
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceOn/off porosity switching and post-assembly modifications of Cu4L4 metal–organic polyhedra†
Witold M.
Bloch
 *a,
Ravichandar
Babarao
bc and
Matthew L.
Schneider
a
*a,
Ravichandar
Babarao
bc and
Matthew L.
Schneider
a
aDepartment of Chemistry, The University of Adelaide, Adelaide, Australia. E-mail: witold.bloch@adelaide.edu.au; Tel: +61 8 8313 5039
bSchool of Science, RMIT University, Melbourne, Victoria 3001, Australia
cCSIRO, Normanby Road, Clayton 3168, Victoria, Australia
First published on 5th March 2020
Abstract
Synthetic porous materials composed of metal–organic polyhedra (MOPs) have found application in topical areas such as gas storage, separation and catalysis. Control over their physical properties (e.g. porosity) has typically been achieved through ligand design or judicious choice of metal ions. Here, we demonstrate pore-size control and on/off porosity in Cu4L4 MOPs by exploiting their structural non-rigidity. We report an aldehyde-functionalised MOP (1) that can be isolated in five distinct solvatomorphs, each exhibiting different structural flexibility. When soaked in MeOH, two of these solvatomorphs undergo a rapid transformation to a thermodynamically favoured phase, whilst in acetone they template the crystallisation of an entirely new crystal packing. We support these findings by single and powder X-ray diffraction and rationalise the observed phase transformations by lattice energy calculations. Of the five solvatomorphs, three can be obtained as solvent-exchanged pseudo-polymorphs with distinct porosities in their activated form (SABET = 35–455 m2 g−1). Further control over the crystal packing of MOPs is achieved through covalent post-assembly modifications, which promote the crystallisation of isoreticular 2-D sheet-like structures.
Introduction
Molecular solids based on cage compounds have recently emerged as an attractive class of porous materials owing to their solution processability and intrinsic porosity.1–3 Unlike metal–organic frameworks (MOFs)4 and covalent–organic frameworks (COFs),5 discrete cage compounds crystallise by virtue of weak intermolecular interactions and thus tend to exhibit a rich crystal-packing behaviour. This is particularly true for purely organic cages, which have been reported to exhibit impressive properties such modular co-crystallisations,6 guest-induced breathing7 and on/off porosity switching.8–10 The crystal-packing behaviour of cage materials formed via metal–ligand bonding, however, has not been examined to the same extent, despite the exciting opportunities that these solids offer for gas adsorption11–13 and catalysis.14Metal–organic polyhedra (MOPs) are a class of discrete nanocages commonly formed by the self-assembly of dicarboxylate linkers and bimetallic paddle-wheels nodes (e.g. Cu2, Rh2 and Cr2).2,12,15 Solvent molecules typically occupy the apical coordination sites of the paddle-wheel nodes, and thus, desolvation allows access to unsaturated metal centres. This feature has motivated extensive studies examining MOPs for selective gas adsorption16 and heterogeneous catalysis.14,17,18 However, the crystal engineering of MOP solids is an area dominated more by serendipity than design; the weak intermolecular interactions that direct the crystal packing of MOPs can depend on the nature of the paddlewheel-bound solvent, making it possible to crystallise different solvatomorphs of the same cage compound.19 To add to the complexity, solvent exchange processes employed en-route to activation can lead to unknown crystalline or amorphous phases,12 making it difficult to control the pore-size and porosity of MOPs. Therefore, gaining control over the crystal packing of MOPs, and hence their physicochemical properties, remains a challenge.
Much of the efforts aiming to design MOP-based solids have focused on immobilising MOP particles in mesoporous supports,20,21 or on approaches that increase intermolecular interactions in the solid-state. For example, introducing ligand-based functionalities such as alkyl substituents,22 bulky hydrophobic groups,23 or coordinative O-donors24 has led to more robust and structurally rigid crystal packings. Owing to the solubility of MOPs, covalent post-assembly modifications (PAM)25 have also been carried out to modify the interior26 or exterior surface functionality.27 However, the development of covalent PAM on paddlewheel-based MOPs is still an emerging field of study, with only a few examples reported.
