
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceUniform nanoporous graphene sponge from natural polysaccharides as a metal-free electrocatalyst for hydrogen generation†
Jinan
Niu
 *ab,
Antonio
Domenech-Carbó
c,
Ana
Primo
a and
Hermenegildo
Garcia
*a
*ab,
Antonio
Domenech-Carbó
c,
Ana
Primo
a and
Hermenegildo
Garcia
*a
aInstituto de Tecnologia Quimica CSIC-UPV, Universitat Politecnica de Valencia, Valencia 46022, Spain. E-mail: hgarcia@qim.upv.es
bSchool of Materials Science and Engineering, China University of Mining and Technology, Xuzhou 221116, P. R. China. E-mail: jinan.niu@cumt.edu.cn
cDepartment of Analytical Chemistry, Faculty of Chemistry, Universitat de Valencia, Av. Doctor Moliner 100, Burjassot, Spain
First published on 21st December 2018
Abstract
Structuring of graphene as graphene sponges in the submicrometric scale has been achieved by using silica spheres (80 nm diameter) as hard templates and chitosan or alginate as precursor of defective N-doped or undoped graphene, respectively. The resulting defective N-doped graphene sponge exhibits a remarkable activity and stability for hydrogen evolution reaction with onset at 203 mV for a current density of 0.5 mA cm−2 with a small Tafel plot slope of 69.7 mV dec−1. In addition, the graphene sponge also exhibits a high double layer capacitance of 11.65 mF cm−2. Comparison with an analogous N-doped graphene sample shows that this electrochemical properties derive from the spatial structuring and large surface area.
Three-dimensional (3D) graphenes have attracted considerable attention due to their unique structure with connected inner pores with graphene walls combining a series of excellent properties such as high surface area, light weight, accessibility of active sites, high mass transfer diffusion coefficients, and adjustable mechanical, thermal and electrical properties by controlling wall thickness and the presence of dopant elements.1,2 Noticeable efforts have been devoted to the development of synthetic procedures for the preparation of 3D graphenes, which can be generally divided into “template-based” and “template-free” approaches.3 Compared to “template-free” protocols, the template-based method can achieve a precise control of pore size and shape of the 3D graphene particles by inverse replication of the hard template.
Chemical vapor deposition (CVD) and graphene self-assembly in solution (GSAS) are two of the most widely used template-based methods to obtain 3D graphenes. CVD methods are based on the in-site graphene synthesis and they can produce 3D graphene on ordered template films with low density of defects and for relatively large areas. However, CVD methods are time-consuming and they are not appropriate for large-scale production. In addition, CVD preparation procedures have only been reported for large pore size from hundreds of nanometers to hundreds of micrometers and the synthesis requires expensive equipment.3
GSAS procedures rely on the assembly of graphene oxide sheets in 3D templates and, although they are suitable for the large-scale production, the resulting material after reduction produces a porous defective graphene. Besides, the common use of graphene oxide as starting material in the GSAS approach4 has the drawback of the prior synthesis of graphene oxide with the associated problems of the use of large volumes of sulfuric acid and permanganate reagents with unacceptable environmental impact.
The templates used in the above two methods include anodic aluminum oxide,5 magnesium oxide,6 nickel coated pyrolyzed photoresist films,7 metallic salts,8 silica spheres,9 polystyrene (PS) spheres,10 melamine foam,11 cellulose12 and natural cotton.13 Among them, silica spheres have been used in both CVD and GSAS procedures to create uniform porous 3D graphene because of the precise control of their size and their high thermal stability. 3D graphene films with uniform pores would hold some unique optical properties similar to photonic crystals and they can also serve to determine the influence of 3D structuring and porosity on the performance of graphene.
In view of the current state of the art regarding the preparation of 3D graphenes, a strategy consisting in the infiltration into template matrix of a solution containing a precursor compound and subsequent conversion of the precursor into graphene, resulting into the in-site formation of graphene can be a convenient alternative to obtain large areas of 3D graphenes with uniform porous structure at competitive costs.
Jang et al. synthesized 3D graphene by applying such infiltration-pyrolysis strategy using polyvinyl alcohol (PVA)–FeCl3 as precursor.14 The presence of FeCl3 was found necessary, since this salt becomes converted into Fe catalyst that promotes the subsequent growth of graphene. Jang's results prove the feasibility of the infiltration-pyrolysis strategy to produce 3D graphene with uniform pores. In addition, Zakhidov et al. following an analogous strategy used phenolic resin as carbon source to obtain amorphous glassy carbon;15 and Stein et al. used phenol–formaldehyde (PF) or resorcinol–formaldehyde (RF) precursors to make 3D ordered mesoporous carbon.16 The resulting materials in the last two studies were denoted as “carbons” due to their low graphitization degree. However, these researches confirm the possibility to perform a good infiltration of the colloid silica template matrix by polymers to form 3D uniform porous materials.
In this context, the present study presents a convenient procedure for the preparation of large areas of 3D defective graphene sponges (GS) based on the use of natural polysaccharides widely available in biomass wastes.
The low-cost natural polysaccharides such as chitosan and alginate have been reported previously as precursors for the synthesis of defective and doped graphenes as well as graphene heterojunctions by our group.17–22 These biopolymers exhibit filmogeneicity, a property consisting in their ability to form defect-free films of subnanometric roughness on arbitrary substrates that are conformal with the support surface. In view of this filmogenic property, it occurred to us that these polysaccharides could be used as well to form 3D GS with uniform pores by using silica templates. It was anticipated that these biopolymers should be able to infiltrate through silica template adopting the shape of the voids, and subsequent pyrolysis of these biopolymers would afford the corresponding undoped or doped defective graphene with a 3D structure.
