
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceElectrochemical urea production using carbon dioxide and nitrate: state of the art and perspectives
Mohsin
Muhyuddin
a,
Giovanni
Zuccante
ab,
Piercarlo
Mustarelli
 a,
Jonathan
Filippi
c,
Alessandro
Lavacchi
c,
Lior
Elbaz
d,
Yu-Han
Chen
e,
Plamen
Atanassov
e and
Carlo
Santoro
*a
a,
Jonathan
Filippi
c,
Alessandro
Lavacchi
c,
Lior
Elbaz
d,
Yu-Han
Chen
e,
Plamen
Atanassov
e and
Carlo
Santoro
*a
aElectrocatalysis and Bioelectrocatalysis Laboratory, Department of Materials Science, University of Milano-Bicocca, U5, Via Roberto Cozzi, 55, 20125, Milano, Italy. E-mail: carlo.santoro@unimib.it
bDepartment of Industrial Engineering, University of Padova, Via Marzolo 9, Padova, 35131, Italy
cIstituto di Chimica Dei Composti OrganoMetallici (ICCOM), Consiglio Nazionale Delle Ricerche (CNR), Via Madonna Del Piano 10, 50019 Sesto Fiorentino, Firenze, Italy
dDepartment of Chemistry and the Institute of Nanotechnology and Advanced Materials, Bar-Ilan University, Ramat-Gan, 5290002, Israel
eDepartment of Chemical and Biomolecular Engineering, University of California, Irvine, CA 92697, USA
First published on 24th April 2024
Abstract
Complete decarbonization of hard-to-abate industrial sectors is critical to reach the carbon neutrality goal set for 2050. The production of nitrogen-containing fertilizers (N-fertilizers) is responsible for 2.1% of the overall global carbon dioxide emissions. Urea is the most common N-fertilizer, and it is currently produced through the Bosch–Meiser process starting from ammonia (NH3) and carbon dioxide (CO2). Electrochemical production of urea can reduce drastically the emission of greenhouse gases and the energy required for the process. Promising results were recently reported using nitrate (NO3−) and CO2 as reagents with increasing production rate and faradaic efficiency. In this mini-review, we summarize the most recent studies, including reaction mechanisms, electrocatalysts, and detection methods, highlighting the challenges in the field. A roadmap for future developments is envisioned with the scope of reaching industrial requirements.
 Mohsin Muhyuddin | Mohsin Muhyuddin is a final year doctoral candidate in the Materials Science and Nanotechnology Department of the University of Milano-Bicocca where he is pursuing his research under the supervision of Prof. Carlo Santoro at the Electrocatalysis and Bioelectrocatalysis laboratory. He completed his Bachelor and Master of Science in Materials Science and Engineering from the Institute of Space Technology (IST), Islamabad, Pakistan in the year 2016 and 2019, respectively. Presently, Mohsin's area of research includes the development of advanced and cost-effective noble metal-free electrocatalysis for sustainable energy applications. |
 Alessandro Lavacchi | Alessandro Lavacchi is a senior researcher at the Institute of Organometallic Chemistry of the National Research Council of Italy, where he is also the head of the Electron Microscopy Facility and the elected head of the Technology Transfer Center. His research focuses on the development and characterization of nanomaterials for electrochemical conversion and storage, with a focus on alkaline membrane electrolysis and biomass conversion in electrochemical devices. He has co-authored more than 130 papers, mostly on energy materials, and the monograph “Nanotechnology in Electrocatalysis for Energy”. |
 Lior Elbaz | Lior Elbaz is a Professor at the Chemistry Department, Bar-Ilan University. He is the head of the Israeli Fuel Cells Consortium (IFCC) and the Director of the Hydrogen Technologies lab at the Israel National Institute for Sustainable Energy (NISE). In his current work, Lior has been focusing on the development of molecular and PGM-free aerogel-based catalysts for hydrogen technologies. |
 Yu-Han Chen | Yu-Han Chen is a second-year PhD student in the Department of Chemical and Biomolecular Engineering at University of California, Irvine. She obtained her BS degree in Chemical Engineering in National Taiwan University. Later, she had 6 years working experience in the petrochemical industry. Presently, she is working in Prof. Plamen Atanassov's group in UC Irvine, where she conducts research on developing electrocatalysts for nitrate reduction to ammonia and co-reduction of nitrate and carbon dioxide to urea. |
 Plamen Atanassov | Plamen Atanassov is a Chancellor's Professor of Chemical & Biomolecular Engineering, University of California Irvine, where his research focuses on the development of non-platinum electrocatalysts for fuel cells, nano-structured platinum-based catalysts and advanced supports, catalysts for CO2 electroreduction and product valorization, ammonia electrosynthesis and new materials and technologies for energy conversion and storage. He is participating in the effort to build California's hydrogen hub by serving on the executive team for Alliance for Renewable Clean Hydrogen Energy Systems (ARCHES) and interfacing with hydrogen technology demonstration and research efforts regionally and Nationwide. Plamen Atanassov was inducted into the National Academy of Inventors, and he is a Fellow of both: The Electrochemical Society and the International Society of Electrochemistry, for which he is the current President-elect. |
 Carlo Santoro | Carlo Santoro got his PhD at the University of Connecticut in 2009, working on microbial fuel cells. He moved to the University of New Mexico in 2013 working on platinum-free electrocatalysts for oxygen reduction reaction and supercapacitive bio-electrochemical systems. Following a spell as a Lecturer at the University of Manchester (2020), he joined the University of Milano-Bicocca in 2021 as Assistant Professor, where he established the Electrocatalysis and Bioelectrocatalysis Lab (EBLab). He is currently Associate Professor and his work focuses on the development of electrocatalysts based on platinum-group metal-free materials for electrochemical systems, pursuing biomimetic and bioinspired approaches. He has published over 130 manuscripts (Hindex = 46) and holds 2 patents. |
Broader contextTo reach the complete decarbonization goal set at 2050, much effort and investment must be pursued in many sectors to decrease their emissions. Chemical industries contribute importantly to greenhouse gas emissions. In particular, nitrogen fertilizers are responsible for 2.1% of the CO2 emissions worldwide. The most used N-fertilizer is urea, synthesized using ammonia and CO2 following the Bosch–Meiser process. Electrifying urea synthesis using electrochemistry is viewed as a potentially disruptive technology for decarbonizing this strategic sector. However, only a few attempts have been carried out using CO2 and a nitrogen containing molecule, e.g. N2, NO, NO2−, or NO3−. The results are promising despite the low activity and selectivity. Attention has recently focused on nitrate due to its high solubility and low bond dissociation energy. This mini-review collects the recent literature and helps to understand the advantages of urea electrosynthesis from CO2 and NO3−, the complex reaction mechanisms and the recent developments in the related electrocatalysis. An in-depth exploration of the in situ and ex situ urea detection methods is presented to help unravel the reaction mechanisms. This work provides a useful overview of the recent achievements in urea electrosynthesis highlighting limitations and challenges and further steps to be pursued for achieving higher technology readiness level. |
Introduction
Manmade greenhouse gas emissions are a significant contributor to climate change, with detrimental consequences that are now becoming more and more visible worldwide. Industry is one of the major contributors to greenhouse gas (GHG) emissions. Particular attention has been given in recent years to the so-called hard-to-abate industrial sectors, where carbon dioxide formation and emission are part of the process. These sectors are responsible for more than 30% of CO2 emissions worldwide. New technologies are required in order to address this issue.The need for fertilizers has been rising throughout the years, along with population growth and an increase in average income per capita, which increases the strain on industrial resources.1 Synthetic nitrogen-containing fertilizer (N-fertilizer) production is responsible for 2.1% of the world's CO2 emissions.2 Ammonia (NH3) is the building block used in most N-fertilizers.
The most common method for ammonia production today is the Haber–Bosch process2,3 which is considered to be energy-intensive and utilizes natural gas to produce hydrogen, a reagent of the process, through steam methane reforming (SMR) and water gas shift reaction (WGSR), as illustrated in Fig. 1(A).4 In this process, 1.9 tons of CO2 are emitted per ton of NH3 produced.2
 | ||
| Fig. 1 (A) Schematic of the Haber–Bosch process for synthesizing ammonia-producing hydrogen through methane. (B) Schematics of a urea production process following the Bosch–Meiser synthetic method. (A) Adapted with permission.3 Royal Society of Chemistry under CC-BY 3.0 (https://creativecommons.org/licenses/by/3.0/). (B) Adapted with permission.12 Copyright 2018, Elsevier LTD. | ||
In 2020 alone, more than 50% of the synthesized NH3 was used for urea synthesis.5 NH3 can be used directly as a fertilizer in its anhydrous form; however, it can be dangerous if not used properly due to its propensity to dehydrate soil, and handling it is also risky for humans.6 NH3 can also be used in compressed liquid form but its storage, transport, handling, and application is energy consuming and dangerous.6
Urea, the main nitrogen-containing fertilizer (N-fertilizer) produced and consumed globally, due to its high nitrogen composition and ease of handling, features low transport and storage costs. Urea is currently produced using the Bosch–Meiser process, which involves the reaction between NH3 with CO2 (Fig. 1(B)).7 The overall production of urea is extremely energy-intensive, costly and is responsible for large emissions of CO2. Urea production is a significant contributor to GHG emissions, as evidenced by a recent life cycle analysis.8 The authors found that at a plant production scale of 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 800000 tons, the ammonia synthesis stage alone contributes to GHG emission with 9.32 × 108 kg CO2 eq., overshadowing the 3.58 × 108 kg CO2 eq. attributed to the urea synthesis stage from CO2 and NH3. Indeed, the cost of urea production is strictly related to the cost of energy and natural gas, which in turn are volatile due to many factors, including geopolitical issues. The cost of urea reached its highest peak of more than 1000 USD per ton in 2022,9 concurrent with the spike in the cost of natural gas skyrocketing due to the Russia–Ukraine war. An increase in the cost of urea increases the cost of crops and the derived processed food. This logical connection is paramount because it underlines how fertilizers are strategically important for every country and their food supply independence. High cost and negative environmental effects are the two main drivers for pursuing more sustainable routes to decarbonize this strategic hard-to-abate sector, while ensuring economic viability.