Herein we show that the crystal-packing and porosity of MOPs can be controlled in the same way as their purely organic analogues; by taking advantage of their rich phase behaviour and amenability to PAM. Thus, we report the synthesis and characterisation of an aldehyde-functionalised Cu4L4 MOP (1, Fig. 1a). By varying the crystallisation conditions, 1 can be obtained in five distinct solvatomorphs with different structural flexibilities. This is demonstrated by phase-dependant transformations which we characterise by X-ray diffraction and rationalise by lattice energy calculations. We show that the activation and porosity of 1 is phase dependant, and non-porous forms can be switched to porous forms by solvent treatment (Fig. 1b). Owing to the solubility of 1, PAM carried out at the cage exterior facilitate control over the crystal packing by favouring the formation of 2-D sheet-like motifs (Fig. 1c).
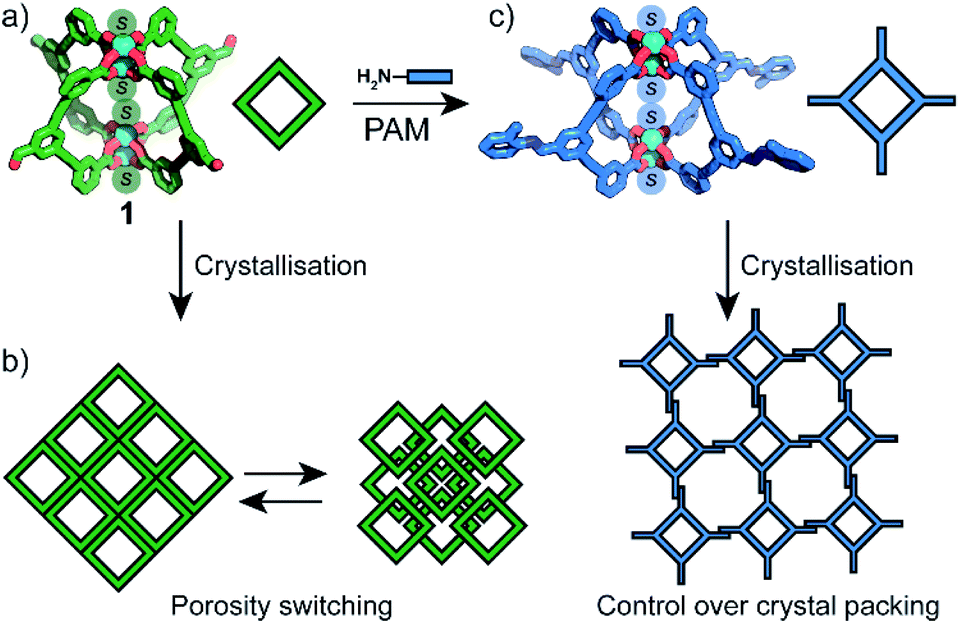 | ||
| Fig. 1 A summary of the work presented herein: (a) the lantern structure of MOP 1 (S = solvent); (b) phase cycling and porosity switching by solvent treatment and recrystallisation; (c) post-assembly modifications (PAM) of 1 with control over the crystal-packing. | ||
Results and discussion
Synthesis and structure of 1a
LH2 (Fig. 2a) was synthesised in two steps, starting with a Sonogashira cross-coupling of 3,5-dibromobenzaldehyde and methyl 3-ethynylbenzoate. The methyl ester compound was then hydrolysed under mild conditions to afford LH2 in an overall yield of 64% (ESI†).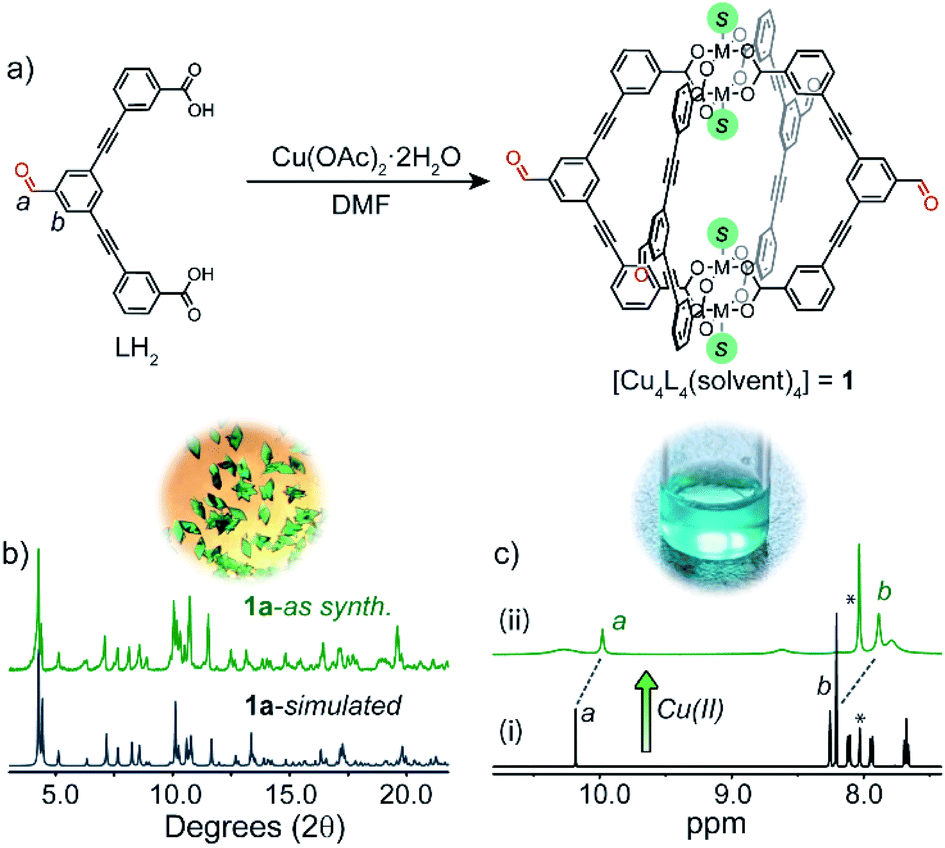 | ||
| Fig. 2 (a) A scheme showing the self-assembly of 1 in solution from LH2 and Cu(OAc)2 (S = solvent); (b) simulated and experimental PXRD patterns of phase 1a; (c) 1H NMR spectrum (500 MHz/DMF-d7, 25 °C): (i) LH2, (ii) LH2 + Cu(OAc)2 (* indicates solvent peak).‡ Above (b) and (c) are photos of crystals of 1a (left) and a DMF solution of 1 (right). | ||
Self-assembly of LH2 with 1.1 equivalents of Cu(OAc)2·2H2O in DMF resulted in the formation of a soluble Cu4L4 lantern cage (1) as indicated by 1H NMR spectroscopy and UV-Vis spectroscopy (Fig. S6†).26,28 Owing to the anomalous magnetic moment of the Cu2 paddlewheel,29 proton resonances not immediately adjacent to the metal nodes could be resolved in the 1H NMR spectrum of 1.‡ For example, the aldehyde resonance of 1 appears upfield shifted relative to the free ligand (ca. −0.2 ppm, Fig. 2c). Slow-vapour diffusion of MeOH into a DMF solution of 1 over a period of 5 days yielded rhombohedral crystals of 1a in 87% yield (based on CHN analysis). Single-crystal X-ray analysis (SCXRD) revealed that 1a crystallises in the triclinic space group P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) with half of two crystallographically independent cages in the asymmetric unit. Closer inspection revealed that the ligand backbones in one cage molecule adopt a flattened conformation when viewing the cage down the axis of the Cu2 paddlewheels. These nodes are coordinated by four carboxylate ligands and two axial DMF ligands. The second crystallographically unique cage adopts a pseudo D4h-symmetric conformation where the ligand backbones arrange in a mutually perpendicular fashion.19 Here, DMF and H2O ligands coordinate to the exterior and interior sites of the paddlewheels, respectively. The two chemically unique cage molecules present in the crystal packing of 1a give an overall formula of [Cu4L4(DMF)4]·[Cu4L4(DMF)2(H2O)2] (Fig. 3a and S33†). The extended packing of 1a is stabilised by multiple intermolecular π–π interactions that exist between the benzaldehyde cores of 1 (closest contact = 3.33 Å). This motif produces ordered channels along the a axis (Fig. 3a) with aperture dimensions measuring 3.8 × 6.1 Å (when omitting coordinating solvents).
with half of two crystallographically independent cages in the asymmetric unit. Closer inspection revealed that the ligand backbones in one cage molecule adopt a flattened conformation when viewing the cage down the axis of the Cu2 paddlewheels. These nodes are coordinated by four carboxylate ligands and two axial DMF ligands. The second crystallographically unique cage adopts a pseudo D4h-symmetric conformation where the ligand backbones arrange in a mutually perpendicular fashion.19 Here, DMF and H2O ligands coordinate to the exterior and interior sites of the paddlewheels, respectively. The two chemically unique cage molecules present in the crystal packing of 1a give an overall formula of [Cu4L4(DMF)4]·[Cu4L4(DMF)2(H2O)2] (Fig. 3a and S33†). The extended packing of 1a is stabilised by multiple intermolecular π–π interactions that exist between the benzaldehyde cores of 1 (closest contact = 3.33 Å). This motif produces ordered channels along the a axis (Fig. 3a) with aperture dimensions measuring 3.8 × 6.1 Å (when omitting coordinating solvents).
