
Polylactic acid macromonomer radical propagation kinetics and degradation behaviour†
Thomas R.
Rooney
a,
Davide
Moscatelli
 b and
Robin A.
Hutchinson
*a
b and
Robin A.
Hutchinson
*a
aDepartment of Chemical Engineering, Dupuis Hall, Queen's University, Kingston, ON K7L 3N6, Canada. E-mail: robin.hutchinson@queensu.ca
bDepartment of Chemistry, Materials and Chemical Engineering “Giulio Natta”, Politecnico di Milano, Via Luigi Mancinelli 7, 20131 Milano, Italy. E-mail: davide.moscatelli@polimi.it
First published on 25th April 2017
Abstract
Polylactic acid ethyl ester methacrylate (PLANEMA) macromonomers are synthesized with N = 1, 5, 7, and 9 average number of polyester units. While propagation rate coefficients (kp) determined by pulsed laser polymerization experiments for bulk PLA1EMA and PLA5EMA are not significantly different over the 40–100 °C temperature range, they are elevated by 60% compared to methyl methacrylate, indicating that the nature of substituents several units beyond the methacrylic group does not decisively impact bulk kp measurements. Compared to bulk PLA5EMA, the apparent kp in 75 wt% n-butanol solution is enhanced due to hydrogen bonding, whereas in 75 wt% dimethylformamide solution it is reduced by 35% because of differences in macromonomer and solvent molar volumes. The PLA5EMA macromonomers are used to produce nanoparticles (NP) by emulsion radical polymerization that degrade almost four times more slowly than NPs produced from their hydroxyl terminated macromonomer counterpart.
Introduction
Polylactic acid (PLA) is a highly versatile material produced from 100% renewable resources with an auspicious outlook for a variety of commodity applications.1 Indeed, packaging, disposable bottles, and biomedical applications benefit from one of PLA's most notable features: hydrolysis of the polyester backbone to yield non-toxic degradation products.2,3 Production of PLA can be accomplished via the polycondensation of lactic acid; however, difficulties associated with efficient removal of the liberated water limit the molecular weight (MW) that can be achieved by this route.4 Thus, most approaches focus on the ring-opening polymerization (ROP) of the cyclic lactide (LA) dimer which allows for better control over PLA's MW characteristics.5 In addition, functional ROP initiators can be implemented to impart customized features onto the final polyester material.6–8When 2-hydroxyethyl methacrylate (HEMA) is used as ROP initiator, the resulting HEMA-PLA2n macromonomers, with average chain length 2n defined by the stoichiometric ratio of LA to HEMA, can be further polymerized via “grafting through” radical polymerization (RP) of the vinyl end-group to produce comb-polymers with well-defined polyester grafts affixed to a higher MW acrylic backbone.9–11 Such HEMA-PLA2n macromonomers have been employed in radical miniemulsion polymerization,12,13 solution polymerization to make hydrogels,14 and emulsion polymerization to produce degradable nanoparticles (NP) for drug delivery applications.15–17 In the case of NPs, degradation time is controllable by the type and average number (typically 1–5 units) of grafted polyesters, a feature that causes distinct changes in material hydrophobicity which can be exploited for various applications.15,18 As bulk erosion can be assumed for PLA hydrolysis of nanoscale materials,19 degradation of the PLA grafts in NPs produced from macromonomers can be compared to the solution hydrolysis of PLA oligomers which is not only influenced by factors such as temperature and pH,20,21 but also chain length due to the difference in reactivity of backbone and terminal esters.22 Terminal ester hydrolysis, via preferential backbiting or chain-end scission mechanisms, is facilitated by the terminal hydroxyl group23 and was found to be chain-length independent.24 For backbiting (or preferential chain-end scission) to be effective, the terminal hydroxyl groups must be accessible to the aqueous environment;20 in general, the balance of water diffusion and PLA degradation behaviour depends on material dimensions and topology (e.g., surface brushes, bulk, solution) which are governed by the intended application and method of production. Thus, to efficiently produce and to better predict degradation performance of polyester macromonomer based comb-(co)polymer materials, an understanding of the underlying macromonomer RP kinetics is required to track both hydroxyl end-group and macromonomer chain length incorporation behaviours.
To date, few works have addressed the RP kinetics of methacrylate type polyester macromonomers. Although studied under limited conditions, typical methacrylate/methacrylate relative reactivity – i.e., equal addition probabilities – was demonstrated for HEMA-PLA2n and polycaprolactone (PCL) based HEMA-PCLn macromonomer copolymerizations with methyl methacrylate (MMA),25–27 while another study detailing the copolymerization of styrene (ST) with polyester methacrylate type macromonomers showed that the relative reactivity depends only on the chemical identity up to several units away from the methacryloyl end-group.28 In terms of propagation rate behaviour, the pulsed laser polymerization coupled with size exclusion chromatography (PLP-SEC) technique is the most accurate and reliable method for determining propagation rate coefficients (kp), as is described in comprehensive detail by Beuermann and Buback.29 The kp is calculated according to eqn (1) where MWi is the MW of the ith inflection point of a low-conversion PLP-generated molar mass distribution (MMD), ϕmon is the volume fraction of monomer in solution, ρmon is the monomer density, and t0 is the time between pulses.
