
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicencePlanar carbenium ions for robust symmetrical all organic redox flow batteries†
Jules
Moutet
 a,
Marko H.
Nowack
b,
David D.
Mills
a,
Diego L.
Lozier
a,
Bo W.
Laursen
b and
Thomas L.
Gianetti
*a
a,
Marko H.
Nowack
b,
David D.
Mills
a,
Diego L.
Lozier
a,
Bo W.
Laursen
b and
Thomas L.
Gianetti
*a
aUniversity of Arizona, Department of Chemistry and Biochemistry, Tucson, AZ, USA. E-mail: tgianetti@arizona.edu
bNano-Science Center and Department of Chemistry, University of Copenhagen, Universitetsparken 5, 2100, Copenhagen, Denmark
First published on 11th September 2023
Abstract
Grid-scale energy storage can benefit from the potential of non-aqueous full organic redox flow batteries (NAORFBs). However, the majority of current NAORFBs rely on the utilization of distinct anolytes and catholytes, separated by a membrane or porous separator. Unfortunately, this setup can lead to crossover of redox active material from one side of the battery to the other, resulting in electrolyte mixing and irreversible fading in energy density and capacity. An attractive solution to tackle this crossover problem is the adoption of symmetric flow batteries, wherein a single bipolar molecule serves as both an anolyte and a catholyte. Herein, we report the use of of a diazatriangulenium ion, a heterocyclic fused carbenium ion, as a bipolar redox active material in such symmetric flow batteries. This redox active molecule exhibits promising characteristics, including a straightforward synthesis, high tunability, non-toxicity, and availability. Evaluation of this molecule through 3-electrode cell testing reveals excellent electrokinetic parameters suitable for NAORFB deployment. The performance is further demonstrated in a prototype of a fully organic symmetrical redox flow battery, exhibiting an large Egap of 2.36 V, energy density exceeding 6 W h L−1, and 99.93% capacity retention over 300 cycles, despite a moderate energetic efficiency.
Introduction
Our modern society increasingly relies on renewable energy sources to transition towards a more sustainable energy system.1 However, even though solar and wind technologies are widely available and mature, their intermittent nature poses a challenge to ensure consistent and reliable power supply.2,3 To overcome this issue, it is crucial to develop efficient, cost-effective, and scalable electrical energy storage (EES) systems.4–6 Redox flow batteries (RFBs) have emerged as an excellent EES option for large-scale stationary applications due to their scalability and engineering flexibility.7–9Currently, commercially developed RFB technologies rely on the redox properties of transition metal ions in aqueous solution, such as iron, chromium, and vanadium (AqRFB).10–14 The use of AqRFB is, however, constrained by a number of factors, such as low Energy Densities (Ed in W h L−1), the narrow thermodynamic electrochemical window of water (1.5 V), high costs of membrane separators, compound toxicity, and the environmental impact of mining ores and handling large amounts of acid.15–17
Non-aqueous RFBs present an attractive alternative due to their potential for higher energy density, large electrochemical windows (>5 V), and mild pH operating conditions.18,19 Additionally, they allow the use of redox-active organic molecules (ROMs), which can be synthesized sustainably and customized to meet particular requirements.20,21 The majority of current non-aqueous fully-organic RFBs (NAORFBs) exhibit an asymmetric design that holds strict selectivity requirements on the exchange membrane due to the use of two distinct ROMs for the posolyte and negolyte tanks. The irreversible capacity loss22 caused by the entropic diffusion of redox-active material throughout the separating interface creates new pathways for chemical deterioration.23 To reduce active material crossover, a number of methods have been suggested, including improved membrane composition,24,25 size discrimination methods,26,27 and also immiscible electrolyte solutions.28
The design of vanadium redox flow batteries (VRFBs) with almost identical tank composition motivated the emergence of symmetrical Organic RFBs (SORFBs), which are now perceived as the most promising approach to reduce membrane crossover and extend the lifespan of the EES.29 In SORFBs, a single bipolar redox-active molecule (BRM) is used on both sides of the cell, allowing the half-reactions of the positive and negative electrodes, respectively.30 Recent advancements in the synthetic flexibility of the ROMs design have enabled the development of different classes of BRMs. A table summarizing all the BRMs reported to date, and their electrochemical properties, is presented in the ESI† (Table S1). Ambipolar molecules, created by bridging a donor redox unit and a compatible acceptor redox moiety, have proven to be a relevant approach to rapidly implement a diverse library of compounds.31–38 On the other hand, open shell molecules, stable organic radicals with multi-event electrochemical properties have shown remarkable results despite their more pronounced reactivity in application.39–43 Our group recently contributed to this field through the use of the helical carbenium ions, dimethoxyquinacridinium (DMQA+) as BRM material in the development of SORFBs.44,45
Triangulenium ions, heterocyclic fused carbocation congener of the DMQA+ ions, have also received attention in recent years due to their photophysical, electrochemical and biological properties.46 DAOTA+ is synthetically available at gram scale in three steps from commercially available starting materials.47 As highlighted by its use as fluorophore in biological systems, DAOTA+ ions are non-toxic and safe to work on large scale.48,49 We recently reported their use as efficient photocatalyst for organic transformation under green light irradiation.50 During our study we observed that, similar to DMQA+, DAOTA+ possess two reversible events and three stable oxidation states. Herein, we report the use of a diazaoxatriangulenium ion (DAOTA+) incorporating two n-propyl arms as new BRM with great potential for large-scale RFB application. This scaffold is the oxygen-bridged analogue of DMQA+, which leads to enhanced stability, solubility and voltage gap (2.3 V for DAOTA+vs. 2.1 V for DMQA+), essential features to reach suitable energy density (Ed) and high cyclability.
