
Covalent modification of franckeite with maleimides: connecting molecules and van der Waals heterostructures†
Julia
Villalva
 a,
Sara
Moreno-Da Silva
a,
Palmira
Villa
b,
Luisa
Ruiz-González
c,
Cristina
Navío
a,
Saül
Garcia-Orrit
a,
Víctor
Vega-Mayoral
a,
Juan
Cabanillas-González
*a,
Andrés
Castellanos-Gomez
d,
Emerson
Giovanelli
a and
Emilio M.
Pérez
*a
a,
Sara
Moreno-Da Silva
a,
Palmira
Villa
b,
Luisa
Ruiz-González
c,
Cristina
Navío
a,
Saül
Garcia-Orrit
a,
Víctor
Vega-Mayoral
a,
Juan
Cabanillas-González
*a,
Andrés
Castellanos-Gomez
d,
Emerson
Giovanelli
a and
Emilio M.
Pérez
*a
aIMDEA Nanociencia, c/Faraday 9, Campus de Cantoblanco, Madrid 28049, Spain. E-mail: juan.cabanillas@imdea.org; emilio.perez@imdea.org; Web: http://www.twitter.com/emiliomperezlab
bInstituto Pluridisciplinar, Universidad Complutense de Madrid, Paseo de Juan XXIII No 1, Madrid 28040, Spain
cDepartamento de Química Inorgánica, Facultad de Ciencias Químicas, Universidad Complutense de Madrid, Av. Complutense s/n, Madrid 28040, Spain
dInstituto de Ciencia de Materiales de Madrid (ICMM-CSIC), Sor Juana Inés de la Cruz, 3, Campus de Cantoblanco, Madrid 28049, Spain
First published on 15th April 2021
Abstract
The building of van der Waals heterostructures and the decoration of 2D materials with organic molecules share a common goal: to obtain ultrathin materials with tailored properties. Performing controlled chemistry on van der Waals heterostructures would add an extra level of complexity, providing a pathway towards 2D–2D–0D mixed-dimensional heterostructures. Here we show that thiol–ene-like “click” chemistry can be used to decorate franckeite, a naturally occurring van der Waals heterostructure with maleimide reagents. ATR-IR and NMR analyses corroborate the Michael addition mechanism via the formation of a S–C covalent bond, while Raman and HR-TEM show that the SnS2–PbS alternating structure of franckeite is preserved, and suggest that SnS2 reacts preferentially, which is confirmed through XPS. We illustrate how this methodology can be used to add functional molecular moieties by decorating franckeite with porphyrins. UV-vis-NIR spectroscopy confirms that the chromophore ground state remains operative, showing negligible ground-state interactions with the franckeite. Excited-state interactions across the hybrid interface are revealed. Time-resolved photoluminescence confirms the presence of excited-state deactivation in the linked porphyrin ascribed to energy transfer to the franckeite.
New conceptsThe heterostructures field is rapidly expanding and requires of new tools for the combination of different materials. To the best of our knowledge, the covalent chemistry of van der Waals heterostructures is unexplored. Franckeite is a naturally occurring van der Waals heterostructure composed of alternate tin(IV) sulfide and lead(II) sulfide layers. We demonstrate that franckeite can be functionalized using maleimides as soft electrophiles using mild enough conditions to preserve the original van der Waals structure. The reaction takes place through a thiol–ene like “click” mechanism confirmed by NMR and occurs preferentially over the SnS2 layer to form S–C bonds. The steady-state photophysical properties of a 2D–2D–0D mixed-dimensional heterostructure obtained through reaction with a porphyrin-decorated maleimide show that the organic chromophore retains its ground state character, whereas an energy transfer process between the porphyrin and the van der Waals heterostructure takes place. These results demonstrate that this reaction is an ideal tool for the synthesis of heterostructures of the 2D–2D–0D type, where 0D are functional organic molecules. |