Due to their large specific surface area, an obvious application of these defective doped 3D GS could be as metal-free electrocatalysts for hydrogen evolution reaction (HER), an electrochemical process of increasing attention in recent decades. Due to low cost and good corrosion resistance, carbon-based materials have become possible alternative HER catalysts to replace traditional Pt-based catalysts. Pristine graphene and some other carbon materials exhibit high electrical conductivity, but they are electrochemically inert. It has been shown that non-metal doping, particularly by N, and incorporation of transition metal nanoparticles are two general strategies to introduce electrocatalytic activity for HER in carbons.23 In addition, complementary strategies to further improve the catalytic activity of doped or metal-containing graphene are based on the increase on the electrochemical surface area of these materials.24 However, so far there have not been studies reporting the electrocatalytic performance of 3D porous graphene prepared with uniform colloid silica spheres.
In this work, the 3D N-doped GS with uniform nanopores was successfully obtained from biomass wastes acting as simultaneous source of carbon and nitrogen, using monodisperse colloid silica spheres (∼100 nm diameter) as template. Fig. 1 illustrates the procedure followed to prepare porous GS from chitosan to produce N-doped defective graphene (GS-CHI). Briefly, infiltration of chitosan solution into close packed silica spheres under reduced pressure results in the almost complete incorporation of the chitosan in the template. Subsequently, the mixture was pyrolyzed at 900 °C under argon atmosphere for 2 h. Then, the silica spheres were selectively etched using hot alkaline solution, as previously reported in the literature.8 Due to the low degree of silica crosslinking for those silica spheres prepared by the Stöber method, these spheres are much easier to dissolve than other types of silicas and can be attacked by NaOH. After washing and drying, the uniform porous GS-CHI was finally obtained. Precedents in the literature have shown that pyrolysis of chitosan at this temperature gives defective graphene samples with a N content about 4.7%.19,20 All the details of the synthesis can be found in the Experimental section. The protocol adopted here has two obvious merits: one is that the properties associated with pore structure, such as surface area, density, thermal and electrical conductivities can be precisely controlled by adjusting template sphere size in a wide dimension range from ten nanometers to several micrometers. The second one is that the in-site graphitization of polysaccharide in the voids of template considerably simplifies the procedure, making unnecessary the use of supernumerary pre-synthesized graphene or to perform subsequent N doping.4,25 The graphene obtained by this method shows much higher specific surface area (366 m2 g−1) than the nonporous reference graphene made from chitosan without template (0.155 m2 g−1); at the same time, it maintains the continuity between different parts of porous N-doped graphene by inheriting the framework formed in filming process of polysaccharide solution. These advantages plus the special properties already determined for defective graphene make the bare GS-CHI material herein exhibiting an excellent electrocatalytic HER activity, as it will be described in the sequel.
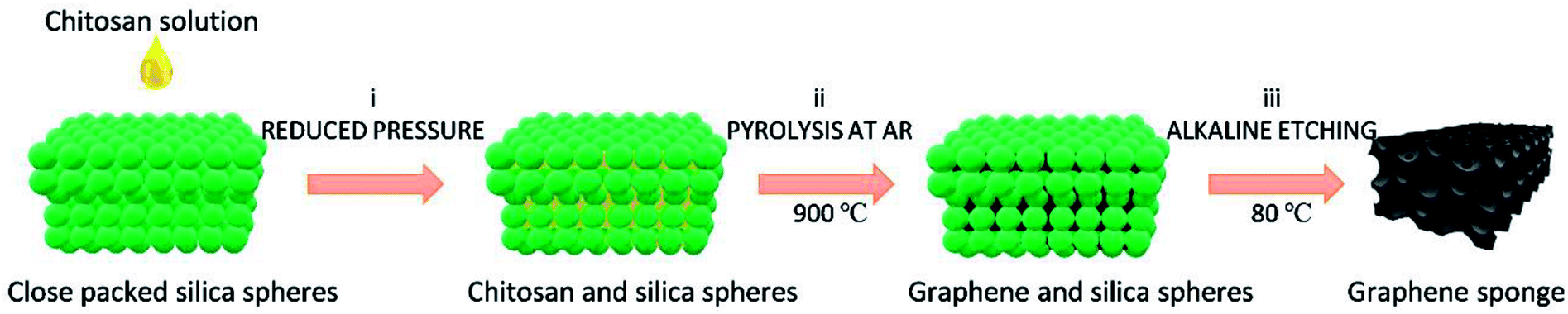 | ||
| Fig. 1 Synthesis procedure of GS using chitosan as starting carbon source. (i) infiltration under reduced pressure; (ii) pyrolysis at 900 °C under Ar; (iii) silica template removal. | ||
The scanning electron microscopy (SEM) images of GS-CHI and nonporous graphene made from chitosan (G-CHI) are compared in Fig. 2. Images of the silica spheres are presented in the ESI (Fig. S1†). In the absence of silica spheres, G-CHI shows the stacking of irregular sheets with various thicknesses (Fig. 2a and S2†). In some regions of the GS-CHI properly focused by electrons, the thin layers can be clearly seen, confirming the successful planarization of chitosan molecules during the pyrolysis (Fig. 2a and b). With silica template, the obtained GS-CHI shows a charming uniform porous structure with a pore size commensurate to the template spheres providing a large surface area (Fig. 2c, d and S3†). As it can be seen in Fig. 2d, the sample exhibiting regular porosity. The precise replica of the original silica template indicates the adequate strength and flexibility of these walls to survive the removal of the silica spheres upon treatment with hot alkaline aqueous solution under intense magnetic stirring. Moreover, the cavities present in GS-CHI are accessible through windows of several nanometers, indicating the interconnected structure of the pores inherited from the geometry of the close-packed silica spheres (inset of Fig. 2d), which can be extremely beneficial for the mass transport of reactive species required for chemical and electrochemical reactions.
