
Hollow nanoparticles as emerging electrocatalysts for renewable energy conversion reactions
Jongsik
Park†
 a,
Taehyun
Kwon†
ab,
Jun
Kim†
ab,
Haneul
Jin†
ab,
Ho Young
Kim
c,
Byeongyoon
Kim
ab,
Sang Hoon
Joo
*c and
Kwangyeol
Lee
*ab
a,
Taehyun
Kwon†
ab,
Jun
Kim†
ab,
Haneul
Jin†
ab,
Ho Young
Kim
c,
Byeongyoon
Kim
ab,
Sang Hoon
Joo
*c and
Kwangyeol
Lee
*ab
aDepartment of Chemistry, Korea University, Seoul 02841, Republic of Korea. E-mail: kylee1@korea.ac.kr
bCenter for Molecular Spectroscopy and Dynamics, Institute for Basic Science (IBS), Seoul 02841, Republic of Korea
cSchool of Energy and Chemical Engineering, Ulsan National Institute of Science and Technology (UNIST), 50 UNIST-gil, Ulsan 44919, Republic of Korea. E-mail: shjoo@unist.ac.kr
First published on 16th July 2018
Abstract
While the realization of clean and sustainable energy conversion systems primarily requires the development of highly efficient catalysts, one of the main issues had been designing the structure of the catalysts to fulfill minimum cost as well as maximum performance. Until now, noble metal-based nanocatalysts had shown outstanding performances toward the oxygen reduction reaction (ORR), oxygen evolution reaction (OER), and hydrogen evolution reaction (HER). However, the scarcity and high cost of them impeded their practical use. Recently, hollow nanostructures including nanocages and nanoframes had emerged as a burgeoning class of promising electrocatalysts. The hollow nanostructures could expose a high proportion of active surfaces while saving the amounts of expensive noble metals. In this review, we introduced recent advances in the synthetic methodologies for generating noble metal-based hollow nanostructures based on thermodynamic and kinetic approaches. We summarized electrocatalytic applications of hollow nanostructures toward the ORR, OER, and HER. We next provided strategies that could endow structural robustness to the flimsy structural nature of hollow structures. Finally, we concluded this review with perspectives to facilitate the development of hollow nanostructure-based catalysts for energy applications.
 Front line: Taehyun Kwon, Jun Kim; Back line: Dr Jongsik Park, Haneul Jin, Byeongyoon Kim (from left to right) | Jongsik Park received his PhD degree in inorganic chemistry from Korea University (South Korea) in 2018. Since March 2018, he has been working as a post-doc researcher at Korea University. His current research interests include the rational synthesis of asymmetric heterostructures and functional nanomaterials for energy conversion. |
Taehyun Kwon, Jun Kim, Haneul Jin, and Byeongyoon Kim are currently pursuing their PhD degree under the supervision of Prof. Kwangyeol Lee in the Department of Chemistry, Korea University. T. K. is interested in the design of nanostructured catalysts and their potential applications in electrolytic water splitting and CO2 conversion. J. K. is interested in designing functional nanomaterials based on non-noble metals for energy conversion/storage and plasmonics. H. J. is interested in the development of synthetic methods for phase-controlled nano-catalysts and investigation of water splitting reaction mechanism on the heterogeneous catalyst surfaces. B. K. focuses on the development of functional nanomaterials for electrocatalytic water splitting and biomedical applications. |
 Ho Young Kim | Ho Young Kim (BS 2015) is currently pursuing his PhD under the supervision of Prof. Sang Hoon Joo at the Department of Energy and Chemical Engineering, UNIST. His research interests include the development of noble metal-based electrocatalysts for energy conversion reactions. |
 Sang Hoon Joo | Professor Sang Hoon Joo is an associate professor at School of Energy and Chemical Engineering of UNIST. He received his BS (1998) and PhD (2004) degrees from KAIST, working with Prof. Ryong Ryoo. Prior to joining UNIST in 2010, he was a research staff member at SAIT, Samsung Electronics (2004–2007), and a postdoctoral associate with Prof. Gabor Somorjai at University of California, Berkeley (2007–2009). His research group focuses on electrocatalysts for energy conversion reactions. He received the TJ Park Junior Faculty Fellowship (2010), the Knowledge Creation Award (2011), and the KCS Award in Materials Chemistry (2016). |
 Kwangyeol Lee | Professor Kwangyeol Lee (born 1971) obtained his PhD degree (1997) in Chemistry from the University of Illinois at Urbana–Champaign. After fulfilling his military obligation, he joined Korea University in 2003 as a chemistry faculty member, before being appointed as a professor. He is the recipient of the 2009 Wiley-KCS Young Scholar Award. His current interests are on the development of synthetic methodologies for nanoscale materials, the application of nanomaterials in biomedical fields, and the development of nanotechnologies to support the environment by creating sustainable energy sources. |
1. Introduction
Global warming and climate change issues arising from fossil fuel usage pose formidable challenges to the preservation of the already fragile global eco-system and the maintenance of human quality of life.1,2 These environmental issues, coupled with the ever-increasing global energy needs, call for the development of innovative and disruptive future energy technologies that can secure a sustainable energy future.3–5 At the forefront of clean and sustainable energy technology development lie fuel cell and water splitting technologies, and the key to their success is the development of active and durable electrocatalysts for the constituent oxygen reduction reaction (ORR), oxygen evolution reaction (OER), and hydrogen evolution reaction (HER).6–9 Thus far, these important electrochemical reactions have been most effectively catalyzed by precious metal-based nanocatalysts whose high cost severely hinders their widespread adoption in commercial applications.10 Recently, intimate collaboration between theoretical and experimental chemists has provided understanding of the detailed thermodynamic aspects of these electrochemical reactions on the surface of electrocatalysts.9,11–14 Based on these efforts, multi-directional research into the design of advanced electrocatalysts has begun.15,16 The most popular approach has been the preparation of noble metal based alloy nanocatalysts, which enable a degree of control over the subtle balance between the binding and desorption of the reaction intermediates via d-band engineering.17–19 Nanoparticle facet control and the structural design concept of core–shell morphology are further tools to fine-tune the surface energy of nanocatalysts.20–22 Facet-controlled nanoparticles exhibit different catalytic activities and selectivities from those of spherical nanoparticles due to the differences in facet-dependent surface energy states.20,21 Additionally, lattice mismatch between the different phases in a core–shell structure creates tensile strain or compressive strain on the surface atoms.22–24 The nature of this strain is critical to the energy states of the surface atoms and thereby their catalytic properties.On the other hand, the catalytic events occurring on the surface of nanocatalysts are intrinsically heterogeneous.25 Therefore, increasing the number of active sites and the overall surface area of a nanocatalyst would be of immense value in boosting the catalytic activity. From this logic, high dispersion of metal atoms on supporting material is crucial for fabricating the efficient heterogeneous electrocatalysts because all catalysis events occurred on the surface of a catalyst particle.16,26 To date, the impregnation reduction method or functionalization of the carbon surface are regarded as effective methods to prepare the noble metal nanoparticles on carbon carriers,27,28 however, these preparation methodologies have been limited when we developed rationally designed complex nanostructures by various different strategies. Alternatively, several groups adopted the incipient-wetness procedure in order to prepare the heterogeneous catalysts with high dispersion.29–31 While the above-described approaches allow for enhanced electrocatalytic activity as well as reduced noble metals content, they are all based on solid nanoparticles, in which a significant fraction of the noble metal atoms are buried under the surface. The past few years have witnessed the development of hollow nanostructures as a novel nanocatalyst design motif.32–34 In general, the term “frames” refers to nanostructures comprised of only edges without facets; such structures can allow external reactants to access the interior of the nanostructures. The term “cages” is used for nanostructures whose side facets have large cavities. These terms have been hardly differentiated, so we have re-defined the term “hollow” to include both “frames” and “cages” in this review. Hollow nanostructures can provide a multitude of advantages for catalysis over solid nanoparticle catalysts. They can expose very high fraction of surface sites, have strain-induced highly reactive surfaces, and allow for increased collision frequency by confining reactants within nanoscale spaces. Furthermore, hollow nanoparticles would require significantly lower amounts of expensive noble metals, representing a noble-metal economic design. Hollow nanostructures can have metal alloy compositions, enabling very high catalytic activities. However, a major drawback of hollow nanostructures, namely, their lack of structural robustness during catalysis, has also been noted.35,36 Intense research efforts have been directed towards fortifying flimsy hollow nanostructures, and some promising outcomes in the development of highly active yet robust electrocatalysts have been reported.35–38
Herein, we aim to summarize recent advances in nanoframe-based electrocatalysts, including related nanocages, for the ORR, OER, and HER. The synthetic concepts for nanoframes and nanocages will be described along with the synthetic toolkits available, followed by a description of the overall performance of the nanoframe and nanocage catalysts toward specific electrocatalytic reactions as compared to those of nanocatalysts of different structural types. Recent research efforts to enhance the structural robustness and catalytic activity of nanoframe and nanocage catalysts will then be described. Finally, a concluding summary and suggestions for further study directions will be presented.
2. Synthetic strategies for hollow nanostructures
The synthesis of hollow nanostructures largely consists of three steps: the synthesis of a template, the formation of over-layers such as shell or frame structures on the template, and the removal of the template. The template, which can be either pre-synthesized or prepared in situ, plays a crucial role in the synthesis of hollow nanostructures in that it determines the final morphology of the hollow nanostructure and affects its atomic composition. While the synthetic methodologies used for hollow nanostructures vary according to the type of template material, the types of reactions used with a pre-synthesized template are different from those used on an in situ formed template. Since the pre-synthesized templates are already thermodynamically stable, heteroepitaxial growth, galvanic replacement, and nanoscale Kirkendall effect processes are feasible with pre-synthesized templates as presented in Scheme 1. However, in situ formed templates require delicate kinetic control of the decomposition of several precursors so that the template will be formed significantly ahead of the deposition of over-layers. Without such delicate kinetic control, only simple alloys containing all the components in the reaction vessel would be formed, and none of the many material design concepts, such as phase mixing/segregation and metastability-empowered etching to produce hollow structures, would be possible.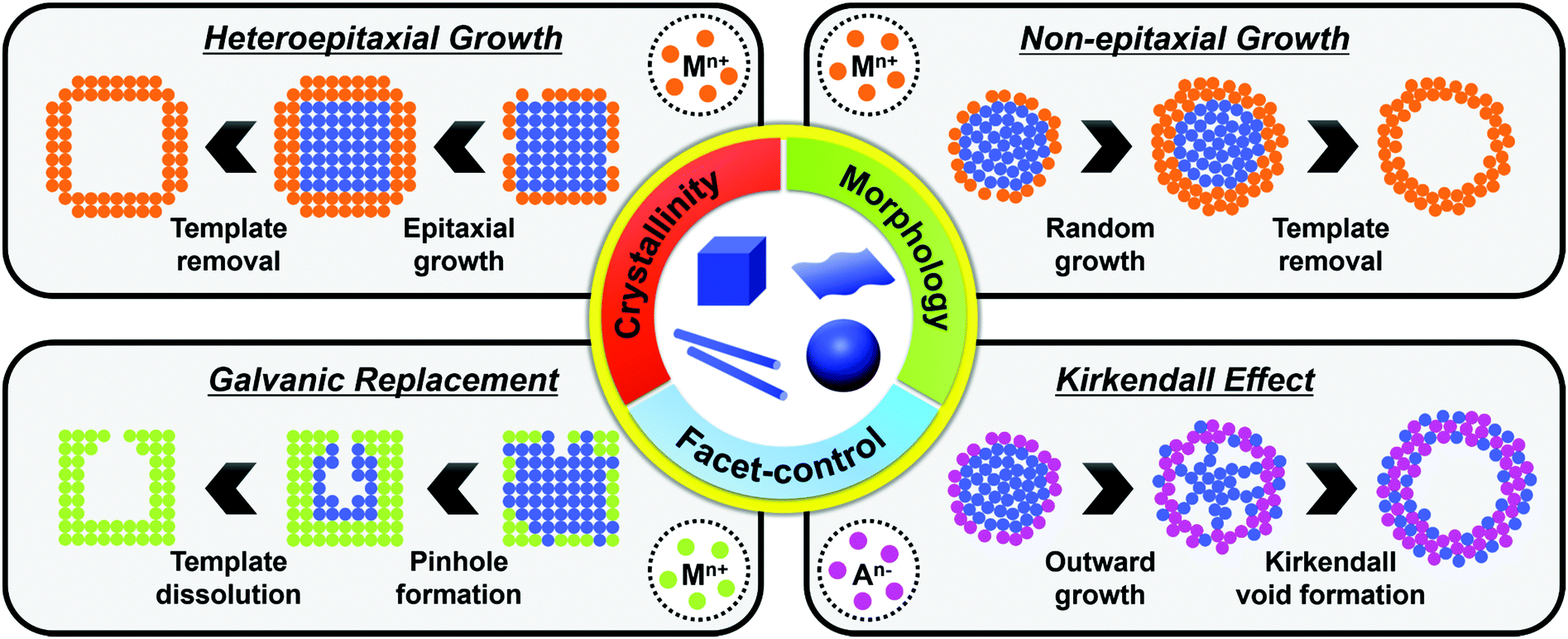 | ||
| Scheme 1 Representative schematic diagram of synthetic methodologies to hollow nanostructures by using various pre-synthesized templates. The physical and chemical properties of resulting hollow nanostructures are heavily dependent on various factors, such as crystallinity, morphology, dimensionality, and exposed facets, of the engaged templates. | ||
2.1. Pre-synthesized template-mediated synthesis: thermodynamic approach
Pre-synthesized templates have a clear advantage over in situ formed templates in that (1) their morphologies are very well defined, (2) the types of material used in these template are much more diverse than those of in situ formed templates, and (3) the types of reactions used to grow the over-layer can be finely tuned. In this section, reactions involving pre-synthesized templates of various materials are reviewed to provide a glimpse of the rich diversity and potential of employing pre-synthesized templates to produce hollow structures.Heteroepitaxial growth on a crystalline template is highly dependent on the crystal structure and lattice parameters of the exposed facets of the template, and minimization of the lattice mismatch between the template and hetero-species is the key to successful hetero-growth on the template.39–43 Facet-controlled Pd nanoparticles with cubic and octahedral morphologies of cubes and octahedrons have been employed in the synthesis of hollow Ru, Rh, Pt, and Ir nanostructures.44–56 Sneed et al. grew Rh on Pd nanocubes and synthesized PdRh nanoboxes or Rh nanoframeworks in reductive or oxidative conditions, respectively (Fig. 1a).47 Similarly, Zhang et al. reported the synthesis of Pt-based nanocages via Pt overgrowth on Pd nanocubes and octahedrons and the subsequent removal of Pd via etching. The growth of Pt on Pd nanocrystal templates occurred epitaxially so that the synthesized Pt-based nanocages inherited the {100} or {111} facets of the cubic or octahedral Pd nanocrystals, respectively (Fig. 1b).50 Ye et al. used truncated Pd octahedral nanocrystals to grow Ru epitaxially (Fig. 1c).53 While Ru itself prefers a hcp structure, the epitaxially grown Ru phase exhibited a fcc structure with {100} and {111} facets due to the presence of the fcc Pd template, and the fcc Ru nanoframe remained intact after the removal of the Pd core via etching. Concave Pt was used for the heteroepitaxial growth of fcc Ru and Rh phases.56 The selective growth of Ru and Rh on the {111} vertices of the Pt nanocube template was induced by capping the {100} facets of the Pt nanocubes with CO molecules. The synthesized octahedral Ru and Rh nanoboxes showed fcc Ru{111} and Rh{111} facets, respectively, whose atom packing motif was inherited from Pt{111}.56
 | ||
| Fig. 1 (a) Schematic diagram of the synthesis of PdRh nanoboxes and Rh nanocages using Pd nanocrystals as templates, and corresponding TEM images for each step. Reproduced with permission from ref. 47. Copyright 2013 American Chemical Society. (b) HAADF-STEM and elemental mapping images of PdPt nanocages. Reproduced with permission from ref. 50. Copyright 2015 AAAS. (c) Schematic diagram of the synthesis of Ru nanocages and TEM images for each step with magnified images in the insets. Reproduced with permission from ref. 53. Copyright 2016 American Chemical Society. | ||
Non-epitaxial growth often occurs on polycrystalline or amorphous template materials, and the crystal structures of the resulting over-layers do not conform to the crystal structure of the underlying template materials.57–65 The growth of the over-layer begins with the random adsorption of hetero-species on the template by surface functionalization/modification or electrostatic interaction.66–71 Since this growth mechanism proceeds regardless of lattice mismatch, a wide range of materials including carbonaceous materials,58–62 silica63–65 and metal compounds66–71 can be used as a template for these hetero-species. Consequently, the morphology of the resulting hollow nanostructures is more closely related to the morphology of the templates employed than their atomic arrangements and lattice parameters. Many research groups have reported hollow nanostructures with various morphologies and compositions by template-mediated non-epitaxial growth as shown in Fig. 2. Yu et al. reported many different forms of hollow micro- and nanostructures with various compositions obtained by this method.57 Recently, they reported a cooperative assembly-directed method to synthesize TiO2 nanoshells from various template materials including metals, metal oxides, polymers, carbon, and metal–organic frameworks (Fig. 2a).72 A mixture of titanium isopropoxide, hexadecylamine surfactants, and ammonia form inorganic–organic composites to coat the template nanoparticles; the coated nanoparticles can then be treated solvothermally to yield either amorphous or crystalline TiO2 nanoshells. In addition to single-shelled hollow nanostructures, the number of reports on multi-shelled hollow nanostructures is increasing in recent years.58,60,61 Wang et al. demonstrated the precisely controlled synthesis of hollow Co3O4 microspheres ranging from single-shelled to quadruple-shelled based on carbonaceous microsphere (CMS) as templates (Fig. 2d).60 By controlling the degree of hydration of the cobalt ions, the reaction temperature, and the porosity of the CMS templates, the diffusion rate and depth of the cobalt ions into the CMS templates can be fine-tuned, which ultimately enabled the controlled synthesis of single-, double-, triple- and quadruple-shelled Co3O4 hollow microspheres. Besides metal oxides and other compounds, synthesis of metallic hollow nanostructures is also feasible by non-epitaxial growth. Zhang et al. reported the synthesis of Au nanoframes by non-epitaxial growth on AgI nanocrystals by non-epitaxial growth (Fig. 2e).71 Unlike many examples of template-mediated growth of metal nanostructures, the as-grown Au nanoframes had no correlation in crystal structure with AgI nanocrystal templates, indicating that the growth of Au occurred in non-epitaxial manner. Furthermore, owing to the significant lattice mismatch between Au and the AgI nanocrystals, the growth of Au proceeded in a way that the resulting Au nanoframes were polycrystalline because each Au domains prefer different growing orientation depending on the lattice structure of underlying AgI nanocrystals.
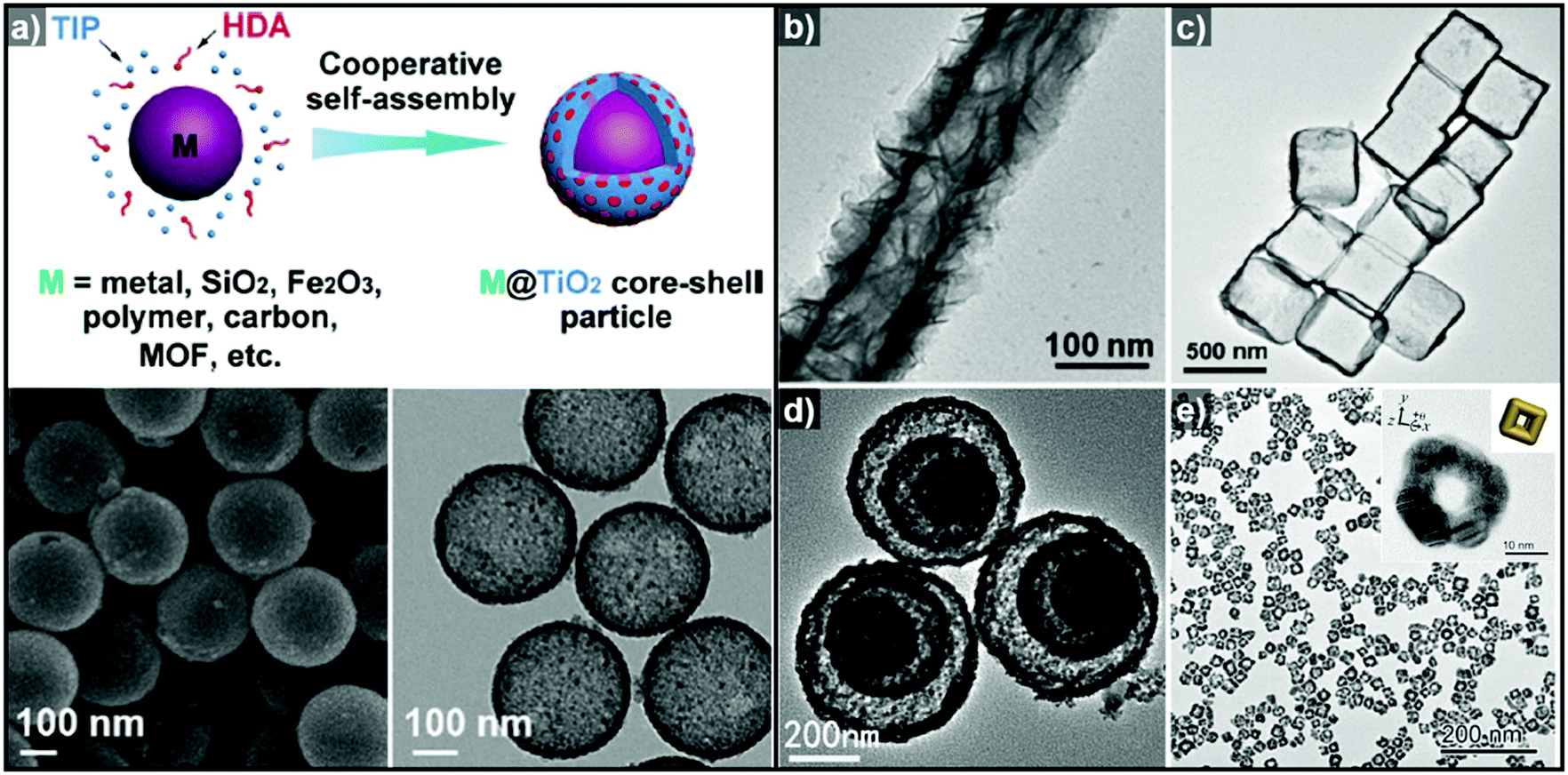 | ||
| Fig. 2 (a) Schematic diagram of the synthesis of M@TiO2 core–shell nanoparticles, and SEM (bottom left) and TEM (bottom right) images of hollow TiO2 nanoparticles. Reproduced with permission from ref. 72. Copyright 2016 AAAS. TEM images of (b) hollow TiO2 nanotube (reproduced with permission from ref. 70. Copyright 2015 – Published by the Royal Society of Chemistry), (c) SnO2 nanoboxes (reproduced with permission from ref. 68. Copyright 2011 American Chemical Society), (d) hollow Co3O4 multi-shells (reproduced with permission from ref. 60. Copyright 2013 Wiley-VCH), and (e) Au nanoframes (reproduced with permission from ref. 71. Copyright 2015 American Chemical Society). | ||
The very first nanoscale Kirkendall effect was demonstrated by Yin et al. who reported the formation of hollow nanocrystals of various phases, including Co3S4, Co9S8, CoO and CoSe, by reacting Co nanoparticles with corresponding main group elements.74,75 Cabot et al. also reported the synthesis of hollow iron oxide nanoparticles by oxidizing iron nanoparticles.76 In the same year, Peng et al. showed a similar result by annealing Fe nanoparticles under oxidative condition to synthesize core–shell–void Fe–Fe3O4 and hollow Fe3O4 nanoparticles (Fig. 3a).77 Chiang et al. reported the Kirkendall effect-induced synthesis of hollow Ni2P and Co2P nanoparticles by reacting corresponding metal nanoparticles with trioctylphosphine.78
 | ||
| Fig. 3 (a) TEM images of Fe@Fe3O4 core–shell and hollow Fe3O4 nanoparticles. Reproduced with permission from ref. 77. Copyright 2007 Wiley-VCH. (b) SEM and TEM images of Cu2S nanocages. Reproduced with permission from ref. 80. Copyright 2011 Wiley-VCH. (c) TEM and elemental mapping images of CoO@CoxSy core–shell nanoparticles and TEM image of Co9S8 nanocages. Reproduced with permission from ref. 81. Copyright 2017 Royal Society of Chemistry. All scale bars are 50 nm. | ||
Alternatively, anion exchange can also be used to form hollow nanoparticles. When metal oxides are used as templates, the exchange with larger anions is often accompanied by the Kirkendall effect. Park et al. reacted ZnO nanoparticles with hexamethyldisilathiane to form ZnO@ZnS yolk–shell and completely hollow ZnS nanoparticles.79 As the anion exchange reaction between oxygen and sulfur proceeded, the outward migration of Zn2+ ions continued until the ZnO core was completely consumed due to the faster diffusion rate of the Zn2+ ions, which are smaller than S2−. Kuo et al. reacted cubic and octahedral Cu2O nanoparticle templates with Na2S to form a Cu2S shell and then removed the Cu2O phase via acid-etching to form Cu2S boxes (Fig. 3b).80 Kim et al. recently reported the synthesis of mixed cobalt–nickel sulfide (CoxNiySz) nanocages using octahedral CoO nanoparticles as templates (Fig. 3c).81 As the surface of the CoO templates reacted with activated sulfur atoms in 1-octadecene, the smaller size of the Co2+ compared to S2− facilitated the outward diffusion of Co2+ to form a Kirkendall void at the interface between the CoO core and the CoxSy shell. The CoxSy nanocages, obtained by acid etching of the CoO core, could further undergo a cation exchange reaction with Ni2+ to form mixed cobalt–nickel sulfide (CoxNiySz) nanocages.