Experimental
General remarks
1H, 13C and 19F NMR spectra were recorded on Bruker Avance III-400 MHz or DRX-500 MHz spectrometers in appropriate solvents using residual solvent signals as standards. The chemical shifts are shown in δ scales. Multiplicities of 1H NMR signals are designated as s (singlet), d (doublet), dd (doublet of doublet), dt (doublet of triplet), t (triplet), quin (quintet), m (multiplet), etc. Compounds were named using ChemDraw and assignments of NMR spectra were done using MestReNova. All glassware or hardware has been dried in an oven at least 24 h prior to introduction in glovebox.Chemicals, reagents, and synthesis of C+
All chemicals and solvents were purchased from Sigma Aldrich, Fisher Scientific, or VWR. Organic solvents used were dried by a standard solvent purification system (MBraun SPS or Vigor Solvent Systems). Propyl-1-amine (nPr-NH2) and pyridinium chloride (Pyr·HCl) were obtained from commercial sources and used without further purification. Tris(2,6 dimethoxyphenyl)carbenium tetrafluoroborate salt was synthetized according to a literature procesdure.51 Supporting electrolyte salts tetrabutylammonium hexafluorophosphate (TBAPF6) was recrystallized three times from ethanol, then dried at 80 °C for three days prior to use in glovebox.UV-visible measurements
Absorption spectra were recorded on an Agilent 8453 UV-Visible spectrophotometer at 25 °C in analytical-grade acetonitrile from 300 nm to 900 nm.Electrochemical studies
Electrochemical analyses were conducted inside an Argon-filled MBraun Unilab glovebox using a BioLogic SP-200 potentiostat/galvanostat and the EC-Lab® software (v11.33). For convenience, potentials are expressed versus the internal reference electrode AgNO3/Ag or versus the cell itself in a 2-electrode setup. Cyclic voltammograms (CV) were measured in a three-electrode electrochemical cell, consisting of a platinum wire counter electrode, a AgNO3/Ag reference electrode (0.01 M AgNO3 in 0.1 M TBAPF6 in CH3CN), and a glassy carbon working electrode (0.071 cm2, CH Instrument, Inc.). The working electrode was polished prior to each record using aluminum oxide on polishing paper and anhydrous CH3CN to remove residual particles. CVs were recorded at different scan rates (10, 25, 75, 100, 250, 400, and 500 mV s−1) in an CH3CN electrolyte solution, containing 1 mM C+ and 0.1 M TBAPF6.Redox flow cell experiments
Flow cell measurements were conducted on a BioLogic SP-200 in galvanostatic mode using the EC-Lab® software (v11.33) in an Argon-filled glovebox. A 5 cm2 single-cell flow battery was used, and, on both sides, a PTFE gasket with a cutout for the exchange surface of 5 cm2 hosted 2 carbon felt electrodes, which were in direct contact with the central exchange membrane. This membrane consists of porous separators placed between the anolyte and catholyte sides of the cell (Fig. S10, ESI†). The components of the flow cell were dried in an oven overnight, assembled outside of the glovebox, and immediately brought into the glovebox through the antechamber. Both anolyte and catholyte tanks were loaded with 4 mL of electrolyte/ROM in 0.1 M TBAPF6 in CH3CN at the desired nPrDAOTA+ concentration. The electrolyte solutions were pumped into and out from the flow cell using a peristaltic pump at a rate of 16 mL min−1. An equilibration period of 4 h was observed before active charging and discharging.Density functional theory calculations
Density functional theory (DFT) calculations were carried out using a long-range corrected Perdew–Burke–Ernzerhof (LC-wPBE) functional with a 6-311G** basis set applied to all atoms. Initial structures were built using Spartan Student v8.0.3 and optimized using Gaussian 16 (see ESI† for details). The optimized structures were determined to be energy minima by lack of imaginary vibrations in the frequency calculations. Solvated models were calculated with the polarizable continuum model (PCM) using the integral equation formalism variant (IEFPCM) as the SCRF method. Acetonitrile was used as the solvent and as defined by G16. Free energies of solvation (ΔGsolv) were determined as the difference in the sum of electronic and thermal free energies between the solvated and gas phase optimized structures.Scanning electron microscope
SEM images were taken on an FEI Inspec-S scanning electron microscope containing a tungsten filament with a gun voltage30 keV. Carbon felt samples were mounted using sticky carbon tape. Carbon felt samples were not subject to any modification.Results and discussion
Synthesis of C+
The synthesis of the 4,8-di-n-propyl-4,8-diaza-12-axatriangulenium hexafluorophosphate salt (nPrDAOTA+), first reported by Laursen et al. in 2000,51 is achieved by successive nucleophilic aromatic substitution reactions (SNAr) on the tris(2,6 dimethoxyphenyl)carbenium tetrafluoroborate salt in CH3CN. In a first step, the 9-(2,6-dimethoxyphenyl)-1,8-dimethoxy-10-n-propylacridinium, nPrDMQA+, is synthesized using n-propylamine as the nucleophile. After an exchange of BF4− counteranions by PF6− facilitating isolation of the intermediate, a second ring-closure SNAr inducing a complete aromatization of the scaffold is obtained using pyridine hydrochloride (Pyr·HCl) as demethylating agent. nPrDAOTA+ is thus obtained in 2 steps with an overall yield of 45% (Scheme 1) and will be denoted C+ in the following.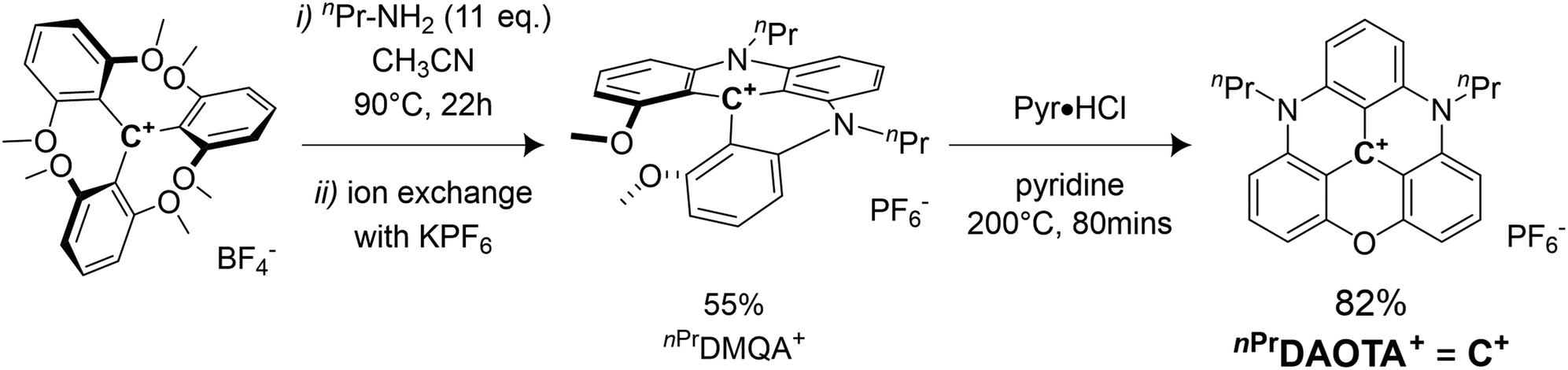 | ||
