et al. compared the performance of Pd-based catalysts with commercial SiO2, SBA-15, and MCM-41 for the DSHP.54 The results showed that the sample with MCM-41 as the support had good activity but poor selectivity and stability. This was due to its small pore size, with only small (approximately 2 nm) metal particles in the channel. In addition, the pore size of the SBA-15 support catalyst was able to accommodate most metals resulting in a high yield of H2O2, and the average particle size of Pd on SBA-15 was suitable to achieve a high metal dispersion. The TEM image taken along the (110) direction of the mesoporous silica shows typical Pd particles in the channel, indicating that most of the Pd is successfully placed in the channel of SBA-15.
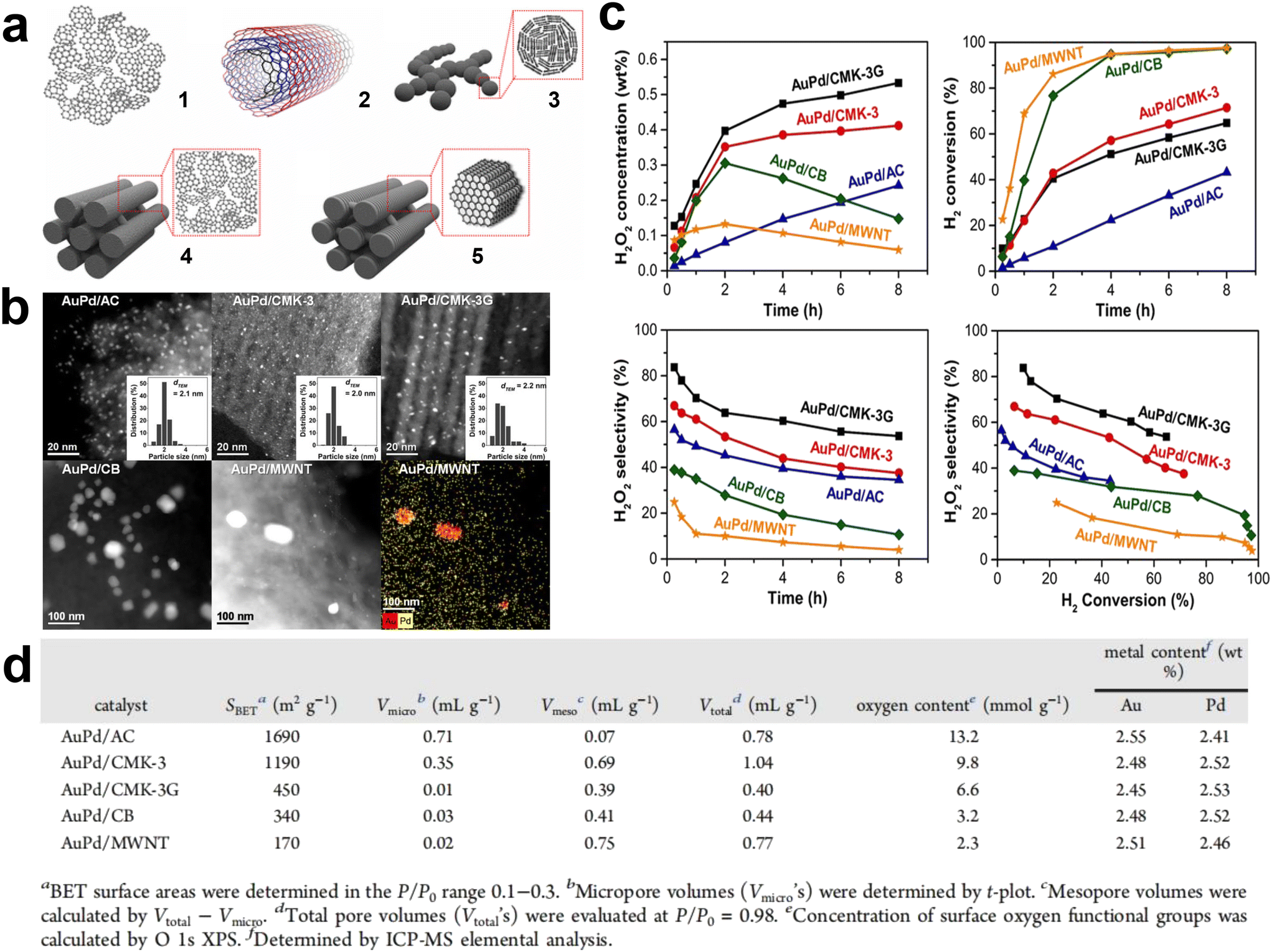 | ||
| Fig. 8 (a) Schematic diagrams of various nanostructured carbon materials: (1) activated carbon (AC), (2) multiwalled carbon nanotubes (MWNTs), (3) carbon black (CB), (4) hexagonally ordered mesoporous carbon (CMK-3), and (5) hexagonally ordered mesoporous carbon with graphitic framework (CMK-3G). (b) High-angle annular dark-field scanning transmission electron microscopy (HAADF-STEM) images. (c) Reaction performance diagram of direct synthesis of hydrogen peroxide. (d) Physical properties of AuPd catalysts.53 Copyright 2017, American Chemical Society. | ||
Lee et al. synthesized Pd core porous SiO2 shell catalysts (Pd@SiO2) of different sizes.55 The size of the catalyst could be adjusted by changing the ratio of polyvinylpyrrolidone (PVP) to Pd. The average particle size of Pd increases as the ratio decreases. The smaller the average particle size of Pd, the easier it was to dissociate the O–O bond and to promote the exposure of the more unsaturated energy centers of by-product water formation, reducing the H2O2 yield and selectivity. The selectivity and activity of the catalyst to generate H2O2 were related to the exposure of energy centers with a higher degree of unsaturation.
3.2 Optimization of the supported metal component
Landon et al. first proposed that Au catalysts have the activity of DSHP.56 Subsequently, Edwards et al. discussed the DSHP from Au, Pd, and Au–Pd catalysts supported on TiO2.57 The results showed that the performance of the Au–Pd catalyst was significantly better than that of the other two single metal catalysts. It had a core–shell structure with Pd distributed on the surface. The uncalcined catalyst had the best catalytic effect, but this type of catalyst was extremely unstable, and the active metal would fall off after the reaction. After high temperature calcination (400 °C), the stability of the catalyst was improved and the active metal was not easily lost and could be reused. This research has laid the foundation for subsequent research on multi-metal catalysts, especially bimetallic catalysts.
The bimetallic catalyst that has been extensively studied is the Pd–Au catalyst. The pathways by which Au doping improves catalyst performance have been extensively investigated and may be due to electronic, structural or segregation effects, and the synergistic effects observed experimentally may be the result of their combined action. Li et al. studied the DSHP on the surface of Pd (111) and Au@Pd (111), respectively.58 The competition between the primary and secondary reactions on the Pd (111) surface was actually between the O–O and O–Pd bonds; on the Au@Pd (111) surface, it was between the O–O and O–Au bonds. On the Au@Pd (111) surface, the weaker O–Au bond inhibited the dissociation of the O–O bond and facilitated the generation of H2O2. Kim et al. reported the experimental results of the DSHP with a Pd@Au core–shell structure catalyst (Fig. 9a–e).59 Pd@Au core–shell NPs induced in-plane compressive strains on the Au shell layer, facilitating charge transfer from Pd to Au. These promoted H2 dissociation but inhibited O–O bond dissociation, thereby increasing the productivity of hydrogen peroxide (Fig. 9f–k). This method of facilitating charge transfer by controlling lattice strain is a means of achieving multi-catalytic functional catalysts.