Introduction
The stacking of different 2D materials is the root of the field of van der Waals heterostructures.1–3 The most widespread method for the synthesis of heterostructures of bidimensional materials is the direct growth of one material on top of the other by CVD, or van der Waals4 or edge5 epitaxy. Alternatively, one can manually stack one material on top of the other, using deterministic transfer methodologies.6,7 This is usually done by using a viscoelastic polymer as transfer medium, on which one of the flakes is adhered and then placed on top of the other using optical microscopy and spectroscopy to monitor the alignment.8 Alternatively, one can obtain ultrathin van der Waals heterostructures from the direct exfoliation of minerals of adequate composition.9–16,47 The motivation behind this field of research is based on the expectation that the combination (or modulation) of properties of nanomaterials is a promising approach towards materials by design.17–23 This exact same motivation fuels the interest in the chemistry of 2D materials:24,25 we expect that the decoration of the nanomaterials with functional molecular fragments will yield superior combined properties.26 Besides this application-oriented point of view, the chemistry of 2D materials presents very interesting fundamental challenges, like obtaining chemical selectivity in materials where most atoms are chemically identical.27,28 To the best of our knowledge, the covalent chemistry of van der Waals heterostructures is still unexplored territory.With these considerations in mind, we decided to explore the chemistry of a naturally occurring van der Waals heterostructure: franckeite. Franckeite (Fk) is composed of the alternate layers of one pseudohexagonal (H) tin(IV) sulfide (SnS2)-like layer and four pseudotetragonal (T) lead(II) sulfide (PbS)-like layers (Fig. 1a).29 Among its attractive features, we can highlight that Fk is an air-stable semiconductor with a very small bandgap (0.5–0.7 eV) and presents p-type doping, mostly due to substitutional Sb(III) in the structure.30
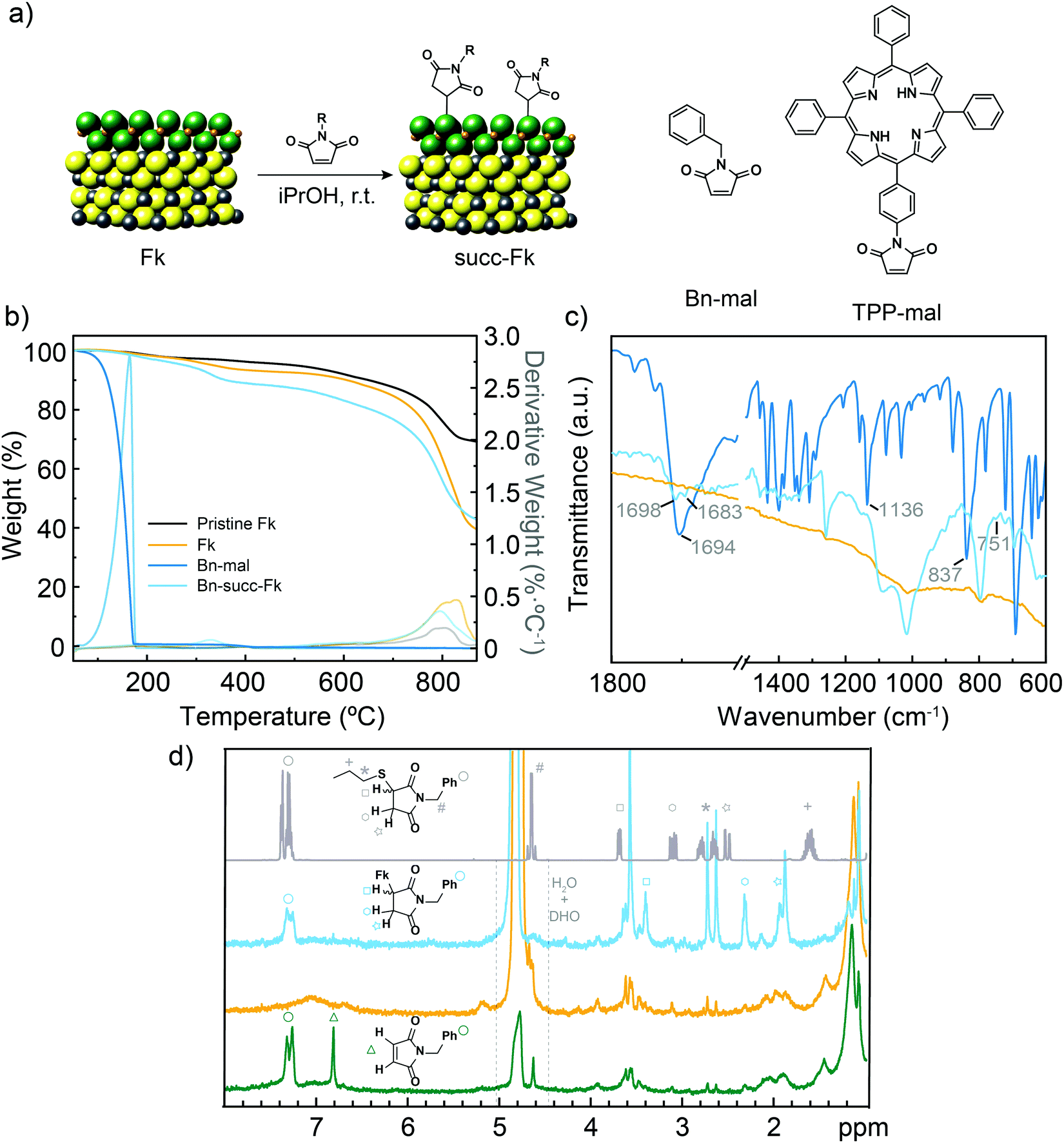 | ||
| Fig. 1 (a) Chemical structure of Fk from its X-ray crystal structure,29 and idealized reaction scheme with maleimides. Based on HR-TEM and XPS data we show SnS2 as the reactive layer. The S atoms in the SnS2-like layers are green, S atoms in the PbS-like layer are yellow, Sn atoms are orange and Pb atoms are gray. The structures of the two maleimide reagents are also shown. (b) TGA (N2, 10 °C min−1) of pristine exfoliated Fk (black), control Fk (yellow), Bn-maleimide (intense blue) and functionalized Bn-succ-Fk (pale blue). The first derivatives are shown in thinner lines. (c) Comparison of the ATR IR spectra of control Fk, Bn-maleimide and functionalized Bn-succ-Fk (same colour code as in b). (d) HR-MAS-1H-NMR (500 MHz) spectra of Bn-succ-Fk (pale blue), control Fk (orange) and a mixture of Fk and Bn-maleimide (dark green) obtained using D2O as the reference. The solution 1H NMR (CDCl3, 400 MHz, r.t.) of the product of the reaction between 1-propanethiol and N-benzylmaleimide is also shown for comparison (gray). | ||
We have very recently reported the use of thiol–maleimide “click” chemistry31,32 to functionalize MoS2 and WS2 under particularly mild conditions.33 We believed this powerful reaction would be an ideal tool to take the first steps towards heterostructures of the 2D–2D–0D type, where the 0D material are functional organic molecules. From the fundamental chemistry point of view, we wanted to address the possibility of performing thiol–maleimide chemistry on a material composed of two different sulfides.