| Scheme 1 Synthesis of 4,8-di-n-propyl-4,8-diaza-12-axatriangulenium (nPrDAOTA+, noted C+). | ||
Electrochemical studies
In this study, we selected acetonitrile as the ideal solvent for our investigations due to its wide electrochemical window of approximately 6.0 V. Acetonitrile not only contributes to enhancing energy density,41,52 but it also has been proven to be suitable for examining the various oxidation states of nPrDAOTA+.50,53 Consistent with previous reports, we observed three distinct single-electron redox events for nPrDAOTA+ (C+) in cyclic voltammetry (CV) experiments conducted in a three-electrode cell containing a 0.1 M TBAPF6 in CH3CN solution (Fig. 1a). The accurate determination of the half-wave potential (E1/2) for these redox processes was achieved by performing Differential Pulse Voltammetry (DPV) experiments (Fig. 1a, pastel). Upon scanning towards the cathodic region, a reversible oxidation event occurred at E1/2Ox = 1.06 V vs. Ag/AgNO3, indicating the formation of the dication radical nPrDAOTA˙++ (C˙++). When scanning the anodic region, we observed a reversible single-electron reduction at a potential of E1/2Red = −1.30 V vs. Ag/AgNO3, which corresponds to the formation of a neutral radical nPrDAOTA˙ (C˙), similar to the previously described DMQA˙.54 A second event in the anodic region was observed at ERed2 = −2.06 V vs. Ag/AgNO3 (shown in grey in Fig. 1a) but was found irreversible and led to the formation of species showing a double oxidation wave between 0.25 V and 0.65 V vs. Ag/AgNO3. This electrochemical behavior is similar to the one observed for DMQA,55 and can be explained by the formation of the neutral nPrDAOTA-H, denoted C–H in the ESI† (Fig. S1), formed upon protonation of the anionic species generated during the second reduction event by acetonitrile.55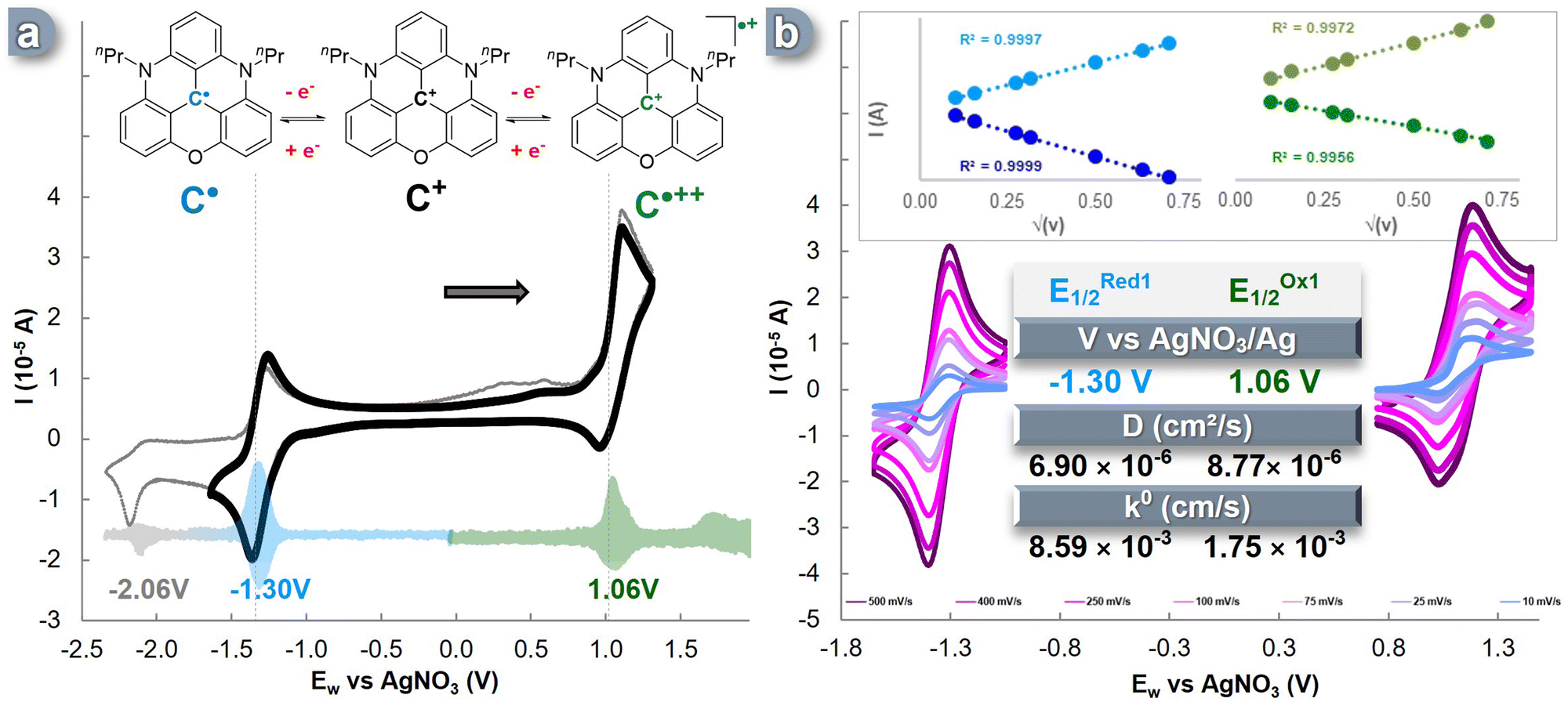 | ||
| Fig. 1 (a) Three stable different oxidation states of C+ and CV of reversible event (black) and non-reversible event (grey) at of 100 mV s−1. DPV in reduction and oxidation in pastel color. (b) CV studies of C+ with scan rates varying from 10 to 500 mV s−1. Inserts show plots of anodic and cathodic peak current densities (I) vs. the square root of the sweep rate (v1/2) for reduction (blue) and oxidation (green) process. Table summarizes E1/2, D and k0 obtained. Study on C+ 1 mM in 0.1 M TBAPF6 CH3CN solution, counter-anions and counter-cations have been omitted for clarity. | ||
To investigate the reversibility of these electronic processes, CV experiments were conducted at various scan rates in the same three-electrode cell (Fig. 1b). A linear relationship was observed between the measured intensity (I) and the square root of the scan rate, indicating unhindered electrolyte diffusion (Fig. 1b, insert). Furthermore, the complete reversibility of the two electronic processes was maintained at each scan rate. The decisive electrochemical kinetic parameters were extracted from these plots. By numerically applying the Randles–Sevcik equation (eqn (S1), ESI†), diffusion coefficient (D) values of 8.77 × 10−6 cm2 s−1 were obtained for the C˙++/C+ pair, and a value of 6.90 × 10−6 cm2 s−1 was obtained for the C+/C˙ couple. These values describe an efficient mass transport of the redox-active species from the bulk solution to the electrode surface and are consistent with, or even superior to, recent ROMs used in ORFB applications.35,38,56–60
The electron transfer rate constant (k0) was determined through the work reported by Lavagnini et al. (eqn (S2) and (S3), ESI†). A high value of k0 = 8.59 × 10−3 cm s−1 was observed for the electronic process of C+ ![[left over right harpoons]](https://www.rsc.org/images/entities/char_21cb.gif) C˙, while the oxidation pathway of C˙++ C+ exhibited a slightly lower value of k0 = 1.75 × 10−3 cm s−1. These parameters are within the range of average transfer rate values for ROMs recently reported in the literature and, thus, remain suitable for application in ORFBs. Furthermore, to assess the robustness of the electronic processes involving C+/C˙ and C˙++/C+, each event was cycled in a three-electrode cell, and minimal loss of CV signal was observed over 300 CV cycles (Fig. S4, ESI†).
C˙, while the oxidation pathway of C˙++ C+ exhibited a slightly lower value of k0 = 1.75 × 10−3 cm s−1. These parameters are within the range of average transfer rate values for ROMs recently reported in the literature and, thus, remain suitable for application in ORFBs. Furthermore, to assess the robustness of the electronic processes involving C+/C˙ and C˙++/C+, each event was cycled in a three-electrode cell, and minimal loss of CV signal was observed over 300 CV cycles (Fig. S4, ESI†).