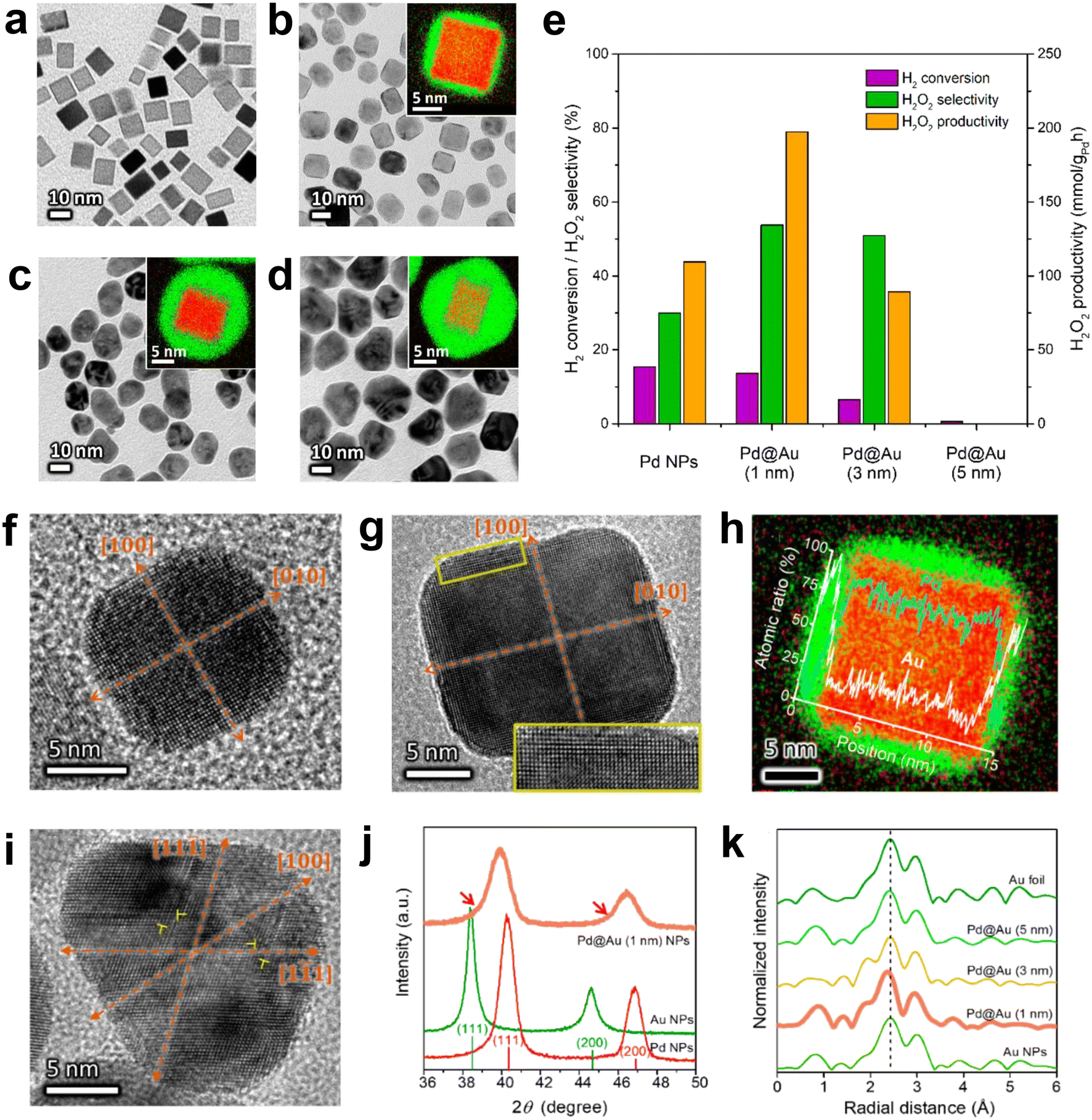 | ||
| Fig. 9 (a) Bright field TEM images of Pd, (b) Pd@Au (1 nm), (c) Pd@Au (3 nm) and (d) Pd@Au (5 nm). The inset is a high-resolution EDS image (green: Au, red: Pd) from each core–shell nanoparticle. (e) Evaluation of the catalytic performance of different NPs to produce hydrogen peroxide. HRTEM images of (f) Pd and (g) Pd@Au(1 nm) NPs on the [001] axis. (h) High-resolution EDS mapping of a Pd@Au (1 nm) NP and (i) an HRTEM image of a Pd@Au (3 nm) NP on the axis of the [011] area. Dislocations can be observed at the position indicated by the symbol ⊥. (j) Low-angle XRD spectra of Pd (red), Au (green) and Pd@Au (1 nm) (pink) NPs. (k) EXAFS radial distances of gold–gold atom pairs measured from different NPs.59 Copyright 2019, American Chemical Society. | ||
Xu et al. found that it was possible to modulate the valence electrons of the Pd atoms by doping with a second metal of suitable electronegativity, thereby improving the activity and selectivity of the catalyst nanoclusters.60 They have concluded that Pd–W, Pd–Pb, Au–Pd–Pb, Au–Pd–Mo and Au–Pd–Ru were all predicted catalysts with better performance. This work has provided a theoretical basis for future experimental testing of catalyst performance and provided a means to better design efficient catalysts. Han et al. used a simple seed-mediated-growth method to design and synthesize Pd-based core–shell nanoparticles (Pd@Pt) partially covered by Pt shells (Fig. 10a).61 The Pd–Pt alloys in Pd@Pt core–shell nanoparticles were located on a platform of nanocubes with high conversion to hydrogen and selectivity to H2O2 (Fig. 10b). In addition, direct seed-mediated growth methods could successfully modulate the geomorphology and composition of bimetallic core–shell nanoparticles more easily than some other routes for the synthesis of core–shell nanoparticles. Gong et al. incorporated a small amount of Pt into the TiO2 supported Pd–Au catalyst to improve the selectivity to H2O2.62 This was attributed to the change in the oxidation state of Pd and the formation of mixed Pd2+–Pd0 domains. Recently, Kim et al. reported that high index crystal planes are generated during the anisotropic growth of Pd@Pt nanocrystals. They provide many active sites for H2 dissociation, significantly facilitating the DSHP.63 This work provided an alternative way of thinking about how to improve the catalytic performance of core@shell nanocrystals by creating high index crystalline planes. Zhang et al. also reported that the addition of Pt single atoms to fully exposed Pd clusters changes the electronic structure and improves the H2O2 yield and selectivity, with traces of Pt single atoms acting as electron promoters.64
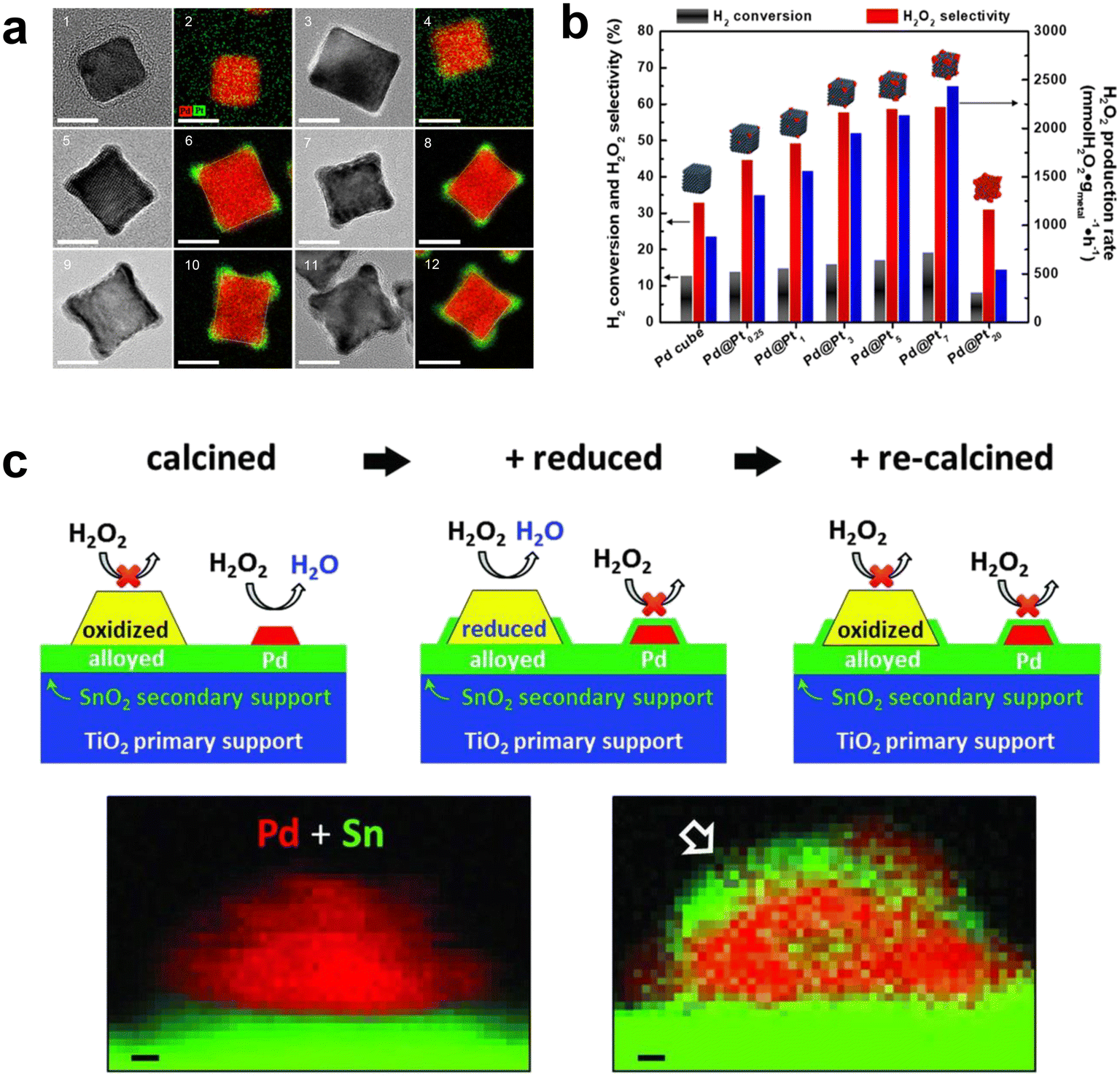 | ||
| Fig. 10 (a) HRTEM and EDS mapping images of Pd@Pt0.25 (1, 2), Pd@Pt1 (3, 4), Pd@Pt3 (5, 6), Pd@Pt5 (7, 8), Pd@Pt7 (9, 10), and Pd@Pt20 concave cubes (11, 12). The scale bar is 10 nm. (b) DSHP results for the Pd@PtN (N = 0, 0.25, 1, 3, 5, 7, and 20) nanoparticles.61 Copyright 2020, American Chemical Society. (c) The role of Sn in improving the activity of Pd particles: forming an alloy with large particles of Pd, and at the same time, SnOx encapsulates small Pd particles that cause H2O2 degradation. STEM-EELS mapping of a model catalyst in the oxidation and O–R–O stages of 5 wt% Pd/SnO2 shows that SnOx (green) partially encapsulates PdNP (red) after the O–R–O heat treatment cycle.66 Copyright 2016, American Association for the Advancement of Science. | ||
Recently, single atom Pd catalysts have been reported to be highly selective and active, with H2O2 yields up to 115 mol g−1 Pd h−1, H2O2 selectivity above 99% and H2O2 concentrations up to 1.07 wt% in a batch.65 Density functional theory calculations further demonstrated that O–O bond cleavage was significantly inhibited at single Pd atoms and that O2 was more readily activated to form OOH*. The OOH* is a key intermediate in the synthesis of H2O2, which effectively inhibits the degradation of H2O2. This report has received widespread attention, and perhaps in the future, single atom catalysts will be a major breakthrough in the field of the DSHP.
In recent years, breakthroughs have been made in some research on non-precious metal Pd-based catalysts. Freakley et al. reported that the Pd–Sn/TiO2 catalyst was more than 95% selective for H2O2 after a special heat treatment cycle.66 Small Pd-rich particles active for H2O2 degradation could be encapsulated in SnOx through a calcination–reduction–calcination heat treatment cycle, while larger Pd–Sn alloy particles were exposed to facilitate H2O2 synthesis (Fig. 10c). More importantly, this method was also applicable to the combination of Pd and other alkali metals (Ni, Zn, Ga, In and Co). Li et al. developed a two-step method to prepare Pd oxide encapsulated on PdSn nanowires, which showed an H2O2 yield of 528 mol kg−1 and H2O2 selectivity of >95% in the DSHP.67 The palladium oxide layer on PdSn nanowires generates doubly coordinated Pd, which leads to different adsorption behaviors of O2, H2, and H2O2 on PdL/PdSn NW, providing new insights into the rational design of efficient catalysts for the DSHP (Fig. 11).