800000 tons, the ammonia synthesis stage alone contributes to GHG emission with 9.32 × 108 kg CO2 eq., overshadowing the 3.58 × 108 kg CO2 eq. attributed to the urea synthesis stage from CO2 and NH3. Indeed, the cost of urea production is strictly related to the cost of energy and natural gas, which in turn are volatile due to many factors, including geopolitical issues. The cost of urea reached its highest peak of more than 1000 USD per ton in 2022,9 concurrent with the spike in the cost of natural gas skyrocketing due to the Russia–Ukraine war. An increase in the cost of urea increases the cost of crops and the derived processed food. This logical connection is paramount because it underlines how fertilizers are strategically important for every country and their food supply independence. High cost and negative environmental effects are the two main drivers for pursuing more sustainable routes to decarbonize this strategic hard-to-abate sector, while ensuring economic viability.
Electrochemical processes have already proven to be instrumental in the transition to sustainable energy and can be utilized also to decarbonize industrial processes and lower geopolitical tensions. In 2013, Bardi et al. showed that the electrification of intensive agriculture has a big potential in climate change mitigation and that, in this context, electrochemical processes might provide a breakthrough route for the synthesis of agrochemicals.10 Electrochemical reactions have recently been shown to be effective in the production of urea starting from nitrate (NO3−) and CO2. This is a remarkable step as the environmental impact of producing ammonia, a building block for urea synthesis, extends significantly beyond its energy consumption. Indeed, the hydrogen needed for the reaction is derived mainly from natural gas, which emits CO2 during the transformation processing. The CO2 emissions from producing hydrogen through the steam methane reforming (SMR) and the water–gas shift reaction (WGSR) represent over half of the total emissions from the entire ammonia production process.11 From the perspective of urea electrosynthesis from NO3− and CO2, the step of hydrogen electrolysis or hydrogen produced from SMR and WGSR would be avoided. In the review, we survey and comment on the most recent developments in this important field of research.
Electrochemical production of urea
Electrochemical devices have proven to be useful in decarbonizing energy-intensive sectors through electrocatalysis of various reactions. This has been demonstrated, for example, by the exponential growth of green H2 production using water electrolyzers for reducing the emission of sectors such as transportation, energy storage and utilization, steel manufacturing, cement production and other chemical industries.12,13 H2 is a critical reagent in the Haber–Bosch process and green H2 produced through water electrolysis could seamlessly substitute grey hydrogen (from SMR and WGSR), and pave the road for a significant reduction in CO2 emissions.3 More recently, electrocatalysis has been used for reducing CO2 into CO or other valuable commodities.14 The co-production of CO and H2, known as syngas, is of particular interest for many industrial processes, which today use methane.15 In the past few years, electrochemical technologies have been used to produce NH3 starting from nitrogen gas (N2)16 or nitrate (NO3−).17 However, for the production of effective fertilizers, NH3 has to be combined with CO2 to form urea through the Bosch–Meiser process. All of the above-mentioned electrochemical processes require electric energy, which in principle could be provided from sustainable sources and thus regarded as fully climate-neutral.Two recent reviews survey urea electrosynthesis from CO2 and N2, CO2 and NO, CO2 and NO2− and CO2 and NO3−.18,19 These routes have significant drawbacks. The selection of NO3− rather than N2 is dictated by the lower bond dissociation energy of N![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O compared to that of N
O compared to that of N![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) N, which is equal to 204 and 941 kJ mol−1, respectively.20 Moreover, N2 has limited solubility in water (0.02 v/v, 298 K, 1 atm) and this impedes its efficient utilization for urea synthesis. NO has a higher solubility than N2; however, at higher concentrations (i.e. >150 ppm), it is toxic.21 NO3− has a known tendency to form solid-state coordination complexes with the metals that are active in the synthesis of urea. Indeed, Cu and Zn tend to form mixed complexes with urea and nitrate, with the latter acting as a secondary ligand.22 The selection of NO3− is also encouraged by its high solubility in water. On the other hand, NO3− is considered as a water pollutant, leading to severe human and environmental issues such as cancer and eutrophication,23 respectively. Thus, nitrate-rich contaminated water derived from industrial processes can be practically used as a reagent for urea production. For example, in aquaculture, the NO3− concentration can reach 500 mg L−1 (∼8 mM),24,25 and in other industrial processes such as cellophane, explosive, pectin and metal finishing, this value can be higher than 1000 mg NO3-N L−1 (>16 mM).26–28 The usage of nitrate-rich wastewater in urea electrochemical production would avoid direct release into the environment or the costly denitrification in wastewater treatment plants.
N, which is equal to 204 and 941 kJ mol−1, respectively.20 Moreover, N2 has limited solubility in water (0.02 v/v, 298 K, 1 atm) and this impedes its efficient utilization for urea synthesis. NO has a higher solubility than N2; however, at higher concentrations (i.e. >150 ppm), it is toxic.21 NO3− has a known tendency to form solid-state coordination complexes with the metals that are active in the synthesis of urea. Indeed, Cu and Zn tend to form mixed complexes with urea and nitrate, with the latter acting as a secondary ligand.22 The selection of NO3− is also encouraged by its high solubility in water. On the other hand, NO3− is considered as a water pollutant, leading to severe human and environmental issues such as cancer and eutrophication,23 respectively. Thus, nitrate-rich contaminated water derived from industrial processes can be practically used as a reagent for urea production. For example, in aquaculture, the NO3− concentration can reach 500 mg L−1 (∼8 mM),24,25 and in other industrial processes such as cellophane, explosive, pectin and metal finishing, this value can be higher than 1000 mg NO3-N L−1 (>16 mM).26–28 The usage of nitrate-rich wastewater in urea electrochemical production would avoid direct release into the environment or the costly denitrification in wastewater treatment plants.
NH3 and CO2 can also be used to produce urea electrochemically. However, as mentioned above, the NH3 synthesized through the Haber Bosch process and used to synthesize urea in the Bosch–Meiser process is responsible for roughly ¾ of the GHG emitted;8 therefore, its utilization for urea electrochemical synthesis is not environmentally friendly compared to the other routes. Hence, the one-step electrosynthesis of urea using NO3− and CO2 as the reagents is extremely appealing because it can be achieved without using H2 produced through SMR and WGSR.29 In addition, the one-step electro-synthesis route also contributes to lowering the overall energy compared to the two steps starting from NO3− or from N2 as below.29
One step (electrochemical, eqn (1)): overall 13.3 GJ per metric ton urea
| CO2 + 2NO3− + 3H2O → CO(NH2)2 + 4O2 + 2OH− | (1) |
Two steps (electrochemical + Bosch–Meiser): overall 20.8 GJ per metric ton urea.
Step 1 (electrochemical, eqn (2)): 13.7 GJ per metric ton urea
| NO3− + 2H2O → NH3 + 2O2 + OH− | (2) |
Step 2 (Bosch–Meiser, eqn (3)): 7.1 GJ per metric ton urea
| 2NH3 + CO2 → CO(NH2)2 + H2O | (3) |
Two steps (Haber–Bosch + Bosch–Meiser): overall 19.7 GJ per metric ton urea.
Step 1 (Haber Bosch, eqn (4)): 12.6 GJ per metric ton urea
| N2 + 3H2 → 2NH3 | (4) |
Step 2 (Bosch–Meiser, eqn (5)): 7.1 GJ per metric ton urea
| 2NH3 + CO2 → CO(NH2)2 + H2O | (5) |
Step 1 + Step 2 (Haber–Bosch + Bosch–Meiser, eqn (6)): 19.7 GJ per metric ton urea
| N2 + 3H2 + CO2 → CO(NH2)2 + H2O | (6) |
The two step traditional process to synthesize urea including step 1 Haber–Bosch (eqn (4)) followed by Bosch–Meiser (eqn (5)). In step 1 (eqn (4)), hydrogen is widely produced through the SMR as described in eqn (7):
| CH4 + H2O → CO + 3H2 | (7) |
Followed by the WGSR presented in eqn (8):
| CO + H2O → CO2 + H2 | (8) |
Therefore, the overall reaction including SMR (eqn (7)) and WGRS (eqn (8)) to produce valuable hydrogen for step 1 is the following:
| CH4 + 2H2O → CO2 + 4H2 | (9) |
Therefore, the overall step 1 can be also extensively written as the following (eqn (10))29 highlighting the usage of methane as a reagent and the emission of carbon dioxide.
 | (10) |
Based on the above calculations, the one-step urea electrosynthesis will decrease the energy consumption by 32–36% with over 50% CO2 reduction.