This electrochemical study demonstrated the reversibility of both oxidation and reduction processes, enabling access to the corresponding neutral radical and dicationic radical species as expected for nPrDAOTA+. These three charge states displayed reversibility and exhibited chemical stability throughout the CV experiments, affirming that C+ satisfies the requirements for its use as a BRM in Symmetrical Organic Redox Flow Batteries (SORFBs).
Maximum Ed
The evaluation of the maximal concentration of C+ in acetonitrile was conducted using UV-Visible spectroscopy. Standards within an established range of concentrations were prepared and measured to determine the molar absorptivity (ε) at three specific wavelengths: λmax = 557, 449 and 359 nm (Fig. 2 and Fig. S6, ESI†). The absorbance of a saturated solution of C+ was then measured using three different dilution ratios (Table S5, ESI†). By applying the previously determined ε-values, the average saturation concentration of C+ was calculated to be 103 mM. The saturation concentration of bis n-propyl DAOTA+ is an order of magnitude higher than its congener bis n-propyl DMQA+ (32.4 mM).44 The main goal of ROMs designed for ORFBs is to maximize the energy density (Ed) achievable, which can be mathematically represented as Ed = nFEcellCactive0.5 (in W h L−1). Within this formula, the variable n represents the quantity of electrons participating in the redox process, F denotes the Faraday constant in A h L−1, Ecell signifies the cell voltage or potential difference between the redox states (E1/2Ox − E1/2Red), and Cactive represents the concentration of the redox-active species, which is equivalent to the electron concentration at both the cathode and anode. The numerical application with for the observed Ecell of 2.36 V yields to a value Ed = 6.5W h L−1 for nPrDAOTA+ (Fig. 2, table), a significant increase compared to Ed = 1.3 W h L−1 for nPrDMQA+.44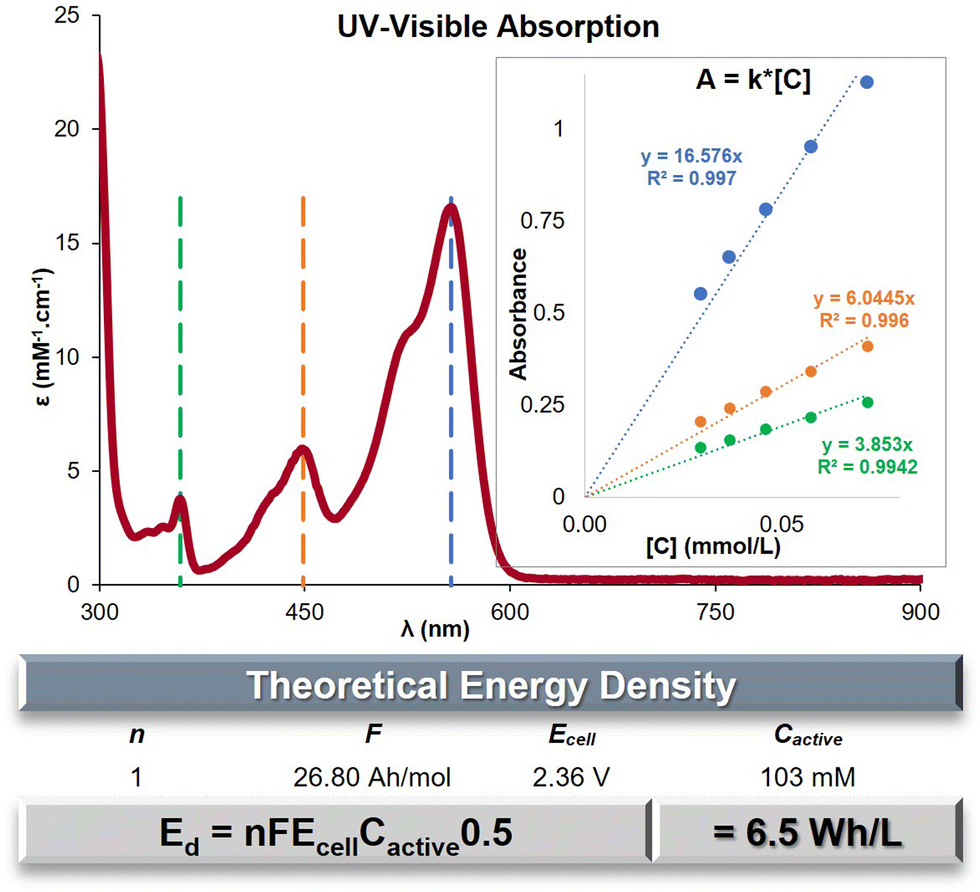 | ||
| Fig. 2 UV-visible absorption spectrum of C+ in CH3CN. Insert: Linear relation measure between concentration and absorption for a range of standards solution. Table: theoretical energy density numerical application. | ||
DFT calculations
In addition to UV-Visible measurement, the evaluation of solubility for the three different redox states in acetonitrile has been conducted through DFT calculations. To gain insights into this characteristic, ΔGsolv values were computed for all redox states of nPrDAOTA+ and eventual side-products (Table S6, ESI†). It is commonly recognized that explicit calculations of compound solubilities are not feasible.61 Nevertheless, computed free energies offer a dependable method to estimate the relative solubilities of diverse compounds.62,63 Particularly, more negative values indicate higher solubilities.64 Regarding the solubility of different redox states, the observed order is C++˙ > C+ > C˙. The solubility of the neutral nPrDAOTA-H, formed during over reduction of the DAOTA platform in CH3CN at ERed2 = −2.06 V vs. Ag/AgNO3, has also been evaluated and found to be the least soluble among the compounds present (Table S6, ESI†).The electronic structure for the various redox states was also investigated. Following the optimization of structures in gas and solvated phases, the molecular orbital (MO) energies [in eV] were plotted for the primary redox species C+, C˙, and C++˙, as well as the electrochemically accessible anionic redox species C− (Fig. S8, ESI†). Spin density analysis reveals that the unpaired electron in C˙ is predominantly localized at the central carbon, while the spin density of C++˙ is mostly localized on the two bridging nitrogen and the aromatic meta positions of the ring located between the two-bridging nitrogen (Fig. S9, ESI†). It was considered that the anionic redox state could have two potential spin states, a singlet (C−) and triplet (C−˙˙), however the computed energy difference between the two spin states suggests the singlet state is more stable by about 28 kcal mol−1 in both gas and solvated phases (Table S7, ESI†).
