
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceA new metallic π-conjugated carbon sheet used for the cathode of Li–S batteries†
Li Zeng ,
Yingxiang Cai*,
Zhihao Xiang,
Yu Zhang and
Xuechun Xu
,
Yingxiang Cai*,
Zhihao Xiang,
Yu Zhang and
Xuechun Xu
Department of Physics, Nanchang University, Jiangxi, Nanchang 330031, P. R. China
First published on 21st December 2018
Abstract
Lithium–sulfur (Li–S) batteries are considered as the most promising next generation high density energy storage devices. However, the commercialization of Li–S batteries is hindered by the shuttle effect of polysulfides, the low electronic conductivity of the sulfur cathode and a large volume expansion during lithiation. Herein, we predict a new two dimensional sp2 hybridized carbon allotrope (PHE-graphene) and prove its thermodynamic and kinetic stability. If it is utilized to encapsulate the cathode of Li–S batteries, not only will the shuttle effect be avoided but also the electronic conductivity of the sulfur cathode will be improved significantly owing to its metallic electronic band structure. The thermal conductivity of PHE-graphene was found to be very high and even comparable with graphene, which is helpful for the heat dissipation of cathodes. In addition, PHE-graphene also exhibited superior mechanical properties including ideal tensile strength and in-plane stiffness.
1 Introduction
With the continuous consumption of finite fossil fuels and serious environmental pollution issues, the demand for renewable and clean energy sources such as solar and wind has increased substantially. Correspondingly, it is necessary to develop efficient rechargeable energy storage systems for these intermittent energy sources.1,2 Rechargeable lithium-ion (Li-ion) batteries are regarded as a suitable choice for energy storage devices.3,4 However, Li-ion batteries can still not meet the rising demands of large scale applications owing to their limited energy density and high cost.3–5 Accordingly, new economical energy storage systems with a high specific capacity are needed. Li–air and Li–S batteries have attracted increasing attention due to their high theoretical capacities of 3000 W h kg−1 and 2600 W h kg−1, respectively.1,6 Unfortunately, there are many insurmountable obstacles existing in Li–air batteries such as a low practical energy density,6 lots of detrimental side reactions,7 instability towards moisture,8 etc. In contrast, it is more practical to deal with the challenges of Li–S batteries, and sulfur is environmentally friendly and abundant in nature.9 Therefore, Li–S batteries are widely viewed as promising energy storage systems to replace the present Li-ion batteries. Nevertheless, there are still a few challenges faced with Li–S batteries. Firstly, sulfur and its reaction products (Li2S and Li2S2) exhibit poor electronic conductivity, which results in a low rate capability and poor cycle performance of Li–S batteries.10–12 Secondly, the dissolved intermediate lithium polysulfides (Li2Sn, 3 ≤ n ≤ 8) can diffuse freely between the cathode and anode during charge/discharge processes. This “shuttle effect” results in lithium anode corrosion, a poor cycle stability of the sulfur cathode, and low coulombic efficiency in Li–S batteries.13–15 Thirdly, the large volumetric expansion of the sulfur cathode during lithiation leads to electrode pulverization and fast capacity fading.16–18To solve these problems faced with Li–S batteries, finding new cathode materials which not only have high electrical conductivity but also are able to confine polysulfides in the cathodes are crucial. Various sulfur-based composites have been proposed, such as carbon–sulfur composites,19–22 polymer–sulfur composites,23–25 and carbon–polymer–sulfur composites.26–28 Among the numerous available cathode materials, graphene has high interest due to its advantageous intrinsic properties for Li–S batteries, such as superior electrical conductivity and high theoretical surface area for sulfur loading.29–31 However, graphene–sulfur composites have failed to eliminate the shuttle effect in Li–S batteries.32,33 To confine polysulfides effectively, methods using graphene oxide to replace graphene34 and encapsulating graphene–sulfur composites with polymers30 have been tried. However, both approaches will deteriorate the electrical conductivity of cathodes.32 Metal oxide–sulfur composites can also alleviate the shuttle effect by transforming soluble polysulfides into insoluble lower-order sulfides,35–37 but the poor electronic conductivity of these metal oxides limits their behavior for improving cell lifetimes.38 A recent study showed that two dimensional (2D) multiporous carbon sheets can be used to encapsulate the cathodes of Li–S batteries.39 Relative to sulfur-based composites, this multiporous carbon has a higher electrical conductivity. For new cathode materials, excellent mechanical properties are essential to bear possible volumetric expansion. In addition, high thermal conductivities are also expected for new cathode materials in order to facilitate the heat dissipation of electrodes.
In this study, we firstly predicted a new 2D sp2 hybridized carbon allotrope (PHE-graphene), and suggested its synthesis by a dehydrogenation reaction of small molecules. Secondly, its thermodynamic and kinetic stability were examined and electronic band structures were investigated. Thirdly, the phonon lifetimes and group velocity in PHE-graphene were calculated. The phonon and electron thermal conductivities were evaluated, then the ideal tensile strength of PHE-graphene was determined. The upper limits of in-plane stiffness were systematically investigated for twenty 2D carbon sheets. Finally, the energy barriers of Li and S atoms diffusing through PHE-graphene were studied and its potential applications in Li–S batteries are discussed.
2 Methods
Our ab initio calculations were performed using the Perdew–Burke–Ernzerhof (PBE) generalized gradient approximation40 and implemented in the VASP code.41,42 Blöchl’s projector augmented wave (PAW) method43,44 was utilized to describe the interactions between the nucleus and the valence electrons of carbon. A plane-wave basis with a cutoff energy of 400 eV was used to expand the wave functions. The geometries of all of the carbon allotropes investigated in this work were fully relaxed including the atomic positions and lattice parameters until the residual forces on each atom were less than 0.0001 eV Å−1. The Brillouin zone sample meshes were dense enough and the k spacing was less than 0.3 Å−1 for all of the structures. The phonon band structure was determined by the direct supercell method as implemented in the Phonopy program.45 Lattice thermal conductivity was calculated by solving the linearized phonon Boltzmann equation within the single-mode relaxation time approximation (RTA) and implemented by the Phono3py code.46 Electron thermal conductivity was calculated according to the rigid-band approximation (RBA) using BoltzTraP2 code.47 The energy barriers of S and Li atoms diffusing were investigated using both PBE and dispersion corrected (DFT-D3)48 density functionals.3 Results and discussion
Many methods can be utilized to prepare graphene, such as top-down exfoliation, bottom-up chemical vapor deposition (CVD) or chemical synthesis methods.49 However, new 2D carbon materials usually cannot be obtained by the exfoliation method due to the lack of bulk layer precursors. CVD is also not suitable for the synthesis of 2D carbon except for graphene. Chemical synthesis from small molecules is a promising route to prepare new 2D carbon sheets, although it is still very challenging. Experimentally, two 2D carbon sheets have been synthesized by the dehydration or dehydrogenation chemical reactions of small molecules as shown in Fig. 1a and b. A 4–6 carbophene (i.e. DHP-graphene)39,50 was synthesized by the dehydration reaction of 1,3,5-trihydroxybenzene molecules and graphdiyne51 was obtained by the dehydrogenation reaction of hexaethynylbenzene molecules. Likewise, a new conjugated π-bonding planar carbon structure might be achieved by the dehydrogenation reaction of acepentalene52 molecules as shown in Fig. 1c. This carbon allotrope has also been found in structure research based on particle swarm optimization for a system with ten carbon atoms. Its structure consists of pentagonal, hexagonal and enneagonal rings, and thus it is named PHE-graphene. PHE-graphene belongs to the space group P![[6 with combining macron]](https://www.rsc.org/images/entities/char_0036_0304.gif) m2 (D3h). Its lattice constant a is 5.738 Å. In the unit cell of PHE-graphene, there are four nonequivalent chemical bonds, whose lengths are 1.385, 1.448, 1.392 and 1.514 Å, respectively. The ratio of chemical bond numbers is 1
m2 (D3h). Its lattice constant a is 5.738 Å. In the unit cell of PHE-graphene, there are four nonequivalent chemical bonds, whose lengths are 1.385, 1.448, 1.392 and 1.514 Å, respectively. The ratio of chemical bond numbers is 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2:1:1. Thus the average bond length is 1.438 Å which is a little higher than that of graphene (1.42 Å).