Reaction mechanisms for urea electrosynthesis using NO3− and CO2
The mechanism of transformation of CO2 and NO3− into urea is still not completely clarified. The reaction pathways are complicated, involving 16 electrons and over 70 intermediate steps. Indeed, it seems that the C–N formation, which in the case of urea takes place twice, is the most critical step(s). It is generally accepted that the first C–N coupling can proceed mainly via two mechanisms: the Eley–Rideal (E–R) mechanism involving an early coupling between (unreduced) CO2 and adsorbed to form the
to form the 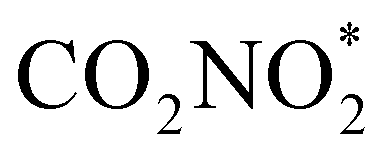 species and the Langmuir–Hinshelwood (L–H) mechanism that proceeds by coupling adsorbed CO* and NH* species to form NHCO*, depending on the electrocatalyst type and morphology. Both mechanisms lead eventually to the hydrogenation of the adsorbed intermediate and subsequent combination with another
species and the Langmuir–Hinshelwood (L–H) mechanism that proceeds by coupling adsorbed CO* and NH* species to form NHCO*, depending on the electrocatalyst type and morphology. Both mechanisms lead eventually to the hydrogenation of the adsorbed intermediate and subsequent combination with another  adsorbed species followed by reduction to form eventually CO(NH2)2.30–32
adsorbed species followed by reduction to form eventually CO(NH2)2.30–32
It was also found that the formation of intermediates such as CO* and  is a key step independent of the nitrogen sources used as reagents (e.g. N2, NO, NO2− and NO3−).18 Remarkably, to date, to our best knowledge, only a scant few studies (around 21) have been published in the past three years related to urea electrosynthesis starting from NO3− and CO2.29–52 These manuscripts provide a solid starting point for the research, but fundamental investigations are needed to clarify the steps that determine the most activity and selectivity: no direct experimental evidence has yet been reported showing the formation of
is a key step independent of the nitrogen sources used as reagents (e.g. N2, NO, NO2− and NO3−).18 Remarkably, to date, to our best knowledge, only a scant few studies (around 21) have been published in the past three years related to urea electrosynthesis starting from NO3− and CO2.29–52 These manuscripts provide a solid starting point for the research, but fundamental investigations are needed to clarify the steps that determine the most activity and selectivity: no direct experimental evidence has yet been reported showing the formation of  intermediate species, but many DFT studies suggest that that pathway is energetically favourable for the formation of urea, at least on some electrocatalytic structures. For example Sargent et al. showed by DFT calculation that the
intermediate species, but many DFT studies suggest that that pathway is energetically favourable for the formation of urea, at least on some electrocatalytic structures. For example Sargent et al. showed by DFT calculation that the  pathway seems to be favoured on Zn, rather than on Cu, in a ZnCu electrocatalyst.29 The same result for copper was published by Chen et al. for a Cu-based MOF.48 Another DFT study suggests the
pathway seems to be favoured on Zn, rather than on Cu, in a ZnCu electrocatalyst.29 The same result for copper was published by Chen et al. for a Cu-based MOF.48 Another DFT study suggests the  pathway on a molybdenum oxide-based electrocatalyst for urea electrosynthesis.49 Some studies suggest instead the reduction of
pathway on a molybdenum oxide-based electrocatalyst for urea electrosynthesis.49 Some studies suggest instead the reduction of  species down to
species down to  species, followed by coupling with CO2 or via separate CO2 reduction to CO* and then coupling (L–H mechanism).43,50,51,53
species, followed by coupling with CO2 or via separate CO2 reduction to CO* and then coupling (L–H mechanism).43,50,51,53
Fig. 2 shows a more detailed schematic of the reported mechanisms that differ mainly in the C-intermediate and N-intermediate that participate in C–N coupling. A comprehensive analysis of the literature summarizes 6 distinct reaction pathways.54 First, the C–N coupling may originate from *CO2 with either *NO2 or *NH2. The early coupling of *CO2 and *NO2 (E–R mechanism) can be effective in mitigating by-products such as NH3 or CO. Second, the C-intermediate *CO could couple with N-intermediate *NO, *NH, *NH2 or *H2NOH (L–H mechanism). In the C–N coupling reactions of *CO and *NH2,  acts as a base and attacks the positively charged center of CO*, promoting the formation a C–N bond and consequently the formation of urea.55 A comprehensive understanding of the potential urea formation mechanisms is imperative for researchers in this field and could aid in the development of rational electrocatalyst designs and enhance urea selectivity.
acts as a base and attacks the positively charged center of CO*, promoting the formation a C–N bond and consequently the formation of urea.55 A comprehensive understanding of the potential urea formation mechanisms is imperative for researchers in this field and could aid in the development of rational electrocatalyst designs and enhance urea selectivity.
 | ||
| Fig. 2 Formation mechanism of organonitrogen molecules including urea through electrochemical C–N coupling. Reproduced with permission.54 Copyright 2024, Elsevier Ltd. | ||
In parallel, in Fig. 2, it is also shown the possibility of synthesizing methylamine (CH3NH2) and ethylamine (C2H7N). Urea electrosynthesis involves the utilization of *CO as the carbon source. Instead, the electrosynthesis of methylamine utilizes another carbon source such as *HCHO.54 Compared to urea electrosynthesis, methylamine electrosynthesis shares a similar pathway on the nitrogen side until *H2NOH (L–H mechanism) and then it reacts with *HCHO, being rapidly attacked by the *NH2OH nucleophile to produce H2C = NOH. The methoxime is further electrochemically reduced to H2CNHOH (N-based methyl hydroxylamine) and then finally to methylamine.
Some examples of the competitive transformation of CO2 and NO3− into compounds other than urea are reported.56 Interestingly, the electrochemical production of methylamine was proven with low faradaic efficiency quantified as 13% over a multiphase molecular cobalt catalyst supported on carbon nanotubes.57 In general, low F. E. is measured. This route can be inhibited by avoiding the electrocatalyst to have efficient selectivity towards CO2RR to formate or formaldehyde.58
Electrocatalysts for urea electrosynthesis using NO3− and CO2