Galvanostatic cycling with potential limitation experiment
Given the promising nature of the measured physicochemical parameters of the C+ as ROM, we then evaluated its electrochemical reversibility and stability through galvanostatic charge–discharge cycles under operational conditions. A complete prototype RFB cell was used to assess the performance of this carbocation, knowing that in this configuration all the hardware elements can have a significant impact on the overall efficiency of the system. The experiment utilized a polymer-based porous membrane exchanger with carbon paper electrodes commonly employed in commercial RFB solutions. However, the process of full-flow battery cycling poses several challenges due to the presence of multiple hardware components, such as tubes, electrodes, bipolar plates, and membranes (Fig. S10, ESI†). These components interact with the redox active molecules carrying a radical charge. Additionally, the flow of electrolyte circulating through the cell and tanks, as well as the potential crossover of “posolyte” and “anolyte” between the two sides of the cell during the flow cycle, introduces the risk of self-discharge and diminished capacity retention.To overcome these challenges, the RFB cell was specifically designed to incorporate bipolar graphite plates with a serpentine flow field. The cell also featured carbon-felt electrodes (Sigracet 29 AA) that were 400 μm thick, had an 80% porosity, and exhibited an electrical resistivity of less than 5 mΩ cm2. The surface chemistry of the electrodes was extensively examined based on existing literature.65 A Daramic-175 porous separator was utilized to separate the two sides of the cell, acting as an exchange membrane for the counter-anion. During galvanostatic cycling, the electrolyte solutions flowed through the cell at a rate of 8 mL min−1 per channel. Recent techno-economic analyses have established a target of achieving an area-specific resistance (ASR) of less than 5 Ω cm−2 to enhance NAqRFB specs.66 To assess the prototype's suitability for scaling up, potential electrochemical impedance spectroscopy (PEIS) was conducted at 0% State of Charge (SOC), which corresponds to the equilibrium state of the SORFB where both negolyte and posolyte tanks contain C+. Charging and discharging were performed at a constant current corresponding to a 2C-rate until reaching a voltage cut-off of +200 mV from the cell's theoretical Ecell = 2.36 V. The lower voltage cut-off was set at 0V (0% SOC). At the interface, the charge accumulated on the BRM was balanced by the movement of the PF6− counter anion through the porous exchange membrane.
A two-electrode setup was assembled with the C+ C˙ process occurring at Ew (Fig. 3a). The electrolyte solution, containing 0.1 M TBAPF6 in acetonitrile, was continuously circulated through the cell at a rate of 8 mL min−1 per tank. Over the course of 300 cycles (i.e. ∼9.5 days), the SORFB exhibited steady capacity retention, with a perfect full charge capacity (Fig. 3b, red circle) and an average discharge capacity value of 67% observed between the 10th and 240th cycle (Fig. 3b, black circle). However, from cycle 240th onwards, the system's charge capacity begins to decrease, with a loss of 0.16%/cycle. This is accompanied by a change in loss regime for the discharge capacity from 0.08%/cycle to 0.05%/cycle. The coulombic efficiency (CE) reached 71% throughout the entire experiment with a point of curvature at cycle 157th (67%) and starting from cycle 240th diverges from alignment with discharge capacity to reach a value of 73% at the 300th cycle (Fig. 3b, green square). This result correlates with the moderate energy efficiency (EE) observed, which was 58% at the 10th cycle, but decreases to an average value of 51% over the course of the experiment. Considering the high charge capacity rate (∼100%) achieved in the first 240 cycles and the minimal decline in discharge rate, some energy must be consumed between charging and discharging, which cannot be explained by self-discharge alone. By monitoring the potential and current data of the RFB cell during cycling, insights were gained about the decrease in charging capacity observed from cycle 240 onwards (Fig. 3c). It was observed that, although the potential did not reach the limit value of −2.56 V as illustrated between the 100th and 105th cycles, from the 245th cycle onwards, the cut-off value was systematically reached (Fig. 3c, 245th cycle). This induces polarity reversal and the start of the discharge sequence, before a full charge is reached, albeit without significantly affecting EE and CE values or cycle times of the system.
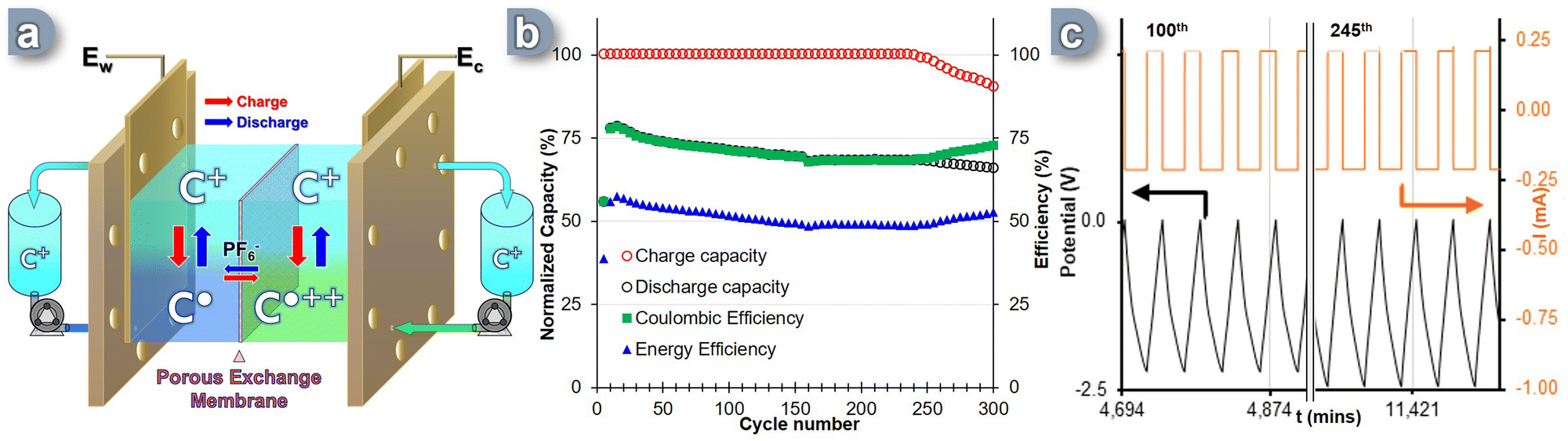 | ||
| Fig. 3 (a) Scheme of the SORFB cell during cycling. (b) Plot of charge, discharge capacity (normalized to theoretical capacity), coulombic and energy voltaic efficiency vs. cycle number for RFB-cell cycling with tanks loaded each with 4 mL of 1 mM C+, flow 8 mL min−1 per channel a 2C-rate for 300 cycles (∼9.5 days). Each data point represents 5 cycles. (c) Monitoring of cell potential and I vs. times for cycle 100th to 105th and 245th to 250th. | ||
