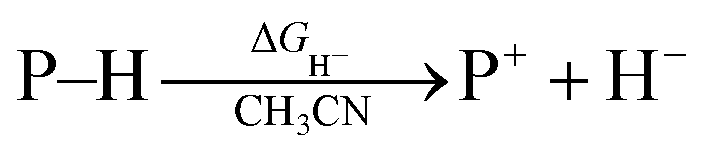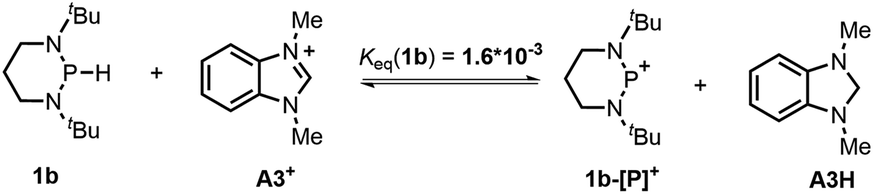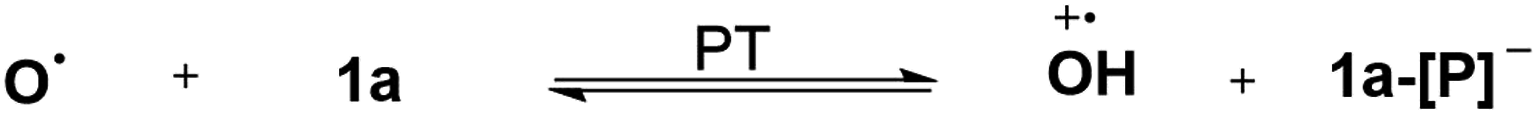DOI:
10.1039/C9SC05883D
(Edge Article)
Chem. Sci., 2020,
11, 3672-3679
Diazaphosphinanes as hydride, hydrogen atom, proton or electron donors under transition-metal-free conditions: thermodynamics, kinetics, and synthetic applications†
Received
20th November 2019
, Accepted 5th March 2020
First published on 5th March 2020
Abstract
Exploration of new hydrogen donors is in large demand in hydrogenation chemistry. Herein, we developed a new 1,3,2-diazaphosphinane 1a, which can serve as a hydride, hydrogen atom or proton donor without transition-metal mediation. The thermodynamics and kinetics of these three pathways of 1a, together with those of its analog 1b, were investigated in acetonitrile. It is noteworthy that, the reduction potentials (Ered) of the phosphenium cations 1a-[P]+ and 1b-[P]+ are extremely low, being −1.94 and −2.39 V (vs. Fc+/0), respectively, enabling corresponding phosphinyl radicals to function as neutral super-electron-donors. Kinetic studies revealed an extraordinarily large kinetic isotope effect KIE(1a) of 31.3 for the hydrogen atom transfer from 1a to the 2,4,6-tri-(tert-butyl)-phenoxyl radical, implying a tunneling effect. Furthermore, successful applications of these diverse P–H bond energetic parameters in organic syntheses were exemplified, shedding light on more exploitations of these versatile and powerful diazaphosphinane reagents in organic chemistry.
Introduction
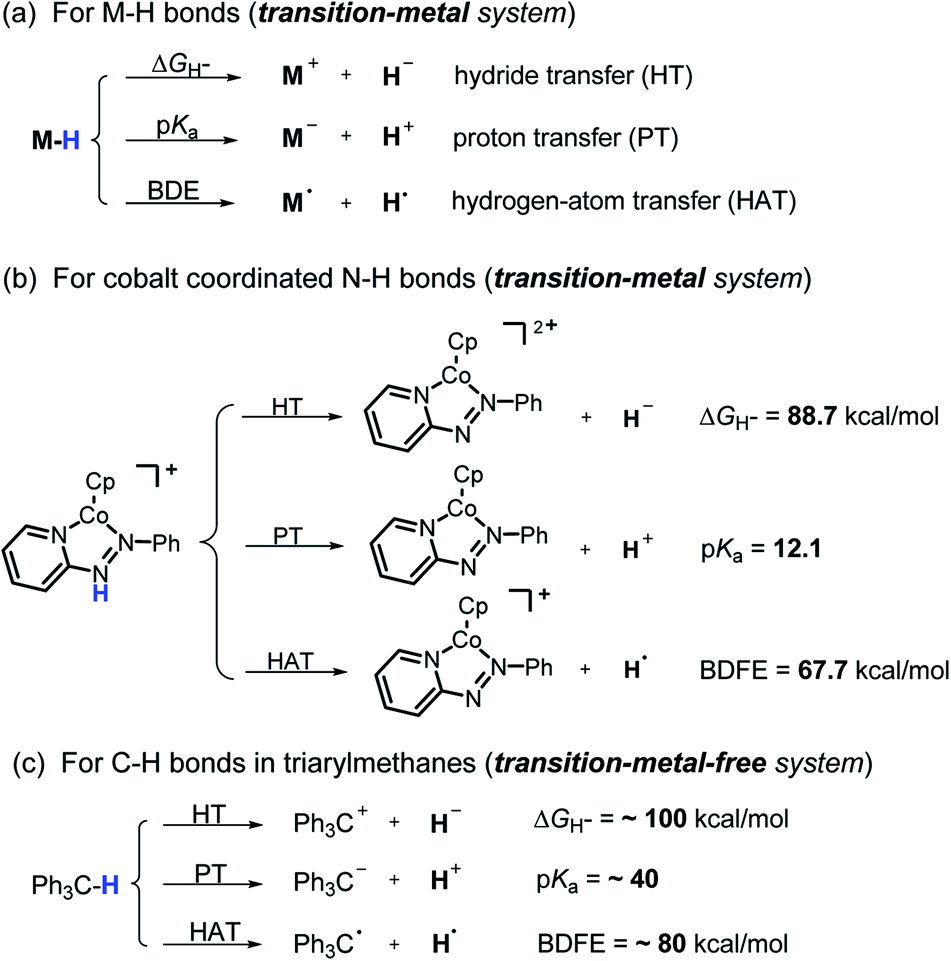
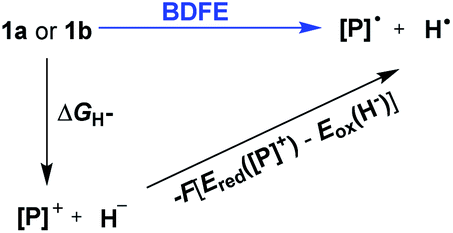
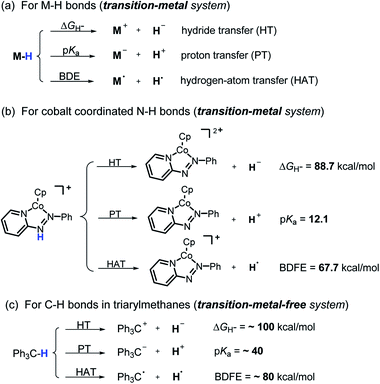
Hydrogen transfer plays a very important role in chemical science and related fields. This process occurs through the cleavage of the targeted R–H bond in three possible pathways (Scheme 1), i.e., hydride (H−),1 hydrogen atom (H˙)2 and proton (H+)3 transfers, which, from a thermodynamic aspect, depends on the R–H bond strength and the nature of the atom bound to the transferred hydrogen atom. Accordingly, knowledge of the relevant thermodynamics of hydricity ΔGH−,1b,c,4 bond-dissociation free-energy, BDFE5 and acidity, pKa3c,6 as well as the kinetics7 relevant to these processes would facilitate rational exploitations of new transformations. In recent years, considerable attention has been paid to the development of new hydrogen sources with versatile hydrogen donor reactivities, with particular interests in transition-metal hydrides M–H,1c such as hydrogenase enzyme analogs8 and hydrogenation catalysts,9 and main-group organic hydrides X–H,1b such as nicotinamide coenzyme models10 and Hantzsch esters,11 and so on.
 |
| | Scheme 1 Possible pathways of hydrogen transfers for transition-metal and transition-metal-free systems as well as corresponding thermodynamic driving forces in acetonitrile. | |
It is well known that metal hydrides (M–H) could serve as H−, H˙ and H+ donors (Scheme 1a).1c,12 Their diverse hydrogen reactivities originate from the readily modulated polarity of M–H bonds through electronic communication between the metal centers and coordinated ligands. Compared to an M–H system, the X–H hydride manifested some advantages in mild reaction conditions, good functional-group compatibility, easy modification, etc. Integration of these three dissociation possibilities into one X–H covalent bond is a substantial challenge, due to the great disparity in the electronegativity of the X and H fragments. Nevertheless, Waymouth et al. very recently reported an excellent new example of such a X–H bond where the N–H bond of the phenylazopyridine ligand in the Co(I) complex can serve as either a H−, H˙ or a H+ donor (Scheme 1b).13 It is noted, however, that the diverse reactivity of this N–H bond may require latent electronic communication between the azopyridine ligand and cobalt atom within the five-membered ring, so it may be viewed as a quasi-M–H system.
