
A colloidal heterostructured quantum dot sensitized carbon nanotube–TiO2 hybrid photoanode for high efficiency hydrogen generation†
Gurpreet Singh
Selopal
 ab,
Mahyar
Mohammadnezhad
b,
Fabiola
Navarro-Pardo
ab,
François
Vidal
b,
Haiguang
Zhao
*c,
Zhiming M.
Wang
*a and
Federico
Rosei
*ab
ab,
Mahyar
Mohammadnezhad
b,
Fabiola
Navarro-Pardo
ab,
François
Vidal
b,
Haiguang
Zhao
*c,
Zhiming M.
Wang
*a and
Federico
Rosei
*ab
aInstitute of Fundamental and Frontier Sciences, University of Electronic Science and Technology of China, Chengdu 610054, P. R. China. E-mail: zhmwang@uestc.edu.cn
bInstitut National de la Recherché Scientifique, Centre Énergie, Matériaux et Télécommunications, 1650 Boul. Lionel Boulet, J3X 1S2 Varennes, Québec, Canada. E-mail: rosei@emt.inrs.ca
cThe Cultivation Base for State Key Laboratory, Qingdao University, No. 308 Ningxia Road, Qingdao 266071, P. R. China. E-mail: hgzhao@qdu.edu.cn
First published on 6th November 2018
Abstract
Solar-driven photoelectrochemical (PEC) hydrogen (H2) generation is a promising approach to harvest solar energy for the production of a clean chemical fuel. However, the low photon-to-fuel conversion efficiency and long-term stability of PEC devices are major challenges to be addressed to enable large-scale commercialization. Here we report a simple, fast and cost-effective approach to fabricate high efficiency and stable PEC devices for H2 generation, by fabricating a hybrid photoanode obtained by incorporating small amounts of multiwall carbon nanotubes (MWCNTs) into a TiO2 mesoporous film and sensitizing with colloidal heterostructured CdSe/(CdSexS1−x)5/(CdS)2 quantum dots (QDs). The latter were specially designed to accelerate the exciton separation through a band engineering approach. The PEC devices based on the TiO2/QD–MWCNT (T/Q–M) hybrid photoanode with an optimized amount of MWCNTs (0.015 wt%) yield a saturated photocurrent density of 15.90 mA cm−2 (at 1.0 VRHE) under one sun illumination (AM 1.5G, 100 mW cm−2), which is 40% higher than that of the reference device based on TiO2/QD (T/Q) photoanodes. This is attributed to a synergistic effect of the promising optoelectronic properties of the colloidal heterostructured QDs and improved electron transport (reduced charge transfer resistance) within the TiO2–MWCNT hybrid anodes enabled by the directional path of MWCNTs for the photo-injected electrons towards FTO. Furthermore, the PEC device based on the T/Q–M hybrid photoanode is more stable (∼19% loss of its initial photocurrent density) when compared with the T/Q photoanode (∼35% loss) after two hours of continuous one sun illumination. Our results provide fundamental insights and a different approach to improve the efficiency and long-term stability of PEC devices and represent an essential step towards the commercialization of this emerging solar-to-fuel conversion technology.
Conceptual insightsHerein, for the first time, we report a novel and cost-effective approach to fabricate highly efficient and long-term stable photoelectrochemical (PEC) devices for hydrogen (H2) generation. At an optimized loading of multiwall carbon nanotubes (MWCNTs) in TiO2 and layered configuration of TiO2/QD–MWCNT (T/Q–M) hybrid photoanodes, the PEC device yields 40% higher photocurrent density than the control device. This is attributed to two features, namely: (i) enhanced electron transport, enabled by the directional path offered to the photo-injected electrons by the MWCNTs and (ii) the promising optoelectronic properties of the specially designed colloidal heterostructured QDs through a band engineering approach. In addition, the PEC devices based on the T/Q–M hybrid photoanodes maintain 81% of the initial value of photocurrent density after two hours of continuous one sun illumination, due to the reduced sensitivity of the T/Q–M hybrid photoanodes in the UV portion of the solar spectrum, combined with the strong conductive and thermally stable network of MWCNTs embedded within the TiO2 nanoparticles. Our findings define a new promising approach to improve the efficiency and long-term stability of PEC devices, which are essential factors for the commercialization of this emerging solar-to-fuel conversion technology. |
Introduction
Solar-driven photoelectrochemical (PEC) hydrogen (H2) generation is an attractive approach for the sustainable production of clean and renewable fuels, to address future global energy demands.1–4 A typical PEC device consists of a semiconductor photoanode and a cathode in an electrolyte solution. An electron–hole pair (exciton) is generated when a semiconductor photoanode absorbs a photon with energy higher than its band gap. A suitable band alignment of the semiconductor–electrolyte interface facilitates exciton separation, leading to hole oxidation and electron transfer through an external circuit to the cathode for H2 generation, as shown in Fig. 1(a).5 Following the pioneering work on PEC water splitting systems based on n-type TiO2 semiconductors,6 significant breakthroughs have been reported in developing high-efficiency photoanode materials based on wide band gap semiconductors including TiO2,7 Cu2O,8 ZnO,9 WO3,10 Fe2O3,11etc. Among these, TiO2 is considered as a low cost, stable, non-toxic and efficient photoanode material to fabricate PEC devices for sustainable H2 generation 7. However, due to its wide band gap (Eg = 3.2 eV), TiO2 can only absorb in the ultraviolet (UV) region of the solar spectrum, which represents a mere 5% of solar photons. Thus, TiO2 is not a suitable semiconductor for efficient solar-to-H2 generation.12,13 The sensitization of the TiO2 mesoporous anode with colloidal chalcogenide quantum dots (QDs) as the light harvesters is an effective approach to extend the photoanode's absorption spectrum toward the visible/NIR region, leading to significant improvement of the PEC performance.14–17 To date, various types of QDs such as PbS, CdS, CdSe and their composite heterostructures (core/shell QDs: PbS/CdS, CdSe/CdS QDs) have been extensively applied as a light harvester to sensitize TiO2 to fabricate high efficiency PEC devices for H2 generation.18–21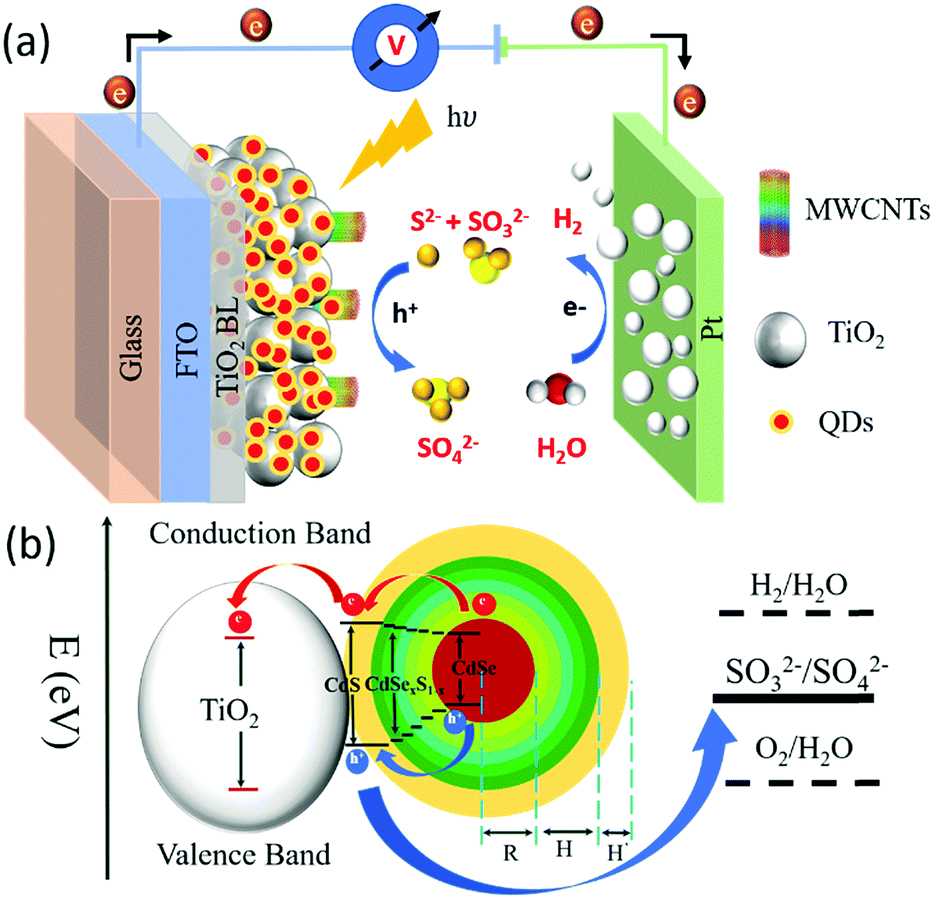 | ||
| Fig. 1 Schematic illustration of the: (a) PEC device working principle for H2 generation; (b) band structure of the graded alloyed core/shell QDs (R = radius of CdSe core; H = thickness of the graded alloyed interfacial layers; H′ = thickness of the outer CdS layer) sensitized TiO2–MWCNT hybrid film applied as the working electrode. The arrows show the electron and hole transfer process. | ||
In parallel, considerable efforts have focused on the design and synthesis of different morphologies of wide band gap semiconductors (e.g. TiO2, ZnO) such as nanorods,22 nanotubes23 and nanowires,24etc., to further increase the solar-to-H2 conversion efficiency. These one-dimensional (1D) nanostructures significantly improve the charge transport properties by directing their flow towards the acceptors. However, the overall efficiency of PEC devices based on these 1D nanostructures is still lower than the expected theoretical value. This is mainly attributed to the lower specific surface area of these 1D nanostructures as compared to nanoparticles.25 Carbon nanomaterials such as multi-walled carbon nanotubes (MWCNTs),26 graphene,27 graphene nanoribbons28 and graphene oxide29 are widely used in various optoelectronic devices due to their unique structural and optoelectronic properties such as excellent conductivity, high mechanical strength and optical transparency. The incorporation of MWCNTs into a TiO2 nanoparticle photoanode is considered as a viable alternative to the use of 1D nanostructures for the purpose of enhancing electron transport, while maintaining a high specific surface area.26,30 In previous work, we demonstrated that the limited addition (0.010 wt%) of MWCNTs in the TiO2 mesoporous layer improves the photovoltaic performance.26 This is mainly due to the enhanced electron transport of the injected electrons from dyes within the photoanode. To date, both colloidal heterostructured QD sensitization and incorporation of carbonaceous materials into wide band gap semiconductors have been explored separately to increase the performance of solar energy conversion devices,14–17,26 while little work has been done on the combination of these two approaches.31 However, to the best of our knowledge, there is no report on the application of MWCNTs in TiO2 mesoporous films together with the sensitization of colloidal heterostructured QDs for PEC devices.