After cycling, the solutions from both tankers were analyzed using a three-electrode CV cell (Fig. 4a). It was immediately apparent that both solutions had undergone significant degradation. The content of the Ec reservoir exhibited a decrease of over 50% in the area corresponding to the C+ C++˙ electronic process, while the content of the Ew reservoir experienced a dramatic degradation, with a 92% reduction in the area associated with the C+ C˙ process, leaving less than 10% of the initial CV's active electroactive material remaining (Fig. 4a, blue trace). This outcome is highly surprising, considering that the SORFB displayed a discharge efficiency of over 65% even after 300 cycles, which contradicts the indications from the CV solution analysis. To gain further insights into these phenomena, additional analyses targeting the materials within the cell were conducted. We first investigated the conductivity of the flow cell through potential electrochemical impedance spectroscopy (PEIS) after cycling compared to the initial state (Fig. 4b, insert). Initially, the ASR value was found to be 1.40 Ω, while after cycling it appeared to be 1.34 Ω, suggesting that after 300 cycles, the system's exchange surface exhibits an improved conductivity (Fig. 4b). This prompted us to examine the physical changes that may have occurred in the carbon electrodes during these cycles. Scanning electron microscope (SEM) analyses were conducted on the carbon felts used as the electron transfer matrix, and the results were conclusive: every electrode image displayed notable deposits (Fig. 4c). While Ec side, the C+ C++˙ phenomenon resulted in few deposits, the Ew side showed a significant impact, with the carbon fibers appearing modified and coated with a highly conductive material (evidenced by higher signal intensity for the same radiation power used). The C+ C˙ process seemed to generate an insoluble compound that was both conductive and capable of storing and releasing energy while remaining absorbed on the electrode. We speculate that the reactive C− species (see DFT calculations, vide-supra), which can be formed by over reduction of the nPrDAOTA scaffold during battery cycling overloads, is a source of electrolyte degradation. Reaction with acetonitrile will generate the poorly soluble nPrDAOTA-H, while radical coupling will form insoluble dimers. Dimerization of a similar planar system has been reported and yields a compound with relatively low solubility.67
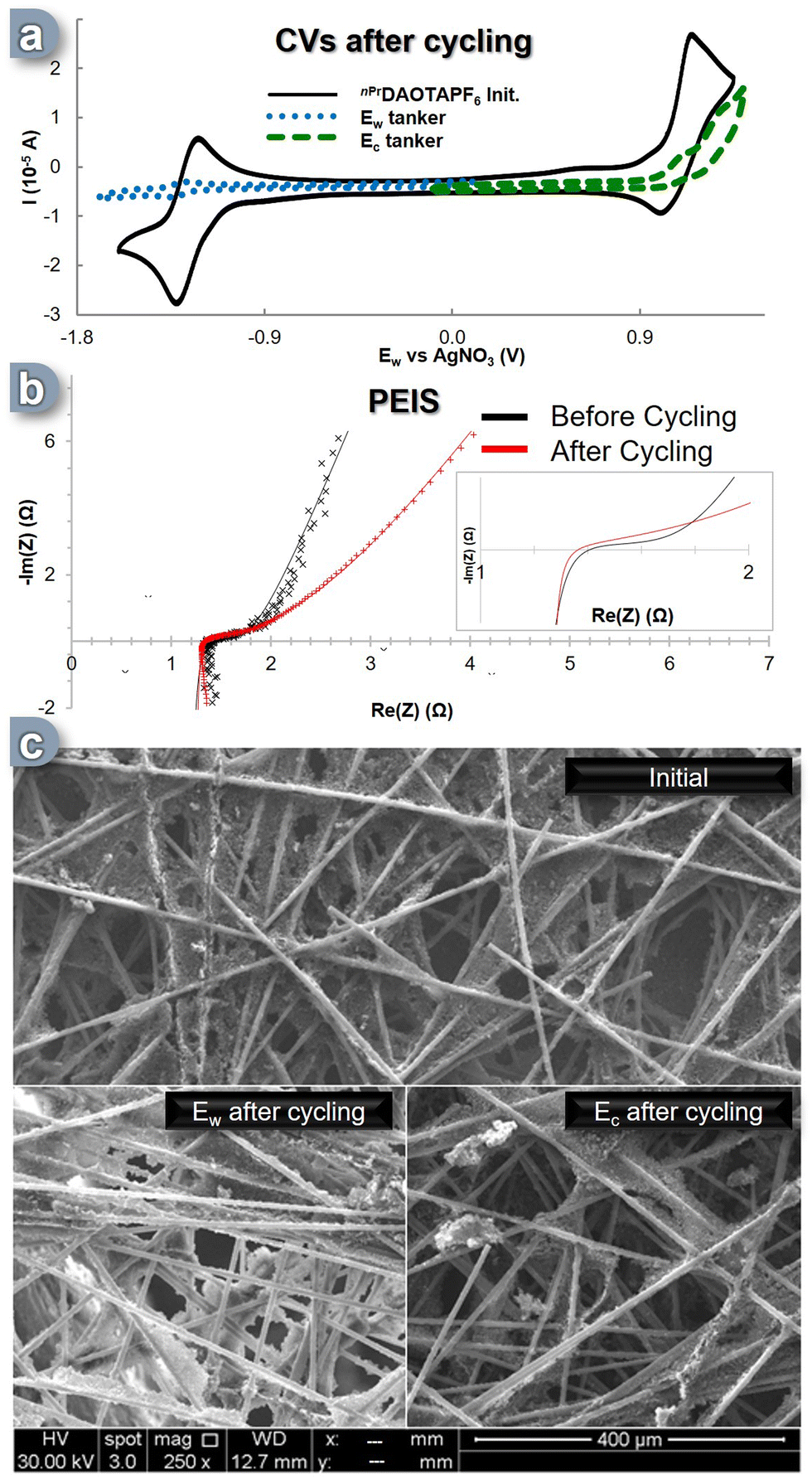 | ||
| Fig. 4 (a) CVs of tank's solutions after 300 cycles (∼9.5 days) and comparison with initial C+ CV. (b) PEIS of the cell in flow before and after 300 cycles (both at 0% SOC) from 1 Ghz to 0.1 Hz with an amplitude of 10 mV. Insert: zoom on the fit intersection with the x-axis (c) SEM pictures of the carbon felt electrodes before and after cycling for both sides of the cell at magnification x250. | ||
Although the in-solution analysis indicated significant loss of electroactive material, PEIS and SEM analysis suggest a deposition of electroactive material on the carbon felt allowing a high charge and discharge efficiency throughout the 9 day period (300 cycles).
Conclusion
In conclusion, we have shown that the 4,8-di-n-propyl-4,8-diaza-12-axatriangulenium carbenium (nPrDAOTA+, C+) combine ease of synthesis, non-toxicity, bipolar electrochemistry, and high chemical stability in all states of charge, making this compound a promising candidate as energy-storage materials for symmetric redox-flow batteries. Cyclic voltammetry and cell cycling studies in CH3CN solution have indicated that these organic BRM exhibit excellent electrochemical reversibility and chemical stability, with a significant 2.36 V gap. Despite a minimal capacity fade during battery cycling, with an overall capacity retention of 99.93% over 300 cycles, post-cycling CV analyses have revealed a notable loss of electroactive material in solution, while PEIS and SEM picture revealed important deposit of material on the electrodes suggesting plausible issues related to insolubility during bulk cycling. However, this deposit on the carbon felt electrodes, although causing a loss of energy efficiency, also demonstrates its capability to store and release electrons efficiently. While it may not be ideally suited for conventional all-liquid RFBs, the emergence of Semi-Solid RFBs (SSRFBs)68,69 offers an elegant solution to overcome the solubility challenges of this specific ROM. Our work represents a foundational step toward the development of innovative all-organic, symmetrical RFB chemistries for EES applications. It lays the groundwork for leveraging the untapped potential of planar carbenium molecules, paving the way for advancements in energy density while effectively addressing solubility challenges within this class of compounds.Author contributions
The manuscript was written through contributions of all authors. All authors have given approval to the final version of the manuscript. J. M. and T. L. G. wrote the manuscript and discussed their conclusions. J. M. planned the experiments and conducted all electrochemical experiments; M. H. N. synthetized and characterized C+; D. D. M. performed the DFT calculations and obtained SEM images, D. L. L. recorded UV-visible spectrums.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We are grateful to the University of Arizona, Salt River Project (Phoenix, AZ), Research Corporation for Science Advancement Cottrell Scholarship 2021 (Award #27536) and by Novo Nordic Foundation award number NNF20OC0062176 for financially supporting this work. All NMR data were collected in the NMR facility of the Department of Chemistry and Biochemistry at the University of Arizona, RRID:SCR_012716. The purchase of the Bruker NEO 500 MHz spectrometer was supported by the National Science Foundation under Grant Number 1920234 and the University of Arizona. All SEM images and data were collected in the W.M. Keck Center for Nano-Scale Imaging in the Department of Chemistry and Biochemistry at the University of Arizona, RRID:SCR_022884, with funding from the W.M. Keck Foundation Grant.References
- M. S. Dresselhaus and I. L. Thomas, Nature, 2001, 414, 332–337 Search PubMed.