 | ||
| Fig. 2 SEM images of nonporous G-CHI (a and b), and porous GS-CHI (c and d). (b) and (d) are the magnification of the rectangular locals in (a) and (c), respectively. | ||
Transmission electron microscopy (TEM) images of GS-CHI particles (Fig. 3a) also prove the presence of regular and uniform pores, evenly distributed in the sample. A high resolution TEM (HRTEM) image shows that the pore wall is composed of less than ten layers graphene with a spacing of about 0.4 nm (Fig. 3b). According to the accepted terminology, these configuration corresponds to few-layers of N-doped defective graphene.20 Many dislocation defects can be also seen from Fig. 3b, which will reduce the crystallinity of obtained graphene, but on the other hand, endows graphene improved catalytic activity due to the presence of defects and more exposed active sites. The porous architecture of GS-CHI is responsible for its large BET surface area compared to that of nonporous thick G-CHI. X-Ray Diffraction (XRD) patterns, with the peak positions close to the standard graphite reference (PDF41-1487), show the formation of graphitic carbon for both of GS-CHI and G-CHI. However, their (001) peaks are shifted towards smaller angles (22.35° and 24.84° for GS-CHI and G-CHI respectively, compared to 26.38° for the reference), indicating the looser stacking of the graphene layers.20 The spacing of GS-CHI calculated by XRD is 0.396 nm that is well consistent with HRTEM measurements. Then the average number of layers in the walls can be estimated for GS-CHI to less than 20 according to the average wall thickness measured by electron microscopy (about 8 nm). In addition, the existence in regions with thinner walls (such as in Fig. 3b and at the tangent of two silica spheres) makes reasonable to consider the porous materials obtained here as a defective 3D few-layers N-doped graphene. For GS-CHI, the (001) peak moves a little more towards lower diffraction angle than G-CHI, which may be caused by the larger spacing between the graphene layers formed in the pyrolysis process for GS-CHI as consequence of the curvature of the walls and the interaction between the chitosan polymer and the hydroxyl groups on silica surface (see Fig. S5† with more detailed explanation). This looser interlayer spacing of graphene sheets in GS-CHI can be beneficial for the deeper penetration of protons into interlayer space from edge, giving rise to larger electrochemical active area beneficial for the HER process.
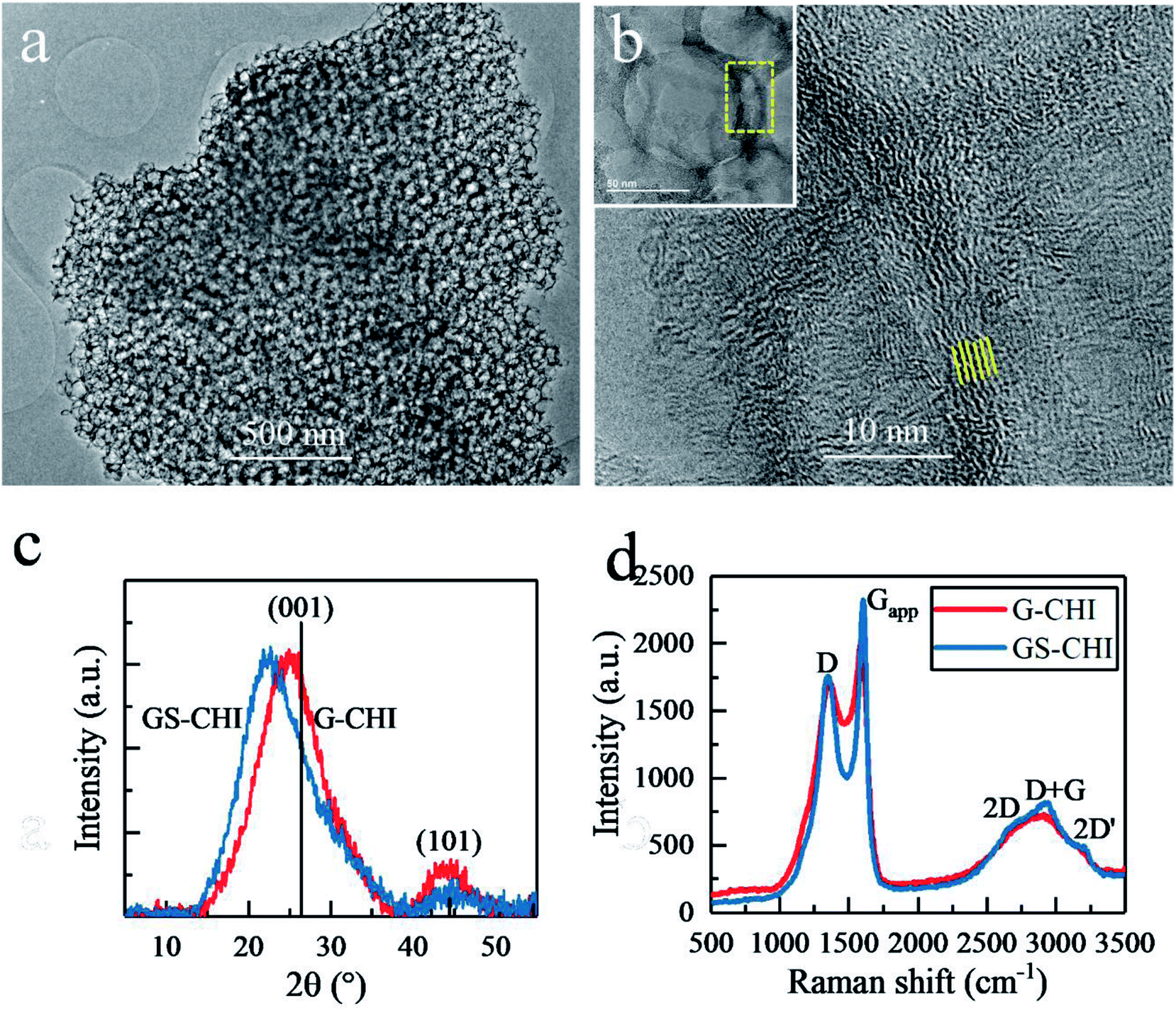 | ||
| Fig. 3 (a) TEM image of a selected GS-CHI particle. (b) HRTEM image of a section of the pore wall marked in the inset. (c) XRD patterns of GS-CHI, G-CHI and a standard reference (PDF41-1487). (d) Raman spectra of GS-CHI and G-CHI. | ||