Recent studies on wide band gap semiconductor sensitization with colloidal heterostructured QDs demonstrated that specially designed “giant” heterostructured QDs, with relatively thick shells (1.5 nm up to 10 nm) around the core were used as a sensitizer for TiO2 to fabricate high efficiency and stable PEC devices for H2 generation32 and other optoelectronic devices.33–35 These “giant” heterostructured QDs exhibit superior optoelectronic properties such as better photophysical/chemical stability, suppressed non-radiative Auger recombination, improved quantum yield (QY), and improved exciton lifetime compared to both pure QDs and core/thin-shell (shell thickness ≤1.5 nm) QDs.36,37 In particular, the formation of quasi-type-II core/shell heterostructures by tailoring the shell thickness/composition as well as the core size is the most promising feature of the “giant” heterostructured QDs. In this architecture, the electrons leak into the shell region, while holes are confined into the core region.38,39
Recently, Bae et al. demonstrated that Auger recombination is significantly suppressed by the addition of an interfacial alloyed layer between the CdSe/CdS core/shell sharp interface.40 In addition, we have observed that the presence of the interfacial layer between the core and shell of the “giant” core/shell QDs generates double color emission41 and enhances the photovoltaic performance.34 The performance of PEC devices based on colloidal QD sensitized wide band gap semiconductors was significantly improved in recent years. For example, in 2010 Lee et al.42 reported a photocurrent density of ∼14.90 mA cm−2 for a CdS/CdSe co-sensitized TiO2 photoelectrode, the highest reported value at the time. In 2013, Seol et al.43 reported a photocurrent density of 13.90 mA cm−2 from a PEC device based on CdSe/CdS/ZnO nanowires modified with IrOx·nH2O. However, the methodologies adopted in these works to improve the performance of PEC devices are quite complex and time-consuming. In addition, the low photon-to-fuel conversion efficiency and long-term stability of the PEC devices based on QD sensitized wide band gap semiconductors are still open issues for this system.
In this work, we report a simple, fast and cost-effective approach for the fabrication of TiO2–MWCNT hybrid anodes sensitized with colloidal heterostructured QDs, to boost the efficiency and long-term stability of PEC cells for H2 generation. The colloidal heterostructured CdSe/(CdSexS1−x)5/(CdS)2 QDs were specially designed to accelerate the exciton separation through a band engineering approach. The highest saturated photocurrent density of PEC devices based on the TiO2/QD–MWCNT (denoted as T/Q–M) hybrid photoanode with the optimized amount of MWCNTs (0.015 wt%) is 15.90 mA cm−2 (at 1.0 VRHE) under one sun illumination (AM 1.5G, 100 mW cm−2), which is 40% higher than the device based on TiO2/QD (denoted as T/Q) photoanodes. This significant improvement is ascribed to the enhanced electron transport properties of the T/Q–M hybrid photoanode with additional spin coated/scattering layers and promising optoelectronic properties of the colloidal heterostructured QDs as light harvesters. In addition, the PEC device based on T/Q–M (M = 0.015 wt%) photoanodes maintains 81% of the initial photocurrent density, while with the T/Q photoanodes it maintains only 65% after two hours of continuous one sun illumination.
Experimental
Materials
Sulfur (100%), oleylamine (OLA) (technical grade, 70%), cadmium oxide (99%), cadmium nitrate tetrahydrate (≥99%), oleic acid (OA), rhodamine 6G and octadecene (ODE), selenium pellets (≥99.999%), trioctyl phosphine oxide (TOPO), trioctyl phosphine (TOP) (97%), hexane, zinc acetate dihydrate (98%), sodium sulfide nonahydrate (≥99.9%), sodium sulfide (Na2S), sodium hydroxide, sodium sulfite (Na2SO3), toluene, methanol, acetone, ethanol, isopropanol (IPA) and MWCNTs with 10 μm average length and 12 nm average diameter (catalog number 698849) were obtained from Sigma-Aldrich Inc. Ti-nanoxide BL/SC was purchased from Solaronix. Titania paste (code18NR-AO) consisting of a blend of active anatase particles (20 nm in diameter) and larger anatase scattering particles (up to 450 nm in diameter) was supplied by Dyesol (Queanbeyan, Australia). Fluorine-doped tin oxide (FTO) coated glass substrates with sheet resistance of 10 Ω (square)−1 were purchased from Pilkington glasses. All chemicals were used as received without any further purification.Electrophoretic deposition (EPD) of the QDs on the TiO2 film and further ZnS/SiO2 coating
As-prepared TiO2–MWCNTs hybrid and TiO2 films grown on FTO substrates were vertically immersed in the QD dispersion in such a way that the deposited films were facing each other. The distance between them was adjusted to around 1 cm and a direct current (DC) bias of 200 V was applied for 120 min. To wash off the unabsorbed QDs after the EPD process, the samples were rinsed several times with toluene and dried under N2 flow at room temperature. Prior to ZnS capping, the photoanodes went through ligand exchange by applying three successive ionic layer adsorption and reaction (SILAR) cycles of methanolic solution cetyl-trimethyl ammonium bromide (CTAB) and toluene for 1 min dipping. After CTAB capping, 1 min dipping in methanol was applied to wash and remove the chemical residuals from the surface, followed by drying under N2. Then finally 1 min dipping in toluene was applied followed by drying under N2 to complete one SILAR cycle. The ZnS capping layer was formed using the SILAR process. In a typical SILAR deposition cycle, Zn2+ ions were deposited from a methanolic 0.1 M solution of Zn(OAc)2. The sulfide precursor was a 0.1 M solution of Na2S in a mixture of methanol/water (1/1 v/v). A single SILAR cycle consists of 1 min dip-coating of the TiO2 photoanode into the cation precursors (Zn2+), and subsequently into the anion solutions (S2−). After each bath, the photoanode was thoroughly rinsed by immersing it into the corresponding solvent (methanol or mixed solution) respectively, to remove the unabsorbed chemical residuals and dried under N2. Two SILAR cycles were applied to form the ZnS capping layer. After ZnS capping, the SiO2 coating was carried out by soaking the photoanode in a 0.01 M ethanolic solution of tetraethylorthosilicate for 1 hour at 35 °C and then rinsed with ethanol and dried under N2.Characterization
X-ray diffraction (XRD) of extensively purified QDs was carried out using a Philips X′pert diffractometer using a Cu Kα radiation source (λ = 0.15418 nm). Transmission electron microscopy (TEM) and high-resolution-TEM (HR-TEM) images of the QDs and T/Q film were collected by using a JEOL 2100F TEM system. Scanning electron microscopy (FE-SEM, JEOL JSM-6900 F) was used to study the morphology of the TiO2 and TiO2–MWCNT hybrid photoanodes. Thermogravimetric analysis (TGA) measurements were performed using a Q500 TGA thermogravimetric analyzer (TA instruments) in the temperature range of 30–700 °C at a heating rate of 15 °C min−1. The chemical composition mapping of the QDs and T/Q photoanodes was carried out using energy dispersive X-ray spectroscopy (EDS). The Raman spectra of the photoanode were recorded using a Renishaw InVia spectrometers coupled with a 514 nm excitation source. The UV-vis absorption spectra were recorded on a Cary 5000 UV-vis-NIR spectrophotometer (Varian) with a scan speed of 600 nm min−1. Fluorescence spectra were recorded using a Fluorolog-3 system (Horiba Jobin Yvon). The PL lifetime of the QDs was measured in the time-correlated single-photon counting (TCSPC) mode using a 444 nm laser.The theoretical wave functions of the electrons and holes were calculated by solving the stationary Schrödinger equation in spherical geometry, in which we used the bulk values for the effective masses of electrons (me*) and holes (mh*), namely me* = 0.44me and mh* = 0.13me for CdSe,44 and me* = 0.2me and mh* = 0.7me for CdS, where me is the electron mass at rest under vacuum.45 The potentials of the electrons and holes as a function of position were approximated as the lowest unoccupied molecular orbital (LUMO) and the highest occupied molecular orbital (HOMO) levels, respectively, for the bulk materials. For CdSe, these levels are −3.71 and −5.81 eV, respectively, while for CdS they are −3.3 and −5.8 eV, respectively. Outside the QDs, the potentials were set to 0 and −9.8 eV for the electrons and holes, respectively. The interactions between the electrons and holes were neglected in the calculations.