For a true transition-metal-free system, till now only triarylmethane analogs, where there exist steric as well as resonance stabilization effects on the incipient radical or charges, were reported to have α-C–H bond-cleavage energies determined for all three pathways (Scheme 1c).14 However, their poor hydrogen donability and severe steric hindrance make these dissociation processes not very useful for synthesis. This, in turn, stimulated our interest to find more useful transition-metal-free X–H systems that promise both the desired energetic measurement and new synthetic applications.
Recently, diazaphospholenium hydrides (P–H) have attracted substantial research interest due to their superior hydricity endowed by the unique diazaphospholene skeleton.15 Many diazaphospholenes were developed and successfully used in various hydridic reductions.16 However, the superior hydricity resulted in their other promising applications, such as serving as hydrogen atom and proton donors, as well as the precursors of strong electron donors, being greatly overlooked. Considering the similar electronegativity of the hydrogen (χAR = 2.20) and phosphorus (χAR = 2.06) atoms,15b we reckoned that the P–H bond, beside being a good hydride donor,17 may have the potential to release proton and hydrogen atoms18 as well, by fine-tuning the electrical properties of the P fragment. And indeed luckily enough, this was realized in the present work.
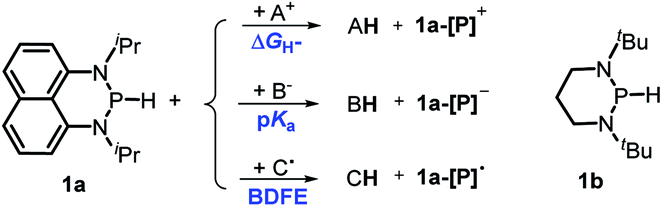
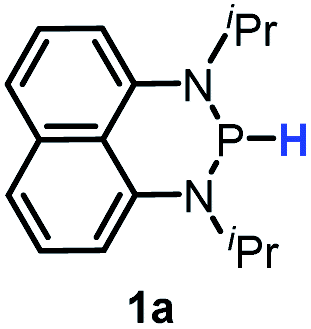
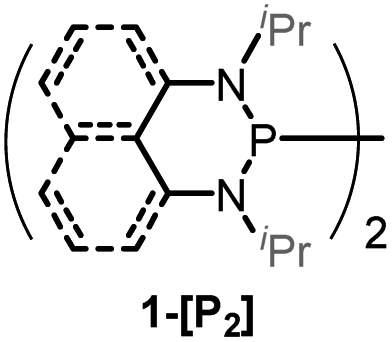
Herein, we reported a new N-heterocyclic phosphine 1a, which could act as a H−, H˙ and H+ donor (Scheme 2). Thermodynamics and kinetics pertinent to these three processes were examined in detail. For comparison, 1b was investigated as well. Based on the P–H bond energetic studies, we also exploited their versatile applications in the reduction of pyridines, synthesis of bisphosphines, H/D exchange, and activation of carbon–halogen bonds as original examples.
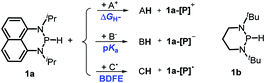 |
| | Scheme 2 P–H reagents 1a and 1b serving as diverse hydrogen donors. A+, B− and C˙ are the hydride, proton and hydrogen atom acceptors, respectively. | |
Results and discussion
Synthesis
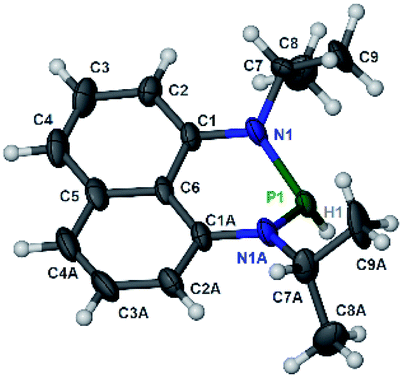
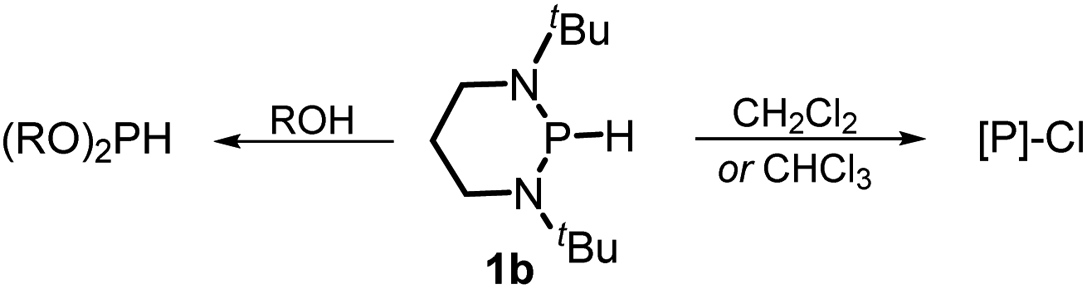
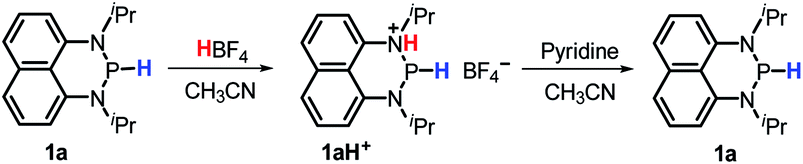
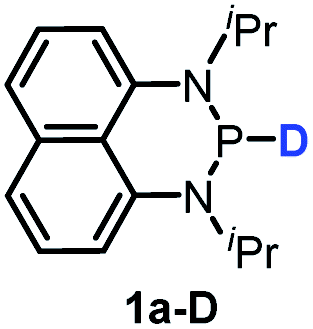
The substrate 1a was prepared via a H/Cl exchange between the corresponding phosphoric chloride and LiAlH4 in THF according to the literature15,17 (see the ESI† for details). LiAlD4 was used for deriving the deuterated substrates 1a-D and 1b-D. The crystal structure of 1a was shown in Fig. 1 and features an envelope configuration with the P atom standing out of the naphthalene plane and the P–H bond adopting a flagpole position, similar to the five-membered ring analogs disclosed previously.15a And a P–H distance of 1.42(2) Å is slightly shorter than that of the five-membered ring analog.1531P NMR spectra of 1a in CD3CN show a dt peak at 25.66 ppm that is split by the hydrogen atoms on the P atom and isopropyl groups. It moves to the higher magnetic field in comparison with other N-heterocyclic phosphines.17 Under an inert atmosphere, 1a is stable enough for over 12 hours in tetrahydrofuran, acetonitrile, alcohols, toluene, dimethylsulfoxide and chloroform. On the other hand, 1b is stable in these solvents as well except in alcohols and chlorinated hydrocarbons, where it decomposes rapidly to yield phosphonites (RO)2PH19 or 2-chloro-1,3,2-diazaphosphinane [P]-Cl (Scheme 3), showing that 1b is more air- and moisture-sensitive than 1a.