The Raman spectra of GS-CHI and G-CHI show coincident peak positions. However, GS-CHI has sharper peak shape than G-CHI (Fig. 3d). By fitting the apparent D peak (Dapp) at ∼1360 cm−1 and G peak (Gapp) at ∼1600 cm−1, the individual D and G peaks were extracted (Fig. S6†). The integrated intensity ratio of D peak to G peak of GS-CHI was calculated to 1.66, which is slightly lower than that of G-CHI (1.69) (Table S1†). And the full width at half maximum (FWHM) of G peak of GS-CHI (95 cm−1) is also lower than that of G-CHI (108 cm−1), suggesting a better quality of the graphene sheets in GS-CHI compared to G-CHI, probably due to the positive catalytic effect of acidic silanol groups in the pyrolysis process, or the improved planarization due to transformation of long chain chitosan molecules into graphene layers in the restricted gaps among silica spheres. The Raman results combined with HRTEM and XRD results confirm the successful synthesis of defective 3D few-layers graphene without other metal cocatalysts by using chitosan as raw material, which can be attributed to the unique filmogeneicity and structure of chitosan.
By incorporating N element into carbon framework, the Gibbs free-energy of hydrogen adsorption (ΔGH*) of graphene will decrease, causing a stronger adsorption between hydrogen (H*) and neighboring C atoms bonded to N. According to theory,26 this stronger H* adsorption is due to the shift of the contribution of the active C atoms in the density of states towards the Fermi level, and the increase in the filling of bonding state of the active C valence orbitals. Therefore, N-doping confers metal-free graphene considerable electrocatalytic activity. N-doped defective graphene produced by pyrolysis of chitosan has been confirmed to be a n-type semiconductor by our group which can more easily provide reductive electrons to facilitate hydrogen generation.27 Based on this prior knowledge and the larger surface area, it was anticipated that GS-CHI should exhibit a notable electrocatalytic activity for HER.
Fig. 4a displays the linear sweep voltammetry (LSV) polarization curves of HER for glass carbon electrodes (GCEs) modified with porous GS-CHI and nonporous G-CHI. For comparison, the blank GCE without modification was also tested under the same conditions. The blank GCE shows the lowest electrochemical activity with a high onset potential (−282 mV at 0.5 mA cm−2) and a small current density (0.67 mA cm−2) at −300 mV potential, attributed to few active sites that could probably be on the smooth surface of the blank GCE. When modifying GCE with nonporous G-CHI, slightly higher current density (2.08 mA cm−2 at 300 mV) and lower onset potential (−203 mV at 0.5 mA cm−2) can be obtained, owing to the increased edge sites of G-CHI. When using porous GS-CHI, an obvious improvement of HER electrocatalytic activity was observed with a low onset potential (−131 mV at 0.5 mA cm−2) and a high current density (15.62 mA cm−2 at 300 mV). The overpotential required to reach the current density of 10 mA cm−2 is a widely used parameter to compare the activity of different electrocatalysts. GS-CHI catalyst could achieve this current density at −267 mV compared to −444 mV for G-CHI and −496 mV for blank GCE. This notable electrocatalytic activity measured for GS-CHI should be due as arising from the combination of the presence of N atoms acting as HER active sites and the large electrochemical area due to the spongy morphology.26
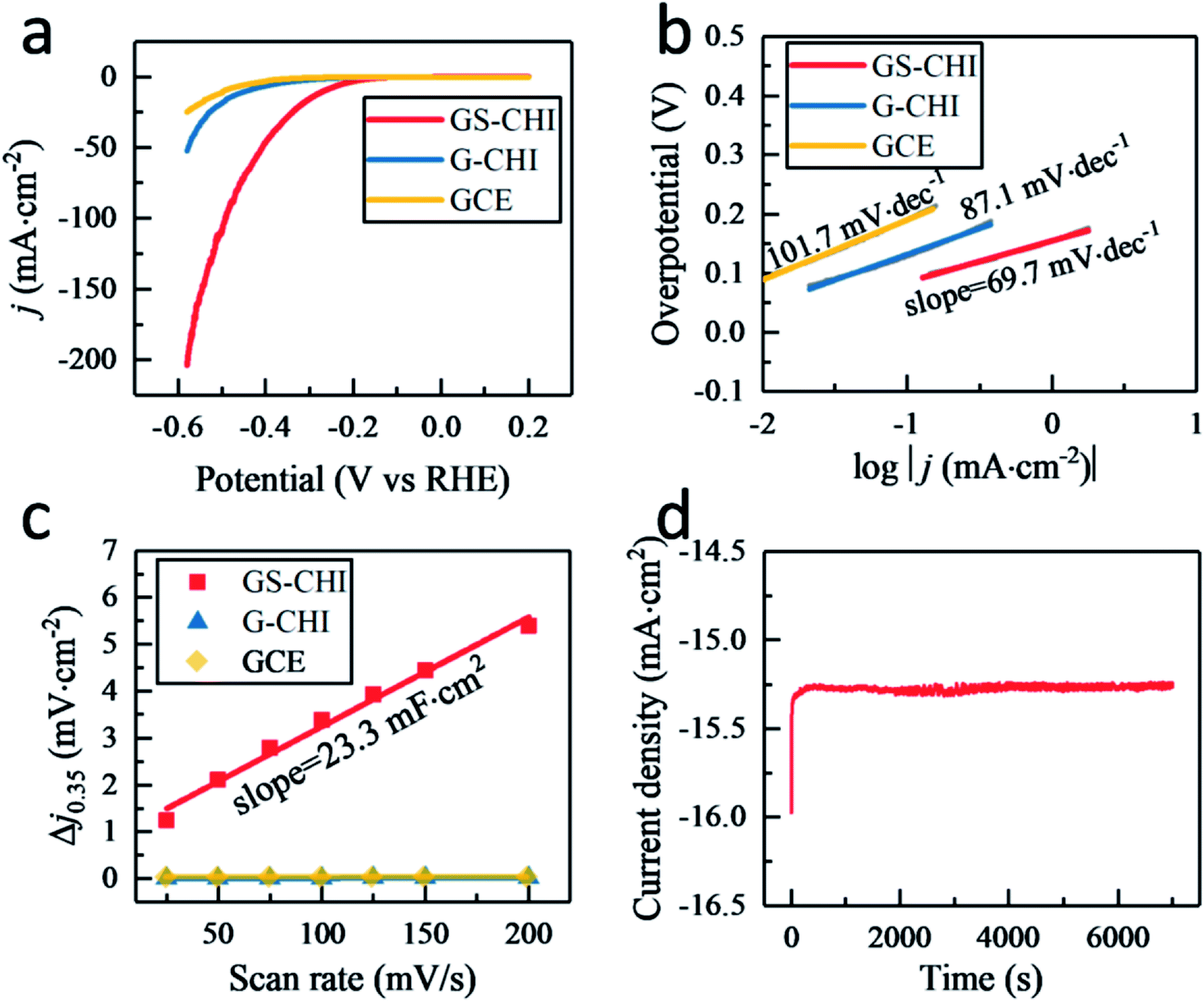 | ||
