
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Nearly all-active-material cathodes free of nickel and cobalt for Li-ion batteries†
Eunryeol
Lee‡
 a,
Dae-Hyung
Lee‡
ab,
Stéphanie
Bessette
c,
Sang-Wook
Park
ab,
Nicolas
Brodusch
c,
Gregory
Lazaris
c,
Hojoon
Kim
ab,
Rahul
Malik
d,
Raynald
Gauvin
c,
Dong-Hwa
Seo
*ab and
Jinhyuk
Lee
*c
a,
Dae-Hyung
Lee‡
ab,
Stéphanie
Bessette
c,
Sang-Wook
Park
ab,
Nicolas
Brodusch
c,
Gregory
Lazaris
c,
Hojoon
Kim
ab,
Rahul
Malik
d,
Raynald
Gauvin
c,
Dong-Hwa
Seo
*ab and
Jinhyuk
Lee
*c
aSchool of Energy and Chemical Engineering, Ulsan National Institute of Science and Technology (UNIST), 50 UNIST-Gil, Ulsan 44919, Republic of Korea
bDepartment of Materials Science and Engineering, Korea Advanced Institute of Science and Technology (KAIST), 291 Daehak-ro, Daejeon 34141, Republic of Korea. E-mail: dseo@kaist.ac.kr
cDepartment of Mining and Materials Engineering, McGill University, Montreal, QC H3A 0C5, Canada. E-mail: jinhyuk.lee@mcgill.ca
dOffice of Energy Research and Development, Natural Resources Canada, Ottawa, ON, Canada
First published on 27th March 2024
Abstract
The global transition to electric vehicles and large-scale energy storage systems requires cost-effective and abundant alternatives to commercial Co/Ni-based cathodes (e.g., LiNi0.6Mn0.2Co0.2O2) for Li-ion batteries (LIBs). Manganese-based disordered rock-salts (Mn-DRXs) can outperform conventional cathodes at lower cost, achieving >900 W h kg-AM−1 (per active material, AM), but such performance has been demonstrated exclusively in cell constructions far removed from commercial viability, namely with diluted electrode films (∼70 wt%-AM) containing excessive carbon and binder. Herein, our work involves a comprehensive study to attain AM-concentrated Mn-DRX cathodes (>95 wt%-AM), covering from inherent material properties to the microstructure of electrodes, to address the formidable challenges in Mn-DRX research. We reveal that Mn-DRXs’ failures in AM-concentrated electrodes originate from their extremely low electrical conductivity (10−10–10−8 S cm−1) and the collapse of the electrical network with volume change over cycling. These failure modes are resolved through electrical percolation engineering and enhancement of electrode mechanical properties, allowing our demonstration of nearly all-AM Mn-DRX cathodes (∼96 wt%-AM) and the highest application-level energy density (∼1050 W h kg-cathode−1) reported to date. This work further unveils the trade-off role of Mn-content on Mn-DRXs’ electrical conductivity and volume change, providing guidelines for material design to advance Co/Ni-free LIBs’ technology readiness.
Broader contextThe quest for lower $ per kWh in Li-ion batteries (LIBs) hinges on the advancement of high-energy cathodes that sidestep supply-constrained elements like Co and Ni. Mn-based disordered rock-salts (Mn-DRXs) emerge as promising contenders, boasting exceptional energy densities (>900 W h kg−1 of active material, AM) and leveraging Mn, an abundant and sustainable resource. Despite their potential, Mn-DRXs have primarily been explored in cathode films with diluted AM content (∼70 wt%-AM), laden with a substantial amount of carbon (15–30 wt%) and binder (5–10 wt%). This configuration diminishes electrode/cell-level energy density and obscures issues in practical cathode films featuring high AM content and loading. This study delves into critical failure mechanisms in Mn-DRX cathodes with elevated AM concentration and tackles these challenges head-on to showcase nearly all-AM Mn-DRX cathodes with the highest application-level energy density (>95 wt%-AM, >1000 W h kg-cathode−1), propelling the advancement of Ni- and Co-free LIBs. |
Introduction
Decades of scientific and technological advancement in Li-ion batteries (LIBs) are now driving the global transition toward electric vehicles and grid energy storage that complements intermittent renewable power generation.1 At the same time, escalating adoption of LIBs is already straining prices of key battery inputs, resulting in the first increase in cell price this year which reverses a decade-long trend of declining prices.2 To meet the growing TWh-scale demand forecast in the immediate future, let alone in decades ahead, LIBs will need to increase their performance while also becoming more inexpensive, as reflected in prominent international battery strategies that specifically isolate the acute need for alternatives to costly Ni and Co in commercial LIB cathodes (e.g., layered oxide LiNi0.6Mn0.2Co0.2O2) that render the cathode cost responsible for nearly ∼50% of the total materials cost for current LIBs.3,4By delivering higher energy density (>900 W h kg-AM−1 per active material, AM) than the layered oxides (∼770 W h kg-AM−1) with the affordability of LiFePO4 (a cheap albeit low energy-density cathode, ∼580 W h kg-AM−1),5 Mn-based cation-disordered rock-salts (Mn-DRXs, e.g., Li2MnO2F,6 Li2Mn1/2Ti1/2O2F,7 and Li1.68Mn1.60O3.7F0.38) made of cheaper metals (Mn, Ti, etc.) than Ni and Co (Fig. 1(a)) are one of only a handful of Co/Ni-free cathode materials with one of the lowest projected cost per energy-stored ($ per W h),9 that can shift the current cathode market-share. However, these materials’ promising performance has been demonstrated in highly diluted cathode films with large amounts of carbon black (15–30 wt%, CB) and binder (5–10 wt%, such as polyvinylidene fluoride, PVDF),7–15 acting as the conductive additive and glue. Testing AM in a diluted film conceals problems evident in practical films and cells with minimized CB/binder content (e.g., ∼2 wt% CB, ∼2 wt% binder), problems that persist and limit materials’ usage in practical LIBs even if AM-level problems are solved.7,9,10,16–18 Moreover, diluting an electrode limits the practical energy density gain at the electrode or cell level (W h kg-electrode/cell−1, W h l-electrode/cell−1) (Fig. 1(b)). For instance, Li1.68Mn1.60O3.7F0.3 could deliver nearly 1100 W h kg-AM−1 but only in a 70 wt%-AM film (AM is only ∼70 wt% of the electrode),8 resulting in cathode energy density of ∼770 W h kg-cathode−1, a marginal improvement over commercial Ni/Co-based layered cathodes (∼740 W h kg-cathode−1, ∼770 W h kg-AM−1) cycling with a small amount of CB and binder.9
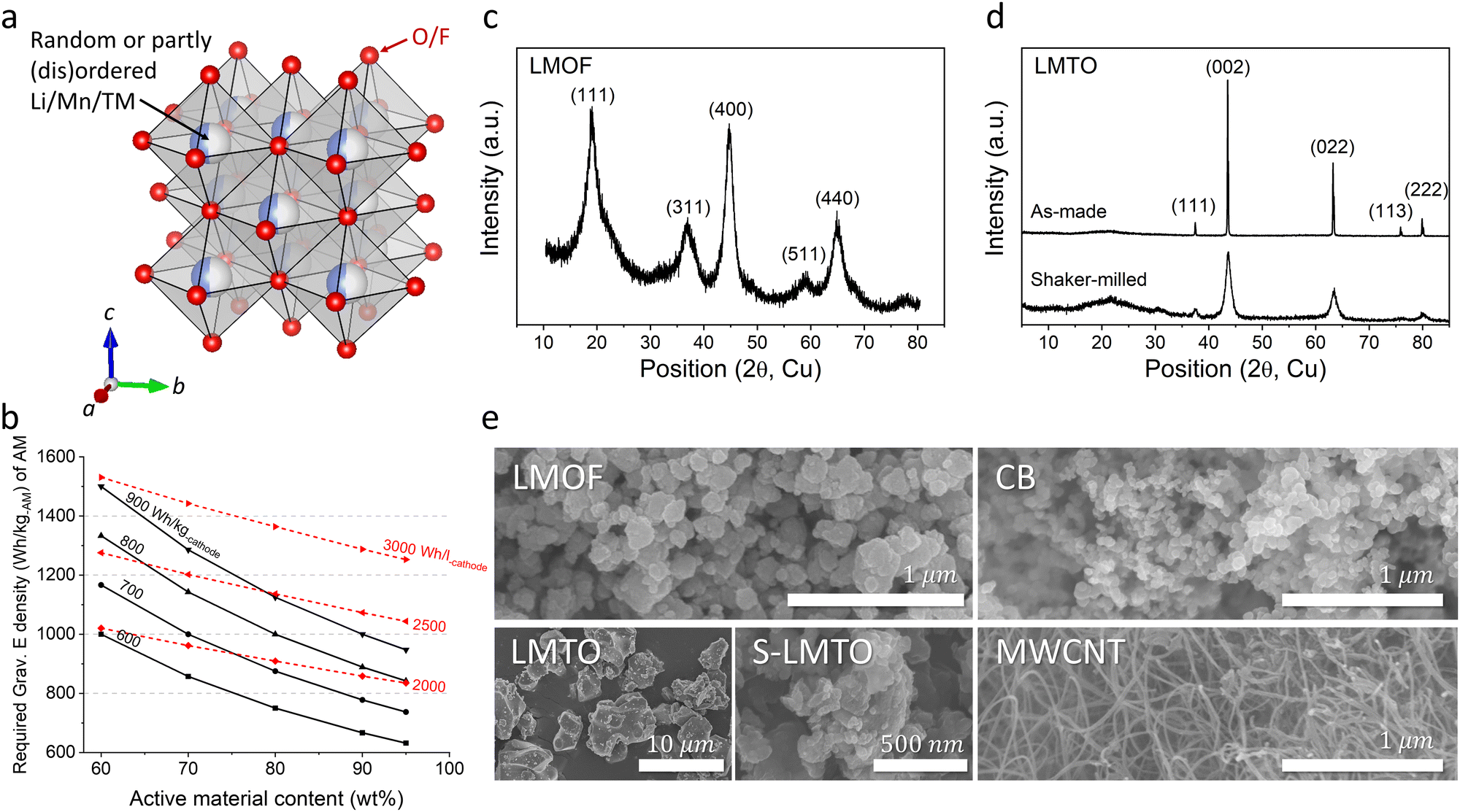 | ||
| Fig. 1 Structure of components in Mn-DRX cathodes. (a) Schematic of the crystal structure of Mn-DRX. (b) The required gravimetric energy density (W h kg-AM−1) in Mn-DRX to achieve various target cathode energy densities (W h kg-cathode−1, W h l-cathode−1) as a function of the AM content. The XRD patterns of (c) LMOF and (d) LMTO (before and after shaker-milling). (e) SEM images of LMOF, LMTO, shaker-milled LMTO (S-LMTO), carbon black (CB), and multiwalled carbon nanotube (MWCNT). | ||
Herein, we investigate the behavior of Mn-DRXs in an AM-concentrated cathode using experiments and computational modeling. The materials’ low electrical conductivity and volume-change-driven electrode disintegration (electrochemical fatigue) limit Mn-DRXs’ capacity and accelerate the capacity fading in an AM-concentrated cathode made with CB as the conductive additive. However, we demonstrate that replacing CB with multiwalled carbon nanotubes (MWCNT) and changing the binder type enable nearly all-AM Mn-DRX cathodes (to 96 wt% AM) that achieve >1050 W h kg-cathode−1, by enhancing the electrical percolation and mechanical properties of the composite electrode. Moreover, we reveal the counteracting role of Mn-content in Mn-DRXs for the electrical conductivity and volume change during cycling to provide further guidelines toward optimized all-AM Mn-DRX cathodes for high-performance Co/Ni-free LIBs.