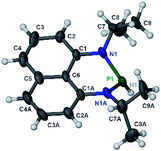 |
| | Fig. 1 The crystal structure of 1a (50% probability thermal ellipsoids).20 Selected bond lengths (in Å): P1–H1 1.42(2), P1–N1 1.747(6), N1–C1 1.329(9), N1–C7 1.440(8), and C1–C6 1.413(8). | |
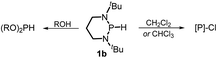 |
| | Scheme 3 Decomposition of 1b in alcohols and chlorinated hydrocarbons. [P] = (CH2)3(NtBu)2P, R = Me or Et. | |
Redox properties
The oxidation potentials of 1a and 1b were examined by cyclic voltammetry (CV), exhibiting a partially reversible oxidation peak of 1a to 1a˙+ at 0.23 V (vs. Fc+/0, Fig. S1a†) and an irreversible oxidation wave of 1b at 0.47 V (vs. Fc+/0, Fig. S1b†) in acetonitrile. The low oxidation potentials of 1a and 1b indicate their good reducing capacity. It is interesting to find that 1a is a better electron donor, in spite of being a far poorer hydride donor than 1b (vide infra). Besides, the redox behaviors of the respective phosphinyl radicals (1a-[P]˙ and 1b-[P]˙) were also examined through the reduction of the corresponding phosphenium cations (1a-[P]+![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 21 and 1b-[P]+), resulting in the CV peaks at −1.94 V and −2.39 V (vs. Fc+/0) in acetonitrile, respectively (Fig. S1c and 1d†). The extremely low potentials, especially for 1b-[P]+, indicate the very high reducing capacity of the corresponding 1a-[P]˙ and 1b-[P]˙ radicals. This suggests that they both have a very promising potential to serve as a super electron donor in organic synthesis to reduce aryl halides.22 And indeed, this was verified in the tentative application of the present work in radical hydrodebromination of bromobenzene (see Application part, vide infra).
21 and 1b-[P]+), resulting in the CV peaks at −1.94 V and −2.39 V (vs. Fc+/0) in acetonitrile, respectively (Fig. S1c and 1d†). The extremely low potentials, especially for 1b-[P]+, indicate the very high reducing capacity of the corresponding 1a-[P]˙ and 1b-[P]˙ radicals. This suggests that they both have a very promising potential to serve as a super electron donor in organic synthesis to reduce aryl halides.22 And indeed, this was verified in the tentative application of the present work in radical hydrodebromination of bromobenzene (see Application part, vide infra).
Thermodynamic driving force and reactivity
Hydride transfer (HT).
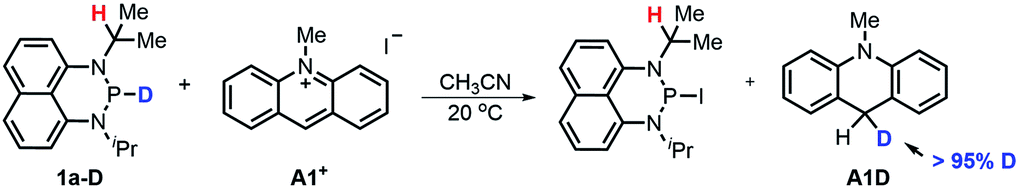
Hydride transfer from 1a or 1b17 to N-methylacridinium iodide A1+ yielded equimolar products (Fig. S3†). To exclude the suspected interruption of the hydridic isopropyl α-C–H, the deuterated substrate 1a-D was employed, and over 95% deuterated A1D was derived (eqn (1) and Fig. S4†). This confirmed that the P–H hydride of 1a is a stronger donor than that of the isopropyl α-C–H.| | 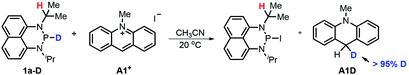 | (1) |
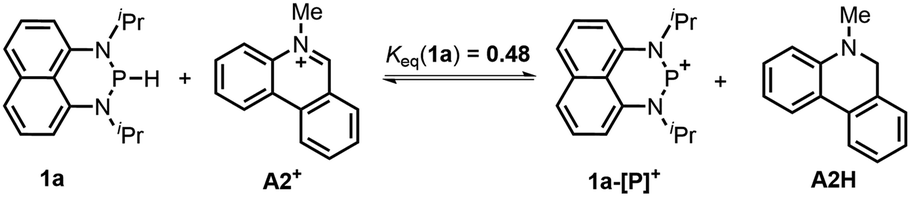
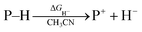
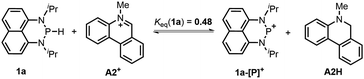
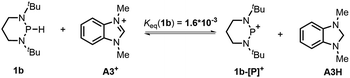
The hydricity (ΔGH−, defined by eqn (2)) of 1a and 1b in acetonitrile was determined by measuring their equilibrium constants with the acceptors of known hydricities (eqn (3)). For example, the measured hydride transfer equilibrium constant Keq(1a) of 0.48 (equivalent to 0.43 kcal mol−1) between 1a and phenanthridinuim trifluoromethanesulfonate A2+ (ΔGH−(A2H) = 61.4 kcal mol−1)1b provided the hydricity ΔGH− of 1a to be 61.8 kcal mol−1 (see Fig. S5 in the ESI† for details) and other equilibria (in Fig. S6 and S7†) verified this value, giving a mean hydricity of 62.2 ± 1.0 kcal mol−1. This demonstrates 1a to be a moderate hydride donor, similar to the hydride-donating ability of the NADH analogs.1b Thus, it should be capable of reducing commonly used hydride acceptors, such as N-methylacridinium cations (ΔGH− = 76 kcal mol−1), 9-phenylxanthylium cations (89 kcal mol−1) and trityl cations (99 kcal mol−1). Similarly, a much lower equilibrium constant Keq (1b) of 1.6 × 10−3 (equivalent to 3.8 kcal mol−1, eqn (5) and Fig. S8†) established between 1b and benzimidazolium perchlorate A3+ (ΔGH−(A3H) = 45.0 kcal mol−1)4b resulted in ΔGH−(1b) being 48.8 ± 1.0 kcal mol−1, which is comparable to that of the commonly used good hydride donor, 2,3-dihydrobenzo[d]imidazoles.1b The good hydridic reactivity of 1b was illustrated in our recent work in the reduction of various unsaturated compounds.17 It is interesting to note that 1b is about 13 kcal mol−1 stronger than 1a in hydricity, but is 0.24 V weaker in electron-donating ability.
| |  | (2) |
| | | ΔGrxn = −RTlnKeq = ΔGH−(1) − ΔGH−(AH) | (3) |
| |  | (4) |
| |  | (5) |
The hydricity can be used to rationalize the disparate reactivity between 1a and some reported diazaphospholenium hydrides. The latter compounds were reported to react with strong acid HBF4 to give H2 and the corresponding phosphenium cations.15b However, when 1a was treated with HBF4 (or HOTf), no gaseous product was generated. Instead, a new phosphorus species with a doublet peak at about 80 ppm was detected in the 31P NMR spectrum (Fig. S9a and S9b†). To identify this newly formed species, a base pyridine was added to the reaction mixture, which rendered 1a the primary product with a small amount of unidentified impurities (Fig. S9c†). Consequently, this mysterious P species was speculated to be protonated 1a (1aH+, Scheme 4). Based on the doublet peak of the phosphorus atom in 31P NMR spectra (if the extraneous proton was connected to the P atom, a triplet 31P peak would be observed) and the unsymmetrical peaks of naphthyl and isopropyl hydrogen atoms in 1H NMR spectra (Fig. S10†), the protonated site was thus assigned to the N atom (Scheme 4). We thought that their dissimilarity in reactivity between 1a and some reported diazaphospholenium hydrides should originate from the relatively weak hydricity of 1a (ΔGH− = 62.2 kcal mol−1). As a consequence, it makes the H2 release unable to compete with its protonation even under the condition of heating (80 °C for 5 hours). The moderate hydricity of 1a endows it with good tolerance to even strong acids, avoiding direct elimination of dihydrogen. This in turn offers a possibility for 1a to act as a hydride donor in acid-catalyzed reduction. Lots of examples in this connection1a can be found, especially in the hydrogenation of imines by hydride donors with comparable hydricity to 1a (∼60–70 kcal mol−1, like Hantzsch ester,23 benzothiazoline24 and cyclohexadiene25) under the catalysis of Brønsted acids (for examples BINOL-phosphoric acid, trifluoroacetic acid and Tf2NH).