| Fig. 4 (a) LSV curves of GS-CHI, G-CHI and blank GCE; (b) Tafel plots of GS-CHI, G-CHI and blank GCE; (c) capacitive current as a function of scan rate for GS-CHI fitted to a linear regression; (d) durability of GS-CHI at −300 mV potential for 7000 s. | ||
To put the electrocatalytic activity of GS-CHI into a broader context, besides comparison with G-CHI and bare GCE, Table 1 summarizes the performance of other related metal-free (co)doped graphenes. As it can be seen in Table 1, the most active material under similar test conditions is, according to the literature, a N,P-codoped graphene,28,29 showing that there is still room for improvement of the electrocatalytic activity of 3D GS-CHI, probably by maximizing its electrochemical surface area, further adjustment of pore size and by multiple codoping with optimal element loading. It is likely that by adjusting the elemental composition and pre/post treatments (as for instance plasma etching reported in other cases30,31), the performance of GS-CHI would be comparable with that of the most efficient graphene-based catalysts. Nevertheless, in spite of the only presence of N as dopant element and the one-step pyrolysis preparation procedure, Table 1 shows that GS-CHI still exhibits lower overpotential at 10 mA cm−2 than most of the reported graphene catalysts, with the lowest Tafel slope of all the materials.32–37 The above results indicate that bare GS-CHI can act as an efficient electrocatalyst, a notable performance that can be attributed to the combination of self N-doping, increased active sites due to the porous spongy structure, larger interlayer spacing, and the continued interconnected framework permitting a fast transport of charges with the electrode with lesser resistance.
| Electrocatalyst (in 0.5 M H2SO4) | Overpotential at 10 mA cm−2 current density (mV) | Tafel slope (mV dec−1) | Year | Ref. |
|---|---|---|---|---|
| a Only metal-free doped graphenes without hetero-phase modifications and pre/post modification treatment are included. | ||||
| N,P-codoped graphene | −420 | 91 | 2014 | 32 |
| B-doped graphene | −440 | 99 | 2014 | 33 |
| N,S-codoped nanoporous graphene | −280 | 81 | 2015 | 34 |
| N,P-codoped nanoporous graphene | −213 | 79 | 2015 | 28 |
| N,S-codoped graphene | −520 | 161 | 2017 | 36 |
| N-doped 3D graphene | −262 | 112 | 2018 | 38 |
| Amine functionalized N doped graphene | −350 | 113 | 2018 | 35 |
| N,P-codoped graphene | ∼−220 | 88 | 2018 | 29 |
| S-doped 3D graphene | ∼−410 | 97 | 2018 | 37 |
| N doped 3D graphene sponge (this work) | −267 | 69.7 | 2018 | This work |
Tafel slope related to the rate-limiting step of the HER process was also investigated. As shown in Fig. 4b, the fitted Tafel slopes for porous GS-CHI, nonporous G-CHI and blank GCE were 69.7, 87.1 and 101.7 mV dec−1, respectively. These Tafel slope values are in the range of 40–120 mV dec−1, indicating that for these materials the HER process follows the Volmer–Heyrovsky mechanism (see equations below).32 In the case of GS-CHI, the rate-limiting step should be the Heyrovsky reaction, because the Tafel slope for this material is closer to 40 mV dec−1, while for blank GCE it should be the Volmer reaction, and for G-CHI the HER mechanism according to the Tafel slope would consist in a combination of Volmer and Heyrovsky reactions.