Here, we describe how to functionalize Fk covalently using maleimides as soft electrophiles. At room temperature in isopropanol (iPrOH), we achieve preferential reactivity of the sulfides in the SnS2 layer to form S–C bonds, with sufficient selectivity to conserve the van der Waals heterostructure. As proof-of-principle application, we describe the functionalization of Fk with a porphyrin-decorated maleimide.
Results and discussion
The structure of Fk and the idealized reaction scheme are shown in Fig. 1. For simplicity, only a minimal repetition unit is drawn. Few-layer Fk colloids were obtained using the liquid-phase exfoliation method previously described.34 Briefly, Fk powder obtained from careful grinding of mineral pieces was bath ultrasonicated in iPrOH (10 mg mL−1) at 20 °C for 1 h. The colloid obtained in the supernatant after centrifugation at 988 g for 30 min presented a homogeneous distribution of flakes, most of them having a few-layer thickness (<7.5 nm from AFM data, S4, ESI†). To this colloid, N-benzylmaleimide (Bn-mal in Fig. 1) was added, and the mixture was ultrasonicated for 5 min then stirred overnight. As for the purification procedure, the suspension was filtered through a polytetrafluoroethylene membrane with a pore size of 0.2 μm, and the solid was washed several times with CHCl3 to remove excess reagents and physisorbed maleimide. CHCl3 was selected after an optimization process as the best washing solvent due to its ability to dissolve Bn-mal. The purification process was monitored by UV-Vis spectroscopy (see S5, ESI†).Using thermogravimetric analysis (TGA) the functionalization was estimated at ca. 7 wt% for the Bn-mal and ca. 4 wt% when the porphyrin-derived maleimide (TPP-mal) was employed (Fig. 1b and Fig. S6, ESI†). For a rigorous analysis, a control Fk sample was stirred overnight and rinsed afterwards with CHCl3 following the exact same procedure described for the functionalization. This control sample presents an initial weight loss at around 300 °C which can correspond to adsorbed solvent; then it is thermally stable until 700 °C, where it starts to decompose. The organic material loss overlaps that of the solvent but is displaced to much higher temperatures (from 155 °C to 310 °C in Bn-succ-Fk and from 460 °C to 560 °C in TPP-succ-Fk). This improved thermal stability is a first indication that the organic material is not simply physisorbed onto the surface but forming covalent bonds. The low degree of functionalization compared to MoS2 and WS2 under similar reaction conditions33 is rationalized as a direct consequence of the structure of Fk (see below).
ATR-FTIR shows the presence of new vibration bands in the Bn-succ-Fk sample (Fig. 1c). The intense carbonyl stretch that appears at 1694 cm−1 in the Bn-mal splits into two new bands in the functionalized sample (1698 and 1683 cm−1). This can be explained by the desymmetrization of the molecule after functionalization. Additionally, the intense alkene C–H bending mode at 837 cm−1 is strongly depleted after functionalization, which is in accordance with the expected Michael addition mechanism. We assign the new 751 cm−1 weak band, absent in the reference Fk material and in Bn-mal, to the new C–S stretching vibration. A similar displacement has been observed for the C–S bond in different 2D materials such as MoS2 and WS2.33 As a control experiment, a mixture of the N-benzylmaleimide derivative, the N-benzylsuccinimide and Fk was stirred under the typical reaction conditions. In this case, no organic signals are observed in FT-IR after performing the same number of washes as for Bn-succ-Fk (see ESI,† S7) indicating that the reaction requires the double bond from the N-benzylmaleimide to work. Moreover, it implies that the washing procedure is efficient enough to remove the physisorbed organic molecules.