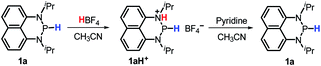 |
| | Scheme 4 Protonation of 1a by HBF4 and recovery of it by adding pyridine in CD3CN. | |
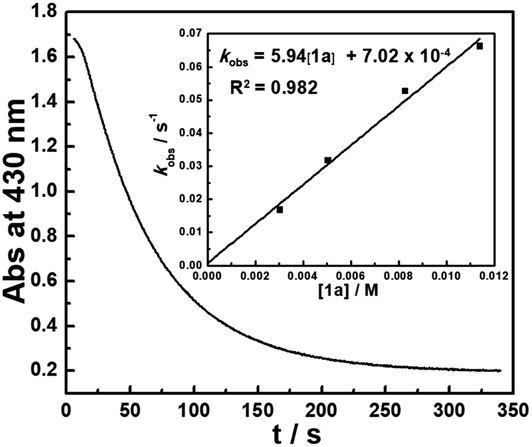
The kinetics of the hydride transfer from 1a to A1+ was conducted by following the decay of the absorption of A1+ in CH3CN at 430 nm with a stopped-flow spectrophotometer (Fig. 2, see the ESI† for details). The second-order rate constant kHT(1a) was found to be 5.94 M−1 s−1 (Inset in Fig. 2 and Table S1†). Moreover, the nucleophilicity of 1a can be estimated by the second-order rate constant of the reaction of 1a with A1+. Because 1a has a similar structure with 1b, based on the nucleophile-specific sensitivity parameter sN of 1b (0.52) and the electrophilicity of A1+ in our recent work,17 the nucleophilicity N, of 1a is estimated to be 8.64. Similarly, the kinetics of 1a-D with A1+kHT(1a-D) was determined to be 2.34 M−1 s−1 (Table S2†). These gave a primary kinetic isotope effect KIE [kHT(1a)/kHT(1a-D)] of 2.5. A comparison with the previously determined kHT(1b) of 5.41 × 102 M−1 s−1 for the reaction of 1b with A1+ in our earlier work17 indicates that 1b is about 100 times more reactive than 1a. This is exactly in accordance with the order of their hydricities (ΔGH−) of 1avs.1b found here.
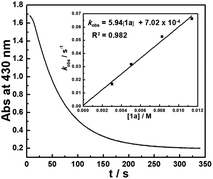 |
| | Fig. 2 Monoexponential decay of the absorbance Abs (at 430 nm) with the time t (s) for the reaction of 1a (3.03 × 10−3 M) with A1+ (5.00 × 10−5 M) in CH3CN at 20 °C. Inset: Correlation of kobs with [1a]. | |
Hydrogen atom transfer (HAT).
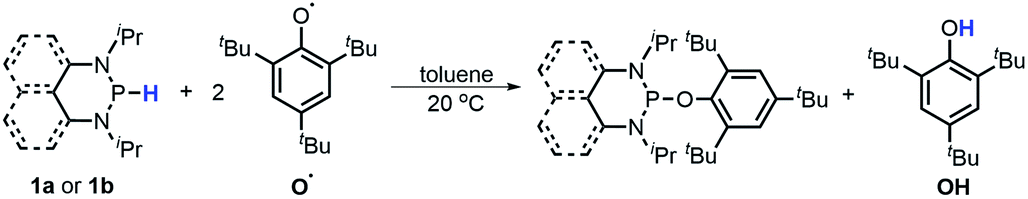
To examine the possibility of a HAT reaction from diazaphosphinane substrates 1, the reactions of 1a and 1b with the 2,4,6-tri-tert-butyl-phenoxyl radical O˙ were conducted, and yielded 2,4,6-tri-tert-butylphenol OH and the corresponding phosphorus species (eqn (6)) without generation of bisphosphines (compared to the reaction of 1a or 1b with AIBN, Table 1). A stoichiometric study found a [1]/[O˙] ratio of 1:2, consistent with the product analysis (Fig. S11 and S12†).| |  | (6) |
Table 1 Applications of 1a and 1b in organic syntheses
| Entry |
Reactant |
Condition |
Product |
Yield |
Type |
|
Isolated yield.
NMR yields determined by the amount of phosphorus species.
NMR yields with 1,3,5-trimethoxybenzene as the internal standard.
|
| (1) |
|
1a-[P]+ (15 mol%), HBpin (1.5 equiv.), CH3CN, 80 °C, 36 h |
|
40%a |
HT |
| (2) |
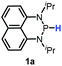
|
t
BuOK, CD3CN, 20 °C, 10 min |
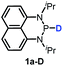
|
Quant.b |
PT |
| (3) |
|
AIBN (1.5 equiv.), C6D6, 80 °C, 3 h |
|
>90%b |
HAT |
| (4) |
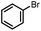
|
1b (1.5 equiv.), AIBN 15 mol%, toluene-d8, 90 °C, 5 h |
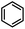
|
>90%c |
HAT&ET |
To evaluate the hydrogen atom donability of diazaphosphinanes (i.e., P–H BDFE), the thermodynamic cycle was applied on the basis of the ΔGH− and Ered([P]+) in hand (Scheme 5 and eqn (7); FEox(H−) is −26.0 kcal mol−11c in acetonitrile vs. Fc0/+).26 Substituting the known values into eqn (7)26a,27 gives P–H BDFEs of 80.9 ± 2.0 and 77.9 ± 2.0 kcal mol−1 for 1a and 1b, respectively, being comparable with those of phenol and thiophenol antioxidants. Therefore, they could transfer hydrogen atoms smoothly to acceptors with X–H BDFEs around or larger than 78 kcal mol−1, such as 2,4,6-tri-tert-butyl-phenoxyl O˙ (77.1 kcal mol−1), CN(CH3)2C˙ (about 88 kcal mol−1) and phenyl radicals (104.7 kcal mol−1).5b,28 Compared to 1b, though the fused aryl groups attenuate the hydricity of 1a by 13 kcal mol−1 thermodynamically, they show a negligible effect on the HAT reactivity.