:2:1:1. Thus the average bond length is 1.438 Å which is a little higher than that of graphene (1.42 Å).
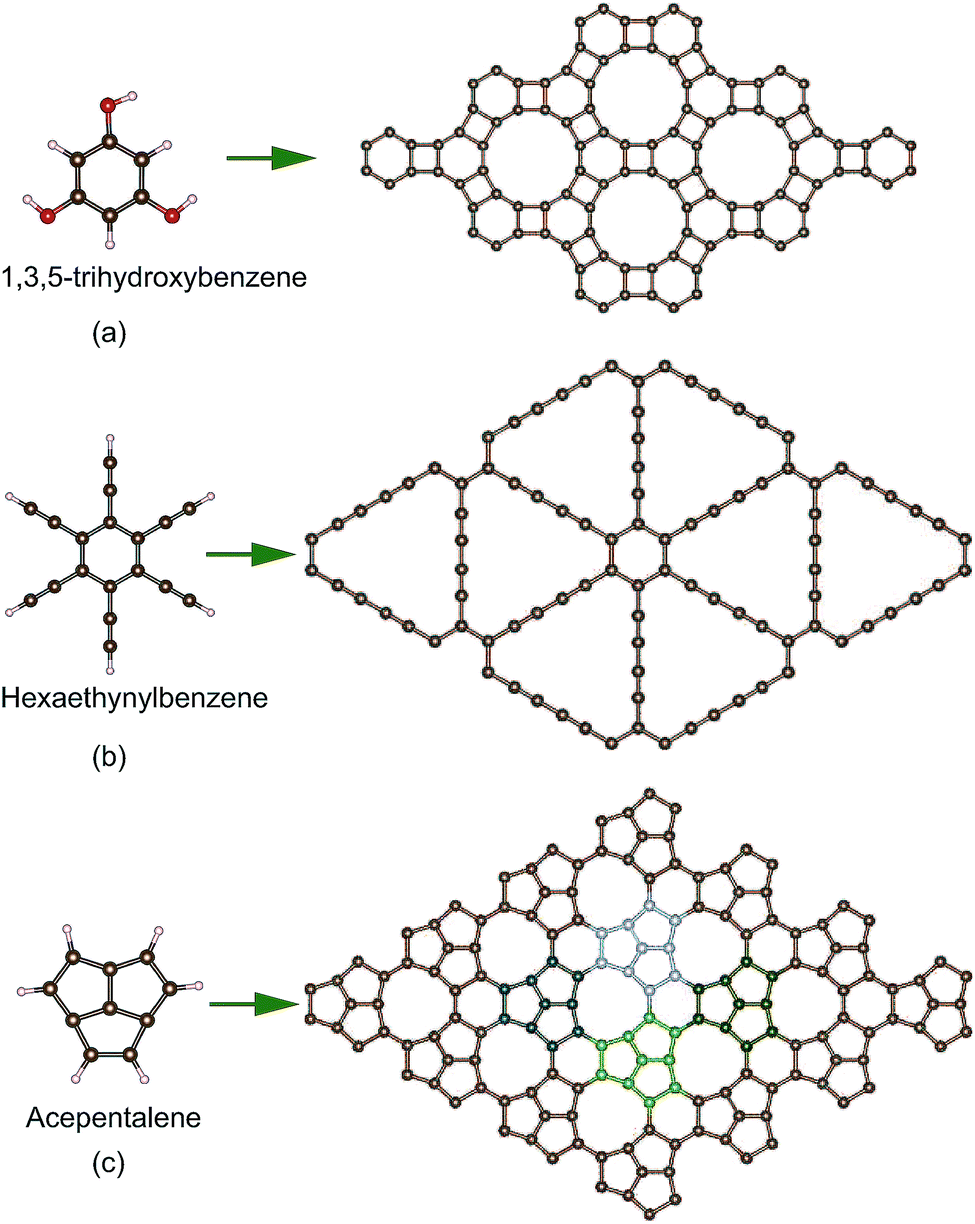 | ||
| Fig. 1 (a) The 4–6 carbophene (i.e. DHP-graphene) prepared by the dehydration reaction of 1,3,5-trihydroxybenzene molecules. (b) Graphdiyne synthesized by the dehydrogenation reaction of hexaethynylbenzene molecules. (c) A possible preparation method for PHE-graphene by the dehydrogenation reaction of acepentalene molecules. | ||
To add PHE-graphene into the carbon material family, we should firstly check its thermodynamic and kinetic stability. Fig. 2a shows the total energies of PHE-graphene and nineteen other 2D carbon allotropes, including DHP-graphene,39 graphdiyne,51 THD-graphene,53 planar T-graphene,54 rectangular heackelite,55 pza-C10,56 phagraphene,57 pentahexoctite,58 pentaheptite,59 OPG-L,60 oblique heackelite,55 HOP graphene,61 hexagonal heackelite,55 graphene,62 γ-graphyne,63 biphenylene,64 β-graphyne,65 α-graphyne65 and 6,6,12-graphyne.65 As expected, graphene is the most stable 2D carbon layer. Five sp + sp2 hybridized structures (including α-graphyne, β-graphyne, γ-graphyne, 6,6,12-graphyne and graphdiyne) were less stable relative to the other sp2 hybridized carbon sheets. Among them, the α-graphyne is the least stable sp + sp2 hybridized structure and graphdiyne is the least stable structure experimentally synthesized. PHE-graphene also exhibited a high thermodynamic stability. At equilibrium geometry, its total energy was lower than not only the sp + sp2 hybridized 2D carbon structures but also sp2 hybridized biphenylene, planar T-graphene and THD-graphene. Fig. 2b shows the phonon dispersion and projected phonon density of states (PhDOS) of PHE-graphene. Since no imaginary phonon frequencies were found, PHE-graphene is kinetically stable. The highest phonon frequency of PHE-graphene was calculated to be 48.24 THz (or 1609 cm−1), which is extremely close to that (around 1600 cm−1 (ref. 66 and 67)) of graphite and graphene. In addition, we could find two phonon bandgaps in the frequency regions of 33–35 and 39–42 THz. From the PhDOS, it can be seen that the high frequency phonons were mainly from the contribution of the C1 atom. Furthermore, a NVT ab initio molecular dynamics simulation was performed, which also confirmed the stability of PHE-graphene (see Fig. S1 in the ESI†).