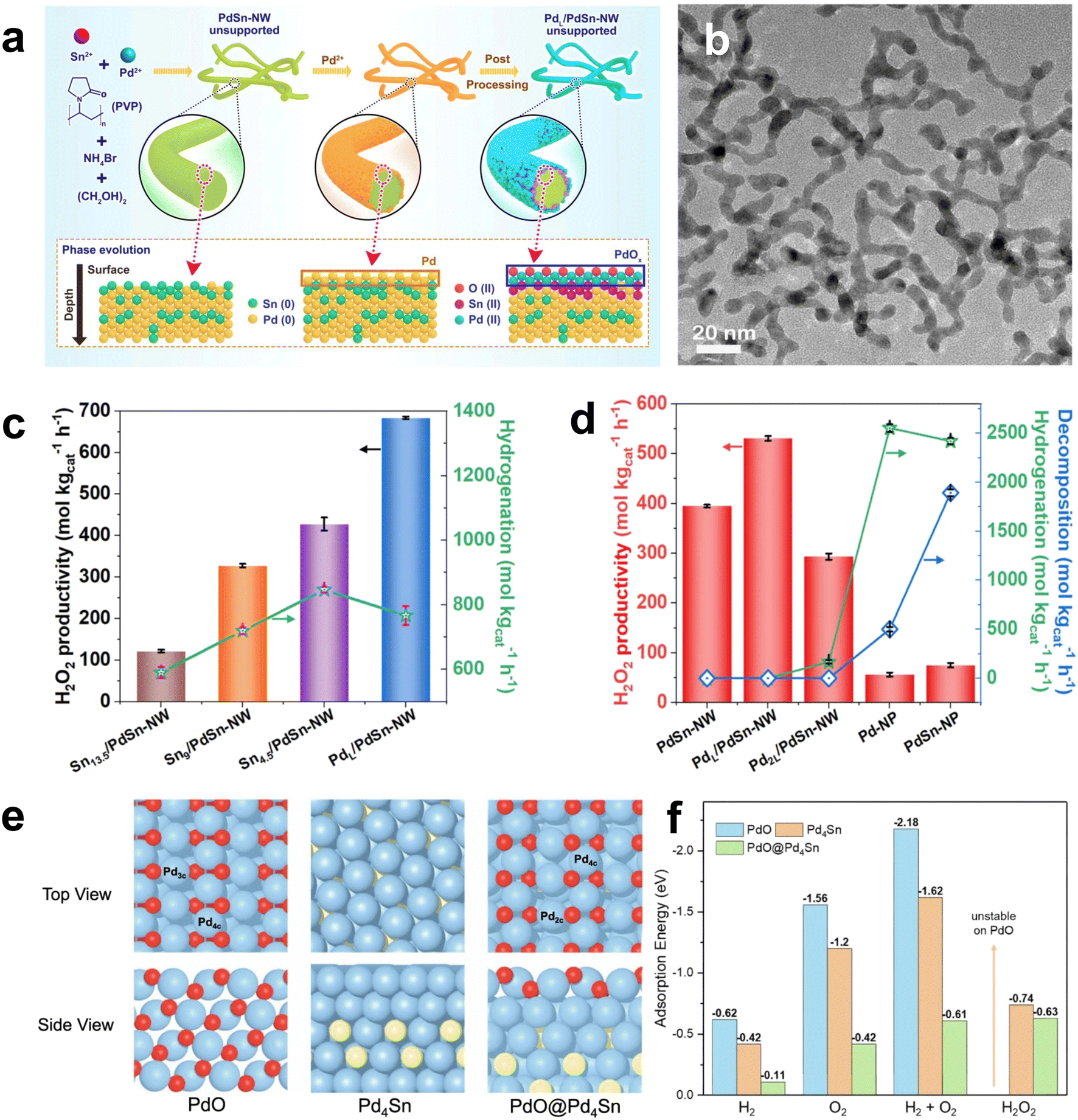 | ||
| Fig. 11 (a) Schematic illustration of the synthesis of the unsupported PdSn-NW and PdL/PdSn-NW. (b) Representative TEM image of the unsupported PdSn nanowire catalyst prepared by two steps (unsupported PdL/PdSn-NW). (c) H2O2 producibility of the supported PdL/PdSn-NW and Snx/PdSn-NW catalysts with different Pd/Sn ratios after annealing in air (350 °C, 8 min), demonstrating that the addition of Sn has a negative effect on the producibility of H2O2. (d) The comparison of the H2O2 producibility, hydrogenation, and decomposition of the supported PdL/PdSn-NW catalyst with other Pd catalysts. (e) DFT optimized structures of PdO (101), Pd4Sn, and PdO@Pd4Sn with (f) adsorption energies of H2, O2, H2 + O2, and H2O2 for O2 reduction by the surface hydrides on these three models.67 Copyright 2022, Springer Nature. | ||
Wang et al. prepared monometallic Pd catalysts and a series of bimetallic PdM (M = Ni, Zn, Ga, Sn, In, Pb) nanocrystalline (NCS) catalysts using acid-pretreated TiO2 as a support, all of which have spherical shapes with similar size distributions.68 There were no reflection features between ordered metals in the XRD pattern, indicating that M (Ni, Zn, Ga, In) is randomly distributed at the (fcc) Pd phase. The PdSn and PdPb bimetallic NCs showed distinct reflections different from the (fcc) Pd phase and were able to form new crystals in a thermally stable manner. It is noteworthy that non-precious metal dopants have successfully modified the electronic/geometric structure of Pd NCs, thus achieving significant results without the use of any reactive additives (acids and halide ions). The PdGa/s-TiO2 and PdSn/s-TiO2 catalyzed H2O2 yields were 2.5–3 times higher than those of single metal Pd catalysts, and the selectivity to H2O2 was significantly improved. In addition, Wang et al. also prepared PdGa/TiO2 and PdIn/TiO2 catalysts for the DSHP.69 Their method of catalyst preparation was an acid-washed sol-immobilisation procedure, which allowed particle size control. They believed that the doping of Ga and Te inhibited the decomposition of H2O2 and improved the catalytic performance by adjusting the ratio of the oxidation state of Pd. Zhang et al. prepared W modified Pd/Al2O3 to improve the synthesis of H2O2. The W was present on the surface of Pd particles in the form of WO3, optimizing the PdO to Pd ratio and increasing the productivity of H2O2.70 They also found a strong interaction with PdAu induced by W, with WO3 species partially enveloping PdAu particles and inducing the formation of surface PdO, improving the catalytic performance for the formation of H2O2.71
Tian and his colleagues reported the DSHP using a Pd–Te bimetallic catalyst (Pd–Te/Al2O3) supported on Al2O3 under mild reaction conditions.72 The yield of the bimetallic catalyst (Pd–Te/Al2O3) catalyzing H2O2 synthesis was significantly improved by doping with metallic Te. In addition, the selectivity for H2O2 has increased with the gradual increase in Te content. They proposed that Te doping blocks the Pd high-energy step center, which has been shown to be highly active for the dissociation of O2, and forms a Pd–Te bimetallic site to facilitate the synthesis of H2O2. Liang et al. synthesized a Pd–Ce/AC catalyst for the DSHP at room temperature.73 The experimental results have shown that the doping effect reduces the particle size of Pd and increases the Pd2+ content, which makes the yield of H2O2 more appreciable. Freakley et al.74 found that 4.5% Ni–0.5% Pd/TiO2 catalysts could achieve higher H2O2 selectivity under favorable reaction conditions. Recently, the Pb doping and nanocrystal shape have been investigated for their influence on the catalytic performance in the DSHP. The results showed that the catalytic efficiency of DSHP is not only influenced by the nanocrystal composition, but also depends on the particle shape.75 Pd3Pb cubes were preferred over other catalysts due to the electronic modification of the Pd surface atoms, which provides a higher potential barrier to O2 dissociation on Pd3Pb, and the lack of larger Pd particles. Tran et al. synthesized FePd catalysts supported on graphite using the microwave irradiation method.76 The geometric and electronic properties of Pd on the carbon surface are affected during the reduction by microwave irradiation, thus affecting the catalytic activity of the DSHP.
Table 1 below compares the performance of Pd-based catalysts doped with various other metals for the DSHP. The introduction of a second metal to enhance the performance of the DSHP is attributed to various structural changes, including the distribution of active sites (single Pd site and continuous Pd–Pd sites), electronic recombination of active sites, lattice strain, and the formation of other special substances that are conducive to the synthesis of H2O2. The rational use of these factors can significantly affect the catalytic activity and provide applicability to multidisciplinary scientific fields.