Additionally, it is quite common for the oxidized template to be removed during a galvanic replacement reaction to form hollow reaction products at the end of the reaction.
| Oxidation: A → An+ + ne− (EA = x) | (1) |
| Reduction: Bn+ + ne− → B (EB = y) | (2) |
| Overall: A + Bn+ → An+ + B (EB–A > 0, y > x) | (3) |
Table 1 shows the reduction potentials of various non-noble and noble metals that are commonly used in galvanic replacement reactions.82 The most extensively studied redox pair is the Au–Ag system.83–89 The first example of the use of a nanoscale galvanic replacement to synthesize hollow nanostructures was demonstrated by Y. Xia et al.83 Using Ag nanoparticles and nanowires as sacrificial templates, they reported a general synthetic route to various hollow nanostructures by introducing Au3+, Pt2+, and Pd2+ salt precursors. Two or three Ag atoms are involved in the reduction of one Pd, Pt, or Au cation, which is conducive to the formation of hollow nanostructures and the complete dissolution of the Ag templates into soluble species. In subsequent works, they used slightly truncated Ag nanocubes as sacrificial templates to form Au nanoboxes.84 It is noteworthy that the galvanic replacement occurred preferentially on the facets with high surface free energy. Because the surface free energy of metals with an fcc crystal structure generally decreases in the order of γ{110} > γ{100} > γ{111},90 the facets, on which the galvanic replacement reaction would occur, for particular metal nanocrystals can be predicted. However, it is also feasible to make the reaction occur only on the desired facets by capping the undesired facets.91
| Reduction reaction | E° (V vs. SHE) |
|---|---|
| Mn2+ + 2e− → Mn | −1.19 |
| Zn2+ + 2e− → Zn | −0.76 |
| Fe2+ + 2e− → Fe | −0.45 |
| Co2+ + 2e− → Co | −0.28 |
| Ni2+ + 2e− → Ni | −0.26 |
| Cu2+ + 2e− → Cu | +0.34 |
| Rh3+ + 3e− → Rh | +0.76 |
| Pd2+ + 2e− → Pd | +0.95 |
| Ag+ + e− → Ag | +0.80 |
| Ir3+ + 3e− → Ir | +1.16 |
| Pt2+ + 2e− → Pt | +1.18 |
| Au3+ + 3e− → Au | +1.50 |
In addition to facet selectivity, various phenomena such as alloying/dealloying and the Kirkendall effect can be coupled with galvanic replacement to add further diversity to the elemental composition and architecture of the hollow nanostructures.92 González et al. reported simultaneous or sequential galvanic replacement and Kirkendall effect using Ag nanocubes to synthesize complex hollow nanostructures of Au–Ag, Ag–Pd, and Au–Ag–Pd (Fig. 4a).93 The initial Au deposition on the Ag nanocube via galvanic replacement reaction results in the formation of pinholes, which enable the dissolution of the inner portion of Ag nanocube. As the reaction continues, Au deposition on the interior surface of the cavity occurs, so that a thin Ag layer is enclosed between two Au shells. At this stage, both Galvanic replacement and Kirkendall effect are in action simultaneously due to the faster diffusion rate of Ag+ ions, leading to the formation of a Kirkendall void between the two Au shells. They also demonstrated a sequential galvanic replacement and Kirkendall effect in which an Ag nanocube first undergoes a galvanic replacement reaction with Pd, and then the addition of Au promotes the formation of cavities, primarily driven by the Kirkendall effect. Lu and Sun et al. demonstrated the fabrication of Au/Ag nanocages and Au nanoframes by combining a galvanic replacement reaction and the selective removal, or dealloying, of Ag (Fig. 4b).88,89 In the initial stage, when Au deposition occurs on the Ag templates, the Au–Ag alloying process is favoured owing to the stronger bonding between Au and Ag as compared to Ag–Ag and Au–Au, and the low energy barrier for interdiffusion between Ag and Au. The overall reaction process can be kinetically controlled to stop at Au–Ag nanocages or to form Au nanoframes via further etching process. In the case of Pt, due to the Pt–Pt bonding being stronger than Pt–Ag bonding, the growth of Pt islands on the Ag template is more favoured than alloying.92
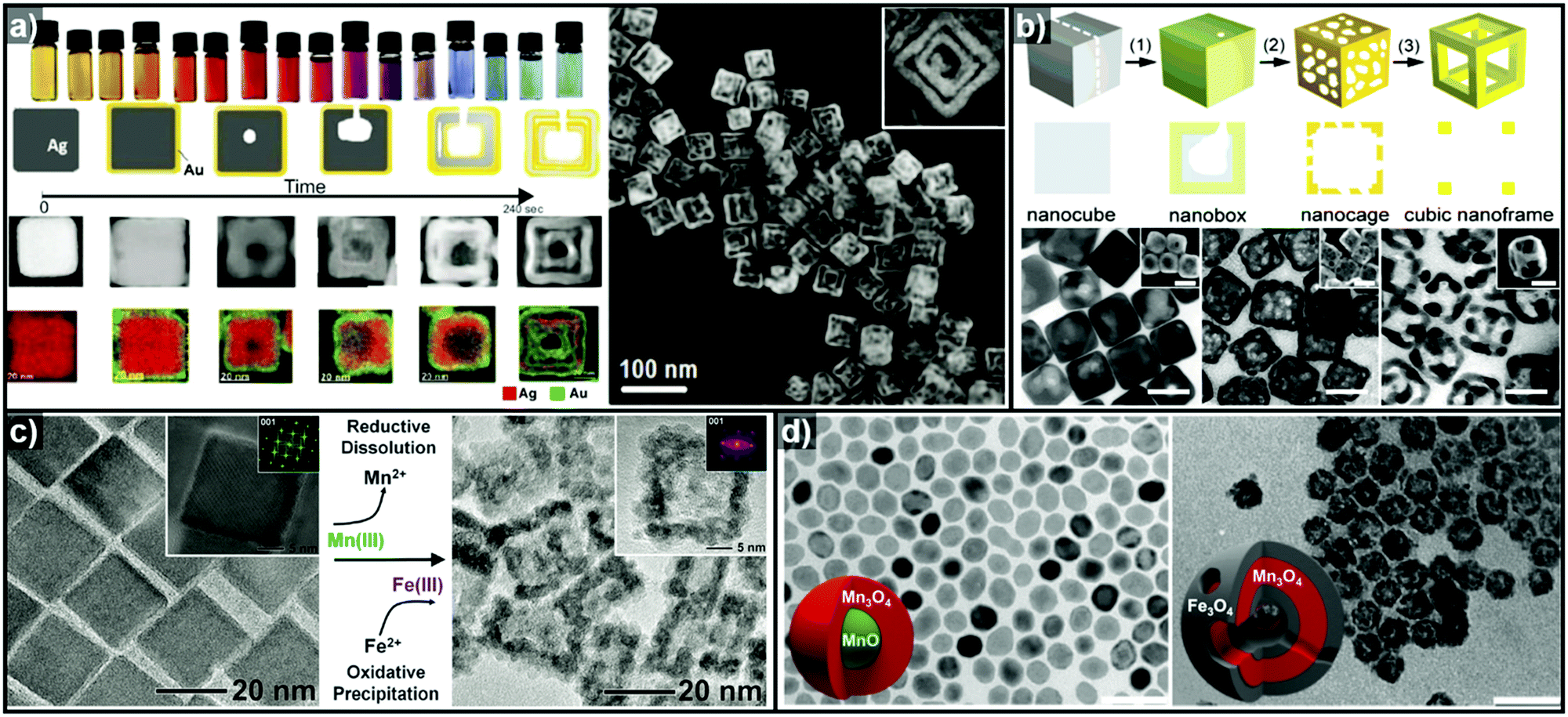 | ||
| Fig. 4 (a) Images of optical and morphological evolution of AuAg double-walled nanoboxes and HAADF-STEM image of AuAg double-walled nanoboxes. Reproduced with permission from ref. 93. Copyright 2011 AAAS. (b) Schematic diagrams of Au–Ag nanocages and Au nanframes from Ag nanocubes and the corresponding TEM images. Reproduced with permission from ref. 88. Copyright 2007 American Chemical Society. (c) TEM images showing Fe2O3 nanocages (right) synthesized from Mn3O4 nanocrystals (left). Reproduced with permission from ref. 98. Copyright 2013 AAAS. (d) TEM images of hollow Mn3O4/Fe3O4 hetero-nanostructures synthesized from MnO@Mn3O4 core–shell nanoparticles. Reproduced with permission from ref. 100. Copyright 2016 American Chemical Society. | ||
In addition to Au and Ag, various other transition metals can be coupled for the galvanic replacement reaction. Hong et al. synthesized Pd–Pt nanocages and hollow dendritic nanostructures based on octahedral and cubic Pd nanocrystals by controlling the concentration of the reducing agent.94 While the formation of hollow nanostructure was attributed to galvanic replacement reaction between the Pd nanocrystals and Pt precursors, the reduction of the Pt precursor also occurs to form Pt branches on the Pd nanocrystals in the presence of increased concentrations of the reductant. Interestingly, the dissolved Pd ions are also reduced to due to the high concentration of reductant, resulting in the eventual formation of hollow Pd–Pt dendritic nanostructures.
Liu et al. used cubic Pd nanocrystals as sacrificial templates to synthesize hollow Pd–Ir alloy nanostructures.95 In the initial stage of the reaction, the galvanic replacement reaction dominates to deposit Ir by preferentially oxidizing Pd atoms on the vertices of the Pd nanocubes to form Pd@Ir core–shell octapod nanostructures. As the reaction continues, the co-reduction of Ir ions and the dissolved Pd ions occurs simultaneously with galvanic replacement, leading to the formation of Pd–Ir alloy nanocages. Cu is another excellent template material with a relatively low reduction potential and high alloying capability.96,97 Recently, Han et al. reported a wet chemistry synthetic route to Cu-based binary hollow nanostructures by controlling the balance between galvanic replacement and the reduction kinetics of the precursors.97 They incorporated a variety of metal species (M = Ru, Rh, Pd, Os, Ir and Pt) into the Cu nanocrystal template to synthesize hollow bimetallic CuM nanostructures with well-defined morphologies. At lower temperature, the galvanic replacement reaction predominates and the original morphology of the Cu template is preserved. However, at higher reaction temperatures, the reduction of the dissolved Cu ions also occurs to facilitate the alloying of Cu and M, which in turn leads to hollow nanostructures with a significantly different morphology from that of the original template.
Thus far, the most used templates for galvanic replacement have been metals with low reduction potentials that can be easily oxidized by other metal ions. However, there are several cases where metal oxides have been used as templates where the metal ion species of the templates possess multiple oxidation states. Oh et al. demonstrated the first galvanic replacement reaction in metal oxide nanocrystals (Fig. 4c), using Mn3O4 nanocrystals as templates for galvanic replacement with Fe2+ ions.98 As the reductive dissolution of Mn3+ to Mn2+ occurs, the oxidative precipitation of Fe3+ from Fe2+ proceeds to yield hollow γ-Fe2O3 nanocages (Fig. 4d). Susman et al. reported the pH-dependent galvanic replacement of Cu2O nanocrystals with Au or Pd cations to synthesize hollow Au or Pd nanocages.99 At lower pH, the separate and independent nucleation processes of Au or Pd are more favoured due to the higher degree of protonation of the Cu2O surface, which changes the surface charge. However, at relatively higher pH, growth process is more favoured than nucleation. This facilitates the deposition of a uniform coating of Au or Pd on the Cu2O templates to form Cu2O@M (M = Au or Pd) core–shell nanocrystals, which yield hollow metal nanocages after etching of the Cu2O core. Recently, López-Ortega et al. also reported the synthesis of hollow Mn3O4/Fe3O4 hetero-nanostructures from MnO@Mn3O4 core–shell seeds using either a trace amount of H2O or an organic oxidizing agent.100 In the presence of an oxidizing agent, the MnO core is oxidized to Mn3O4, and simultaneously, Mn3+ is reduced to Mn2+ as Fe2+ is deposited to form Fe3O4 as shown in Fig. 4d.
2.2. In situ formed template-mediated synthesis: kinetic approach
Recently, the synthesis of hollow structures from in situ formed templates has undergone great advances. While the obvious advantage of using in situ formed templates would be the ease of synthesis, the intricate play between alloying and dealloying also makes this synthetic approach very enticing. In order to ensure the selective in situ formation of the template, fine control of the decomposition kinetics of the multiple metal precursors is required.Yoon et al. reported the sequential decomposition of Cu and Ir precursors, to form CuIr octahedral nanocrystals with a Cu-rich core and Ir-rich shell.101 It is tempting to explain the core–shell structure by the fact that the thermal decomposition of the Cu precursor occurs faster than that of the Ir precursor. However, the immiscibility between Ir and Cu also aided the formation of the core–shell structure in this case. In general, the final compositional distribution and morphology of the nanoparticles cannot be solely attributed to kinetic considerations, since kinetically-formed nanoparticles can undergo changes in compositional distribution and morphology due to thermodynamic factors such as mixing enthalpy, phase segregation, surface energy, and reduction potentials.36–38,102–104 One of the most important thermodynamic factors in the synthesis of multicomponent alloy nanoparticles is the enthalpy of mixing, which represents the miscibility of the multiple components.105,106 The enthalpy of mixing values are available for various solid solution phases, and can be used to explain the tendencies of miscibility and composition changes in bulk materials. However, the experimental results for many nanoscale materials do not agree with the miscibility predicted from the enthalpy of mixing.104,105,107–109 For example, Huang et al. reported the solid–solution alloying of Ru and Cu in nanoscale, which are completely immiscible in bulk (Fig. 5a).107 While an hcp structure is favored for Ru, which is immiscible with Cu, fcc structured Ru can be stabilized at the nanoscale and can mix with Cu, unlike the bulk materials. Due to the distinctly different mixing behavior at the nanoscale and in the bulk state, great efforts have been made to understand the compositional stability of multicomponent nanoparticles.110–112
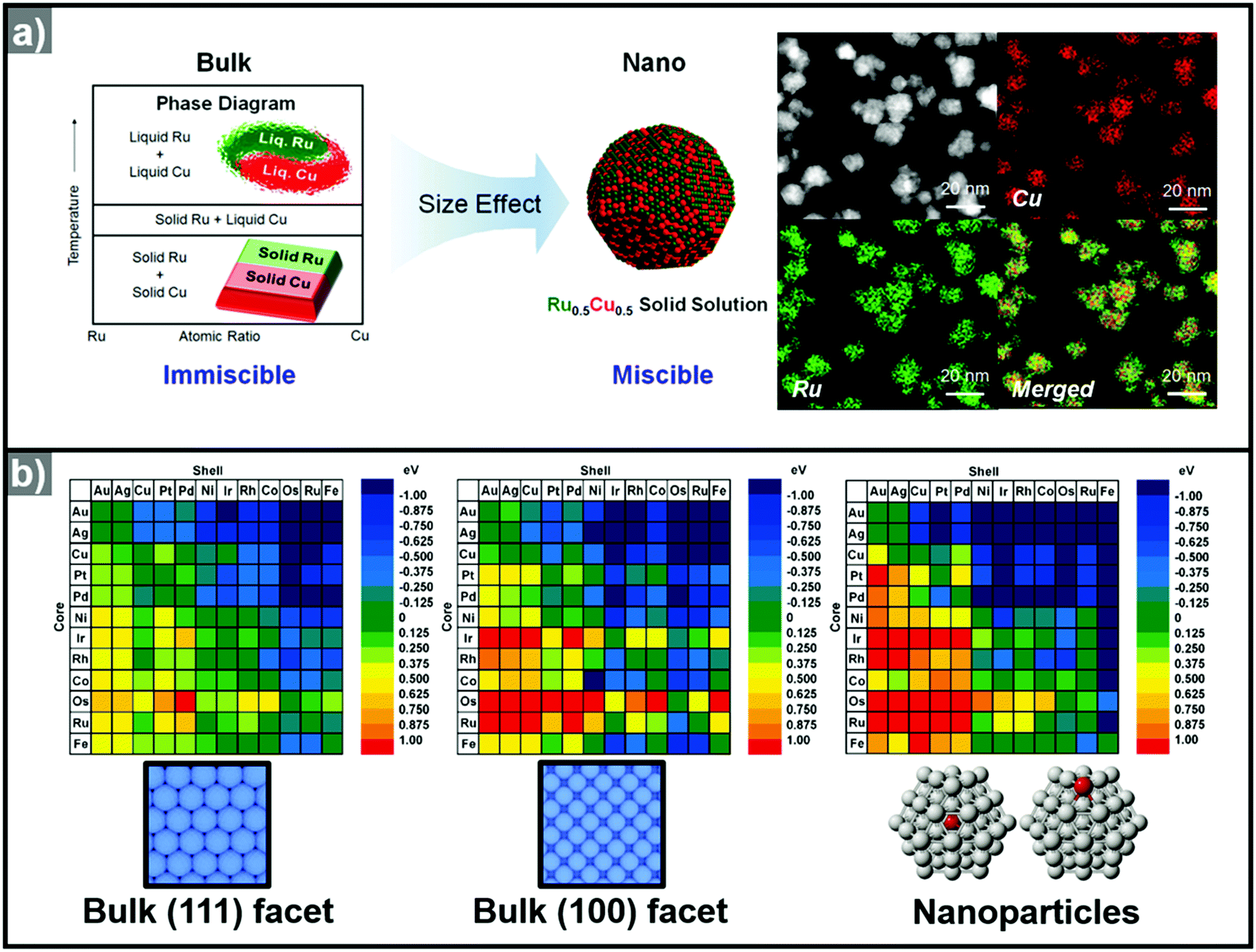 | ||
| Fig. 5 (a) (left) Schematic representation of Ru–Cu solid solution alloy NPs within the nano-size dimension. Ru and Cu are immiscible in bulk state. (right) HAADF-STEM image and its corresponding EDX elemental mapping analysis of RuCu solid solution alloy NPs. Reproduced with permission from ref. 107. Copyright 2017 American Chemical Society. (b) Color-coded matrix of segregation energies for impurities in 55-atom NPs composed of 12 late-transition metals. Color towards the blue end indicates a stable core/shell structure, as depicted in the matrix. Conversely, color towards the red end indicates that the core/shell structure is reversed. The matrix located at left and center related to the (111) and (100) surfaces, the matrix at the right is related to the cluster. Reproduced with permission from ref. 112 Copyright 2009, American Chemical Society. | ||
Many researchers also have made a large effort to predict the phase segregation behavior in nanoscale, which largely depends on nanoparticle size, shape and composition.113–116 Xiong et al. reported the calculation of alloying ability bimetallic system such as Cu–Ag, Au–Ni, Ag–Pt and Au–Pt, all of which are immiscible in bulk.113 Based on their calculation result, Cu–Ag system, for example, showed remarkable drop down of formation enthalpy with decreasing size. For Cu0.5Ag0.5 nanoparticles, the formation enthalpy turns positive (immiscible) to negative (miscible) when the particle size reduces below 3.1 nm.117–119 On the contrary, initially-formed nanoscale alloy undergo phase segregation behavior by various parameters such as temperature, atmosphere, nanoparticle size etc.117,120–123 For instance, Guisbiers et al. reported theoretical calculation and corresponding experimental results of alloy formation and phase segregation behaviors of bimetallic Cu–Ni nanoparticles, which is completely miscible system in bulk phase.117 Under elevated temperature with appropriate heat treatment, Cu and Ni easily forms solid solution alloy nanoparticles. However, by decreasing the temperature, the miscibility gap between Cu and Ni was generated, and subsequent phase segregation of Ni toward nanoparticle surface occurred. Their calculation results also showed that the miscibility temperature of Cu–Ni pair is increased with larger particle size and increased number of facets. By their calculation results phase segregation behavior of Cu–Ni solid solution nanoparticle can be predicted, which is transformed to core–shell or Janus structure by nanoparticle size or shape.