The pursuit of developing efficient electrocatalysts for C–N coupling started in the late 90s. In fact, Shibata et al. probed the concurrent reduction of nitrates and CO2 on the gas-diffusion electrodes (GDEs) based on various transition metals and observed the highest faradaic efficiency of urea formation (around 35%) on Zn-containing GDEs at −1.75 V vs. SHE.59 However, different families of electrocatalysts for urea electrosynthesis using NO3− and CO2 have evolved over the past 3 years. Quite interestingly, contrary to the initial aforementioned findings of Shibata et al., very recently, metallic Ag displayed a urea selectivity and faradaic efficiency close to 100% at −1.25 V vs. RHE, thus becoming an outstanding electrocatalyst for urea production.40 Such a remarkable ability of the Ag electrocatalyst was justified by easy initial and secondary C–N coupling steps with an endergonic formation of formamide from *CONH2, which improves the selectivity and kinetics when the NO3− concentration is adequately high. Lately, Shin and coworkers created an atomic spacing of ∼6 Å between the Cu facets via lithiation and realized an encouraging urea yield of 7541.9 mg h−1 mgcat−1 due to a considerable reduction in the energy barrier for C–N coupling, kinetically and thermodynamically favoring the C–N bond formation for urea synthesis.39 On the other hand, an oxygen-coordinated cobalt single-atom electrocatalyst (Co–O–C) achieved a urea yield rate of 2704.2 ± 183.9 mg h−1 mgcat−1 due to reliable interactions between neighboring Co–(O–C2)4 sites promoted the C–N coupling for urea electrosynthesis.46 Moreover, atomically dispersed nitrogen-coordinated single transition metal sites (e.g., Cu–Nx–C)35,37,60 (Fig. 3(A)) and diatomic (e.g., Fe–Ni)43 electrocatalysts have also been studied. In contrast to single-atom and isolated diatomic electrocatalysts, the bonded Fe–Ni pairs provided efficacious active sites for adsorption and activation of multiple reactants and hence uplifted the urea yield.43 However, Zn–Mn diatomic electrocatalysts axially coordinated with chlorine exhibited high resilience against CO poisoning and realized the faradaic efficiency of 63.5%.38 While approaching it from another viewpoint, Zhang et al. have recently demonstrated that Re–Mn electrocatalysts favor the generation of *C entities due to higher CO binding energies resulting in ammonia production.61 Whereas the lower binding of CO on Zn–Mn sites fosters CO migration and its coupling with adsorbed nitrogen and hence urea formation with up to 89% N-selectivity can be realized.61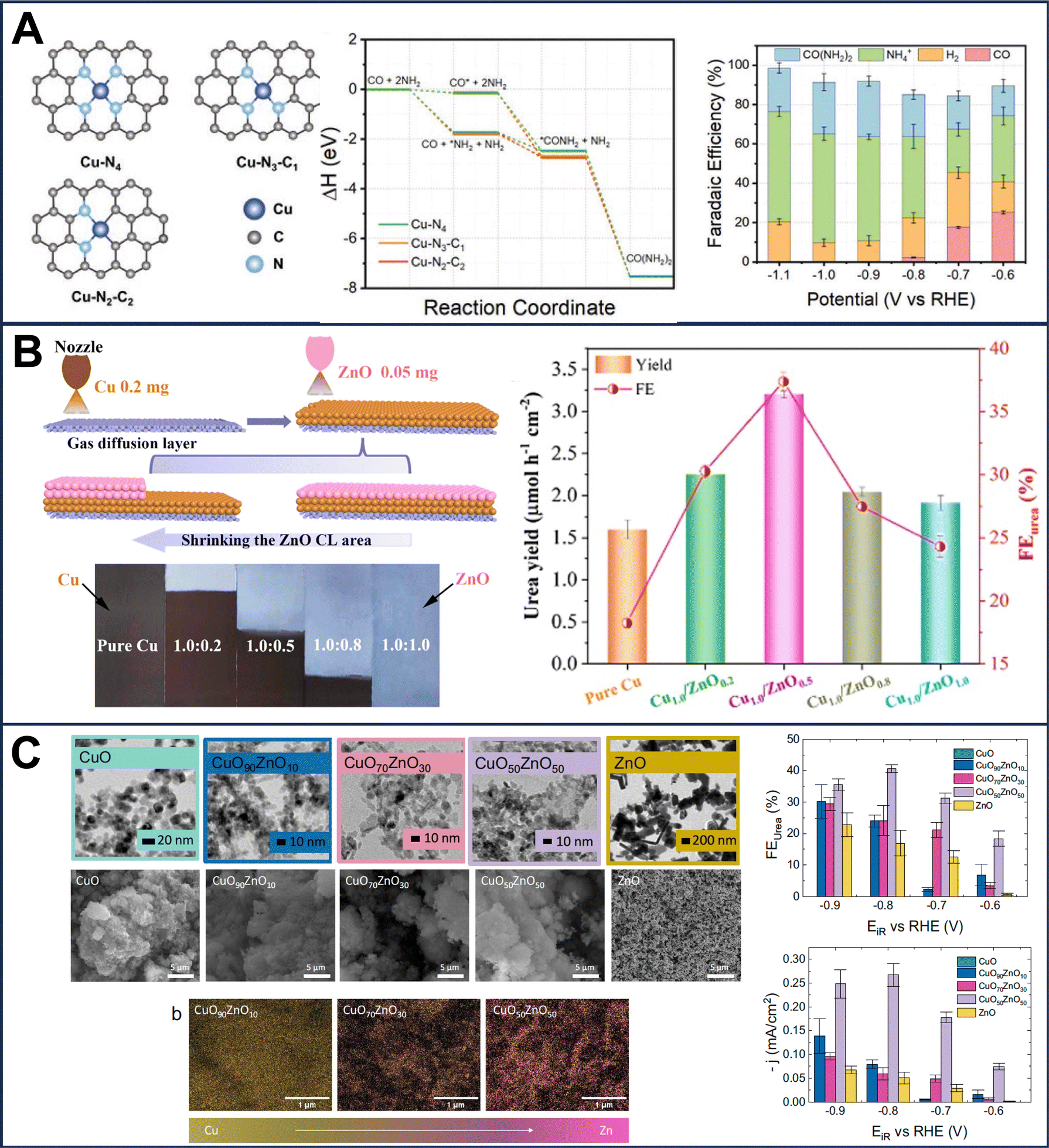 | ||
| Fig. 3 (A) Schematic of a single atom containing Cu coordinated with nitrogen, DFT reaction coordinates with this specific electrocatalyst and related faradaic efficiency at different potentials. (B) Schematic of a tandem electrocatalyst based on Cu and ZnO at different ratios with related urea yield and faradaic efficiency. (C) TEM, SEM and SEM/EDX images of a CuOxZny electrocatalyst with different metal ratios and the related faradaic efficiency are reported indicating also the partial current densities towards urea. (A) Reproduced with permission.37 Copyright 2022, Wiley-VCH GmbH. (B) Reproduced with permission.36 Copyright 2023, American Chemical Society. (C) Reproduced with permission.41 Nature Portfolio under CC-BY 4.0 (https://creativecommons.org/licenses/by/4.0/). | ||
Furthermore, metal oxides containing oxygen vacant sites such as ZnO33 or coupled with Cu such as Cu–ZnO (Fig. 3(B)),36 CuOx–ZnOy (Fig. 3(C))41 and Cu–TiO255 were also used as electrocatalysts. In these cases, the oxygen vacancies are attacked by the nitrite–nitrate ions and through a multi-step proton-coupled electron transfer reaction, an  intermediate is formed. In one case, Nafion™-modified TiO2 without Cu was used as the electrocatalyst.62 Mimicking the high-valence Mo-based reaction center of nitrate reductase, the CuWO4 catalyst with the high valence of the W catalytic center reached an outstanding 98.5 ± 3.2 μg h−1 mgcat−1 urea yield and 70.1 ± 2.4% at −0.2 V vs. RHE.42 Zn–Cu nanoparticles were also proposed with a maximum faradaic efficiency of 75%.29 In another recent work, symbiotic carbon-encapsulated amorphous iron (Fe(a)@C) and iron oxide nanoparticles (Fe3O4 NPs) on carbon nanotubes (denoted as Fe(a)@C-Fe3O4/CNTs) were proposed.34 By the same token, FeOOH electrodeposited on carbon paper delivered 512 μg h−1 cm−2 urea production rate.63 Combining the theoretical calculations with experimentation revealed that low spin states in Fe act an electron acceptors and facilitate the electron pair donation from the filled σ orbitals of the *NO intermediates to vacant Fe d orbitals. This electronic transfer contributes to a decrease in the rate-determining step's energy barrier and enhances the urea yield. Recently Ru/Pt/Pd nanoparticles were deposited over Cu foam (CF) showing promising urea productivity with Ru–CF being more effective compared to Pt–CF and Pd–CF.44 In(OH)3 was also studied successfully as an electrocatalyst for urea electrosynthesis.45
intermediate is formed. In one case, Nafion™-modified TiO2 without Cu was used as the electrocatalyst.62 Mimicking the high-valence Mo-based reaction center of nitrate reductase, the CuWO4 catalyst with the high valence of the W catalytic center reached an outstanding 98.5 ± 3.2 μg h−1 mgcat−1 urea yield and 70.1 ± 2.4% at −0.2 V vs. RHE.42 Zn–Cu nanoparticles were also proposed with a maximum faradaic efficiency of 75%.29 In another recent work, symbiotic carbon-encapsulated amorphous iron (Fe(a)@C) and iron oxide nanoparticles (Fe3O4 NPs) on carbon nanotubes (denoted as Fe(a)@C-Fe3O4/CNTs) were proposed.34 By the same token, FeOOH electrodeposited on carbon paper delivered 512 μg h−1 cm−2 urea production rate.63 Combining the theoretical calculations with experimentation revealed that low spin states in Fe act an electron acceptors and facilitate the electron pair donation from the filled σ orbitals of the *NO intermediates to vacant Fe d orbitals. This electronic transfer contributes to a decrease in the rate-determining step's energy barrier and enhances the urea yield. Recently Ru/Pt/Pd nanoparticles were deposited over Cu foam (CF) showing promising urea productivity with Ru–CF being more effective compared to Pt–CF and Pd–CF.44 In(OH)3 was also studied successfully as an electrocatalyst for urea electrosynthesis.45
As shown above, different families of electrocatalysts have been reported in the literature in recent years.18 These electrocatalysts contain diverse atomically dispersed (single or diatomic) metal nanoparticles as alloys or oxides or nanoparticles anchored over the support (Fig. 2). All these materials can be fabricated using different synthetic processes resulting in distinct morphologies, and structures and in turn, possess different activities and selectivity towards urea formation as highlighted by a few examples presented in Fig. 2.