Conclusive evidence about the functionalization and its mechanism is obtained when the powder sample is analysed using high-resolution magic angle spinning (HR-MAS) NMR. HR-MAS 1H NMR spectroscopy permits the direct analysis of heterogeneous samples, such as tissue35 or materials,36 and has previously been used to track organic reactions in the solid state.37 Using deuterated water as the reference, the control Fk sample (orange in Fig. 1d) shows only weak signals in the alkane region, probably due to adsorbed hydrocarbons and residual iPrOH, and a broad and very weak signal centered around 7 ppm (most likely physisorbed iPrOH). In contrast, the benzylsuccinimide signals are clearly observed in the functionalized Bn-succ-Fk (blue in Fig. 1d), which can be unambiguously assigned to covalent functionalization of Fk through Michael addition by comparison with a Fk and N-benzylmaleimide mixture (green in Fig. 1d): the total depletion of the alkene signals at around 6.8 ppm (green triangle, ca. 6.8 ppm) is accompanied by the appearance of three new signals (marked with a square at ca. 3.4 ppm, an hexagon at ca. 2.3 ppm, and a star at ca. 1.9 ppm, in Fig. 1d) in the alkane region. The new signals correspond to the new aliphatic diastereotopic protons of the newly formed succinimide. Comparison with the experimental 1H solution NMR spectrum of the product of the reaction between 1-propanethiol and N-benzylmaleimide (CDCl3, 400 MHz, r.t., gray in Fig. 1d), where the signals can be unambiguously assigned, further confirms this observation: a consistent upfield shift is observed for all the succinimide signals due to the presence of Fk, while the aromatic protons, initially oriented away from Fk, are hardly affected. Unfortunately, the benzylic –CH2 cannot be reliably identified in the different Fk samples, as spectra were acquired with water suppression.
With the reaction effectiveness and mechanism clearly established, we went on to investigate if/how the Fk structure and electronic properties were affected by the covalent modification. To this end, Raman spectroscopy provided valuable information. Both control and functionalized samples exhibit very similar spectra, with the characteristic Fk Raman peaks (Fig. S8 and Table S9, ESI†). In particular, we observe a band centered at 86 cm−1, which results from the spheroidal and transverse acoustic phonon modes of the PbS layers. The band at 141 cm−1 is assigned to a combination of the 2nd order effect of the SnS2 layers and the transverse optical and acoustic phonon modes of the PbS layers. The outcome of the longitudinal optical phonon mode of the PbS layers and the atomic orbital Eg of the SnS2 layers is the band at 201 cm−1. The band at 253 cm−1 is the combination of the phonon modes of both layers, PbS and SnS2, and therefore its relative intensity is a good indication that the thickness of the unique 2D–2D heterostructure is preserved upon functionalization, which is as well proven by AFM (see ESI,† S10). The A1g mode of SnS2 results in the formation of the band at 318 cm−1. Finally, the shoulder from ∼400 to 650 cm−1 shows the longitudinal optical phonon modes of PbS and a 2nd order effect from the SnS2 layers.9
In agreement with the Raman measurements, high-resolution transmission electron microscopy HRTEM (200 kV) shows no major changes in the van der Waals heterostructure during functionalization. Fig. 2a and b show the comparison of characteristic micrographs for Fk and Bn-succ-Fk along the [100] direction (see inset).29 In both micrographs, the same Moiré pattern is observed, which originates from the superposition of the H (SnS2) and Q (PbS) layers (see ESI,† S11 for additional measurements). Fig. 2c and d, correspond to an edge-on perspective (image plane perpendicular to the basal plane of Fk) of both samples. The measured periodicities, as better observed in the enhanced details (Fig. 2e and f), are in agreement with the stacking of the H and Q layers of the Fk lattice. A closer look at the final layer (Fig. 2e and f) suggests that the H layer, i.e., SnS2, as proved by Velicky et al.21 is the one exposed to ambient and is therefore most probably the one that reacts preferentially. This is consistent with the chemical intuition based on the lower electronegativity of Sn compared to Pb, and the smaller radius and higher charge of Sn(IV) compared to Pb(II): the combination of these properties should render SnS2 more purely ionic compared to PbS38 and, as a consequence, the sulfur atoms directly attached to Sn more anionic and more nucleophilic. This hypothesis also explains the low degree of functionalization observed, as for every potentially reactive SnS2 layer, there are four PbS layers that remain fundamentally intact. Additionally, the amorphous layer due to organic material appears thicker in Bn-succ-Fk, in accordance with functionalization, although this last observation is far from being conclusive.