- J. Rugolo and M. J. Aziz, Energy Environ. Sci., 2012, 5, 7151–7160 Search PubMed.
- M. S. Ziegler, J. M. Mueller, G. D. Pereira, J. Song, M. Ferrara, Y. Chiang and J. E. Trancik, Joule, 2019, 3, 2134–2153 Search PubMed.
- Z. Yang, J. Zhang, M. C. W. Kintner-Meyer, X. Lu, D. Choi, J. P. Lemmon and J. Liu, Chem. Rev., 2011, 111, 3577–3613 Search PubMed.
- S. P. S. Badwal, S. S. Giddey, C. Munnings, A. I. Bhatt and A. F. Hollenkamp, Front. Chem., 2014, 2, 1–28 Search PubMed.
- T. M. Gür, Energy Environ. Sci., 2018, 11, 2696–2767 Search PubMed.
- B. Dunn, H. Kamath and J.-M. Tarascon, Science, 2011, 334, 928–935 Search PubMed.
- P. Alotto, M. Guarnieri and F. Moro, Renewable Sustainable Energy Rev., 2014, 29, 325–335 Search PubMed.
- E. Sánchez-Díez, E. Ventosa, M. Guarnieri, A. Trovò, C. Flox, R. Marcilla, F. Soavi, P. Mazur, E. Aranzabe and R. Ferret, J. Power Sources, 2021, 481, 228804 Search PubMed.
- Y. K. Zeng, T. S. Zhao, L. An, X. L. Zhou and L. Wei, J. Power Sources, 2015, 300, 438–443 Search PubMed.
- J. A. Suttil, J. F. Kucharyson, I. L. Escalante-Garcia, P. J. Cabrera, B. R. James, R. F. Savinell, M. S. Sanford and L. T. Thompson, J. Mater. Chem. A, 2015, 3, 7929–7938 Search PubMed.
- E. S. Beh, D. De Porcellinis, R. L. Gracia, K. T. Xia, R. G. Gordon and M. J. Aziz, ACS Energy Lett., 2017, 2, 639–644 Search PubMed.
- K. Lourenssen, J. Williams, F. Ahmadpour, R. Clemmer and S. Tasnim, J. Energy Storage, 2019, 25, 100844 Search PubMed.
- M. Park, E. S. Beh, E. M. Fell, Y. Jing, E. F. Kerr, D. Porcellinis, M. Goulet, J. Ryu, A. A. Wong, R. G. Gordon, J. Cho and M. J. Aziz, Adv. Energy Mater., 2019, 9, 1900694 Search PubMed.
- DOE Office of ARPR-E. ARPA-E, GRIDS Program Overview, https://arpa-e.energy.gov/sites/default/files/documents/files/GRIDS_ProgramOverview.pdf.
- U.S. DEPARTMENT OF HEALTH AND HUMAN SERVICES Public Health Service Agency for Toxic Substances and Disease Registry, in ATSDR's Toxicological Profiles, CRC Press, 2012.
- R. M. Wittman, M. L. Perry, T. N. Lambert, B. R. Chalamala and Y. Preger, J. Electrochem. Soc., 2020, 167, 090545 Search PubMed.
- Z. Rhodes, J. R. Cabrera-Pardo, M. Li and S. D. Minteer, Isr. J. Chem., 2021, 61, 101–112 Search PubMed.
- L. Kortekaas, S. Fricke, A. Korshunov, I. Cekic-Laskovic, M. Winter and M. Grünebaum, Batteries, 2022, 9, 4 Search PubMed.
- J. Winsberg, T. Hagemann, T. Janoschka, M. D. Hager and U. S. Schubert, Angew. Chem., Int. Ed., 2017, 56, 686–711 Search PubMed.
- Y. Ding, C. Zhang, L. Zhang, Y. Zhou and G. Yu, Chem. Soc. Rev., 2018, 47, 69–103 Search PubMed.
- M. L. Perry, J. D. Saraidaridis and R. M. Darling, Curr. Opin. Electrochem., 2020, 21, 311–318 Search PubMed.
- A. Shrestha, K. H. Hendriks, M. S. Sigman, S. D. Minteer and M. S. Sanford, Chem. – A Eur. J., 2020, 26, 5369–5373 Search PubMed.
- M. T. Tsehaye, G. Mourouga, T. J. Schmidt, J. O. Schumacher, S. Velizarov, B. Van der Bruggen, F. Alloin and C. Iojoiu, Renewable Sustainable Energy Rev., 2023, 173, 113059 Search PubMed.
- B. H. Robb, T. Y. George, C. M. Davis, Z. Tang, C. H. Fujimoto, M. J. Aziz and M. P. Marshak, J. Electrochem. Soc., 2023, 170, 030515 Search PubMed.
- S. E. Doris, A. L. Ward, A. Baskin, P. D. Frischmann, N. Gavvalapalli, E. Chénard, C. S. Sevov, D. Prendergast, J. S. Moore and B. A. Helms, Angew. Chem., Int. Ed., 2017, 56, 1595–1599 Search PubMed.
- K. H. Hendriks, S. G. Robinson, M. N. Braten, C. S. Sevov, B. A. Helms, M. S. Sigman, S. D. Minteer and M. S. Sanford, ACS Cent. Sci., 2018, 4, 189–196 Search PubMed.
- P. Navalpotro, N. Sierra, C. Trujillo, I. Montes, J. Palma and R. Marcilla, ACS Appl. Mater. Interfaces, 2018, 10, 41246–41256 Search PubMed.
- R. A. Potash, J. R. McKone, S. Conte and H. D. Abruña, J. Electrochem. Soc., 2016, 163, A338–A344 Search PubMed.
- M. Li, J. Case and S. D. Minteer, ChemElectroChem, 2021, 8, 1215–1232 Search PubMed.
- T. Janoschka, C. Friebe, M. D. Hager, N. Martin and U. S. Schubert, ChemistryOpen, 2017, 6, 216–220 Search PubMed.