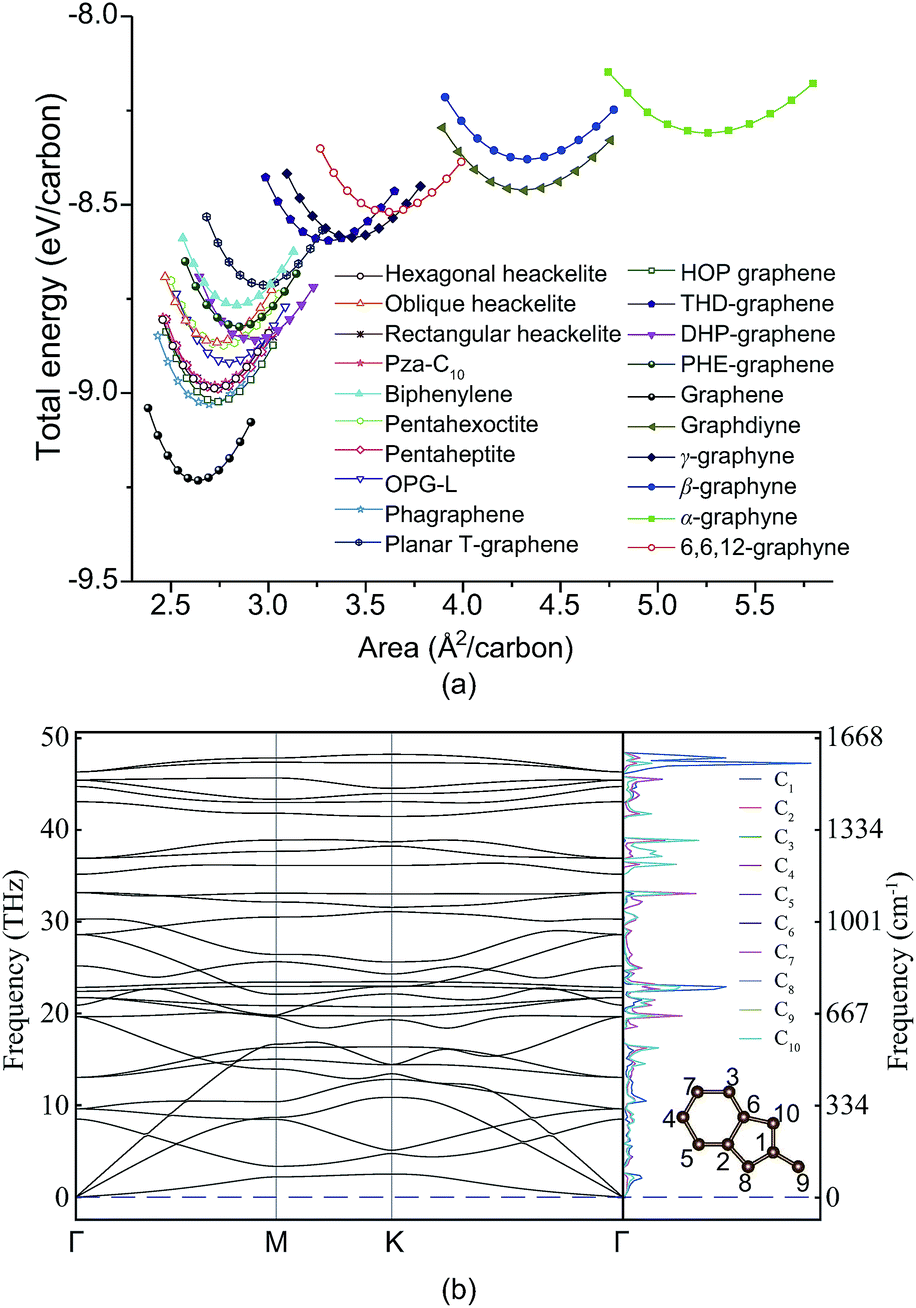 | ||
| Fig. 2 (a) Total energy as a function of volume for the 2D carbon allotropes. (b) Phonon dispersion of PHE-graphene and projected phonon density of states (PhDOS). The insert shows the ten non-equivalent carbon atoms in the unit cell. | ||
PHE-graphene has ten carbon atoms and forty 2s22p2 electrons in its unit cell. If PHE-graphene is semiconductive, the 20th band should be below the Fermi level (EF) and the 21st band should be above the EF. From Fig. 3a, we can find that both the 20th and 21st bands were partially occupied and crossed the EF. Therefore, PHE-graphene is a metallic carbon material. Fig. 3b shows the projected density of states (PDOS: s, px, py and pz). The PDOS near EF completely results from the pz electrons, which discloses the π-conjugated nature of PHE-graphene. If the pz DOS is projected on each carbon atom, it can be seen that the PDOS near the EF is mainly ascribed to the contributions of the C8, C9 and C10 atoms as shown in Fig. 3c. Based on the DOS analysis, we can conclude that PHE-graphene is a metallic π-bonding carbon structure with excellent electrical conductivity. Fig. 3d shows the charge densities of the 20th and 21st bands. It can be seen that the electrons in PHE-graphene are delocalized especially for the 20th band, in which the electrons behave just like the free electrons in metals.