The presence of multiple metal precursors could easily lead to the formation of alloy nanoparticles, thwarting attempts to form multi-component structures with a well-defined radial distribution. Therefore, the phase segregation phenomena of alloy nanoparticles are often exploited in the synthesis of hollow nanostructures based on in situ formed template nanoparticles.37,124,125 Phase segregation at the surface of a multimetallic nanoparticle is caused by the difference in the surface energies of the metal components.120 Numerous parameters contribute to the driving force which affects the degree of segregation or the alloying process. The metal–metal binding energies, for example, the energies of the A–A, B–B, and A–B bonds in an AB bimetallic nanoparticle system, are linked to the phase segregation phenomenon.120–123 When the A–B bonds are stronger than any other bonds, the nanoparticles gradually form the AB alloy phase. Second, the discrepancy between the surface energies of different metals can also affect the phase segregation.120,122,123,126,127 The element with lower surface energy migrates to the surface to gain thermodynamic stability.120 Third, the strain caused by the lattice mismatch driven by the difference in the atomic radii of the elements can induce phase segregation.23,128,129 The mismatch causes an increase in the strain energy, resulting in phase segregation in the nanoparticles. The presence of adsorbents or surface binding moieties can also induce phase segregation behavior.120,128–132 Core elements can be drawn to the surface when adsorbents and surface binding moieties favor binding with them. Tao et al. reported a notable example of phase segregation induced by surrounding gas conditions in a PdRh nanoparticle.133 Because Pd atoms prefer to bind to gaseous CO, the Pd@Rh core shell structure can be transformed into Rh@Pd. On the other hand, without CO (in the presence of only NO), the Rh@Pd nanoparticles revert to the Pd@Rh composition because of the surface energy of Rh is lower than that of Pd.
The use of computational methods to understand the phase segregation behavior of multi-component nanoparticles has also been explored recently.112,134 Wang et al. predicted the trends of core–shell preferences for binary alloy nanoparticles.112 Based on a DFT method, they calculated the segregation energies of each metal combination. Fig. 5b shows that the surface segregation tendencies depend on the exposed atomic arrangements or regio-specific active sites. Therefore, the phase segregation phenomena in the three-dimensional nanostructures would be different from those in conventional zero-dimension nanoparticles. In short, the migration and the change in surface composition could be controlled through understanding of the phase segregation phenomena.
Moreover, facet control of alloy nanoparticles can add further diversity to phase mixing and segregation. Facet-controlled nanoparticles exhibit distinctive surface structural features, namely, vertices, edges, and facets, which have different surface energies.20,135,136 While the thermodynamics of atom migration within an alloy nanoparticle is governed by the expression ΔG = ΔH − TΔS, the ΔS terms are notably different among these surface structural features and therefore the surface alloying can be different from one surface structure to another.120,121,126,137 For example, large atoms within an alloy nanoparticle would migrate to the vertices and edges where the free space can best accommodate larger atoms.120,126,128,129 When the vertices and edges have a very robust composition and the facet center consists of a phase that is unstable in a caustic medium, nanoframe can be obtained by etching. Furthermore, when phase segregation occurs in an alloy nanoparticle, new interfaces are generated between the segregated phases.120,121 These grain boundaries are intrinsically unstable due to lattice mismatch and structural defects, and can serve as effective passages for atom movement, thus greatly facilitating intra-nanoparticle diffusion processes.37,102,125 While phase segregation confined to the surface gives rise to single-layered nanoframes, the presence of intra-nanoparticle diffusion passages can produce structurally-fortified nanoframes.37,38
Thus, hollow nanostructures such as nanocages and nanoframes with well-defined morphologies can be fabricated only when the initial formation of template nanoparticles occurs quickly and the final multicomponent nanoparticle has a compositional arrangement that allows easy etching of the template phase. Cu and Ni are excellent template materials for the one-pot, wet-chemical synthesis of hollow nanostructures, since the decomposition temperatures of these metal precursors are quite low, allowing the fast formation of the template nanoparticles, and they can easily be removed by etching. For example, Yoon et al. reported the one-step synthesis of octahedral Cu-doped Ru nanocages via both kinetic and thermodynamic approaches.103 The co-decomposition of the Cu and Ru precursors resulted in the formation of Cu-rich template nanoparticles during the initial stage and the subsequent growth of Ru on the Cu-rich nanoparticles. However, the unstable nature of the Cu phase in the presence of bromide ions resulted in the in situ dissolution of the Cu-rich phase at the core to give a hollow nanocage structure. In short, the fast decomposition kinetics of the Cu precursor and the metastable nature of the Cu nanoparticles enabled a one-pot approach to hollow Ru nanostructures. Other hollow nanostructures based on noble-metals such as Pt, Pd, Ir, and Rh could also be prepared by exploiting the metastable nature of Cu as a template.104,138–141 A similar strategy using metastable Ni is also applicable to the formation of hollow noble metal nanostructures.37,142–146 For example, Hwang et al. reported the thermal co-decomposition of Ni and Ru precursors to give Ni@Ru core–shell nanosandwiches, which can be subsequently treated with acid to yield hollow nanobox structures.145 The faster decomposition kinetics of the Ni precursor enabled the formation of a Ni core and a Ru shell. Also, many researchers exploited in situ generated Ni nanoparticles prepared from Bönnemann synthesis or polyol methods for sacrificial templates for Pt-based alloy hollow nanostructures.147–154 For instance, Bae et al. reported one-pot synthesis of carbon-supported PtNi hollow nanoparticles via fast reduction of NiCl2 precursor by NaBH4, followed by galvanic replacement of Pt.147 Moreover, Chattot et al. also reported atomic-scale snapshots of the formation, growth and hollowing out mechanisms via scanning transmission electron microscopy (STEM), energy-dispersive X-ray spectroscopy (EDX), operando wide-angle and small-angle X-ray scattering (WAXS and SAXS) techniques.154,155 Their study revealed that Ni-rich nanoparticles embedded in a NixByOz shell were formed first, and the combined reaction of galvanic replacement and nanoscale Kirkendall effect resulted the formation of Ni-rich core@Pt-rich shell nanoparticles, and finally formed PtNi hollow nanoparticles. More than Cu and Ni, other first-raw transition metal such as Co, Fe, and Zn also can conduct similar role as an in situ formed sacrificial templates.150,156–158
As stated previously, the presence of adsorbents or surface binding moieties can also induce phase segregation behavior in multicomponent nanoparticles, which in turn enables the fabrication of hollow nanostructures. For example, Oh et al. demonstrated the phase segregation behavior of octahedral Pt@Ni core@shell nanoparticles under a CO atmosphere.37 The CO atmosphere induces the outward migration of Pt from the core towards the 〈100〉 (vertex) direction rather than the 〈111〉 (facet) and 〈110〉 (edge) directions. The site-specific energy level difference was induced by the adsorption of CO molecules on the surface, resulting in geometrically precise phase segregation in the octahedral PtNi nanoparticles. The resulting dealloyed nanoparticle has Pt-rich edges and core with a Cartesian coordinate-like morphology. The removal of the Ni component via acid treatment resulted in an octahedral nanoframe structure with Cartesian coordinates.
In summary, the initially formed multicomponent nanoparticles can undergo further compositional and morphological changes due to a number of thermodynamic parameters, and the selective destabilization of certain components and the removal of metastable phases can lead to the formation of hollow nanostructures (Fig. 6).
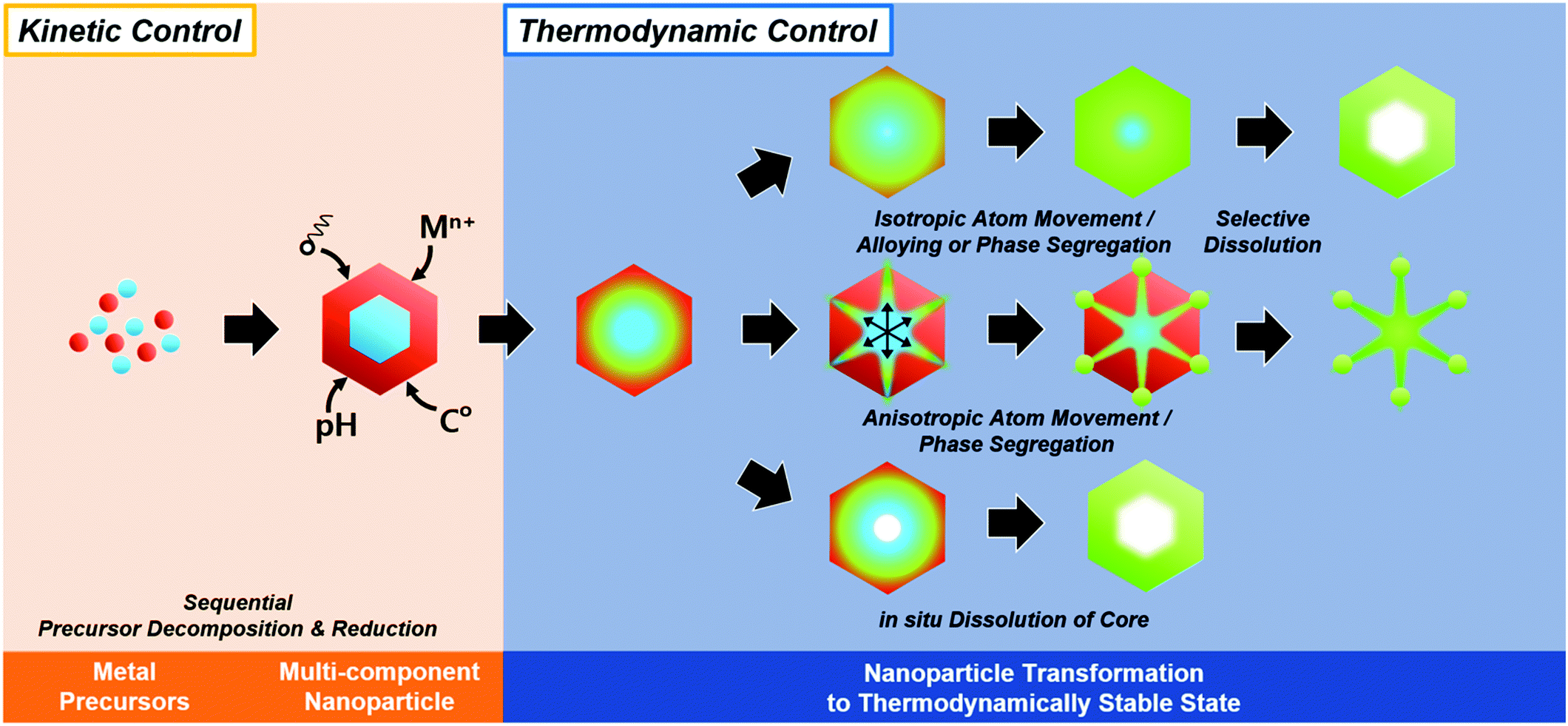 | ||
| Fig. 6 Schematic illustration of in situ formed template-mediated synthetic approach toward hollow nanostructures with kinetic and thermodynamic control. | ||
3. Electrocatalytic application of hollow nanostructures
Clean energy sources and sustainable energy production methods are urgently needed to prevent severe environmental problems. Hydrogen has received a great deal of attention as a promising energy source in fuel cells due to its high energy density. Hydrogen fuel cells are operated by two complementary redox reactions: the hydrogen oxidation reaction (HOR) at the anode and the ORR at the cathode, and produce electricity and pure water without any pollutants.17,159,160 On the other hand, water electrolyzers, in which the electrode reactions comprise the OER at the anode and HER at the cathode, represent an attractive method to produce pure hydrogen in a carbon-neutral manner when coupled with renewable energy sources.4,161,162 While there is great potential for these technologies to provide at least a partial solution to rapidly worsening environmental problems, the cost of electricity for the production of hydrogen and the efficiency of the fuel cells are formidable obstacles that must be overcome in order for these technologies to penetrate the mainstream energy market.15,161,163–165 Both technologies require the development of highly active and durable catalysts that far surpass the performance of state-of-the-art catalysts; accordingly, great research efforts are being focused upon the development of catalysts with new design concepts and excellent performances.162,166Catalytic activity can be enhanced by (1) increasing the number of catalytic events on a given catalyst and (2) enhancing the intrinsic catalytic activity of the catalyst. Increasing the surface area has been an effective strategy to increase the number of catalytic events, and can be accomplished by decreasing the particle size of the heterogeneous catalysts and by forming highly open catalyst morphologies such as hollow nanocages and nanoframes. On the other hand, the intrinsic activity can be improved by controlling the surface energy, which depends on the chemical composition, structural disorders, defects and coordination nature of the surface and exposed facets. Furthermore, several recent researches underlined the calculation results of the relationship between catalytic activity and surface coordination number.11,167 By calculation results from Calle-Vallejo et al., concave surface geometry with higher coordination number than convex surface is predicted to show better oxygen reduction reaction.167 Unlike solid nanoparticle surface, hollow nanostructures are prone to have concave surface in their interior, which can show higher electrocatalytic activity than solid nanoparticles, which have only convex surface. Asset et al. reported experimental identification method of concave and convex catalytic surface via electrochemical CO-stripping method.155,168,169 Also, they experimentally demonstrated specific activity of concave catalytic surface is more active than convex catalytic surface.155 Moreover, hollow nanoparticles easily tend to have structural disorders or defects in their structure, which are usually generated during the dissolution of the core component.170–172 Combining these together, hollow nanoparticles have high intrinsically active catalytic surface as well as increased catalytic surface area. Therefore, there has been intense research interest in the fabrication of hollow nanostructured catalysts with controlled surface compositions and crystal facets.47,50,81
3.1. Fuel cell electrode reactions
3.1.1.1. Brief summary of recent advances in Pt-based ORR electrocatalyst. Pt-based ORR electrocatalysts have a long history, and therefore numerous excellent reviews are available in this field (Fig. 7).6,17,159,160,173–175 In this section, a brief overview of the developments in Pt-based electrocatalysts during the last decade is given. Investigation of the ORR activity of single crystalline Pt electrodes, such as Pt(111), Pt(100), Pt(110), etc., clearly revealed a strong relationship between the catalytic activity and the surface energy.176,177 Motivated by this model catalyst study, the synthesis of facet-controlled Pt nanoparticles with particular shapes was attempted in order to exploit the structural effects of nanoparticle catalysts. For instance, octahedral Pt nanoparticles enclosed by (111) facets were found to be more active for the ORR than cubic Pt nanoparticles with (100) facets using a perchloric acid electrolyte, which is consistent with the results from bulk single crystalline Pt electrodes.177,178 Moreover, enhanced activity was observed in Pt nanoparticles enclosed by high-index facets, which have a high density of low coordinated sites on surface features such as steps, kinks, and ledges, leading to a high surface energy.179–190 For example, Yu et al. reported Pt concave nanocubes, enclosed with high-index facets such as {510}, {720} and {830} showed 3.1 to 4.1-fold improved ORR specific activity compared to Pt nanocubes enclosed with {100} facets.190
 | ||
| Fig. 7 Schematic illustration of brief history of the development of Pt-based ORR electrocatalysts (modified from ref. 5. Copyright 2016 AAAS). | ||
Another mainstream approach to fabricate superior ORR catalysts has been the development of Pt-based alloy nanoparticles, because the incorporation of a transition metal into a Pt-based catalyst can result in high ORR activity.191–194 Alloying Pt with other transition metals such as Ni, Co, Fe, Cu, Ag, Au, and Pd induces compressive strain on the nanoparticle surface due to shorter Pt–Pt distances, thereby downshifting the d-band center of Pt.17,178,182,195,196 The downshift of the Pt d-band weakens the adsorption of OH species, which are intermediates of the ORR, thereby facilitating the dissociation of H2O molecules from the catalyst surface to eventually increase the ORR reaction rate.18,192,196–199 Stamenkovic et al. demonstrated that alloying Pt with transition metals can decrease the adsorption energy of oxygen on the top Pt layer of Pt3M (M = Ti, V, Fe, Co, Ni) alloys. The ORR activities followed the order Pt < Pt3Ti < Pt3V < Pt3Ni < Pt3Fe ≒ Pt3Co, demonstrating a volcano relationship.18 However, the transition metal atoms on the surface of a Pt-based alloy catalyst usually undergo dissolution, resulting in low chemical stability for the catalyst.200,201 More specifically, the dissolution of the transition metal from a Pt–M alloy results in a change in the surface composition and an increase in the surface roughness of the catalyst, therefore altering the initial ORR activity of the alloy electrocatalyst.17,200,202–204 In 2009, Strasser et al. demonstrated that the electrochemical treatment of PtCu alloy nanoparticles results in preferential dissolution of the Cu component, forming dealloyed nanoparticles with a thin Pt skin over the PtCu alloy.202 Wang et al. also reported the electrochemical dealloying of PtCu3 intermetallic alloy nanoparticles. Fine tuning of the electrochemical cycling parameters resulted in dealloyed nanoparticles with various morphologies, such as solid or sponge-like nanoparticles with a Pt-rich shell, which showed enhanced ORR activity compared to the initial PtCu alloy nanoparticles.203
Whether the dissolution process improves or deteriorates the catalyst performance, the stability of the catalyst surface represents an important challenge in ORR electrocatalysts. In order to reinforce the surface of Pt-based alloy electrocatalysts, thermal treatments in which the non-noble metal atoms migrate to the sublayers of the catalyst have been used to form Pt-skin type surfaces.130,205–207 The outward migration of the Pt atoms and inward migration of the transition metals create a reinforced surface, which can overcome the problems caused by surface structure changes induced by transition metal dissolution.17,131,208 Wang et al. reported the evolution of a Pt skin surface from PtNi alloy nanoparticles due to phase segregation by acid and thermal treatment.209 The acid treatment of the PtNi alloy nanoparticles results in the formation of a Pt-skeleton on the nanoparticles. Subsequent thermal annealing of the acid-treated nanoparticles results in the formation of a multilayered Pt-skin on the nanoparticles, which showed enhanced ORR activity compared to Pt/C and PtNi alloy nanoparticles. In 2016, the in situ TEM observation of thermal facet healing of octahedral PtNi was reported.210 Gan et al. characterized the in situ annealing of Pt1.5Ni nanoparticles from room temperature to 800 °C using transition electron microscopy (TEM). They observed the diffusion of Pt atoms from the corners toward the concave Ni-rich {111} facet. Based on these studies, the thermal treatment of Pt–M alloy nanoparticles results in outward atomic diffusion and phase segregation of Pt atoms.
The phase segregation between Pt and other metal atoms in Pt-based alloys can be also induced by acid leaching,120,211,212 the binding of small molecules or ligands,37,130,213,214 and applied potentials during electrochemical process142,202,204 among other methods. Since the reversible core–shell phase reconstruction of RhPd binary nanoparticles by an oxidative or reductive atmosphere was reported in 2008,133 the phase conversion of Pt-based alloy nanoparticles by changing the surrounding atmosphere has been widely studied. Ahmadi et al. reported the phase segregation behavior of Pt0.5Ni0.5 octahedral nanoparticles during thermal annealing under vacuum, H2, and O2 atmospheres via AFM and XPS.213 The vacuum and O2 atmospheres induced outward Ni migration at lower temperatures (<300 °C), resulting in the formation of Ni oxides at the surface. On the other hand, the H2 atmosphere restricted Ni segregation to the surface at low temperatures due to the reduction of surface oxide species. Furthermore, they also demonstrated the effect of a CO atmosphere on the atomic diffusion behavior in octahedral PtNi nanoparticles during thermal treatment.214 CO treatment at room temperature before annealing enhanced the thermal stability of the PtNi nanoparticles against coarsening. On the other hand, the presence of CO during annealing resulted the formation of Ni(CO)4 species, which induced an initial phase segregation of Ni at temperatures below 400 °C. In the case of PtCo nanoparticles, the adsorbed CO molecules also induced phase segregation of Pt atoms at the nanoparticle surface.130
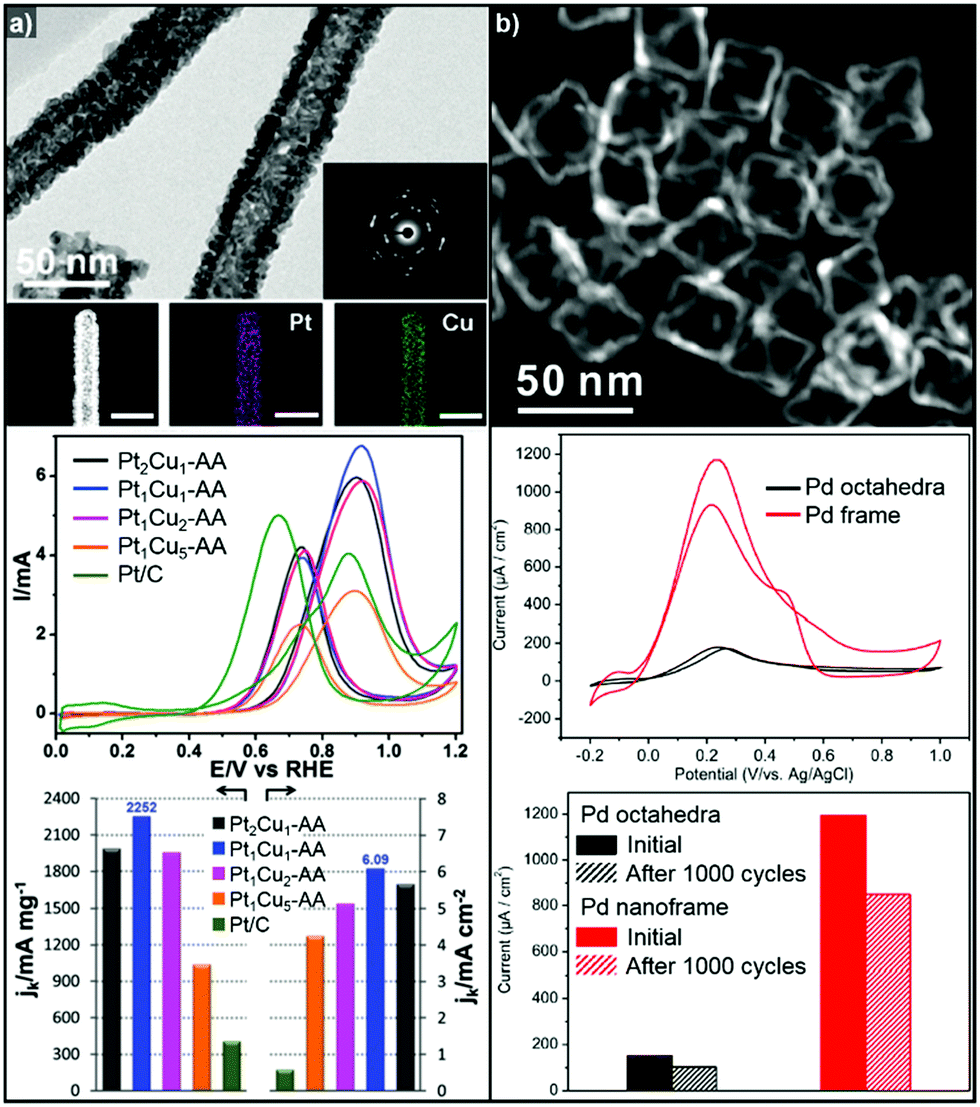
3.1.1.2. Hollow Pt-based electrocatalysts via pre-synthesized templates. In addition to the efforts to enhance their intrinsic catalytic properties, attempts to improve the inherent surface area of Pt-based electrocatalysts have also been carried out. One effective strategy to maximize the surface area of the catalyst is the formation of nanostructured electrocatalysts with a hollow interior. In general, the formation of hollow Pt-based catalysts is accomplished by the template-mediated growth of Pt atoms and subsequent removal of the template by acid etching or thermal annealing.51,52,54,215 Wang et al. reported the synthesis of hollow dendridic Pt nanoparticles by chemical etching of dendritic Pd@Pt core–shell nanoparticles.215 Wang et al. reported the synthesis of icosahedral nanocages with an ultrathin Pt wall via the selective removal of the Pd component from Pd@Pt core–shell icosahedra utilizing FeCl3 and KBr (Fig. 8a).51 The resulting nanocages exhibited a multiply twinned structure and a high surface area, and showed a 4-fold enhancement in ORR mass activity over the commercial catalyst Pt/C. Additionally, Park et al. demonstrated the synthesis of cubic Pt nanoframes utilizing cubic Pd nanoparticles as a template (Fig. 8b).52 The site selective coverage of Br− ions induces the selective deposition of Pt atoms on the edges and corners, and the Pt cubic nanoframes can then be obtained by selective removal of the Pd cubes using acid and H2O2. The resulting Pt cubic nanoframe showed an 8-fold enhancement in ORR mass activity over 15
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 cycles compared to Pt/C.