Results and discussions
Performance of Mn-DRXs under different electrode compositions
We chose Li1.68Mn1.60O3.7F0.38 (LMOF, made by mechanochemistry) and Li1.2Mn0.4Ti0.4O219 (LMTO, by solid-state synthesis and pulverization) for our preliminary test. X-ray diffraction (XRD) patterns in Fig. 1(c) and (d) characterize LMOF as partially ordered spinel (DRX with spinel-like local cation order) and LMTO as a completely cation-disordered DRX. Shaker milling on the as-made LMTO particles broadens the XRD peaks, indicating particle pulverization and consistent with our scanning electron microscopy (SEM) observation (Fig. 1(e)). Both LMOF and LMTO after shaker milling show polycrystalline particles (diameter, d < 300 nm) made with smaller size grains, consistent with previous Mn-DRX research. Fig. 1(e) also shows the SEM images of CB and MWCNT that were used to make cathode films. CB particles have a spherical shape with d < 50 nm, while MWCNT possesses the typical 1-D nanotube morphology.Mn-DRXs have been exclusively cycled in diluted electrodes with excessive conductive additives and binder.7,9,10,20 To examine their behavior in an AM-concentrated electrode, we prepared LMOF and LMTO films with different electrode compositions by using CB and PVDF as the conductive additives and binder: 70![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :20:10 (AM:CB:PVDF, by wt%), 80:10:10, and 90:5:5. Increasing the AM content in the electrode decreases the electrode thickness upon achieving a similarly loaded (i.e., mg-cathode cm−2) electrode for a higher density (∼3.96 kg l−1) of LMOF (or any Mn-DRX) than CB (∼1.8 kg l−1) (Fig. S2, ESI†), which would appear beneficial if one only considers transport distance of Li+ or electrons. Fig. 2(a)–(c) show LMOF's voltage profiles in the 70:20:10, 80:10:10, and 90:5:5 films (25 mA g-AM−1, 1.5–4.8 V, room temperature). The mass loading of these electrodes was consistently maintained at ∼5 mg-cathode cm−2. In the 70:20:10 film, LMOF delivers 370 mA h g-AM−1 upon 1st discharging, and its capacity decreases to 327 and 268 mA h g-AM−1 after 5 and 25 cycles. Meanwhile, the LMOF 80:10:10 electrode delivers 335, 239, and 38 mA h g-AM−1 in the 1st, 5th, and 25th cycle, showing smaller capacity and drastically faster capacity fading. Also, LMOF in the 90:5:5 film achieves only 77, 39, and 23 mA h g-AM−1 in the 1st, 5th, and 25th cycle. Finally, LMOF's capacity at a higher rate of 250 mA g-AM−1 also decreases significantly without electrode dilution, demonstrating 325 mA h g-AM−1 in the 70:20:10 film but no capacity at all in the 90:5:5 film (Fig. S3, ESI†). Note that the achieved capacity and its retention of these LMOF:CB:PVDF electrodes are sensitive to the calendering (electrode roll pressing) process and become significantly worse without it (Fig. S4, ESI†), which is necessary for intimate contact between AM, CB, and PVDF particles and reduced electrode porosity.21
:20:10 (AM:CB:PVDF, by wt%), 80:10:10, and 90:5:5. Increasing the AM content in the electrode decreases the electrode thickness upon achieving a similarly loaded (i.e., mg-cathode cm−2) electrode for a higher density (∼3.96 kg l−1) of LMOF (or any Mn-DRX) than CB (∼1.8 kg l−1) (Fig. S2, ESI†), which would appear beneficial if one only considers transport distance of Li+ or electrons. Fig. 2(a)–(c) show LMOF's voltage profiles in the 70:20:10, 80:10:10, and 90:5:5 films (25 mA g-AM−1, 1.5–4.8 V, room temperature). The mass loading of these electrodes was consistently maintained at ∼5 mg-cathode cm−2. In the 70:20:10 film, LMOF delivers 370 mA h g-AM−1 upon 1st discharging, and its capacity decreases to 327 and 268 mA h g-AM−1 after 5 and 25 cycles. Meanwhile, the LMOF 80:10:10 electrode delivers 335, 239, and 38 mA h g-AM−1 in the 1st, 5th, and 25th cycle, showing smaller capacity and drastically faster capacity fading. Also, LMOF in the 90:5:5 film achieves only 77, 39, and 23 mA h g-AM−1 in the 1st, 5th, and 25th cycle. Finally, LMOF's capacity at a higher rate of 250 mA g-AM−1 also decreases significantly without electrode dilution, demonstrating 325 mA h g-AM−1 in the 70:20:10 film but no capacity at all in the 90:5:5 film (Fig. S3, ESI†). Note that the achieved capacity and its retention of these LMOF:CB:PVDF electrodes are sensitive to the calendering (electrode roll pressing) process and become significantly worse without it (Fig. S4, ESI†), which is necessary for intimate contact between AM, CB, and PVDF particles and reduced electrode porosity.21
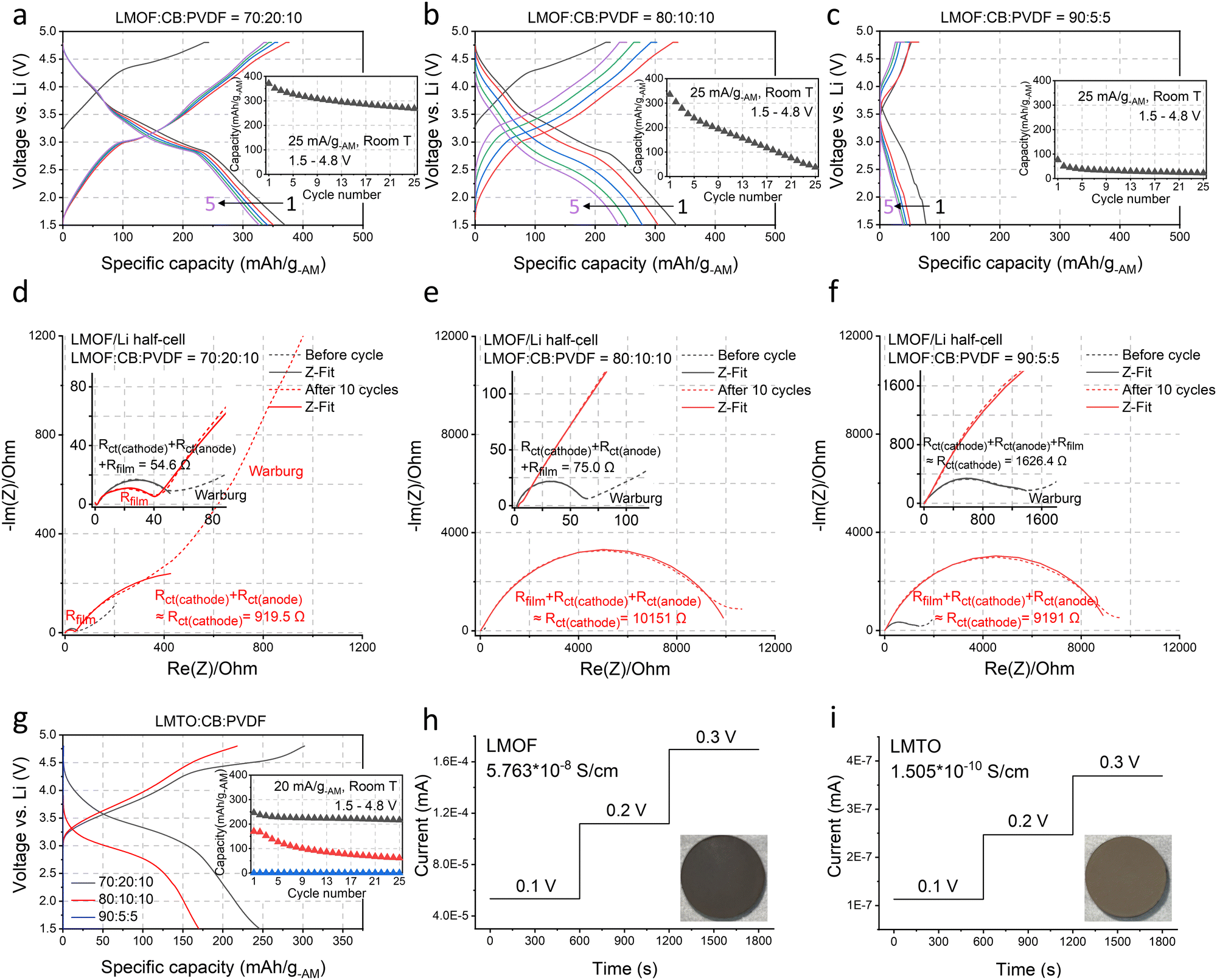 | ||
| Fig. 2 The electrochemical properties of LMOF and LMTO (under different electrode compositions) and their electrical conductivities. (a)–(c) The voltage profiles and the capacity retention of LMOF when cycled at 25 mA g-AM−1 between 1.5–4.8 V in (a) a 70:20:10 (AM:CB:PVDF, by weight), (b) 80:10:10, and (c) 90:5:5 electrode film. (d)–(f) The Nyquist plots of LMOF/Li-metal half-cells before cycle (black) and after 10 cycles (red) at 25 mA g-AM−1 between 1.5–4.8 V in a (d) 70:20:10, (e) 80:10:10, and (f) 90:5:5 film (short dash: experimental values, line: fitted values): cathode/electrolyte and solid/electrolyte interfacial resistance (Rfilm), charge-transfer resistance of cathode (Rct(cathode)) or anode (Rct(anode)), and Warburg resistance. Z-Fitting details are explained in Methods and Fig. S5 (ESI†). The ‘≈’ notation represents the dominant resistance in the mixed contribution of various resistances, supported by DRT analysis in Fig. S6 and S7 (ESI†). (g) The first cycle voltage profiles and the capacity retention of LMTO when cycled at 20 mA g-AM−1 between 1.5–4.8 V in a 70:20:10, 80:10:10, and 90:5:5 film. The DC polarization test results of (h) LMOF and (i) LMTO for electrical conductivity measurements: insets show the image of LMOF and LMTO pellets used in this test. | ||