The PEC performance of the photoanodes was evaluated in a typical three-electrode configuration, consisting of a T/Q–M photoanode as a working electrode, a Pt counter electrode, and a saturated Ag/AgCl reference electrode. An insulating epoxy resin was used to cover the sample's surface except for the active area, to avoid any direct contact between the electrolyte and the conducting back-contact and/or the connecting wire. Subsequently, the sample was fully immersed in the electrolyte containing 0.25 M Na2S and 0.35 M Na2SO3 (pH ∼ 13) as the sacrificial hole scavenger. All potentials, measured with respect to the reference electrode of Ag/AgCl during the PEC measurements, were converted to scale according to the following equation VRHE = VAg/AgCl + 0.197 + pH × (0.059). The photoresponse was measured by using a 150 W xenon lamp as the light source with an AM 1.5G filter. The sample was placed at a distance of 2 cm from the lamp case (7 cm away from the actual bulb). Prior to each measurement, light intensity was monitored by a thermopile and adjusted to 100 mW cm−2. All the current versus potential measurements were carried out at a 20 mV s−1 sweep rate. The wavelength-dependent incident photon-to-current efficiency (IPCE) was calculated according to the following equation:46

Electrochemical impedance spectroscopy (EIS) was carried out under dark conditions by using a SOLARTRON 1260 A Impedance/Gain-Phase Analyzer with a bias voltage from 550 to 750 mV. The tests were recorded over a frequency range of 10 mHz and 300 kHz, and the AC signal was 10 mV in amplitude. All the impedance measurements were analysed using an appropriate equivalent circuit model with Z-View software (v3.5, Scribner Associate, Inc.).
Results and discussion
Structural characterizations of the hybrid anode film
Fig. 2(a–f) shows the SEM images of the TiO2 and TiO2–MWCNTs hybrid mesoporous films at different concentrations of MWCNTs deposited on the ultrasonically cleaned FTO glass substrate. However, it was not possible to see the MWCNTs in the SEM images of the TiO2–MWCNT hybrid mesoporous film due to the low concentration of MWCNTs (0.015 wt%) and their complete conformal coverage by the TiO2 nanoparticles. The surface of the TiO2–MWCNT hybrid mesoporous film at a high concentration of MWCNTs (0.030 wt%) shows deep cracks [shown by white arrows in Fig. 2(f) and rectangular circles in the inset of Fig. 2(f)], which are absent in the TiO2 (Fig. 2(a and b)) and TiO2–MWCNT hybrid mesoporous film with an optimized concentration of MWCNTs (0.015 wt%) (Fig. 2(c and d)). Such cracks lead to electron trapping, which hinders efficient electron transport, and also act as recombination sites for back-electron transfer at the electrolyte/FTO interface. This reduces the overall PEC performance, as discussed later.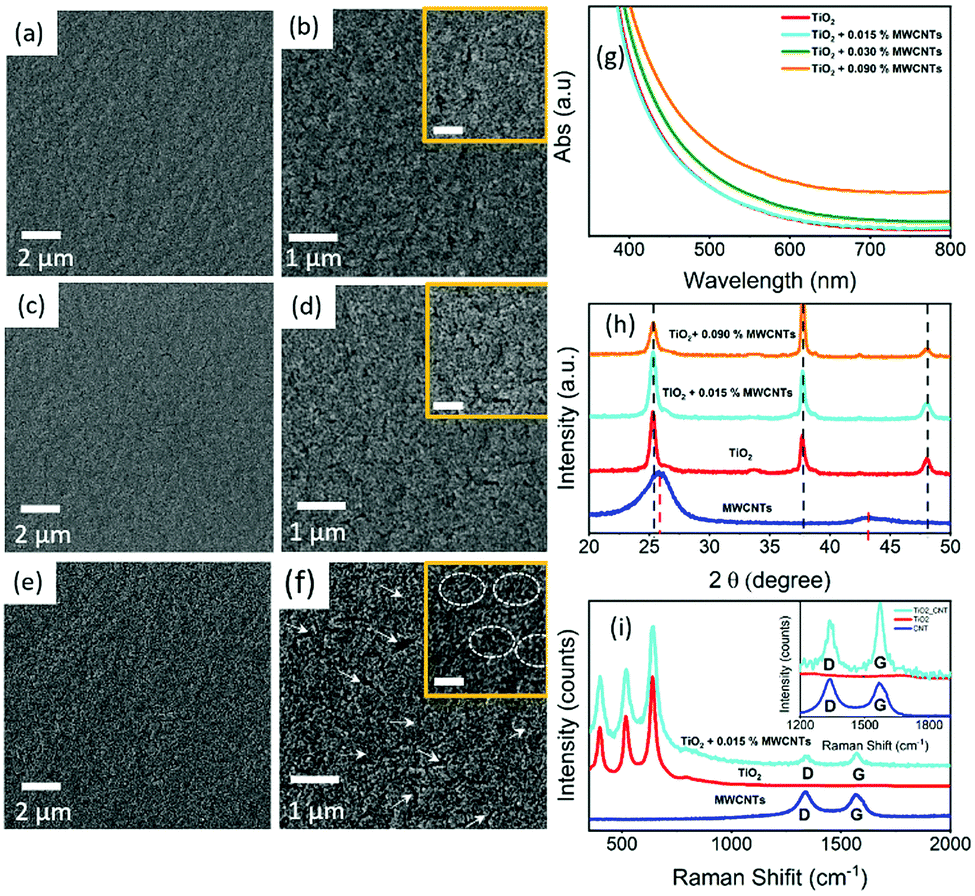 | ||
| Fig. 2 SEM images of mesoporous TiO2 films with different content of MWNCTs: (a and b) 0 wt %; (c and d) 0.015 wt% and (e and f) 0.030 wt% at different magnifications. The inset of b, d, and f are high-resolution SEM images of corresponding mesoporous films. The presence of deep cracks induced at higher concentrations of the MWCNTs is clearly visible in (f) (white arrows) and the inset (dotted circles highlights a crack zone area). A systematic comparison of the patterns of MWCNTs, bare TiO2 and TiO2–MWCNT mesoporous films at different concentrations of MWNCTs: (g) absorption spectra; (h) XRD; (i) Raman spectra, the inset highlights the D and G bands of the MWCNTs and TiO2–MWCNTs. | ||
The optical properties of the TiO2 mesoporous film were modified by the addition of MWCNTs. Fig. 2(g) shows the absorption spectra of TiO2 and TiO2–MWNCT hybrid mesoporous films at different concentrations of MWCNTs in the UV-visible region. The results show that the transparency of the TiO2–MWNCT hybrid film in the UV-visible region is not affected by the addition of MWNCTs up to 0.015 wt%. However, on addition of higher concentrations of MWCNTs above 0.015 wt%, light absorption by the MWNCTs increases, which reduces the transparency of the TiO2–MWNCT hybrid film [Fig. 2(g)]. This direct light absorption by the MWCNTs results in loss of part of the solar radiation available for exciton generation, which in turn may reduce the overall PEC performance.
Fig. 2(h) displays the XRD pattern of the MWNCT powder, TiO2 and TiO2–MWCNT hybrid anode at different concentrations of MWCNTs. The characteristic peaks of the XRD pattern of the MWNCTs are observed at 2θ = 26.0° and 43.0°.47 The presence of XRD pattern peaks at 2θ = 25.3°, 37.8° and 48.1° in both the TiO2 and TiO2–MWCNT hybrid anode, confirms the anatase phase of TiO2. The peaks at 2θ = 25.3° and 48.1° correspond to the (101) and (200) anatase crystal planes of TiO2 (JCPDS No. 21-1272) consistent with the previous literature reported on the same system.48,49 However, there is no characteristic peak of MWCNTs observed in the TiO2–MWCNTs hybrid films at different concentrations of MWCNTs. This is mainly due to the overlap of the characteristic peak of MWCNTs (at 2θ = 26.0°) and the anatase TiO2 (at 2θ = 25.3°).
In addition, the concentration of MWCNTs in the TiO2–MWCNT hybrid film is below the XRD detection limit.
Fig. 2(i) depicts the comparison of the Raman spectra of MWCNTs, TiO2 and the TiO2–MWCNT hybrid anode with an optimized concentration of MWCNTs. The pristine MWCNTs display the presence of the D and G bands at 1350 cm−1 and 1580 cm−1 respectively, which are the specific peaks of the carbonaceous materials.50 The Raman spectra of the TiO2–MWCNT hybrid film clearly show the presence of D and G bands at 1350 cm−1 and 1580 cm−1 respectively (see inset of Fig. 2(i)) even at a very low concentration of MWCNTs (0.015 wt%). This confirms the presence of MWNCTs without any structural changes in the TiO2–MWCNT hybrid photoanodes after 30 min annealing at 500 °C under ambient conditions. The stability of the MWCNTs at 500 °C under ambient conditions is further confirmed by the TGA of MWCNTs in the temperature range of 30–500 °C (see Fig. S1, ESI†). However, the relative intensity of the D and G bands is lower, which may be due to the conformal coverage of MWCNTs by the TiO2 nanoparticles.