- P. W. Antoni, T. Bruckhoff and M. M. Hansmann, J. Am. Chem. Soc., 2019, 141, 9701–9711 Search PubMed.
- N. H. Attanayake, J. A. Kowalski, K. V. Greco, M. D. Casselman, J. D. Milshtein, S. J. Chapman, S. R. Parkin, F. R. Brushett and S. A. Odom, Chem. Mater., 2019, 31, 4353–4363 Search PubMed.
- V. V. Sentyurin, O. A. Levitskiy and T. V. Magdesieva, Curr. Opin. Electrochem., 2020, 24, 6–14 Search PubMed.
- P. Geysens, Y. Li, I. Vankelecom, J. Fransaer and K. Binnemans, ACS Sustainable Chem. Eng., 2020, 8, 3832–3843 Search PubMed.
- J. S. Tracy, E. S. Horst, V. A. Roytman and F. D. Toste, Chem. Sci., 2022, 13, 10806–10814 Search PubMed.
- Y. Liu, G. Dai, Y. Chen, R. Wang, H. Li, X. Shi, X. Zhang, Y. Xu and Y. Zhao, ACS Energy Lett., 2022, 7, 1274–1283 Search PubMed.
- B. I. Loomans, S. E. Bottle and J. P. Blinco, Batteries Supercaps, 2023, 6, e202200561 Search PubMed.
- T. Hagemann, J. Winsberg, B. Häupler, T. Janoschka, J. J. Gruber, A. Wild and U. S. Schubert, NPG Asia Mater., 2017, 9, e340–e340 Search PubMed.
- C. G. Armstrong and K. E. Toghill, Electrochem. Commun., 2018, 91, 19–24 Search PubMed.
- A. Korshunov, M. J. Milner, M. Grünebaum, A. Studer, M. Winter and I. Cekic-Laskovic, J. Mater. Chem. A, 2020, 8, 22280–22291 Search PubMed.
- V. V. Sentyurin, O. A. Levitskiy and T. V. Magdesieva, Curr. Opin. Electrochem., 2020, 24, 15–23 Search PubMed.
- J. S. Steen, J. L. Nuismer, V. Eiva, A. E. T. Wiglema, N. Daub, J. Hjelm and E. Otten, J. Am. Chem. Soc., 2022, 144, 5051–5058 Search PubMed.
- J. Moutet, J. M. Veleta and T. L. Gianetti, ACS Appl. Energy Mater., 2021, 4, 9–14 Search PubMed.
- J. Moutet, D. Mills, M. M. Hossain and T. L. Gianetti, Mater. Adv., 2022, 3, 216–223 Search PubMed.
- J. Bosson, J. Gouin and J. Lacour, Chem. Soc. Rev., 2014, 43, 2824–2840 Search PubMed.
- B. W. Laursen and F. C. Krebs, Chemistry, 2001, 7, 1773–1783 Search PubMed.
- B. P. Maliwal, R. Fudala, S. Raut, R. Kokate, T. J. Sørensen, B. W. Laursen, Z. Gryczynski and I. Gryczynski, PLoS One, 2013, 8, e63043 Search PubMed.
- M. Rosenberg, K. R. Rostgaard, Z. Liao, A. Ø. Madsen, K. L. Martinez, T. Vosch and B. W. Laursen, Chem. Sci., 2018, 9, 3122–3130 Search PubMed.
- M. H. Nowack, J. Moutet, P. B. W. Laursen and T. L. Gianetti, Synlett, 2023, 327–331 Search PubMed.
- B. W. Laursen and F. C. Krebs, Angew. Chem., 2000, 39, 3432–3434 Search PubMed.
- K. Gong, Q. Fang, S. Gu, S. F. Y. Li and Y. Yan, Energy Environ. Sci., 2015, 8, 3515–3530 Search PubMed.
- I. H. Delgado, S. Pascal, C. Besnard, S. Voci, L. Bouffier, N. Sojic and J. Lacour, Chem. – A Eur. J., 2018, 24, 10186–10195 Search PubMed.
- A. C. Shaikh, J. Moutet, J. M. Veleta, M. M. Hossain, J. Bloch, A. V. Astashkin and T. L. Gianetti, Chem. Sci., 2020, 11, 11060–11067 Search PubMed.
- T. J. Sørensen, M. F. Nielsen and B. W. Laursen, ChemPlusChem, 2014, 79, 1030–1035 Search PubMed.
- Y. Yan, S. G. Robinson, M. S. Sigman and M. S. Sanford, J. Am. Chem. Soc., 2019, 141, 15301–15306 Search PubMed.
- X. Xing, Q. Liu, W. Xu, W. Liang, J. Liu, B. Wang and J. P. Lemmon, ACS Appl. Energy Mater., 2019, 2, 2364–2369 Search PubMed.
- J. Chai, A. Lashgari, Z. Cao, C. K. Williams, X. Wang, J. Dong and J. Jimmy Jiang, ACS Appl. Mater. Interfaces, 2020, 12, 15262–15270 Search PubMed.
- Y. Yan, T. P. Vaid and M. S. Sanford, J. Am. Chem. Soc., 2020, 0c07464 Search PubMed.
- L. Zhang, Y. Qian, R. Feng, Y. Ding, X. Zu, C. Zhang, X. Guo, W. Wang and G. Yu, Nat. Commun., 2020, 11, 3843 Search PubMed.
- D. S. Palmer, A. Llinàs, I. Morao, G. M. Day, J. M. Goodman, R. C. Glen and J. B. O. Mitchell, Mol. Pharm., 2008, 5, 266–279 Search PubMed.
- L. Cheng, R. S. Assary, X. Qu, A. Jain, S. P. Ong, N. N. Rajput, K. Persson and L. A. Curtiss, J. Phys. Chem. Lett., 2015, 6, 283–291 Search PubMed.
- G. Duarte Ramos Matos, D. Y. Kyu, H. H. Loeffler, J. D. Chodera, M. R. Shirts and D. L. Mobley, J. Chem. Eng. Data, 2017, 62, 1559–1569 Search PubMed.
- S. Dasari and B. S. Mallik, J. Mol. Liq., 2020, 301, 112449 Search PubMed.
- K. V. Greco, A. Forner-Cuenca, A. Mularczyk, J. Eller and F. R. Brushett, ACS Appl. Mater. Interfaces, 2018, 10, 44430–44442 Search PubMed.
- J. D. Milshtein, J. L. Barton, T. J. Carney, J. A. Kowalski, R. M. Darling and F. R. Brushett, J. Electrochem. Soc., 2017, 164, A2487–A2499 Search PubMed.
- A. C. Shaikh, J. M. Veleta, J. Moutet and T. L. Gianetti, Chem. Sci., 2021, 68, 42–61 Search PubMed.
- X. Wang, J. Chai and J. Jimmy Jiang, Nano Mater. Sci., 2021, 3, 17–24 Search PubMed.
- E. Ventosa, Curr. Opin. Chem. Eng., 2022, 37, 100834 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3ma00417a |
| This journal is © The Royal Society of Chemistry 2023 |