000 cycles compared to Pt/C.
 | ||
| Fig. 8 Examples on hollow Pt-based nanostructures utilizing pre-synthesized templates. (a) PdPt icosahedral nanocage. (upper) HAADF-STEM image and its corresponding EDX elemental mapping image of Pt icosahedral nanocages. (lower) ORR polarization curves and mass activity comparison of Pt icosahedral nanocages and commercial Pt/C catalysts. Reproduced with permission from ref. 51. Copyright 2016 American Chemical Society. (b) Pt cubic nanoframe. (upper) Schematic illustration of the formation process of Pt cubic nanoframes. (lower) TEM image of Pt cubic nanoframes and ORR mass activity change of catalysts after different numbers of cycles of repeated potential sweeping during an accelerated durability test. Reproduced with permission from ref. 52. Copyright 2016 Wiley-VCH. | ||
Galvanic replacement has also been successfully exploited for the preparation of hollow Pt-based nanostructures.50,94,97,151,216,217 Hong et al. demonstrated fine control of the galvanic replacement reaction between Pd and Pt to form Pd–Pt nanoparticles with core–shell, dendritic, and cage morphologies.94 They utilized octahedral and cubic Pd nanocrystals as templates, and showed that the concentration of the reducing reagent dictates the exchange behavior between Pd and Pt. The octahedral Pd–Pt nanocages showed the greatest improvement in ORR performance among the various Pd–Pt nanocrystals. Moreover, Zhang et al. reported fine control of the thickness of Pt nanocages fabricated using Pd nanoparticles as a template.50 The number of the Pt atoms grown on the Pd nanoparticles was tuned from 1 to 6 atomic layers by controlling the reaction conditions that affect the growth, diffusion, and dissolution behavior of the Pt and Pd atoms.
3.1.1.3. Hollow Pt-based electrocatalysts via in situ formed templates. The usage of pre-synthesized templates such as Pd and Ag nanoparticles in the synthesis of hollow nanostructures has several limitations: (1) the synthetic process inevitably requires multiple steps of template synthesis, shell growth, and template removal, which limits its practical use. (2) The majority of strategies utilizing pre-synthesized templates are based on noble metals such as Pd and Ag as sacrificial templates, wasting expensive materials. (3) The final morphology of the hollow nanocage and nanoframe structures is limited to the initial morphology of the pre-synthesized template. In order to overcome these disadvantages and to further explore the possibility of fine-tuning the surface energies and the nanoscale phase mixing and segregation, the in situ formation of nanotemplates has been investigated for the synthesis of novel hollow nanostructures.
The one-pot synthesis of binary PtNi nanoparticles has led to a wide variety of nanoparticle morphologies. Interestingly, these binary nanocrystals are metastable, allowing the formation of the nanocrystals in situ, phase segregation, and even the removal of the template to give rise to the hollow nanostructures to be accomplished in one step. In 2014, Chen et al. demonstrated a one-pot process to prepare PtNi nanoframes that involved the formation of a PtNi3 polyhedron, the in situ erosion of the core into PtNi, and finally, the formation of a rhombic dodecahedral Pt3Ni nanoframe with an extraordinarily high surface area (Fig. 9a).143 The open-framework structure showed a phase-segregated Pt-skin structure, and exhibited a 36-fold enhancement in ORR mass activity as compared to commercial Pt/C. Ding et al. reported the preparation of PtNi nanoframes with tetrahexahedral (THH) or rhombic dodecahedral (RD) morphologies.142 The final morphology of the PtNi nanoparticles was controlled by the ratio of oleylamine and oleic acid, with higher oleic acid concentration resulting in the THH morphology. The THH and RD PtNi nanoframes were obtained by simple acetic acid treatment to remove the Ni template, and the PtNi THH nanoframe showed enhanced ORR activity, with 20.9 times higher mass activity than commercial Pt/C.
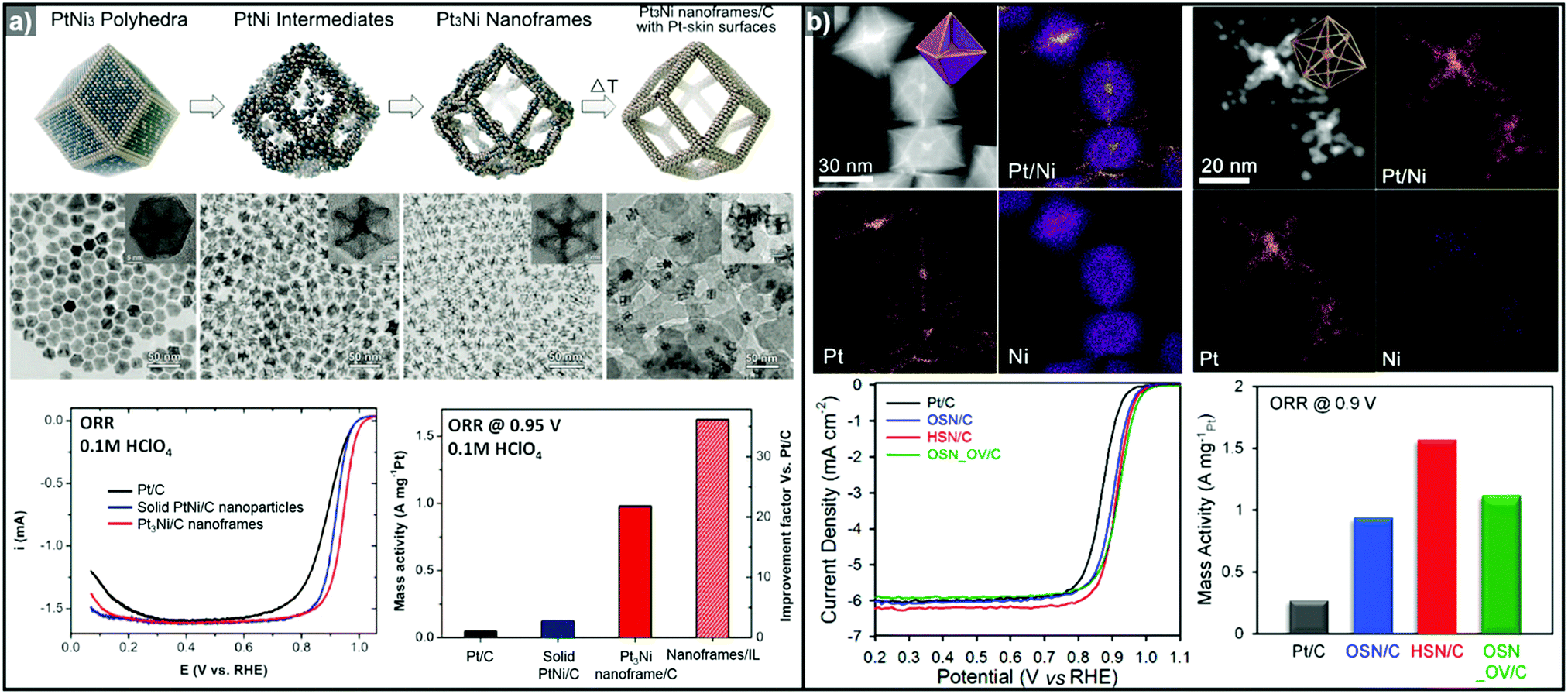 | ||
| Fig. 9 Examples of PtNi nanoframes via in situ template-mediated methods. (a) Pt3Ni rhombic dodecahedral nanoframes with thin Pt skin. (upper) Schematic illustration, TEM image of the formation process of Pt3Ni rhombic dodecahedral nanoframes with thin Pt skin. (lower) ORR polarization curves and mass activities at 0.95 V vs. RHE of catalysts in 0.1 M HClO4 electrolyte. Reproduced with permission from ref. 143. Copyright 2014 AAAS. (b) PtNi octahedral nanoframe with Cartesian coordinates. (upper) HAADF-STEM image and corresponding EDX elemental mapping of PtNi octahedral nanoparticles and nanoframes with cartesian coordinates. (lower) ORR polarization curves and mass activities of catalysts at 0.9 V vs. RHE in 0.1 M HClO4 electrolyte. Reproduced with permission from ref. 37. Copyright 2015 American Chemical Society. | ||
As mentioned above, three-dimensional polyhedral nanoparticles consist of vertexes, edges, and facets, each of which has a different energy state due to the difference in the number of coordinated adjacent atoms.218,219 Therefore, phase segregation in three dimensional morphologies is more complicated than in a simple spherical nanoparticle. The complexity of the energy states in polyhedral nanoparticles enables site-specific phase segregation in Pt-based nanoparticles, and structurally advanced Pt-based hollow nanocages and nanoframes can be obtained by utilizing this phenomenon. In 2015, Oh et al. demonstrated the phase segregation behavior of an octahedral Pt@Ni core@shell nanoparticle under a CO atmosphere (Fig. 9b).37 The presence of CO induces anisotropic phase segregation of Pt along 〈100〉 directions, which forms Pt-based core with Cartesian coordinate-like morphology in Pt@Ni octahedral nanoparticle. The removal of the Ni component via acid treatment yielded an octahedral nanoframe structure with Cartesian coordinates, and this unique nanoframe showed improved ORR activity compared to commercial Pt/C. The anisotropic diffusion and growth of the Pt atoms in PtNi alloy nanoparticles with different morphologies were also reported by other groups. Niu et al. reported the anisotropic phase segregation and migration of Pt atoms during the synthesis of rhombic dodecahedral PtNi nanoframe structures.102,143 During the formation process of the PtNi rhombic dodecahedra, the anisotropic growth and diffusion of Pt atoms were observed. In the initial stage of the synthesis, Pt-rich tetradecapods were formed in the core with their arms oriented towards the 14 vertices of the PtNi rhombic dodecahedra. The Pt atoms diffused outward and were relocated to the edges of PtNi rhombic dodecahedra, which could be further converted to rhombic dodecahedral nanoframe structures via the selective removal of Ni. Additionally, Wang et al. reported the preferential removal of the Ni component of tetrahexahedral (THH) PtNi nanoparticles using CO.144 They suggested that Ni was preferentially removed from the 〈100〉 direction using CO molecules; this process was facilitated by the lattice strain between the Pt thin shell and PtNi alloy core. The annealing of the PtNi THH nanoparticles in the presence of CO resulted in Pt3Ni THH nanoframe structures that showed 8-fold greater ORR mass activity than commercial Pt/C.
In situ formed templates have also been used in PtCu systems. For instance, Zhang et al. reported the synthesis of five-fold-twinned PtCu nanoframes by a one-pot process.220 The reaction process consisted of the in situ formation of a Cu-based five-fold twinned template and a subsequent Galvanic replacement process. The PtCu nanoframe catalysts showed higher ORR activity than commercial Pt/C and solid PtCu nanoparticles. Binary compositions such as PtCu221,222 and PtCo124 can also produce nanoframe structures with a rhombic dodecahedral morphology. The ORR activities of representative Pt-based electrocatalysts with and without hollow morphologies are summarized in Fig. 10 and Table 2.
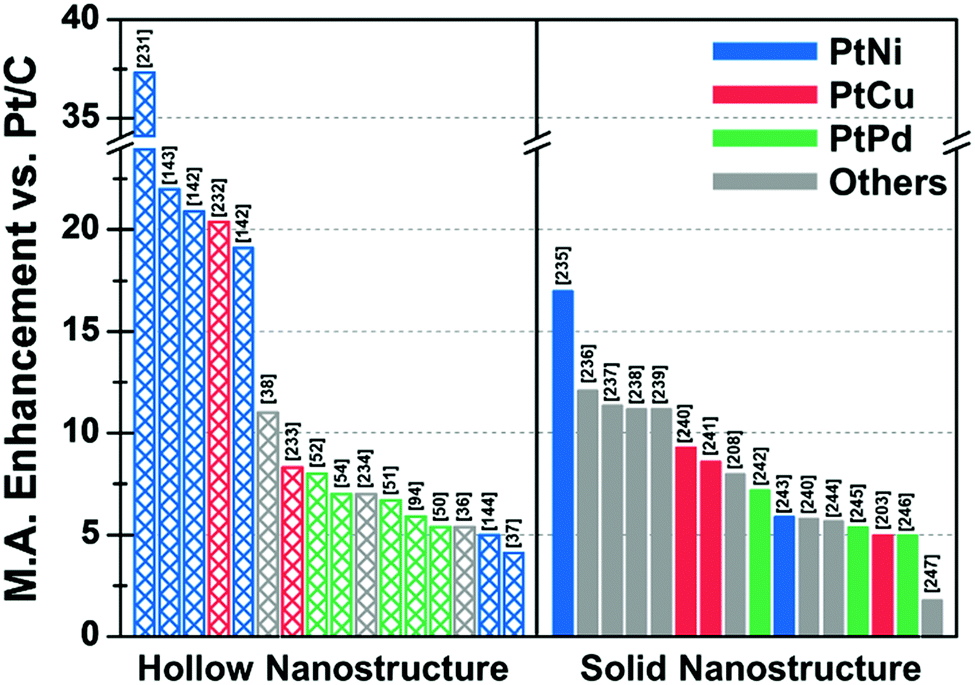 | ||
| Fig. 10 Comparison of ORR mass activity enhancement vs. commercial Pt/C of various Pt-based multicomponent electrocatalysts. Detailed values of various catalysts are described in Table 2. References are denoted in the brackets. | ||
| Structure | Catalyst | ECSA (Hupd) (m2 gPt−1) | Mass activity @ 0.9 V vs. RHE (A mgPt−1) | Mass activity enhancement vs. Pt/C | Specific activity @ 0.9 V vs. RHE (mA cmPt−2) | Specific activity enhancement vs. Pt/C | Ref. |
|---|---|---|---|---|---|---|---|
| a Measured by CO-stripping method. b Measured by Cu under potential deposition method. c Mass and specific activity @ 0.85 V vs. RHE. d Compared with Pt black. | |||||||
| Hollow | Porous Pt3Ni6 NW/C-AA | 66.8 | 5.6 | 37.3 | 8.38 | 38.4 | 231 |
| Pt3Ni RDH nanoframe/C | 66a | 5.7 | 22 | 8.6 | 16 | 143 | |
| PtNi THH nanoframe/C | 45.3 | 2.95 | 20.9 | 6.37 | 25.9 | 142 | |
| PtCu octopod nanoframe/C | 54.8 | 3.26 | 20.4 | 5.98 | 21.4 | 232 | |
| PtNi RDH nanoframe/C | 54.6 | 2.69 | 19.1 | 4.93 | 19.9 | 142 | |
| PtCu@PtCuNi dendrite@frame/C | 33.8 | 2.48 | 11 | 7.33 | 16 | 38 | |
| PtCu RDH nanoframe/C | 53.9 | 1.08 | 8.3 | 1.7 | 2.3 | 233 | |
| Pt cubic nanoframe/C (after 15k cycle) | 70.1 | 0.82 | 8 | 1.17 | 2.9 | 52 | |
| Pt icosahedral nanocage/C | 45 | 1.12 | 7 | 2.48 | 10 | 54 | |
| porous PtCuBiMn nanosheet/C | 28.6a | 0.69 | 7 | 2.41 | 10 | 234 | |
| Pt icosahedral nanocage/C | 36.6 | 1.28 | 6.7 | 3.5 | 10 | 51 | |
| PdPt octahedral nanocage/C | 40.3 | 1.2c | 5.9 | 2.98c | 6.7 | 94 | |
| Pt octahedral nanocage/C | 37.9 | 0.75 | 5.4 | 1.98 | 7.9 | 50 | |
| Co–PtCu nanoframe/C | 31 | 1.56 | 5.4 | 5.03 | 16.8 | 36 | |
| PtNi THH nanoframe/C | 18.4 | 0.47 | 5 | 2.55 | 8 | 144 | |
| PtNi OSN_OV/C | 28 | 1.12 | 4.1 | 4 | 11.1 | 37 | |
| Solid | 9 nm Pt2.5Ni octahedra/C | 31b | 3.3 | 17 | 10.6 | 81 | 235 |
| PtNi0.56Pd1.42 NWs/C | 55.5 | 1.93 | 12.1 | 3.48 | 13.4 | 236 | |
| PtPb1.12Ni0.14 octahedra/C | 37.2 | 1.92 | 11.4 | 5.16 | 19.6 | 237 | |
| Pd0.42Ni0.58@Pt/C | 42.1b | 1.45 | 11.2 | 0.61 | 2.7 | 238 | |
| Pt2CuNi Oh/C | 35.3 | 2.35 | 11.2 | 6.65 | 28.9 | 239 | |
| Pt3Cu icosahedra/C | 34.4 | 0.746 | 9.3d | 2.17 | 5.7d | 240 | |
| PtCu octahedra/C | 20a | 1.2 | 8.6 | 4.25 | 21.3 | 241 | |
| Co@Pt nanoparticle/C | 54 | 1.76 | 8 | 2.24 | 6.2 | 208 | |
| Pd@Pt2.7L icosahedra/C | 47.1 | 0.64 | 7.2 | 1.36 | 7.8 | 242 | |
| PtNi-Cl-2/C | 37 | 1.01 | 5.9 | 2.69 | 13.5 | 243 | |
| Pt3Cu octahedra/C | 31.5 | 0.461 | 5.8d | 1.47 | 3.9d | 240 | |
| PtFe1.2/C | 58.9 | 0.765 | 5.7 | 1.298 | 6.7 | 244 | |
| Pd@Pt2–3L/C | 79 | 0.48 | 5.4 | 0.73 | 4.3 | 245 | |
| Dealloyed PtCu3 NP/C (after 100k cycle) | 51.1 | 0.46 | 5 | 0.9 | 6 | 203 | |
| Pd@Pt concave decahedra/C | 95.9 | 1.6 | 5 | 1.66 | 4.6 | 246 | |
| Ir@Pt nanoparticle/C | 37 | 0.35 | 1.8 | 0.9 | 2.6 | 247 | |
3.1.1.4. Limitations in Pt-based binary hollow nanostructures. In general, Pt-based hollow nanostructures have shown much greater ORR catalytic activity in acidic electrolytes than commercial Pt/C. These catalysts benefit from both the high surface area and d-band center optimization of the PtM alloy composition. However, the ORR mass activity of hollow nanostructures is inferior to that of the world record catalyst, ultrafine jagged platinum nanowires (13.6 A mgPt−1).223 Moreover, due to their intrinsically high surface energies, nanoframe and nanocage structures are prone to collapse under the harsh acidic conditions used during electrocatalytic cycling.11,36,218,224 Alloyed transition metal species such as Ni and Cu in the hollow nanostructures can dissolve during catalytic cycles, further complicating the issue of catalyst durability.201,224–226 In short, although hollow-structured Pt-based ORR catalysts showed significant activity enhancement as compared to commercial Pt/C, there are still great barriers to the further practical use of PEMFCs in terms of the performance and the structural and compositional stability of the catalysts. Further efforts are necessary to overcome the limitations of Pt-based hollow-structured ORR catalysts.