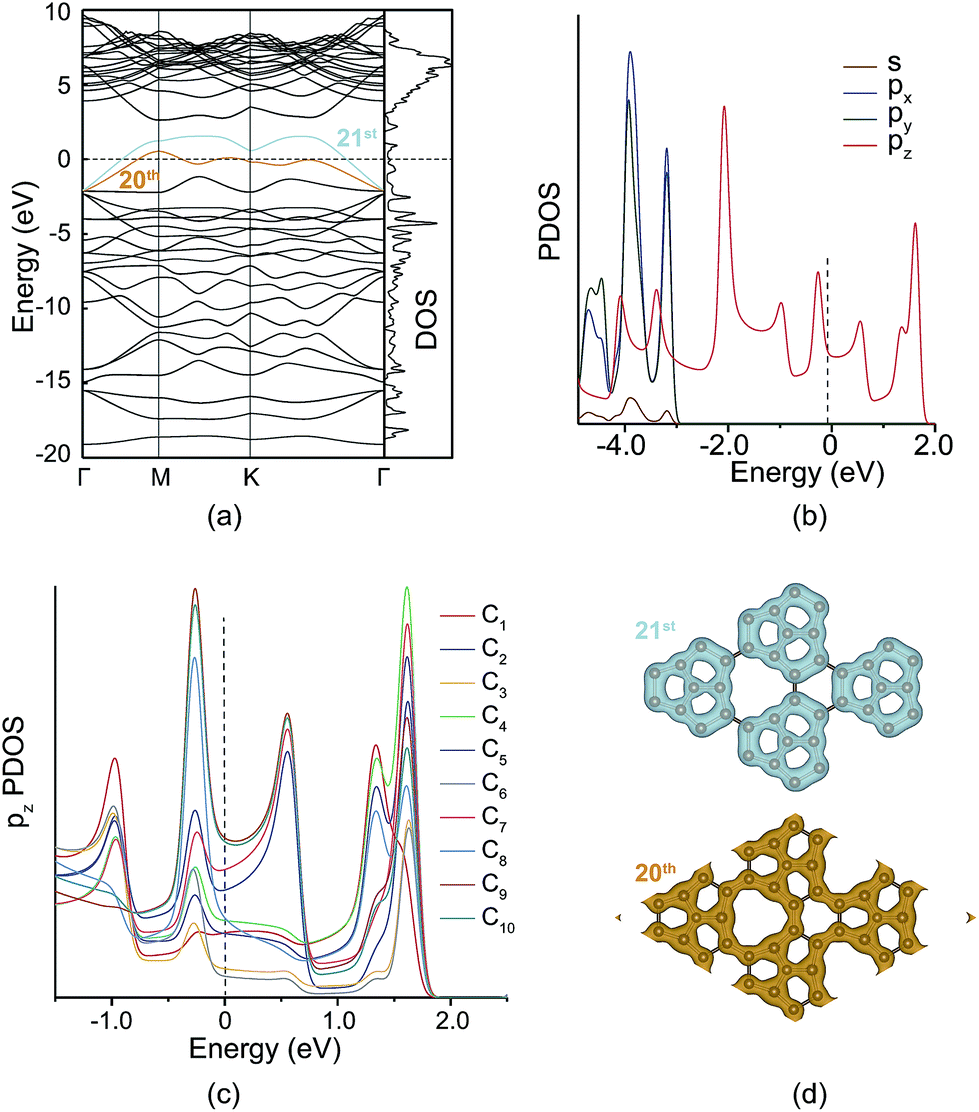 | ||
| Fig. 3 (a) Band structures and the density of states (DOS) of PHE-graphene. (b) Partial density of states (PDOS) of s, px, py and pz. (c) The pz PDOS of the C1–C10 atoms. (d) Charge densities of the 20th and 21st bands. | ||
The thermal conductivity of a material results from the transportation of electrons and phonons. Fig. 4a shows the phonon lifetimes calculated from the imaginary part of phonon self energy.46 We find that most phonons have short lifetimes, especially the high frequency phonons. The longest phonon lifetime around 20 THz is still less than 7 ps. Fig. 4b shows the phonon group velocity. Only a few low frequency phonons have group velocities of more than 10 km s−1 and most phonons’ group velocities are less than 5 km s−1. Two phonon bandgaps around 33–35 and 39–42 THz can also be found from Fig. 4a and b. Due to low phonon lifetimes and group velocity, the lattice thermal conductivity (κL) of PHE-graphene is quite low. At room temperature (300 K), the κL is only 4.8 W m−1 K−1. Along the [100] and [210] directions, thermal transport is almost isotropic as shown in Fig. 4c. With increasing temperature, κL decreases drastically due to the enhancement of phonon dispersion. The electron thermal conductivity (κe) is the function of both temperature and chemical potential (μ). Owing to the metallic electronic band structure, κe should dominate the thermal transport of PHE-graphene just like other metals. Fig. 4d shows κe/τ as functions of temperature for five μ − EF, where μ is chemical potential. It can be seen that κe/τ increases with rising temperature. While μ − EF is −1 eV, κe/τ is 2.986 × 1014 W m−1 K−1 s−1. Accurate electron relaxation time of PHE-graphene is not available at present. However, a few studies of 2D carbon materials have confirmed that τ is usually tens of picoseconds (ps). For instance, τ is 14.6 ps for THD-graphene,53 13–14 ps for graphene68 and 16–19 ps for graphdiyne.69 Supposing the τ of PHE-graphene is 14 ps, close to sp2 hybridized THD-graphene and graphene, κe is estimated to be up to 4180 W m−1 K−1 while μ − EF is −1.0 eV. Such a high thermal conductivity of PHE-graphene is even comparable with single-layer graphene (5300 W m−1 K−1),70 and superior to few-layer graphene (1300–2800 W m−1 K−1),71 bulk graphite (2000 W m−1 K−1),72 carbon nanotubes (3000 W m−1 K−1)73 and diamond (1000–2200 W m−1 K−1).74
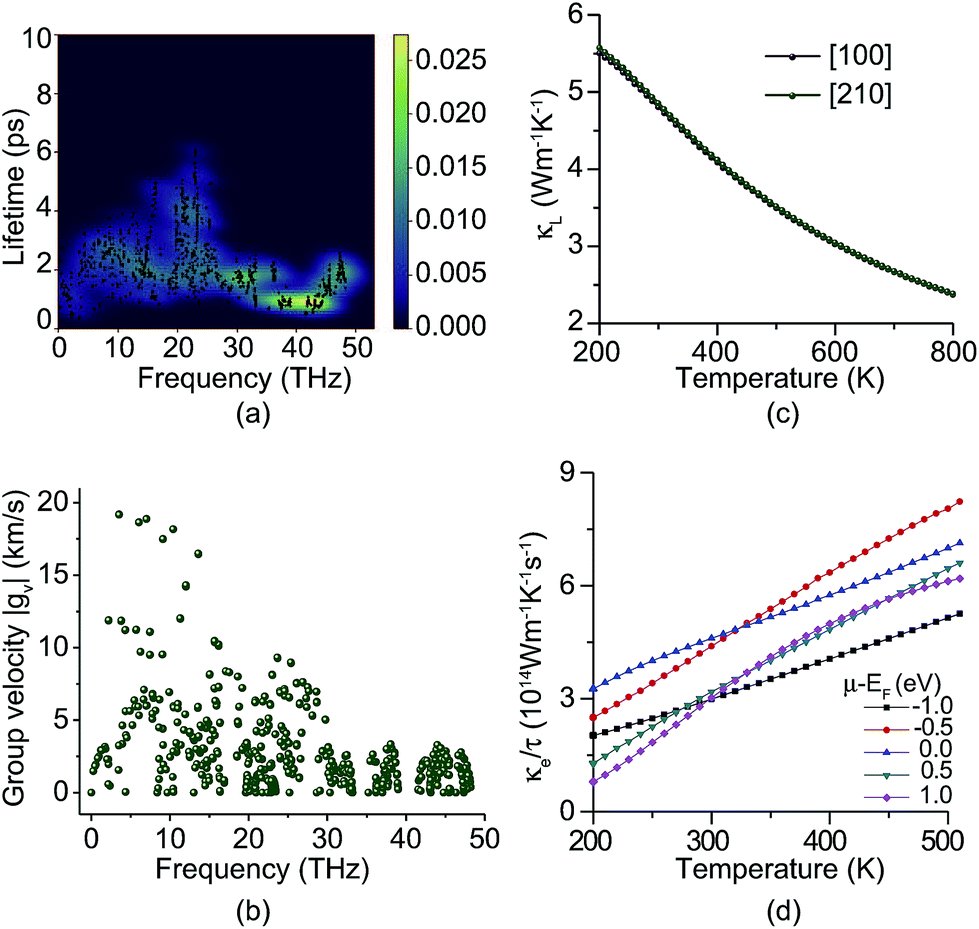 | ||
| Fig. 4 (a) Phonon lifetimes. (b) Phonon group velocity. (c) Lattice thermal conductivity (κL) of PHE-graphene as a function of temperature. (d) Electron thermal conductivity (κe) divided by relaxation time (τ) as a function of temperature. | ||