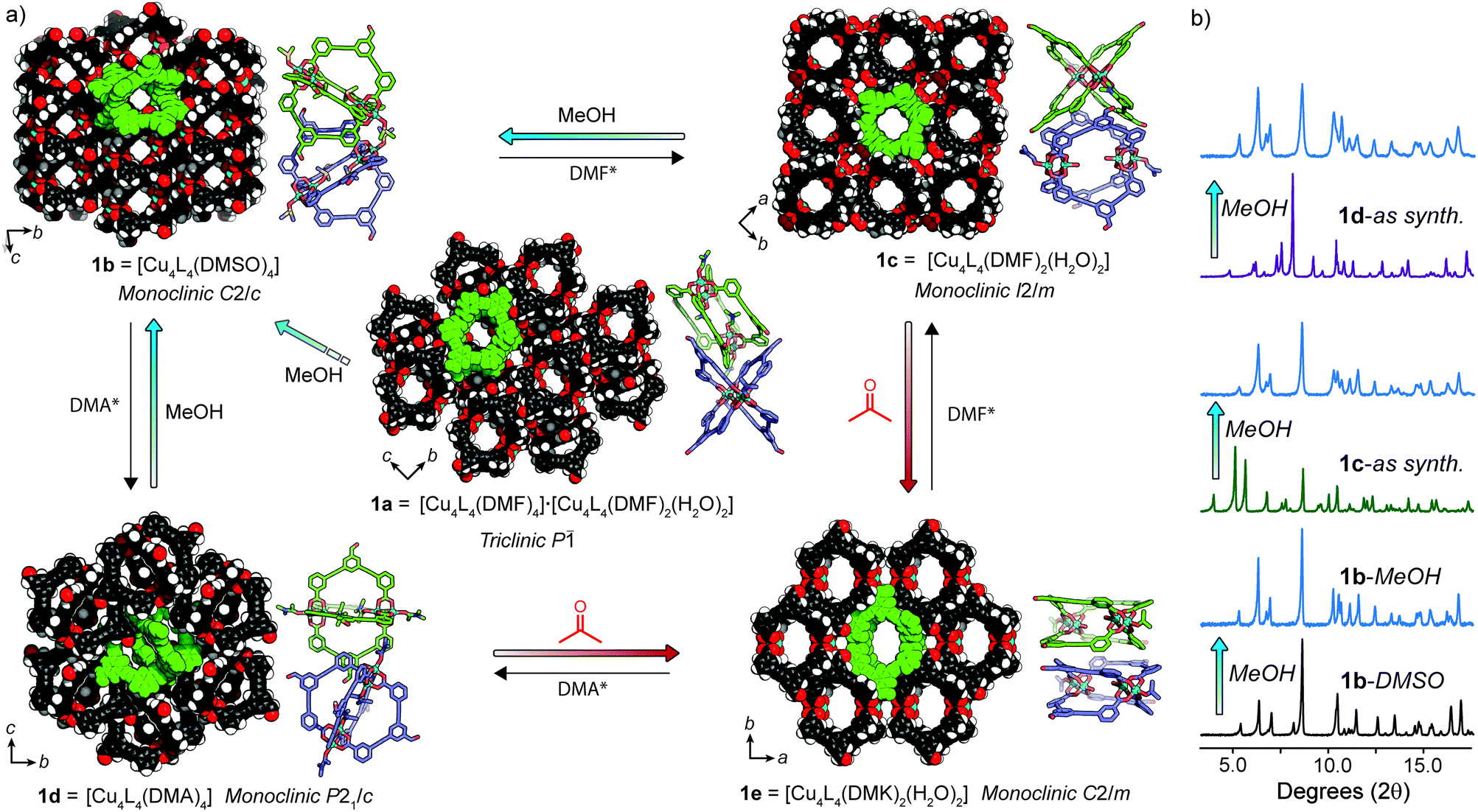 | ||
| Fig. 3 (a) Single crystal X-ray structures of solvatomorphs 1a–e in a scheme portraying their structural transformations. The crystal packing of 1a–e is represented as space-filling (coordinating solvents removed for clarity) and alongside are depictions of the orientations of cage molecules within each phase. Each phase can be regenerated through recrystallisation (*co-solvent = H2O); (b) PXRD patterns highlighting the solid-state transformations of 1c and 1d → 1b upon exposure to MeOH. Full details of the transformations shown in (a) can be found in the ESI (Fig. S11–S16†). | ||
X-ray structures of solvatomorphs 1b–d
We found that 1 could be reproducibly crystallised in three other distinct solvatomorphs by varying the solvent and precipitant of the slow-vapour diffusion (SVD) crystallisation (Table S1†). The crystals of each phase were suitable for SCXRD analysis and powder X-ray diffraction (PXRD) confirmed that the single crystals were representative of their bulk phases (Fig. 2b and S11–S16†).Needle-shaped crystals of 1b were obtained when a mixture of LH2 and Cu(OAc)2·2H2O was heated in DMSO at 85 °C for 16 h. The same crystal packing could also be obtained by SVD of MeOH into a DMA solution of 1 (1b-DMA, Fig. S13c†), although only the former conditions produced crystals suitable for SCXRD. 1b-DMSO crystallises in the monoclinic space group C2/c with half of the lantern cage in the asymmetric unit. Both interior and exterior paddlewheel sites are occupied by disordered DMSO ligands to give an overall formula of [Cu4L4(DMSO)4]. The crystal packing of 1b is stabilised by multiple intermolecular π–π interactions between the phenyl carboxylate and benzaldehyde moieties of 1 (shortest distance = 3.32 Å). This bilateral π-stacking motif results in molecules of 1 arranging in an edge-to-edge fashion. When viewing the extended structure along hkl = 111 plane, open channels intrinsic to the cage structure are evident and measure approximately 4.4 × 4.6 Å in diameter (Fig. 3a).
Slow diffusion of atmospheric moisture into a DMF solution of 1 resulted in the overnight formation of large block-shaped crystals of a new phase (1c), as indicated by SCXRD and PXRD analysis. We postulate that under these conditions, 1c forms in favour of 1a due to the rapid rate of crystallisation (24 h) compared to DMF/MeOH (5 days).91c crystallises in the monoclinic space group I2/m, with a quarter of two crystallographically independent cages in the asymmetric unit. In contrast to 1a, both cage molecules have identical paddlewheel compositions; DMF and H2O ligands occupy the exterior and interior apical sites, respectively. The overall formula of 1c is therefore [Cu4L4(DMF)2(H2O)2] (Fig. 3a and S33†). Interestingly, the extended structure of 1c comprises of a mutually perpendicular face-to-face cage packing. This arrangement creates open channels along the a and c axis, with the largest pore dimensions measuring 5.4 × 6.0 Å. However, this packing motif lacks defined π–π interactions and is stabilised by weak hydrogen bonding between the coordinated DMF ligands and the aldehyde group of 1 (C–H⋯O = 2.31 Å, 119°).
Another distinct crystalline phase could be obtained by slow diffusion of atmospheric moisture into a DMA solution of 1. 1d crystallises in the monoclinic space group P21/c, with half of the lantern cage in the asymmetric unit. In this solvatomorph, both apical coordination sites of the Cu2 paddlewheel are occupied by DMA ligands (Fig. S33†). In the crystal packing of 1d, cage molecules arrange in an edge-to-edge fashion and exhibit intermolecular π–π interactions between the benzaldehyde and phenyl carboxylate moieties. Despite possessing a similar crystal density to that of 1b (Table S2†) the arrangement of cage molecules in 1d does not result in defined solvent-accessible channels (Fig. 3a).
Owing to the solubility of 1 in DMF and DMA, crystals of each phase could be recrystallised to recycle between 1a–d (Fig. 3a).