Methanol oxidation reaction (MOR):
| CH3OH + H2O → CO2 + 6H+ + 6e− |
Formic acid oxidation reaction (FAOR):
| HCOOH → CO2 + 2H+ + 2e− |
Despite the advantages described above, the practical use of the DMFC and DFAFC presents several challenges. The major problem is the partial oxidation of the liquid fuels, which results in sluggish oxidation reaction kinetics and also reduces the number of transferred electrons.228,230 In the DMFC, less than 30% of the energy can be exploited as electricity.248 Another major concern is the crossover of the fuel molecules from the anode to the cathode via diffusion through the proton exchange membrane in the cell.249–251 This crossover phenomenon not only reduces the overall cell voltage, but also contaminates the cathode electrocatalysts. The partial oxidation of liquid fuels produces carbon monoxide (CO), which strongly adsorbs onto the surface of cathode catalyst, deteriorating the ORR performance of the cathode during cell operation. This phenomenon is called CO-poisoning, and many studies have addressed this issue.230,250
Binary Pt-based alloy phases such as PtRu and PtCu are known to have CO-tolerant surfaces and thus show promising activity for the MOR.230,252–257 The aforementioned strategies to fabricate hollow nanostructures have also been exploited to generate high-performance MOR catalysts.222,258 Li et al. also demonstrated the preparation of a crystalline PtCu alloy with a porous nanotube structure with highly open structured surfaces using Cu nanowires as the template (Fig. 11a).259 The porous PtCu nanotubes showed 5.5 and 10.3 times greater mass activity and specific activity, respectively, toward the MOR compared to commercial Pt/C, due to their three dimensional accessible surface. Xia et al. reported a one-pot synthetic method to prepare cubic PtCu3 nanocages, which showed a higher oxidation current density (14.1 mA cm−2) than solid PtCu nanoparticles (8.4 mA cm−2) and commercial Pt catalysts (12.8 mA cm−2).260 Moreover, the PtCu3 nanocage catalysts generated less CO-poisoning surface energy states compared to commercial Pt catalysts, which is derived from electrochemical dealloying of PtCu3 phase in initial stage of electrocatalysis. The MOR activities of representative Pt-based electrocatalysts with hollow interiors are summarized in Table 3.
 | ||
| Fig. 11 Examples of MOR and FAOR electrocatalysts with hollow structures. (a) PtCu porous nanotube. (upper) TEM, HAADF-STEM image and corresponding EDX elemental mapping of PtCu porous nanotubes. (middle) Polarization curves of MOR of PtCu nanotube catalysts. (lower) MOR mass and specific activity comparison of catalytsts. Reproduced with permission from ref. 259. Copyright 2017 Royal Society of Chemistry. (b) Pd octahedral nanoframes. (upper) TEM image of Pd octahedral nanoframes. (middle) Polarization curves of FAOR of Pd octahedra and nanoframes. (lower) FAOR current density comparison of Pd octahedra and nanoframes. Reproduced with permission from ref. 268. Copyright 2016 American Chemical Society. | ||
| Catalyst | Electrolyte | ECSA (Hupd) (m2 gPt−1) | Mass activity @ 0.9 V vs. RHE (A mgPt−1) | Mass activity enhancement vs. Pt/C | Specific activity @ 0.9 V vs. RHE (mA cmPt−2) | Specific activity enhancement vs. Pt/C | Ref. |
|---|---|---|---|---|---|---|---|
| a Compared with Pt black. | |||||||
| Concave PtCu2 nanoframes/C | 0.5 M H2SO4 + 1.0 M CH3OH | 44.5 | 3.36 | 7 | 7.5 | 7.2 | 247 |
| Pt1Cu1 nanotubes/C | 0.5 M H2SO4 + 1.0 M CH3OH | 38 | 2.25 | 5.5 | 6.09 | 10.3 | 259 |
| PtPd dendritic nanocages/C | 0.5 M H2SO4 + 1.0 M CH3OH | — | 0.58 | 5.27a | 1.36 | 2.67a | 215 |
| Hollow-PtCu/C | 0.5 M H2SO4 + 1.0 M CH3OH | — | 0.89 | 3.74 | 1.77 | 4.46 | 258 |
| Co–PtCu RDH nanoframes/C | 0.1 M HClO4 + 1.0 M CH3OH | 31 | 4.11 | 3.7 | 13.3 | 9.7 | 36 |
| PtNi RDH nanoframes/C | 0.1 M HClO4 + 0.2 M CH3OH | 54.6 | 1.04 | 3.25 | 1.9 | 3.39 | 142 |
| PtNi THH nanoframes/C | 0.1 M HClO4 + 0.2 M CH3OH | 45.3 | 0.84 | 2.63 | 2.19 | 3.91 | 142 |
| PtCu RDH nanoframes/C | 0.1 M HClO4 + 1.0 M CH3OH | 41.3 | 0.98 | 1.72 | 2.35 | 2.77 | 222 |
On the other hand, Pd-based electrocatalysts are widely known to give the best performance for the FAOR.261,262 DFAFC devices exploiting Pd-based catalysts can generate energy density performances comparable to those of DMFCs.227,263,264 Therefore, the development of Pd-based nanocatalysts to enhance the catalytic activity of the FAOR has been actively pursued.265–267 In particular, hollow nanostructures of Pd and Pd-based alloys have demonstrated improved FAOR activity compared to commercially available Pd/C catalysts.268–273 For example, Wang et al. reported the synthesis of well-defined Pd nanoframes by controlling the rates of etching and the regrowth process (Fig. 11b).268 The resulting octahedral Pd nanoframe catalyst showed a 7.5-fold increase in peak current density compared to a solid Pd octahedral nanoparticle catalyst. Liu et al. also reported the synthesis of hollow cubic PdAg alloy nanocages by galvanic exchange utilizing Ag nanocubes as the template.269 The cubic PdAg nanocages showed improved electrocatalytic FAOR activity and enhanced CO-tolerance, which stem from the high surface area and significant change in the adsorption energy of the intermediates in the PdAg alloy system. Huang et al. also reported one-pot synthesis of hollow PdPt alloy nanocubes via galvanic replacement reaction of Pt toward in situ formed Pd nanocubes.274 The reduction rate of Pt and diffusion rate of Pd atoms were fine-controlled by halide ligands and reaction temperature, which resulted hollow nanocube structure with PdPt alloy phase. The hollow PdPt nanocube showed two-fold enhancement of FAOR current density compared to PdPt solid nanocube.
3.2. Electrolytic water splitting reactions
Only a limited number of catalysts can operate stably in water electrolysis using acidic electrolytes, which are relevant to PEM water electrolyzers (PEMWEs). IrO2 and RuO2 have shown the most pronounced catalytic activity for the OER.287–291 Similar to other catalytic reactions, the OER benefits from increased surface area of catalysts. Accordingly, a few OER catalysts with hollow structures have been developed. Wang et al. synthesized hollow NiIr nanocrystals with Ir:Ni ratios of Ni2.76Ir, Ni2.53Ir, Ni1.43Ir, and Ni0.93Ir via Ni template mediated Ir growth.292 The as-synthesized NiIr nanocrystals were oxidized electrochemically to form Ni-doped IrOx nanocrystals.293 The Ni2.53Ir nanocrystal contained the greatest amount of active Ir sites among the prepared samples and exhibited the best OER activity, in part due to the large active surface area and the Ni dopant, which optimized surface d-band structure for oxygen evolution. Jin et al. reported the synthesis of IrNi alloyed nanoframe structures by growing Ir on Ni template, with the aid of a lanthanide metal complex as a passivation agent on the facets of the Ni template (Fig. 12a).294 The site-selective growth of Ir on the edge and corner regions of the template induced the formation of an Ir-based frame structure, which is obtained by acid etching of the Ni template. The synthesized IrNi nanoframe showed twice the surface area of commercial Ir/C as informed by CO stripping analysis, and demonstrated much better OER activity than Ir/C.
 | ||
| Fig. 12 (a) TEM image of IrNi nanoframe with graphical model in the inset and comparison of its OER activity with Ir/C (right; up). Specific activity and mass activity of IrNi nanoframe and commercial Ir/C are compared at 1.53 V (right; down). Reproduced with permission from ref. 294. Copyright 2017 Elsevier. (b) Scheme for the synthesis of IrCoNi multimetallic porous hollow nanocrystals and comparison of their OER activities (down; left) and turnover frequency (TOF) values and current densities of OER normalized to the mass of Ir (down right). Reproduced with permission from ref. 296. Copyright 2017 Wiley-VCH. | ||
Cu can also serve as a good template material for the formation of hollow structured OER catalysts due to its metastable nature.55,103 Park et al. reported the synthesis of a truncated octahedral RhCu nanoframe using Cu as the template material.104 The truncated octahedral RhCu nanoframes had three times the surface area of Cu@Rh core–shell nano-octahedrons, and showed more than twice the mass activity for the OER. Furthermore, Wang et al. reported the OER activities of a series of IrCu nanocages with various Ir:Cu ratios (Cu0.66Ir, Cu0.86Ir, Cu0.95Ir, Cu1.11Ir, Cu1.41Ir, and Cu2.04Ir).295 The Cu1.11Ir nanocage showed the best OER performance among the compared catalysts in 0.05 M H2SO4, with 1.2 and 2.65 times higher mass activity than Cu1.41Ir and Ir black, respectively. Evaluation of the Ir(III–IV) redox peak current indicated that the Cu1.11Ir nanocage had the highest number of active Ir sites among the composition-controlled samples. This was responsible for the enhanced OER activity, thereby demonstrating the effectiveness of a hollow morphology in boosting catalytic activity for the OER.
Ternary alloy systems based on hollow/frame structures have also been developed by template-mediated syntheses. Feng et al. synthesized IrCo, IrNi, and IrCoNi hollow nanoparticles by etching solid particles of IrCo, IrNi, and IrCoNi using Fe3+ ions (Fig. 12b).296 They demonstrated transition-metal dependent OER activity enhancement by comparing the calculated binding energies of OH, O, and OOH on the IrCoNi and Ir surfaces. When Ir is alloyed with transition metals, the stable binding of oxygen on pure Ir becomes weaker, with the binding energies following the order IrCoNi < IrCo < IrNi < Ir. The hollow IrCoNi structures showed the best OER activity. Kwon et al. demonstrated that a transition metal dopant can serve as a morphology engineering agent as well as a catalysis promoter.297 The Cu core component of an octahedral Cu@Ir core–shell nanoparticle could not be leached due to the impermeable nature of the Ir shell.101 This impermeable shell could be made permeable via the introduction of cobalt as a dopant in the synthesis, which allowed the deposition of Ir as well as Co in the shell layer on the Cu core.297 Upon etching, the majority of the Co atoms in the shell layer were removed, and consequently the Cu core could be removed by the penetration of the etchant into the core. The resulting Co–IrCu nanocages showed a large active surface area and high OER activity. Park et al. also synthesized double nanoframe and single nanoframe structures of IrNiCu via precise control of the reduction rates of multiple precursors.298 The synthesized double and single nanoframe structures exhibited surface areas almost three-fold and two-fold larger than that of Ir/C, respectively. The IrNiCu double nanoframe exhibited better OER activity than the single nanoframe and Ir/C, which demonstrates the straightforward advantage of increased surface area in driving high OER activity. The OER activities of representative Ir-based electrocatalysts with and without hollow morphologies are summarized in Table 4 and Fig. 13.
| Structure | Catalyst | Electrolyte | Ir loading (μg cm−2) | Overpotential (mV @ 10 mA cm−2) | Mass activity (A mgPt−1 @ 300 mV) | Ref. |
|---|---|---|---|---|---|---|
| Hollow | Co–IrCu ONC/C | 0.1 M HClO4 | 20 | 293 | 0.64 | 297 |
| IrNiCu DNF/C | 0.1 M HClO4 | 20 | 303 | 0.46 | 298 | |
| IrNiCu SNF/C | 0.1 M HClO4 | 20 | 306 | 0.4 | 298 | |
| IrNi-RF/C | 0.1 M HClO4 | 15.8 | 313.6 | 0.47 | 294 | |
| IrCoNi PHNC/C | 0.1 M HClO4 | 10 | 303 | 0.72 | 296 | |
| Cu1.11Ir | 0.05 M H2SO4 | 140 | 286 | 0.1 | 295 | |
| Ni2.53Ir NC | 0.05 M H2SO4 | 10 | 302 | 0.11 | 292 | |
| Solid | Ir–Ni TL/C | 0.05 M H2SO4 | 2 | 350 (@ 5 mA cm−2) | 1.2 (@ 280 mV) | 299 |
| IrNi NP | 0.05 M H2SO4 | 2 | 370 (@ 5 mA cm−2) | 0.49 (@ 280 mV) | 299 | |
| IrNiOx/Meso-ATO-180 | 0.05 M H2SO4 | 10.2 | 330 | 0.1 | 293 | |
| Cu–IrO2 | 0.1 M HClO4 | 140 | 351 | 0.05 (@ 350 mV) | 300 | |
| Ir-ND/ATO (at 280 mV) | 0.05 M H2SO4 | 10.2 | 400 | 0.18 | 301 | |
| IrOx/ATO | 0.05 M H2SO4 | 10.2 | 360 | 0.1 | 279 | |
| DO-IrNi3.3 | 0.1 M HClO4 | 10.2 | 310 | 0.62 | 302 | |
| IrOx–Ir | 0.5 M H2SO4 | 133 | 290 | 0.1 | 303 | |
 | ||
| Fig. 13 Comparison of overpotential and mass activity of Ir based OER catalysts in terms of morphology (hollow or solid) in acidic electrolyte. Detailed values of various catalysts are described in Table 4. References are denoted in the brackets. | ||
Noble metal based hollow or frame-like nanocrystals with HER activity comparable to that of Pt/C in an acidic electrolyte have also been reported.125,275,276 Zhang et al. demonstrated the synthesis of necklace-like hollow NixRuy nanoalloys by the galvanic replacement of Ni nanochain template nanoparticles with Ru and the Kirkendall effect.315 Among a number of studied compositions, Ni43Ru57 nanoalloys required the lowest overpotential, with 41 mV of overpotential at a current density of 10 mA cm−2 and a Tafel slope value (31 mV dec−1) comparable to commercial Pt/C. Furthermore, Yoon et al. showed the promising HER activity of metallic Ru supported on copper sulfide nanoplates in alkaline electrolyte.316 In addition to the synthesis of non-Pt metal catalysts, the strategy of using Pt based alloys to lower the Pt content can be used to achieve cost reduction for catalyst synthesis while meeting catalyst performance requirements. Oh et al. reported the preparation and catalytic activities of PtNiCo and PtNi nano-hexapods, which showed lower overpotentials and 7 and 6 times higher exchange current densities than commercial Pt/C, respectively.125 The PtNiCo hexapods showed better performance than the PtNi hexapods due to the synergistic effect of Ni and Co in tuning the surface formation energy of Pt-OHad. Also, Cao et al. reported the preparation and catalytic activity of monocrystalline PtNi branched nanocages, which showed lower overpotentials and 2.5 times higher mass current density than commercial Pt/C at 100 mV overpotential.317 The enhanced alkaline HER activity of PtNi branched nanocage stems from large surface area and synergistic effect of Pt–Ni(OH)2, which facilitates the dissociation of H2O and the formation of Had species on the catalyst.
4. The issue of catalyst stability during the electrocatalysis
As described in the preceding sections, hollow nanoparticles with cage and frame structures can provide an avenue to highly active nanocatalysts. However, prolonged electrochemical operation can cause serious stability issues in hollow nanocatalysts, leading to unreliable catalytic performance.18,218,318,319 It can also induce unwanted phase segregation in the alloy nanoparticle, exposing certain phases with higher stability under electrocatalysis conditions. This can potentially lead to the leaching of certain constituent metal species from the dealloyed nanoparticle, leading to the formation of an electrochemically inactive nanocatalyst.As a representative example of the nanocatalyst stability issue, in 2013, Cui et al. discovered that shape-controlled PtNi alloy nanoparticles form Pt-rich domains along their active sites such as corners and edges during electrocatalysis, whereas their Ni atoms are segregated to the facets.218 The selectively leached Ni phases at the facets induced the formation of a concave facet structure, providing more active sites than the solid PtNi alloy nanoparticles. However, the authors also suggested that the collapse of the near-surface alloy structure in the Ni-rich octahedrons affects the surface adsorption of oxygen in a detrimental way and thus finally degrades the ORR catalytic activity. Escudero-Escribano et al. suggested that Pt overlayers are formed on the alloy nanoparticle surface during electrocycling tests even in Pt-based alloys synthesized through the sputter cleaning method.318 With repeated cycling, the structural features of nanocatalysts can be altered because leachable metal phases become segregated in the core due to their dissolution at the surface. Furthermore, this structural degradation can cause a decrease in the electrochemically active surface area (ECSA), resulting in catalytic activities inferior to those of measured at an initial stage. Mayrhofer and Stamenkovic et al. reported that the long-term stability of Pt-based bimetallic nanoparticles could affect the agglomeration of the particles and the leaching of the non-noble metal components.18,319 They compared the surface compositions of used and unleached Pt3Co nanoparticles after the cycling test. The dynamic oxidizing conditions induced the segregation of Co at the surface, while Pt was segregated at the surface under a reducing CO atmosphere, which is used in the synthetic conditions.
The unit cell transformation of noble metal catalysts during electrocatalysis is also regarded as a critical factor in the decrease of their structural stability, and is mainly observed in the OER.279,298 Among the noble metal based water splitting electrocatalysts, Ir- and Ru-based oxide catalysts exhibit the highest electrocatalytic activities toward the OER.9,280,320 Recently, electrochemically oxidized Ir nanoparticles were demonstrated to have superior stability and activity to metallic Ir and thermally oxidized IrO2 nanoparticles.321 The difference in catalytic activity was ascribed to the altered corrosion mechanism. The thermally oxidized IrO2 nanoparticles showed drastic dissolution in acidic media before 0.9 V vs. RHE. At OER relevant potentials, both air-oxidized and thermally oxidized Ir nanoparticles were dissolved, predominantly through the formation of unstable Ir(>IV) species. In contrast, the electrochemical oxidation of Ir nanoparticles formed the stable Ir(III) species, which has ability to be reduced. The inevitable oxidation from Ir to IrOx during the oxidation process poses a serious obstacle to attempts to utilize Ir or Ru-based metal nanoframes as OER catalysts. The three-dimensional morphologies of Ir or Ru alloy-based nanoframes might be destroyed during long-term stability test, because the unit cell structures of Ir(cubic) and IrOx (rutile) are drastically different.
While there are cases in which electrochemical operation actually improves the catalytic performance of alloy nanocatalysts, the surface composition changes of alloy systems and the deformation of hollow or cage nanostructures are, in general, formidable obstacles to the development of active and durable catalysts. In order to solve the problems associated with the structural and compositional changes of nanocatalysts, several ingenuous strategies have emerged, as described in the following sections.
4.1. Multimetallic nanocrystals
Preservation of the structural integrity of innately unstable nanoframes during electrochemical operation is a daunting challenge. In order to endow such materials with stability, the introduction of an additional metal dopant has recently been attempted.35,288,322 The introduction of a dopant into noble metal-based alloy nanoparticles not only obstructs atomic rearrangement during electrocatalysis but also produces robust ternary or quaternary alloy phases that are remarkably stable toward fuel cell and water splitting applications.000 cycles, while the reference PtNi octahedral particles completely lost their shape under the same conditions. Oh et al. reported that Co atoms could also suppress the atomic migration of Pt atoms by alloying with Ni phase on the surface of PtNi octahedra.125 Consequently, the Cartesian-coordinate-like hexapod shaped PtNiCo alloy nanostructures showed high electrocatalytic activity and stability toward the HER and ORR in alkaline media. Kwon et al. synthesized Co-doped IrCu hollow nanoframes that showed excellent electrocatalytic activity and long-term durability for the OER in acidic media (Fig. 14c).297 The introduced Co dopant provides structural integrity to the surface iridium oxide under OER conditions. Similarly, Seitz et al. reported that the formation of highly active surface layers such as IrO3 or anatase IrO2 phases during strontium leaching produced electrocatalysts that were highly efficient and robust toward the OER.288
 | ||
| Fig. 14 Examples of multicomponent nanoparticles with high electrocatalytic stability, enabled by doping or alloying process. (a) TEM image of Mo-doped Pt3Ni octahedra and its mass activity after electrocatalytic cycling test toward ORR. Reproduced with permission from ref. 322. Copyright 2015 AAAS. (b) TEM image and corresponding elemental mapping analysis of Rh-doped PtNi octahedra. The bar graphs indicate the mass activity and specific activity of Rh-doped PtNi octahedral and PtNi octahedra. Reproduced with permission from ref. 35. Copyright 2016 American Chemical Society. (c) HAADF-STEM and corresponding elemental mapping analysis of Co-doped IrCu nanocage. (below) Bar graphs indicate the comparison of overpotentials to drive 10 mA cm−2 and current densities at 1.53 V (vs. RHE) between the samples. Reproduced with permission from ref. 297. Copyright 2017 Wiley-VCH. (d) TEM images of Pt2CuNi octahedra before and after electrocatalysis. The enhanced electrocatalytic stability driven from the ternary alloy phase is described below the TEM images. Reproduced with permission from ref. 239. Copyright 2015 American Chemical Society. (e) STEM image and corresponding elemental mapping analysis of tetrametallic PtPdRhAg nanocages. Reproduced with permission from ref. 325. Copyright 2017 Wiley-VCH. | ||
000 cycles, respectively, while PtNi octahedral nanocrystals retained only 52.9% and 35.0% of their initial mass activity (Fig. 14d).239 They attributed this improved stability to the uniform ternary alloy composition inhibiting the leaching of Ni and Cu. Saleem and Ud Din et al. demonstrated that tetrametallic PtPdRhAg nanoframes and porous PtCuBiMn nanosheets show highly durable and active electrocatalytic performance toward the FAOR/MOR and ORR (Fig. 14e).234,325 They suggested that the leaching or dealloying process was passivated due to the unique tetrametallic compositions of the nanoframes. Furthermore, in the case of the PtCuBiMn nanosheets, the introduction of the metal Bi improved the methanol tolerance of the catalyst, which showed only a 3.8% loss of mass activity at 0.9 V after 10000 cycles. These results suggest the great potential of ternary and quaternary alloy nanoframeworks as robust electrocatalysts.
Despite a few successful demonstrations of multicomponent alloys as robust electrocatalysts, theoretical predictions of alloy phase stability and catalytic activity are currently lacking. Also, rational synthetic routes to multicomponent alloy nanoparticles are seriously underdeveloped due to the complexity and difficulty in achieving the desired composition distributions within a single nanoparticle. Therefore, further study of the theoretical prediction of alloy phase stability and catalytic activity, as well as the development of rational synthetic routes to tailored multicomponent alloy nanostructures, is required.