Fig. 5a shows the stress–strain relations. We found that the ideal tensile strength of PHE-graphene is up to 71.6 GPa which is far less than that (130 ± 10 GPa) of graphene.75 However, PHE-graphene still has a higher tensile strength relative to most 2D materials, such as MoS2 (25 GPa),76 silicene (12.5 GPa)77 and phosphorene (4.45 GPa).78 Before yielding, PHE-graphene can endure 15% tensile strain. As for 2D PHE-graphene, its elastic behavior under strain should be evaluated by in-plane stiffness C rather than Young’s modulus. If S0 is defined as the area of PHE-graphene at equilibrium geometry, the in-plane stiffness can be given by C = (1/S0) × (∂2E/∂ε2), where E is the strain energy calculated by subtracting the total energy of the strained system from the equilibrium total energy and ε is the strain. In this study, we applied strain ε on both the [100] and [010] directions at the same time, which meant that the 2D lattice constants or the unit cell were enlarged (1 + ε) times. Thus, we calculated the upper limit of the stiffness, i.e. Cmax. Fig. 5b shows the Cmax of the twenty carbon allotropes. It can be found that the Cmax significantly depends on the number of carbon atoms per unit area, Nσ. For sp + sp2 hybridized carbon sheets (α-graphyne, β-graphyne, graphdiyne, 6,6,12-graphyne, and γ-graphyne), they usually have a lower Cmax due to their small Nσ. In contrast, Cmax is usually higher than 300 N m−1 for sp2 hybridized carbon allotropes except for THD-graphene. Graphene has the highest Nσ (0.379 Å−1) and its Cmax is up to 422 N m−1, which is very close to the experimental upper limit (about 419 N m−1) of graphene.75 The Cmax of PHE-graphene was also up to 353 N m−1 and thus it possesses excellent mechanical properties.
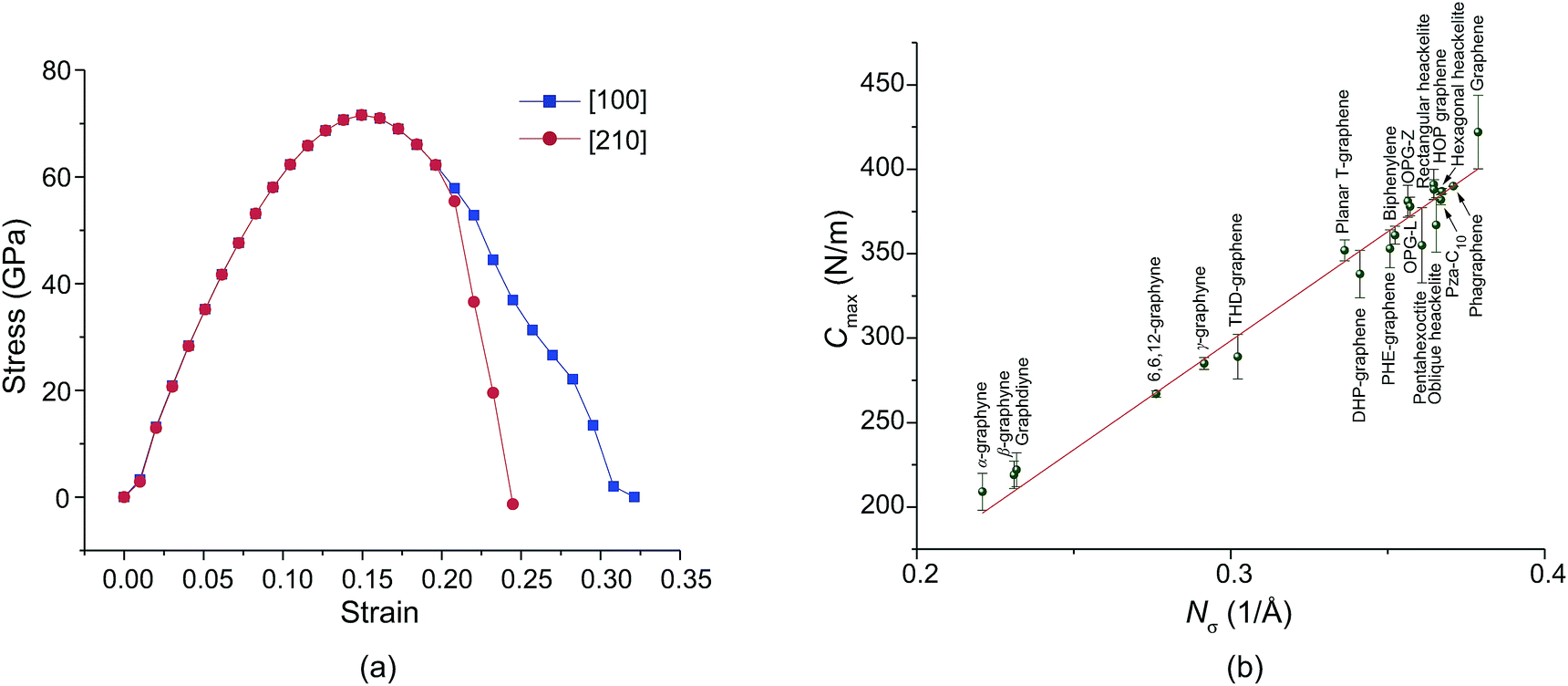 | ||
| Fig. 5 (a) The stress–strain relations of PHE-graphene in the [100] and [210] directions. (b) The upper limit of in-plane stiffness (Cmax) as a function of the number (Nσ) of carbon atoms per unit area. | ||
Fig. 6a shows the energy barrier of sulfur atoms passing through the enneagonal rings of PHE-graphene. It can be seen that a very high barrier prevents the sulfur atoms from penetrating through PHE-graphene. When one sulfur atom gradually approaches PHE-graphene, the total energy drops by 0.5 eV. This indicates that PHE-graphene has weak adsorption for sulfur atoms. The effects of the PBE functional and the DFT-D3 dispersion corrected functional on energy barriers are negligible. Surprisingly, we find that PHE-graphene strongly adsorbs Li atoms as shown in Fig. 6b. For the most stable adsorption geometry, the distance between Li and PHE-graphene was about 1.08 Å and the adsorption energy was 1.97 (1.90) eV at the PBE (DFT-D3) level. If one Li atom passes through the PHE-graphene, only an energy barrier around 0.47–0.48 eV needs to be overcome. In view of PHE-graphene preventing sulfur passing through it, PHE-graphene could be used to encapsulate the cathodes of Li–S batteries, by which the “shuttle effect” could be effectively avoided. The adsorption geometries and energies of Li2Sn (n ≤ 8) and S8 on PHE-graphene were also evaluated as shown in Table. S1 and Fig. S2 (see the ESI†). The adsorption energy was the highest for Li2S but was relatively weak for Li2Sn (3 ≤ n ≤ 8) and S8 species. In addition, the low conductivity faced in Li–S batteries at present can also be significantly improved because PHE-graphene is a metallic