| H3O+ + e− + * → H* + H2O (Volmer reaction, Tafel slope ≈ 120 mV dec−1) |
| H3O+ + e− + H* → H2 + H2O (Heyrovsky reaction, Tafel slope ≈ 40 mV dec−1) |
| H* + H* → H2 (Tafel reaction, Tafel slope ≈ 30 mV dec−1) |
Thus, the slope of the Tafel plots indicates much faster proton discharge kinetics for GS-CHI due to the simultaneous existence on the surface of this material of both hydrated protons (H3O+) and adsorbed hydrogens (H*), as required in the Heyrovsky reaction equation. The coexistence of both hydrated protons and adsorbed hydrogens on GS-CHI can be attributed to the presence of two types of accessible sites (basic and H adsorption centers), in agreement with the composition of GS-CHI containing oxygen, nitrogen and edges and the large interlayer distance and surface area of this material. The absorbed H* on GS-CHI surface would form H2 by first combining with hydrated proton H3O+ and then receiving an electron e− from the electrode through the conductive defective graphene walls. However, for G-CHI and blank GCE, the surface active sites are not enough to adsorb simultaneously sufficient density of H3O+ and adsorb H*, therefore, the Volmer reaction is in those materials the rate-limiting step. Compared with other reported metal-free codoped graphenes (Table 1), it should be noted that GS-CHI exhibits the smallest Tafel slope, a fact that can be attributed to its more appropriate porous spongy structure conferring it with large area and electrical connectivity.
Other electrochemical parameter, the double layer capacitance (Cdl) related to the effective electrochemical surface area, was also measured by the cyclic voltammetry (CV) method in a potential range of 0.3–0.4 V (vs. RHE) (Fig. S7†). Capacitive current at 0.35 V was plotted against scan rate from 25–200 mV s−1 in Fig. 4c and GS-CHI shows a Cdl of 11.65 mF cm−2 that is 542 and 1148 times higher than that of G-CHI (21.5 μF cm−2) and blank GCE (10.15 μF cm−2), respectively, confirming experimentally the higher effective electrochemical surface area for charge accumulation in GS-CHI.
In addition, besides activity, the electrochemical stability was also evaluated. After 1000 cycles, GS-CHI showed nearly overlaying LSV polarization curve with the initial one of the fresh GS-CHI material (Fig. S8†). At −300 mV potential, an almost constant current was observed in the 7000 s electrolysis test, indicating the excellent stability of GS-CHI in HER process (Fig. 4d).
The above electrochemical data provide a good indication of the remarkable activity improvement that can be achieved by applying the concept of 3D GS using close-packed silica spheres as hard template and a natural polysaccharide with filmogeneicity as simultaneous carbon and nitrogen source to synthesize a structured 3D sponge with surprisingly high HER catalytic activity.
To show the general applicability of the method, similar 3D uniform porous graphene structure was also obtained using another polysaccharide, alginate (sample denoted as GS-ALG) by applying the same infiltration–pyrolysis strategy. The SEM and TEM images of GS-ALG show even better and clearer porous spongy structure which can be attributed to the good penetrative ability of alginate aqueous solution due to its lower viscosity compared to more viscous chitosan solution (Fig. 5). Noteworthy, the lower viscosity of alginate solution is responsible for the fact that under optimal conditions, even thinner graphene walls approaching the single-layer configuration can be achieved.
 | ||
| Fig. 5 SEM (a) and TEM (b) images of GS-ALG. | ||
In conclusion, the present manuscript reports that submicrometric silica templates can be used for the preparation of doped and undoped few-layers graphene sponges that exhibit enhanced electrocatalytic activity. This activity enhancement is a consequence of the spatial structuring resulting in large area, interconnected macroporosity, and electrical continuity of the 3D particles. Considering the flexibility that filmogenic natural biopolymers offer for the preparation of graphene heterojunctions and as support of metal and metal oxide nanoparticles, it can be easily anticipated that the methodology presented here can be also used to prepare a vast type of different related materials with improved catalytic and electrocatalytic activity.
Experimental
Preparation of silica templates
Silica template spheres were synthesized following our previous work.39,40 Briefly, 43 ml deionized water, 10 ml ammonia water and 56 ml TEOS were added into 390 ml ethanol and stirred at 500 rpm for 24 h at 50 °C. A translucent solid was prepared by centrifugation of the reaction solution (10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 rpm for 30 min). The solid was sintered at 500 °C for 30 min to obtain the final close-packed template.
000 rpm for 30 min). The solid was sintered at 500 °C for 30 min to obtain the final close-packed template.