 | ||
| Fig. 2 HR-TEM micrographs obtained for pristine Fk (a, c and e) and Bn-succ-Fk (b, d and f). The insets show the orientations that originate the Moiré pattern in (a and b), and the edge-on perspective of (c and d).29 Scale bars are 10 nm for a and b and 5 nm for (c and d). (e) and (f) show a zoom in on the edge areas highlighted with white squares in (c) and (d), respectively. | ||
To further investigate this point, we have performed X ray photoelectron spectroscopy (XPS) measurements under ultra high vacuum (UHV) conditions (see ESI† for experimental details). The C 1s core level centered in 284.6 eV was used as a binding energy reference and the selected regions of S 2p, Pb 4f and Sn 3d are shown in Fig. 3. With the known structure of Fk, we would expect for the valence states of the pristine material to show two different chemical environments for S (one related to the Q type and another to the H type), one for the Pb (related to the Q layer) and two for the Sn (one related to the Q type and another to the H type). In Fig. 3a–c we show the core levels for the S 2p, Pb 4f and Sn 3d for the Fk control sample. The fit of the S 2p region shows two clear doublets with the 2p3/2 level centered at 160.5 eV and 161.5 eV. The proportion of the area for each doublet can help on the labeling of the components: as the Q layer is composed by four atomic layers with S, the intensity of this component in the S 2p core level is expected to be larger than for the H layer. We therefore assign the component at 160.5 eV to the Q layer and the one centered in 161.5 eV to the H layer. For the Pb 4f case (Fig. 3b) we observe two chemical states instead of one, the Pb2+ expected for the Q layer and the appearance of a Pb4+ component that can be attributed to the appearance of lead oxide.9 For the Sn 3d we only observe one chemical state for the Fk control, which shows the Sn 3d5/2 centered at 485.8 eV.
 | ||
| Fig. 3 XPS spectra of S, Pb and Sn core levels for (a–c) the control Fk material and (d–f) Bn-succ-Fk. | ||
After functionalization we observe some changes on the XPS spectra, shown in Fig. 3d–f. Due to the formation of a C–S bond, a third component could be expected in the S 2p region, but the low degree of functionalization together with limitations on the resolution of XPS make this new chemical state overlap with the components for the Fk control.39 This is reflected as a change in the relative atomic percentage in the area of the S 2p. The evidence of the functionalization in the XPS spectra (apart from the appearance of a very weak N 1s peak) is the new component in the Sn 3d region (Fig. 3f) located in 487.4 eV, which amounts to 7% of the total area, in good agreement with the degree of functionalization determined by TGA, and is again in support of preferential reaction of the SnS2 layers.
As proof-of-principle application of our methodology, a maleimide (TPP-mal) bearing a tetraphenylporphyrin TPP group as a chromophore was employed in the reaction. In this case, a clear sign of functionalization is observed in the UV-Vis spectra of the TPP-succ-Fk sample (Fig. 4a). The Soret band of the porphyrin core (415 nm in TPP-mal) is without a doubt responsible for the new absorption band observed in TPP-succ-Fk at 417 nm. Furthermore, perfect match of the Soret band resonances is seen in the 2D PL excitation–emission contour plots depicted in Fig. 4c and d and in the PLE spectra (Fig. S13, ESI† and Fig. 1), suggesting the lack of ground-state interactions across the hybrid interface. The Q band resonances present in the 470–600 nm spectral window of the control sample are not so evident in the TPP-succ-Fk excitation spectra. This is most likely a consequence of the low amount of TPP-succ-Fk dispersed in solution rather than a change in electronic structure, as those resonances can be guessed just above the measurement noise level. The lack of ground state interactions is explained by the physical separation between the chromophore and Fk and the truncated p-conjugation caused by the maleimide bridge preventing electronic overlap between the 0D and 2D entities. In line with TPP-mal, TPP-succ-Fk PL exhibits two bands at 650 and 750 nm. The absence of spectral shifts in the PL spectra (Fig. 4b) of TPP-mal and TPP-succ-Fk also seems to point towards negligible dielectric constant changes in the porphyrin surrounding upon its anchoring to Fk.
 | ||
| Fig. 4 (a) UV-Vis spectra of the control Fk sample, tetraphenylporphyrin-derived maleimide (TPP-mal) and TPP-succ-Fk. (b) Normalized PL spectra of TPP-mal and TPP-succ-Fk. (c) and (d) PLE intensity maps of (c) TPP-mal and (d) TPP-succ-Fk, respectively. Rayleigh scattering has not been filtered and the excitation wavelength second order diffraction can be seen as the straight line starting at 700 nm in (c) and (d) x axes. This is a fingerprint of highly scattering samples measured with single grating spectrometers. (e) TRPL dynamics of TPP-mal and TPP-succ-Fk photoexcited at 405 nm and detected at 650 nm together with bi-exponential fits (straight lines). (f) Summary sketch depicting the decay lifetime values and corresponding statistical weights of TPP-mal and TPP-succ-Fk. All spectra were recorded in iPrOH at room temperature. | ||