| Active phase | Support | Solvent | T (°C) | P (bar) | Time (h) | H2O2 selectivity (%) | mol H2O2 kgPd−1 h−1 |
|---|---|---|---|---|---|---|---|
| Description: Ti-NT titanate nanotubes, AC activated carbon, OAC oxidized activated carbon, SZ sulfated zirconia, Rb2.5H0.5PW12O40, Cs2.8H0.2PW12O40 heteropolyacids. | |||||||
| 0.1%O–Pd/TiO2 (ref. 65) | TiO2 | MeOH/H2O | 2 | 42 | 30 | >99 | 115![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 000 |
| 1%Pd–IPr (1:1)/TiO2 (ref. 79) |
TiO2 | MeOH/H2O | 2 | 40 | 0.5 | — | 16000 |
| Pd/Ag 40/1 (ref. 104) | AC | MeOH/H2SO4 | 2 | 30 | 0.25 | 71 | 7022 |
| 1.1%Pd + 0.4%Au (ref. 105) | Ti-NT | MeOH/H2O | 5 | 40 | 0.66 | — | 15818 |
| 1.25%Pd + 1.25%Au (ref. 106) | SZ | MeOH/H2SO4 | 20 | 1 | 3 | 61 | 1270 |
| 2%Pd + 1%Au (ref. 41) | TiO2 | EtOH/H2SO4 | 10 | — | — | 48 | 3495 |
| 2.5%Pd + 2.5%Au (ref. 44) | AC | MeOH/H2O | 2 | 37 | 0.5 | >95 | 320 |
| 2.5%Pd + 2.5%Au (ref. 107) | Carbon | MeOH/H2O | 2 | 37 | 0.5 | 80 | 4400 |
| 2.5%Pd + 2.5%Au (ref. 108) | Rb2.5H0.5PW12O40 | MeOH/H2O | 2 | 40 | 0.5 | — | 10080 |
| 2.5%Pd + 2.5%Au (ref. 109) | Cs2.8H0.2PW12O40 | MeOH/H2O | 2 | 40 | 0.5 | — | 7920 |
| 1%Pd + 2%Au (ref. 110) | OAC | MeOH | 0 | 38 | 3 | 63 | 51400 |
| 3.3%Pd + 1.7%Au (ref. 29) | SiO2 | MeOH/H2SO4 | 25 | 20 | 1 | 35 | 864 |
| 3.3Pd% + 5.3%Au (ref. 111) | SiO2 | EtOH/HCl | 10 | 1 | 5 | 59 | 1890 |
| 1.3%Pd + 0.2%Pt (ref. 112) | SZ | MeOH/H2SO4 | 20 | 1 | 5 | 70 | 1236 |
| 3.3%Pd + 0.2%Pt (ref. 113) | SiO2 | EtOH/HCl | 10 | 1 | 5 | 63 | 1770 |
| 1%Pd + 4%Sn (ref. 66) | TiO2 | MeOH/H2O | 2 | 40 | 0.5 | >95 | 5000 |
| 1%Pd + 5%Zn (ref. 114) | Al2O3 | MeOH/H2SO4 | 2 | 30 | 0.25 | 78.5 | 25431 |
| 0.2%Au + 4.6%Pd + 0.2%Pt (ref. 115) | TiO2 | MeOH/H2O | 2 | 33 | 0.5 | — | 4000 |
| 4.5%Pd + 0.45%Ru + 0.05%Au (ref. 116) | TiO2 | MeOH/H2O | 2 | 40 | 0.5 | — | 3400 |
| Pt0.006Pd/TiO2 NCs64 | TiO2 | MeOH/HCl | 0 | 40 | 0.5 | 86.5 | 37300 |
3.3 Heteroatom doping
To solve the H2O2 yield problem, the addition of other elements is also a more economical option.It is well known that controlling the interaction of the metal–support is one of the most effective ways of optimizing supported catalysts. Liu et al. effectively improved the yield and selectivity of DSHP by doping B atoms between Pd and TiO2 supports.77 The in-depth research results show that the doped B atoms change the proportion of Pd2+, providing more active sites for O2 non-dissociative activation. In addition, the electronic effect between B and Pd increases the adsorption and activation of H2, leading to the improvement of the DSHP performance. This work provides an effective and economical way to improve the performance of supported catalysts through interface doping. Moreover, Wang et al. developed S-doped Pd/C catalysts where the sulfur species promoted the in situ formation of Pd nanoparticles. This enhanced their dispersion on the support, significantly improving the catalytic performance of the DSHP.78
In addition, Lewis et al. found that the supported Pd catalyst modified with N-heterocyclic carbene (NHC) had significant effects on the synthesis of H2O2 from DSHP.79 And by carefully selecting the N-substituents, the catalytic performance can be greatly improved compared to unmodified catalysts, and a significant concentration of H2O2 can be achieved. The ability of this NHC ligand as an electronic modifier for Pd is similar to modifying Pd species electronically by introducing secondary metals. It is worth noting that this is an effective new method for synthesizing H2O2.
4 Application of H2O2 as an in situ oxidant
4.1 Propylene epoxidation to propylene oxide
Propylene oxide (PO) is one of the most important raw materials for industrial chemistry. It is mainly used in the production of various non-ionic surfactants, polyurethanes, resins and other chemicals. It is important in the petroleum, chemical, pesticide, and textile industries. At present, the main methods of industrial production of PO are the co-oxidation method and chlorohydrin method,80 but the chlorohydrin method could consume a lot of water resources and produce a lot of wastewater and waste residues. The equipment investment of the co-oxidation method is relatively large, and the process flow is complicated.81 However, the hydrogen peroxide direct oxidation method (HPPO) is a new process in which H2O2 catalyzes the epoxidation of propylene to PO. Only propylene oxide and water are produced in the production process. Therefore, the process is simple, the product yield is high, and it is basically pollution-free, showing significant environmental and economic advantages (Fig. 12a).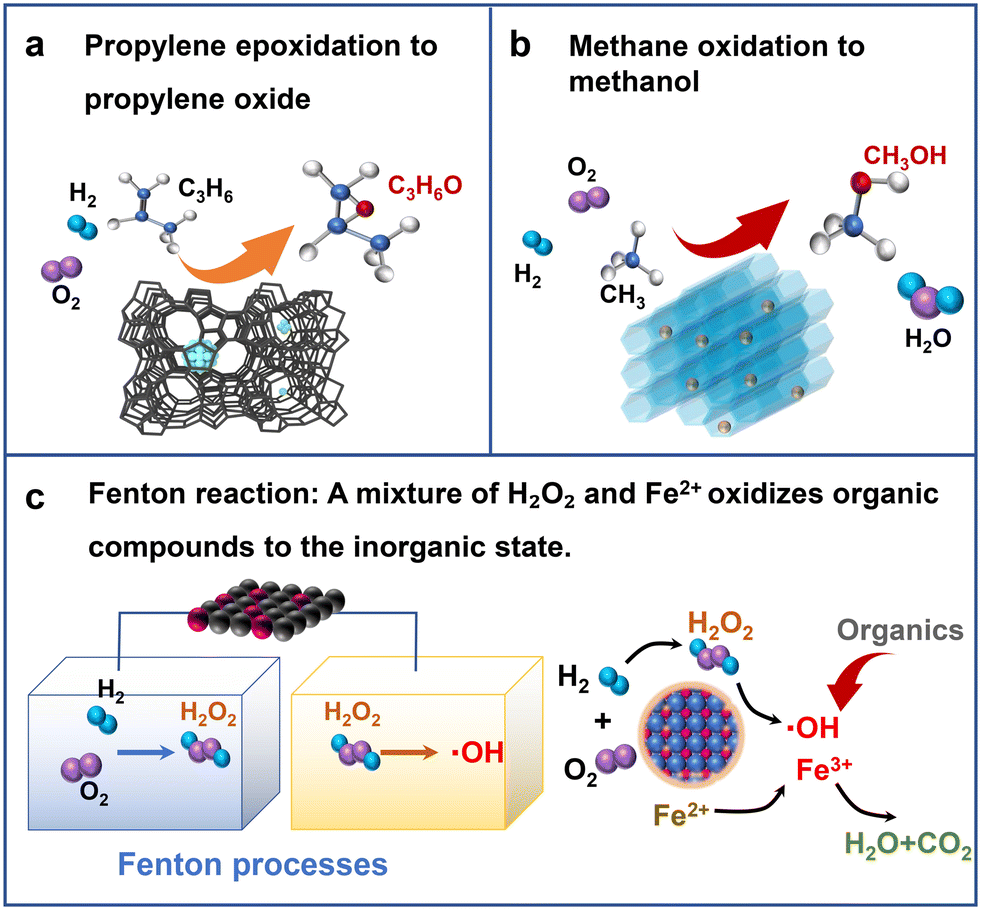 | ||
| Fig. 12 Schematic diagram of (a) propylene epoxidation to epoxypropane, (c) Fenton reaction, and (b) methane oxidation to methane. | ||
At present, the industrial HPPO process uses H2O2 solution and propylene (C3H5) gas as raw materials, and selects a suitable titanium–silicon catalyst to perform the oxidation reaction in water or methanol reaction solution. However, a nearby H2O2 production plant is required to avoid transportation costs. Therefore, exploring the in situ oxidation of propylene using H2O2 as a feedstock with H2, O2 and C3H5 has advantages in terms of economy, environmental protection and future development. Reports have shown that TS-1,82–87 Ti-MCM-41,88–90 and their oxide supports91–93 loaded with precious metals as active sites, in particular Pd, Au, Pt or their combination, form a bifunctional catalyst.
However, the catalytic selectivity to propylene oxide is still a common problem. Chen et al. found that when ammonium acetate is used as the inhibitor, the selectivity to PO can reach 81.8% by using 0.2% Pd/TS-1 and 0.02 Pt%/TS-1 catalysts in a compressed carbon dioxide medium.94 In addition, the addition of inorganic salts such as Cs2CO3,95 CsNO3,96 and CsCl (ref. 97) to the reaction system could inhibit propylene hydrogenation, PO hydrolysis, and PO with methanol and improve the yield of PO. The focus of future research is to achieve high selectivity for PO without a promoter.
4.2 Fenton reaction
The Fenton reaction refers to passing organic pollutants through a mixed solution of H2O2 and Fe2+, and then H2O2 undergoes disproportionation to generate hydroxyl radicals and other active oxygen species with strong oxidizing ability (Fig. 12c). Then, chemical methods are used to oxidize and decompose organic matter under mild conditions. It is suitable for the degradation of organic pollutants in low to medium concentration wastewater and is a high potential advanced catalytic oxidation technology.Esplugas et al. studied and compared the effects of different catalytic systems, such as ozone, ultraviolet light, H2O2, Fe2+ and photocatalysis, on the oxidative degradation of phenol.98 The Fenton reagent showed the greatest degradation rate for phenol. When H2O2 was selected as the oxidant, the only oxidation products were environmentally friendly species such as oxygen and water. This process displayed the advantages of simple operation, non-toxicity, fast reaction, and complete degradation. Commercially, it is mostly high concentration H2O2 produced by the anthraquinone method, and its transportation and dilution costs are relatively high. Therefore, exploring the technology of direct in situ generation of H2O2 for Fenton wastewater treatment is important and has great advantages. Osegueda et al. prepared a membrane catalytic reactor with Pd as the active species for the DSHP.99 They found that the reaction system showed good phenol oxidation activity after the addition of Fe2+. Therefore, it was proved that the in situ H2O2 generation technology directly used the feasibility of the Fenton reaction. Underhill et al. used a loaded Pd–Fe bimetallic catalyst for in situ phenol degradation compared to commercial H2O2 addition and found the former to be more effective and active.100 However, the intermediate products of phenol oxidation have caused the loss of some of the active ingredient Fe, affecting its stability and longevity. In future studies, the catalyst life and stability can be improved by optimizing the catalyst or improving the reaction system, thus maintaining the high degradation activity of phenol.