Phase transformations of 1
Understanding transformations in porous materials is crucial in relating structure to function. For flexible MOFs, solid-state transformations often involve preservation of metal–ligand connectivity and framework topology, making their characterisation straightforward by PXRD methods.30,31 In contrast, phase transformations in MOP solids are difficult to elucidate since the weak intermolecular interactions that govern their crystal packing allow for dramatic reorientations of the cage molecules without preservation of single crystallinity. As a result, assigning the structure of solvent exchanged MOP solids is often omitted.The crystallisation of 1a–d indicated to us that the solvent and precipitant both contribute in determining the extended packing of the MOP structure. As such, we posited that in the four structures of 1, the apical paddlewheel solvent stabilises the crystal packing to different degrees. In order to examine this hypothesis, we subjected solvatomorphs 1a–d to solvent exchange with MeOH and acetone, as these solvents are commonly used en-route to activation.32,33 With the X-ray structures of 1a–d in hand, we were able to use powder diffraction to map out the solvent-induced transformations in bulk samples of 1 (Fig. 3b and S11–S16†).
PXRD analysis revealed that both 1a and 1b retain their overall crystal packing after solvent exchange with MeOH (Fig. 3b, S11 and S13†). Whilst the methanol exchanged form of 1b was stable indefinitely, a time-dependant PXRD experiment of 1a revealed that after 9 h, a slow transformation to 1b begins to occur and is complete within ∼24 h (Fig. 3a and S12†). In contrast, washing 1c and 1d in MeOH resulted in a rapid (<5 minutes) and quantitative transformation to 1b as indicated by PXRD (Fig. 3b, S14 and S15†). Notably, these conversions proceed entirely in the solid-state as 1 is insoluble in MeOH and DMF/MeOH mixtures (Fig. S8†).§ Although the mechanism of these types of transformations is difficult to elucidate,8 we speculate that the relative cage re-orientations that occur during this process are different for each phase (Fig. S34†).
Interestingly, we observed a different transformation when solvatomorphs of 1 were immersed in acetone. Whilst 1a and 1b retained their crystal packing even after 3 months (Fig. S11 and S13†), 1c and 1d both transformed to a new phase (1e) over a period of 3 weeks (Fig. S14 and S15†). The crystals of the former sample were of sufficient quality for structural determination by synchrotron SCXRD. 1e crystallises in the monoclinic space group C2/m and contains a quarter of the cage in the asymmetric unit. Here, the apical sites of the Cu2 paddlewheel nodes are occupied by exterior acetone (DMK) and interior water ligands to give the formula [Cu4L4(DMK)2(H2O)2]. The lantern cage adopts an extremely flattened conformation that facilitates close intermolecular π-stacking between the benzaldehyde and phenyl carboxylate moieties (shortest distance = 3.29 Å). The eclipsed face-to-face cage packing results in open channels along the c axis with apertures of 6.0 × 8.9 Å (Fig. 3a). Unsurprisingly, we found that the PXRD pattern of 1e remains unchanged upon solvent exchange with MeOH (Fig. S16†).
To further investigate the formation of 1e, we soaked the as-synthesised crystals of 1c and 1d in acetone-d6. Indeed, we observed trace quantities of 1 in the 1H NMR spectrum of the suspension, albeit only in the presence of DMA or DMF; washing the samples further with acetone-d6 (in order to exchange out the remaining solvent) led to the disappearance of resonances originating from 1 (Fig. S8†). As the crystals of 1c and 1d do not completely dissolve during the transformation, we suggest that the conversion to 1e is solvent assisted,34,35 although a traditional recrystallisation cannot be ruled out. It is noteworthy that 1e could not be obtained through conventional crystallisation methods or by the conversion of other solvatomorphs (Fig. S17, Table S1†), suggesting that 1c and 1d act as crystalline templates for this transformation.
To rationalise the observed transformations we performed lattice energy calculations on the set of five structures based on periodic density functional theory (DFT) calculations as implemented in the software package VASP 5.4.4.36 We note that lattice energy comparisons are commonly made for purely organic polymorphs (where solvent does not play a structural role).37,38 Nevertheless, we expected that the relative lattice energies of the desolvated forms of 1a–e should give insight into their transformations despite their experimental accessibility. In our DFT calculations, electron exchange and correlation were described using the generalised gradient approximation Perdew, Burke, and Ernzerhof (PBE) form39 and the projector-augmented wave potentials were used to treat core and valence electrons.40 To account for the long-range dispersion interactions, we included the dispersion corrections using DFT-D3 method.41 A full optimisation of the structures was performed allowing both the atomic and cell parameters to relax in the absence of the apical paddlewheel solvents. We observed no substantial change in the structural conformation and the lattice parameters for each of the five structures in the DFT optimisation (full details are given in the ESI†). The results of the DFT calculations were consistent with the expected correlation of higher density structures (e.g.1a, 1b and 1e) being lower in energy compared to structures with lower density (e.g.1c and 1d, Table S6†).37 As observed experimentally, 1b is indeed the lowest energy structure within the set of five phases (Table S4†), closely followed by 1e (+15.3 kJ mol−1, Fig. 4). The fact that these two phases are energetically similar yet do not interconvert through solvent exchange suggests that their crystal packing is stabilised predominantly by intermolecular cage interactions rather than the apical paddlewheel solvent. Relative to 1b, the lattice energy of 1a differs by only +22.3 kJ mol−1 which is in agreement with its metastability and slow transformation to 1b in MeOH. On the other hand, the lattices energies of 1c and 1d are comparatively higher (+106 kJ mol−1 and +69 kJ mol−1, respectively) which also supports their transformation to the low energy structures 1b (in MeOH) or 1e (in acetone). Therefore, compared to the other phases, 1c and 1d are largely stabilised by intermolecular interactions involving the coordinated paddlewheel and pore-bound solvents.