To gain insights into excited-state interactions across the hybrid interface, time-resolved PL measurements were deployed in TPP-succ-Fk and TPP-mal, observing a clear acceleration of the PL dynamics in the former. The PL dynamics of TPP-mal detected at 650 nm displays two decay components of 1 ns (5%) and 10 ns (95%) (Fig. 4e and f) whilst a new decay component of 0.5 ps (12%) is resolved in TPP-succ-Fk. The possible influence of porphyrin aggregation in the observed PL dynamics changes was foreseen and ruled out based on the identical decay times measured in solutions with lower TPP concentrations decreased by a two decades range (Fig. S10–S13, ESI†). Thus, the fast decay component observed in TPP-succ-Fk is ascribed to excited-state deactivation taking place at the porphyrin/Fk interface. Given the low energy bandgap of Fk (0.5 eV)20 and its p-type character, a type I heterojunction is expected at the interface, with the Fk conduction and valence bands located within the TPP bandgap (2.49 eV HOMO–LUMO energy).40 This scenario would favor a porphyrin-to-Fk energy transfer as the most plausible interaction. Finally, fs-Transient Absorption Spectroscopy (TAS) measurements upon predominant Fk photoexcitation (387 nm) were conducted (Fig. S14, ESI†). In these measurements, the differential transmission (DT/T) spectra and dynamics of TPP-succ-Fk (Fig. S14, ESI† and Fig. 1c, f) are dominated by a broad photoinduced absorption band ascribed to Fk (Fig. S14a and d, ESI†), finding no significant difference between Fk and TPP-succ-Fk (neither in spectral shape nor in temporal dynamics). Global fit analysis of the TAS Fk data applied following a linear cascade model yielded three different relaxation processes with lifetimes τ1 = 200 fs, τ2 = 56 ps, and τ3 = 5.4 ns. Similar components are often found in other semiconducting 2D materials such as MoS2 or WS2, being the sub-ps relaxation ascribed to thermalization of hot excitons,41,42 while slower processes are assigned to diffusion,43 defect trapping42,44 or band-to-band recombination.45 The lack of significant changes in the TAS spectra and dynamics of Fk and TPP-succ-Fk upon predominant Fk photoexcitation together with the evidences for TPP-succ-Fk PL quenching aforementioned are both compatible with a TPP-to-Fk energy transfer mechanism.
In conclusion, we describe how the naturally occurring van der Waals heterostructure Fk can be functionalized covalently making use of the inherent soft electrophilicity of maleimides.33 The good quality of the 1H HR-MAS NMR signals allows us to confirm the proposed thiol–maleimide “click” functionalization mechanism beyond reasonable doubt, and serves as an encouraging example for the use of NMR techniques for the characterization of 2D materials.46 HR-TEM and XPS confirm that the functionalization is sufficiently mild to respect the native van der Waals heterostructure, occurring preferentially at the SnS2 layer.
Finally, we prove that this reaction can be used to connect functional organic molecules by successfully attaching a porphyrin derivative. The steady-state photophysical properties of the 2D–2D–0D mixed-dimensional heterostructure obtained show that the organic chromophore retains its ground state character, whereas an energy transfer process between the porphyrin and the van der Waals heterostructure takes place.
Conflicts of interest
The authors declare no competing financial interest.Acknowledgements
We acknowledge funding from the European Union (ERC-PoC-842606), MINECO (CTQ2014-60541-P and CTQ2017-86060-P), and the Comunidad de Madrid and the European Structural Funds for their financial support through FotoArt-CM project (S2018/NMT-4367) and MAD2D-CM S2013/MIT-3007. L. R. G. acknowledges the support of the Spanish Ministry of Science and Innovation through Research Project MAT2017-82252-R and to the National Centre for Electron Microscopy (ELECMI National Singular Scientific Facility) for provision of corrected aberration microscopy. J. C.-G. acknowledges financial support from the Spanish Ministry of Science, Innovation and Universities (RTI2018-097508-B-I00), and from the Regional Government of Madrid (S2018/NMT4511). V. V.-M. acknowledges financial support from the regional government of Madrid “Atracción del talento” program (2019-T2/IND-12737). IMDEA Nanociencia acknowledges support from the “Severo Ochoa” Programme for Centres of Excellence in R&D (MINECO, grant SEV-2016-0686).References
- Y. Liu, N. O. Weiss, X. Duan, H.-C. Cheng, Y. Huang and X. Duan, Nat. Rev. Mater., 2016, 1, 16042 CrossRef CAS.
- K. S. Novoselov, A. Mishchenko, A. Carvalho and A. H. Castro Neto, Science, 2016, 353, aac9439 CrossRef CAS.
- A. K. Geim and I. V. Grigorieva, Nature, 2013, 499, 419–425 CrossRef CAS PubMed.
- J. A. Robinson, ACS Nano, 2016, 10, 42–45 CrossRef CAS PubMed.
- P. K. Sahoo, S. Memaran, Y. Xin, L. Balicas and H. R. Gutierrez, Nature, 2018, 553, 63–67 CrossRef CAS PubMed.
- A. Castellanos-Gomez, M. Buscema, R. Molenaar, V. Singh, L. Janssen, H. S. J. van der Zant and G. A. Steele, 2D Mater., 2014, 1, 011002 CrossRef CAS.
- R. Frisenda, E. Navarro-Moratalla, P. Gant, D. Perez De Lara, P. Jarillo-Herrero, R. V. Gorbachev and A. Castellanos-Gomez, Chem. Soc. Rev., 2018, 47, 53–68 RSC.