carbon material. Fig. 6c shows the vertical distance (Δd) between Li and PHE-graphene during Li atom diffusion on the PHE-graphene surface. The minimum Δd occurred at the enneagonal hollow sites and was around 1.08 Å. In contrast, the Δd was at a maximum on the top of C, which was about 1.97 Å. The Li atom diffusion barriers on the PHE-graphene surface are presented in Fig. 6d. The diffusion barriers between adjacent carbon rings were 0.589, 0.548, 0.301, and 0.302 eV for enneagon⇀hexagon, enneagon⇀pentagon, hexagon⇀pentagon, and pentagon⇀pentagon, and 0.395, 0.339, 0.286, and 0.302 eV for the reverse processes, respectively. Therefore, Li atoms shift readily along the path of ⋯hexagon⇀pentagon⇀pentagon⇀hexagon⋯ and the corresponding barriers were close to that (0.327 eV) of Li atom diffusion on the graphene surface.79
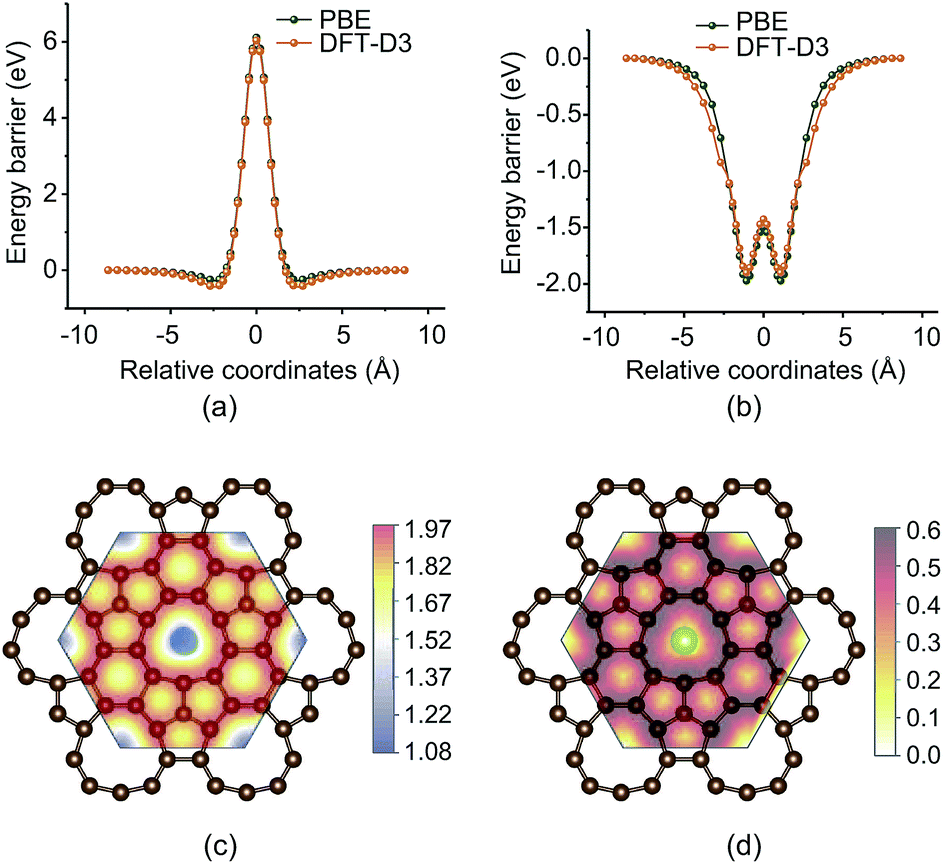 | ||
| Fig. 6 (a) and (b) show the energy barriers of S and Li atoms diffusing through the enneagon rings, respectively. (c) The vertical distance (Δd) between Li and the PHE-graphene during Li atom diffusion on the surface of PHE-graphene. (d) A 2D contour plot of the energy barrier for Li atom diffusion on the surface of PHE-graphene. | ||
4 Conclusions
In summary, we proposed a new carbon sheet (PHE-graphene) and suggested its preparation by a dehydrogenation reaction of small molecules. The thermodynamic and kinetic stability of PHE-graphene were examined by means of ab initio calculations. Electronic band structure calculations showed that PHE-graphene is a metallic carbon material. The density of states near the Fermi energy level completely result from the contributions of delocalized pz electrons. Similar to graphene, PHE-graphene is a typical π-conjugated system. Anharmonic lattice dynamics calculations revealed the low phonon lifetimes and group velocity in PHE-graphene, which directly results in its low lattice thermal conductivity. However, the electron thermal conductivity was found to be very high and even comparable with graphene. The enneagonal rings in PHE-graphene provide a passage for Li atoms but prevent S atoms from passing through. If PHE-graphene is utilized in cathodes, the major challenges faced by Li–S batteries, including low conductivity and the shuttle effect can be solved. Its high thermal conductivity is also helpful for the heat dissipation of cathodes. In addition, PHE-graphene also exhibits superior mechanical properties. Its ideal tensile strength was up to 71.6 GPa and the upper limit of its in-plane stiffness was 353 N m−1.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported financially by the National Natural Science Foundation of China (Grant No. 11464028), the Natural Science Foundation of Jiangxi Province, China (Grant No. 20171ACB21007) and the Department of Education of Jiangxi Province, China (Grant No. GJJ150025).Notes and references
- M. Arumugam, C. Sheng-Heng and Z. Chenxi, Adv. Mater., 2015, 27, 1980–2006 CrossRef.
- J.-Q. Huang, T.-Z. Zhuang, Q. Zhang, H.-J. Peng, C.-M. Chen and F. Wei, ACS Nano, 2015, 9, 3002–3011 CrossRef CAS.