LMOF's poorer performance in the AM-concentrated electrodes arises from significantly increased charge-transfer resistance.22,23Fig. 2(d)–(f) show the Nyquist plots of LMOF/Li-metal half-cells before cycling and after 10 cycles (1.5–4.8 V, 25 mA g-AM−1) with the 70:20:10, 80:10:10, and 90:5:5 LMOF:CB:PVDF electrodes. With the 70:20:10 electrode, the (depressed) semi-circle in the high- and medium-frequency range, corresponding to the sum of charge-transfer resistances of the cathode and anode (Rct(cathode) & Rct(anode)) and electrode/electrolyte film resistances (Rfilm from cathode/electrolyte interface, CEI, and solid–electrolyte interface, SEI) has ∼55 Ω diameter before cycling and slightly increases to ∼920 Ω after 10 cycles dominantly by the Rct(cathode) changes. We provide more detailed interpretations of the results in the ESI† by using the distribution of relaxation times (DRT) measurement (Fig. S6 and S7, ESI†). Before cycling, the LMOF/Li cell using the 80:10:10 LMOF film also shows a small diameter of ∼75 Ω at the same frequency range, which increases to nearly ∼10000 Ω after 10 cycles (vs. ∼920 Ω in the 70:20:10 case), primarily due to the substantially greater LMOF charge-transfer degradation, elucidating the origin of LMOF's smaller capacity and accelerated capacity loss in the 80:10:10 film (Fig. 2(b)). For the 90:5:5 film, the resistance already starts much higher at ∼1600 Ω and reaches ∼9200 Ω after 10 cycles (Fig. 2(f)). In the ESI,† we present Nyquist plots of these LMOF/Li cells obtained at different states of charge, illustrating the consistent trend of significantly higher resistances from the LMOF film with a higher AM content (Fig. S8, ESI†). LMTO exhibits similar behavior to LMOF. LMTO's capacity decreases from ∼250 mA h g-AM−1 to ∼170 and 0 mA h g-AM−1 as the electrode composition changes from 70:20:10 to 80:10:10 and 90:5:5, indicating even severer performance degradation in a concentrated film than LMOF (Fig. 2(g)). Also, LMTO's capacity retention declines drastically with higher AM content.
Using the DC polarization method, we evaluated the electrical conductivity of LMOF and LMTO pellets as low AM conductivities can limit the charge-transfer reaction in a concentrated film with low CB content (Fig. 2(h) and (i)). Our numbers here serve as reference points instead of the most accurate values, as the electrical conductivity can be measured slightly differently by the measurement method and particle microstructure (Fig. S9, ESI†). We measured ∼5.76 × 10−8 S cm−1 for LMOF and ∼1.51 × 10−10 S cm−1 for LMTO, noticeably smaller values than conventional layered oxides like LiNi0.8Mn0.1Co0.1O2 (∼1.1 × 10−3 S cm−1, Fig. S10, ESI†) and even lower for LMTO than LiFePO4 (∼5 × 10−8 S cm−1) notorious for its poor electrical conductivity.24 In particular, LMTO has ∼380 times lower conductivity than LMOF, consistent with the lighter color of LMTO than LMOF (insets in Fig. 2(h) and (i)), explaining why LMTO's performance depends more on the CB content than LMOF.
Note that in addition to the poor electrical conductivity of the AM particles limiting electron transfer, suppressed Li+ transport along the electrode pores may also contribute to increased charge-transfer resistance by reducing the number of Li+–e− recombination sites at the electrode/electrolyte interface. However, our use of the typical carbonate electrolyte (1 M LiPF6 in EC:DMC = 1:1) with low viscosity and relatively thin electrodes (30–40 μm, Fig. S2, ESI†) for the impedance tests facilitates electrolyte soaking into the electrode to mitigate the Li+ accessibility problem. Consequently, the observed increase in charge-transfer resistance and lower capacities in the electrode with a smaller CB content (higher AM content) are most likely attributed to the poor electrical conductivity of the AM. This is also supported by our galvanostatic intermittent titration technique (GITT) test, demonstrating that the difference in the overpotential of charging the LMOF:CB:PVDF electrodes with varying ratios is predominantly attributed to non-Warburg-related resistances, rather than Li-transport-related Warburg resistances (Fig. S11, ESI†).
Electrochemical fatigue in Mn-DRX cathodes upon cycling
Cycling poorly electrically conductive AM requires robust percolation of conductive additives like CB within the electrode that also forms intimate contact with AM particles.25,26 SEM-energy dispersive spectroscopy (SEM-EDS) analysis confirms that CB percolation becomes limited in an AM-concentrated film. Fig. 3(a)–(c) show the distribution of LMOF (red), CB/PVDF (blue), and pores (white) in the cross-section of pristine 70:20:10, 80:10:10, and 90:5:5 electrodes. Unlike the 70:20:10 or 80:10:10 electrode, the CB percolation (blue) is disturbed by LMOF particles (red) in the 90:5:5 electrode, explaining the significantly higher charge-transfer resistance in the 90:5:5 electrode.
 | ||
| Fig. 3 The microstructure of the LMOF/CB/PVDF electrode before and after cycling. (a)–(c) The SEM-EDS maps of LMOF (red), CB/PVDF (blue), and pore (white) in the (a) 70:20:10, (b) 80:10:10, and (c) 90:5:5 electrodes before cycling. The cross-sectional samples were prepared with Argon ion milling. (d)–(f) The SEM images of the LMOF 80:10:10 electrode (d) before cycle, (e) after 5 cycles, and (f) after 20 cycles [1.5–4.8 V, 25 mA g-AM−1]: LMOF (white), CB/PVDF (gray), and pores (black). | ||
While low AM's electrical conductivities paired with limited CB percolation explain smaller capacities of LMOF and LMTO in an AM-concentrated electrode, we find their faster capacity loss in the concentrated electrode mainly stems from electrode disintegration during cycling (i.e., electrochemical fatigue), degrading the AM/CB and CB/CB contacts more significantly in the AM-concentrated film (thus damaging the electrical percolation in the electrode) and allowing CEI layers to form. Fig. 3(d)–(f) are SEM images of the cross-section of the 80:10:10 LMOF electrode before cycling and after 5 and 20 cycles (1.5–4.8 V, 25 mA g-AM−1), respectively. The white, gray, and black regions correspond to LMOF, CB/PVDF, and pores. Intimate contacts are seen between LMOF and CB/PVDF in the electrode before cycling (Fig. 3(d)), but many pores appear (already) after 5 cycles that loosen the LMOF/CB and CB/CB contacts (Fig. 3(e)). After 20 cycles, significant pore accumulation occurs in the electrode on certain regions (Fig. 3(f)). Also, we observe crack-generation and propagation upon cycling (Fig. 3(d)–(f), bottom images), making the 20-cycled 80:10:10 electrode highly fragile (Fig. S12, ESI†). Note that electrode porosity or cracks may develop by electrolyte soaking to the electrode alone,27 but our electrolyte soaking test suggests that this should not be the main cause of the electrode disintegration during our cycling tests (Fig. S13, ESI†).