4.2. Structurally-fortified nanocrystals
Well-defined three-dimensional nanoparticle structures show distinct structural features such as corners, edges, and facets, but they are prone to degrade under harsh conditions. The preservation of the structural integrity of catalytically active surface structures has been attempted by (1) the fortification of the most inherently vulnerable sites, namely the vertices, via alloying or heterostructure formation and (2) the insertion of supporting material into the nanoframework to maintain the overall high surface area of the structure without seriously sacrificing the exposed surface area on the extremities of the nanostructure.36–38,226,298 | ||
| Fig. 15 Schematic illustration of synthetic routes to structurally-fortified nanoparticles and their electrocatalytic performance. (a) TEM image and loss of peak current density of Au islands on Pt–Ni nanoframe dependent on the number of cycling test toward MOR. Reproduced with permission from ref. 226. Copyright 2014 American Chemical Society. (b) TEM image of Co-doped PtCu nanoframe (inset: STEM image) and its electrocatalytic stability compared to PtCu and commercial Pt/C after 10000 cycles toward ORR. Reproduced with permission from ref. 36. Copyright 2018 Wiley-VCH. (c) TEM image of PtCuNi@PtCu (PC@PCN) dendrite@frame and ORR polarization curves of PC@PCN and commercial Pt/C before and after the cycling test for 5000 cycles. Reproduced with permission from ref. 38. Copyright 2017 American Chemical Society. (d) TEM image of IrNiCu double-layered nanoframe and OER polarization cures of IrNiCu double layered nanoframe and single layered nanoframe before and after durability test of 2500 potential cycles. Reproduced with permission from ref. 298. Copyright 2017 American Chemical Society. | ||
5. Outlook
Thus far, a number of hollow nanostructures with novel compositions have demonstrated superb electrocatalytic activities toward the ORR, HER, and OER, benefitting from the increased number of active sites provided by the inherently high surface area structure. Accordingly, the number of hollow nanocatalysts is increasing rapidly due to the intense synthetic efforts and resulting accumulation of synthetic knowledge for this unique class of materials. However, rational synthetic routes to these nanostructures are still underdeveloped, and the true origins of catalytic the activity and stability of multicomponent materials are not well understood. Therefore, in order to facilitate the development of hollow nanostructure-based catalysts for energy applications, the following points must be addressed.5.1. Enhancing the intrinsic catalytic activity of hollow nanostructures by modulating the energy states of the active sites
The open structures of hollow nanostructures provide access to numerous active sites and remarkably enhance electrocatalytic performance. While this strategy for increasing the number of active sites is undoubtedly beneficial for catalysis, a more important issue in catalyst development is enhancement of the intrinsic activity of catalytically active sites.288 Recent advances in this area show that it is feasible to modulate the electronic state of the active sites at which the reactants are adsorbed and desorbed, through nanostructural engineering.151,171,326 For example, the strain on the nanoparticle surface induced by the interface lattice mismatch between the core and the shell can greatly affect the surface energy states.202,326 The strain can be further modulated by controlling the alloy composition of the core or the thickness of the shell.242,245,327 Most recently, Bu et al. reported the synthesis of biaxially strained PtPb/Pt core/shell nanoplates with enhanced oxygen reduction catalysis performance.326 They found that the core–shell structural features lead to a large tensile strain that optimizes the Pt–O bond strength, which is crucial for the ORR. In addition, Li et al. synthesized ultrafine jagged platinum nanowires with state-of-the-art mass activity at 0.9 V toward the ORR.223 They suggested that the superior electrocatalytic activity originated from the atomic stress in the nanowires, based on a reactive molecular dynamics study. Considering these examples, the synthesis of hollow nanostructures featuring surface strain is an appealing strategy for the development of active catalysts. Furthermore, studies show that ternary or even quaternary alloy systems can exhibit greatly enhanced catalytic activity and material stability.234,325,328 Therefore, hollow nanostructures with exotic alloy compositions might usher in the widespread usage of electrochemical water splitting and fuel cell technologies.5.2. Development of theoretical tools to understand the true origin of the catalytic activity of multicomponent hollow nanostructures
Theoretical studies using the optimal simulation models for nanocrystals are crucial for understanding the reaction mechanism, and they should involve the assessment of the surface energy of the exposed nanoparticle or the binding energy between reactant intermediates and the catalyst surface. Armed with this knowledge, it would be feasible to design next generation nanocatalysts with an acceptable level of catalytic activity and stability. Recently, Bacq et al. reported the atomic vacancies or defects generating during the surface restructuring of hollow nanostructures are pivotal in determining high electrocatalytic performance of hollow nanostructure after durability test and proved the true origin of the sustained catalytic activity by DFT calculation.172 However, current studies are mostly confined to facet-controlled bimetallic nanoparticles, and the understanding of the atom arrangements of ternary or quaternary alloy phases during catalysis and their interaction with reaction intermediates is virtually non-existent, due to the difficulties encountered with numerous plausible surface atom configurations. Therefore, the development of methods to greatly simplify the theoretical calculation of complex atomistic arrangements might greatly aid our understanding of the origin of catalytic activity and the development of nanoalloy particles with optimal compositions.5.3. Understanding the dynamics of atomic rearrangement within nanocatalysts during the electrocycling test
As discussed in Section 4, the surface migration of atoms during the electrocycling test might induce enhanced electrocatalytic performance, but it can deteriorate the catalytic stability at the same time. In the case of a few bimetallic alloy systems, it was feasible to identify certain factors affecting the phase segregation phenomena by observing the changes in their atomic arrangement with ex situ TEM techniques. Recently, intra-particle atom movements have been followed using in situ TEM techniques.329–332 However, in this case, the condition that led to the real-time atom movement, i.e. simple heating, is not relevant to the catalytic setting. Therefore, the development of in situ cell designs and the observation of real time atom movements in catalyst nanoparticles during catalysis would great elevate the level of our understanding of nanoparticle phase conversion and catalyst stability as well as the origin of catalytic activity.5.4. Considering the actual catalysis operating condition which is to be practically used
The thermodynamically stable alloy phase is a little bit different between during the synthesis and during the electrocatalytic test as already discussed in previous chapter. Until now, some efforts have raised to consider the electrochemical performance measured at high electrochemical potential or elevated temperature, which are similar conditions compared to practical PEMFC application or water electrolysers. Dubau et al. the stability test of hollow nanostructures at elevated temperatures greatly affected the structural degradation of the hollow nanostructure.150,333,334 The hollow PtNi/C nanoparticles retained their morphologies and compositions during accelerated stress testing at 25 °C, however, the dissolution of Ni atoms was extremely accelerated with increasing the temperature (>80 °C). Although the specific dissolution of the alloying transition metal could be observed, the electrocatalytic performance of hollow PtNi/C nanoparticles toward the ORR still exhibits higher than that of solid PtNi/C nanoparticles. They proposed that the origin of retained electrocatalytic activity is driven from the atomic vacancies contained in the PtNi shell and grain boundaries.170In addition, mass-transport limitations of hollow nanostructures in real membrane electrode assemblies are also critical issue. Snyder, Erlebacher et al. discovered that the ECSA of hollow nanostructures could be remarkably different dependent on the range of overpotential.335 At low overpotential, the oxygen molecules could be diffused into the inner surfaces of hollow nanoparticles, however, they are suppressed to be diffused into the inner surfaces at higher overpotential. Due to this result, we expected that the large surface area of hollow nanostructures might be diminished in actual operating condition. Several recently reported documents proposed that using ionic liquid instead of aqueous solution could offer higher oxygen solubility by filling the inner surface area of hollow nanostructures.38,143,207,336 This concept also proved the advantage of hollow structural feature, but it is important to take into account when we used in actual condition. Therefore, profound understanding of dealloying procedure and limitation of mass-transport in the actual condition is recommended in order to make sustainable electrocatalysts.
6. Conclusion
As described in this review, the structural advantage of hollow nanostructures as electrocatalysts for the HER, OER, and ORR does not originate only from their inherent high surface area. A detailed understanding of the mechanistic aspects of the catalytic events suggests that facet control and alloy composition control can also greatly improve the chance of widespread societal penetration of these energy conversion technologies. The design concept of hollow nanostructures can evolve further to incorporate ternary or quaternary alloy systems into the nanoframeworks as well as the additional structural features of lattice-mismatched interfaces or twinning. Despite the great potential benefits of multicomponent nanoframe structures as electrocatalysts, we have only recently embarked on the journey of nanoscale alloy formation and phase segregation, and thus our synthetic tools for the fabrication of alloy nanoframes are still primitive. Therefore, there is a huge research potential in the area of multicomponent hollow nanostructures with greater complexity than binary or ternary systems as electrocatalysts. Additionally, examples of hollow nanostructures of many catalytically important metal compounds, such as metal oxides, sulfides, and phosphides, are very rare. Eventually, catalysts should be extremely atom efficient, and the recent development of single atom catalysts is certainly a promising research direction. It is utterly intriguing to envisage hollow nanostructures featuring the novel design concepts of single atom catalysis and nanofeatures that modify the surface energy, such as lattice-mismatch or twinning. Therefore, it is not difficult to imagine the advent of next generation hollow electrocatalysts incorporating some of these exciting structural features in the near future.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by IBS-R023-D1, NRF-2017R1A2B3005682, and Korea University Future Research Grant. S. H. J. and H. Y. K. were supported by the NRF of Korea (NRF-2017R1A2B2008464).References
- H. B. Gray, Nat. Chem., 2009, 1, 7 CrossRef PubMed
.
- M. G. Walter, E. L. Warren, J. R. McKone, S. W. Boettcher, Q. Mi, E. A. Santori and N. S. Lewis, Chem. Rev., 2010, 110, 6446–6473 CrossRef PubMed
- T. R. Cook, D. K. Dogutan, S. Y. Reece, Y. Surendranath, T. S. Teets and D. G. Nocera, Chem. Rev., 2010, 110, 6474–6502 CrossRef PubMed
- J. Qi, W. Zhang and R. Cao, Adv. Energy Mater., 2018, 8, 1701620 CrossRef
- I. E. L. Stephens, J. Rossmeisl and I. Chorkendorff, Science, 2016, 354, 1378–1379 CrossRef PubMed
- Y. Jiao, Y. Zheng, M. Jaroniec and S. Z. Qiao, Chem. Soc. Rev., 2015, 44, 2060–2086 RSC
- J. H. Montoya, L. C. Seitz, P. Chakthranont, A. Vojvodic, T. F. Jaramillo and J. K. Nørskov, Nat. Mater., 2017, 16, 70–81 CrossRef PubMed
- S. Anantharaj, S. R. Ede, K. Sakthikumar, K. Karthick, S. Mishra and S. Kundu, ACS Catal., 2016, 6, 8069–8097 CrossRef
- T. Reier, H. N. Nong, D. Teschner, R. Schlögl and P. Strasser, Adv. Energy Mater., 2017, 7, 1601275 CrossRef
- Y. Xia and X. Yang, Acc. Chem. Res., 2017, 50, 450–454 CrossRef PubMed
- F. Calle-Vallejo, M. D. Pohl, D. Reinisch, D. Loffreda, P. Sautet and A. S. Bandarenka, Chem. Sci., 2017, 8, 2283–2289 RSC
- J. K. Nørskov, T. Bligaard, J. Rossmeisl and C. H. Christensen, Nat. Chem., 2009, 1, 37–46 CrossRef PubMed
- K. S. Exner and H. Over, Acc. Chem. Res., 2017, 50, 1240–1247 CrossRef PubMed
- M. M. Montemore, M. A. van Spronsen, R. J. Madix and C. M. Friend, Chem. Rev., 2017, 118, 2816–2862 CrossRef PubMed
- V. R. Stamenkovic, D. Strmcnik, P. P. Lopes and N. M. Markovic, Nat. Mater., 2017, 16, 57–69 CrossRef PubMed
- W. Gao, Z. D. Hood and M. Chi, Acc. Chem. Res., 2017, 50, 787–795 CrossRef PubMed
- I. E. L. Stephens, A. S. Bondarenko, U. Grønbjerg, J. Rossmeisl and I. Chorkendorff, Energy Environ. Sci., 2012, 5, 6744 RSC
- V. R. Stamenkovic, B. S. Mun, M. Arenz, K. J. J. Mayrhofer, C. A. Lucas, G. Wang, P. N. Ross and N. M. Markovic, Nat. Mater., 2007, 6, 241–247 CrossRef PubMed
- J. Greeley and M. Mavrikakis, Nat. Mater., 2004, 3, 810 CrossRef PubMed
- A. R. Tao, S. Habas and P. Yang, Small, 2008, 4, 310–325 CrossRef
- M. Chen, B. Wu, J. Yang and N. Zheng, Adv. Mater., 2012, 24, 862–879 CrossRef PubMed
- M. B. Gawande, A. Goswami, T. Asefa, H. Guo, A. V. Biradar, D.-L. Peng, R. Zboril and R. S. Varma, Chem. Soc. Rev., 2015, 44, 7540–7590 RSC
- B. T. Sneed, A. P. Young and C.-K. Tsung, Nanoscale, 2015, 7, 12248–12265 RSC
- R. Ghosh Chaudhuri and S. Paria, Chem. Rev., 2012, 112, 2373–2433 CrossRef PubMed
- R. Schlögl, Angew. Chem., Int. Ed., 2015, 54, 3465–3520 CrossRef PubMed
- S. Schauermann, N. Nilius, S. Shaikhutdinov and H.-J. Freund, Acc. Chem. Res., 2013, 46, 1673–1681 CrossRef PubMed
- S. M. S. Kumar, N. Hidyatai, J. S. Herrero, S. Irusta and K. Scott, Int. J. Hydrogen Energy, 2011, 36, 5453–5465 CrossRef
- J. Cho, L. Xu, C. Jo and R. Ryoo, Chem. Commun., 2017, 53, 3810–3813 RSC
- S. H. Joo, S. J. Choi, I. Oh, J. Kwak, Z. Liu, O. Terasaki and R. Ryoo, Nature, 2001, 412, 169 CrossRef PubMed
- Y. Keereeta, S. Thongtem and T. Thongtem, Powder Technol., 2015, 284, 85–94 CrossRef
- S. S. Park, S.-W. Chu, C. Xue, D. Zhao and C.-S. Ha, J. Mater. Chem., 2011, 21, 10801–10807 RSC
- W. Wang, M. Dahl and Y. Yin, Chem. Mater., 2013, 25, 1179–1189 CrossRef
- B. D. Anderson and J. B. Tracy, Nanoscale, 2014, 6, 12195–12216 RSC
- H. J. Fan, U. Gösele and M. Zacharias, Small, 2007, 3, 1660–1671 CrossRef PubMed
- V. Beermann, M. Gocyla, E. Willinger, S. Rudi, M. Heggen, R. E. Dunin-Borkowski, M. G. Willinger and P. Strasser, Nano Lett., 2016, 16, 1719–1725 CrossRef PubMed
- T. Kwon, M. Jun, H. Y. Kim, A. Oh, J. Park, H. Baik, S. H. Joo and K. Lee, Adv. Funct. Mater., 2018, 28, 1706440 CrossRef
- A. Oh, H. Baik, D. S. Choi, J. Y. Cheon, B. Kim, H. Kim, S. J. Kwon, S. H. Joo, Y. Jung and K. Lee, ACS Nano, 2015, 9, 2856–2867 CrossRef PubMed
- J. Park, M. Kanti Kabiraz, H. Kwon, S. Park, H. Baik, S.-I. Choi and K. Lee, ACS Nano, 2017, 11, 10844–10851 CrossRef PubMed
- K. Lee, M. Kim and H. Kim, J. Mater. Chem., 2010, 20, 3791–3798 RSC
- B. Lim, J. Wang, P. H. Camargo, M. Jiang, M. J. Kim and Y. Xia, Nano Lett., 2008, 8, 2535–2540 CrossRef PubMed
- B. Lim, M. Jiang, P. H. Camargo, E. C. Cho, J. Tao, X. Lu, Y. Zhu and Y. Xia, Science, 2009, 324, 1302–1305 CrossRef PubMed
- F. R. Fan, D. Y. Liu, Y. F. Wu, S. Duan, Z. X. Xie, Z. Y. Jiang and Z. Q. Tian, J. Am. Chem. Soc., 2008, 130, 6949–6951 CrossRef PubMed
- T. Kim, J. Park, H. Jin, A. Oh, H. Baik, S. H. Joo and K. Lee, Nanoscale, 2018, 10, 9845–9850 RSC
- M. Zhao, L. Figueroa-Cosme, A. O. Elnabawy, M. Vara, X. Yang, L. T. Roling, M. Chi, M. Mavrikakis and Y. Xia, Nano Lett., 2016, 16, 5310–5317 CrossRef PubMed
- M. Zhao, A. O. Elnabawy, M. Vara, L. Xu, Z. D. Hood, X. Yang, K. D. Gilroy, L. Figueroa-Cosme, M. Chi, M. Mavrikakis and Y. Xia, Chem. Mater., 2017, 29, 9227–9237 CrossRef
- S. Xie, N. Lu, Z. Xie, J. Wang, M. J. Kim and Y. Xia, Angew. Chem., Int. Ed., 2012, 51, 10266–10270 CrossRef PubMed
- B. T. Sneed, C. N. Brodsky, C. H. Kuo, L. K. Lamontagne, Y. Jiang, Y. Wang, F. F. Tao, W. Huang and C. K. Tsung, J. Am. Chem. Soc., 2013, 135, 14691–14700 CrossRef PubMed
- W. Ye, S. Kou, X. Guo, F. Xie, H. Sun, H. Lu and J. Yang, Nanoscale, 2015, 7, 9558–9562 RSC
- N. Fan, Y. Yang, W. Wang, L. Zhang, W. Chen, C. Zou and S. Huang, ACS Nano, 2012, 6, 4072–4082 CrossRef PubMed
- L. Zhang, L. T. Roling, X. Wang, M. Vara, M. Chi, J. Liu, S. I. Choi, J. Park, J. A. Herron, Z. Xie, M. Mavrikakis and Y. Xia, Science, 2015, 349, 412–416 CrossRef PubMed
- X. Wang, L. Figueroa-Cosme, X. Yang, M. Luo, J. Liu, Z. Xie and Y. Xia, Nano Lett., 2016, 16, 1467–1471 CrossRef PubMed
- J. Park, H. Wang, M. Vara and Y. Xia, ChemSusChem, 2016, 9, 2855–2861 CrossRef PubMed
- H. Ye, Q. Wang, M. Catalano, N. Lu, J. Vermeylen, M. J. Kim, Y. Liu, Y. Sun and X. Xia, Nano Lett., 2016, 16, 2812–2817 CrossRef PubMed
- D. S. He, D. He, J. Wang, Y. Lin, P. Yin, X. Hong, Y. Wu and Y. Li, J. Am. Chem. Soc., 2016, 138, 1494–1497 CrossRef PubMed
- S. Bang, D. Yoon, J. Kim, H. Baik, H. Yang and K. Lee, CrystEngComm, 2015, 17, 4084–4088 RSC
- H. Jin, K. W. Lee, N. T. Khi, H. An, J. Park, H. Baik, J. Kim, H. Yang and K. Lee, Small, 2015, 11, 4462–4468 CrossRef PubMed
- L. Yu, H. Hu, H. B. Wu and X. W. D. Lou, Adv. Mater., 2017, 29, 1604563 CrossRef PubMed
- X. Lai, J. Li, B. A. Korgel, Z. Dong, Z. Li, F. Su, J. Du and D. Wang, Angew. Chem., Int. Ed., 2011, 50, 2738–2741 CrossRef PubMed
- X. Lai, J. E. Halpert and D. Wang, Energy Environ. Sci., 2012, 5, 5604–5618 RSC
- J. Wang, N. Yang, H. Tang, Z. Dong, Q. Jin, M. Yang, D. Kisailus, H. Zhao, Z. Tang and D. Wang, Angew. Chem., Int. Ed., 2013, 52, 6417–6420 CrossRef PubMed
- G. Zhang and X. W. D. Lou, Angew. Chem., Int. Ed., 2014, 53, 9041–9044 CrossRef PubMed
- Y. Guo, L. Yu, C.-Y. Wang, Z. Lin and X. W. D. Lou, Adv. Funct. Mater., 2015, 25, 5184–5189 CrossRef
- P. M. Arnal, M. Comotti and F. Schüth, Angew. Chem., Int. Ed., 2006, 45, 8224–8227 CrossRef PubMed
- X. W. Lou, C. M. Li and L. A. Archer, Adv. Mater., 2009, 21, 2536–2539 CrossRef
- R. Jin, Y. Yang, Y. Xing, L. Chen, S. Song and R. Jin, ACS Nano, 2014, 8, 3664–3670 CrossRef PubMed
- X. W. Lou, C. Yuan and L. A. Archer, Adv. Mater., 2007, 19, 3328–3332 CrossRef
- X. W. Lou and L. A. Archer, Adv. Mater., 2008, 20, 1853–1858 CrossRef
- Z. Wang, D. Luan, F. Y. Boey and X. W. Lou, J. Am. Chem. Soc., 2011, 133, 4738–4741 CrossRef PubMed
- Z. Wang and X. W. D. Lou, Adv. Mater., 2012, 24, 4124–4129 CrossRef PubMed
- H. Hu, L. Yu, X. Gao, Z. Lin and X. W. Lou, Energy Environ. Sci., 2015, 8, 1480–1483 RSC
- L. Zhang, T. Liu, K. Liu, L. Han, Y. Yin and C. Gao, Nano Lett., 2015, 15, 4448–4454 CrossRef PubMed
- B. Y. Guan, L. Yu, J. Li and X. W. Lou, Sci. Adv., 2016, 2, e1501554 Search PubMed
- H. J. Fan, M. Knez, R. Scholz, D. Hesse, K. Nielsch, M. Zacharias and U. Gosele, Nano Lett., 2007, 7, 993–997 CrossRef PubMed
- Y. Yin, R. M. Rioux, C. K. Erdonmez, S. Hughes, G. A. Somorjai and A. P. Alivisatos, Science, 2004, 304, 711–714 CrossRef PubMed
- Y. Yin, C. K. Erdonmez, A. Cabot, S. Hughes and A. P. Alivisatos, Adv. Funct. Mater., 2006, 16, 1389–1399 CrossRef
- A. Cabot, V. F. Puntes, E. Shevchenko, Y. Yin, L. Balcells, M. A. Marcus, S. M. Hughes and A. P. Alivisatos, J. Am. Chem. Soc., 2007, 129, 10358–10360 CrossRef PubMed
- S. Peng and S. Sun, Angew. Chem., Int. Ed., 2007, 46, 4155–4158 CrossRef PubMed
- R.-K. Chiang and R.-T. Chiang, Inorg. Chem., 2007, 46, 369–371 CrossRef PubMed
- J. Park, H. Zheng, Y. W. Jun and A. P. Alivisatos, J. Am. Chem. Soc., 2009, 131, 13943–13945 CrossRef PubMed
- C.-H. Kuo, Y.-T. Chu, Y.-F. Song and M. H. Huang, Adv. Funct. Mater., 2011, 21, 792–797 CrossRef
- J. Kim, H. Jin, A. Oh, H. Baik, S. H. Joo and K. Lee, Nanoscale, 2017, 9, 15397–15406 RSC
- S. G. Bratsch, J. Phys.