| | | BDFE = ΔGH− − F(Ered([P]+) − Eox(H−)) = ΔGH− − 23.06Ered([P]+) − 26.0 (kcal mol−1) | (7) |
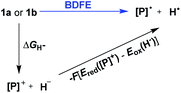 |
| | Scheme 5 Thermodynamic cycle for deriving the P–H BDFEs of 1a and 1b. | |
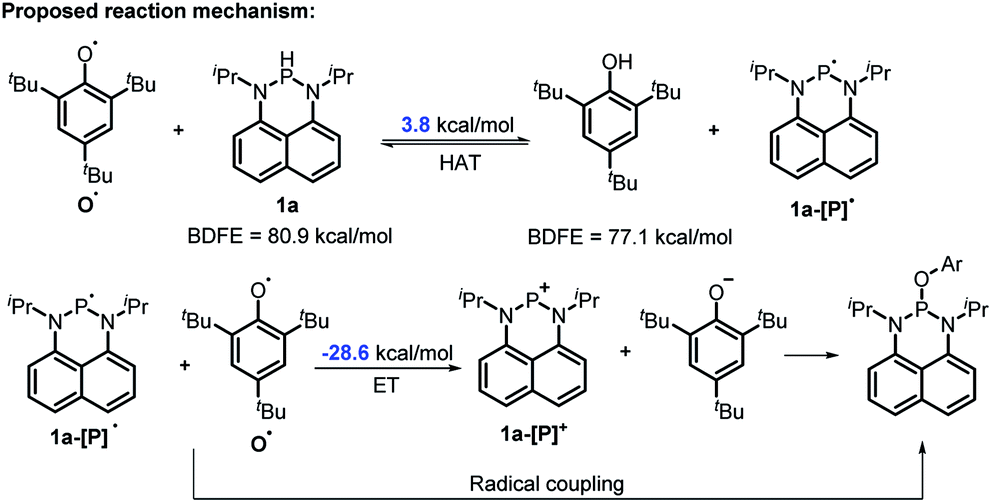
The reaction mechanism of 1a with radical O˙ (eqn (6)) is proposed in Scheme 6 based on the derived thermodynamic data (the reaction of 1b with O˙ is similar). Since hydrogen atom transfer from 1a to O˙ is slightly endothermic (∼4 kcal mol−1), it implies the possibility for a reversible HAT. This reversible HAT could be further driven to the far right by the largely exothermic electron transfer, ET of 1a-[P]˙ radical (Eox(1a-[P]˙) = −1.94 V) or a follow-up radical coupling. The observed stoichiometric ratio of 1:2 for the reactants [1] vs. [O˙] is in exact accordance with the HAT mechanism proposed. On the other hand, a suspected proton transfer between 1a and the O˙ radical can be easily ruled out due to the very weak acidity of 1a (vide infra) and strong acidity (pKa of ∼−3)5b reported for the phenol radical cations (eqn (8)). The other suspected possibility, i.e. the ET path between 1a and the O˙ radical (Ered = −0.70 V5b) can also be excluded by a similar strategy on the basis of their respective thermodynamics determined, that indicates that the ET from 1a to O˙ is remarkably uphill (ΔGET = 21.4 kcal mol−1, eqn (9)).13,29 It is thus safe to conclude that the initial HAT should be the rate-determining step (RDS) for the reaction expressed in eqn (6).
| |  | (8) |
| |  | (9) |
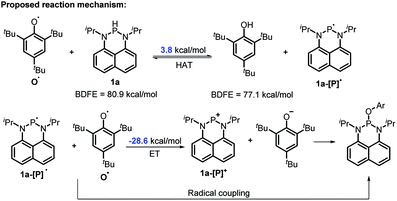 |
| | Scheme 6 The possible mechanism for the reaction of 1a with O˙. Ar = 2,4,6-tri-tert-butyl-phenyl group. | |
The rate constants of this hydrogen atom transfer from 1a and 1a-D to radical O˙ were measured to be a kHAT(1a) of 0.70 M−1 s−1 and a kHAT(1a-D) of 2.24 × 10−2 M−1 s−1, respectively (Tables S3 and S4†). This intermediately points out an extremely large KIE(1a) of 31.3, implying a tunneling effect in this HAT process. Such a large KIE has been found in biochemical30 and transition-metal systems,31 but seldom observed in organic HAT processes. Similarly, the rate of the analogous HAT of 1bkHAT(1b) was determined to be 0.74 M−1 s−1 (Table S5†). The quite close kHAT for 1a and 1b agrees well with their comparable P–H BDFEs (80.9 vs. 77.9 kcal mol−1, vide supra), verifying their similar HAT reactivity. However, the same kinetic study for 1b-D gave a kHAT(1b-D) of 0.12 M−1 s−1 (Table S6†), resulting in a primary KIE(1b) of 6.2 with no tunneling phenomenon detected.
To understand this discrepancy, temperature-dependent kinetics were further performed (Tables S7–S10†). The ratio of Arrhenius pre-factors [A(1a)/A(1a-D)] was obtained to be 0.021, and the activation energy difference [Ea(1a) − Ea(1a-D)] was −5.53 kcal mol−1 (Fig. S2 and Table S11†). These kinetic characteristics [A(1a)/A(1a-D) ≪ 1 and Ea(1a) − Ea(1a-D) ≪ 0] are in fact in line with the tunneling model illustrated by Klinman,30a which features a “through-the-barrier” transition state near the top of the classical barrier. In the 1b system, on the other hand, the normal primary KIE of 6.2, A(1b)/A(1b-D) of 3.39 and [Ea(1b) − Ea(1b-D)] of −0.38 kcal mol−1 all support a classical “over-the-barrier” transition state. A more comprehensive investigation of the tunneling issue is presently underway.
Proton transfer (PT).
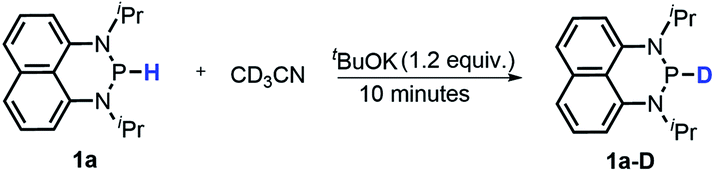
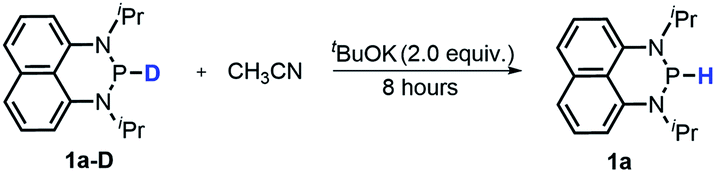
It's known that a good hydride donor usually exhibits poor acidity. This explains why the acidity of widely-used X–H type hydride donors, e.g. 2,3-dihydrobenzo[d]-imidazoles, has never been determined in the absence of metal-coordination. The hydricity of the present N-heterocyclic phosphines is also quite good (vide supra), hence an investigation of their acidic reactivity cannot be expected to be too straightforward. Nevertheless, as we learned from the initial failures in deprotonating 1a or 1b with some ordinary strong bases, such as 1,8-diazabicyclo[5,4,0]-7-undecene (DBU, pKa = 24.34 in CH3CN), 1,3,4,6,7,8-hexahydro-2H-pyrimido[1,2-a]pyrimidine (TBD, pKa = 26.03) and (tert-butylimino)tris(pyrrolidino)-phosphorane (BTPP, pKa = 28.42)28 (see the ESI† for details), we finally found that the H/D exchange of the 1a P–H proton in the presence of stoichiometric tBuOK in CD3CN (eqn (10) and Fig. S13†) was accomplished in about 10 minutes at room temperature,32 whereas the same operation for 1a-D in CH3CN was finished in 8 hours at room temperature (eqn (11) and Fig. S14†). The reversible H/D exchange of 1a in acetonitrile under the promotion of tBuOK suggested that the acidity of 1a reaches the measurable acidity limitation in acetonitrile (see the ESI† for details).33 As for 1b with much stronger hydricity, its acidity is, unfortunately, too weak to examine in solution by any existing experiment. However, it is worth noting that the acidity of a P–H bond can be significantly enhanced upon activation by coordination with transition metals34 or other Lewis acids.35| | 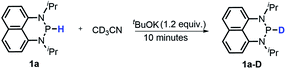 | (10) |
| | 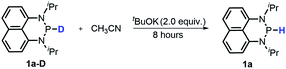 | (11) |
Applications in syntheses
Based on the above thermodynamic and kinetic investigations, the potential of applying 1a and 1b in organic syntheses, encompassing the diverse hydrogen (H+, H˙ and H−) or electron transfer, was preliminarily explored and showcased herein.