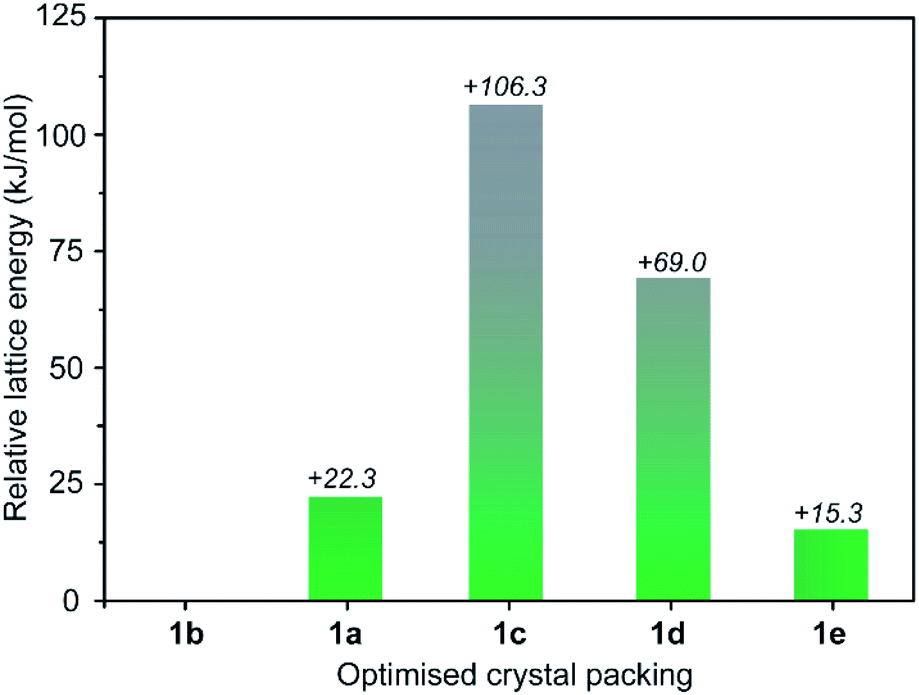 | ||
| Fig. 4 Energy landscape of the experimentally observed phases of 1a–e, relative to 1b. The calculated lattice energies are based on DFT optimisation including dispersion interactions with apical paddlewheel solvents omitted. | ||
Porosity control and switching in pseudo-polymorphs of 1
Although the crystallisation of MOP solvatomorphs is not unique to our system,17,19,42,43 their solvent exchange behaviour and porosity have scarcely been compared.24,32 We were interested whether different adsorbing forms of 1 are produced when MeOH-exchanged pseudo-polymorphs are activated under identical conditions. Therefore, prior to adsorption analysis, 1a-MeOH, 1b-MeOH and 1e-MeOH were subjected to high vacuum at 100 °C for 6 h (ESI†). The N2 isotherm of 1a at 77 K displays a maximum uptake 26.7 cm3 g−1 and a Brunauer–Emmett–Teller surface area (SABET) of 35 m2 g−1. The uptake of N2 by 1b and 1e, however, is markedly different, with both displaying type I adsorption isotherms and SABET of 421 m2 g−1 and 455 m2 g−1 at 77 K, respectively. It is worth noting that the latter represents the highest BET surface area for M4L4 lantern MOPs. Interestingly, the porosity of 1a could be ‘switched on’ by soaking the sample in MeOH. PXRD revealed that a conversion to a crystalline solid had occurred that perfectly matches the PXRD profile of 1b. Indeed, activation of this sample yielded a type I isotherm with a SABET = 392 m2 g−1 (Fig. 5d). Furthermore, the on/off porosity can be cycled; after washing the activated form of 1a with MeOH (to obtain 1b), the metastable phase 1a can be regenerated through recrystallisation from DMF/MeOH.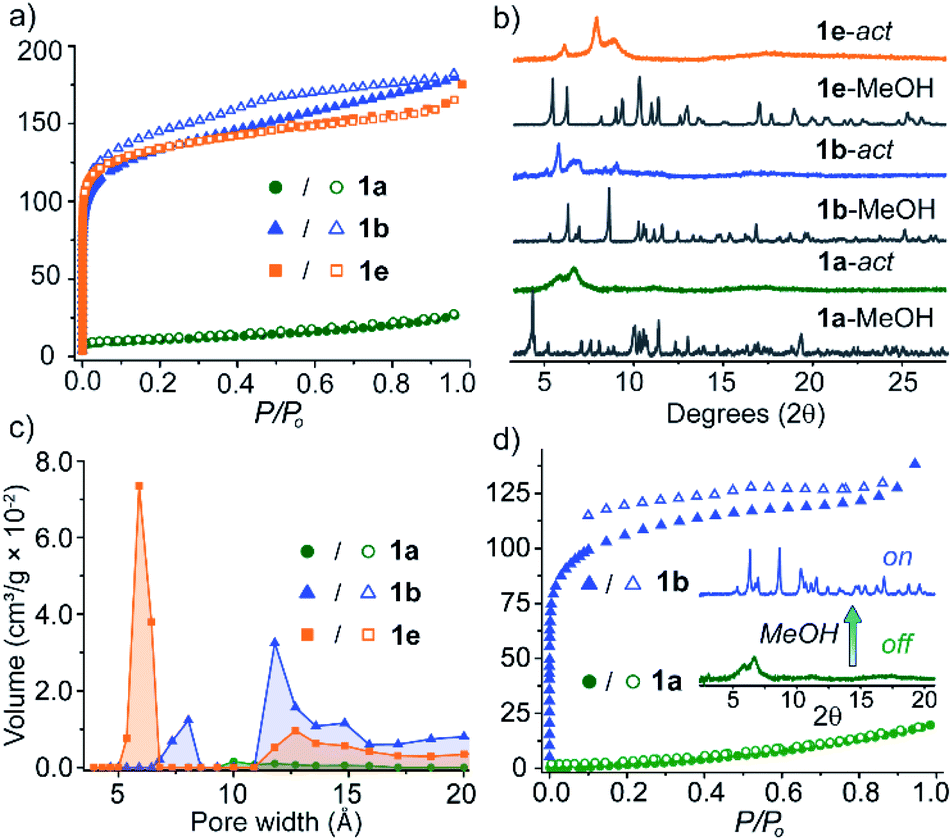 | ||
| Fig. 5 (a) N2 adsorption isotherms (in cm3 g−1) of 1a, 1b and 1e measured at 77 K; (b) PXRD patterns of pseudo-polymorphs as well as their activated forms; (c) pore-size distributions of 1a, 1b and 1e; (d) 77 K N2 isotherms (in cm3 g−1) showing on/off porosity switching between the activated form of 1a and its solvent treated form (1b). | ||
The structure of the activated phases was investigated by deriving pore-size distributions (PSD) from the low-pressure region of the isotherm data. PSD analysis revealed that indeed, 1a, 1b and 1e display distinct pore-sizes in the microporous region, despite possessing the same chemical structure. 1a shows a broad pore size centered around 10 Å, whilst 1b and 1e display much sharper distributions. 1b has pore volume maxima at 8.0 Å and 11.8 Å, whilst 1e possesses a sharp maximum at 5.9 Å (Fig. 5c).
Due to the weak, non-covalent interactions that stabilise their crystal packings, the PXRD patterns of the desolvated and activated samples showed shifting and broadening of the low-angle peaks with a reduction of long-range order compared to their parent MeOH-exchanged phases (Fig. 5b and S21†). This is common for carboxylate paddlewheel MOPs and general strategies to totally prevent lattice changes upon desolvation remain scarce.42,44 Nevertheless, the low angle diffraction peaks of these samples suggest that the activated forms of 1a, 1b and 1e possess unique crystal packings, which is also supported by their pore-size distributions. Therefore, we infer that the structural changes accompanying activation are different for each pseudo-polymorph,32 especially since different adsorbing forms of 1 are the end result. Upon re-solvating the activated forms of 1b and 1e in MeOH, we observed the appearance of sharp PXRD peaks consistent with 1b-MeOH. Resolvation of the activated samples of 1a, 1b, and 1e in their mother liquor, however, resulted in the regeneration of their crystalline, as-synthesised forms (Fig S20†). Thus, the resolvation experiments indicate that the activated pseudo-polymorphs retain structural flexibility and further support 1b as the lowest energy structure in the group of five solvatomorphs.