In Table 1, the applied potentials, obtained selectivity and faradaic efficiency of the electrocatalysts used for synthesizing urea electrochemically are reported.
| Material | Applied potential [V vs. RHE] | Urea production rate | Selectivity | Faradaic efficiency [%] | Ref. | |
|---|---|---|---|---|---|---|
| 1 | ZnO–V nanosheets | −0.79 | 16.56 μmol h−1 | 23.26 | 33 | |
| 2 | Zn/Cu | −0.8 | 16 μmol h−1 | 75 | 29 | |
| 3 | Fe(a)@CFe3O4/CNTs | −0.65 | 1341.3 ± 112.6 μg h−1 mgcat−1 | 16.5 ± 6.1 | 34 | |
| 4 | Nitrogen-doped carbon (NC) | −0.5 | 596.1 μg mg−1 h−1 | 62 | 35 | |
| 5 | Cu/ZnO stacked tandem gas-diffusion electrode (GDE) | −0.3 | 3.2 μmol h−1 cm−2 | 37.4 | 36 | |
| 6 | Cu-GS-800 | −0.9 | 4.3 nmol s−1 cm−2 | 28 | 37 | |
| 7 | ZnMn-N,Cl | −0.3 | 4.13 mmol g−1 h−1 | (N-selectivity 100%) | 63.5 | 38 |
| 8 | Cu with atomic-scale spacing | −0.41 | 7541.9 mg h−1 mgcat−1 | 51.97 ± 0.8 | 39 | |
| 9 | Ag | −1.25 | (N-selectivity ∼100%) | ∼100 | 40 | |
| 10 | Ru/Pt/Pd–Cu CF | 0.13 | 151.6 μg h−1 cm−2 | 25.4 | 44 | |
| 11 | B-FeNi-DASC | −1.5 | 20.2 mmol h−1 g−1 | 17.8 | 43 | |
| 12 | CuO50ZnO50 | −0.8 | 41 | 41 | ||
| 13 | CuWO4 nanoparticles | −0.2 | 98.5 ± 3.2 μg h−1 mgcat−1 | (N-selectivity 76.2 ± 1.7%) | 70.1 ± 2.4 | 42 |
| 14 | In(OH)3 | −0.6 | 533.1 μg h−1 mgcat−1 | (N-selectivity 82.9%) | 53.4 | 45 |
| 15 | Co–O–C | −1.5 | 2704.2 ± 183.9 mg h−1 mgcat−1 | 31.4 ± 2.1 | 46 | |
| 16 | FeNi/NC loaded with FeNi3 nanoparticles | −0.9 | 496.5 μg h−1 mgcat−1 | 16.58 | 47 | |
| 17 | Cu-HATNA | −0.6 | 1.46 g h−1 gcat−1 | 25 | 48 | |
| 18 | MoOx/C | −0.6 | 1431.5 μg h−1 mgcat−1 | (NH4+ selectivity over 90%) | 27.7 |
49 |
| 19 | AuPd nanoalloy | −0.5 |
204.2 μg mg−1 h−1 | 15.6 | 50 | |
| 20 | Core–shell Cu@Zn nanowires | −1.02 | 7.29 μmol cm−2 h−1 | 9.28 | 51 |
As can be seen, all these quantities are very scattered in the cited literature, indicating that the route pursued is promising but more investigations need to be conducted for a deeper understanding of the reaction mechanisms and, consequently, for synthesizing the optimized electrocatalysts.
Detection methods
In order to avoid “false positive” results of urea formation, isotope-labelled (e.g.13C or 15N) experiments can be performed to produce and verify clear results regarding the source and the mechanism of urea formation. For example, NOx as contaminants are predominantly present in chemicals and in the atmosphere, so it is extremely important to validate and prove that urea produced through the electrocatalysis technique arises due to the reduction reactions of the reactants and not due to the presence of unwanted contaminants. Detection methodologies involving isotope assignments are efficaciously applied to avoid any misleading indications of urea production, while allowing the identification of the origin of the produced urea with reasonable sureness. State-of-the-art characterization tools such as Fourier-transformed infrared spectroscopy (FTIR), UV-Vis spectroscopy and nuclear magnetic resonance (NMR) can be employed to qualitatively and, to some extent, quantitatively assess the urea formation while determining the chemical functionalities and isotope assignments, respectively.18It is noteworthy that most of the developed electrocatalysts for urea electrosynthesis lack product selectivity as the reaction may undergo various inappropriate pathways, leading to the formation of sub-products and unreacted intermediates.64 Such sub-products could be of both gaseous and liquid natures and hence their accurate detection for the precise estimation of faradaic efficiency becomes a complicated and laborious task. The sub-products of gaseous nature can involve H2, CO and unreacted CO2 whereas the liquid sub-products could be HCOO−, CH3NH2 and NH3, NO2− and unreacted NO3−.18 Therefore, different in situ and ex situ methods involving spectroscopic and colorimetric techniques have been used to identify the targeted products.
One of the established methods used for colorimetric quantification of urea is the diacetyl monoxime method.65 This method involves the mixing of the standard urea solution, diacetyl monoxime and thiosemicarbazide solution and acid-ferric solution in definite proportions and taking absorbance readings at a particular wavelength, i.e. λ = 525 nm through UV-Vis spectroscopy followed by driving the calibration curves.35 It should be noted, the by-product NO2− is a major interferent to this method and could largely affect its reliability.66 Yet, another paper suggests that the concentration of urea may be affected by NH4+.67,68
Another colorimetric way to quantify the produced urea could be the decomposition of urea into CO2 and ammonia using urease and then by analyzing the ammonia content prior to and after the enzymatic decomposition, the quantity of the produced urea can be estimated.43 However, this method should not be applied in the electroproduction routes of urea where ammonia is present as a by-product. Therefore, the indirect urease method should be coupled with additional direct methods for urea identification and quantification.38,43 Recently, Huang et al. employed high-performance liquid chromatography–mass spectrometry (HPLC–MS) for urea determination by getting its benefits of superior resolution, reproducibility and lower impurity disruption.69 HPLC can effectively separate the products considering the variance in the adsorption capacity and affinity, the molecular sizes and partition coefficient between the mobile and stationary phases.
NMR is an advanced tool able to not only detect the produced urea and by-products by identifying their signature peaks but also indicate the nitrogen origin as various nitrates are present as common contaminants in the environment.181H-NMR spectra can help in effectively figuring out the nitrogen origin whereas the 13C-NMR spectral examination can identify the carbon origin in the produced urea. However, it must be pointed out that the signal-to-noise ratio of an NMR experiment is proportional to B3/20, where B0 is the magnetic field.70 For this reason, in the case of urea small concentration, i.e. below 5 ppm, the right practice is to use spectrometers equipped with magnets stronger than 14 T (corresponding to 600 MHz 1H Larmor frequency) which, today, are accessible to most users.
Another important method for the detection of urea is FTIR. Lately, Li et al. employed operando synchrotron radiation-FTIR (SR-FTIR) to probe the bending, wagging and rocking modes of –NH2 in the urea at 1635, 1307 and 1107 cm−1 along with the stretching mode of C–N at 1419 cm−1, evidencing successful C–N coupling in the urea formation during electrocatalytic reactions.35 Similarly, Geng and coworkers verified the mechanism of C–N coupling via in situ FTIR that categorically confirmed the C–N coupling via the coupling reaction of CO2 and NO3−. They further confirmed the formation of the C–N stretching mode with A1 symmetry (at 1000 cm−1) via in situ Raman spectroscopy34 since the in situ Raman can effectively screen the superficially located species during electrocatalytic reactions. By using this technique, Y. Zhao et al. found that the C–N bond formation, i.e. the rate-determining step according to the authors, comes from the coupling between the intermediate *NO2 and *CO on the bridging configuration, whose peaks were observed at 1428 and 1979 cm−1 Raman shift, respectively.42 Along with urea detection, the identification and quantification of the by-products and intermediates is also an important task since their involvement not only influences the reaction mechanism but also affects the faradaic efficiency. In addition to the aforementioned techniques, ammonia can be effectively determined via the robust indophenol blue or Nessler's methods using color reagents,38,71,72 whereas hydrazine can be detected by the Watt and Chrisp method.38 The gaseous products for instance CO, H2, N2 and methane can be analyzed through gas chromatography (GC) whereas to quantify NO2− ion exchange chromatography (IC) can be used.40 Moreover, the Griess method is also a suitable way to examine the nitrite content using UV-Vis spectroscopy.36 Moreover, a very modern technique of operando attenuated total reflection surface-enhanced FTIR (ATR-SEIRAS) has an outstanding potential to critically allow the understanding of the NO3− and CO2 co-reduction mechanism on the electrocatalyst surface in a definite potential window.35 In the achieved ATR-SEIRAS spectrum, the consumption/depletion of particular species or functional groups can be identified by negative peaks whereas the positive peaks signify the production or increase in the concentration of specific species or functionalities. The detection methods used in some of the recent studies are summarized in Table 2.