Structural/optical characterizations of the colloidal heterostructured QDs
The diameter of the starting CdSe core QDs is around 3.30 ± 0.29 nm, as calculated from the TEM image (see Fig. S2, ESI†). The size distribution of the as-synthesized CdSe core QDs is shown in Fig. S2(b) (ESI†). The increased size of the QDs after graded alloyed shell growth was confirmed by TEM imaging (see Fig. 3(a)). After the growth of five interfacial layers of CdSexS1−x (x = 0.9–0.1) and two monolayers of CdS, the final diameter reaches 7.6 ± 1.3 nm (H = 2.15 nm). The size distribution of the as-synthesized graded alloyed core/shell QDs is shown in Fig. S2(c) (ESI†). The high crystallinity with lattice fringes of the QDs is clearly visible in the HRTEM image (see Fig. 3(b)). The calculated lattice parameter of the QDs is 3.61 Å which corresponds to (101) lattice spacing of the wurtzite (WZ) crystal structure consistent with the SAED and XRD patterns. An HRTEM image of the QD sensitized TiO2 film is shown in Fig. 3(c). | ||
| Fig. 3 TEM images: (a) (Cdse)/(CdSexS1−x)5/(CdS)1 heterostructured QDs; (b) HRTEM image; (c) HRTEM of the QD sensitized TiO2 mesoporous film, the dotted orange circles highlight the presence of QDs. Optical properties of QDs: (d) absorption spectrum in the UV-visible region of the as synthesized QDs in toluene; (e) PL spectrum of the QDs in toluene; (f) XRD pattern of the QDs. The Joint Committee on Powder Diffraction Standards (JCPDS) card files for CdSe (00190191, green dashed for ZB and 08-459, dark green for WZ) and CdS (01-077-2306, cyan for WZ) are shown for identification. | ||
The homogeneous dispersion of the QDs (orange dotted circles) on the surface of the TiO2 mesoporous film without any aggregation during the deposition of QDs via EPD is confirmed by TEM imaging [see Fig. 3(c)]. This is consistent with the measurements of the optical properties of the as-synthesized QDs and QDs deposited onto TiO2 mesoporous films [Fig. S2(e), ESI†]. The EDS spectra of the colloidal heterostructured QDs and T/Q photoanode coated with ZnS and SiO2 are shown in Fig. S3 [ESI†]. The EDS spectra of the QDs confirm the presence of Cd, Se and S elements (Fig. S3(a), ESI†). Similarly, the EDS spectra of the T/Q photoanode coated with ZnS and SiO2 confirms the presence of Ti, Cd, Se, S, Zn and Si (Fig. S3(b), ESI†). In addition, the EDS spectrum of T/Q is also acquired by SEM. Fig. S4 (ESI†) shows the EDS spectra of T/Q and the corresponding elemental wt% ratios reported in Table S1 (ESI†), which confirms the presence of Ti, O, Cd, Se and S.
The absorption spectrum of the as-synthesized QDs is shown in Fig. 3(d). The results show that the first excitonic absorption peak is observed at around 627 nm and the absorption spectrum from 400 to 700 nm. The PL spectrum of the corresponding QDs in toluene is shown in Fig. 3(e). The PL peak of the as-synthesized QDs in toluene is observed at 650 nm.
The crystal structure of the QDs deposited on a silicon substrate was characterized by XRD. Fig. 3(f) displays the XRD patterns of the colloidal heterostructured QDs. The peak positions of the XRD pattern of the QDs shows the combined reflection of the WZ crystal structure of CdS and the WZ crystal structure of CdSe, which confirms the formation of alloyed QDs and is consistent with our previous reports.34 The SAED pattern of a QD shown in Fig. S2(d) (ESI†) is ascribed to (111), (211), (220), (310), and (311) planes of the WZ phase respectively. These findings are consistent with XRD analysis.
Carrier dynamics of T/Q
The band structure and carrier dynamics of the colloidal heterostructured QD sensitized TiO2 photoanodes is shown in Fig. 1(b). An exciton is generated upon the absorption of a photon, which is dissociated at the T/Q interface. Although the conduction band (CB) of CdS is higher than that of CdSe, the presence of a gradient band alignment offered by the CdSexS1−x alloyed interfacial layers facilitates the electrons to overcome the original high energy barrier and improves the leakage of electrons from the CdSe core to the CdS shell (quasi type-II).38,39 The formation of the quasi-type-II core/shell heterostructure band alignment of the gradient CdSe/(CdSexS1−x)5/(CdS)2 heterostructure QDs is confirmed by the red shift in PL34 and calculated electron/hole spatial overlap from theoretical simulations [see Fig. 4(c)]. In this band alignment, the electrons and holes leak into the shell region and then from the CdS shell to the CB of TiO2. Similarly, the holes leak from the CdSe core region to the shell region and are then extracted by the electrolyte (Na2S/Na2SO3, pH ∼ 13), which serves as a hole scavenger.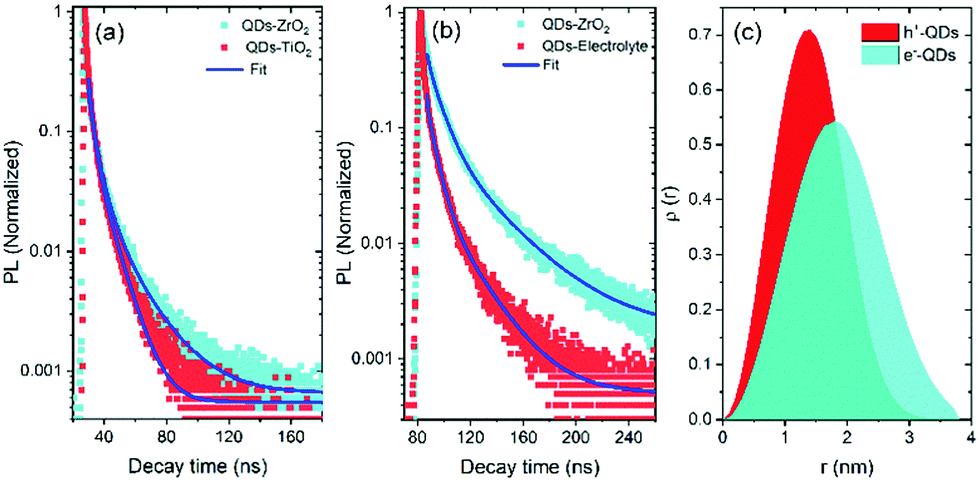 | ||
| Fig. 4 Comparison of the transient PL curves of heterostructured QDs: (a) deposited onto a ZrO2 and TiO2 mesoporous film; (b) deposited onto a ZrO2 mesoporous film and QDs with electrolyte. The excitation wavelength is λex = 444 nm. (c) Comparison of the spatial probability distribution value ρ(r) of the electron and hole as a function of QD radius r (nm) (H = 2.15 nm, red for hole and cyan for an electron). | ||
Transient PL spectroscopy was applied to investigate the charge dynamics of the graded alloyed core/shell QDs coupled with carrier scavengers including TiO2 and the electrolyte. The absorption of a photon by the QDs generates an exciton which dissociates at the QD/metal oxide and the QD/redox couple electrolyte interface. The optimized band alignment of the CB of the QDs with the CB of the metal oxide and the valence band (VB) of the QDs with the redox potential of the electrolyte, leads to an efficient transfer of the photogenerated electron and hole. The electron lifetime and carrier transfer rate of the QDs with carrier scavengers (metal oxide and electrolyte) were measured under an excitation wavelength λ = 444 nm. All the PL decay curves were well fitted using an exponential decay. The intensity-weighted average lifetime (〈τ〉) was calculated by using the following equation:
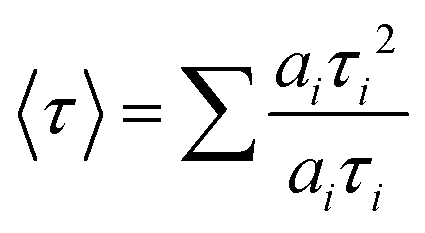 | (1) |
In this measurement, the TiO2 mesoporous film was used as an electron scavenger, while the electrolyte containing 0.25 M Na2S and 0.35 M Na2SO3 (pH ∼ 13) serves as a hole scavenger. We already ruled out the possibility of energy transfer from the QDs to the metal oxide films due to the lack of spectral overlap between the PL of the QDs and the absorption of the metal oxide.32 Hence the variation of PL decay rate is dependent on the electron transfer from the QDs to the metal oxide. These results demonstrate that PL decay is faster in the case of QDs coupled with a TiO2 mesoporous film than the QDs coupled with a ZrO2 mesoporous film [see Fig. 4(a)], confirming that the electron transfer from the CB of the QDs to the CB of TiO2 is more efficient than that to the CB of ZrO2. This is mainly due to the difference in the electronic band alignment between ZrO2 (Eg = 5 eV)51 and TiO2, which is not suitable for electron transfer from QDs. Similarly, a hole transfer from the VB of the QDs to the electrolyte is more efficient than that to the VB of ZrO2.
The carrier lifetime values for QDs coupled with the TiO2 mesoporous film (an electron scavenger) and the electrolyte (a hole scavenger) are shorter than that of the QDs coupled with a ZrO2 mesoporous film, as shown in Fig. 4(a and b).52 We calculated the carrier transport rate (Ket![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) orht) from the differences in the carrier lifetime values, by using the following equation:
orht) from the differences in the carrier lifetime values, by using the following equation:
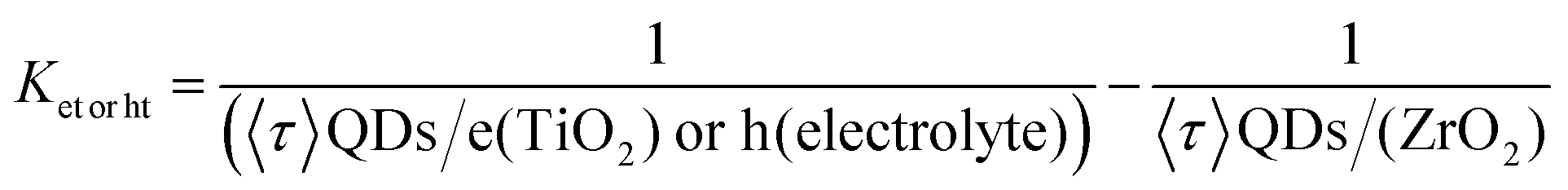 | (2) |
The calculated electron transport rate (Ket = 2.6 ± 0.1 × 107 s−1) and hole transport rate (Kht = 5.1 ± 0.1 × 107 s−1) values52,53 confirm efficient carrier transfer from the QDs to the carrier scavengers (TiO2 and electrolyte).