- X. Yu, Z. Bi, F. Zhao and A. Manthiram, ACS Appl. Mater. Interfaces, 2015, 7, 16625–16631 CrossRef CAS.
- A. Manthiram, Y. Fu and Y.-S. Su, Acc. Chem. Res., 2013, 46, 1125–1134 CrossRef CAS.
- J. Xiao, J. Z. Hu, H. Chen, M. Vijayakumar, J. Zheng, H. Pan, E. D. Walter, M. Hu, X. Deng, J. Feng, B. Y. Liaw, M. Gu, Z. D. Deng, D. Lu, S. Xu, C. Wang and J. Liu, Nano Lett., 2015, 15, 3309–3316 CrossRef CAS.
- A. Kraytsberg and Y. Ein-Eli, J. Power Sources, 2011, 196, 886–893 CrossRef CAS.
- L. Li, S.-H. Chai, S. Dai and A. Manthiram, Energy Environ. Sci., 2014, 7, 2630–2636 RSC.
- L. Jang-Soo, T. K. Sun, C. Ruiguo, C. Nam-Soon, L. Meilin, L. K. Tae and C. Jaephil, Adv. Energy Mater., 2010, 1, 34–50 CrossRef.
- Q. Wang, N. Yan, M. Wang, C. Qu, X. Yang, H. Zhang, X. Li and H. Zhang, ACS Appl. Mater. Interfaces, 2015, 7, 25002–25006 CrossRef CAS PubMed.
- D. Bing, Y. Changzhou, S. Laifa, X. Guiyin, N. Ping and Z. Xiaogang, Chem.–Eur. J., 2013, 19, 1013–1019 CrossRef PubMed.
- P. G. Bruce, S. A. Freunberger, L. J. Hardwick and J.-M. Tarascon, Nat. Mater., 2011, 11, 19 CrossRef PubMed.
- X. Ji and L. F. Nazar, J. Mater. Chem., 2010, 20, 9821–9826 RSC.
- B. Zhang, X. Qin, G. R. Li and X. P. Gao, Energy Environ. Sci., 2010, 3, 1531–1537 RSC.
- K. Kumaresan, Y. Mikhaylik and R. E. White, J. Electrochem. Soc., 2008, 155, A576–A582 CrossRef CAS.
- Y. V. Mikhaylik and J. R. Akridge, J. Electrochem. Soc., 2004, 151, A1969–A1976 CrossRef CAS.
- Y. Wei, Y. Tao, C. Zhang, J. Wang, W. Qiao, L. Ling and D. Long, Electrochim. Acta, 2016, 188, 385–392 CrossRef CAS.
- G. Zheng, Y. Yang, J. J. Cha, S. S. Hong and Y. Cui, Nano Lett., 2011, 11, 4462–4467 CrossRef CAS.
- J. Zang, T. An, Y. Dong, X. Fang, M. Zheng, Q. Dong and N. Zheng, Nano Res., 2015, 8, 2663–2675 CrossRef CAS.
- H. Wang, Y. Yang, Y. Liang, J. T. Robinson, Y. Li, A. Jackson, Y. Cui and H. Dai, Nano Lett., 2011, 11, 2644–2647 CrossRef CAS.
- X. Ji, K. T. Lee and L. F. Nazar, Nat. Mater., 2009, 8, 500 CrossRef CAS PubMed.
- X. Li, Y. Cao, W. Qi, L. V. Saraf, J. Xiao, Z. Nie, J. Mietek, J.-G. Zhang, B. Schwenzer and J. Liu, J. Mater. Chem., 2011, 21, 16603–16610 RSC.
- S. Wei, H. Zhang, Y. Huang, W. Wang, Y. Xia and Z. Yu, Energy Environ. Sci., 2011, 4, 736–740 RSC.
- Y. Fu and A. Manthiram, J. Phys. Chem. C, 2012, 116, 8910–8915 CrossRef CAS.
- Y. Zhang, Z. Bakenov, Y. Zhao, A. Konarov, T. N. L. Doan, M. Malik, T. Paron and P. Chen, J. Power Sources, 2012, 208, 1–8 CrossRef CAS.
- L. Xiao, Y. Cao, J. Xiao, B. Schwenzer, M. H. Engelhard, L. V. Saraf, Z. Nie, G. J. Exarhos and J. Liu, Adv. Mater., 2012, 24, 1176–1181 CrossRef CAS PubMed.
- Y. Bu, J. Wu, X. Zhao, K. Ding, Q. Liu, Y. Huang, J. Lv and Y. Wang, RSC Adv., 2016, 6, 104591–104596 RSC.
- G. Zheng, Q. Zhang, J. J. Cha, Y. Yang, W. Li, Z. W. Seh and Y. Cui, Nano Lett., 2013, 13, 1265–1270 CrossRef CAS PubMed.
- X. Li, M. Rao and W. Li, J. Solid State Electrochem., 2016, 20, 153–161 CrossRef CAS.
- G. Zhou, L.-C. Yin, D.-W. Wang, L. Li, S. Pei, I. R. Gentle, F. Li and H.-M. Cheng, ACS Nano, 2013, 7, 5367–5375 CrossRef CAS PubMed.
- Z. Chenxi and M. Arumugam, Adv. Energy Mater., 2013, 3, 1008–1012 CrossRef.
- T. Lin, Y. Tang, Y. Wang, H. Bi, Z. Liu, F. Huang, X. Xie and M. Jiang, Energy Environ. Sci., 2013, 6, 1283–1290 RSC.
- X. Yang, L. Zhang, F. Zhang, Y. Huang and Y. Chen, ACS Nano, 2014, 8, 5208–5215 CrossRef CAS PubMed.
- Z. Li, Y. Huang, L. Yuan, Z. Hao and Y. Huang, Carbon, 2015, 92, 41–63 CrossRef CAS.
- L. Ji, M. Rao, H. Zheng, L. Zhang, Y. Li, W. Duan, J. Guo, E. J. Cairns and Y. Zhang, J. Am. Chem. Soc., 2011, 133, 18522–18525 CrossRef CAS PubMed.
- Z. Wei Seh, W. Li, J. J. Cha, G. Zheng, Y. Yang, M. T. Mcdowell, P.-C. Hsu and Y. Cui, Nat. Commun., 2013, 4, 1331 CrossRef.
- X. Tao, J. Wang, Z. Ying, Q. Cai, G. Zheng, Y. Gan, H. Huang, Y. Xia, C. Liang, W. Zhang and Y. Cui, Nano Lett., 2014, 14, 5288–5294 CrossRef CAS.
- Q. Pang, D. Kundu, M. Cuisinier and L. F. Nazar, Nat. Commun., 2014, 5, 4759 CrossRef CAS PubMed.
- T. Zeng, X. Hu, P. Ji and G. Zhou, Solid State Ionics, 2016, 291, 47–68 CrossRef CAS.
- Y. Cai, Y. Guo, B. Jiang and Y. Lv, Sci. Rep., 2017, 7, 14948 CrossRef PubMed.