| Product type | Detection method | Used for quantification purposes? | Advantage | Limitations | In situ/Ex situ | Ref. |
|---|---|---|---|---|---|---|
| Urea | UV-Vis spectrophotometry by urease method | Yes | High accessibility | Strong interference by NH4+, NO2− and metal ions | Ex situ | 34, 41 and 43 |
| UV-Vis spectrophotometry by diacetylmonoxime method | Yes | Low limit of detection (LOD ∼ 0.15 ppm), high accessibility | Strong interference by NO2− or NH4+ | Ex situ | 29, 35–38, 40, 44 and 45 | |
| NMR | Yes | High anti-interference ability. | Very close position of the urea peak at 5.6 ppm to the water peak at 4.7 ppm affecting the signal-to-noise ratio. | Ex situ | 38, 39 and 43 | |
| 13C NMR and 15N NMR can be used for isotope labeling | Relatively high LOD | |||||
| HPLC/LC | Yes | High anti-interference ability. | Relatively high LOD | Ex situ | 33 | |
| Infrared reflection–absorption spectroscopy (IRRAS) | No | Detection of adsorbed intermediates selectively due to selection rules (if PM-IRRAS) | Sensitive to matrices | In situ | 29 and 73 | |
| Surface-enhanced Raman spectroscopy (SERS) | Semi-quantitative | High sensitivity, selective, identification of adsorbed intermediate due to selection rules | May need immobilization of specific urea-sensing enzymes on the substrate to increase selectivity | In situ | 29 and 74–76 | |
| Attenuated total internal reflectance Fourier transform infrared spectroscopy (ATR-FTIR) | Semi-quantitative | Specification of intermediate functionalities and C–N coupling, sensitivity, compatible with various matrices | Detects product in the liquid interface and not adsorbed ones | In situ | 34, 43, 73 and 77 | |
| Raman | No | In situ | 34, 36 and 42 | |||
| HCOO− | 1H NMR | Yes | Minimal sample preparation | Lower accessibility and complex expertise for instrumentation | Ex situ | 29 and 36 |
| Isotope labelling for carbon source verification | ||||||
| HPLC | Yes | Easier for analysis of large amount of samples due to automation of most instruments | Toxic and flammable organic solvents are used for the eluent of mobile phase, flushing and cleaning | Ex situ | 40, 44 and 45 | |
| Ion chromatogram instrument (IC) | Yes | High sensitivity (LOD ∼ 0.1 μM) | Cost of buffer eluent, columns and consumables | Ex situ | 45 | |
| NO2− | Griess test | Yes | High sensitivity (LOD ∼ 1 μM) | Dilutions may be needed for the absorbance to fall within the range of calibration | Ex situ | 34–36, 44 and 78 |
| Ion exchange chromatography (IC) | Yes | Simultaneous detection of nitrite and nitrate ions | Variations between instruments | Ex situ | 29, 40, 42, 45 and 78 | |
| NO, NO2, NOx | Chemiluminescence | Yes | Very high sensitivity (LOD ∼ 1 nM) | Specificity (detects other NOx species) | Ex situ | 33 |
| NO–NO2–NOx analyzer | Variations between instruments | |||||
| Frequent calibration required | ||||||
| NH3 | UV-Vis spectrophotometry by indophenol blue method | Yes | High accessibility | Long time for preparation | Ex situ | 33–38, 40, 42–45, 79 and 80 |
| Good sensitivity (LOD ∼ 0.01 ppm) | Good accuracy only under neutral or alkaline conditions | |||||
| 1H NMR | Yes | High sensitivity | Lower accessibility and complex expertise for instrumentation | Ex situ | 29, 79 and 80 | |
| High stability in different electrolyte pH | ||||||
| NMR can be used for isotope labelling to exclude contaminant ammonia | ||||||
| DEMS | No | In situ | 33 | |||
| N2H4 | UV-Vis spectrophotometry by Watt and Chrisp methods | Yes | High sensitivity (LOD ∼ 0.06 ppm) | Interference by semicarbazide or urea | Ex situ | 36, 38, 42, 45 and 81 |
| Liquid product such as C2H5OH, CH3COOH, n-propanol, CH3NH2 | 1H NMR | Yes | Capability of providing chemical information and allowing for quantification of most liquid phase products | Lower accessibility and complex expertise for instrumentation | Ex situ | 29, 36, 42 and 82 |
| Gas product such as CO, CH4, C2H4, N2 and H2 | GC | Yes | Most commonly used and available method for quantification of gas species | CO2 as the main species in the gas phase due to a low conversion needs to be quantified to determine the reaction progress and/or the extent of CO2 dissolution in the liquid phase | Ex situ | 29, 33, 34, 36–40, 42, 45 and 82 |
| DEMS | Semi-quantitative | Possibility to identify hydrocarbon products | Difficulty to discriminate CO ion fragmentation from CO2 | In situ | 33, 83 and 84 |
A combination of more than one method is anyway strongly suggested to the operators in order to double-check the results, have additional confirmations and overcome the limitation of a specific detection method.
Challenges to overcome and future research directions
Urea electrosynthesis has freshly captured the interest of the scientific community with the important goal of finding alternatives to the energy-intensive and heavily polluting chemical industries producing fertilizers such as urea. Electrochemical urea production might be a resolutive solution to replace this hard-to-abate process with positive economic, environmental, and social implications. However, despite the fact that urea electro-synthesis seems to be advantageous, several important issues and bottlenecks need to be faced and overcome:(1) The reaction mechanisms are still under debate and may change between different families of electrocatalysts.
(2) The selectivity is still relatively low, with a few cases above 70%, with the formation of undesired by-products.
(3) Higher productivity and improved reaction rates need to be pursued by investigating novel electrocatalyst combinations.
Although a few manuscripts have shown high selectivity (Table 1), a value up to 100% is needed for industrial applications. The generation and separation of intermediates and byproducts necessitate additional installations, posing challenges to economic viability and system stability. Moreover, to meet the requirements of the technological process, higher productivity and improved reaction is also required to be competitive with the state-of-the-art Bosch–Meiser process.
Ideally, an efficient electrocatalyst should promote C–N coupling while restricting or suppressing the side reactions, including hydrogen evolution,72,85,86 carbon dioxide reduction87 and nitrate reduction reactions.30 However, it is not yet clear which family of electrocatalysts is the most selective for urea electrosynthesis from the perspective of the aforementioned considerations. Nevertheless, recent scientific endeavors on a variety of electrocatalysts including transition metal-based, single-atom, heteroatomic and bimetallic systems are intuitive, but still rational design and proper benchmarking are primarily missing. Strategies to enhance mass transport, charge delocalization, defect inducement and engineering of the local coordination of the metal-based active moieties can augment the activation and durable ultimate transformation of the intermediates. Therefore, it is essential to elucidate the exact origin of the electrocatalytic activities during C–N coupling and corresponding urea formation while considering the physiochemical features and surface chemistry of the given electrocatalysts. To date, apparently, the presence of two metals in a bimetallic configuration can boost C–N formation and the following hydrogenation. However, optimization of reactor configuration such as the application of a GDE in flow cells instead of typical H-type cells to provide CO2 in gaseous form other than dissolved form and increasing the nitrate concentration may also help improve the efficacy of the single metal-based electrocatalysts.40,88 But again, the reaction mechanisms carried out among bifunctional, tandem, co-catalysis or cascade reactions need to be analyzed thoroughly and well-understood in order to enhance conversion and selectivity. Moreover, up to now, urea electrosynthesis has been carried out simply by applying a fixed electrochemical potential. This might not be the most suitable route to be pursued, as each step of the reaction might have its optimum at different potentials; therefore, alternating the potential might be pursued to effectively transform the intermediates and improve the overall faradaic efficiency. Similarly, pulsed potentials within the designated window may help locally increase the concentration of CO2 and NO3− while reducing the pH to help C–N coupling enriched by *CO and *NH2 intermediates.52
Detection of urea ex situ might encounter issues related to the presence of by-products that might not be detected. NMR is a strong solution to this aim. Consolidated methods for studying in situ urea formation and detection during electrochemical operation are required and needed. The recognition of intermediates and other by-products is not fully developed and not yet accurate. In situ detection methods are paramount to decipher the intermediates and product formation during the reaction and more importantly, to understand the reaction mechanisms and their coordinates. Having insights into the reaction mechanisms, in turn, would be beneficial for improving the electrocatalyst's behaviors during operation.
Alongside the detection techniques, we highlight the need for the introduction of model systems that, by a precise control of surface termination, element distribution, and morphology, may allow the deconvolution of the contributions to reactivity. By simulating various scenarios and parameters, model systems would provide invaluable insights into designing more effective electrocatalyst solutions.
Development of novel electrocatalysts and understanding of the electrocatalysis to be used run in parallel with the need for a robust method for detecting urea in situ during electrochemical operations. Many questions remain unanswered and thus many challenges need to be overcome before this critical electrochemical reaction will be selective towards urea production. However, these initial results seem to be promising and encouraging for further investigations of the electrochemical community.
Conflicts of interest
The authors declare no conflicts of interest.Acknowledgements
C. S. and L. E. would like to acknowledge the Italian Ministry of Foreign Affairs and International Cooperation – Directorate General for Cultural and Economic Promotion and Innovation (Italian Republic) and the Israel Ministry of Science and Technology within the bilateral project Italy-Israel (WE-CAT). C. S., A. L. and J. F. would like to acknowledge also the Cariplo Foundation, Call for Circular Economy through the project “Transformation of Plastic Waste in Electrocatalysts, Supported by exhausted Gases Recovery Layout” (TESLA). G. Z. acknowledges a PhD scholarship from the Italian National PhD program “Scientific, technological and Social Methods Enabling Circular Economy”, Curriculum “Technical Materials for Circularity” funded by Italy's Recovery and Resilient Plan and EU Recovery Plan. This work is supported in part by a subcontract from the U.S. Department of Energy (DOE), Office of Energy Efficiency and Renewable Energy (EERE), Bioenergy Technology Office (DE-EE0008923) led by Colorado State University.References
- C. I. Ludemann, A. Gruere, P. Heffer and A. Dobermann, Sci. Data, 2022, 9, 501 CrossRef PubMed.