Electrode disintegration via electrochemical fatigue is commonly observed in layered oxide cathodes due to stress and strain built within the electrode, induced by the AM's 2–3% volume change upon cycling.28,29 However, volume change can reach nearly 10% for some Mn-DRXs. In particular, LMOF exhibits nearly ∼8.4% volume change during cycling according to our XRD analysis (Fig. S14–S16, ESI†), consistent with the number (∼8.3%) from Ceder et al.:8 note that volume change analysis of Mn-DRXs based on XRD may not be most accurate due to broad XRD peaks from the Mn-DRXs’ pulverized particle morphology and structure amorphization upon mixed Mn- and O-redox processes during cycling, leaving a certain degree of arbitrariness in XRD refinement. Yet, lattice parameters and thus volume are among the most reliable information from XRD analysis, and we report these volume change numbers (%) as particle-ensemble-averaged reference numbers, not as the most precise values, obtaining which deserves dedicated research. When the electrode is diluted, AM volume change has a mitigated effect on AM performance and electrode integrity. Excessive CB and binder buffer the stress, provide strong adhesion between components, maintain electrical percolation even if some AM/CB or CB/CB contacts are lost, and slow down the CEI resistance growth during cycling, but these benefits disappear in AM-concentrated electrodes. This explains our observation of (i) large cracks for the 80:10:10 LMOF electrode after cycling (Fig. 3(f) and Fig. S17, ESI†) but not for the 70:20:10 electrode (Fig. S17, ESI†) and (ii) the faster capacity loss from non-Warburg-related (e.g., charge-transfer) resistances with smaller CB/PVDF content (Fig. 2).
Electrical percolation engineering and mechanical property enhancement of nearly all-AM Mn-DRX cathodes
To overcome these issues, we engineered highly AM-concentrated Mn-DRX cathodes by replacing CB with MWCNT and applying more adhesive binders. Unlike CB with a spherical nanoparticle shape (d ∼ 50 nm), MWCNT is 1-D carbon (Fig. 1(e)) with higher electrical conductivity (103–105 S cm−1)30 than CB (10−2–10 S cm−1).31 The nanotube structure allows electrons to flow along the tube direction (length >1 μm) without disruption, in contrast to CB, which requires long-range electron transfer through CB/CB contacts that are easily loosened or detached (Fig. 3(f)). Also, the greater volume per gram for MWCNT (∼4.54 cm3 g−1) than CB (0.48–0.56 cm3 g−1) allows less wt% of MWCNT to span out the electrode for electrical percolation. Finally, “vine-like” MWCNT can entangle AM and binder particles to limit electrode disintegration, leading to more robust electrical percolation in the AM-concentrated electrode.28 In this regard, MWCNT was used as a conductive additive or a coating material for other electrode materials as well.28
Fig. 4(a) and (b) show the EDS maps and SEM images of the 90:5:5 electrode made with LMOF, MWCNT, and PVDF before and after 5 cycles (1.5–4.8 V, 25 mA g-AM−1). The blue area in the EDS map is from MWCNT and PVDF. Unlike the case of CB (Fig. 3(c)), we observe percolating MWCNT/PVDF throughout the 90:5:5 electrode, which facilitates electron transport and improves the electrode's mechanical strength. In turn, the LMOF electrodes with MWCNT show remarkably high capacities and energy density of >360 mA h g-AM−1 and ∼1090 W h kg-AM−1 (∼4330 W h l-AM−1: calculated based on the theoretical crystal density of LMOF, 3.96 kg l−1) even as the AM content increases to 96 wt% (Fig. 4(c)) without any noticeable difference in the capacity retention (∼65%) over 30 cycles (1.5–4.8 V, 25 mA g-AM−1), strikingly better than the 80:10(CB):10 LMOF electrode showing <50 mA h g-AM−1 (∼11% retention) after 25 cycles (Fig. 2(b)). This improved capacity retention with MWCNT (vs. CB) is due to much-suppressed impedance growth upon cycling (Fig. S22, ESI†). Also, with MWCNT, the rate capability of even the 96 wt%-AM LMOF electrode in Fig. S23 (ESI†) is impressive, delivering 244 mA h g-AM−1 and 194 mA h g-AM−1 at 2 A g-AM−1 and 4 A g-AM−1, respectively. Note that the as-made LMOF(90):MWCNT(5):PVDF(5) and LMOF(96):MWCNT(2):PVDF(2) electrodes discussed above have the electrode porosity of 13.53% and 16.65%, respectively, and their loading density was ∼5 mg-cathode cm−2 (Fig. S24, ESI†). Many electrode-processing factors, such as calendering, loading (mg cm−2), electrode thickness, and the amount of residual basic Li species (e.g., LiOH, Li2CO3 generated upon air exposure or non-fully reacted Li precursors), can further affect the cycling performance, which is discussed in the ESI†(Fig. S2, S4, and S25, S26).21,32–34 Of note, we find that the MWCNT-based electrode's performance is much less sensitive to the calendering process than the CB-based electrode (Fig. S4, ESI†). Finally, MWCNT's morphology influences the enhancement in performance (Fig. S27, ESI†). MWCNTs with lower tap density appear more effective than those with higher tap density, facilitating intimate mixing between the AM and carbon and resulting in a larger AM/carbon contact area during electrode preparation. These morphology effects observed with different carbons, such as MWCNT and graphene, warrant dedicated studies in the future.
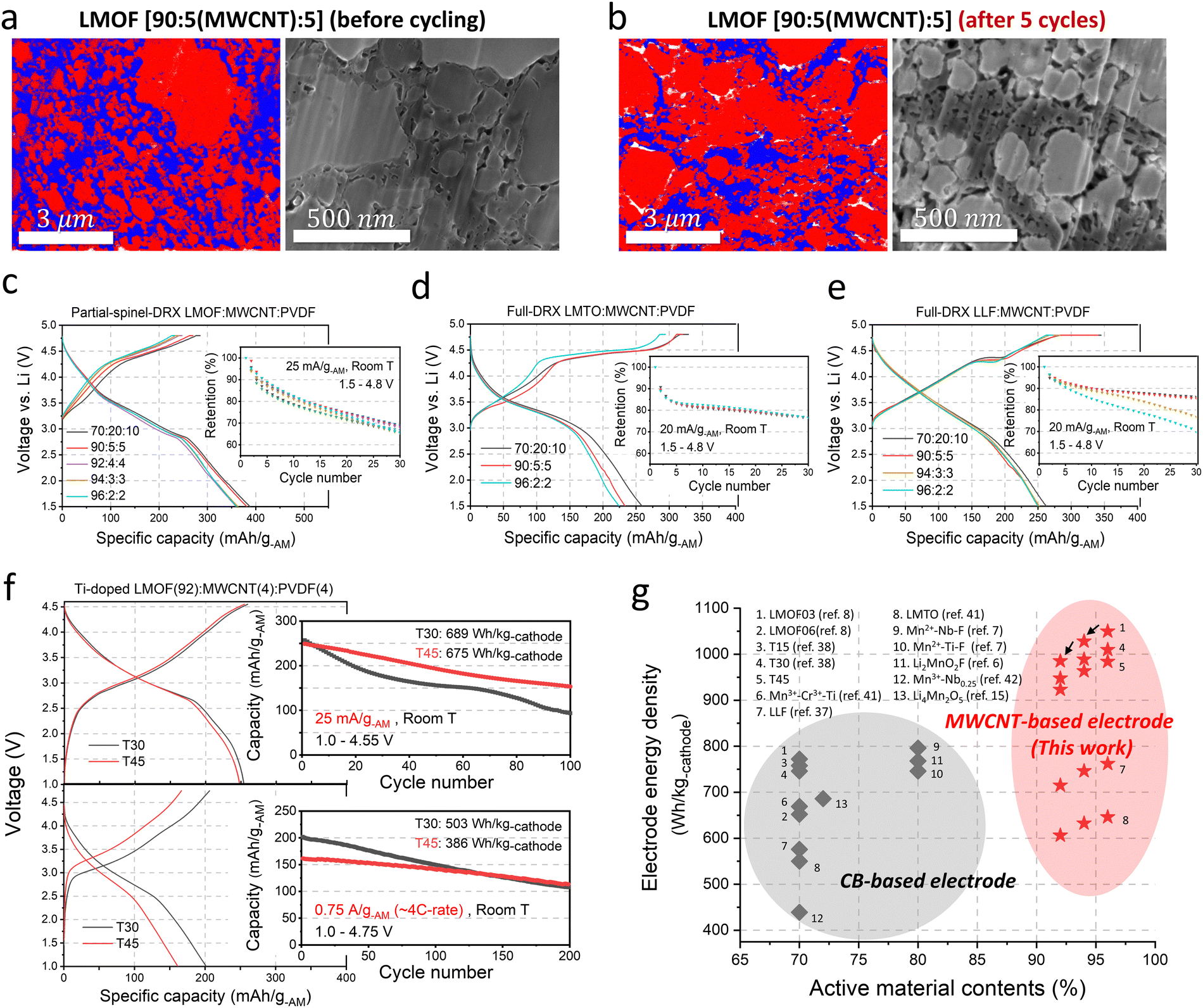 | ||
| Fig. 4 The microstructure and performance of Mn-DRX/MWCNT/PVDF electrodes. (a) and (b) The SEM-EDS mapping (left) of LMOF (red), MWCNT/PVDF (blue), and pore (white) electrode and the SEM images (right) of the LMOF (light gray), MWCNT/PVDF (dark gray), and pores (black) in the 90:5:5 (LMOF:MWCNT:PVDF) electrode (a) before cycling and (b) after 5 cycles [1.5–4.8 V, 25 mA g-AM−1]. (c)–(e) The first cycle voltage profiles and the capacity retention of (c) LMOF in the 70:20:10, 90:5:5, 92:4:4, 94:3:3, and 96:2:2 electrodes with MWCNT [1.5–4.8 V, 25 mA g-AM−1, anode: Li metal], and those of (d) LMTO and (e) LLF in the 70:20:10, 90:5:5, 94(LLF):3:3 and 96:2:2 electrodes with MWCNT [1.5–4.8 V, 20 mA g-AM−1, anode: Li metal]. (f) The first cycle voltage profiles and the capacity retention of full-cells (anode: graphite) using Ti-doped LMOF (T30 and T45) cathodes at (top) 25 mA g-AM−1 [N/P ratio = 1.2, 1.0–4.55 V] and (bottom) 0.75 A g-AM−1 (∼4C-rate) [N/P ratio = 1.2, 1.0–4.75 V] in the 92:4:4 highly-concentrated electrodes with MWCNT. (g) The reported conventional (CB-based) electrode energy density (W h kg-cathode−1) and the MWCNT-based electrode energy density (this work) of various Mn-DRXs as a function of AM contents (%). | ||
We still observe some pore generation for the 90:5(MWCNT):5 LMOF electrode (1.5–4.8 V, 25 mA g-AM−1, after 5 cycles) during cycling, seen as white regions in the EDS map or black areas in the SEM image (Fig. 4(a) and (b)). Such residual porosity increase may constitute remaining capacity fading along with LMOF's AM-level problems. Meanwhile, unlike the highly fragile 80:10(CB):10 LMOF electrode after 20 cycles, the 92:4(MWCNT):4 electrode does not display such fragility, indicating improved mechanical property even with less binder (Fig. S12, ESI†). The cross-sectional SEM analysis (Fig. S28, ESI†) confirms the improved mechanical properties of the MWCNT-based electrode, in which crack-generation and pore-propagation are barely seen even in the 96:2(MWCNT):2 electrode after 20 cycles, likely because the vine-like MWCNT entangles with other components to hold them together, as in CNT metal matrix composites.35,36 Moreover, our electrode peeling test shows ∼70% higher load needed to peel off the 96:2(MWCNT):2 electrode than the 96:2(CB):2 electrode, confirming improved electrode mechanical properties with MWCNT (Fig. S26, ESI†).