4.3 Methane oxidation to methanol
Methane is widely distributed and abundant in nature. The selective conversion of methane molecules into liquid fuels and other chemical products allows for the efficient use of natural gas. This is important for the restructuring of energy sources and the realization of green chemical production. Among these, the direct partial oxidation of methane to methanol is always a challenging subject. The current production conditions for the industrial conversion of methane to methanol are relatively harsh, and the process requirements are high. Although a certain selectivity can be achieved, the cost is high and pollutants are generated, which is not conducive to the development of long-term green chemical production.Therefore, research into new and friendly green oxidants (such as H2O2) to replace common oxidants such as concentrated sulfuric acid or O2 to realize the selective oxidation of methane to produce methanol under mild reaction conditions is of great economic and environmental significance (Fig. 12b). Rahim et al. first proposed that the supported Au–Pd/TiO2 bimetallic catalyst still exhibited good catalytic performance for the DSHP without a promoter.101 Subsequently, Rahim et al. used it for in situ methane oxidation to methanol.102 The in situ synthesis of H2O2 was more conducive to the capture of O2 and the generation of reactive oxygen species in the liquid phase than the addition of commercial H2O2. Therefore, in situ oxidation of methane to methanol could display effective oxidation efficiency. Recently, Jin et al. encapsulated Pd–Au alloy nanoparticles inside silicate molecular sieves by hydrophobic treatment of the support with organosilanes.103 H2 and O2 entered the zeolite crystals through mass transfer to form H2O2. The hydrophobic groups in the outer layer of the zeolite could hinder the diffusion of H2O2 and increase the local concentration of H2O2. At this point, the methane molecules can pass through the hydrophobic layer into the interior of the zeolite to react with the H2O2. This method significantly improved the reaction efficiency, resulting in a methanol yield of 91.6 mmol h−1 g−1.
At present, some progress has been made in the research on the production of methanol from the original oxidized methane, but the yield of methanol is still very low and far from what is required for industrial applications. Therefore, it is still a major challenge for future research to increase the methanol yields by designing and developing highly active catalysts or improving reactors and process routes.
5 Conclusions and future perspectives
The direct synthesis of hydrogen peroxide (DSHP) is more economical and environmentally friendly than other commercial methods for producing H2O2. However, the process of the DSHP has a long distance to realize industrialization. So far, the research in this field has made great progress. This review focused on optimizing Pd catalysts from the following aspects: firstly, the catalytic mechanism of Pd-based catalysts was analyzed, and the catalytically active sites were summarized. Secondly, at the macro level of catalyst design, including the optimization of the support, the introduction of other metals and the incorporation of heteroatoms, the performance of the DSHP can be optimized and improved. By adjusting the support of the catalyst, the dispersion and electronic state of Pd could be changed to adjust the performance of the catalyst. The introduction of other metals can adjust the electronic structure and geometric structure of the active component to inhibit the side reaction through a synergistic effect. Finally, the reaction effect of the DSHP could be further optimized by selecting a suitable solvent and carrying out the reaction under appropriate reaction conditions.However, in order to realize the industrialization of DSHP technology, the selectivity and activity of the catalyst still need to be further improved, and comprehensive issues such as cost must be considered. In the future, it will be necessary to develop new Pd-based catalysts that meet the needs of industrialization and have high economic benefits. Through the research of this review, we may start from the following aspects: (1) strengthening the research on the active sites of Pd-based catalysts and fundamentally clarifying the reaction mechanism of DSHP will provide important guidance for the future design of new Pd-based catalysts. (2) In order to achieve a higher level of economic benefits, doping with non-noble metals is an important direction. The electronic structure and geometric structure are adjusted by synergy to optimize the reaction performance. In addition, single-atom catalysts may lead to unexpected breakthroughs. (3) The reaction performance can also be effectively optimized through the adjustment and treatment of the support. (4) Minimize the use of additives to avoid excessive costs and pollution in the industrial process.
Finally, this work also reviewed the in situ oxidation reactions of H2O2, including propylene epoxidation to propylene oxide, the Fenton reaction, and methane oxidation to methanol. Compared with the processes commonly used in modern industry, the reaction conditions for the selective in situ oxidation of H2O2 generated by H2 and O2 are mild and environmentally friendly. The reaction process could be effectively controlled, significantly reducing production and equipment costs. Through the development and design of high-efficiency multifunctional catalysts, technological processes and equipment, the exploration of in situ oxidation systems by using H2, O2, and organic materials as raw materials displays important research significance from the perspective of both the chemical market and safety production.
Conflicts of interest
The authors declare no competing financial interest.Acknowledgements
This work is supported by the National Key R&D Program of China (2021YFB3801600) and the National Natural Science Foundation of China (22078005).References
- Y. Jiao, D. Shao, S. Chang, C. Xu and D. J. C. Hinks, Pilot-plant investigation on low-temperature bleaching of cotton fabric with TBCC-activated peroxide system, Cellulose, 2017, 24, 1–9 CrossRef.
- A. Babuponnusami and K. Muthukumar, A review on Fenton and improvements to the Fenton process for wastewater treatment, J. Environ. Chem. Eng., 2014, 2, 557–572 CrossRef CAS.
- S. Mohammadi, A. Kargari, H. Sanaeepur, K. Abbassian, A. Najafi and E. Mofarrah, Phenol removal from industrial wastewaters: A short review, Desalin. Water Treat., 2015, 53, 2215–2234 CrossRef CAS.
- Y. Jiang, P. Ni, C. Chen, Y. Lu, P. Yang, B. Kong, A. Fisher and X. Wang, Selective electrochemical H2O2 production through two-electron oxygen electrochemistry, Adv. Energy Mater., 2018, 8, 1801909 CrossRef.
- S. Fukuzumi, Y. Yamada and K. D. Karlin, Hydrogen peroxide as a sustainable energy carrier: Electrocatalytic production of hydrogen peroxide and the fuel cell, Electrochim. Acta, 2012, 82, 493–511 CrossRef CAS PubMed.
- A. B. Sorokin and E. V. Kudrik, Phthalocyanine metal complexes: Versatile catalysts for selective oxidation and bleaching, Catal. Today, 2011, 159, 37–46 CrossRef CAS.
- R. V. Ottenbacher, E. P. Talsi and K. P. Bryliakov, Recent progress in catalytic oxygenation of aromatic C–H groups with the environmentally benign oxidants H2O2 and O2, Appl. Organomet. Chem., 2020, 34, e5900 CrossRef CAS.
- B. Lu, N. Cai, J. Sun, X. Wang, X. Li, J. Zhao and Q. Cai, Solvent-free oxidation of toluene in an ionic liquid with H2O2 as oxidant, Chem. Eng. J., 2013, 225, 266–270 CrossRef CAS.
- T. T. Ponduru, C. Qiu, J. X. Mao, A. Leghissa, J. Smuts, K. A. Schug and H. V. R. Dias, Copper(I)-based oxidation of polycyclic aromatic hydrocarbons and product elucidation using vacuum ultraviolet spectroscopy and theoretical spectral calculations, New J. Chem., 2018, 42, 19442–19449 RSC.
- Y. Fang, J. Li, N. Sheng, X. Wang, D. Chen, M. Cai, Y. An, Y. Chen and L. Dai, Enhanced catalytic oxidation of anthracene by deposition of MoO3 and WO3 nanoparticles on MCM-41, Mol. Catal., 2020, 497, 111209 CrossRef CAS.
- R. Ciriminna, L. Albanese, F. Meneguzzo and M. J. C. Pagliaro, Inside back cover: Hydrogen peroxide: A key chemical for today's sustainable development, ChemSusChem, 2016, 9, 3527–3527 CrossRef CAS.
- G. Mamba, P. J. Mafa, V. Muthuraj, A. Mashayekh-Salehi, S. Royer, T. I. T. Nkambule and S. Rtimi, Heterogeneous advanced oxidation processes over stoichiometric ABO3 perovskite nanostructures, Mater. Today Nano, 2022, 18, 100184 CrossRef CAS.
- J. M. Campos-Martin, G. Blanco-Brieva and J. L. G. Fierro, Hydrogen peroxide synthesis: An outlook beyond the anthraquinone process, Angew. Chem., Int. Ed., 2006, 45, 6962–6984 CrossRef CAS PubMed.
- J. K. Edwards, S. J. Freakley, R. J. Lewis, J. C. Pritchard and G. J. Hutchings, Advances in the direct synthesis of hydrogen peroxide from hydrogen and oxygen, Catal. Today, 2015, 248, 3–9 CrossRef CAS.
- S. Anantharaj, S. Pitchaimuthu and S. Noda, A review on recent developments in electrochemical hydrogen peroxide synthesis with a critical assessment of perspectives and strategies, Adv. Colloid Interface Sci., 2021, 287, 102331 CrossRef CAS PubMed.
- J. An, Y. Feng, Q. Zhao, X. Wang, J. Liu and N. Li, Electrosynthesis of H2O2 through a two-electron oxygen reduction reaction by carbon based catalysts: From mechanism, catalyst design to electrode fabrication, Environ. Sci. Ecotechnology, 2022, 11, 100170 CrossRef CAS PubMed.
- R. J. Lewis and G. J. Hutchings, Recent advances in the direct synthesis of H2O2, ChemCatChem, 2019, 11, 298–308 CrossRef CAS.
- M. G. Seo, H. J. Kim, S. S. Han and K. Y. Lee, Direct synthesis of hydrogen peroxide from hydrogen and oxygen using tailored Pd nanocatalysts: a review of recent findings, Catal. Surv. Asia, 2017, 21, 1–12 CrossRef CAS.