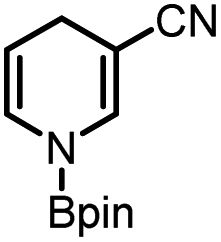
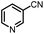
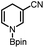
Catalytic reduction of pyridines is a prevailing strategy for the synthesis of dihydropyridines. Recently, Kinjo et al.16gand Speed et al.36 exploited 1,3,2-diazaphosphenium-catalyzed hydroboration of pyridines with good regio- and chemo-selectivity.16g However, in their system, the substrate 3-CN-pyridine gave a mixture of three dihydropyridine isomers. To our delight, we found that under the catalysis of 15 mol% of our 1a-[P]+,37 the sole 1,4-hydroborated isomer of this reaction was selectively produced in a moderate yield of 40% (entry 1 in Table 1 and Fig. S15†). The good regio-selectivity may be ascribed to the steric factors of the isopropyl group on the nitrogen atom of 1a based on Kinjo's DFT calculations.16g This good regio-selectivity exhibits the catalytic potential of 1a in the reduction of unsaturated compounds.
Moreover, the acidic character of 1a (vide supra) offers a more economic method for the synthesis of deuterated reagent 1a-D (entry 2 in Table 1) by employing the much cheaper deuterated solvent CD3CN instead of expensive LiAlD4 in an almost quantitative yield (see the ESI† for details about the synthetic method). Meanwhile, it also provides a new approach for isotope labeling of many other hydridic species. The generation of the hydridic hydrogen from a “protic” solvent as illustrated here may serve as an example of Umpolung which was also proposed by Gudat et al. for the P–H bond.15a
Since bisphosphines can easily decompose into phosphinyl radicals,38 they could serve as radical reservoirs for relevant studies and applications. Based on thermodynamic analysis, CN(CH3)2C˙ (C–H BDFE ≈ 85 kcal mol−1), generated from the homolysis of azo-bis-isobutyronitrile (AIBN), could abstract the hydrogen atoms from 1a (P–H BDFE = 80.9 kcal mol−1). Then, the generated phosphinyl radical 1a-[P]˙ coupled quickly with each other to yield bisphosphines (entry 3 in Table 1, Fig. S16† for 1a and Fig. S17† for 1b). The targeted dimers39 are readily extracted from reaction mixtures in a high yield. This transformation, together with Gudat's photochemical dehydrocoupling,18 offers easy access to bisphosphines, avoiding complicated and harsh operations and workups required in other synthetic processes using metals such as sodium38d and magnesium38a–c.
Furthermore, we attempted to design a new radical transformation by an integrated use of hydrogen atoms and electron transfers from N-heterocyclic phosphines. This was inspired by the reactions of AIBN with 1a and 1b, where phosphinyl radicals can be easily generated from their P–H precursors through hydrogen atom release. The highly negative oxidation potentials of phosphinyl radicals imply their competency in the reduction of challenging substrates. If a substrate in the reaction system is sufficiently oxidative, its single-electron capture from phosphinyl radicals may be able to compete with radical coupling. In such cases, subsequent transformations may be triggered. Based on the redox potentials of 1b-[P]˙ (Eox = −2.39 V vs. Fc+/0, the same with the Ered(1b-[P]+)) and bromobenzene (Ered = −2.8 V),40 hydrodehalogenation of bromobenzene by 1b was performed in toluene with 15 mol% AIBN as the initiator (entry 4 in Table 1, Fig. S18†). After 5 hours, the expected reduction product, benzene was obtained in over 90% yield. Due to the poor reduction ability of 1a-[P]˙, the reaction could not proceed when 1a was used.41 Therefore, it could be anticipated that these N-heterocyclic phosphines may open up a promising avenue for the development of super electron donors.
Conclusions
Hydrogen and electron transfers from phosphines 1a and 1b were thermodynamically and kinetically investigated in acetonitrile. The P–H bond of 1a can participate in reactions involving formal transfer of a hydride/hydrogen atom/proton to a substrate under respective conditions in the absence of the usually required transition-metal- or Lewis acid-mediation. It was observed that 1a is a moderate hydride and hydrogen atom donor, but a poor Brønsted acid. In comparison, 1b is a good hydride donor and a moderate hydrogen atom donor; but did not show a detectable acidic reactivity even under the strongest basic solution. With regard to electron donability, 1a (Eox(1a) = 0.23 V) is superior to 1b (Eox(1b) = 0.47 V), contrasting with the order of their hydridic reactivity. Their corresponding phosphinyl radicals are very strong electron donors as reflected by their extremely negative redox potentials. Kinetic studies revealed a tunneling contribution (KIE = 31.3) to the hydrogen atom transfer from 1a to the phenoxy radical. Based on the derived energetic parameters, their synthetic applications were tentatively exploited. Preliminary results from the synthesis attempts by using the new reagents 1a and 1b confirmed their competence as effective hydrogen and electron donors in various redox transformations. The effectiveness of applying the relevant physicochemical parameters in the analysis and design of hydrogen transfer reactions was also shown.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
We are grateful for the financial grants from the National Natural Science Foundation of China (No. 21973052, 21933008, 21602116, and 91745101), the National Science & Technology Fundamental Resource Investigation Program of China (No. 2018FY201200), and the Tsinghua University Initiative Scientific Research Program (No. 20181080083).
Notes and references
-
(a) J.-D. Yang, J. Xue and J.-P. Cheng, Chem. Soc. Rev., 2019, 48, 2913–2926 RSC;
(b) S. Ilic, A. Alherz, C. B. Musgrave and K. D. Glusac, Chem. Soc. Rev., 2018, 47, 2809–2836 RSC;
(c) E. S. Wiedner, M. B. Chambers, C. L. Pitman, R. M. Bullock, A. J. M. Miller and A. M. Appel, Chem. Rev., 2016, 116, 8655–8692 CrossRef CAS PubMed.
-
(a) A. Wu and J. M. Mayer, J. Am. Chem. Soc., 2008, 130, 14745–14754 CrossRef CAS PubMed;
(b) T. Matsuo and J. M. Mayer, Inorg. Chem., 2005, 44, 2150–2158 CrossRef CAS PubMed;
(c) J. M. Mayer, Acc. Chem. Res., 2011, 44, 36–46 CrossRef CAS PubMed.
-
(a) Z. Wang, F. Gao, P. Ji and J.-P. Cheng, Chem. Sci., 2018, 9, 3538–3543 RSC;
(b) K. Kaupmees, N. Tolstoluzhsky, S. Raja, M. Rueping and I. Leito, Angew. Chem., Int. Ed., 2013, 52, 11569–11572 CrossRef CAS PubMed;
(c) J.-D. Yang, P. Ji, X.-S. Xue and J.-P. Cheng, J. Am. Chem. Soc., 2018, 140, 8611–8623 CrossRef CAS PubMed.
-
(a) S. Ilic, U. P. Kadel, Y. Basdogan, J. A. Keith and K. D. Glusac, J. Am. Chem. Soc., 2018, 140, 4569–4579 CrossRef CAS PubMed;
(b) X.-Q. Zhu, M.-T. Zhang, A. Yu, C.-H. Wang and J.-P. Cheng, J. Am. Chem. Soc., 2008, 130, 2501–2516 CrossRef CAS PubMed.
-
(a) M. J. Bezdek and P. J. Chirik, Angew. Chem., Int. Ed., 2018, 57, 2224–2228 CrossRef CAS PubMed;
(b) J. J. Warren, T. A. Tronic and J. M. Mayer, Chem. Rev., 2010, 110, 6961–7001 CrossRef CAS PubMed;
(c) G. Ali, P. E. VanNatta, D. A. Ramirez, K. M. Light and M. T. Kieber-Emmons, J. Am. Chem. Soc., 2017, 139, 18448–18451 CrossRef CAS PubMed.
-
(a) Z. Li, X. Li and J.-P. Cheng, J. Org. Chem., 2017, 82, 9675–9681 CrossRef CAS PubMed;
(b) C. Mao, Z. Wang, Z. Wang, P. Ji and J.-P. Cheng, J. Am. Chem. Soc., 2016, 138, 5523–5526 CrossRef CAS PubMed.