We also applied MWCNT to LMTO and Li1.25Mn0.75O1.33F0.67 (LLF,37 full DRX, Fig. S29, ESI†). Even the 96:2(MWCNT):2 LMTO electrode delivers ∼220 mA h g-AM−1 (∼673 W h kg-AM−1, ∼2560 W h l-AM−1) with similar capacity retention as the 70:20(MWCNT):10 and 90:5(MWCNT):5 electrodes (1.5–4.8 V, 20 mA g-AM−1), contrary to the 90:5(CB):5 electrode showing zero capacity (Fig. 4(d) and 2(g)). Meanwhile, the 1st-discharge-capacity reduction with higher AM content (less MWCNT) is more pronounced for LMTO than LMOF, considering that LMOF's capacity reduction is ∼7% in the 70:20(MWCNT):10 and 96:2(MWCNT):2 electrodes but ∼14% for LMTO (Fig. S30, ESI†). This trend correlates well with the ∼380 times poorer electrical conductivity of LMTO than LMOF, making its capacity more sensitive to the carbon amount (here MWCNT). In this regard, LMTO's rate capability in the MWCNT electrode also depends more on the MWCNT amount than LMOF (Fig. S23, ESI†).
Similarly, LLF delivers ∼250 mA h g-AM−1 (∼786 W h kg-AM−1, ∼3000 W h l-AM−1) even in the 96:2(MWCNT):2 electrode (Fig. 4(e)), which is impossible with CB (Fig. S31, ESI†). However, unlike LMOF and LMTO, LLF's capacity retention becomes notably poorer with 94 wt%-AM or 96 wt%-AM (Fig. 4(e)). The relative capacity retention after 30 cycles of the LLF electrode with 96 wt%-AMvs. 70 wt%-AM is only ∼81%, while that of LMOF and LMTO electrodes are ∼100% and ∼98%, respectively (Fig. S30, ESI†). This appears contradictory as LLF (∼1.52 × 10−9 S cm−1) has 10 times higher electrical conductivity than LMTO, but LLF shows a much larger volume change (∼7%) than LMTO (∼4.3%) during cycling to promote the electrode disintegration more significantly. Interestingly, the volume change of LMOF (∼8.4%) is slightly larger than LLF, but the MWCNT-based LMOF electrode exhibits nearly the same capacity retention between the 96 wt%-AM and 70 wt%-AM cases. This difference is most likely due to the much higher electrical conductivity of LMOF than LLF (by ∼38 times) and LMTO (by ∼380 times), enabling LMOF to better carry currents itself (in addition to MWCNT) to maintain electrical conduction in the electrode even with some porosity increase. Thus, for stable capacity in the AM-concentrated electrode, Mn-DRX must be either more electrically conductive, have a small volume change, or be assembled in a highly stable electrode composite matrix such that a given AM-volume change leads to smaller degradation of the electrode and electrical percolation. In this regard, binders with higher adhesive strength, such as styrene–butadiene rubber (SBR)/sodium salt of carboxymethyl cellulose (CMC) and high-molecular-weight PVDF, can further improve the capacity retention of a highly AM-concentrated Mn-DRX electrode by improving the mechanical stability of the composite electrode. For instance, after 50 cycles (1.5–4.8 V, 25 mA g-AM−1), LMOF's capacity retention in the 96:2(MWCNT):2 electrode improves from 47% to 64% as the PVDF5130 binder (that we used to make all electrodes except for this test) is replaced with SBR/CMC binder (Fig. S32, ESI†). Also, this stronger adhesion with SBR/CMC binder leads to a nearly two times higher load needed to peel off the 96:2(MWCNT):2(SBR/CMC) electrode than the 96:2(MWCNT):2(PVDF) electrode (Fig. S26, ESI†).
Using the 92:4(MWCNT):4(PVDF) electrode, we evaluated the performance of Mn-DRX cathodes in a full cell with a graphite anode. (Fig. 4(f)) The Li1.68Mn1.30Ti0.30O3.7F0.3 (T30) and Li1.68Mn1.15Ti0.45O3.7F0.3 (T45), Ti-doped versions of partially ordered LMOF, were used in this test, as they exhibit better capacity/voltage-retention, reduced transition-metal (TM) dissolution, and higher coulombic efficiency than LMOF, rendering them more suitable for the full-cell tests.38 The half-cell performance of T45 in the 92:4(MWCNT):4(PVDF) electrode is shown in the ESI,† demonstrating similar performance across different cathode loading up to ∼17 mg-cathode cm−2 (Fig. S25, ESI†). When cycled in a full cell between 1.0–4.55V (in Fig. 4(f), or 1.0–4.75 V in Fig. S35, ESI†) at 25 mA g-AM−1, T30 and T45 achieve 689 W h kg-cathode−1 (770 W h kg-cathode−1) and 675 W h kg-cathode−1 (737 W h kg-cathode−1), which are comparable to the energy density of the LiNi0.8Co0.1Mn0.1O2 cathode in a full cell (699–732 W h kg-cathode−1, depending on the upper cut-off voltage, Fig. S35, ESI†): the specific capacity and energy density of T30 and T45 reported here are based on the weight of the T30 and T45 after pre-lithiation of the compounds required to achieve a high capacity in a full cell, where (unlike in a half-cell) over-lithiation of the partially ordered (initially Li-vacancy containing) compounds during the first discharging is not possible due to the use of a non-Li-containing graphite anode. Furthermore, 92:4(MWCNT):4 T30 and T45 electrodes show 202 mA h g-AM−1 and 162 mA h g-AM−1 discharge capacity in a full cell with 53% and 70% capacity retention over 200 cycles at a high current of 0.75 A g-AM−1 (∼4C rate, 1.0–4.75 V, Fig. 4(f), bottom), respectively. This corresponds to energy densities of 503 W h kg-cathode−1 (power density of 670 W kg-cathode−1) for T30 and 386 W h kg-cathode−1 (power density of 673 W kg-cathode−1) for T45. In the ESI,† we also show the full-cell performance of other Mn-DRX-MWCNT electrodes (Fig. S36, ESI†).