To further investigate the optoelectronic properties of the QDs, we calculated the electron (ψe(r)) and hole (ψh(r)) wave functions by solving the stationary Schrödinger wave equation in a spherical geometry44 (more details are reported in the Experimental section). Fig. 4(c) displays the variation of calculated spatial probability distribution (ρ(r)) of ψe(r) and ψh(r) as a function of QD radius (r (nm)). The ρ(r) versus r (nm) curves show that the electrons leak into the shell region, while the holes are confined in the CdSe core. This confirms the possible formation of a quasi-type II core/shell band alignment38,39 with the electron–hole spatial overlap area of 72%.
PEC measurements
As a proof of concept, colloidal heterostructured sensitized TiO2 and TiO2–MWCNTs hybrid mesoporous films at different concentrations of MWCNTs were applied as a working electrode in a PEC system, to highlight the effects of MWCNTs. The thickness of the TiO2–MWCNT hybrid and bare TiO2 mesoporous films is 12 ± 0.5 micrometres.The PEC cell measurements were carried out in the dark, under continuous and chopped illumination (AM 1.5G, 100 mW cm−2) by using a typical three electrode configuration system. The current density vs. applied potential curves of the T/Q–M photoanode at different concentrations of MWCNTs and the T/Q photoanode as a benchmark are shown in Fig. 5(a–f). The photocurrent density for all samples gradually increases with the increase in the concentration of MWCNTs in TiO2. The variation of saturated photocurrent density values versus the concentration of MWCNTs in the T/Q–M photoanode at 1.0 VRHE is summarized in Fig. 5(g). The highest saturated photocurrent density of the PEC system based on the T/Q photoanode is 9.05 mA cm−2 under one sun illumination [Fig. 5(a)]. With the addition of 0.010 wt% of MWCNTs into the TiO2 mesoporous film, the saturated photocurrent density increases to 9.80 mA cm−2 under one sun illumination [Fig. 5(b)], then reaches a maximum value of 10.87 mA cm−2 at 0.015 wt% MWCNTs addition, which is 20% higher than that of the PEC system based on the T/Q photoanode [Fig. 5(c)]. This improvement in photocurrent density with the inclusion of 0.015 wt% MWCNTs in T/Q–M is mainly attributed to enhanced electron transport and reduced charge transfer resistance (see discussion in the ESI† section). The high electrical conductivity of the MWCNTs provides a direct pathway to the photoinjected electrons towards the FTO without passing through the numerous grain boundaries of TiO2 nanoparticles, thereby enhancing electron collection. Moreover, this small concentration of MWCNTs (0.015 wt%) in mesoporous TiO2 does not affect the optical transparency and structural morphology, as confirmed by UV-visible and SEM measurements respectively. For further increases in the concentration of MWCNTs, the photocurrent density reduces from 10.87 to 8.38 mA cm−2 (0.020 wt%) [Fig. 5(d)], 6.33 mA cm−2 (0.025 wt% MWCNTs) [Fig. 5(e)] and 5.23 mA cm−2 (0.030 wt%) [Fig. 5(f)]. This is mainly due to the detrimental effects of a high concentration of MWCNTs, such as the formation of cracks, which act as the recombination centers during carrier transport, and also due to the reduced optical transparency of the hybrid photoanode, as aforementioned.
 | ||
| Fig. 5 Photocurrent density-potential curves of PEC devices based on colloidal heterostructured QD sensitized TiO2–MWCNTs hybrid photoanodes with different concentrations of MWCNTs under dark, continuous and chopped illumination (AM 1.5G, 100 mW cm−2): (a) 0.0 wt%; (b) 0.010 wt%; (c) 0.015 wt%; (d) 0.020 wt%; (e) 0.025 wt% and (f) 0.030 wt%. (g) Variation of current density at 1.0 V vs. RHE under 100 mW cm−2 illumination with the concentration of MWCNTs. | ||
Effect of the spin-coated TiO2 layer
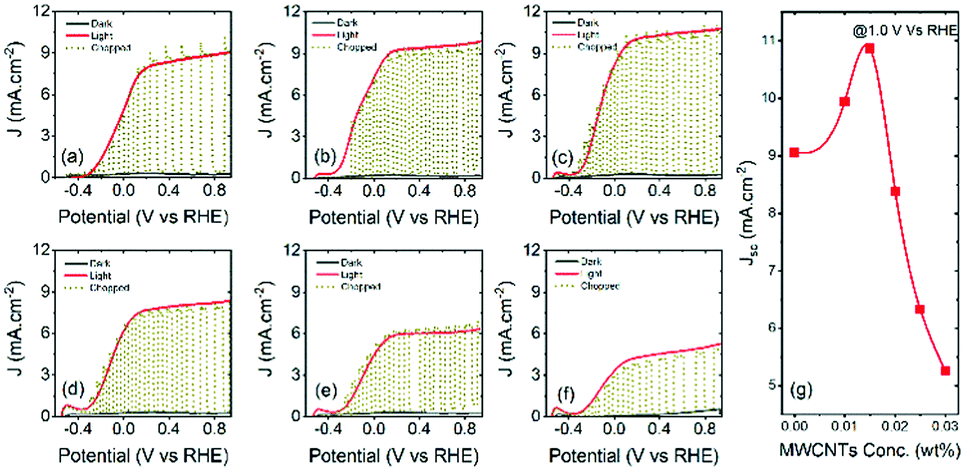
After the systematic analysis of effects of MWCNTs on the photocurrent density of PEC devices, the best performing T/Q–M (M = 0.015 wt%) photoanodes were further optimized by adding an additional spin-coated TiO2 layer composed of 20 nm sized particles between the blocking and active layers. The total thickness of the TiO2–MWCNT hybrid and bare TiO2 mesoporous films is 13 ± 0.1 micrometres including a 300 nm spin-coated layer after annealing at 500 °C for 30 min. The PEC devices based on the T/Q and T/Q–M (M = 0.015 wt%) photoanodes with a spin-coated TiO2 layer exhibit a higher photocurrent density as compared to the respective configuration of the photoanodes without the spin-coated TiO2 layer. A systematic comparison of photocurrent density under one sun illumination is shown in Fig. S5 (ESI†) and the corresponding values at 1.0 VRHE are reported in Table 1. The photocurrent density increases from 9.05 to 9.60 mA cm−2 for the bare T/Q photoanode (see Fig. S5(a), ESI†), whereas in the case of the T/Q–M (M = 0.015 wt%) photoanode the photocurrent density increases significantly from 10.87 to 11.94 mA cm−2 (see Fig. S5(b), ESI†). This is mainly attributed to an improved adhesion between the compact blocking and the mesoporous active layer of TiO2 particles with an additional spin coated TiO2 nanoparticle layer. A significant improvement in the PEC performance of the T/Q–M (M = 0.015 wt%) photoanode is due to the synergetic effect of enhanced electron transport and improved adhesion.| Photoanode structure | Spin-coated layer | Scattering layer | J (mA cm−2) |
|---|---|---|---|
| T/Q | No | No | 9.05 |
| Yes | No | 9.60 | |
| Yes | Yes | 11.30 | |
| T/Q–M | No | No | 10.87 |
| Yes | No | 11.94 | |
| Yes | Yes | 15.90 | |
Effect of the TiO2 scattering layer
The performance of the PEC system based on T/Q–M (M = 0.015 wt%) photoanodes was further enhanced by adding a 4–5 μm thick scattering layer of 150–200 nm sized particles over the mesoporous active layer. The latter system with the added scattering layer shows significantly enhanced photocurrent density as compared to the respective configuration of photoanodes without MWCNTs [shown in Fig. 6(a and b)]. The total thickness of the TiO2–MWCNT hybrid and bare TiO2 mesoporous films including the scattering layer is 16 ± 0.5 micrometres. The saturated photocurrent density values at 1.0 V vs. RHE of the respective PEC devices under continuous and chopped illumination (AM 1.5G, 100 mW cm2) are reported in Table 1.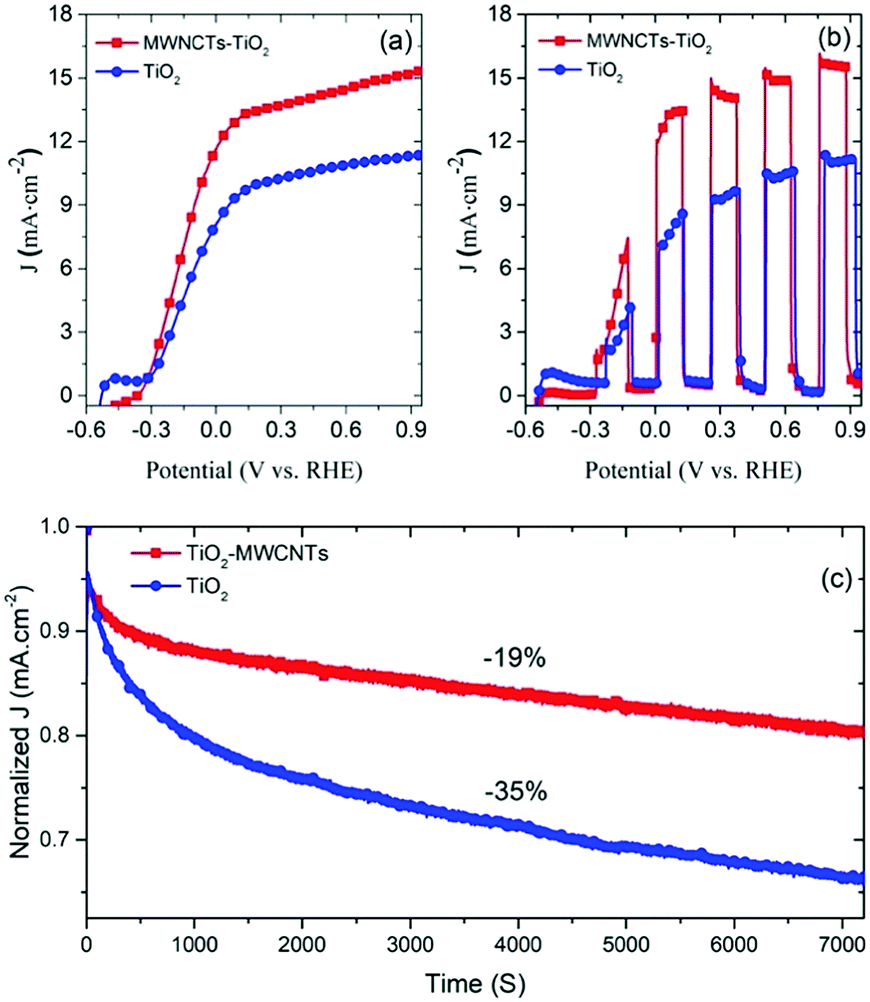 | ||
| Fig. 6 Photocurrent density versus bias potential (versus RHE) of PEC systems based on the colloidal alloyed core/shell QDs sensitized pristine TiO2 (blue circle) and TiO2–MWCNT hybrid (red square) photoanodes using three electrode configurations under one sun illumination (AM 1.5G, 100 mW cm−2): (a) under continuous illumination; (b) chopped illumination. Comparison of the long-term stability of PEC devices based on TiO2–MWCNT (red square) hybrid and bare TiO2 (blue circle) photoanodes: (c) normalized current density (mW cm−2) versus time at 0.6 V (versus RHE) under one sun continuous illumination (AM 1.5G, 100 mW cm−2). | ||
The photocurrent density increases from 11.30 to 15.90 mA cm−2. This is mainly attributed to the effect of the scattering layer that acts as a light scattering barrier. Light scattering extends the time spent by light inside the active layer. Consequently, the possibility of light captured by the QDs is higher for a photoanode with a scattering layer.