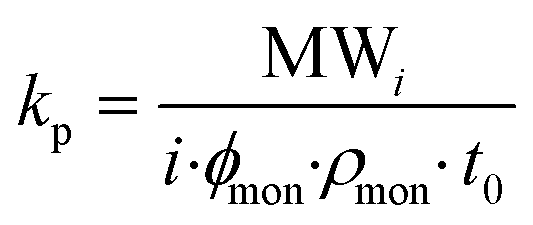 | (1) |
The IUPAC subcommittee on “Modeling of Polymerization Kinetics and Processes” has established family type behaviour for MMA, ethyl methacrylate (EMA), n-butyl methacrylate (BMA), and dodecyl methacrylate (DMA),30,31 where an increase in the length of the linear alkyl ester group correlates to an increase in the value of kp measured for bulk monomer using the PLP-SEC technique; this trend is reported more recently to extend even up to behenyl methacrylate.32 A similar increase in bulk kp with increasing ester side chain length was reported for polyethylene glycol ethyl ether methacrylate (PEGEEMA, 3 PEG units) compared to EEMA (1 PEG unit),33 although another study found no additional increase in bulk kp for polyethylene glycol methyl ether methacrylate (PEGMA, 7–8 PEG units) at similar temperatures.34 For the polyester macromonomer systems of interest in this study, the estimation of kp could not be completed for hydroxyl-terminated HEMA-PCL3 because of the poor solubility of the resulting comb-polymer in tetrahydrofuran (THF), the SEC eluent; nonetheless, bulk copolymerizations with up to 50 wt% MMA indicated no significant differences in copolymer propagation rate coefficient (kp,cop) for HEMA-PCLn with average chain length n = 2 and n = 3.27
Since macromonomers are inherently viscous (some even solid at room temperature) their solution propagation behaviour is also of high practical and technical importance. The influences of solvent on propagation kinetics are extensive and can arise from both specific and non-specific interactions between monomer and solvent.35 For example, at a monomer concentration of 0.8 mol L−1 in n-butanol (BuOH), the kp of BMA is enhanced by as much as 85% because of the well-documented hydrogen bond formation between hydroxyl and methacryloyl carbonyl which reduces the electron density at the double bond, making BMA more reactive towards radical addition.36 On the other hand, differences between the molar volumes of monomer and solvent (Vmon − Vsol) can manifest as a competition for positions at the radical chain-end leading to a lower than or greater than analytical local monomer concentration and corresponding increase or decrease in apparent kp measured by PLP-SEC, respectively.35 Beuermann and Garcia substantiated this concept by establishing a linear relationship between Vmon − Vsol and the ratio of kp at infinite dilution to bulk kp (i.e., kp,∞/kp,bulk) for a variety of monomer/solvent pairs contained within −100 cm3 mol−1 < Vmon − Vsol < 150 cm3 mol−1.37 In terms of macromonomers, this relationship was extended to PEGEEMA solution homopolymerizations in toluene (Vmon − Vsol = 139 cm3 mol−1) and THF (Vmon − Vsol = 164 cm3 mol−1), for which the apparent kp at 25 °C in 80 vol% solvent was reduced by 32% and 53% compared to bulk, respectively.33
In this work, polylactic acid ethyl ester methacrylate (PLANEMA, where N average PLA units corresponds to 2n + 1 cyclic LA monomers in the ROP step because the ethyl 2-hydroxypropionate initiator fragment contributes 1 PLA unit to each macromonomer chain), was synthesized according to Scheme 1. As the alkyl end-group of PLANEMA ensures the resulting comb-polymer is THF soluble, the determination of macromonomer kp (an important parameter required to efficiently produce comb-polymers with tailored properties) is not hindered by solubility limitations, as is the case for hydroxyl-terminated HEMA-PCLn or HEMA-PLA2n macromonomers. Another benefit of the alkyl end-group is that PLANEMA can be copolymerized with HEMA-PLA2n to yield PLA-grafted comb-polymers with tunable hydroxyl group densities (an important design parameter for post-polymerization modifications such as esterification reactions); however, the influence of the ethyl ester end-group on the hydrolytic degradability of PLA must be assessed. Therefore, this work investigates both the production and hydrolysis performance of PLANEMA-based comb-polymers. Firstly, PLANEMA homopropagation kinetics are studied in BuOH, dimethylformamide (DMF) and xylenes solutions in addition to bulk, to examine the effects of hydrogen bonding and molar volumes on polyester macromonomer kp. Secondly, PLANEMA macromonomers are used to produce NPs by emulsion radical polymerization in order to evaluate the influence of PLANEMA's ethyl ester end-group on the rate of NP hydrolytic degradation in comparison to NPs produced from hydroxyl-terminated HEMA-PLA2n macromonomers.