Electrical conductivity of Mn-DRXs primarily determined by the Mn-content
We showed that electrical percolation engineering with MWCNT can restore Mn-DRX's capacity in the nearly all-AM cathode, achieving the highest cathode energy density of ∼1000 W h kg-cathode−1 reported to date for Mn-DRXs (Fig. 4(g)). However, Mn-DRX's volume change still contributes to the residual capacity loss, whose detrimental effect is less if the AM has higher electrical conductivity (e.g., LMOF vs. LLF). Also, AM electrical conductivity dictates the rate capability in AM-concentrated electrodes. Hence, improving the electrical conductivity while limiting the volume change of Mn-DRXs is critical for developing practical all-AM Mn-DRX cathodes.We explore this further and find that Mn-content correlates well with the Mn-DRX's electrical conductivity; thus, keeping the content high would be beneficial. Fig. 5(a) shows the electrical conductivities of Li+–Mn3+–Ti4+–O2− and Li+–Mn3+–Nb5+–O2− pellets: note the trend is more important here than the absolute measured values as they can slightly change with different measurement techniques and particle morphology (Fig. S9, ESI†). As the Li-excess level increases from 10% to 20% and 30%, electrical conductivity decreases from 2.62 × 10−8 S cm−1 to 1.51 × 10−10 S cm−1 and 3.60 × 10−11 S cm−1 for Li+–Mn3+–Ti4+–O2−; and 2.29 × 10−8 S cm−1 to 7.35 × 10−9 S cm−1 and 4.12 × 10−10 S cm−1 for Li+–Mn3+–Nb5+–O2−, thus showing a lower electrical conductivity with higher Li-excess (reduced Mn-content). Interestingly, Li+–Mn3+–Nb5+–O2− exhibits higher conductivities than Li+–Mn3+–Ti4+–O2− when highly Li-excessed, with ∼50 times greater value at the 20% Li-excess level. Ti4+ and Nb5+ are d0 TM species without d electrons, making them not oxidizable upon cathode charging to participate in charging-induced hole polaron conduction (to be discussed below). Also, they barely constitute valence or conduction bands of Mn3+-DRXs (Fig. S45, ESI†), limiting their participation in thermally excited charge-carriers conduction as well. Therefore, such a difference in electrical conductivity is most likely due to a higher Mn-content in Li+–Mn3+–Nb5+–O2− than Li+–Mn3+–Ti4+–O2− at the same Li-excess level. For instance, at 20% Li-excess level, Mn-content is Mn0.6 (Li1.2Mn0.6Nb0.2O2) for Li+–Mn3+–Nb5+–O2−; whereas it is Mn0.4 (Li1.2Mn0.4Ti0.4O2) for Li+–Mn3+–Ti4+–O2−.
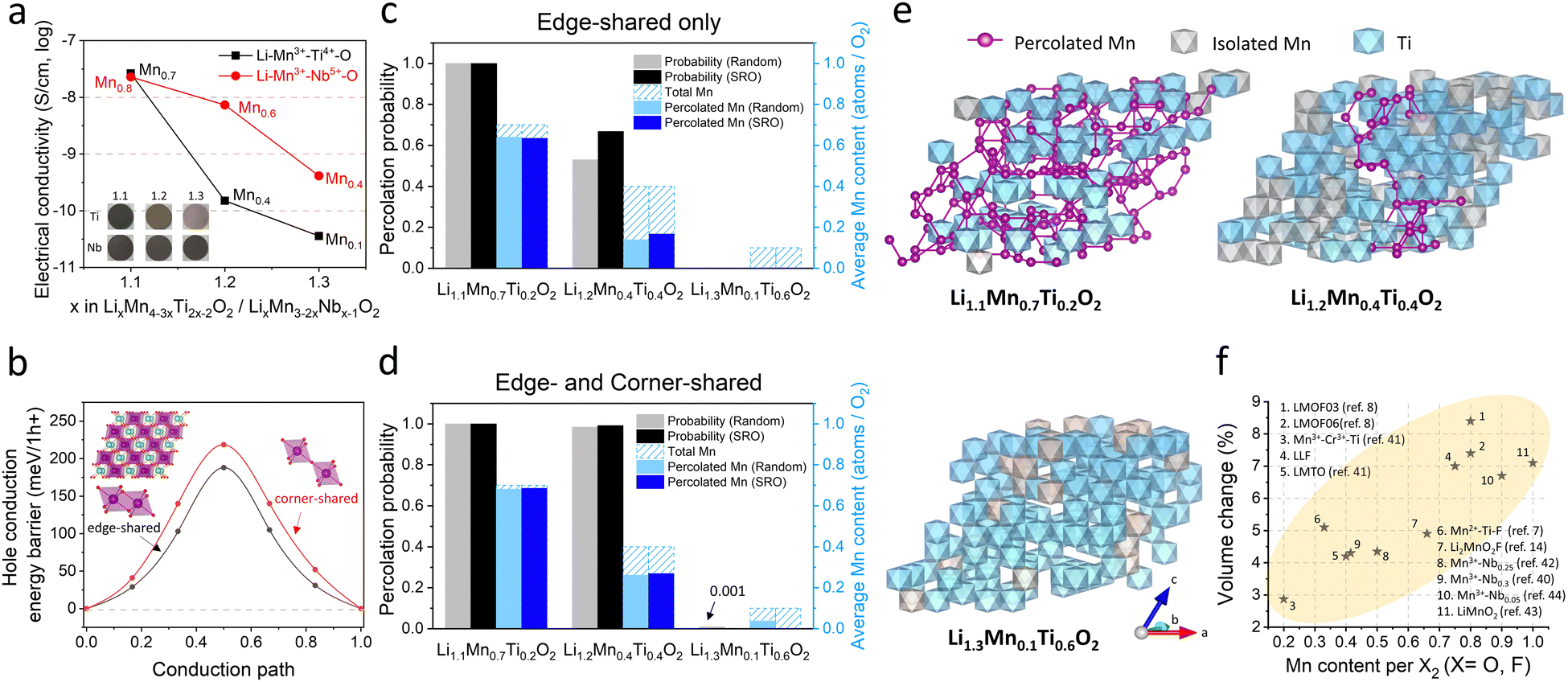 | ||
| Fig. 5 Electrical conductivities and volume change of Mn-DRXs. (a) Apparent electrical conductivities (S cm−1, log) of Li+–Mn3+–Ti4+–O2− and Li+–Mn3+–Nb5+–O2− as a function of the Li-excess level (the Mn-content level). The inset shows the pellets used in this measurement. (b) Energy landscape upon hole polaron hopping through edge-shared or corner-shared Mn–Mn path. (c), (d) Mn percolation probability (∼electron percolation probability) and the average percolated Mn-content per stoichiometric O2 in the DRX Li+–Mn3+–Ti4+–O2− compounds through (c) only edge-shared path and (d) both edge- and corner-shared paths, obtained by random-based Monte Carlo (MC) and SRO-considered Markov Chain MC (MCMC) percolation simulations. (e) Representative MC structures for Li1.1Mn0.7Ti0.2O2, Li1.2Mn0.4Ti0.4O2 and Li1.3Mn0.1Ti0.6O2. Mn ions included in the electron percolating network through edge- and corner-shared paths are labeled with purple spheres, and connected Mn sites are bridged with purple bonds. (f) The reported 1st charge volume change (%, the absolute value of (volume (1st charge) − volume (pristine))/volume (pristine) × 100%) of various Mn-DRX cathodes as a function of Mn-content per X2 (X = O, F), except for partially ordered Mn-DRXs (LMOF03, LMOF06) for which the 2nd charge volume change was considered due to the over-lithiation in the 1st discharge. | ||
This trend is consistent with our theoretical study on electrical conduction in Mn-DRXs using DFT calculations and Monte Carlo (MC) percolation simulation. Electrical conduction can stem from ionic and electronic (electron, hole) transport. Here we consider charging-induced hole transport as the primary conductivity source, as holes are naturally introduced upon charging the cathode. Ionic conduction should be a minor source for our samples. Otherwise, the electrical conductivity would rather increase with higher Li-excess due to improved Li+ diffusion through the 0-TM percolation network.11
As the Fermi level of Mn-DRXs lies within the Mn 3d band in the discharge state, we calculated the hole polaron hopping barrier through connected Mn sites (Fig. 5(b)). For this calculation, we prepared a cation-mixed LiMnO2 model structure containing symmetric edge-shared and corner-shared Mn–Mn octahedra. We find that the hole polaron hopping barrier is lower when the Mn–Mn octahedra are edge-shared (∼190 meV per h+) rather than corner-shared (∼220 meV per h+), implying that hole conduction would be faster in Mn-DRXs if the crystal structure had more edge-shared Mn-octahedra. The corner-shared path may also contribute to the conduction, as the energy gap (30 meV per h+) between the edge-shared and corner-shared Mn–Mn paths is not high.
Since hole polaron conduction in Mn-DRX benefits from continuously connected Mn-sites, we studied the percolation property of Mn-sites in the two types of DRX model structures using MC percolation simulation for random cation order and Markov Chain MC (MCMC) percolation simulation considering short-range order (SRO). Fig. 5(c) shows the percolation probability of “edge-shared” Mn-sites in Li+–Mn3+–Ti4+–O2− and the average percolated Mn-content per stoichiometric O2 within the percolation network. Regarding the structure with a random cation order, the percolation probability drastically decreases from 100% to 50%, and 0% as the Li-excess level increases from 10% (Li1.1Mn0.7Ti0.2O2) to 20% (Li1.2Mn0.4Ti0.4O2) and 30% (Li1.3Mn0.1Ti0.6O2). Moreover, the average percolated Mn-content per O2 within the percolation network also significantly decreases with higher Li-excess (lower Mn-content). Adding the “corner-shared” Mn octahedra in the simulation (Fig. 5(d)), the percolation probability remains near 100% for both Li1.1Mn0.7Ti0.2O2 and Li1.2Mn0.4Ti0.4O2. Nevertheless, the fraction of Mn in the percolation network decreases from 98% to 63%, suggesting that many holes on Mn generated upon charging cannot directly participate in the electrical conduction in Li1.2Mn0.4Ti0.4O2 even after accounting for both edge-sharing and corner-sharing Mn–Mn hole hopping, as visualized with representative MC structures in Fig. 5(e). In the same vein, the structure considering SRO shows the equivalent trend (Fig. 5(c) and (d)). The SRO (Mn-clustering; agglomerating Mn ions) in Li+–Mn3+–Ti4+–O2− slightly improves the Mn-percolation probability and accessible Mn-content within the network, showing an ambivalent effect of SRO that was shown to bring a “negative” impact on 0-TM percolation for Li diffusion in Mn-DRXs.19 Overall, these results support our experimental finding that the Mn-content correlates well with the electrical conductivity of Mn-DRXs.