The PEC system based on the T/Q–M (M = 0.015 wt%) photoanode with a scattering layer shows better performance compared to the T/Q photoanodes due to the combined effect of the improved electron transport and enhanced light absorption.
A systematic comparison of the performance of the PEC system based on the T/Q and T/Q–M (M = 0.015 wt%) photoanodes with and without a scattering layer is shown in Fig. S6 (ESI†). The photocurrent density increases from 9.60 to 11.30 mA cm−2 for the T/Q photoanode, whereas in the case of T/Q–M (M = 0.015 wt%), the photocurrent density increases significantly from 11.94 to 15.90 mA cm−2. Hence the champion PEC device based on the T/Q–M (M = 0.015 wt%) photoanode with a spin-coated/scattering layer yields a saturated photocurrent density of 15.9 mA cm−2 (at 1.0 VRHE) under one sun illumination (AM 1.5G, 100 mW cm−2), which is 40% higher than the device based on the TiO2/QD photoanodes. This is a record photocurrent density for PEC devices based on core/thick shell QD sensitized wide band gap semiconductors18,32,37 (see Table S3, ESI†). In addition, the IPCE measurements of the PEC devices based on the T/Q–M (M = 0.015 wt%) hybrid and T/Q bare photoanodes were carried out under one sun illumination (AM 1.5 G, 100 mW cm−2). A systematic comparison of the IPCE values calculated at 1.0 V vs. RHE for a PEC device with the T/Q–M (M = 0.015 wt%) hybrid and T/Q bare photoanodes is shown in Fig. S7 (ESI†). The IPCE values of a PEC device with a T/Q–M (M = 0.015 wt%) hybrid photoanode are higher than those of a PEC device with a T/Q bare photoanode over the whole visible spectrum. These results are consistent with the obtained differences in the photocurrent density values for the PEC devices with the T/Q–M (M = 0.015 wt%) hybrid and T/Q bare photoanodes.
Based on the measured saturated photocurrent density, the H2 evolution rate was calculated for the T/Q and T/Q–M (M = 0.015 wt%) photoanode based PEC devices, yielding 100 mL cm−2 per day and 140 mL cm−2 per day, respectively.
Long-term stability measurements
The long-term stability of the PEC device is a critical factor towards commercialization. The stability measurements were carried out under continuous one sun illumination (AM 1.5G, 100 mW cm−2).To highlight the effect of MWCNTs on long-term stability, we fabricated two types of PEC devices with the T/Q and T/Q–M (M = 0.015 wt%) photoanodes. Fig. 6(c) displays the variation of the normalized photocurrent density of the PEC devices under continuous one sun illumination as a function of time. Our results demonstrate that the PEC cell based on the T/Q–M photoanode shows only 19% loss of the initial photocurrent density after two hours of continuous one sun illumination, whereas in the case of the PEC cell with a T/Q photoanode it shows a 35% loss of the initial photocurrent density under the same experimental conditions. This significant difference in the long-term stability of the PEC devices with and without MWNCTs is mainly attributed to the presence of MWNCTs in the photoanode as all the other components were the same. To understand the long-term stability degradation mechanism, we studied the structure of TiO2. In general, non-stoichiometry in TiO2 occurs due to the reduction of Ti4+ to Ti3+, which creates shallow energy levels.54,55 The latter are approximately 1 eV below the CB and do not participate in the electron transport.55 Upon illumination, the UV portion of the solar spectrum generates an electron–hole pair in TiO2. A hole in the VB recombines with an electron at the shallow energy levels and leaves behind a free electron in the CB. The latter rapidly recombines with a hole in the electrolyte.56 After electron–hole recombination, these empty shallow energy levels tend to trap another photo-injected electron from the CB, which further recombines with the hole in the electrolyte. Hence, these energy levels create a cascade electronic path, which enhances the recombination of the photo-injected electrons in the CB of TiO2 with the holes of the electrolytes. These structural defects in the TiO2 mesoporous films affect the electronic transport properties57 that reduce the overall long-term stability.
To address this issue, several methods have been reported such as using UV filters, replacing TiO2 with UV inactive materials and doping of TiO2.57,58 However, the performance of solar energy conversion devices based on these approaches is lower due to the reduced electron injection/extraction efficiency as well as absorption of light available for exciton generation. In this work, the addition of small amounts (0.015 wt%) of MWCNTs in the TiO2 mesoporous films improves the long-term stability as carbon nanomaterials are widely applied as the UV light absorbers and blocking agents.59 In addition, the temperature of the PEC photoanode increases under continuous one sun illumination (AM 1.5G, 100 mW cm−2), which creates thermal stress. The latter tends to break the TiO2 particle interconnections and reduces the electron transport rate, hence degrading the overall long-term stability of the device. However, the presence of MWCNTs in the TiO2 mesoporous film enhances the thermal stability as MWCNTs have high thermal conductivity and provide a better connectivity between the TiO2 particles.60 The difference in the long-term stability of the PEC device based on T/Q–M with respect to the control device is mainly due to the highly conductive and the thermally stable network of MWCNTs embedded within the TiO2 particles that improve the mechanical strength and prevent the degradation of the T/Q–M photoanode under thermal stress. Fig. S8(a and b) (ESI†) displays the systematic comparison of the photocurrent density of fresh samples and after two hours of continuous one sun illumination for T/Q and T/Q–M respectively. We consequently demonstrated a simple, fast and cost-effective approach to enhance the long-term stability of the PEC H2 generation devices.
Electrochemical impedance spectroscopy analysis
To understand the effect of the incorporation of small amounts of MWCNTs into the TiO2 mesoporous film on the carrier transport properties, EIS measurements were carried out in a three-electrode configuration in the dark. To retrieve the carrier transport properties of the working electrode, we fitted the spectra in the mid-frequency range by using the equivalent circuit of the transmission model composed of series resistance (Rs) followed by the parallel of the charge transfer resistance (Rct) and a constant phase element (CPE). Fig. 7 shows the systematic comparison of EIS measurements as a function of the bias voltage of the PEC devices based on the TiO2–MWCNTs hybrid (0.015 wt%) and bare TiO2 photoanode. From the Nyquist plots, the semicircles define the carrier transport properties at the photoanode/electrolyte interface.61 Clearly, the diameter of the semicircle for a PEC device based on the TiO2–MWCNT hybrid photoanode is smaller than that of the bare TiO2 photoanode, indicating a reduced Rct for TiO2–MWCNTs. This confirms that the presence of small amounts of MWCNTs in TiO2 significantly reduces the Rct and leads to a better electron transport for the TiO2–MWCNT hybrid photoanode than the bare TiO2 photoanode.26 | ||
| Fig. 7 Electrochemical impedance spectroscopy analysis of the photoanodes with MWCNTs (blue square) and without MWCNTs (red circles) in three-electrode configuration and the electrolyte solution: (a) Nyquist plot at 0.6 V applied bias; (b) charge transfer resistance (Rct) as a function of applied bias. | ||
The calculated Rct from the Nyquist plot fitting at different values of the bias potential for PEC devices based on the TiO2–MWCNT hybrid and bare TiO2 photoanode is shown in Fig. 7(b). A lower value of Rct is obtained for the PEC based on the TiO2–MWCNT hybrid photoanode than the PEC based on TiO2 photoanodes. This lower Rct value clearly demonstrates the efficient electronic transport properties of the TiO2–MWCNT hybrid photoanode, which reduces carrier recombination and leads to a high photocurrent density, consistent with the obtained trend of photocurrent density values of the respective PEC devices.