Synthesis of GS
Porous graphene sponge was synthesized by a facile hard template-associated pyrolysis method. Typically, 2 ml 2 wt% chitosan solution (including 400 mg chitosan, 400 μl glacial acetic acid and 20 ml water) and 2 g of silica template were mixed and extracted for 30 min under reduced pressure. After centrifugation at 6000 rpm for 10 min and washing with 40 ml water for two times, the obtained solids were dried at 60 °C overnight and then pyrolyzed under an argon gas flow (200 sccm, atmospheric pressure) at 900 °C for 2 h with a ramping rate of 10 °C min−1,18 leading to the formation of silica–graphene composite. The silica template was selectively etched in aqueous 2 M NaOH at 80 °C for 2 h. The remaining graphene was collected by vacuum filtration and washed with excessive water and dried at 60 °C. The nonporous graphene reference, G-CHI was directly prepared using chitosan powder under the same pyrolysis conditions to GS-CHI. When synthesizing GS-ALG, we just used an alginate sodium aqueous solution (including 200 mg sodium alginate, 40 ml water) according to our previous work41 instead of chitosan solution.Characterization
The crystal structures of the synthesized samples were characterized by XRD (Shimadzu XRD-7000 with Cu Kα radiation at 40 kV and 40 mA). The morphology was examined by using field emission SEM (Zeiss Ultra 55) and TEM (JEOL2100F). Raman spectra were collected with a Horiba Jobin Yvon-Labram HR UV-Visible-NIR (200–1600 nm) Raman Microscope Spectrophotometer, using 514 nm excitation wavelength. The BET value of samples was measured by Accelerated Surface Area and Porosimetry System (ASAP 2420, Micromeritics) under liquid nitrogen temperature (77.3 K).Electrochemical measurements
The electrochemical measurements were performed in a three electrode system on a VersaSRAT3 (AMETEK) electrochemical workstation. A GCE (3 mm in diameter) was used as the substrate of the working electrode. When preparing working electrode, 1 mg of graphene was dispersed in 250 μL of water/ethanol (4:1 volume/volume) mixed solvent containing 20 μL of 5 wt% Nafion solution (Sigma) by sonication for 1 h to form homogeneous ink. Then 5.0 μL of the graphene ink (containing 0.02 mg catalyst) was carefully loaded onto the cylindrical end surface of GCE (loading ∼0.065 mg cm−2). LSV polarization curves were obtained with scan rate of 5 mV s−1 in 0.5 M H2SO4 by sweeping the potential from −0.80 to 0 V (vs. SCE). The current density was normalized by electrode geometric area (0.07 cm2). For electrochemical capacitance, the potential was swept from 0.3 to 0.4 V (vs. RHE) and back to 0.3 V at each of seven different scan rates (25, 50, 75, 100, 125, 150 and 200 mV s−1). A Pt disk electrode was used as counter electrode. The Ag/AgCl electrode (3.5 M KCl) was used as the reference. All the potentials reported here were referenced to the reversible hydrogen electrode (RHE) by adding a value of 0.205 V.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
Financial support from the Spanish Ministry of Economy and Competitiveness (Severo Ochoa and CTQ2015-698153-COP2-R1) and Generalitat Valenciana (Prometeo 2017-083) is gratefully acknowledged. A. P. thanks the Spanish Ministry for a Ramón y Cajal research associate contract. J. Niu also gratefully acknowledges financial support from the China Scholarship Council (201706425026) and the National Natural Science Foundation of China (41502032).References
- S. Mao, G. Lu and J. Chen, Nanoscale, 2015, 7, 6924–6943 RSC.
- Y. Ito, Y. Tanabe, K. Sugawara, M. Koshino, T. Takahashi, K. Tanigaki, H. Aoki and M. Chen, Phys. Chem. Chem. Phys., 2018, 20, 6024–6033 RSC.
- B. Qiu, M. Xing and J. Zhang, Chem. Soc. Rev., 2018, 47, 2165–2216 RSC.
- X. Huang, K. Qian, J. Yang, J. Zhang, L. Li, C. Yu and D. Zhao, Adv. Mater., 2012, 24, 4419–4423 CrossRef CAS.
- M. Zhou, T. Lin, F. Huang, Y. Zhong, Z. Wang, Y. Tang, H. Bi, D. Wan and J. Lin, Adv. Funct. Mater., 2013, 23, 2263–2269 CrossRef CAS.
- G. Ning, Z. Fan, G. Wang, J. Gao, W. Qian and F. Wei, Chem. Commun., 2011, 47, 5976–5978 RSC.
- X. Xiao, T. E. Beechem, M. T. Brumbach, T. N. Lambert, D. J. Davis, J. R. Michael, C. M. Washburn, J. Wang, S. M. Brozik, D. R. Wheeler, D. B. Burckel and R. Polsky, ACS Nano, 2012, 6, 3573–3579 CrossRef CAS.
- S. Mao, Z. Wen, H. Kim, G. Lu, P. Hurley and J. Chen, ACS Nano, 2012, 6, 7505–7513 CrossRef CAS.
- G. H. Moon, Y. Shin, D. Choi, B. W. Arey, G. J. Exarhos, C. Wang, W. Choi and J. Liu, Nanoscale, 2013, 5, 6291–6296 RSC.
- J. S. Lee, S. I. Kim, J. C. Yoon and J. H. Jang, ACS Nano, 2013, 7, 6047–6055 CrossRef CAS.
- X. L. Huang, D. Xu, S. Yuan, D. L. Ma, S. Wang, H. Y. Zheng and X. B. Zhang, Adv. Mater., 2014, 26, 7264–7270 CrossRef CAS PubMed.