- S. Ranganathan and V. Sieber, Recent advances in the direct synthesis of hydrogen peroxide using chemical catalysis-a review, Catalysts, 2018, 8(9), 379 CrossRef.
- N. M. Wilson and D. W. Flaherty, Mechanism for the direct synthesis of H2O2 on Pd clusters: Heterolytic reaction pathways at the liquid–solid interface, J. Am. Chem. Soc., 2016, 138, 574–586 CrossRef CAS PubMed.
- T. Deguchi, H. Yamano and M. Iwamoto, Kinetic and mechanistic studies on direct H2O2 synthesis from H2 and O2 catalyzed by Pd in the presence of H+ and Br− in water: A comprehensive paper, Catal. Today, 2015, 248, 80–90 CrossRef CAS.
- D. W. Flaherty, Direct synthesis of H2O2 from H2 and O2 on Pd catalysts: Current understanding, outstanding questions, and research needs, ACS Catal., 2018, 8, 1520–1527 CrossRef CAS.
- N. M. Wilson, J. Schröder, P. Priyadarshini, D. T. Bregante, S. Kunz and D. W. Flaherty, Direct synthesis of H2O2 on PdZn nanoparticles: The impact of electronic modifications and heterogeneity of active sites, J. Catal., 2018, 368, 261–274 CrossRef CAS.
- J. S. Adams, A. Chemburkar, P. Priyadarshini, T. Ricciardulli, Y. Lu, V. Maliekkal, A. Sampath, S. Winikoff, A. M. Karim, M. Neurock and D. W. Flaherty, Solvent molecules form surface redox mediators in situ and cocatalyze O2 reduction on Pd, Science, 2021, 371, 626–632 CrossRef CAS.
- A. Akram, G. Shaw, R. J. Lewis, M. Piccinini, D. J. Morgan, T. E. Davies, S. J. Freakley, J. K. Edwards, J. A. Moulijn and G. J. Hutchings, The direct synthesis of hydrogen peroxide using a combination of a hydrophobic solvent and water, Catal. Sci. Technol., 2020, 10, 8203–8212 RSC.
- V. R. Choudhary, S. D. Sansare and A. G. Gaikwad, Direct oxidation of H2 to H2O2 and decomposition of H2O2 over oxidized and reduced Pd-containing zeolite catalysts in acidic medium, Catal. Lett., 2002, 84, 81–87 CrossRef CAS.
- X. Song, K. Sun, X. Hao, H.-Y. Su, X. Ma and Y. Xu, Facet-dependent of catalytic selectivity: The case of H2O2 direct synthesis on Pd surfaces, J. Phys. Chem. C, 2019, 123, 26324–26337 CrossRef CAS.
- F. Wang, C. Xia, S. P. de Visser and Y. Wang, How does the oxidation state of palladium surfaces affect the reactivity and selectivity of direct synthesis of hydrogen peroxide from hydrogen and oxygen gases? A Density Functional Study, J. Am. Chem. Soc., 2019, 141, 901–910 CrossRef CAS.
- S. Kanungo, L. van Haandel, E. J. M. Hensen, J. C. Schouten and M. F. Neira d'Angelo, Direct synthesis of H2O2 in AuPd coated micro channels: An in-situ X-Ray absorption spectroscopic study, J. Catal., 2019, 370, 200–209 CrossRef CAS.
- Q. Liu, K. K. Gath, J. C. Bauer, R. E. Schaak and J. H. Lunsford, The active phase in the direct synthesis of H2O2 from H2 and O2 over Pd/SiO2 catalyst in a H2SO4/ethanol system, Catal. Lett., 2009, 132, 342–348 CrossRef CAS.
- D. P. Dissanayake and J. H. Lunsford, Evidence for the role of colloidal palladium in the catalytic formation of H2O2 from H2 and O2, J. Catal., 2002, 206, 173–176 CrossRef CAS.
- P. Priyadarshini and D. W. Flaherty, Form of the catalytically active Pd species during the direct synthesis of hydrogen peroxide, AIChE J., 2019, 65, e16829 CrossRef CAS.
- L. Chen, J. W. Medlin and H. Grönbeck, On the reaction mechanism of direct H2O2 formation over Pd catalysts, ACS Catal., 2021, 11, 2735–2745 CrossRef CAS.
- L. Ouyang, P. F. Tian, G. J. Da, X. C. Xu, C. Ao, T. Y. Chen, R. Si, J. Xu and Y. F. Han, The origin of active sites for direct synthesis of H2O2 on Pd/TiO2 catalysts: Interfaces of Pd and PdO domains, J. Catal., 2015, 321, 70–80 CrossRef CAS.
- C. Ao, P. Tian, L. Ouyang, G. Da, X. Xu, J. Xu and Y.-F. Han, Dispersing Pd nanoparticles on N-doped TiO2: A highly selective catalyst for H2O2 synthesis, Catal. Sci. Technol., 2016, 6, 5060–5068 RSC.
- T.-T. Huynh, W.-H. Huang, M.-C. Tsai, M. Nugraha, S.-C. Haw, J.-F. Lee, W.-N. Su and B. J. Hwang, Synergistic hybrid support comprising TiO2–Carbon and ordered PdNi alloy for direct hydrogen peroxide synthesis, ACS Catal., 2021, 11, 8407–8416 CrossRef CAS.
- F. Li, Q. Shao, M. Hu, Y. Chen and X. Huang, Hollow Pd–Sn nanocrystals for efficient direct H2O2 synthesis: The critical role of sn on structure evolution and catalytic performance, ACS Catal., 2018, 8, 3418–3423 CrossRef CAS.
- M. Ledendecker, E. Pizzutilo, G. Malta, G. V. Fortunato, K. J. J. Mayrhofer, G. J. Hutchings and S. J. Freakley, Isolated Pd sites as selective catalysts for electrochemical and direct hydrogen peroxide synthesis, ACS Catal., 2020, 10, 5928–5938 CrossRef CAS.
- T. Yang, C. Yang, J. Le, Z. Yu, L. Bu, L. Li, S. Bai, Q. Shao, Z. Hu, C.-W. Pao, J. Cheng, Y. Feng and X. Huang, Atomically isolated Pd sites within Pd-S nanocrystals enable trifunctional catalysis for direct, electrocatalytic and photocatalytic syntheses of H2O2, Nano Res., 2022, 15, 1861–1867 CrossRef CAS.
- D. Ding, X. Xu, P. Tian, X. Liu, J. Xu and Y.-F. Han, Promotional effects of Sb on Pd-based catalysts for the direct synthesis of hydrogen peroxide at ambient pressure, Chin. J. Catal., 2018, 39, 673–681 CrossRef CAS.
- L. Ouyang, G.-J. Da, P.-F. Tian, T.-Y. Chen, G.-D. Liang, J. Xu and Y.-F. Han, Insight into active sites of Pd-Au/TiO2 catalysts in hydrogen peroxide synthesis directly from H2 and O2, J. Catal., 2014, 311, 129–136 CrossRef CAS.
- T. Ricciardulli, S. Gorthy, J. S. Adams, C. Thompson, A. M. Karim, M. Neurock and D. W. Flaherty, Effect of Pd coordination and isolation on the catalytic reduction of O2 to H2O2 over PdAu bimetallic nanoparticles, J. Am. Chem. Soc., 2021, 143, 5445–5464 CrossRef CAS PubMed.
- J. K. Edwards, S. F. Parker, J. Pritchard, M. Piccinini, S. J. Freakley, Q. He, A. F. Carley, C. J. Kiely and G. J. Hutchings, Effect of acid pre-treatment on AuPd/SiO2 catalysts for the direct synthesis of hydrogen peroxide, Catal. Sci. Technol., 2013, 3, 812–818 RSC.
- J. K. Edwards, B. Solsona, E. N. N, A. F. Carley, A. A. Herzing, C. J. Kiely and G. J. Hutchings, Switching off hydrogen peroxide hydrogenation in the direct synthesis process, Science, 2009, 323, 1037–1041 CrossRef CAS PubMed.
- W. Liang, J. Dong, M. Yao, J. Fu, H. Chen and X. Zhang, Enhancing the selectivity of Pd/C catalysts for the direct synthesis of H2O2 by HNO3 pretreatment, New J. Chem., 2020, 44, 18579–18587 RSC.
- H. T. Thuy Vu, V. L. Nam Vo and Y.-M. Chung, Geometric, electronic, and synergistic effect in the sulfonated carbon-supported Pd catalysts for the direct synthesis of hydrogen peroxide, Appl. Catal., A, 2020, 607, 117867 CrossRef CAS.
- F. Alotaibi, S. Al-Mayman, M. Alotaibi, J. K. Edwards, R. J. Lewis, R. Alotaibi and G. J. Hutchings, Direct synthesis of hydrogen peroxide using Cs-containing heteropolyacid-supported palladium–copper catalysts, Catal. Lett., 2019, 149, 998–1006 CrossRef CAS.
- E. N. Ntainjua, M. Piccinini, S. J. Freakley, J. C. Pritchard, J. K. Edwards, A. F. Carley and G. J. Hutchings, Direct synthesis of hydrogen peroxide using Au-Pd-exchanged and supported heteropolyacid catalysts at ambient temperature using water as solvent, Green Chem., 2012, 14, 170–181 RSC.
- D. Gudarzi, W. Ratchananusorn, I. Turunen, T. Salmi and M. Heinonen, Preparation and study of Pd catalysts supported on activated carbon cloth (ACC) for direct synthesis of H2O2 from H2 and O2, Top. Catal., 2013, 56, 527–539 CrossRef CAS.
- A. Rodriguez-Gomez, F. Platero, A. Caballero and G. Colon, Improving the direct synthesis of hydrogen peroxide from hydrogen and oxygen over Au-Pd/SBA-15 catalysts by selective functionalization, Mol. Catal., 2018, 445, 142–151 CrossRef CAS.
- I. Yuranov, L. Kiwi-Minsker, P. Buffat and A. Renken, Selective synthesis of Pd nanoparticles in complementary micropores of SBA-15, Faraday Discuss., 2004, 16, 760–761 CAS.