Conclusions and perspectives
In summary, we prepared TiO2–MWCNT hybrid anodes at different concentrations (wt%) of MWCNTs. Then the TiO2–MWCNT hybrid anodes were sensitized with specially designed CdSe/(CdSexS1−x)5/(CdS)2 colloidal heterostructured QDs to fabricate high efficiency and stable PEC devices. An optimized concentration of MWCNTs (0.015 wt%) in a TiO2 mesoporous film as well as the addition of spin-coated and scattering layers yielded the highest photocurrent density of 15.90 mW cm−2 under one sun illumination (AM 1.5G, 100 mW cm−2), which is 40% higher than the device based on T/Q photoanodes of similar configuration and also higher than the photocurrent density reported for the PEC devices based on the colloidal “giant” core/shell heterostructured QD sensitized wide band gap semiconductors. This highlights that a small amount of MWCNTs (0.015 wt%) in TiO2 improves the electron transport (reducing the Rct) within the photoanode, enabled by the directional path of MWCNTs for the photo-injected electrons towards FTO, without affecting the optical/structural properties, as indicated by EIS, UV-visible and SEM measurements. In addition, PEC devices based on a T/Q–M photoanode show better long-term stability as compared to the T/Q photoanode. These findings provide fundamental insights and define a cost-effective approach to improve the photocurrent density and long-term stability of PEC devices, which are crucial factors to commercialize this promising solar energy conversion technology. Future directions will focus on the application of functionalized MWCNTs and metal nanoparticle decorated MWCNTs in TiO2 anodes as well as the optimization of the interfacial alloyed shell thickness/composition of heterostructured colloidal QDs to further improve the performance of PEC devices.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors acknowledge the funding from the Natural Science and Engineering Research Council of Canada (NSERC, Discovery Grants) and the Canada Foundation for Innovation (CFI) for infrastructure and its operating funds. F. R. acknowledges partial salary support and funding from the Canada Research Chairs program. G. S. S. and F. N. P. acknowledge the UNESCO Chair in MATECSS for a PDF Excellence Scholarship. G. S. S. and F. N. P. acknowledge the funding from the University of Electronic Science and Technology of China and the China Postdoctoral Foundation (Grant No. Y02006023607941). F. N. P. acknowledges the funding from the National Natural Science Foundation of China (Grant No. 5171101224). M. M. is grateful to FRQNT for a PhD Scholarship (B2X). H. Zhao acknowledges the start-up funds from Qingdao University and the funding from the Natural Science Foundation of Shandong province (ZR2018MB001). F. R. is grateful to the government of China for a Chang Jiang Scholar short-term award and to Sichuan province for a 1000 Talents Plan short-term award.Notes and references
- Y. Tachibana, L. Vayssieres and J. R. Durrant, Nat. Photonics, 2012, 6, 511–518 CrossRef CAS.
- S. Kamimura, M. Higashi, R. Abe and T. J. Ohno, J. Mater. Chem. A, 2016, 4, 6116–6123 RSC.
- S. C. Warren, K. Voitchovsky, H. Dotan, C. M. Leroy, M. Cornuz, F. Stellacci, C. Hebert, A. Rothschild and M. Graetzel, Nat. Mater., 2013, 12, 842–849 CrossRef CAS PubMed.
- X. Zou and Y. Zhang, Chem. Soc. Rev., 2015, 44, 5148–5180 RSC.
- T. Bak, J. Nowotny, M. Rekas and C. C. Sorrell, Int. J. Hydrogen Energy, 2002, 27, 991–1022 CrossRef CAS.
- A. Fujishima and K. Honda, Nature, 1972, 238, 37–38 CrossRef CAS PubMed.
- M. Ni, M. K. H. Leung, D. Y. C. Leung and K. Sumathy, Renewable Sustainable Energy Rev., 2007, 11, 401–425 CrossRef CAS.
- J. Luo, L. Steier, M.-K. Son, M. Schreier, M. T. Mayer and M. Grätzel, Nano Lett., 2016, 16, 1848–1857 CrossRef CAS PubMed.
- G. Wang, X. Yang, F. Qian, J. Z. Zhang and Y. Li, Nano Lett., 2010, 10, 1088–1092 CrossRef CAS PubMed.
- X. Yang, A. Wolcottt, G. Wang, A. Sobo, R. C. Fitzmorris, F. Qian and J. Z. Zhang, Nano Lett., 2009, 9, 2331–2336 CrossRef CAS PubMed.
- (a) B. Klahr, S. Gimenez, F. Fabregat-Santiago, T. Hamann and J. Bisquert, J. Am. Chem. Soc., 2012, 134, 4294–4302 CrossRef CAS PubMed; (b) K. Sivula, F. Le Formal and M. Gratzel, ChemSusChem, 2011, 4, 432–449 CrossRef CAS PubMed.
- A. Wolcott, W. A. Smith, T. R. Kuykendall, Y. Zhao and J. Z. Zhang, Small, 2009, 5, 104–111 CrossRef CAS PubMed.
- A. Hagfeldt and M. Gratzel, Chem. Rev., 1995, 95, 49–68 CrossRef CAS.
- H.-J. Ahn, M.-J. Kim, K. Kim, M.-J. Kwak and J.-H. Jang, Small, 2014, 10, 2325–2330 CrossRef CAS PubMed.
- Q. Kang, S. Liu, L. Yang, Q. Cai and C. A. Grimes, ACS Appl. Mater. Interfaces, 2011, 3, 746–749 CrossRef CAS PubMed.
- R. Trevisan, P. Rodenas, V. Gonzalez-Pedro, C. Sima, R. S. Sanchez, E. M. Barea, I. Mora-Sero, F. Fabregat-Santiago and S. Gimenez, J. Phys. Chem. Lett., 2013, 4, 141–146 CrossRef CAS PubMed.
- J. Luo, S. K. Karuturi, L. Liu, L. T. Su, A. I. Y. Tok and H. Fan, Sci. Rep., 2012, 2, 451 CrossRef PubMed.
- L. Jin, G. Sirigu, X. Tong, A. Camellini, A. Parisini, G. Nicotra, C. Spinella, H. Zhao, S. Sun, V. Morandi, M. Zavelani Rossi, F. Rosei and A. Vomiero, Nano Energy, 2016, 30, 531–541 CrossRef CAS.
- H. J. Lee, J. H. Yum, H. C. Leventis, S. M. Zakeeruddin, S. A. Haque, P. Chen, S. Seok, M. Gratzel and M. K. Nazeeruddin, J. Phys. Chem. C, 2008, 112, 11600–11608 CrossRef CAS.
- R. Vogel, K. Pohl and H. Weller, Chem. Phys. Lett., 1990, 174, 241–246 CrossRef CAS.
- L. Li, H. T. Dai, L. F. Feng, D. Luo, S. G. Wang and X. W. Sun, Nanoscale Res. Lett., 2015, 10, 418 CrossRef PubMed.
- (a) I. Cho, Z. Chen, A. Forman, D. Kim, P. Rao, T. Jaramillo and X. Zheng, Nano Lett., 2011, 11, 4978–4984 CrossRef CAS PubMed; (b) B. Sun, T. Shi, Z. Peng, W. Sheng, T. Jiang and G. Liao, Nanoscale Res. Lett., 2013, 8, 462 CrossRef PubMed.
- (a) S. Palmas, A. D. Pozzo, F. Delogu, M. Mascia, A. Vacca and G. J. Guisbiers, J. Power Sources, 2012, 204, 265–272 CrossRef CAS; (b) S. K. Mohapatra, M. Misra, V. K. Mahajan and K. S. Raja, J. Phys. Chem. C, 2007, 111, 8677–8685 CrossRef CAS; (c) M. Paulose, K. Shankar, S. Yoriya, H. E. Prakasam, O. K. Varghese, G. K. Mor, T. A. Latempa, A. Fitzgerald and C. A. Grimes, J. Phys. Chem. B, 2006, 110, 16179–16184 CrossRef CAS PubMed; (d) Slamet, D. Tristantini, Valentina and M. Ibadurrohman, Photocatalyst. Int. J. Energy Res., 2013, 37, 1372–1381 CrossRef CAS.
- (a) M. Liu, N. d. L. Snapp and H. Park, Chem. Sci., 2011, 2, 80–87 RSC; (b) S. Hoang, S. Guo, N. T. Hahn, A. J. Bard and C. B. Mullins, Nano Lett., 2012, 12, 26–32 CrossRef CAS PubMed; (c) G. Wang, H. Wang, Y. Ling, Y. Tang, X. Yang, R. C. Fitzmorris, C. Wang, J. Z. Zhang and Y. Li, Nano Lett., 2011, 11, 3026–3033 CrossRef CAS PubMed.
- M. Ge, C. Cao, J. Huang, S. Li, Z. Chen, K.-Q. Zhang, S. S. Al-Deyab and Y. Lai, J. Mater. Chem. A, 2016, 4, 6772–6801 RSC.
- (a) K. T. Dembele, G. S. Selopal, C. Soldano, R. Nechache, J. C. Rimada, I. Concina, G. Sberveglieri, F. Rosei and A. Vomiero, J. Phys. Chem. C, 2013, 117, 14510–14517 CrossRef CAS; (b) M. Mohammadnezhad, G. S. Selopal, Z. M. Wang, B. Stansfield and F. Rosei, ChemPlusChem, 2018, 83, 682–690 CrossRef CAS.