We want to point out that at high states of charge, electron polaron conduction will prevail over hole polaron conduction due to the changes in the majority of charge carriers from holes to electrons. However, our Mn-connectivity analysis remains applicable within the states of charge where Mn3+ and Mn4+ coexist, as both holes and electrons must pass through the connected Mn-sites. One catch is when high states of charge involve O oxidation, which tends to form O–O dimers.9,39 In this case, polaron conduction would become slower, continuously reforming O–O dimers along the conduction pathways. In this regard, limiting the usage of O-redox to avoid O–O dimer formation would improve the electrical conductivity, which also suggests that higher Mn-content (thus higher Mn-redox capacity to use before O-redox) in Mn-DRXs would be beneficial.
Note that along with the cycling-induced polaronic conduction discussed above, thermally-excited conduction-band electrons and valence-band holes may also contribute to electrical conduction (although their contribution will be orders of magnitude smaller due to the large band gap in Mn-DRXs), whose degree depends on the material's band gap, band structure, and temperature. In the ESI,† we additionally discuss this thermally-excited charge-carrier conduction (Fig. S45 and S46, ESI†), showing that Mn-DRXs’ band gap decreases with a higher Mn-content to lead to the same conclusion that increasing Mn content improves the electrical conductivity of Mn-DRXs as the case of the charging-induced hole-polaronic electrical conduction.
Trade-off role of Mn-content for the Mn-DRX cathode design
While increasing the Mn-content improves the electrical conductivity, we recognize that a higher Mn-content generally increases Mn-DRX's volume change during cycling (Fig. 5(f)), likely due to more pronounced Jahn–Teller (JT) distortion resulting from a greater content of Mn3+.40 For instance, while Li1.2Mn0.4Ti0.4O2 (Mn0.4),41 Li1.25Mn0.5Nb0.25O2 (Mn0.5),42 and Li1.30Mn0.4Nb0.3O2 (Mn0.4)40 exhibit volume change of ∼4.3% (after ∼280 mA h g-AM−1 charge), ∼4.3% (∼300 mA h g-AM−1 charge), and ∼4.2% (∼270 mA h g-AM−1 charge), LiMnO2 (Mn1.0)43 and Li1.05Mn0.9Nb0.05O2 (Mn0.9)44 show larger volume change of ∼7.1% (∼200 mA h g-AM−1 charge) and ∼6.7% (∼180 mA h g-AM−1 charge). Thus, Mn-DRXs with a higher Mn-content undergo larger volume change, which is exacerbated when cycling a large amount of Li, as in the case of LMOF (Mn0.8 per O1.85F0.15, cycling 360 mA h g−1) or LLF (Mn0.75 per O1.33F0.67, cycling 250 mA h g−1) that undergo >7% volume change. Recently, Ceder et al. have shown that DRXs using TM eg electrons as the electron source (which is the case of Mn-DRXs using Mn2+/Mn3+/Mn4+ redox) exhibit larger volume change than the ones using TM t2g electrons (e.g., Cr3+, V3+).40 Substituting “t2g” TM species for Mn in Mn-DRXs may reduce volume change at a given total TM content. In particular, doping Cr3+, which (when holes are generated on them, e.g., Cr4+) can also participate in electrical conduction in Mn-DRX by having a similar electron energy level as Mn3+ and give similarly high operating potential as Mn, may simultaneously limit the volume change while enhancing or preserving the electrical conductivity in Mn-DRXs without losing energy density. Meanwhile, it is also possible for Cr3+ to disproportionate into Cr3+ and Cr6+ upon charging for their generally preferred chemical stability over Cr4+ or Cr5+ if the local structure around the species energetically favors the disproportionation.45 This does not necessarily mean that polaronic hole transport through a mixed Mn-Cr network in Cr-doped Mn-DRXs will be disturbed: for example, the following hole (h+) transport can occur: 3 Mn4+(3 h+) − 3 Cr3+ − 3 Mn3+ → 3 Mn3+ − 2 Cr3+ & 1 Cr6+(3 h+) − 3 Mn3+ → 3 Mn3+ − 3 Cr3+ − 3 Mn4+(3 h+). However, more study on this would be needed as such collective polaron transport involving multiple TM cations may increase the polaron hopping barrier.Supposing that simultaneously improving the electrical conductivity while limiting the volume change appears impossible, we suggest prioritizing sacrificing intrinsic electrical conductivity and instead minimizing AM volume change since Mn-DRX's poor electrical conductivity can be overcome at the cathode film level by using advanced conductive additives like MWCNT, as demonstrated in this work, or carbon coating of AM. Also, in this way, one does not need to maximize the Mn-content, accompanying minimizing the Li-excess level needed to endow 0-TM percolation for facile Li diffusion in Mn-DRX, which will become progressively more important when cycling large-size Mn-DRX particles for which long-range Li diffusion is critical.44
Finally, addressing the capacity fading observed in Mn-DRX cathodes and improving their performance requires attention to additional material-level issues, such as the JT distortion around Mn3+ ions, which can promote Mn dissolution and aggravate volume changes,38,40 irreversible cation rearrangement in the bulk structure,46 and the degradation of the DRX particle surface due to side reactions with the electrolyte or oxygen loss, both of which can degrade the Li diffusion (Li+ conductivity) in Mn-DRX and cause voltage fading.9 Therefore, alongside electrode-level engineering, it is essential to incorporate additional material-level strategies to effectively mitigate these challenges. Furthermore, while this study primarily addressed electrical percolation engineering within electrodes to enhance electron transfer, additional attention should be given to engineering of the electrode's microstructure (e.g., pore size/distribution, electrode thickness) since it can affect the Li+ ion conduction (beside electrical conduction) within the electrode and the degree of side reactions with the electrolyte, which can affect both the overall capacity and capacity retention of a cathode film by modulating charge- and mass-transfer resistances and their evolution during cycling.
Conclusions
This study showcased Mn-DRX cathodes with nearly all-AM content (>95 wt%-AM), a significant advancement beyond the 70–80 wt%-AM range reported in previous literature, achieving the highest application-level energy density reported to date. This achievement was made possible by a comprehensive understanding of the failure mechanisms in AM-concentrated Mn-DRX cathodes, which encompassed both inherent material properties and electrode microstructure. The failures observed in AM-concentrated Mn-DRX cathodes can be attributed to the remarkably low intrinsic electrical conductivity of Mn-DRXs (10−10–10−8 S cm−1) and electrochemical fatigue phenomena such as porosity growth and crack propagation within the electrode. These issues arise due to the substantial volume change (>8%) and pulverized morphology of Mn-DRXs, ultimately leading to the collapse of the electrical percolation network within the electrode, thereby limiting their electrochemical performance. To address these issues, an electrode engineering strategy was introduced, utilizing multi-functional carbon and adhesive binders to build a robust electrical percolation network. This approach enabled the realization of nearly all-AM (96 wt%-AM) Mn-DRX cathodes with an energy density of ∼1050 W h kg-cathode−1. Furthermore, it was discovered that while increasing the Mn-content in Mn-DRXs improves electrical conductivity, it exacerbates volume change during cycling. This trade-off underscores the importance of addressing intricate electro-chemo-mechanical failures in highly AM-concentrated Mn-DRX cathodes through complementary electrode-level engineering, as demonstrated in this study, rather than relying solely on the composition engineering of Mn-DRXs. Overall, this work advances Mn-DRX research to a higher technology readiness level, paving the way for developing high-energy, nearly all-AM Co/Ni-free Li-ion cathodes.Author contributions
E. L., D.-H. S. and J. L., planned the project. D.-H. S. and J. L. supervised all aspects of the research. E. L. performed overall experimental and computational parts with the interpretation of the results. D.-H. L. contributed to the experiments and the Monte Carlo simulation. S. B., N. B. and R. G. performed a characterization and an interpretation via SEM analysis. S.-W. P., H. K. and G. L. assisted experiments. E. L., D.-H. S. and J. L. wrote the manuscript and the manuscript was revised by R. M.Conflicts of interest
There authors declare no conflict of interest.Acknowledgements
Discovery Grant (RGPIN-2020-04463) and Discovery Accelerator Supplements (RGPAS-2020-00115) given to J. L. by the Natural Sciences and Engineering Research Council of Canada (NSERC); Korea Institute of Energy Technology Evaluation and Planning (KETEP) grant funded by the Korean government (MOTIE) (20214000000660) and the National Research Foundation of Korea funded by the Ministry of Science and Information & Communication Technology (2023R1A2C2008242, RS-2023-00282389 and RS-2023-00283902) given to D.-H. S.; the Natural Sciences and Engineering Research Council of Canada (NSERC) operating grant given to R. G.; the computational work was supported by the Supercomputing Center/Korea Institute of Science and Technology Information with supercomputing resources, including technical support (KSC-2023-CRE-0199 to D.-H. S.).References
- J. Liu, J. G. Zhang, Z. Yang, J. P. Lemmon, C. Imhoff, G. L. Graff, L. Li, J. Hu, C. Wang and J. Xiao, Adv. Funct. Mater., 2013, 23, 929–946 CrossRef CAS.
- D. Chung, T. James, E. Elgqvist, A. Goodrich and S. Santhanagopalan, Automotive Lithium-ion Battery (LIB) supply chain and U.S. Competitiveness Considerations, National Renewable Energy Lab.(NREL), Golden, CO (United States), 2015 Search PubMed.
- J. Xu, F. Lin, M. M. Doeff and W. Tong, J. Mater. Chem. A, 2017, 5, 874–901 RSC.