- W. Ouyang, J. Sun, J. Memon, C. Wang, J. Geng and Y. Huang, Carbon, 2013, 62, 501–509 CrossRef CAS.
- H. Bi, Z. Yin, X. Cao, X. Xie, C. Tan, X. Huang, B. Chen, F. Chen, Q. Yang, X. Bu, X. Lu, L. Sun and H. Zhang, Adv. Mater., 2013, 25, 5916–5921 CrossRef CAS.
- J. C. Yoon, J. S. Lee, S. I. Kim, K. H. Kim and J. H. Jang, Sci. Rep., 2013, 3, 1788 CrossRef.
- A. A. Zakhidov, R. H. Baughman, Z. Iqbal, C. Cui, I. Khayrullin, S. O. Dantas, J. Marti and V. G. Ralchenko, Science, 1998, 282, 897–901 CrossRef CAS.
- A. Vu, X. Li, J. Phillips, A. Han, W. H. Smyrl, P. Bühlmann and A. Stein, Chem. Mater., 2013, 25, 4137–4148 CrossRef CAS.
- D. Mateo, I. Esteve-Adell, J. Albero, A. Primo and H. García, Appl. Catal., B, 2017, 201, 582–590 CrossRef CAS.
- D. Mateo, I. Esteve-Adell, J. Albero, J. F. S. Royo, A. Primo and H. Garcia, Nat. Commun., 2016, 7, 1–8 Search PubMed.
- H. G. Baldoví, F. Albarracín, M. Álvaro, B. Ferrer and H. García, ChemPhysChem, 2015, 16, 2094–2098 CrossRef PubMed.
- A. Primo, E. Sánchez, J. M. Delgado and H. García, Carbon, 2014, 68, 777–783 CrossRef CAS.
- M. Latorre-Sánchez, A. Primo and H. García, Angew. Chem., Int. Ed., 2013, 52, 11813–11816 CrossRef PubMed.
- A. Primo, P. Atienzar, E. Sanchez, J. M. Delgado and H. García, Chem. Commun., 2012, 48, 9254–9256 RSC.
- W. Zhou, J. Jia, J. Lu, L. Yang, D. Hou, G. Li and S. Chen, Nano Energy, 2016, 28, 29–43 CrossRef CAS.
- X. Huang, Y. Zhao, Z. Ao and G. Wang, Sci. Rep., 2015, 4, 7557 CrossRef PubMed.
- B. G. Choi, M. Yang, W. H. Hong, J. W. Choi and Y. S. Huh, ACS Nano, 2012, 6, 4020–4028 CrossRef CAS PubMed.
- Y. Jiao, Y. Zheng, K. Davey and S.-Z. Qiao, Nat. Energy, 2016, 1, 16130 CrossRef CAS.
- M. Latorre-Sánchez, A. Primo, P. Atienzar, A. Forneli and H. García, Small, 2015, 11, 970–975 CrossRef.
- H. Jiang, Y. Zhu, Y. Su, Y. Yao, Y. Liu, X. Yang and C. Li, J. Mater. Chem. A, 2015, 3, 12642–12645 RSC.
- T. Li, D. Tang, M. Wang, Q. Song and C. M. Li, ChemistrySelect, 2018, 3, 6814–6820 CrossRef CAS.
- A. Dey, A. Chroneos, N. S. J. Braithwaite, R. P. Gandhiraman and S. Krishnamurthy, Appl. Phys. Rev., 2016, 3, 021301 Search PubMed.
- R. Yang, L. Zhang, Y. Wang, Z. Shi, D. Shi, H. Gao, E. Wang and G. Zhang, Adv. Mater., 2010, 22, 4014–4019 CrossRef CAS PubMed.
- A. Ambrosi, C. K. Chua, N. M. Latiff, A. H. Loo, C. H. A. Wong, A. Y. S. Eng, A. Bonanni and M. Pumera, Chem. Soc. Rev., 2016, 45, 2458–2493 RSC.
- B. R. Sathe, X. Zou and T. Asefa, Catal. Sci. Technol., 2014, 4, 2023–2030 RSC.
- Y. Ito, W. Cong, T. Fujita, Z. Tang and M. Chen, Angew. Chem., Int. Ed., 2015, 54, 2131–2136 CrossRef CAS PubMed.
- Z. Jiang, Z. J. Jiang, X. Tian and W. Chen, J. Mater. Chem. A, 2014, 2, 441–450 RSC.
- J. Zhou, H. Yue, F. Qi, H. Wang and Y. Chen, Int. J. Hydrogen Energy, 2017, 42, 27004–27012 CrossRef CAS.
- J. Zhou, F. Qi, Y. Chen, Z. Wang, B. Zheng and X. Wang, J. Mater. Sci., 2018, 53, 7767–7777 CrossRef CAS.
- Y. Tian, Y. Ye, X. Wang, S. Peng, Z. Wei, X. Zhang and W. Liu, Appl. Catal., A, 2017, 529, 127–133 CrossRef CAS.
- J. Niu, S. Shen, S. He, Z. Liu, P. Feng, S. Zhang, X. Ou, Y. Qiang and Z. Zhu, Ceram. Int., 2015, 41, 11936–11944 CrossRef CAS.
- J. Niu, S. Shen, L. Zhou, Z. Liu, P. Feng, X. Ou and Y. Qiang, RSC Adv., 2016, 6, 62907–62910 RSC.
- D. Mateo, J. Albero and H. García, Energy Environ. Sci., 2017, 10, 2392–2400 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c8ra08745h |
| This journal is © The Royal Society of Chemistry 2019 |