- S. Menegat, A. Ledo and R. Tirado, Sci. Rep., 2022, 12, 14490 CrossRef CAS PubMed.
- C. Smith, A. K. Hill and L. Torrente-Murciano, Energy Environ. Sci., 2020, 13, 331–344 RSC.
- M. H. Vu, M. Sakar and T. O. Do, Catalysts, 2018, 8, 621 CrossRef.
- Ammonia Technology Roadmap, IEA, 2021, Paris, https://www.iea.org/reports/ammonia-technology-roadmap.
- D. A. Shea, L. J. Schierow and S. D. Szymendera, CRS Report for Congress, 2013, https://nationalaglawcenter.org/wp-content/uploads/assets/crs/R43070.pdf.
- J. Meessen, Chem. Ing. Tech., 2014, 86, 2180–2189 CrossRef CAS.
- S. K. Masjedi, A. Kazemi, M. Moeinnadini, E. Khaki and S. I. Olsen, Sci. Total Environ., 2024, 908, 168225 CrossRef CAS PubMed.
- Trading economics, https://tradingeconomics.com/commodity/urea, (accessed January 2024).
- U. Bardi, T. El Asmar and A. Lavacchi, J. Cleaner Prod., 2013, 53, 224–231 CrossRef.
- Chemical and Engineering News, https://cen.acs.org/environment/green-chemistry/Industrial-ammonia-production-emits-CO2/97/i24, (accessed January 2024).
- A. Rafiee, K. Rajab Khalilpour, D. Milani and M. Panahi, J. Environ. Chem. Eng., 2018, 6, 5771–5794 CrossRef CAS.
- A. Franco and C. Giovannini, Energies, 2023, 16, 6098 CrossRef CAS.
- Q. Wang, C. Cai, M. Dai, J. Fu, X. Zhang, H. Li, H. Zhang, K. Chen, Y. Lin, H. Li, J. Hu, M. Miyauchi and M. Liu, Small Science, 2021, 1, 2000028 CrossRef CAS.
- T. Lieuwen, Y. Vigor and R. Yetter, Synthesis Gas Combustion: Fundamentals and Applications, CRC Press, Boca Raton, 2009 Search PubMed.
- C. Lee and Q. Yan, Curr. Opin. Electrochem., 2021, 29, 100808 CrossRef CAS.
- E. Murphy, Y. Liu, I. Matanovic, Y. Huang, A. Ly, S. Guo, W. Zang, X. Yan, A. Martini, J. Timoshenko, B. R. Cuenya, I. V. Zenyuk, X. Pan, E. D. Spoerke and P. Atanassov, Nat. Commun., 2023, 14, 4554 CrossRef CAS PubMed.
- S. Paul, A. Adalder and U. K. Ghorai, Mater. Chem. Front., 2023, 7, 3820–3854 RSC.
- C. Chen, N. He and S. Wang, Small Science, 2021, 1, 2100070 CrossRef CAS.
- M. Duca and M. T. M. Koper, Energy Environ. Sci., 2012, 5, 9726–9742 RSC.
- M. Gorguner and M. Akgun, Eurasian J. Med., 2010, 42, 28–35 CrossRef PubMed.
- T. Prior and R. Kift, J. Chem. Crystallogr., 2009, 39, 558–563 CrossRef CAS.
- R. Picetti, M. Deeney, S. Pastorino, M. R. Miller, A. Shah, D. A. Leon, A. D. Dangour and R. Green, Environ. Res., 2022, 210, 112988 CrossRef CAS PubMed.
- L. Cao, W. Wang, Y. Yang, C. Yang, Z. Yuan, S. Xiong and J. Diana, Environ. Sci. Pollut. Res., 2007, 14, 452–462 CrossRef CAS PubMed.
- M. C. J. Verdegem, Rev. Aquacult., 2013, 5, 158–171 CrossRef.
- D. Bilanovic, P. Battistoni, F. Cecchi, P. Pavan and J. Mata-Alvarez, Water Res., 1999, 33, 3311–3320 CrossRef CAS.
- T. Watanabe, H. Motoyama and M. Kuroda, Water Res., 2001, 35, 4102–4110 CrossRef CAS PubMed.
- S. Zala, A. Nerurkar, A. Desai, J. Ayyer and V. Akolkar, Biotechnol. Lett., 1999, 21, 481–485 CrossRef CAS.
- Y. Luo, K. Xie, P. Ou, C. Lavallais, T. Peng, Z. Chen, Z. Zhang, N. Wang, X. Y. Li, I. Grigioni, B. Liu, D. Sinton, J. B. Dunn and E. H. Sargent, Nat. Catal., 2023, 6, 939–948 CrossRef CAS.
- J. Liu, X. Guo, T. Frauenheim, Y. Gu and L. Kou, Adv. Funct. Mater., 2023, 34, 2313420 CrossRef.
- G. L. Yang, C. T. Hsieh, Y. S. Ho, T. C. Kuo, Y. Kwon, Q. Lu and M. J. Cheng, ACS Catal., 2022, 12, 11494–11504 CrossRef CAS.
- H. C. Li, Y. S. Ho, G. L. Yang, R. H. Li, C. T. Kuo, C. T. Hsieh, Y. Kwon and M. J. Cheng, J. Phys. Chem. C, 2024, 128, 1058–1067 CrossRef CAS.
- N. Meng, Y. Huang, Y. Liu, Y. Yu and B. Zhang, Cell. Rep. Phys. Sci., 2021, 2, 100378 CrossRef CAS.
- J. Geng, S. Ji, M. Jin, C. Zhang, M. Xu, G. Wang, C. Liang and H. Zhang, Angew. Chem., Int. Ed., 2023, 62, e202210958 CrossRef CAS PubMed.
- Y. Li, S. Zheng, H. Liu, Q. Xiong, H. Yi, H. Yang, Z. Mei, Q. Zhao, Z. W. Yin, M. Huang, Y. Lin, W. Lai, S. X. Dou, F. Pan and S. Li, Nat. Commun., 2024, 15, 176 CrossRef CAS PubMed.
- Y. Wang, S. Xia, J. Zhang, Z. Li, R. Cai, C. Yu, Y. Zhang, J. Wu and Y. Wu, ACS Energy Lett., 2023, 8, 3373–3380 CrossRef CAS.
- J. Leverett, T. Tran-Phu, J. A. Yuwono, P. Kumar, C. Kim, Q. Zhai, C. Han, J. Qu, J. Cairney, A. N. Simonov, R. K. Hocking, L. Dai, R. Daiyan and R. Amal, Adv. Energy Mater., 2022, 12, 2201500 CrossRef CAS.
- X. Zhang, X. Zhu, S. Bo, C. Chen, K. Cheng, J. Zheng, S. Li, X. Tu, W. Chen, C. Xie, X. Wei, D. Wang, Y. Liu, P. Chen, S. P. Jiang, Y. Li, Q. Liu, C. Li and S. Wang, Angew. Chem., Int. Ed., 2023, 62, e202305447 CrossRef CAS PubMed.
- S. Shin, S. Sultan, Z. X. Chen, H. Lee, H. Choi, T. U. Wi, C. Park, T. Kim, C. Lee, J. Jeong, H. Shin, T. H. Kim, H. Ju, H. C. Yoon, H. K. Song, H. W. Lee, M. J. Cheng and Y. Kwon, Energy Environ. Sci., 2023, 16, 2003–2013 RSC.
- N. C. Kani, I. Goyal, S. Olusegun, R. R. Bhawnani, J. A. Gauthier and M. R. Singh, ChemRxiv, 2023, preprint DOI:10.26434/chemrxiv-2023-h1lfq.
- D. Anastasiadou, B. Ligt, Y. He, R. C. J. van de Poll, J. F. M. Simons and M. C. Figueiredo, Commun. Chem., 2023, 6, 199 CrossRef CAS PubMed.
- Y. Zhao, Y. Ding, W. Li, C. Liu, Y. Li, Z. Zhao, Y. Shan, F. Li, L. Sun and F. Li, Nat. Commun., 2023, 14, 4491 CrossRef CAS PubMed.
- X. Zhang, X. Zhu, S. Bo, C. Chen, M. Qiu, X. Wei, N. He, C. Xie, W. Chen, J. Zheng, P. Chen, S. P. Jiang, Y. Li, Q. Liu and S. Wang, Nat. Commun., 2022, 13, 5337 CrossRef CAS PubMed.
- J. Qin, N. Liu, L. Chen, K. Wu, Q. Zhao, B. Liu and Z. Ye, ACS Sustainable Chem. Eng., 2022, 10, 15869–15875 CrossRef CAS.