- G. Blanco-Brieva, F. Desmedt, P. Miquel, J. M. Campos-Martin and J. L. G. Fierro, Direct synthesis of hydrogen peroxide without the use of acids or halide promoters in dissolution, Catal. Sci. Technol., 2020, 10, 2333–2336 RSC.
- S. Yook, H. C. Kwon, Y.-G. Kim, W. Choi and M. Choi, Significant roles of carbon pore and surface structure in AuPd/C catalyst for achieving high chemoselectivity in direct hydrogen peroxide synthesis, ACS Sustainable Chem. Eng., 2017, 5, 1208–1216 CrossRef CAS.
- E. Ghedini, F. Menegazzo, M. Signoretto, M. Manzoli, F. Pinna and G. Strukul, Mesoporous silica as supports for Pd-catalyzed H2O2 direct synthesis: Effect of the textural properties of the support on the activity and selectivity, J. Catal., 2010, 273, 266–273 CrossRef CAS.
- S. Kim, D.-W. Lee, K.-Y. Lee and E. A. Cho, Effect of Pd particle size on the direct synthesis of hydrogen peroxide from hydrogen and oxygen over Pd core–porous SiO2 shell catalysts, Catal. Lett., 2014, 144, 905–911 CrossRef CAS.
- P. Landon, P. J. Collier, A. J. Papworth, C. J. Kiely and G. J. Hutchings, Direct formation of hydrogen peroxide from H2/O2 using a gold catalyst, Chem. Commun., 2002, 2058–2059 RSC.
- J. K. Edwards, B. E. Solsona, P. Landon, A. F. Carley, A. Herzing, C. J. Kiely and G. J. Hutchings, Direct synthesis of hydrogen peroxide from H2 and O2 using TiO2-supported Au–Pd catalysts, J. Catal., 2005, 236, 69–79 CrossRef CAS.
- J. Li, T. Ishihara and K. Yoshizawa, Theoretical revisit of the direct synthesis of H2O2 on Pd and Au@Pd surfaces: A comprehensive mechanistic study, J. Phys. Chem. C, 2011, 115, 25359–25367 CrossRef CAS.
- J.-S. Kim, H.-K. Kim, S.-H. Kim, I. Kim, T. Yu, G.-H. Han, K.-Y. Lee, J.-C. Lee and J.-P. Ahn, Catalytically active Au layers grown on Pd nanoparticles for direct synthesis of H2O2: Lattice strain and charge-transfer perspective analyses, ACS Nano, 2019, 13, 4761–4770 CrossRef CAS PubMed.
- H. X. Xu, D. J. Cheng and Y. Gao, Design of high-performance Pd-based alloy nanocatalysts for direct synthesis of H2O2, ACS Catal., 2017, 7, 2164–2170 CrossRef CAS.
- G.-H. Han, X. Xiao, J. Hong, K.-J. Lee, S. Park, J.-P. Ahn, K.-Y. Lee and T. Yu, Tailored palladium–platinum nanoconcave cubes as high performance catalysts for the direct synthesis of hydrogen peroxide, ACS Appl. Mater. Interfaces, 2020, 12, 6328–6335 CrossRef CAS PubMed.
- X. Gong, R. J. Lewis, S. Zhou, D. J. Morgan, T. E. Davies, X. Liu, C. J. Kiely, B. Zong and G. J. Hutchings, Enhanced catalyst selectivity in the direct synthesis of H2O2 through Pt incorporation into TiO2 supported AuPd catalysts, Catal. Sci. Technol., 2020, 10, 4635–4644 RSC.
- M.-C. Kim, G.-H. Han, X. Xiao, J. Song, J. Hong, E. Jung, H.-K. Kim, J.-P. Ahn, S. S. Han, K.-Y. Lee and T. Yu, Anisotropic growth of Pt on Pd nanocube promotes direct synthesis of hydrogen peroxide, Appl. Surf. Sci., 2021, 562, 150031 CrossRef CAS.
- Y. Zhang, Q. Sun, G. Guo, Y. Cheng, X. Zhang, H. Ji and X. He, Trace Pt atoms as electronic promoters in Pd clusters for direct synthesis of hydrogen peroxide, Chem. Eng. J., 2023, 451, 138867 CrossRef CAS.
- S. Yu, X. Cheng, Y. Wang, B. Xiao, Y. Xing, J. Ren, Y. Lu, H. Li, C. Zhuang and G. Chen, High activity and selectivity of single palladium atom for oxygen hydrogenation to H2O2, Nat. Commun., 2022, 13, 4737 CrossRef CAS.
- S. J. Freakley, Q. He, J. H. Harrhy, L. Lu, D. A. Crole, D. J. Morgan, E. N. Ntainjua, J. K. Edwards, A. F. Carley, A. Y. Borisevich, C. J. Kiely and G. J. Hutchings, Palladium-tin catalysts for the direct synthesis of H2O2 with high selectivity, Science, 2016, 351, 965–968 CrossRef CAS.
- H.-C. Li, Q. Wan, C. Du, J. Zhao, F. Li, Y. Zhang, Y. Zheng, M. Chen, K. H. L. Zhang, J. Huang, G. Fu, S. Lin, X. Huang and H. Xiong, Layered Pd oxide on PdSn nanowires for boosting direct H2O2 synthesis, Nat. Commun., 2022, 13, 6072 CrossRef CAS PubMed.
- S. Wang, D. E. Doronkin, M. Hähsler, X. Huang, D. Wang, J.-D. Grunwaldt and S. Behrens, Palladium-based bimetallic nanocrystal catalysts for the direct synthesis of hydrogen peroxide, ChemSusChem, 2020, 13, 3243–3251 CrossRef CAS PubMed.
- S. Wang, R. J. Lewis, D. E. Doronkin, D. J. Morgan, J.-D. Grunwaldt, G. J. Hutchings and S. Behrens, The direct synthesis of hydrogen peroxide from H2 and O2 using Pd-Ga and Pd-In catalysts, Catal. Sci. Technol., 2020, 10, 1925–1932 RSC.
- M. Zhang, Y. Luo, D. Wu, Q. Li, H. Xu and D. Cheng, Promoter role of tungsten in W-Pd/Al2O3 catalyst for direct synthesis of H2O2: Modification of Pd/PdO ratio, Appl. Catal., A, 2021, 628, 118392 CrossRef CAS.
- M. Zhang, H. Xu, Y. Luo, J. Zhu and D. Cheng, Enhancing the catalytic performance of PdAu catalysts by W-induced strong interaction for the direct synthesis of H2O2, Catal. Sci. Technol., 2022, 12, 5290–5301 RSC.
- P. Tian, F. Xuan, D. Ding, Y. Sun, X. Xu, W. Li, R. Si, J. Xu and Y.-F. Han, Revealing the role of tellurium in palladium-tellurium catalysts for the direct synthesis of hydrogen peroxide, J. Catal., 2020, 385, 21–29 CrossRef CAS.
- W. Liang, J. Fu, H. Chen, X. Zhang and G. Deng, Pd-Ce/AC catalyst with high productivity for direct synthesis of H2O2 at ambient temperature, Mater. Lett., 2021, 283, 128857 CrossRef CAS.
- D. A. Crole, R. Underhill, J. K. Edwards, G. Shaw, S. J. Freakley, G. J. Hutchings and R. J. Lewis, The direct synthesis of hydrogen peroxide from H2 and O2 using Pd-Ni/TiO2 catalysts, Philos. Trans. R. Soc., A, 2020, 378, 20200062 CrossRef CAS PubMed.
- V. R. Naina, S. Wang, D. I. Sharapa, M. Zimmermann, M. Hähsler, L. Niebl-Eibenstein, J. Wang, C. Wöll, Y. Wang, S. K. Singh, F. Studt and S. Behrens, Shape-selective synthesis of intermetallic Pd3Pb nanocrystals and enhanced catalytic properties in the direct synthesis of hydrogen peroxide, ACS Catal., 2021, 11, 2288–2301 CrossRef CAS.
- X. T. Tran, V. L. N. Vo and Y.-M. Chung, Fast microwave-assisted synthesis of iron–palladium catalysts supported on graphite for the direct synthesis of H2O2, Catal. Today, 2023, 411–412, 113821 CrossRef CAS.
- S. Liu, Y. Deng, L. Fu, L. Huang, L. Ouyang and S. Yuan, Understanding the role of boron on the interface modulation of the Pd/TiO2 catalyst for direct synthesis of H2O2, ACS Sustainable Chem. Eng., 2022, 10, 3264–3275 CrossRef CAS.
- S. Wang, G. Jiang, Z. Yang, L. Mu, T. Ji, X. Lu and J. Zhu, Boosted H2O2 productivity by sulfur-doped Pd/carbon catalysts via direct synthesis from H2 and O2 at atmospheric pressure, ACS Sustainable Chem. Eng., 2022, 10, 13750–13758 CrossRef CAS.
- R. J. Lewis, M. Koy, M. Macino, M. Das, J. H. Carter, D. J. Morgan, T. E. Davies, J. B. Ernst, S. J. Freakley, F. Glorius and G. J. Hutchings, N-heterocyclic carbene modified palladium catalysts for the direct synthesis of hydrogen peroxide, J. Am. Chem. Soc., 2022, 144, 15431–15436 CrossRef CAS PubMed.
- G. Blanco-Brieva, M. C. Capel-Sanchez, M. P. de Frutos, A. Padilla-Polo, J. M. Campos-Martin and J. L. G. Fierro, New two-step process for propene oxide production (HPPO) based on the direct synthesis of hydrogen peroxide, Ind. Eng. Chem. Res., 2008, 47, 8011–8015 CrossRef CAS.
- T. A. Nijhuis, M. Makkee, J. A. Moulijn and B. M. Weckhuysen, The production of propene oxide: Catalytic processes and recent developments, Ind. Eng. Chem. Res., 2006, 45, 3447–3459 CrossRef CAS.
- Q. Chen and E. J. Beckman, One-pot green synthesis of propylene oxide using in situ generated hydrogen peroxide in carbon dioxide, Green Chem., 2008, 10, 934 RSC.
- B. Taylor, J. Lauterbach and W. N. Delgass, Gas-phase epoxidation of propylene over small gold ensembles on TS-1, Appl. Catal., A, 2005, 291, 188–198 CrossRef.