-
(a) D. Richter and H. Mayr, Angew. Chem., Int. Ed., 2009, 48, 1958–1961 CrossRef CAS PubMed;
(b) V. Morozova, P. Mayer and G. Berionni, Angew. Chem., Int. Ed., 2015, 54, 14508–14512 CrossRef CAS PubMed;
(c) M. Horn, L. H. Schappele, G. Lang-Wittkowski, H. Mayr and A. R. Ofial, Chem.–Eur. J., 2013, 19, 249–263 CrossRef CAS PubMed;
(d) H. Mayr, G. Lang and A. R. Ofial, J. Am. Chem. Soc., 2002, 124, 4076–4083 CrossRef CAS PubMed.
-
(a) R. H. Morris, Chem. Rev., 2016, 116, 8588–8654 CrossRef CAS PubMed;
(b) M. E. Ahmed, S. Chattopadhyay, L. Wang, D. Brazzolotto, D. Pramanik, D. Aldakov, J. Fize, A. Morozan, M. Gennari, C. Duboc, A. Dey and V. Artero, Angew. Chem., Int. Ed., 2018, 57, 16001–16004 CrossRef CAS PubMed.
-
(a) R. Zhong, Z. Wei, W. Zhang, S. Liu and Q. Liu, Chem, 2019, 5, 1552–1566 CrossRef CAS;
(b) W. Ai, R. Zhong, X. Liu and Q. Liu, Chem. Rev., 2019, 119, 2876–2953 CrossRef CAS PubMed.
-
(a) Y. Zu, R. J. Shannon and J. Hirst, J. Am. Chem. Soc., 2003, 125, 6020–6021 CrossRef CAS PubMed;
(b) A. Bucci, S. Dunn, G. Bellachioma, G. M. Rodriguez, C. Zuccaccia, C. Nervi and A. Macchioni, ACS Catal., 2017, 7, 7788–7796 CrossRef CAS.
-
(a) Q.-A. Chen, M.-W. Chen, C.-B. Yu, L. Shi, D.-S. Wang, Y. Yang and Y.-G. Zhou, J. Am. Chem. Soc., 2011, 133, 16432–16435 CrossRef CAS PubMed;
(b) V. N. Wakchaure, P. S. J. Kaib, M. Leutzsch and B. List, Angew. Chem., Int. Ed., 2015, 54, 11852–11856 CrossRef CAS PubMed.
-
(a) D. E. Berning, B. C. Noll and D. L. DuBois, J. Am. Chem. Soc., 1999, 121, 11432–11447 CrossRef CAS;
(b) J. R. Norton and J. Sowa, Chem. Rev., 2016, 116, 8315–8317 CrossRef PubMed.
- E. A. McLoughlin, K. M. Waldie, S. Ramakrishnan and R. M. Waymouth, J. Am. Chem. Soc., 2018, 140, 13233–13241 CrossRef CAS PubMed.
-
(a) W. S. Matthews, J. E. Bares, J. E. Bartmess, F. G. Bordwell, F. J. Cornforth, G. E. Drucker, Z. Margolin, R. J. McCallum, G. J. McCollum and N. R. Vanier, J. Am. Chem. Soc., 1975, 97, 7006–7014 CrossRef CAS;
(b) X.-M. Zhang, J. W. Bruno and E. Enyinnaya, J. Org. Chem., 1998, 63, 4671–4678 CrossRef CAS;
(c) M. Finn, R. Friedline, N. K. Suleman, C. J. Wohl and J. M. Tanko, J. Am. Chem. Soc., 2004, 126, 7578–7584 CrossRef CAS PubMed;
(d) K. Xia, G.-B. Shen and X.-Q. Zhu, Org. Biomol. Chem., 2015, 13, 6255–6268 RSC.
-
(a) D. Gudat, A. Haghverdi and M. Nieger, Angew. Chem., Int. Ed., 2000, 39, 3084–3086 CrossRef CAS;
(b) S. Burck, D. Gudat, M. Nieger and W.-W. Du Mont, J. Am. Chem. Soc., 2006, 128, 3946–3955 CrossRef CAS PubMed.
-
(a) C. C. Chong, B. Rao and R. Kinjo, ACS Catal., 2017, 7, 5814–5819 CrossRef CAS;
(b) M. R. Adams, C. H. Tien, B. S. N. Huchenski, M. J. Ferguson and A. W. H. Speed, Angew. Chem., Int. Ed., 2017, 56, 6268–6271 CrossRef CAS PubMed;
(c) C. C. Chong, H. Hirao and R. Kinjo, Angew. Chem., Int. Ed., 2014, 53, 3342–3346 CrossRef CAS PubMed;
(d) C. C. Chong, H. Hirao and R. Kinjo, Angew. Chem., Int. Ed., 2015, 54, 190–194 CrossRef CAS PubMed;
(e) C. C. Chong and R. Kinjo, Angew. Chem., Int. Ed., 2015, 54, 12116–12120 CrossRef CAS PubMed;
(f) M. R. Adams, C. T. Tien, R. McDonald and A. W. H. Speed, Angew. Chem., Int. Ed., 2017, 56, 16660–16663 CrossRef CAS PubMed;
(g) B. Rao, C. C. Chong and R. Kinjo, J. Am. Chem. Soc., 2018, 140, 652–656 CrossRef CAS PubMed;
(h) T. Lundrigan, E. N. Welsh, T. Hynes, C.-H. Tien, M. R. Adams, K. R. Roy, K. N. Robertson and A. W. H. Speed, J. Am. Chem. Soc., 2019, 141, 14083–14088 CrossRef CAS PubMed.
- J. Zhang, J.-D. Yang and J.-P. Cheng, Angew. Chem., Int. Ed., 2019, 58, 5983–5987 CrossRef CAS PubMed.
- O. Puntigam, L. Könczöl, L. Nyulászi and D. Gudat, Angew. Chem., Int. Ed., 2015, 54, 11567–11571 CrossRef CAS PubMed.
- R. B. King and P. M. Sundaram, J. Org. Chem., 1984, 49, 1784–1789 CrossRef CAS.
- A single crystal of 1a was obtained by volatilization of solution of 1a in hexane at −30 °C. Due to the air- and moisture- sensitivity of this compound, the single crystal could not be fine sealed when X-ray analyzing in the cool air and the minor compound 1a (about 10%) was oxidized. And in case of the 10% by product, the obtained crystals are of insufficient quality despite several attempts. Thus the quality of the acquired diffraction data was slightly below the average level (see the attached CIF file for details). Despite this, the present X-ray diffraction data confirmed the chemical structure of 1a unambiguously.
- H. A. Spinney, I. Korobkov, G. A. DiLabio, G. P. A. Yap and D. S. Richeson, Organometallics, 2007, 26, 4972–4982 CrossRef CAS.
-
(a) S. S. Hanson, E. Doni, K. T. Traboulsee, G. Coulthard, J. A. Murphy and C. A. Dyker, Angew. Chem., Int. Ed., 2015, 54, 11236–11239 CrossRef CAS PubMed;
(b) J. A. Murphy, T. A. Khan, S.-z. Zhou, D. W. Thomson and M. Mahesh, Angew. Chem., Int. Ed., 2005, 44, 1356–1360 CrossRef CAS PubMed;
(c) J. A. Murphy, S.-z. Zhou, D. W. Thomson, F. Schoenebeck, M. Mahesh, S. R. Park, T. Tuttle and L. E. A. Berlouis, Angew. Chem., Int. Ed., 2007, 46, 5178–5183 CrossRef CAS PubMed;
(d) L. Zhang and L. Jiao, Chem. Sci., 2018, 9, 2711–2722 RSC.
-
(a) S. Hoffmann, A. M. Seayad and B. List, Angew. Chem., Int. Ed., 2005, 44, 7424–7427 CrossRef CAS PubMed;
(b) R. I. Storer, D. E. Carrera, Y. Ni and D. W. C. MacMillan, J. Am. Chem. Soc., 2006, 128, 84–86 CrossRef CAS PubMed;
(c) Q.-A. Chen, K. Gao, Y. Duan, Z.-S. Ye, L. Shi, Y. Yang and Y.-G. Zhou, J. Am. Chem. Soc., 2012, 134, 2442–2448 CrossRef CAS PubMed.