Post-assembly modifications of 1
Due to the difficulty in predicting the crystal packing of MOP solids (as observed for 1a–e), we assessed whether phase control can be achieved by covalent Post-Assembly Modifications (PAM).25 We rationalised that extending the cage architecture along its edges should modulate the inter-cage distance in the solid-state via π–π interactions. We note that covalent PAM of Cu2 paddlewheel MOPs are rare17,26 and the crystal packing of modified MOPs is rarely investigated.27Indeed, the deliberate instalment of the exterior aldehyde groups in 1 enabled us to carry out Lewis-acid catalysed imine condensation. Heating a DMF-d7 solution of 1 at 45 °C in the presence of 0.1 equivalents of Sc(OTf)3 (ref. 45) and 16 equivalents of o-toluidine resulted in the formation of 2 after 16 h. The 1H NMR spectrum of the reaction mixture revealed a characteristic imine resonance at 8.6 ppm and an absence of the aldehyde resonance normally observed at 9.9 ppm (Fig. 6b). Subjecting the reaction mixture to slow-vapour diffusion with diethyl ether yielded rod-like crystals of 2 after three days. SCXRD analysis confirmed that the complete conversion of all four aldehyde substituents had occurred without interfering with the Cu4L4 connectivity (Fig. 6c). 2 crystallises in the triclinic space group P with half of the formula 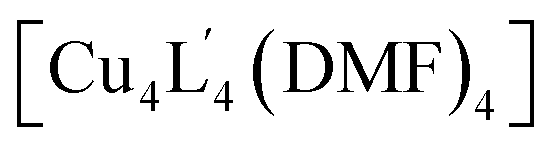 in the asymmetric unit. In the extended structure, the newly formed cage packs in an interdigitated edge-to-edge fashion. This arrangement is stabilised by numerous intermolecular π–π interactions and results in an overall 2D sheet-like packing (Fig. 6d). Within this motif, the average Cu⋯Cu distance between cage units measures 18.7 Å, which is an increase of 3.7 Å compared to the furthest Cu⋯Cu distance observed in the crystal packing of 1 (1c = 15.0 Å).
in the asymmetric unit. In the extended structure, the newly formed cage packs in an interdigitated edge-to-edge fashion. This arrangement is stabilised by numerous intermolecular π–π interactions and results in an overall 2D sheet-like packing (Fig. 6d). Within this motif, the average Cu⋯Cu distance between cage units measures 18.7 Å, which is an increase of 3.7 Å compared to the furthest Cu⋯Cu distance observed in the crystal packing of 1 (1c = 15.0 Å).
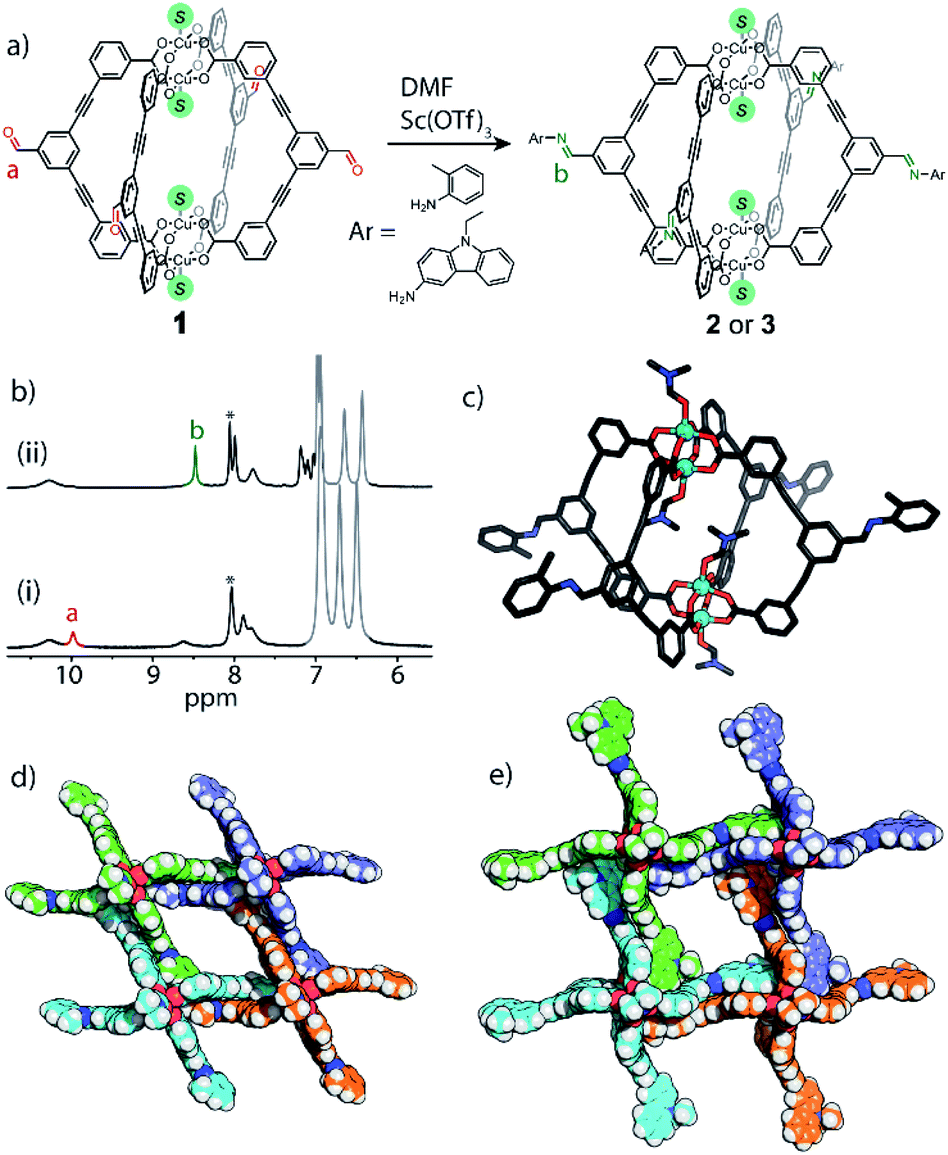 | ||
| Fig. 6 (a) Scheme illustrating the covalent PAM of 1 to give 2 or 3; (b) 1H NMR (500 MHz/DMF-d7) of (ii) the reaction mixture of 2 after heating and (i) before heating.‡ Excess aniline shown in grey, *DMF solvent peak; (c) X-ray structure of 2; (d) perspective view of the crystal packing of 2; (e) perspective view of the crystal packing of 3. | ||
We also performed covalent PAM of 1 with 9-ethyl-9H-carbazol-3-amine due to its solubility and extended aromatic structure. This allowed us to thus further examine extensions to the cage architecture and their subsequent effect on the crystal packing. Once again, 1H NMR spectroscopy revealed a >98% conversion of 1 to 3 under the similar reaction conditions to that of 2 (Fig. S9a†). Slow vapour diffusion of diethyl ether into the reaction mixture yielded X-ray quality crystals of a monoclinic phase with the space group P21/n. Interestingly, in the extended structure of 3, molecules of 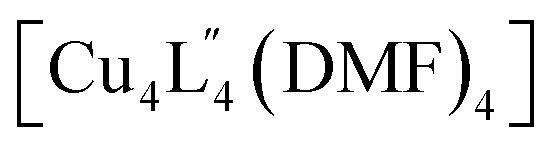 interdigitate in an identical manner to that of 2. The bulkier carbazole moiety pushes the cages further apart within the 2D layers to give an average inter-cage Cu⋯Cu distance of 21.1 Å (Fig. 6e).
interdigitate in an identical manner to that of 2. The bulkier carbazole moiety pushes the cages further apart within the 2D layers to give an average inter-cage Cu⋯Cu distance of 21.1 Å (Fig. 6e).