Chem. Ref. Data, 1989, 18, 1–21 CrossRef
- Y. G. Sun, B. T. Mayers and Y. N. Xia, Nano Lett., 2002, 2, 481–485 CrossRef
- Y. Sun and Y. Xia, Science, 2002, 298, 2176–2179 CrossRef PubMed
- X. Sun, J. Kim, K. D. Gilroy, J. Liu, T. A. F. König and D. Qin, ACS Nano, 2016, 10, 8019–8025 CrossRef PubMed
- Y. Choi, S. Hong, L. Liu, S. K. Kim and S. Park, Langmuir, 2012, 28, 6670–6676 CrossRef PubMed
- J. Chen, B. Wiley, Z. Y. Li, D. Campbell, F. Saeki, H. Cang, L. Au, J. Lee, X. Li and Y. Xia, Adv. Mater., 2005, 17, 2255–2261 CrossRef
- X. Lu, L. Au, J. McLellan, Z. Y. Li, M. Marquez and Y. Xia, Nano Lett., 2007, 7, 1764–1769 CrossRef PubMed
- Y. Sun and Y. Xia, J. Am. Chem. Soc., 2004, 126, 3892–3901 CrossRef PubMed
- L. Vitos, A. V. Ruban, H. L. Skriver and J. Kollar, Surf. Sci., 1998, 411, 186–202 CrossRef
- C. M. Cobley and Y. Xia, Mater. Sci. Eng., R, 2010, 70, 44–62 CrossRef PubMed
- X. Xia, Y. Wang, A. Ruditskiy and Y. Xia, Adv. Mater., 2013, 25, 6313–6333 CrossRef PubMed
- E. González, J. Arbiol and V. F. Puntes, Science, 2011, 334, 1377–1380 CrossRef PubMed
- J. W. Hong, S. W. Kang, B.-S. Choi, D. Kim, S. B. Lee and S. W. Han, ACS Nano, 2012, 6, 2410–2419 CrossRef PubMed
- M. Liu, Y. Zheng, S. Xie, N. Li, N. Lu, J. Wang, M. J. Kim, L. Guo and Y. Xia, Phys. Chem. Chem. Phys., 2013, 15, 11822 RSC
- S. Xie, M. Jin, J. Tao, Y. Wang, Z. Xie, Y. Zhu and Y. Xia, Chem. – Eur. J., 2012, 18, 14974–14980 CrossRef PubMed
- L. Han, P. Wang, H. Liu, Q. Tan and J. Yang, J. Mater. Chem. A, 2016, 4, 18354–18365 RSC
- M. H. Oh, T. Yu, S. H. Yu, B. Lim, K. T. Ko, M. G. Willinger, D. H. Seo, B. H. Kim, M. G. Cho, J. H. Park, K. Kang, Y. E. Sung, N. Pinna and T. Hyeon, Science, 2013, 340, 964–968 CrossRef PubMed
- M. D. Susman, R. Popovitz-Biro, A. Vaskevich and I. Rubinstein, Small, 2015, 11, 3942–3953 CrossRef PubMed
- A. López-Ortega, A. G. Roca, P. Torruella, M. Petrecca, S. Estradé, F. Peiró, V. Puntes and J. Nogués, Chem. Mater., 2016, 28, 8025–8031 CrossRef
- D. Yoon, S. Bang, J. Park, J. Kim, H. Baik, H. Yang and K. Lee, CrystEngComm, 2015, 17, 6843–6847 RSC
- Z. Niu, N. Becknell, Y. Yu, D. Kim, C. Chen, N. Kornienko, G. A. Somorjai and P. Yang, Nat. Mater., 2016, 15, 1188–1194 CrossRef PubMed
- D. Yoon, S. Park, J. Park, J. Kim, H. Baik, H. Yang and K. Lee, Nanoscale, 2014, 6, 12397–12402 RSC
- J. Park, J. Kim, Y. Yang, D. Yoon, H. Baik, S. Haam, H. Yang and K. Lee, Adv. Sci., 2016, 3, 1500252 CrossRef PubMed
- H. Kobayashi, K. Kusada and H. Kitagawa, Acc. Chem. Res., 2015, 48, 1551–1559 CrossRef PubMed
- G. Bruni, Chem. Rev., 1925, 1, 345–375 CrossRef
- B. Huang, H. Kobayashi, T. Yamamoto, S. Matsumura, Y. Nishida, K. Sato, K. Nagaoka, S. Kawaguchi, Y. Kubota and H. Kitagawa, J. Am. Chem. Soc., 2017, 139, 4643–4646 CrossRef PubMed
- Z. Swiatkowska-Warkocka, A. Pyatenko, F. Krok, B. R. Jany and M. Marszalek, Sci. Rep., 2015, 5, 9849 CrossRef PubMed
- K. Kusada, H. Kobayashi, R. Ikeda, Y. Kubota, M. Takata, S. Toh, T. Yamamoto, S. Matsumura, N. Sumi, K. Sato, K. Nagaoka and H. Kitagawa, J. Am. Chem. Soc., 2014, 136, 1864–1871 CrossRef PubMed
- D. Srikanth, A. Gargi, K. Sanket Ranjan, K. Sanjeet and C. Abhijit, Modell. Simul. Mater. Sci. Eng., 2017, 25, 085011 CrossRef
- M. Cui, H. Lu, H. Jiang, Z. Cao and X. Meng, Sci. Rep., 2017, 7, 41990 CrossRef PubMed
- L. L. Wang and D. D. Johnson, J. Am. Chem. Soc., 2009, 131, 14023–14029 CrossRef PubMed
- S. Xiong, W. Qi, B. Huang and M. Wang, ChemPhysChem, 2011, 12, 1317–1324 CrossRef PubMed
- W. H. Qi, B. Y. Huang and M. P. Wang, Phys. B, 2009, 404, 1761–1765 CrossRef
- W. H. Qi, M. P. Wang and Q. H. Liu, J. Mater. Sci., 2005, 40, 2737–2739 CrossRef
- A. Christensen, P. Stoltze and J. K. Norskov, J. Phys.: Condens. Matter, 1995, 7, 1047 CrossRef
- G. Guisbiers, S. Khanal, F. Ruiz-Zepeda, J. Roque de la Puente and M. Jose-Yacaman, Nanoscale, 2014, 6, 14630–14635 RSC
- M. Cui, H. Lu, H. Jiang, Z. Cao and X. Meng, Sci. Rep., 2017, 7, 41990 CrossRef PubMed
- G. Guisbiers, R. Mendoza-Pérez, L. Bazán-Díaz, R. Mendoza-Cruz, J. J. Velázquez-Salazar and M. José-Yacamán, J. Phys. Chem. C, 2017, 121, 6930–6939 CrossRef
- H. Liao, A. Fisher and Z. J. Xu, Small, 2015, 11, 3221–3246 CrossRef PubMed
- R. Ferrando, J. Jellinek and R. L. Johnston, Chem. Rev., 2008, 108, 845–910 CrossRef PubMed
- A. Zaleska-Medynska, M. Marchelek, M. Diak and E. Grabowska, Adv. Colloid Interface Sci., 2016, 229, 80–107 CrossRef PubMed
- B.-J. Hwang, L. S. Sarma, J.-M. Chen, C.-H. Chen, S.-C. Shih, G.-R. Wang, D.-G. Liu, J.-F. Lee and M.-T. Tang, J. Am. Chem. Soc., 2005, 127, 11140–11145 CrossRef PubMed
- N. Becknell, C. D. Zheng, C. Chen and P. D. Yang, Surf. Sci., 2016, 648, 328–332 CrossRef
- A. Oh, Y. J. Sa, H. Hwang, H. Baik, J. Kim, B. Kim, S. H. Joo and K. Lee, Nanoscale, 2016, 8, 16379–16386 RSC
- F. Calvo, Phys. Chem. Chem. Phys., 2015, 17, 27922–27939 RSC
- J. Suntivich, Z. Xu, C. E. Carlton, J. Kim, B. Han, S. W. Lee, N. Bonnet, N. Marzari, L. F. Allard, H. A. Gasteiger, K. Hamad-Schifferli and Y. Shao-Horn, J. Am. Chem. Soc., 2013, 135, 7985–7991 CrossRef PubMed
- S.-Y. Lee, N. Jung, J. Cho, H.-Y. Park, J. Ryu, I. Jang, H.-J. Kim, E. Cho, Y.-H. Park, H. C. Ham, J. H. Jang and S. J. Yoo, ACS Catal., 2014, 4, 2402–2408 CrossRef
- K. J. Andersson, F. Calle-Vallejo, J. Rossmeisl and I. Chorkendorff, J. Am. Chem. Soc., 2009, 131, 2404–2407 CrossRef PubMed
- K. J. J. Mayrhofer, V. Juhart, K. Hartl, M. Hanzlik and M. Arenz, Angew. Chem., Int. Ed., 2009, 48, 3529–3531 CrossRef PubMed
- G. E. Ramírez-Caballero, Y. Ma, R. Callejas-Tovar and P. B. Balbuena, Phys. Chem. Chem. Phys., 2010, 12, 2209 RSC
- R. Mu, X. Guo, Q. Fu and X. Bao, J. Phys. Chem. C, 2011, 115, 20590–20595 CrossRef
- F. Tao, M. E. Grass, Y. Zhang, D. R. Butcher, J. R. Renzas, Z. Liu, J. Y. Chung, B. S. Mun, M. Salmeron and G. A. Somorjai, Science, 2008, 322, 932–934 CrossRef PubMed
- A. J. Logsdail, L. O. Paz-Borbon and C. A. Downing, J. Phys. Chem. C, 2018, 122, 5721–5730 CrossRef
- A. Lopes, G. Tréglia, C. Mottet and B. Legrand, Phys. Rev. B: Condens. Matter Mater. Phys., 2015, 91, 035407 CrossRef
- W. Niu, L. Zhang and G. Xu, ACS Nano, 2010, 4, 1987–1996 CrossRef PubMed
- X. He, S.-E. Zhang, F. Cheng and Z.-X. Chen, Chem. Commun., 2018, 54, 638–641 RSC
- Y. G. Chen, Z. J. Yu, Z. Chen, R. A. Shen, Y. Wang, X. Cao, Q. Peng and Y. D. Li, Nano Res., 2016, 9, 2632–2640 CrossRef
- C. Li, K. Eid, H. J. Wang, Y. Y. Deng, S. L. Lu, X. N. Li, L. Wang and H. W. Gu, New J. Chem., 2018, 42, 798–801 RSC
- F. Nosheen, Z. C. Zhang, J. Zhuang and X. Wang, Nanoscale, 2013, 5, 3660–3663 RSC
- J. Pei, J. Mao, X. Liang, C. Chen, Q. Peng, D. Wang and Y. Li, Chem. Commun., 2016, 52, 3793–3796 RSC
- J. Ding, L. Bu, S. Guo, Z. Zhao, E. Zhu, Y. Huang and X. Huang, Nano Lett., 2016, 16, 2762–2767 CrossRef PubMed
- C. Chen, Y. Kang, Z. Huo, Z. Zhu, W. Huang, H. L. Xin, J. D. Snyder, D. Li, J. A. Herron, M. Mavrikakis, M. Chi, K. L. More, Y. Li, N. M. Markovic, G. A. Somorjai, P. Yang and V. R. Stamenkovic, Science, 2014, 343, 1339–1343 CrossRef PubMed
- C. Wang, L. Zhang, H. Yang, J. Pan, J. Liu, C. Dotse, Y. Luan, R. Gao, C. Lin, J. Zhang, J. P. Kilcrease, X. Wen, S. Zou and J. Fang, Nano Lett., 2017, 17, 2204–2210 CrossRef PubMed
- H. Hwang, T. Kwon, H. Y. Kim, J. Park, A. Oh, B. Kim, H. Baik, S. H. Joo and K. Lee, Small, 2018, 14, 1702353 CrossRef PubMed
- G. Chen, S. Desinan, R. Rosei, F. Rosei and D. Ma, Chem. Commun., 2012, 48, 8009 RSC
- S. J. Bae, S. J. Yoo, Y. Lim, S. Kim, Y. Lim, J. Choi, K. S. Nahm, S. J. Hwang, T.-H. Lim, S.-K. Kim and P. Kim, J. Mater. Chem., 2012, 22, 8820–8825 RSC
- H. Bönnemann, G. Braun, W. Brijoux, R. Brinkmann, A. S. Tilling, K. Seevogel and K. Siepen, J. Organomet. Chem., 1996, 520, 143–162 CrossRef
- B. Helmut, B. Werner, B. Rainer, J. Thomas, K. Barbara and D. Eckard, Angew. Chem., Int. Ed. Engl., 1991, 30, 1312–1314 CrossRef
- T. Asset, R. Chattot, M. Fontana, B. Mercier-Guyon, N. Job, L. Dubau and F. Mailard, ChemPhysChem, 2018, 19, 1552–1567 CrossRef PubMed
- J. X. Wang, C. Ma, Y. Choi, D. Su, Y. Zhu, P. Liu, R. Si, M. B. Vukmirovic, Y. Zhang and R. R. Adzic, J. Am. Chem. Soc., 2011, 133, 13551–13557 CrossRef PubMed
- L. Dubau, M. Lopez-Haro, J. Durst, L. Guetaz, P. Bayle-Guillemaud, M. Chatenet and F. Maillard, J. Mater. Chem. A, 2014, 2, 18497–18507 RSC
- L. Dubau, T. Asset, R. Chattot, C. Bonnaud, V. Vanpeene, J. Nelayah and F. Maillard, ACS Catal., 2015, 5, 5333–5341 CrossRef
- R. Chattot, T. Asset, J. Drnec, P. Bordet, J. Nelayah, L. Dubau and F. Maillard, Nano Lett., 2017, 17, 2447–2453 CrossRef PubMed
- T. Asset, R. Chattot, J. Drnec, P. Bordet, N. Job, F. Maillard and L. Dubau, ACS Appl. Mater. Interfaces, 2017, 9, 25298–25307 CrossRef PubMed
- L. Han-Pu, Z. Hui-Min, H. Jin-Song, G. Yu-Guo, W. Li-Jun and B. Chun-Li, Angew. Chem., Int. Ed., 2004, 43, 1540–1543 CrossRef PubMed
- L. Dubau, J. Durst, F. Maillard, L. Guétaz, M. Chatenet, J. André and E. Rossinot, Electrochim. Acta, 2011, 56, 10658–10667 CrossRef
- M. Lopez-Haro, L. Dubau, L. Guétaz, P. Bayle-Guillemaud, M. Chatenet, J. André, N. Caqué, E. Rossinot and F. Maillard, Appl. Catal., B, 2014, 152–153, 300–308 CrossRef
- M. K. Debe, Nature, 2012, 486, 43–51 CrossRef PubMed
- M. Shao, Q. Chang, J.-P. Dodelet and R. Chenitz, Chem. Rev., 2016, 116, 3594–3657 CrossRef PubMed
- M. Carmo, D. L. Fritz, J. Merge and D. Stolten, Int. J. Hydrogen Energy, 2013, 38, 4901–4934 CrossRef
- N. S. Lewis, Science, 2016, 351, aad1920 CrossRef PubMed
- F. Gutierrez-Martin and I. Guerrero-Hernandez, Int. J. Hydrogen Energy, 2012, 37, 1151–1161 CrossRef
- K. Zeng and D. K. Zhang, Prog. Energy Combust. Sci., 2010, 36, 307–326 CrossRef
- P. C. K. Vesborg, B. Seger and I. Chorkendorff, J. Phys. Chem. Lett., 2015, 6, 951–957 CrossRef PubMed
- Z. W. Seh, J. Kibsgaard, C. F. Dickens, I. Chorkendorff, J. K. Nørskov and T. F. Jaramillo, Science, 2017, 355, eaad4998 CrossRef PubMed
- F. Calle-Vallejo, J. Tymoczko, V. Colic, Q. H. Vu, M. D. Pohl, K. Morgenstern, D. Loffreda, P. Sautet, W. Schuhmann and A. S. Bandarenka, Science, 2015, 350, 185–189 CrossRef PubMed
- A. Tristan, C. Raphaël, N. Jaysen, J. Nathalie, D. Laetitia and M. Frédéric, ChemElectroChem, 2016, 3, 1591–1600 CrossRef
- T. Asset, N. Job, Y. Busby, A. Crisci, V. Martin, V. Stergiopoulos, C. Bonnaud, A. Serov, P. Atanassov, R. Chattot, L. Dubau and F. Maillard, ACS Catal., 2018, 8, 893–903 CrossRef
- L. Dubau, J. Nelayah, S. Moldovan, O. Ersen, P. Bordet, J. Drnec, T. Asset, R. Chattot and F. Maillard, ACS Catal., 2016, 6, 4673–4684 CrossRef
- R. Chattot, T. Asset, P. Bordet, J. Drnec, L. Dubau and F. Maillard, ACS Catal., 2017, 7, 398–408 CrossRef
- L. B. Olivier, P. Alain, C. Raphaël, P. Bruno, N. Jaysen, A. Tristan, D. Laetitia and M. Frédéric, ChemCatChem, 2017, 9, 2324–2338 CrossRef
- W. Wang, B. Lei and S. Guo, Adv. Energy Mater., 2016, 6, 1600236 CrossRef
- M. C. Luo and S. J. Guo, Nat. Rev. Mater., 2017, 2, 17059 CrossRef
- S. Sui, X. Y. Wang, X. T. Zhou, Y. H. Su, S. Riffatc and C. J. Liu, J. Mater. Chem. A, 2017, 5, 1808–1825 RSC
- N. M. Markovic, R. R. Adzic, B. D. Cahan and E. B. Yeager, J. Electroanal. Chem., 1994, 377, 249–259 CrossRef
- N. M. Markovic, T. J. Schmidt, V. Stamenkovic and P. N. Ross, Fuel Cells, 2001, 1, 105–116 CrossRef
- V. R. Stamenkovic, B. Fowler, B. S. Mun, G. Wang, P. N. Ross, C. A. Lucas and N. M. Markovic, Science, 2007, 315, 493–497 CrossRef PubMed
- N. Hoshi, M. Nakamura and A. Hitotsuyanagi, Electrochim. Acta, 2013, 112, 899–904 CrossRef
- Z. Quan, Y. Wang and J. Fang, Acc. Chem. Res., 2012, 46, 191–202 CrossRef PubMed
- B. Y. Xia, H. B. Wu, X. Wang and X. W. D. Lou, Angew. Chem., Int. Ed., 2013, 52, 12337–12340 CrossRef PubMed
- J. Wu and H. Yang, Acc. Chem. Res., 2013, 46, 1848–1857 CrossRef PubMed
- T. Sheng, N. Tian, Z.-Y. Zhou, W.-F. Lin and S.-G. Sun, ACS Energy Lett., 2017, 2, 1892–1900 CrossRef
- X. Xu, X. Zhang, H. Sun, Y. Yang, X. Dai, J. Gao, X. Li, P. Zhang, H. H. Wang, N. F. Yu and S. G. Sun, Angew. Chem., Int. Ed., 2014, 53, 12522–12527 Search PubMed
- X. Huang, Z. Zhao, J. Fan, Y. Tan and N. Zheng, J. Am. Chem. Soc., 2011, 133, 4718–4721 CrossRef PubMed
- Q. Chen, Y. Jia, S. Xie and Z. Xie, Chem. Soc. Rev., 2016, 45, 3207–3220 RSC
- Y. Jia, Y. Jiang, J. Zhang, L. Zhang, Q. Chen, Z. Xie and L. Zheng, J. Am. Chem. Soc., 2014, 136, 3748–3751 CrossRef PubMed
- Q. Chen, Z. Cao, G. Du, Q. Kuang, J. Huang, Z. Xie and L. Zheng, Nano Energy, 2017, 39, 582–589 CrossRef
- J. Zhang, Q. Kuang, Y. Jiang and Z. Xie, Nano Today, 2016, 11, 661–677 CrossRef
- T. Yu, Y. Kim, H. Zhang and Y. Xia, Angew. Chem., 2011, 123, 2825–2829 CrossRef
- T. Toda, H. Igarashi, H. Uchida and M. Watanabe, J. Electrochem. Soc., 1999, 146, 3750–3756 CrossRef
- J. Greeley, I. E. L. Stephens, A. S. Bondarenko, T. P. Johansson, H. A. Hansen, T. F. Jaramillo, J. Rossmeisl, I. Chorkendorff and J. K. Nørskov, Nat. Chem., 2009, 1, 552–556 CrossRef PubMed
- V. Čolić and A. S. Bandarenka, ACS Catal., 2016, 6, 5378–5385 CrossRef
- Y. Bing, H. Liu, L. Zhang, D. Ghosh and J. Zhang, Chem. Soc. Rev., 2010, 39, 2184 RSC
- V. R. Stamenkovic, B. S. Mun, M. Arenz, K. J. J. Mayrhofer, C. A. Lucas, G. Wang, P. N. Ross and N. M. Markovic, Nat. Mater., 2007, 6, 241 CrossRef PubMed
- E. Toyoda, R. Jinnouchi, T. Hatanaka, Y. Morimoto, K. Mitsuhara, A. Visikovskiy and Y. Kido, J. Phys. Chem. C, 2011, 115, 21236–21240 CrossRef
- V. Viswanathan, H. A. Hansen, J. Rossmeisl and J. K. Nørskov, ACS Catal., 2012, 2, 1654–1660 CrossRef
- H. F. Lv, D. G. Li, D. Strmcnik, A. P. Paulikas, N. M. Markovic and V. R. Stamenkovic, Nano Energy, 2016, 29, 149–165 CrossRef
- F. H. B. Lima, J. Zhang, M. H. Shao, K. Sasaki, M. B. Vukmirovic, E. A. Ticianelli and R. R. Adzic, J. Phys. Chem. C, 2007, 111, 404–410 CrossRef
- P. Strasser and S. Kuhl, Nano Energy, 2016, 29, 166–177 CrossRef
- L. Gan, C. Cui, M. Heggen, F. Dionigi, S. Rudi and P. Strasser, Science, 2014, 346, 1502–1506 CrossRef PubMed
- P. Strasser, S. Koh, T. Anniyev, J. Greeley, K. More, C. Yu, Z. Liu, S. Kaya, D. Nordlund, H. Ogasawara, M. F. Toney and A. Nilsson, Nat. Chem., 2010, 2, 454–460 CrossRef PubMed
- D. Wang, Y. Yu, J. Zhu, S. Liu, D. A. Muller and H. D. Abruna, Nano Lett., 2015, 15, 1343–1348 CrossRef PubMed
- S. Koh and P. Strasser, J. Am. Chem. Soc., 2007, 129, 12624–12625 CrossRef PubMed
- S. Koh, N. Hahn, C. Yu and P. Strasser, J. Electrochem. Soc., 2008, 155, B1281–B1288 CrossRef
- J. Durst, M. Lopez-Haro, L. Dubau, M. Chatenet, Y. Soldo-Olivier, L. Guétaz, P. Bayle-Guillemaud and F. Maillard, J. Phys. Chem. Lett., 2014, 5, 434–439 CrossRef PubMed
- H. Kwon, M. K. Kabiraz, J. Park, A. Oh, H. Baik, S.-I. Choi and K. Lee, Nano Lett., 2018, 18, 2930–2936 CrossRef PubMed
- L. Wang, W. Gao, Z. Liu, Z. Zeng, Y. Liu, M. Giroux, M. Chi, G. Wang, J. Greeley, X. Pan and C. Wang, ACS Catal., 2017, 8, 35–42 CrossRef
- C. Wang, M. Chi, D. Li, D. Strmcnik, D. van der Vliet, G. Wang, V. Komanicky, K. C. Chang, A. P. Paulikas, D. Tripkovic, J. Pearson, K. L. More, N. M. Markovic and V. R. Stamenkovic, J. Am. Chem. Soc., 2011, 133, 14396–14403 CrossRef PubMed
- L. Gan, M. Heggen, C. Cui and P. Strasser, ACS Catal., 2015, 6, 692–695 CrossRef
- S. Lu and Z. Zhuang, J. Am. Chem. Soc., 2017, 139, 5156–5163 CrossRef PubMed