- C. Lv, L. Zhong, H. Liu, Z. Fang, C. Yan, M. Chen, Y. Kong, C. Lee, D. Liu, S. Li, J. Liu, L. Song, G. Chen, Q. Yan and G. Yu, Nat. Sustain., 2021, 4, 868–876 CrossRef.
- S. Zhang, M. Jin, H. Xu, X. Zhang, T. Shi, Y. Ye, Y. Lin, L. Zheng, G. Wang, Y. Zhang, H. Yin, H. Zhang and H. Zhao, Energy Environ. Sci., 2024, 17, 1950–1960 RSC.
- T. Hou, J. Ding, H. Zhang, S. Chen, Q. Liu, J. Luo and X. Liu, Mater. Chem. Front., 2023, 7, 4952–4960 RSC.
- M. D. Zhang, J. R. Huang, P. Q. Liao and X. M. Chen, Chem. Commun., 2024, 60, 3669–3672 RSC.
- M. Sun, G. Wu, J. Jiang, Y. Yang, A. Du, L. Dai, X. Mao and Q. Qin, Angew. Chem., Int. Ed., 2023, 62, e202301957 CrossRef CAS PubMed.
- H. Wang, Y. Jiang, S. Li, F. Gou, X. Liu, Y. Jiang, W. Luo, W. Shen, R. He and M. Li, Appl. Catal., B, 2022, 318, 121819 CrossRef CAS.
- N. Meng, X. Ma, C. Wang, Y. Wang, R. Yang, J. Shao, Y. Huang, Y. Xu, B. Zhang and Y. Yu, ACS Nano, 2022, 16, 9095–9104 CrossRef CAS PubMed.
- Q. Hu, W. Zhou, S. Qi, Q. Huo, X. Li, M. Lv, X. Chen, C. Feng, J. Yu, X. Chai, H. Yang and C. He, Nat. Sustain., 2024, 7, 442–451 CrossRef.
- Y. Zhang, Y. Zhao, M. G. Sendeku, F. Li, J. Fang, Y. Wang, Z. Zhuang, Y. Kuang, B. Liu and X. Sun, Small Methods, 2023, 8, 2300811 CrossRef PubMed.
- K. Yu, H. Wang, W. Yu, S. Li, X. Zhang and Z. Bian, Appl. Catal., B, 2024, 341, 123292 CrossRef CAS.
- N. Cao, Y. Quan, A. Guan, C. Yang, Y. Ji, L. Zhang and G. Zheng, J. Colloid Interface Sci., 2020, 577, 109–114 CrossRef CAS PubMed.
- Z. Tao, C. L. Rooney, Y. Liang and H. Wang, J. Am. Chem. Soc., 2021, 143, 19630–19642 CrossRef CAS PubMed.
- Y. S. Wu, Z. Jiang, Z. C. Lin, Y. Y. Liang and H. L. Wang, Nat. Sustain., 2021, 4, 725–730 CrossRef.
- D. Ewis, M. Arsalan, M. Khaled, D. Pant, M. M. Ba-Abbad, A. Amhamed and M. H. El-Naas, Sep. Purif. Technol., 2023, 316, 123811 CrossRef CAS.
- M. Shibata, K. Yoshida and N. Furuya, J. Electrochem. Soc., 1998, 145, 2348 CrossRef CAS.
- J. Akther, C. Song, K. Fatih and P. G. Pickup, ECS Adv., 2023, 2, 030503 CrossRef CAS.
- X. Zhang, X. Zhu, S. Bo, C. Chen, Q. Zhai, S. Li, X. Tu, J. Zheng, D. Wang, X. Wei, W. Chen, T. Wang, Y. Li, Q. Liu, S. P. Jiang, L. Dai and S. Wang, Chem, 2024 DOI:10.1016/j.chempr.2024.01.025.
- D. Saravanakumar, J. Song, S. Lee, N. H. Hur and W. Shin, ChemSusChem, 2017, 10, 3999–4003 CrossRef CAS PubMed.
- C. Wang, C. Yu, B. Qian, Y. Ren, L. Wang, Y. Xie, X. Tan, X. He and J. Qiu, Small, 2023, 20, 2307349 CrossRef PubMed.
- Z. Liang, C. Lee, J. Liu, Y. Hu, D. Han, L. Niu and Q. Yan, Mater. Today Catal., 2023, 2, 100011 CrossRef.
- M. Rahmatullah and T. R. C. Boyde, Clin. Chim. Acta, 1980, 107, 3–9 CrossRef CAS PubMed.
- Y. Huang, Y. Wang, Y. Liu, A. Ma, J. Gui, C. Zhang, Y. Yu and B. Zhang, Chem. Eng. J., 2023, 453, 139836 CrossRef CAS.
- X. Wei, X. Wen, Y. Liu, C. Chen, C. Xie, D. Wang, M. Qiu, N. He, P. Zhou, W. Chen, J. Cheng, H. Lin, J. Jia, X. Z. Fu and S. Wang, J. Am. Chem. Soc., 2022, 144, 11530–11535 CrossRef CAS PubMed.
- S. Zhang, J. Geng, Z. Zhao, M. Jin, W. Li, Y. Ye, K. Li, G. Wang, Y. Zhang, H. Yin, H. Zhang and H. Zhao, EES Catal., 2023, 1, 45–53 RSC.
- A. Abragam, The Principles of Nuclear Magnetism, Clarendon Press, Oxford, 1961 Search PubMed.
- Y. Huang, R. Yang, C. Wang, N. Meng, Y. Shi, Y. Yu and B. Zhang, ACS Energy Lett., 2022, 7, 284–291 CrossRef CAS.
- A. Aminot, D. S. Kirkwood and R. Krouel, Mar. Chem., 1997, 56, 59–75 CrossRef CAS.
- J. Y. Fang, Q. Z. Zheng, Y. Y. Lou, K. M. Zhao, S. N. Hu, G. Li, O. Akdim, X. Y. Huang and S. G. Sun, Nat. Commun., 2022, 13, 7899 CrossRef CAS PubMed; Y. Ren, C. Yu, X. Tan, H. Huang, Q. Wei and J. Qiu, Energy Environ. Sci., 2021, 14, 1176–1193 RSC.
- M. A. Ramin, G. Le Bourdon, N. Daugey, B. Bennetau, L. Vellutini and T. Buffeteaud, Langmuir, 2011, 27, 6076–6084 CrossRef CAS PubMed.
- X. X. Han, R. S. Rodriguez, C. L. Haynes, Y. Ozaki and B. Zhao, Nat. Rev. Methods Primers, 2022, 1, 87 CrossRef.
- A. Saini, R. Medwal, S. Bedi, B. Metha, R. Gupta, T. Maurer, J. Plain and S. Annapoorni, Plasmonic, 2015, 10, 617–623 CrossRef CAS.
- C. Leordean, V. Canpean and S. Astilean, Spectrosc. Lett., 2012, 45, 550–555 CrossRef CAS.
- E. Tan, N. B. Julmohammad, W. Y. Koh, M. S. A. Sani and B. Rasti, Foods, 2023, 12, 2855 CrossRef CAS PubMed.
- L. Bellavia, D. B. Kim-Shapiro and S. B. King, Future Sci., 2015, 1(1) DOI:10.4155/fso.15.36.
- Y. Zhao, R. Shi, X. Bian, C. Zhou, S. Zhang, F. Wu, G. I. N. Waterhouse, L. Wu, C. Tung and T. Zhang, Adv. Sci., 2019, 6, 1802109 CrossRef PubMed.
- A. J. Martín, F. L. P. Veenstra, J. Lüthi, R. Verel and J. Pérez-Ramírez, Chem. Catal., 2021, 1, 1505–1518 CrossRef.
- G. W. Watt and J. D. Chrisp, Anal. Chem., 1952, 24, 2006–2008 CrossRef CAS.
- J. Hong, W. Zhang, J. Ren and R. Xu, Anal. Methods, 2013, 5, 1086–1097 RSC.
- A. Javier, B. Chmielowiec, J. Sanabria-Chinchilla, Y. G. Kim, J. H. Baricuatro and M. P. Soriaga, Electrocatalysis, 2015, 6, 127–131 CrossRef CAS.
- C. J. Bondue and M. T. M. Koper, J. Electroanal. Chem., 2020, 875, 113842 CrossRef CAS.
- J. M. Barlow, J. W. Ziller and J. Y. Yang, ACS Catal., 2021, 11, 8155–8164 CrossRef CAS.
- P. An, L. Wei, H. Li, B. Yang, K. Liu, J. Fu, H. Li, H. Liu, J. Hu, Y. R. Lu, H. Pan, T. S. Chan, N. Zhang and M. Liu, J. Mater. Chem. A, 2020, 8, 15936–15941 RSC.
- P. Li, Z. Zhang, X. Yang, Y. Zhu, Z. Zhou, X. Jiang, Q. Wang, X. Gao, X. Yang, Y. Shen and M. Wang, ChemCatChem, 2024, 16, e202301302 CrossRef CAS.
- Q. Zhao, Y. Zhang, D. Cao and M. Shao, Curr. Opin. Electrochem., 2024, 45, 101479 CrossRef.
| This journal is © The Royal Society of Chemistry 2024 |