We confirmed the bulk purity of 2 and 3 by IR spectroscopy and PXRD (Fig. S9b and S18†). The former revealed new C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N stretches at 1619 cm−1 and 1630 cm−1 for 2 and 3 respectively, as well as the absence of the aldehyde stretch (1698 cm−1). Attempting other crystallisation conditions to obtain solvatomorphs of 2 and 3 led to the same crystal packing or the isolation of poorly crystalline precipitates that were not further investigated. By providing more opportunities for π–π interactions, it is evident that the range of solvatomorphs may be limited compared to 1. However, this may be advantageous for the crystal engineering of MOP solids; other functionalities (e.g. H-bonding) could be introduced through covalent PAM to control the structural rigidity, thus limiting the effect that coordinating solvent plays in determining the crystal packing.
N stretches at 1619 cm−1 and 1630 cm−1 for 2 and 3 respectively, as well as the absence of the aldehyde stretch (1698 cm−1). Attempting other crystallisation conditions to obtain solvatomorphs of 2 and 3 led to the same crystal packing or the isolation of poorly crystalline precipitates that were not further investigated. By providing more opportunities for π–π interactions, it is evident that the range of solvatomorphs may be limited compared to 1. However, this may be advantageous for the crystal engineering of MOP solids; other functionalities (e.g. H-bonding) could be introduced through covalent PAM to control the structural rigidity, thus limiting the effect that coordinating solvent plays in determining the crystal packing.
Conclusions
In summary, we have demonstrated the remarkable propensity of an aldehyde-functionalised metal–organic polyhedron to form a range of unique crystal packings. In our system, five different solvatomorphs were isolated, which allowed us to compare their solvent exchange behaviour, phase transformations and porosity. Notably, one phase (1e) could only be obtained through the conversion of 1c or 1d in acetone, suggesting that certain solvatomorphs can act as templates for the formation of otherwise inaccessible crystal packings. A comparison of the N2 adsorption profiles of pseudo-polymorphs of 1 revealed that different adsorbing forms are produced under analogous activation conditions. We also showed that the porosity of a non-porous phase can be switched on by solvent treatment. This demonstrates that the rich crystal-phase behaviour commonly observed for purely organic cages is also inherent to paddlewheel-based MOPs. Given their interest as functional materials, exploiting their solvatomorphism, structural flexibility and phase-dependant porosity may have implications for modulating their gas separation and catalytic properties.Finally, we demonstrated that 1 can be covalently modified in solution via imine condensation. In the solid-state, the covalent extensions of 1 led to an interdigitated 2D sheet-like motif, which was observed in both of the modified samples. We think that this approach may open up new avenues for the design and crystal engineering of porous MOP materials. Indeed, the assembly of hierarchical materials is an emerging field of research,46–48 and the exterior modifications of 1 show promise for assembling COF-like materials from MOP building blocks. Efforts towards this goal are currently underway in our laboratory.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
We thank Dr John Denman (University of South Australia) for assistance with TGA measurements. W. M. B. gratefully acknowledges the Australian Research Council for funding (DE190100327). Preliminary results of this research were supported by a University of Adelaide Ramsay Fellowship. Aspects of this work were undertaken on the MX1 beamline49 at the Australian Synchrotron, Victoria, Australia. R. B. acknowledges the Australian Research Council (DE160100987) and Alexander von Humboldt Foundation for financial support, and the National Computing Infrastructure (NCI), CSIRO Pearcey cluster, and Pawsey supercomputing facilities.Notes and references
- T. Hasell and A. I. Cooper, Nat. Rev. Mater., 2016, 1, 16053 CrossRef CAS.
- D. J. Tranchemontagne, Z. Ni, M. O'Keeffe and O. M. Yaghi, Angew. Chem., Int. Ed., 2008, 47, 5136–5147 CrossRef CAS PubMed.
- F. Beuerle and B. Gole, Angew. Chem., Int. Ed., 2018, 57, 4850–4878 CrossRef CAS PubMed.
- H. Furukawa, K. E. Cordova, M. O'Keeffe and O. M. Yaghi, Science, 2013, 341, 1230444 CrossRef PubMed.
- X. Feng, X. Ding and D. Jiang, Chem. Soc. Rev., 2012, 41, 6010–6022 RSC.
- J. T. A. Jones, T. Hasell, X. Wu, J. Bacsa, K. E. Jelfs, M. Schmidtmann, S. Y. Chong, D. J. Adams, A. Trewin, F. Schiffman, F. Cora, B. Slater, A. Steiner, G. M. Day and A. I. Cooper, Nature, 2011, 474, 367–371 CrossRef CAS PubMed.
- Z. Wang, N. Sikdar, S.-Q. Wang, X. Li, M. Yu, X.-H. Bu, Z. Chang, X. Zou, Y. Chen, P. Cheng, K. Yu, M. J. Zaworotko and Z. Zhang, J. Am. Chem. Soc., 2019, 141, 9408–9414 CrossRef PubMed.
- J. T. A. Jones, D. Holden, T. Mitra, T. Hasell, D. J. Adams, K. E. Jelfs, A. Trewin, D. J. Willock, G. M. Day, J. Bacsa, A. Steiner and A. I. Cooper, Angew. Chem., Int. Ed., 2011, 50, 749–753 CrossRef CAS PubMed.
- A. Avellaneda, P. Valente, A. Burgun, J. D. Evans, A. W. Markwell-Heys, D. Rankine, D. J. Nielsen, M. R. Hill, C. J. Sumby and C. J. Doonan, Angew. Chem., Int. Ed., 2013, 52, 3746–3749 CrossRef CAS PubMed.
- S. Bera, K. Dey, T. K. Pal, A. Halder, S. Tothadi, S. Karak, M. Addicoat and R. Banerjee, Angew. Chem., Int. Ed., 2019, 58, 4243–4247 CrossRef CAS PubMed.
- A. C. Sudik, A. R. Millward, N. W. Ockwig, A. P. Côté, J. Kim and O. M. Yaghi, J. Am. Chem. Soc., 2005, 127, 7110–7118 CrossRef CAS PubMed.
- J. Park, Z. Perry, Y. P. Chen, J. Bae and H. C. Zhou, ACS Appl. Mater. Interfaces, 2017, 9, 28064–28068 CrossRef CAS PubMed.
- W.-H. Xing, H.-Y. Li, X.-Y. Dong and S.-Q. Zang, J. Mater. Chem. A, 2018, 6, 7724–7730 RSC.
- L. Chen, T. Yang, H. Cui, T. Cai, L. Zhang and C.-Y. Su, J. Mater. Chem. A, 2015, 3, 20201–20209 RSC.
- S. Furukawa, N. Horike, M. Kondo, Y. Hijikata, A. Carné-Sánchez, P. Larpent, N. Louvain, S. Diring, H. Sato, R. Matsuda, R. Kawano and S. Kitagawa, Inorg. Chem., 2016, 55, 10843–10846 CrossRef CAS PubMed.