- C. Zhu, K. Saito, M. Yamanaka and T. Akiyama, Acc. Chem. Res., 2015, 48, 388–398 CrossRef CAS PubMed.
-
(a) R. P. Thummel, W. E. Cravey and D. B. Cantu, J. Org. Chem., 1980, 45, 1633–1637 CrossRef CAS;
(b) I. Chatterjee and M. Oestreich, Org. Lett., 2016, 18, 2463–2466 CrossRef CAS PubMed.
-
(a) V. D. Parker, K. L. Handoo, F. Roness and M. Tilset, J. Am. Chem. Soc., 1991, 113, 7493–7498 CrossRef CAS;
(b) M. Tilset and V. D. Parker, J. Am. Chem. Soc., 1989, 111, 6711–6717 CrossRef CAS.
- F. G. Bordwell, J.-P. Cheng and J. A. Harrelson, J. Am. Chem. Soc., 1988, 110, 1229–1231 CrossRef CAS.
-
J.-P. Cheng, et al., Internet Bond-energy Databank (iBonD) Home Page, http://ibond.nankai.edu.cn, accessed at Oct. 2019 Search PubMed.
-
(a) The energetics of the electron transfer (ET) from 1a (Eox = 0.23 V) to the O˙ radical (Ered = −0.70 V) shows that its thermodynamic driving force is remarkably uphill (ΔGET = 21.4 kcal mol−1), rendering its equilibrium constant close to 0. Such an unfavorable thermodynamic driving force prohibits electron transfer to be a feasible path;
(b) J.-P. Cheng, Y. Lu, X.-Q. Zhu and L. Mu, J. Org. Chem., 1998, 63, 6108–6114 CrossRef CAS PubMed.
-
(a) J. P. Klinman and A. R. Offenbacher, Acc. Chem. Res., 2018, 51, 1966–1974 CrossRef CAS PubMed;
(b) J. C. Price, E. W. Barr, T. E. Glass, C. Krebs and J. M. Bollinger, J. Am. Chem. Soc., 2003, 125, 13008–13009 CrossRef CAS PubMed;
(c) J. P. Layfield and S. Hammes-Schiffer, Chem. Rev., 2014, 114, 3466–3494 CrossRef CAS PubMed;
(d) M. A. Ehudin, D. A. Quist and K. D. Karlin, J. Am. Chem. Soc., 2019, 141, 12558–12569 CrossRef CAS PubMed.
-
(a) E. J. Klinker, S. Shaik, H. Hirao and L. Que Jr, Angew. Chem., Int. Ed., 2009, 48, 1291–1295 CrossRef CAS PubMed;
(b) S. H. Bae, X.-X. Li, M. S. Seo, Y.-M. Lee, S. Fukuzumi and W. Nam, J. Am. Chem. Soc., 2019, 141, 7675–7679 CrossRef CAS PubMed.
- When the same reaction was performed in MeOH and toluene solutions (with other conditions identical), none of the H/D exchange was observed.
- A. Kütt, S. Selberg, I. Kaljurand, S. Tshepelevitsh, A. Heering, A. Darnell, K. Kaupmees, M. Piirsalu and I. Leito, Tetrahedron Lett., 2018, 59, 3738–3748 CrossRef.
-
(a) J.-J. Brunet, R. Chauvin, O. Diallo, B. Donnadieu, J. Jaffart and D. Neibecker, J. Organomet. Chem., 1998, 570, 195–200 CrossRef CAS;
(b) P. K. Majhi, A. W. Kyri, A. Schmer, G. Schnakenburg and R. Streubel, Chem.–Eur. J., 2016, 22, 15413–15419 CrossRef CAS PubMed.
-
(a) A. Longeau and P. Knochel, Tetrahedron Lett., 1996, 37, 6099–6102 CrossRef CAS;
(b) M. Blum, J. Kappler, S. H. Schlindwein, M. Nieger and D. Gudat, Dalton Trans., 2018, 47, 112–119 RSC.
- T. Hynes, E. N. Welsh, R. McDonald, M. J. Ferguson and A. W. H. Speed, Organometallics, 2018, 37, 841–844 CrossRef CAS.
- Although phosphenium triflate 1a-[P]+ (as hydride acceptor) is used as a catalyst in the hydride transfer reaction (entry 1 in Table 1), generation of P–H 1a from 1a-[P]+ is the initial step of the catalytic cycle. Then, the pyridine is reduced by in situ generated 1a. The hydride donor 1a and acceptor 1a-[P]+ coexist in the reaction system. Their hydride-donating/accepting abilities (hydricity) are crucial in the catalytic cycle.
-
(a) O. Puntigam, D. Förster, N. A. Giffin, S. Burck, J. Bender, F. Ehret, A. D. Hendsbee, M. Nieger, J. D. Masuda and D. Gudat, Eur. J. Inorg. Chem., 2013, 2013, 2041–2050 CrossRef CAS;
(b) R. Edge, R. J. Less, E. J. L. McInnes, K. Müther, V. Naseri, J. M. Rawson and D. S. Wright, Chem. Commun., 2009, 1691–1693 RSC;
(c) D. Förster, H. Dilger, F. Ehret, M. Nieger and D. Gudat, Eur. J. Inorg. Chem., 2012, 2012, 3989–3994 CrossRef;
(d) N. A. Giffin, A. D. Hendsbee, T. L. Roemmele, M. D. Lumsden, C. C. Pye and J. D. Masuda, Inorg. Chem., 2012, 51, 11837–11850 CrossRef CAS PubMed;
(e) J.-P. Bezombes, K. B. Borisenko, P. B. Hitchcock, M. F. Lappert, J. E. Nycz, D. W. H. Rankin and H. E. Robertson, Dalton Trans., 2004, 1980–1988 RSC;
(f) A. Dumitrescu, V. L. Rudzevich, V. D. Romanenko, A. Mari, W. W. Schoeller, D. Bourissou and G. Bertrand, Inorg. Chem., 2004, 43, 6546–6548 CrossRef CAS PubMed;
(g) M. Blum, O. Puntigam, S. Plebst, F. Ehret, J. Bender, M. Nieger and D. Gudat, Dalton Trans., 2016, 45, 1987–1997 RSC.
- E. E. Nifantiev, N. S. Vyazankin, S. F. Sorokina, L. A. Vorobieva, O. A. Vyazankina, D. A. Bravo-Zhivotovsky and A. R. Bekker, J. Organomet. Chem., 1984, 277, 211–225 CrossRef.
-
(a) C. Costentin, M. Robert and J.-M. Savéant, J. Am. Chem. Soc., 2004, 126, 16051–16057 CrossRef CAS PubMed;
(b) L. Pause, M. Robert and J.-M. Savéant, J. Am. Chem. Soc., 1999, 121, 7158–7159 CrossRef CAS.
- DFT calculations showed that 1a-[P]˙ and 1b-[P]˙ should have a comparable ability (with an energy difference of 1.3 kcal mol−1, see the ESI† for details) in abstracting the bromine atom. This failed to explain the disparate yields of <10% for 1a-[P]˙ and 90% for 1b-[P]˙. Besides, according to the redox potentials of 1b-[P]˙ (Eox = −2.39 V vs. Fc in MeCN) and bromobenzene (Ered = −2.8 V), the electron transfer from 1b-[P]˙ to bromobenzene is a feasible reversible process, while that for 1a-[P]˙ (Eox = −1.94 V) is thermodynamically prohibited. These findings preferentially support an ET-initiated mechanism rather than a direct bromine abstraction.
Footnote |
| † Electronic supplementary information (ESI) available: Details of syntheses and experimental determinations. CCDC 1947293. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c9sc05883d |
|
| This journal is © The Royal Society of Chemistry 2020 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a  *a and
Jin-Pei
Cheng
*ab
*a and
Jin-Pei
Cheng
*ab