- (a) Q. He, S. Wu, Z. Yin and H. Zhang, Chem. Sci., 2012, 3, 1764–1772 RSC; (b) D. Benetti, K. T. Dembele, J. Benavides, H. Zhao, S. Cloutier, I. Concina, A. Vomiero and F. Rosei, J. Mater. Chem. C, 2016, 4, 3555–3562 RSC.
- R. Akilimali, G. S. Selopal, D. Benetti, I. Serrano-Esparza, P. A. Algarabel, J. Maria De Teresa, Z. M. Wang, B. Stansfield, H. Zhao and F. Rosei, J. Power Sources, 2018, 396, 566–573 CrossRef CAS.
- (a) G. Eda, G. Fanchini and M. Chhowalla, Nat. Nanotechnol., 2008, 3, 270–274 CrossRef CAS PubMed; (b) Z. Y. Yin, S. X. Wu, X. Z. Zhou, X. Huang, Q. C. Zhang, F. Boey and H. Zhang, Small, 2010, 6, 307–312 CrossRef CAS PubMed.
- K.-M. Lee, C.-W. Hu, H.-W. Chen and K.-C. Ho, Sol. Energy Mater. Sol. Cells, 2008, 92, 1628–1633 CrossRef CAS.
- J. Hensel, G. Wang, Y. Li and J. Z. Zhang, Nano Lett., 2010, 10, 478–483 CrossRef CAS PubMed.
- (a) R. Adhikari, L. Jin, F. Navarro-Pardo, D. Benetti, B. AlOtaibi, S. Vanka, H. Zhao, H. Z. T. Mi, A. Vomiero and F. Rosei, Nano Energy, 2016, 27, 265–274 CrossRef CAS; (b) H. Zhao, G. Liu, F. Vidal, Y. Wang and A. Vomiero, Nano Energy, 2018, 53, 116–124 CrossRef CAS; (c) X. Tong, X.-T. Kong, C. Wang, Y. Zhou, F. Navarro-Pardo, D. Barba, D. Ma, S. Sun, A. O. Govorov, H. Zhao, Z. M. Wang and F. Rosei, Adv. Sci., 2018, 5, 1800656 CrossRef PubMed.
- B. N. Pal, Y. Ghosh, S. Brovelli, R. Laocharoensuk, V. I. Klimov, J. A. Hollingsworth and H. Htoon, Nano Lett., 2012, 12, 331–336 CrossRef CAS PubMed.
- G. S. Selopal, H. Zhao, X. Tong, D. Benetti, F. Navarro-Pardo, Y. Zhou, D. Barba, F. Vidal, Z. M. Wang and F. Rosei, Adv. Funct. Mater., 2017, 27, 1701468 CrossRef.
- H. Zhao and F. Rosei, Chem, 2017, 3, 229–258 CAS.
- (a) B. Lee, C. C. Stoumpos, N. Zhou, F. Hao, C. Malliakas, C. Y. Yeh, T. J. Marks, M. G. Kanatzidis and R. P. Chang, J. Am. Chem. Soc., 2014, 136, 15379–15385 CrossRef CAS PubMed; (b) H. McDaniel, N. Fuke, N. S. Makarov, J. M. Pietryga and V. I. Klimov, Nat. Commun., 2013, 4, 2887 CrossRef PubMed; (c) B. Mahler, P. Spinicelli, S. Buil, X. Quelin, J. P. Hermier and B. Dubertret, Nat. Mater., 2008, 7, 659–664 CrossRef CAS PubMed.
- F. Navarro-Pardo, H. Zhao, Z. M. Wang and F. Rosei, Acc. Chem. Res., 2018, 51, 609–618 CrossRef CAS PubMed.
- S. Kim, B. Fisher, H. J. Eisler and M. Bawendi, J. Am. Chem. Soc., 2003, 125, 11466–11467 CrossRef CAS PubMed.
- (a) Y. Chen, J. Vela, H. Htoon, J. L. Casson, D. J. Werder, D. A. Bussian, V. I. Klimov and J. A. Hollingsworth, J. Am. Chem. Soc., 2008, 130, 5026–5027 CrossRef CAS PubMed; (b) B. Blackman, D. M. Battaglia, T. D. Mishima, M. B. Johnson and X. Peng, Chem. Mater., 2007, 19, 3815–3821 CrossRef CAS.
- W. K. Bae, Y.-S. Park, J. Lim, D. Lee, A. L. Padilha, H. McDaniel, I. Robel, C. Lee, J. M. Pietryga and V. I. Klimov, Nat. Commun., 2013, 4, 2661 CrossRef PubMed.
- H. Zhao, G. Sirigu, A. Parisini, A. Camellini, G. Nicotra, F. Rosei, V. Morandi, M. Zavelani-Rossi and A. Vomiero, Nanoscale, 2016, 8, 4217–4226 RSC.
- Y. L. Lee, C. F. Chi and S. Y. Liau, Chem. Mater., 2010, 22, 922–927 CrossRef CAS.
- M. Seol, J. W. Jang, S. Cho, J. S. Lee and K. Yong, Chem. Mater., 2013, 25, 184–189 CrossRef CAS.
- Q. Shen, K. Katayama, T. Sawada, M. Yamaguchi and T. Toyoda, Jpn. J. Appl. Phys., 2006, 45, 5569–5574 CrossRef CAS.
- S. Brovelli, R. D. Schaller, S. A. Crooker, F. Garcia-Santamaria, Y. Chen, R. Viswanatha, J. A. Hollingsworth, H. Htoon and V. I. Klimov, Nat. Commun., 2011, 2, 1–8 Search PubMed.
- (a) R. van de Krol and M. Grätzel, Photoelectrochemical hydrogen production, Springer, 2011 Search PubMed; (b) L. Jin, B. AlOtaibi, D. Benetti, S. Li, H. Zhao, Z. Mi, A. Vomiero and F. Rosei, Adv. Sci., 2016, 3, 1500345 CrossRef PubMed.
- W. C. Oh, F. J. Zhang and M. L. Chen, J. Ind. Eng. Chem., 2010, 16, 321–326 CrossRef CAS.
- J. Yu, J. Fan and B. Cheng, J. Power Sources, 2011, 196, 7891–7898 CrossRef CAS.
- Y.-F. Chan, C.-C. Wang, B.-H. Chen and C.-Y. Chen, Prog. Photovoltaics, 2013, 21, 47–57 CAS.
- A. Saha, A. Moya, A. Kahnt, D. Iglesias, S. Marchesan, R. Wannemacher, M. Prato, J. Vilatela and D. M. Guldi, Nanoscale, 2017, 9, 7911–7921 RSC.
- X. Chen and W. Shagguan, Front. Energy, 2013, 7, 111 CrossRef.
- (a) Y.-L. Lee and C.-H. Chang, J. Power Sources, 2008, 185, 584–588 CrossRef CAS; (b) K. Tvrdy, P. A. Frantsuzov and P. V. Kamat, PNAS, 2011, 108, 29–34 CrossRef CAS PubMed.
- R. Adhikari, K. Basu, Y. Zhoua, F. Vetrone, D. Ma, S. Sun, F. Vidala, H. Zhao and F. Rosei, J. Mater. Chem. A, 2018, 6, 6822–6829 RSC.
- S. Wendt, P. T. Sprunger, E. Lira, G. K. H. Madsen, Z. Li, J. O. Hansen, J. Matthiesen, A. Bleking-Rasmussen, E. Laegsgaard, B. Hammer and F. Besenbacher, Science, 2008, 320, 1755 CrossRef CAS PubMed.
- S. K. Pathak, A. Abate, P. Ruckdeschel, B. Roose, K. C. Godel, Y. Vaynzof, A. Santhala, S.-I. Watanabe, D. J. Hollman, N. Noel, A. Sepe, U. Wiesner, R. Friend, H. J. Snaith and U. Steiner, Adv. Funct. Mater., 2014, 24, 6046–6055 CrossRef CAS.
- Y. Yu, K. Wu and D. Wang, Appl. Phys. Lett., 2011, 99, 192104 CrossRef.
- (a) I. Nakamura, N. Negishi, S. Kutsuna, T. Ihara, S. Sugihara and K. Takeuchi, J. Mol. Catal. A: Chem., 2000, 161, 205–212 CrossRef CAS; (b) T. Leijtens, G. E. Eperon, S. Pathak, A. Abate, M. M. Lee and H. J. Snaith, Nat. Commun., 2013, 4, 1–9 Search PubMed.
- R. Asahi, T. Morikawa, T. Ohwaki and Y. Taga, Science, 2001, 293, 269–271 CrossRef CAS PubMed.
- (a) S. Zhang, H. Niu, Y. Lan, C. Cheng, J. Xu and X. Wang, J. Phys. Chem. C, 2011, 115, 22025–22034 CrossRef CAS; (b) W. Wang, P. Serp, P. Kalck and J. L. Faria, J. Mol. Catal. A: Chem., 2005, 235, 194–199 CrossRef CAS.
- C. Y. Yen, Y. F. Lin, S. H. Liao, C. C. Weng, C. C. Huang, Y. H. Hsiao, C. C. M. Ma, M. C. Chang and H. Shao, Nanotechnology, 2008, 19, 045604 CrossRef PubMed.
- (a) N. Yang, J. Zhai, D. Wang, Y. Chen and L. Jiang, ACS Nano, 2010, 4, 887–894 CrossRef CAS PubMed; (b) M. Y. Yen, M. C. Hsiao, S. H. Liao, P. I. Liu, H. M. Tsai, C. C. M. Ma, N. W. Pu and M. Der Ger, Carbon, 2011, 49, 3597–3606 CrossRef CAS; (c) S. Sun, L. Gao and Y. Liu, Appl. Phys. Lett., 2010, 96, 83113 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c8nh00227d |
| This journal is © The Royal Society of Chemistry 2019 |