- G. R. Lorzing, B. A. Trump, C. M. Brown and E. D. Bloch, Chem. Mater., 2017, 29, 8583–8587 CrossRef CAS.
- W. Lu, D. Yuan, A. Yakovenko and H.-C. Zhou, Chem. Commun., 2011, 47, 4968–4970 RSC.
- H. Vardhan, M. Yusubov and F. Verpoort, Coord. Chem. Rev., 2016, 306, 171–194 CrossRef CAS.
- M. Jaya Prakash, M. Oh, X. Liu, K. N. Han, G. H. Seong and M. S. Lah, Chem. Commun., 2010, 46, 2049–2051 RSC.
- Y. H. Kang, X. D. Liu, N. Yan, Y. Jiang, X. Q. Liu, L. B. Sun and J. R. Li, J. Am. Chem. Soc., 2016, 138, 6099–6102 CrossRef CAS PubMed.
- Y.-H. Kang, N. Yan, Z.-Y. Gao, P. Tan, Y. Jiang, X.-Q. Liu and L.-B. Sun, J. Mater. Chem. A, 2017, 5, 5278–5282 RSC.
- O. Barreda, G. Bannwart, G. P. A. Yap and E. D. Bloch, ACS Appl. Mater. Interfaces, 2018, 10, 11420–11424 CrossRef CAS PubMed.
- T.-F. Liu, Y.-P. Chen, A. A. Yakovenko and H.-C. Zhou, J. Am. Chem. Soc., 2012, 134, 17358–17361 CrossRef CAS PubMed.
- Z. Niu, S. Fang, X. Liu, J.-G. Ma, S. Ma and P. Cheng, J. Am. Chem. Soc., 2015, 137, 14873–14876 CrossRef CAS PubMed.
- D. A. Roberts, B. S. Pilgrim and J. R. Nitschke, Chem. Soc. Rev., 2018, 47, 626–644 RSC.
- V. Brega, M. Zeller, Y. He, H. Peter Lu and J. K. Klosterman, Chem. Commun., 2015, 51, 5077–5080 RSC.
- A. Carné-Sánchez, J. Albalad, T. Grancha, I. Imaz, J. Juanhuix, P. Larpent, S. Furukawa and D. Maspoch, J. Am. Chem. Soc., 2019, 141, 4094–4102 CrossRef PubMed.
- R. W. Larsen, J. Am. Chem. Soc., 2008, 130, 11246–11247 CrossRef CAS PubMed.
- M. Kato, H. B. Jonassen and J. C. Fanning, Chem. Rev., 1964, 64, 99–128 CrossRef CAS.
- W. M. Bloch, R. Babarao, M. R. Hill, C. J. Doonan and C. J. Sumby, J. Am. Chem. Soc., 2013, 135, 10441–10448 CrossRef CAS PubMed.
- A. Schneemann, V. Bon, I. Schwedler, I. Senkovska, S. Kaskel and R. A. Fischer, Chem. Soc. Rev., 2014, 43, 6062–6096 RSC.
- G. A. Craig, P. Larpent, S. Kusaka, R. Matsuda, S. Kitagawa and S. Furukawa, Chem. Sci., 2018, 9, 6463–6469 RSC.
- J. M. Teo, C. J. Coghlan, J. D. Evans, E. Tsivion, M. Head-Gordon, C. J. Sumby and C. J. Doonan, Chem. Commun., 2016, 52, 276–279 RSC.
- W. M. Bloch and C. J. Sumby, Chem. Commun., 2012, 48, 2534–2536 RSC.
- T. M. Muzioł, N. Tereba, R. Podgajny, D. Kędziera and G. Wrzeszcz, Dalton Trans., 2019, 48, 11536–11546 RSC.
- G. Kresse and J. Hafner, Phys. Rev. B: Condens. Matter Mater. Phys., 1993, 48, 13115–13118 CrossRef CAS PubMed.
- S. L. Price, Acc. Chem. Res., 2009, 42, 117–126 CrossRef CAS PubMed.
- E. O. Pyzer-Knapp, H. P. G. Thompson, F. Schiffmann, K. E. Jelfs, S. Y. Chong, M. A. Little, A. I. Cooper and G. M. Day, Chem. Sci., 2014, 5, 2235–2245 RSC.
- S. Grimme, S. Ehrlich and L. Goerigk, J. Comput. Chem., 2011, 32, 1456–1465 CrossRef CAS PubMed.
- J. P. Perdew, K. Burke and M. Ernzerhof, Phys. Rev. Lett., 1996, 77, 3865–3868 CrossRef CAS PubMed.
- G. Kresse and D. Joubert, Phys. Rev. B: Condens. Matter Mater. Phys., 1999, 59, 1758–1775 CrossRef CAS.
- S. Mollick, S. Mukherjee, D. Kim, Z. Qiao, A. V. Desai, R. Saha, Y. D. More, J. Jiang, M. S. Lah and S. K. Ghosh, Angew. Chem., Int. Ed., 2019, 58, 1041–1045 CrossRef CAS PubMed.
- Y. Gong, Y. Zhang, C. Qin, C. Sun, X. Wang and Z. Su, Angew. Chem., Int. Ed., 2019, 58, 780–784 CrossRef CAS PubMed.
- S. Mollick, S. Fajal, S. Mukherjee and S. K. Ghosh, Chem.–Asian J., 2019, 14, 3096–3108 CrossRef CAS PubMed.
- M. Matsumoto, R. R. Dasari, W. Ji, C. H. Feriante, T. C. Parker, S. R. Marder and W. R. Dichtel, J. Am. Chem. Soc., 2017, 139, 4999–5002 CrossRef CAS PubMed.
- S. Datta, M. L. Saha and P. J. Stang, Acc. Chem. Res., 2018, 51, 2047–2063 CrossRef CAS PubMed.
- G. Lal, M. Derakhshandeh, F. Akhtar, D. M. Spasyuk, J.-B. Lin, M. Trifkovic and G. K. H. Shimizu, J. Am. Chem. Soc., 2019, 141, 1045–1053 CrossRef CAS PubMed.
- A. Carné-Sánchez, G. A. Craig, P. Larpent, T. Hirose, M. Higuchi, S. Kitagawa, K. Matsuda, K. Urayama and S. Furukawa, Nat. Commun., 2018, 9, 2506 CrossRef PubMed.
- N. P. Cowieson, D. Aragao, M. Clift, D. J. Ericsson, C. Gee, S. J. Harrop, N. Mudie, S. Panjikar, J. R. Price, A. Riboldi-Tunnicliffe, R. Williamson and T. Caradoc-Davies, J. Synchrotron Radiat., 2015, 22, 187–190 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available: Full experimental details and supporting analysis. CCDC 1969619–1969625. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/d0sc00070a |
| ‡ Owing to the paramagnetic Cu2 paddlewheel node of 1–3, proton resonances in close proximity to the metal node are significantly broadened.26 |
| § During the transformation of 1c/d to 1b, the crystals of the parent phase fragment rapidly. Despite the insolubility of 1 in MeOH and even mixtures of DMF/MeOH, we cannot completely rule out the possibility of an extremely rapid recrystallisation. |
| This journal is © The Royal Society of Chemistry 2020 |