- F. Lin, D. Nordlund, I. M. Markus, T.-C. Weng, H. L. Xin and M. M. Doeff, Energy Environ. Sci., 2014, 7, 3077–3085 RSC.
- G. Chen, X. Song and T. J. Richardson, Electrochem. Solid-State Lett., 2006, 9, A295 CrossRef CAS.
- R. Sharpe, R. A. House, M. J. Clarke, D. Förstermann, J.-J. Marie, G. Cibin, K.-J. Zhou, H. Y. Playford, P. G. Bruce and M. S. Islam, J. Am. Chem. Soc., 2020, 142, 21799–21809 CrossRef CAS PubMed.
- J. Lee, D. A. Kitchaev, D.-H. Kwon, C.-W. Lee, J. K. Papp, Y.-S. Liu, Z. Lun, R. J. Clement, T. Shi and B. D. McCloskey, Nature, 2018, 556, 185–190 CrossRef CAS PubMed.
- H. Ji, J. Wu, Z. Cai, J. Liu, D.-H. Kwon, H. Kim, A. Urban, J. K. Papp, E. Foley and Y. Tian, Nat. Energy, 2020, 5, 213–221 CrossRef CAS.
- H. Li, R. Fong, M. Woo, H. Ahmed, D.-H. Seo, R. Malik and J. Lee, Joule, 2022, 6, 53–91 CrossRef CAS.
- N. Yabuuchi, M. Takeuchi, M. Nakayama, H. Shiiba, M. Ogawa, K. Nakayama, T. Ohta, D. Endo, T. Ozaki and T. Inamasu, Proc. Natl. Acad. Sci. U. S. A., 2015, 112, 7650–7655 CrossRef CAS PubMed.
- J. Lee, A. Urban, X. Li, D. Su, G. Hautier and G. Ceder, Science, 2014, 343, 519–522 CrossRef CAS PubMed.
- L. Li, B. Ouyang, Z. Lun, H. Huo, D. Chen, Y. Yue, C. Ophus, W. Tong, G. Chen and G. Ceder, Nat. Commun., 2023, 14, 7448 CrossRef PubMed.
- J. Qian, Y. Ha, K. P. Koirala, D. Huang, Z. Huang, V. S. Battaglia, C. Wang, W. Yang and W. Tong, Adv. Funct. Mater., 2023, 33, 2205972 CrossRef CAS.
- A. Kanno, Y. Ugata, I. Ikeuchi, M. Hibino, K. Nakura, Y. Miyaoka, I. Kawamura, D. Shibata, T. Ohta and N. Yabuuchi, ACS Energy Lett., 2023, 8, 2753–2761 CrossRef CAS.
- M. Freire, N. V. Kosova, C. Jordy, D. Chateigner, O. Lebedev, A. Maignan and V. Pralong, Nat. Mater., 2016, 15, 173–177 CrossRef CAS PubMed.
- T. Y. Huang, Z. Cai, M. J. Crafton, L. A. Kaufman, Z. M. Konz, H. K. Bergstrom, E. A. Kedzie, H. M. Hao, G. Ceder and B. D. McCloskey, Adv. Energy Mater., 2023, 2300241 CrossRef CAS.
- J. Huang, B. Ouyang, Y. Zhang, L. Yin, D.-H. Kwon, Z. Cai, Z. Lun, G. Zeng, M. Balasubramanian and G. Ceder, Nat. Mater., 2023, 22, 353–361 CrossRef CAS PubMed.
- Y. Yue, Y. Ha, R. Giovine, R. Clément, W. Yang and W. Tong, Chem. Mater., 2022, 34, 1524–1532 CrossRef CAS.
- H. Ji, A. Urban, D. A. Kitchaev, D.-H. Kwon, N. Artrith, C. Ophus, W. Huang, Z. Cai, T. Shi and J. C. Kim, Nat. Commun., 2019, 10, 1–9 CrossRef PubMed.
- N. Yabuuchi, M. Nakayama, M. Takeuchi, S. Komaba, Y. Hashimoto, T. Mukai, H. Shiiba, K. Sato, Y. Kobayashi, A. Nakao, M. Yonemura, K. Yamanaka, K. Mitsuhara and T. Ohta, Nat. Commun., 2016, 7, 13814 CrossRef CAS PubMed.
- R. Sim, S. Lee, W. Li and A. Manthiram, ACS Appl. Mater. Interfaces, 2021, 13, 42898–42908 CrossRef CAS PubMed.
- G. Chen, C. C. Waraksa, H. Cho, D. D. Macdonald and T. E. Mallouka, J. Electrochem. Soc., 2003, 150, E423 CrossRef CAS.
- C. Park, E. Lee, S. H. Kim, J.-G. Han, C. Hwang, S. H. Joo, K. Baek, S. J. Kang, S. K. Kwak and H.-K. Song, J. Power Sources, 2022, 521, 230923 CrossRef CAS.
- Y.-N. Xu, S.-Y. Chung, J. T. Bloking, Y.-M. Chiang and W. Ching, Electrochem. Solid-State Lett., 2004, 7, A131 CrossRef CAS.
- Y. Itou, N. Ogihara and S. Kawauchi, J. Phys. Chem. C, 2020, 124, 5559–5564 CrossRef CAS.
- M. Indrikova, S. Grunwald, F. Golks, A. Netz, B. Westphal and A. Kwade, J. Electrochem. Soc., 2015, 162, A2021 CrossRef CAS.
- J. Qian, C. G. Wiener, Y. Zhu and B. D. Vogt, Polymer, 2018, 143, 237–244 CrossRef CAS.
- K. Sheem, Y. H. Lee and H. S. Lim, J. Power Sources, 2006, 158, 1425–1430 CrossRef CAS.
- B. Song, T. Sui, S. Ying, L. Li, L. Lu and A. M. Korsunsky, J. Mater. Chem. A, 2015, 3, 18171–18179 RSC.
- Q. Cao, Q. Yu, D. W. Connell and G. Yu, Clean Technol. Environ. Policy, 2013, 15, 871–880 CrossRef CAS.
- L.-J. Her, J.-L. Hong and C.-C. Chang, J. Power Sources, 2006, 161, 1247–1253 CrossRef CAS.
- K.-Y. Park, J.-W. Park, W. M. Seong, K. Yoon, T.-H. Hwang, K.-H. Ko, J.-H. Han, Y. Jaedong and K. Kang, J. Power Sources, 2020, 468, 228369 CrossRef CAS.
- C. Heubner, A. Nickol, J. Seeba, S. Reuber, N. Junker, M. Wolter, M. Schneider and A. Michaelis, J. Power Sources, 2019, 419, 119–126 CrossRef CAS.
- C.-x Zhou, P.-b Wang, B. Zhang, L.-b Tang, H. Tong, Z.-j He and J.-c Zheng, ACS Appl. Mater. Interfaces, 2019, 11, 11518–11526 CrossRef CAS PubMed.
- X.-M. Liu, Z. Dong Huang, S. Woon Oh, B. Zhang, P.-C. Ma, M. M. Yuen and J.-K. Kim, Compos. Sci. Technol., 2012, 72, 121–144 CrossRef CAS.
- X.-M. Liu, Z.-D. Huang, S. Oh, P.-C. Ma, P. C. Chan, G. K. Vedam, K. Kang and J.-K. Kim, J. Power Sources, 2010, 195, 4290–4296 CrossRef CAS.
- Z. Lun, B. Ouyang, Z. Cai, R. J. Clément, D.-H. Kwon, J. Huang, J. K. Papp, M. Balasubramanian, Y. Tian and B. D. McCloskey, Chem, 2020, 6, 153–168 CAS.
- E. Lee, T. U. Wi, J. Park, S. W. Park, M. H. Kim, D. H. Lee, B. C. Park, C. Jo, R. Malik and J. H. Lee, Adv. Mater., 2023, 35, 2208423 CrossRef CAS PubMed.
- K. McColl, R. A. House, G. J. Rees, A. G. Squires, S. W. Coles, P. G. Bruce, B. J. Morgan and M. S. Islam, Nat. Commun., 2022, 13, 5275 CrossRef CAS PubMed.
- X. Zhao, Y. Tian, Z. Lun, Z. Cai, T. Chen, B. Ouyang and G. Ceder, Joule, 2022, 6, 1654–1671 CrossRef CAS.
- J. Huang, P. Zhong, Y. Ha, D.-H. Kwon, M. J. Crafton, Y. Tian, M. Balasubramanian, B. D. McCloskey, W. Yang and G. Ceder, Nat. Energy, 2021, 6, 706–714 CrossRef CAS.
- R. Wang, X. Li, L. Liu, J. Lee, D.-H. Seo, S.-H. Bo, A. Urban and G. Ceder, Electrochem. Commun., 2015, 60, 70–73 CrossRef CAS.
- T. Sato, K. Sato, W. Zhao, Y. Kajiya and N. Yabuuchi, J. Mater. Chem. A, 2018, 6, 13943–13951 RSC.
- J. Lee, C. Wang, R. Malik, Y. Dong, Y. Huang, D. H. Seo and J. Li, Adv. Energy Mater., 2021, 11, 2100204 CrossRef CAS.
- W. E. Gent, I. I. Abate, W. Yang, L. F. Nazar and W. C. Chueh, Joule, 2020, 4, 1369–1397 CrossRef CAS.
- Y. Shirazi Moghadam, Y. Hu, A. El Kharbachi, S. Belin, T. Diemant, J. Chen, R. A. House, P. G. Bruce and M. Fichtner, Chem. Mater., 2023, 35, 8922–8935 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4ee00551a |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2024 |