- K. Kim, M. Yankovitz, B. Fallahazad, S. Kang, H. C. P. Movva, S. Huang, S. Larentis, C. M. Corbet, T. Taniguchi, K. Watanabe, S. K. Banerjee, B. J. LeRoy and E. Tutuc, Nano Lett., 2016, 16, 1989–1995 CrossRef CAS PubMed.
- A. J. Molina-Mendoza, E. Giovanelli, W. S. Paz, M. A. Nino, J. O. Island, C. Evangeli, L. Aballe, M. Foerster, H. S. J. van der Zant, G. Rubio-Bollinger, N. Agrait, J. J. Palacios, E. M. Perez and A. Castellanos-Gomez, Nat. Commun., 2017, 8, 14409 CrossRef CAS PubMed.
- M. Velický, P. S. Toth, A. M. Rakowski, A. P. Rooney, A. Kozikov, C. R. Woods, A. Mishchenko, L. Fumagalli, J. Yin, V. Zólyomi, T. Georgiou, S. J. Haigh, K. S. Novoselov and R. A. W. Dryfe, Nat. Commun., 2017, 8, 14410 CrossRef PubMed.
- E. Burzuri, M. Vera-Hidalgo, E. Giovanelli, J. Villalva, A. Castellanos-Gomez and E. M. Perez, Nanoscale, 2018, 10, 7966–7970 RSC.
- R. Gusmão, Z. Sofer, J. Luxa and M. Pumera, J. Mater. Chem. A, 2018, 6, 16590–16599 RSC.
- H. Chen, J. He, C. D. Malliakas, C. C. Stoumpos, A. J. E. Rettie, J.-K. Bao, D. Y. Chung, W.-K. Kwok, C. Wolverton and M. G. Kanatzidis, J. Am. Chem. Soc., 2019, 141, 7544–7553 CrossRef CAS PubMed.
- Y. Niu, J. Villalva, R. Frisenda, G. Sanchez-Santolino, L. Ruiz-González, E. M. Pérez, M. García-Hernández, E. Burzurí and A. Castellanos-Gomez, 2D Mater., 2019, 6, 035023 CrossRef CAS.
- S. Gan, Y. Zhao, X. Dai and Y. Xiang, Results Phys., 2019, 13, 102320 CrossRef.
- B. Wang, S. P. Zhong, Z. B. Zhang, Z. Q. Zheng, Y. P. Zhang and H. Zhang, Appl. Mater. Today, 2019, 15, 115–138 CrossRef.
- K. Zhang, T. Zhang, G. Cheng, T. Li, S. Wang, W. Wei, X. Zhou, W. Yu, Y. Sun, P. Wang, D. Zhang, C. Zeng, X. Wang, W. Hu, H. J. Fan, G. Shen, X. Chen, X. Duan, K. Chang and N. Dai, ACS Nano, 2016, 10, 3852–3858 CrossRef CAS PubMed.
- H. Zhang, X. Zhang, C. Liu, S.-T. Lee and J. Jie, ACS Nano, 2016, 10, 5113–5122 CrossRef CAS PubMed.
- M. Massicotte, P. Schmidt, F. Vialla, K. G. Schadler, A. Reserbat-Plantey, K. Watanabe, T. Taniguchi, K. J. Tielrooij and F. H. L. Koppens, Nat. Nanotechnol., 2016, 11, 42–46 CrossRef CAS PubMed.
- M. Long, E. Liu, P. Wang, A. Gao, H. Xia, W. Luo, B. Wang, J. Zeng, Y. Fu, K. Xu, W. Zhou, Y. Lv, S. Yao, M. Lu, Y. Chen, Z. Ni, Y. You, X. Zhang, S. Qin, Y. Shi, W. Hu, D. Xing and F. Miao, Nano Lett., 2016, 16, 2254–2259 CrossRef CAS PubMed.
- G. W. Mudd, S. A. Svatek, L. Hague, O. Makarovsky, Z. R. Kudrynskyi, C. J. Mellor, P. H. Beton, L. Eaves, K. S. Novoselov, Z. D. Kovalyuk, E. E. Vdovin, A. J. Marsden, N. R. Wilson and A. Patane, Adv. Mater., 2015, 27, 3760–3766 CrossRef CAS PubMed.
- L. Wang, X. Zheng, L. Chen, Y. Xiong and H. Xu, Angew. Chem., Int. Ed., 2018, 57, 3454–3458 CrossRef CAS PubMed.
- S. Jiang, J. Shan and K. F. Mak, Nat. Mater., 2018, 17, 406–410 CrossRef CAS PubMed.
- A. Hirsch and F. Hauke, Angew. Chem., Int. Ed., 2017, 57, 4338–4354 CrossRef.
- S. Bertolazzi, M. Gobbi, Y. Zhao, P. Samori and C. Backes, Chem. Soc. Rev., 2018, 47, 6845–6888 RSC.
- E. Coronado, C. Martí-Gastaldo, E. Navarro-Moratalla, E. Burzurí, A. Camón and F. Luis, Adv. Mater., 2011, 23, 5021–5026 CrossRef CAS PubMed.