- J. P. Perdew, K. Burke and M. Ernzerhof, Phys. Rev. Lett., 1996, 77, 3865–3868 CrossRef CAS PubMed.
- G. Kresse and J. Furthmüller, Comput. Mater. Sci., 1996, 6, 15–50 CrossRef CAS.
- G. Kresse and J. Furthmüller, Phys. Rev. B: Condens. Matter Mater. Phys., 1996, 54, 11169–11186 CrossRef CAS.
- P. E. Blöchl, Phys. Rev. B: Condens. Matter Mater. Phys., 1994, 50, 17953–17979 CrossRef.
- G. Kresse and D. Joubert, Phys. Rev. B: Condens. Matter Mater. Phys., 1999, 59, 1758–1775 CrossRef CAS.
- A. Togo and I. Tanaka, Scr. Mater., 2015, 108, 1–5 CrossRef CAS.
- A. Togo, L. Chaput and I. Tanaka, Phys. Rev. B: Condens. Matter Mater. Phys., 2015, 91, 094306 CrossRef.
- G. K. Madsen, J. Carrete and M. J. Verstraete, Comput. Phys. Commun., 2018, 231, 140–145 CrossRef CAS.
- S. Grimme, J. Antony, S. Ehrlich and H. Krieg, J. Chem. Phys., 2010, 132, 154104 CrossRef.
- X. Cai, Y. Luo, B. Liu and H.-M. Cheng, Chem. Soc. Rev., 2018, 47, 6224–6266 RSC.
- Q.-S. Du, P.-D. Tang, H.-L. Huang, F.-L. Du, K. Huang, N.-Z. Xie, S.-Y. Long, Y.-M. Li, J.-S. Qiu and R.-B. Huang, Sci. Rep., 2017, 7, 40796 CrossRef CAS.
- M. Long, L. Tang, D. Wang, Y. Li and Z. Shuai, ACS Nano, 2011, 5, 2593–2600 CrossRef CAS.
- A. de Meijere, P. R. Schreiner, F.-M. Schüngel, F. Gerson and P. Merstetter, Chem. Commun., 1999, 2189–2190 RSC.
- X. Xu, B. Jiang, Y. Guo and Y. Cai, Mater. Chem. Phys., 2017, 200, 50–56 CrossRef CAS.
- H. Huang, Y. Li, Z. Liu, J. Wu and W. Duan, Phys. Rev. Lett., 2013, 110, 029603 CrossRef PubMed.
- H. Terrones, M. Terrones, E. Hernández, N. Grobert, J.-C. Charlier and P. M. Ajayan, Phys. Rev. Lett., 2000, 84, 1716–1719 CrossRef CAS PubMed.
- X. Luo, L.-M. Liu, Z. Hu, W.-H. Wang, W.-X. Song, F. Li, S.-J. Zhao, H. Liu, H.-T. Wang and Y. Tian, J. Phys. Chem. Lett., 2012, 3, 3373–3378 CrossRef CAS.
- Z. Wang, X.-F. Zhou, X. Zhang, Q. Zhu, H. Dong, M. Zhao and A. R. Oganov, Nano Lett., 2015, 15, 6182–6186 CrossRef CAS PubMed.
- B. R. Sharma, A. Manjanath and A. K. Singh, Sci. Rep., 2014, 4, 7164 CrossRef CAS PubMed.
- V. H. Crespi, L. X. Benedict, M. L. Cohen and S. G. Louie, Phys. Rev. B: Condens. Matter Mater. Phys., 1996, 53, R13303–R13305 CrossRef CAS.
- C. Su, H. Jiang and J. Feng, Phys. Rev. B: Condens. Matter Mater. Phys., 2013, 87, 075453 CrossRef.
- B. Mandal, S. Sarkar, A. Pramanik and P. Sarkar, Phys. Chem. Chem. Phys., 2013, 15, 21001–21006 RSC.
- J. C. Meyer, A. K. Geim, M. I. Katsnelson, K. S. Novoselov, T. J. Booth and S. Roth, Nature, 2007, 446, 60 CrossRef CAS PubMed.
- N. Narita, S. Nagai, S. Suzuki and K. Nakao, Phys. Rev. B: Condens. Matter Mater. Phys., 1998, 58, 11009–11014 CrossRef CAS.
- M. A. Hudspeth, B. W. Whitman, V. Barone and J. E. Peralta, ACS Nano, 2010, 4, 4565–4570 CrossRef CAS PubMed.
- D. Malko, C. Neiss, F. Viñes and A. Görling, Phys. Rev. Lett., 2012, 108, 086804 CrossRef PubMed.
- J. Maultzsch, S. Reich, C. Thomsen, H. Requardt and P. Ordejón, Phys. Rev. Lett., 2004, 92, 075501 CrossRef CAS PubMed.
- O. Dubay and G. Kresse, Phys. Rev. B: Condens. Matter Mater. Phys., 2003, 67, 035401 CrossRef.
- J. Xi, M. Long, L. Tang, D. Wang and Z. Shuai, Nanoscale, 2012, 4, 4348–4369 RSC.
- M. Long, L. Tang, D. Wang, Y. Li and Z. Shuai, ACS Nano, 2011, 5, 2593–2600 CrossRef CAS PubMed.
- A. A. Balandin, S. Ghosh, W. Bao, I. Calizo, D. Teweldebrhan, F. Miao and C. N. Lau, Nano Lett., 2008, 8, 902–907 CrossRef CAS.
- S. Ghosh, W. Bao, D. L. Nika, S. Subrina, E. P. Pokatilov, C. N. Lau and A. A. Balandin, Nat. Mater., 2010, 9, 555 CrossRef CAS PubMed.
- A. A. Balandin, Nat. Mater., 2011, 10, 569 CrossRef CAS PubMed.
- H.-Y. Chiu, V. V. Deshpande, H. W. C. Postma, C. N. Lau, C. Mikó, L. Forró and M. Bockrath, Phys. Rev. Lett., 2005, 95, 226101 CrossRef PubMed.
- A. Sukhadolau, E. Ivakin, V. Ralchenko, A. Khomich, A. Vlasov and A. Popovich, Diamond Relat. Mater., 2005, 14, 589–593 CrossRef CAS.
- C. Lee, X. Wei, J. W. Kysar and J. Hone, Science, 2008, 321, 385–388 CrossRef CAS PubMed.
- T. Li, Phys. Rev. B: Condens. Matter Mater. Phys., 2012, 85, 235407 CrossRef.
- Q.-X. Pei, Z.-D. Sha, Y.-Y. Zhang and Y.-W. Zhang, J. Appl. Phys., 2014, 115, 023519 CrossRef.
- Z.-D. Sha, Q.-lX. Pei, Z. Ding, J.-W. Jiang and Y.-W. Zhang, J. Appl. Phys., 2015, 48, 395303 Search PubMed.
- C. Uthaisar and V. Barone, Nano Lett., 2010, 10, 2838–2842 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c8ra07074a |
| This journal is © The Royal Society of Chemistry 2019 |