- N. Yap, R. P. Andres and W. N. Delgass, Reactivity and stability of Au in and on TS-1 for epoxidation of propylene with H2 and O2, J. Catal., 2004, 226, 156–170 CrossRef CAS.
- B. S. Uphade, S. Tsubota, T. Hayashi and M. J. C. L. Haruta, Selective oxidation of propylene to propylene oxide or propionaldehyde over Au supported on titanosilicates in the presence of H2 and O2, Chem. Lett., 1998, 12, 1277–1278 CrossRef.
- B. Chowdhury, J. Bravo-Suárez, M. Daté, S. Tsubota and M. J. A. C. Haruta, Trimethylamine as a gas-phase promoter: Highly efficient epoxidation of propylene over supported gold catalysts, Angew. Chem., Int. Ed., 2005, 45(3), 412–415 CrossRef PubMed.
- W. S. Lee, M. C. Akatay, E. A. Stach, F. H. Ribeiro and W. N. Delgass, Reproducible preparation of Au/TS-1 with high reaction rate for gas phase epoxidation of propylene, J. Catal., 2012, 287, 178–189 CrossRef CAS.
- B. S. Uphade, M. Okumura, S. Tsubota and M. General, Effect of physical mixing of CsCl with Au/Ti-MCM-41 on the gas-phase epoxidation of propene using H2 and O2: Drastic depression of H2 consumption, Appl. Catal., A, 2000, 109(1–2), 43–50 CrossRef.
- A. K. Sinha, S. Seelan, T. Akita, S. Tsubota and M. Haruta, Vapor phase propylene epoxidation over Au/Ti-MCM-41 catalysts prepared by different Ti incorporation modes, Appl. Catal., A, 2003, 240(1–2), 243–252 CrossRef CAS.
- B. S. Uphade, Y. Yamada, T. Akita, T. Nakamura and M. Haruta, Synthesis and characterization of Ti-MCM-41 and vapor-phase epoxidation of propylene using H2 and O2 over Au/Ti-MCM-41, Appl. Catal., A, 2001, 215, 137–148 CrossRef CAS.
- G. Mul, A. Zwijnenburg, B. V. D. Linden, M. Makkee and J. A. Moulijn, Stability and selectivity of Au/TiO2 and Au/TiO2/SiO2 catalysts in propene epoxidation: An in situ FT-IR study, J. Catal., 2001, 201, 128–137 CrossRef CAS.
- T. Hayashi, K. Tanaka and M. Haruta, Selective vapor-phase epoxidation of propylene over Au/TiO2 catalysts in the presence of oxygen and hydrogen, J. Catal., 1998, 178, 566–575 CrossRef CAS.
- A. Ruiz, B. Linden, M. Makkee and G. Mul, Acrylate and propoxy-groups: Contributors to deactivation of Au/TiO2 in the epoxidation of propene, J. Catal., 2009, 266, 286–290 CrossRef CAS.
- Q. Chen, Direct synthesis of hydrogen peroxide from oxygen and hydrogen using carbon dioxide as an environmentally benign solvent and its application in green oxidation, PhD, University of Pittsburgh, America, 2008, pp. 50–58 Search PubMed.
- R. Wang, X. Guo, X. Wang and J. J. C. T. Hao, Gas-phase epoxidation of propylene over Ag/Ti-containing catalysts, Catal. Today, 2004, 93, 217–222 CrossRef.
- F. Wang, C. Qi and J. J. C. C. Ma, The study of the uncalcined Au catalyst and inorganic salts on direct gas-phase epoxidation of propylene, Catal. Commun., 2007, 8, 1947–1952 CrossRef CAS.
- B. S. Uphade, M. Okumura, S. Tsubota and M. Haruta, Effect of physical mixing of CsCl with Au/Ti-MCM-41 on the gas-phase epoxidation of propene using H2 and O2: Drastic depression of H2 consumption, Appl. Catal., A, 2000, 109(1–2), 43–50 CrossRef.
- S. Esplugas, J. Giménez, S. Contreras, E. Pascual and M. J. W. R. Rodrguez, Comparison of different advanced oxidation processes for phenol degradation, Water Res., 2002, 36, 1034–1042 CrossRef CAS PubMed.
- O. Osegueda, A. Dafinov, J. Llorca, F. Medina and J. Suerias, In situ generation of hydrogen peroxide in catalytic membrane reactors, Catal. Today, 2012, 193, 128–136 CrossRef CAS.
- R. Underhill, R. J. Lewis, S. J. Freakley, M. Douthwaite, P. Miedziak, O. Akdim, J. K. Edwards and G. J. Hu Tchings, Oxidative degradation of phenol using in situ generated hydrogen peroxide combined with Fenton's process, Johnson Matthey Technol. Rev., 2018, 62, 417–425 CrossRef CAS.
- M. H. A. Rahim, M. M. Forde, C. Hammond, R. L. Jenkins, N. Dimitratos, J. A. Lopez-Sanchez, A. F. Carley, S. H. Taylor, D. J. Willock and G. J. Hutchings, Catalysis, Systematic study of the oxidation of methane using supported gold palladium nanoparticles under mild aqueous conditions, Top. Catal., 2013, 56, 1843–1857 CrossRef.
- M. H. Ab Rahim, M. M. Forde, R. L. Jenkins, C. Hammond, Q. He, N. Dimitratos, J. A. Lopez-Sanchez, A. F. Carley, S. H. Taylor and D. J. J. A. C. Willock, Systematic study of the oxidation of methane using supported gold palladium nanoparticles under mild aqueous conditions, Angew. Chem., 2013, 125, 1318–1322 CrossRef.
- Z. Jin, L. Wang, E. Zuidema, K. Mondal, M. Zhang, J. Zhang, C. Wang, X. Meng, H. Yang, C. Mesters and F.-S. Xiao, Hydrophobic zeolite modification for in situ peroxide formation in methane oxidation to methanol, Science, 2020, 367, 193–197 CrossRef CAS PubMed.
- J. J. Gu, S. L. Wang, Z. Y. He, Y. Han and J. L. Zhang, Direct synthesis of hydrogen peroxide from hydrogen and oxygen over activated-carbon-supported Pd–Ag alloy catalysts, Catal. Sci. Technol., 2016, 6, 809–817 RSC.
- L. Torrente-Murciano, Q. He, G. J. Hutchings, C. J. Kiely and D. Chadwick, Enhanced Au-Pd activity in the direct synthesis of hydrogen peroxide using nanostructured titanate nanotube supports, ChemCatChem, 2014, 6, 2531–2534 CrossRef CAS.
- F. Menegazzo, P. Burti, M. Signoretto, M. Manzoli, S. Vankova, F. Boccuzzi, F. Pinna and G. Strukul, Effect of the addition of Au in zirconia and ceria supported Pd catalysts for the direct synthesis of hydrogen peroxide, J. Catal., 2008, 257, 369–381 CrossRef CAS.
- E. N. N, J. K. Edwards, A. F. Carley, J. A. Lopez-Sanchez, J. A. Moulijn, A. A. Herzing, C. J. Kiely and G. J. Hutchings, The role of the support in achieving high selectivity in the direct formation of hydrogen peroxide, Green Chem., 2008, 10, 1162–1169 RSC.
- S. J. Freakley, R. J. Lewis, D. J. Morgan, J. K. Edwards and G. J. Hutchings, Direct synthesis of hydrogen peroxide using Au–Pd supported and ion-exchanged heteropolyacids precipitated with various metal ions, Catal. Today, 2015, 248, 10–17 CrossRef CAS.
- E. N. Ntainjua, M. Piccinini, S. J. Freakley, J. C. Pritchard, J. K. Edwards, A. F. Carley and G. J. Hutchings, Direct synthesis of hydrogen peroxide using Au–Pd-exchanged and supported heteropolyacid catalysts at ambient temperature using water as solvent, Green Chem., 2012, 14, 170–181 RSC.
- D. Gudarzi, W. Ratchananusorn, I. Turunen, M. Heinonen and T. Salmi, Promotional effects of Au in Pd–Au bimetallic catalysts supported on activated carbon cloth (ACC) for direct synthesis of H2O2 from H2 and O2, Catal. Today, 2015, 248, 58–68 CrossRef CAS.
- Y. F. Han, Z. Y. Zhong, K. Ramesh, F. X. Chen, L. W. Chen, T. White, Q. L. Tay, S. N. Yaakub and Z. Wang, Au promotional effects on the synthesis of H2O2 directly from H2 and O2 on supported Pd−Au alloy catalysts, J. Phys. Chem. C, 2007, 111, 8410–8413 CrossRef CAS.
- G. Bernardotto, F. Menegazzo, F. Pinna, M. Signoretto, G. Cruciani and G. Strukul, New Pd–Pt and Pd–Au catalysts for an efficient synthesis of H2O2 from H2 and O2 under very mild conditions, Appl. Catal., A, 2009, 358, 129–135 CrossRef CAS.
- J. Xu, L. Ouyang, G.-J. Da, Q.-Q. Song, X.-J. Yang and Y.-F. Han, Pt promotional effects on Pd–Pt alloy catalysts for hydrogen peroxide synthesis directly from hydrogen and oxygen, J. Catal., 2012, 285, 74–82 CrossRef CAS.
- S. Wang, K. Gao, W. Li and J. Zhang, Effect of Zn addition on the direct synthesis of hydrogen peroxide over supported palladium catalysts, Appl. Catal., A, 2017, 531, 89–95 CrossRef CAS.
- J. K. Edwards, J. Pritchard, P. J. Miedziak, M. Piccinini, A. F. Carley, Q. He, C. J. Kiely and G. J. Hutchings, The direct synthesis of hydrogen peroxide using platinum promoted gold–palladium catalysts, Catal. Sci. Technol., 2014, 4, 3244–3250 RSC.
- E. N. Ntainjua, S. J. Freakley and G. J. Hutchings, Direct synthesis of hydrogen peroxide using ruthenium catalysts, Top. Catal., 2012, 55, 718–722 CrossRef CAS.
| This journal is © Institute of Process Engineering of CAS 2024 |