- B. Han, C. E. Carlton, A. Kongkanand, R. S. Kukreja, B. R. Theobald, L. Gan, R. O'Malley, P. Strasser, F. T. Wagner and Y. Shao-Horn, Energy Environ. Sci., 2015, 8, 258–266 RSC
- M. Ahmadi, F. Behafarid, C. Cui, P. Strasser and B. R. Cuenya, ACS Nano, 2013, 7, 9195–9204 CrossRef PubMed
- M. Ahmadi, C. Cui, H. Mistry, P. Strasser and B. Roldan Cuenya, ACS Nano, 2015, 9, 10686–10694 CrossRef PubMed
- L. Wang and Y. Yamauchi, J. Am. Chem. Soc., 2013, 135, 16762–16765 CrossRef PubMed
- T. Fu, J. Fang, C. S. Wang and J. B. Zhao, J. Mater. Chem. A, 2016, 4, 8803–8811 RSC
- H. Zhang, M. Jin, H. Liu, J. Wang, M. J. Kim, D. Yang, Z. Xie, J. Liu and Y. Xia, ACS Nano, 2011, 5, 8212–8222 CrossRef PubMed
- C. Cui, L. Gan, M. Heggen, S. Rudi and P. Strasser, Nat. Mater., 2013, 12, 765–771 CrossRef PubMed
- Y. Xia, X. Xia and H. C. Peng, J. Am. Chem. Soc., 2015, 137, 7947–7966 CrossRef PubMed
- Z. Zhang, Z. Luo, B. Chen, C. Wei, J. Zhao, J. Chen, X. Zhang, Z. Lai, Z. Fan, C. Tan, M. Zhao, Q. Lu, B. Li, Y. Zong, C. Yan, G. Wang, Z. J. Xu and H. Zhang, Adv. Mater., 2016, 28, 8712–8717 CrossRef PubMed
- L. Han, H. Liu, P. L. Cui, Z. J. Peng, S. J. Zhang and J. Yang, Sci. Rep., 2014, 4, 6414 CrossRef PubMed
- J. Ding, X. Zhu, L. Bu, J. Yao, J. Guo, S. Guo and X. Huang, Chem. Commun., 2015, 51, 9722–9725 RSC
- M. Li, Z. Zhao, T. Cheng, A. Fortunelli, C. Y. Chen, R. Yu, Q. Zhang, L. Gu, B. V. Merinov, Z. Lin, E. Zhu, T. Yu, Q. Jia, J. Guo, L. Zhang, W. A. Goddard, 3rd, Y. Huang and X. Duan, Science, 2016, 354, 1414–1419 CrossRef PubMed
- S. Luo and P. K. Shen, ACS Nano, 2016, 11, 11946–11953 CrossRef PubMed
- E. Antolini, J. R. C. Salgado and E. R. Gonzalez, J. Power Sources, 2006, 160, 957–968 CrossRef
- Y. Wu, D. Wang, G. Zhou, R. Yu, C. Chen and Y. Li, J. Am. Chem. Soc., 2014, 136, 11594–11597 CrossRef PubMed
- X. W. Yu and P. G. Pickup, J. Power Sources, 2008, 182, 124–132 CrossRef
- A. S. Arico, S. Srinivasan and V. Antonucci, Fuel Cells, 2001, 1, 133–161 CrossRef
- S. Wasmus and A. Küver, J. Electroanal. Chem., 1999, 461, 14–31 CrossRef
- J. N. Tiwari, R. N. Tiwari, G. Singh and K. S. Kim, Nano Energy, 2013, 2, 553–578 CrossRef
- K. Jiang, Q. Shao, D. Zhao, L. Bu, J. Guo and X. Huang, Adv. Funct. Mater., 2017, 27, 1700830 CrossRef
- S. Luo, M. Tang, P. K. Shen and S. Ye, Adv. Mater., 2017, 29, 1601687 Search PubMed
- L.-M. Lyu, Y.-C. Kao, D. A. Cullen, B. T. Sneed, Y.-C. Chuang and C.-H. Kuo, Chem. Mater., 2017, 29, 5681–5692 CrossRef
- M. A. Ud Din, F. Saleem, B. Ni, Y. Yong and X. Wang, Adv. Mater., 2017, 29, 1604994 CrossRef PubMed
- S. I. Choi, S. Xie, M. Shao, J. H. Odell, N. Lu, H. C. Peng, L. Protsailo, S. Guerrero, J. Park, X. Xia, J. Wang, M. J. Kim and Y. Xia, Nano Lett., 2013, 13, 3420–3425 CrossRef PubMed
- N. Zhang, Y. Feng, X. Zhu, S. Guo, J. Guo and X. Huang, Adv. Mater., 2017, 29, 1603774 CrossRef PubMed
- L. Bu, Q. Shao, E. Bin, J. Guo, J. Yao and X. Huang, J. Am. Chem. Soc., 2017, 139, 9576–9582 CrossRef PubMed
- L. Luo, F. Zhu, R. Tian, L. Li, S. Shen, X. Yan and J. Zhang, ACS Catal., 2017, 7, 5420–5430 CrossRef
- C. Zhang, W. Sandorf and Z. Peng, ACS Catal., 2015, 5, 2296–2300 CrossRef
- X. Sun, K. Jiang, N. Zhang, S. Guo and X. Huang, ACS Nano, 2015, 9, 7634–7640 CrossRef PubMed
- B. A. Lu, T. Sheng, N. Tian, Z. C. Zhang, C. Xiao, Z. M. Cao, H. B. Ma, Z. Y. Zhou and S. G. Sun, Nano Energy, 2017, 33, 65–71 CrossRef
- X. Wang, S.-I. Choi, L. T. Roling, M. Luo, C. Ma, L. Zhang, M. Chi, J. Liu, Z. Xie, J. A. Herron, M. Mavrikakis and Y. Xia, Nat. Commun., 2015, 6, 7594 CrossRef PubMed
- J. Gu, G. X. Lan, Y. Y. Jiang, Y. S. Xu, W. Zhu, C. H. Jin and Y. W. Zhang, Nano Res., 2015, 8, 1480–1496 CrossRef
- J. H. Jang, E. Lee, J. Park, G. Kim, S. Hong and Y. U. Kwon, Sci. Rep., 2013, 3, 2872 CrossRef PubMed
- J. Park, L. Zhang, S.-I. Choi, L. T. Roling, N. Lu, J. A. Herron, S. Xie, J. Wang, M. J. Kim, M. Mavrikakis and Y. Xia, ACS Nano, 2015, 9, 2635–2647 CrossRef PubMed
- X. Wang, M. Vara, M. Luo, H. Huang, A. Ruditskiy, J. Park, S. Bao, J. Liu, J. Howe, M. Chi, Z. Xie and Y. Xia, J. Am. Chem. Soc., 2015, 137, 15036–15042 CrossRef PubMed
- A. L. Strickler, A. Jackson and T. F. Jaramillo, ACS Energy Lett., 2016, 2, 244–249 CrossRef
- S. K. Kamarudin, F. Achmad and W. R. W. Daud, Int. J. Hydrogen Energy, 2009, 34, 6902–6916 CrossRef
- A. Heinzel and V. M. Barragan, J. Power Sources, 1999, 84, 70–74 CrossRef
- S. Gottesfeld and J. Pafford, J. Electrochem. Soc., 1988, 135, 2651–2652 CrossRef
- T. Tamaki, A. Yamauchi, T. Ito, H. Ohashi and T. Yamaguchi, Fuel Cells, 2011, 11, 394–403 CrossRef
- P. Xinsheng, K. Kallum and C. Aicheng, Nanotechnology, 2007, 18, 305605 CrossRef
- C. Dai, Y. Yang, Z. Zhao, A. Fisher, Z. Liu and D. Cheng, Nanoscale, 2017, 9, 8945–8951 RSC
- S. J. Yoo, T.-Y. Jeon, K. S. Kim, T.-H. Lim and Y.-E. Sung, Phys. Chem. Chem. Phys., 2010, 12, 15240 RSC
- S. Şen, F. Şen and G. Gökağaç, Phys. Chem. Chem. Phys., 2011, 13, 6784 RSC
- S. Stolbov, M. A. Ortigoza, R. Adzic and T. S. Rahman, J. Chem. Phys., 2009, 130, 124714 CrossRef PubMed
- L. Guo, L. B. Huang, W. J. Jiang, Z. D. Wei, L. J. Wan and J. S. Hu, J. Mater. Chem. A, 2017, 5, 9014–9021 RSC
- X. Q. Huang, Y. Chen, E. B. Zhu, Y. X. Xu, X. F. Duan and Y. Huang, J. Mater. Chem. A, 2013, 1, 14449–14454 RSC
- H. H. Li, Q. Q. Fu, L. Xu, S. Y. Ma, Y. R. Zheng, X. J. Liu and S. H. Yu, Energy Environ. Sci., 2017, 10, 1751–1756 RSC
- B. Y. Xia, H. B. Wu, X. Wang and X. W. Lou, J. Am. Chem. Soc., 2012, 134, 13934–13937 CrossRef PubMed
- K. Jiang, H.-X. Zhang, S. Zou and W.-B. Cai, Phys. Chem. Chem. Phys., 2014, 16, 20360–20376 RSC
- V. Mazumder and S. Sun, J. Am. Chem. Soc., 2009, 131, 4588–4589 CrossRef PubMed
- X. W. Yu and P. G. Pickup, J. Appl. Electrochem., 2011, 41, 589–597 CrossRef
- S. Ha, R. Larsen and R. I. Masel, J. Power Sources, 2005, 144, 28–34 CrossRef
- M. Jin, H. Zhang, Z. Xie and Y. Xia, Energy Environ. Sci., 2012, 5, 6352–6357 RSC
- V. Mazumder, M. Chi, M. N. Mankin, Y. Liu, O. Metin, D. Sun, K. L. More and S. Sun, Nano Lett., 2012, 12, 1102–1106 CrossRef PubMed
- Z. Yin, L. Lin and D. Ma, Catal. Sci. Technol., 2014, 4, 4116–4128 RSC
- Z. Wang, H. Wang, Z. Zhang, G. Yang, T. He, Y. Yin and M. Jin, ACS Nano, 2016, 11, 163–170 CrossRef PubMed
- D. Liu, M. L. Xie, C. M. Wang, L. W. Liao, L. Qiu, J. Ma, H. Huang, R. Long, J. Jiang and Y. J. Xiong, Nano Res., 2016, 9, 1590–1599 CrossRef
- R. Li, H. Hao, W.-B. Cai, T. Huang and A. Yu, Electrochem. Commun., 2010, 12, 901–904 CrossRef
- M. J. Ren, J. Chen, Y. Li, H. F. Zhang, Z. Q. Zou, X. M. Li and H. Yang, J. Power Sources, 2014, 246, 32–38 CrossRef
- D. Chen, P. Cui, H. Liu and J. Yang, Electrochim. Acta, 2015, 153, 461–467 CrossRef
- D. Chen, P. Cui, H. He, H. Liu and J. Yang, J. Power Sources, 2014, 272, 152–159 CrossRef
- H. Xiaoqing, Z. Huihui, G. Changyou, Z. Zhiyou and Z. Nanfeng, Angew. Chem., Int. Ed., 2009, 48, 4808–4812 CrossRef PubMed
- C. Spöri, J. T. H. Kwan, A. Bonakdarpour, D. P. Wilkinson and P. Strasser, Angew. Chem., Int. Ed., 2017, 56, 5994–6021 CrossRef PubMed
- L. Giordano, B. Han, M. Risch, W. T. Hong, R. R. Rao, K. A. Stoerzinger and Y. Shao-Horn, Catal. Today, 2016, 262, 2–10 CrossRef
- S. M. Alia, S. Shulda, C. Ngo, S. Pylypenko and B. S. Pivovar, ACS Catal., 2018, 8, 2111–2120 CrossRef
- Y.-H. Fang and Z.-P. Liu, J. Am. Chem. Soc., 2010, 132, 18214–18222 CrossRef PubMed
- H.-S. Oh, H. N. Nong, T. Reier, A. Bergmann, M. Gliech, J. Ferreira de Araújo, E. Willinger, R. Schlögl, D. Teschner and P. Strasser, J. Am. Chem. Soc., 2016, 138, 12552–12563 CrossRef PubMed
- S. Cherevko, S. Geiger, O. Kasian, N. Kulyk, J.-P. Grote, A. Savan, B. R. Shrestha, S. Merzlikin, B. Breitbach, A. Ludwig and K. J. J. Mayrhofer, Catal. Today, 2016, 262, 170–180 CrossRef
- X. Xu, F. Song and X. Hu, Nat. Commun., 2016, 7, 12324 CrossRef PubMed
- J. Xie, X. Zhang, H. Zhang, J. Zhang, S. Li, R. Wang, B. Pan and Y. Xie, Adv. Mater., 2017, 29, 1604765 CrossRef PubMed
- E. Antolini, ACS Catal., 2014, 4, 1426–1440 CrossRef
- D. Friebel, M. W. Louie, M. Bajdich, K. E. Sanwald, Y. Cai, A. M. Wise, M.-J. Cheng, D. Sokaras, T.-C. Weng, R. Alonso-Mori, R. C. Davis, J. R. Bargar, J. K. Nørskov, A. Nilsson and A. T. Bell, J. Am. Chem. Soc., 2015, 137, 1305–1313 CrossRef PubMed
- F. Song and X. Hu, Nat. Commun., 2014, 5, 4477 CrossRef PubMed
- B. Zhang, X. Zheng, O. Voznyy, R. Comin, M. Bajdich, M. Garcia-Melchor, L. Han, J. Xu, M. Liu, L. Zheng, F. P. Garcia de Arquer, C. T. Dinh, F. Fan, M. Yuan, E. Yassitepe, N. Chen, T. Regier, P. Liu, Y. Li, P. De Luna, A. Janmohamed, H. L. Xin, H. Yang, A. Vojvodic and E. H. Sargent, Science, 2016, 352, 333–337 CrossRef PubMed
- K. A. Stoerzinger, L. Qiao, M. D. Biegalski and Y. Shao-Horn, J. Phys. Chem. Lett., 2014, 5, 1636–1641 CrossRef PubMed
- L. C. Seitz, C. F. Dickens, K. Nishio, Y. Hikita, J. Montoya, A. Doyle, C. Kirk, A. Vojvodic, H. Y. Hwang, J. K. Norskov and T. F. Jaramillo, Science, 2016, 353, 1011–1014 CrossRef PubMed
- Y. Ping, R. J. Nielsen and W. A. Goddard, J. Am. Chem. Soc., 2017, 139, 149–155 CrossRef PubMed
- M. Escudero-Escribano, A. F. Pedersen, E. A. Paoli, R. Frydendal, D. Friebel, P. Malacrida, J. Rossmeisl, I. E. L. Stephens and I. Chorkendorff, J. Phys. Chem. B, 2018, 122, 947–955 CrossRef PubMed
- Y. Lee, J. Suntivich, K. J. May, E. E. Perry and Y. Shao-Horn, J. Phys. Chem. Lett., 2012, 3, 399–404 CrossRef PubMed
- C. Wang, Y. Sui, M. Xu, C. Liu, G. Xiao and B. Zou, ACS Sustainable Chem. Eng., 2017, 5, 9787–9792 CrossRef
- H. N. Nong, H.-S. Oh, T. Reier, E. Willinger, M.-G. Willinger, V. Petkov, D. Teschner and P. Strasser, Angew. Chem., Int. Ed., 2015, 54, 2975–2979 CrossRef PubMed
- H. Jin, Y. Hong, J. Yoon, A. Oh, N. K. Chaudhari, H. Baik, S. H. Joo and K. Lee, Nano Energy, 2017, 42, 17–25 CrossRef
- C. Wang, Y. M. Sui, G. J. Xiao, X. Y. Yang, Y. J. Wei, G. T. Zou and B. Zou, J. Mater. Chem. A, 2015, 3, 19669–19673 RSC
- J. Feng, F. Lv, W. Zhang, P. Li, K. Wang, C. Yang, B. Wang, Y. Yang, J. Zhou, F. Lin, G.-C. Wang and S. Guo, Adv. Mater., 2017, 29, 1703798 CrossRef PubMed
- T. Kwon, H. Hwang, Y. J. Sa, J. Park, H. Baik, S. H. Joo and K. Lee, Adv. Funct. Mater., 2017, 27, 1604688 CrossRef
- J. Park, Y. J. Sa, H. Baik, T. Kwon, S. H. Joo and K. Lee, ACS Nano, 2017, 11, 5500–5509 CrossRef PubMed
- J. Lim, S. Yang, C. Kim, C.-W. Roh, Y. Kwon, Y.-T. Kim and H. Lee, Chem. Commun., 2016, 52, 5641–5644 RSC
- W. Sun, Y. Song, X.-Q. Gong, L.-m. Cao and J. Yang, Chem. Sci., 2015, 6, 4993–4999 RSC
- H.-S. Oh, H. N. Nong, T. Reier, M. Gliech and P. Strasser, Chem. Sci., 2015, 6, 3321–3328 RSC
- H. N. Nong, L. Gan, E. Willinger, D. Teschner and P. Strasser, Chem. Sci., 2014, 5, 2955–2963 RSC
- P. Lettenmeier, L. Wang, U. Golla-Schindler, P. Gazdzicki, N. A. Cañas, M. Handl, R. Hiesgen, S. S. Hosseiny, A. S. Gago and K. A. Friedrich, Angew. Chem., Int. Ed., 2016, 55, 742–746 CrossRef PubMed
- A. B. Laursen, K. R. Patraju, M. J. Whitaker, M. Retuerto, T. Sarkar, N. Yao, K. V. Ramanujachary, M. Greenblatt and G. C. Dismukes, Energy Environ. Sci., 2015, 8, 1027–1034 RSC
- J. Kibsgaard, T. F. Jaramillo and F. Besenbacher, Nat. Chem., 2014, 6, 248 CrossRef PubMed
- J. Kibsgaard, Z. Chen, B. N. Reinecke and T. F. Jaramillo, Nat. Mater., 2012, 11, 963 CrossRef PubMed
- M. A. Lukowski, A. S. Daniel, F. Meng, A. Forticaux, L. Li and S. Jin, J. Am. Chem. Soc., 2013, 135, 10274–10277 CrossRef PubMed
- L. Lin, N. Miao, Y. Wen, S. Zhang, P. Ghosez, Z. Sun and D. A. Allwood, ACS Nano, 2016, 10, 8929–8937 CrossRef PubMed
- B. Seo, G. Y. Jung, Y. J. Sa, H. Y. Jeong, J. Y. Cheon, J. H. Lee, H. Y. Kim, J. C. Kim, H. S. Shin, S. K. Kwak and S. H. Joo, ACS Nano, 2015, 9, 3728–3739 CrossRef PubMed
- D. Yoon, B. Seo, J. Lee, K. S. Nam, B. Kim, S. Park, H. Baik, S. H. Joo and K. Lee, Energy Environ. Sci., 2016, 9, 850–856 RSC
- M. Liu and J. Li, ACS Appl. Mater. Interfaces, 2016, 8, 2158–2165 CrossRef PubMed
- E. J. Popczun, C. G. Read, C. W. Roske, N. S. Lewis and R. E. Schaak, Angew. Chem., Int. Ed., 2014, 53, 5427–5430 CrossRef PubMed
- J. F. Callejas, J. M. McEnaney, C. G. Read, J. C. Crompton, A. J. Biacchi, E. J. Popczun, T. R. Gordon, N. S. Lewis and R. E. Schaak, ACS Nano, 2014, 8, 11101–11107 CrossRef PubMed
- J. Xu, J. Cui, C. Guo, Z. Zhao, R. Jiang, S. Xu, Z. Zhuang, Y. Huang, L. Wang and Y. Li, Angew. Chem., Int. Ed., 2016, 55, 6502–6505 CrossRef PubMed
- C. Zhang, Y. Liu, Y. Chang, Y. Lu, S. Zhao, D. Xu, Z. Dai, M. Han and J. Bao, ACS Appl. Mater. Interfaces, 2017, 9, 17326–17336 CrossRef PubMed
- D. Yoon, J. Lee, B. Seo, B. Kim, H. Baik, S. H. Joo and K. Lee, Small, 2017, 13, 1700052 CrossRef PubMed
- Z. Cao, H. Li, C. Zhan, J. Zhang, W. Wang, B. Xu, F. Lu, Y. Jiang, Z. Xie and L. Zheng, Nanoscale, 2018, 10, 5072–5077 RSC
- M. Escudero-Escribano, P. Malacrida, M. H. Hansen, U. G. Vej-Hansen, A. Velazquez-Palenzuela, V. Tripkovic, J. Schiotz, J. Rossmeisl, I. E. Stephens and I. Chorkendorff, Science, 2016, 352, 73–76 CrossRef PubMed
- K. J. J. Mayrhofer, J. C. Meier, S. J. Ashton, G. K. H. Wiberg, F. Kraus, M. Hanzlik and M. Arenz, Electrochem. Commun., 2008, 10, 1144–1147 CrossRef
- T. Reier, M. Oezaslan and P. Strasser, ACS Catal., 2012, 2, 1765–1772 CrossRef
- P. Jovanovic, N. Hodnik, F. Ruiz-Zepeda, I. Arcon, B. Jozinovic, M. Zorko, M. Bele, M. Sala, V. S. Selih, S. Hocevar and M. Gaberscek, J. Am. Chem. Soc., 2017, 139, 12837–12846 CrossRef PubMed
- X. Huang, Z. Zhao, L. Cao, Y. Chen, E. Zhu, Z. Lin, M. Li, A. Yan, A. Zettl, Y. M. Wang, X. Duan, T. Mueller and Y. Huang, Science, 2015, 348, 1230–1234 CrossRef PubMed
- D. Mocatta, G. Cohen, J. Schattner, O. Millo, E. Rabani and U. Banin, Science, 2011, 332, 77–81 CrossRef PubMed
- L. Cao and T. Mueller, Nano Lett., 2016, 16, 7748–7754 CrossRef PubMed
- F. Saleem, B. Ni, Y. Yong, L. Gu and X. Wang, Small, 2016, 12, 5261–5268 CrossRef PubMed
- L. Bu, N. Zhang, S. Guo, X. Zhang, J. Li, J. Yao, T. Wu, G. Lu, J. Y. Ma, D. Su and X. Huang, Science, 2016, 354, 1410–1414 CrossRef PubMed
- S. Xie, S.-I. Choi, N. Lu, L. T. Roling, J. A. Herron, L. Zhang, J. Park, J. Wang, M. J. Kim, Z. Xie, M. Mavrikakis and Y. Xia, Nano Lett., 2014, 14, 3570–3576 CrossRef PubMed
- A. Mahmood, N. Xie, M. A. Ud Din, F. Saleem, H. Lin and X. Wang, Chem. Sci., 2017, 8, 4292–4298 RSC
- C. W. Han, T. Choksi, C. Milligan, P. Majumdar, M. Manto, Y. Cui, X. Sang, R. R. Unocic, D. Zemlyanov, C. Wang, F. H. Ribeiro, J. Greeley and V. Ortalan, Nano Lett., 2017, 17, 4576–4582 CrossRef PubMed
- T. Avanesian, S. Dai, M. J. Kale, G. W. Graham, X. Pan and P. Christopher, J. Am. Chem. Soc., 2017, 139, 4551–4558 CrossRef PubMed
- A. P. LaGrow, D. C. Lloyd, P. L. Gai and E. D. Boyes, Chem. Mater., 2018, 30, 197–203 CrossRef
- M. Vara, L. T. Roling, X. Wang, A. O. Elnabawy, Z. D. Hood, M. Chi, M. Mavrikakis and Y. Xia, ACS Nano, 2017, 11, 4571–4581 CrossRef PubMed
- L. Dubau, M. Lopez-Haro, J. Durst and F. Maillard, Catal. Today, 2016, 262, 146–154 CrossRef
- L. Dubau, J. Nelayah, T. Asset, R. Chattot and F. Maillard, ACS Catal., 2017, 7, 3072–3081 CrossRef
- J. Snyder and J. Erlebacher, ECS Trans., 2011, 41, 1021–1030 Search PubMed
- J. Snyder, T. Fujita, M. W. Chen and J. Erlebacher, Nat. Mater., 2010, 9, 904 CrossRef PubMed
Footnote |
| † These authors contributed equally. |
| This journal is © The Royal Society of Chemistry 2018 |