- J. J. Navarro, F. Calleja, R. Miranda, E. M. Perez and A. L. Vazquez de Parga, Chem. Commun., 2017, 53, 10418–10421 RSC.
- J. J. Navarro, S. Leret, F. Calleja, D. Stradi, A. Black, R. Bernardo-Gavito, M. Garnica, D. Granados, A. L. Vazquez de Parga, E. M. Perez and R. Miranda, Nano Lett., 2016, 16, 355–361 CrossRef CAS PubMed.
- E. Makovicky, M. Dušek, V. Petříček and D. Topa, Am. Mineral., 2011, 96, 1686–1702 CrossRef CAS.
- K. Ray, A. E. Yore, T. Mou, S. Jha, K. K. H. Smithe, B. Wang, E. Pop and A. K. M. Newaz, ACS Nano, 2017, 11, 6024–6030 CrossRef CAS PubMed.
- C. E. Hoyle and C. N. Bowman, Angew. Chem., Int. Ed., 2010, 49, 1540–1573 CrossRef CAS PubMed.
- M. J. Kade, D. J. Burke and C. J. Hawker, J. Polym. Sci., Part A: Polym. Chem., 2010, 48, 743–750 CrossRef CAS.
- M. Vera-Hidalgo, E. Giovanelli, C. Navio and E. M. Perez, J. Am. Chem. Soc., 2019, 141, 3767–3771 CrossRef CAS PubMed.
- A. J. Molina-Mendoza, E. Giovanelli, W. S. Paz, M. Angel Nino, J. O. Island, C. Evangeli, L. Aballe, M. Foerster, H. S. J. van der Zant, G. Rubio-Bollinger, N. Agrait, J. J. Palacios, E. M. Perez and A. Castellanos-Gomez, Nat. Commun., 2017, 8, 14409 CrossRef CAS.
- O. Beckonert, M. Coen, H. C. Keun, Y. Wang, T. M. D. Ebbels, E. Holmes, J. C. Lindon and J. K. Nicholson, Nat. Protoc., 2010, 5, 1019 CrossRef CAS PubMed.
- T. M. Alam and J. E. Jenkins, HR-MAS NMR Spectroscopy in Material Science, in Advanced Aspects of Spectroscopy, ed. M. A. Farrukh, IntechOpen, 2012 DOI:10.5772/48340. Available from https://www.intechopen.com/books/advanced-aspects-of-spectroscopy/hr-mas-nmr-spectroscopy-in-material-science.
- R. Warrass and G. Lippens, J. Org. Chem., 2000, 65, 2946–2950 CrossRef CAS PubMed.
- L. Pauling, J. Am. Chem. Soc., 1932, 54, 988–1003 CrossRef CAS.
- In fact, adding a third component for the S 2p peak improves the fitting (Fig. S12) but considering our experimental resolution we do not think it is sufficiently justified .
- P. Zhao, Y. Huang, J. Chen, S. Shao, H. Miao, J. Xia, C. Jia and M. Hua, J. Photochem. Photobiol., 2020, 3–4, 100014 CrossRef.
- T. Borzda, C. Gadermaier, N. Vujicic, P. Topolovsek, M. Borovsak, T. Mertelj, D. Viola, C. Manzoni, E. A. A. Pogna, D. Brida, M.-R. Antognazza, F. Scotognella, G. Lanzani, G. Cerullo and D. Mihailovic, Adv. Funct. Mater., 2015, 25, 3351–3358 CrossRef CAS.
- V. Vega-Mayoral, D. Vella, T. Borzda, M. Prijatelj, I. Tempra, E. A. A. Pogna, S. Dal Conte, P. Topolovsek, N. Vujicic, G. Cerullo, D. Mihailovic and C. Gadermaier, Nanoscale, 2016, 8, 5428–5434 RSC.
- N. Kumar, J. He, D. He, Y. Wang and H. Zhao, J. Appl. Phys., 2013, 113, 133702 CrossRef.
- V. Vega-Mayoral, T. Borzda, D. Vella, M. Prijatelj, E. A. A. Pogna, C. Backes, J. N. Coleman, G. Cerullo, D. Mihailovic and C. Gadermaier, 2D Mater., 2017, 5, 015011 CrossRef.
- D. Kozawa, R. Kumar, A. Carvalho, K. K. Amara, W. Zhao, S. Wang, M. Toh, R. M. Ribeiro, A. H. Castro Neto, K. Matsuda and G. Eda, Nat. Commun., 2014, 5, 4543 CrossRef CAS PubMed.
- I. A. Vacchi, C. Spinato, J. Raya, A. Bianco and C. Menard-Moyon, Nanoscale, 2016, 8, 13714–13721 RSC.
- R. Frisenda, Y. Niu and P. Gant, npj 2D Mater. Appl., 2020, 4(1), 38 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental details, characterization and supporting figures. See DOI: 10.1039/d1nh00147g |
| This journal is © The Royal Society of Chemistry 2021 |