
Catalysis and photocatalysis by metal organic frameworks

Abstract
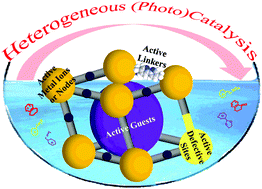
Metal organic frameworks (MOFs) are a class of porous crystalline materials that feature a series of unique properties, such as large surface area and porosity, high content of transition metals, and possibility to be designed and modified after synthesis, that make these solids especially suitable as heterogeneous catalysts. The active sites can be coordinatively unsaturated metal ions, substituents at the organic linkers or guest species located inside the pores. The defects on the structure also create these open sites. The present review summarizes the current state of the art in the use of MOFs as solid catalysts according to the type of site, making special emphasis on the more recent strategies to increase the population of these active sites and tuning their activity, either by adapting the synthesis conditions or by post-synthetic modification. This review highlights those reports illustrating the synergy derived from the presence of more than one of these types of sites, leading to activation of a substrate by more than one site or to the simultaneous activation of different substrates by complementary sites. This synergy is frequently the main reason for the higher catalytic activity of MOFs compared to homogeneous catalysts or other alternative solid materials. Besides dark reactions, this review also summarizes the use of MOFs as photocatalysts emphasizing the uniqueness of these materials regarding adaptation of the linkers as light absorbers and metal exchange at the nodes to enhance photoinduced electron transfer, in comparison with conventional inorganic photocatalysts. This versatility and flexibility that is offered by MOFs to optimize their visible light photocatalytic activity explains the current interest in exploiting these materials for novel photocatalytic reactions, including hydrogen evolution and photocatalytic CO2 reduction.
- This article is part of the themed collection: New catalytic materials for energy and chemistry in transition
 Please wait while we load your content...
Please wait while we load your content...