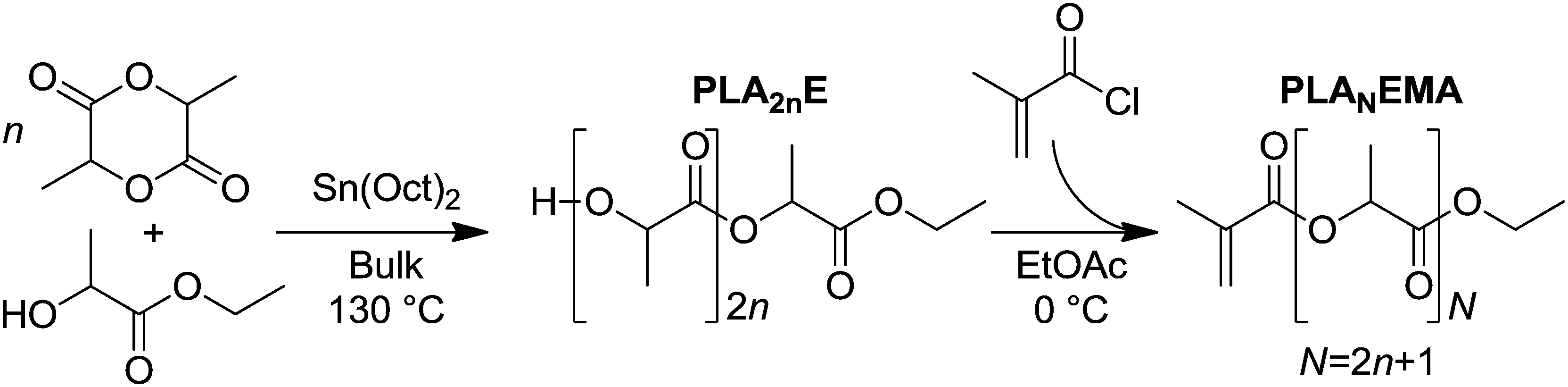 | ||
| Scheme 1 Synthetic route for production of PLANEMA. | ||
Experimental
Materials
Methyl methacrylate (MMA, 99%), (3S)-cis-3,6-dimethyl-1,4-dioxane-2,5-dione (LA, 98%), ethyl 2-hydroxypropionate (ETL, ≥98%), 2-hydroxyethyl methacrylate (HEMA, 97%), tin(II) 2-ethylhexanoate (Sn(oct)2, 92.5–100.0%), triethylamine (TEA, ≥99.5%), basic alumina (Brockmann 1), 2,2-dimethoxy-2-phenylacetophenone (DMPA, 99%), ammonium persulfate (APS, >98%), n-butanol (BuOH, 99%), and dimethylformamide (DMF, 99.8%) were purchased from Sigma Aldrich and used as received. Tetrahydrofuran (THF, >99%, ACP Chemicals), hexanes (reagent grade, ACP Chemicals), methanol (reagent grade, ACP Chemicals), ethyl acetate (EtOAc, reagent grade, ACP Chemicals), hydroquinone (reagent grade, Fisher Scientific), xylenes (99.9%, Fisher Chemical), dichloromethane (DCM, 99.9%, Fisher Chemical), sodium dodecyl sulfate (SDS, ∼99%, MP Biomedicals), chloroform-d (CDCl3, 99.8% D, Sigma Aldrich), and dimethylsulfoxide-d6 (DMSO-d6, 99.9% D, Cambridge Isotope Laboratories) were used as received. Methacryloyl chloride (MACl, 97%, Sigma Aldrich) was distilled immediately before use. All water used in this work was in-house distilled water that was further purified (18.2 MΩ cm) using a Millipore Synergy water purification system equipped with SynergyPak purification cartridges.Macromonomer syntheses
All (macro)monomer and poly(macromonomer) 1H NMR characterizations were performed on a Bruker Avance instrument operating at 400 MHz using the peak assignments and calculations presented in our previous copolymerization study.28![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :400 was prepared separately (1:200 for the N = 9 synthesis), then 2.17 g of this mixture (corresponding to 18.3 mmol ETL) was added to the LA by syringe and allowed to react for 4 hours at 130 °C to afford polylactic acid ethyl ester (PLA2nE) with number average 2n = 5.5 and LA conversion ≈94% (as determined by 1H NMR).
:400 was prepared separately (1:200 for the N = 9 synthesis), then 2.17 g of this mixture (corresponding to 18.3 mmol ETL) was added to the LA by syringe and allowed to react for 4 hours at 130 °C to afford polylactic acid ethyl ester (PLA2nE) with number average 2n = 5.5 and LA conversion ≈94% (as determined by 1H NMR).
Next, in a sealed 3 neck 100 mL round bottom flask, PLA2nE (7.44 g, 18.3 mmol –OH) was dissolved in 38 mL EtOAc to which 7 mL TEA (50.6 mmol) was then added. The solution was cooled to 0 °C using an ice bath, bubbled with nitrogen for 10 min, and then 2.3 mL freshly distilled MACl (23.6 mmol) was fed over 20 minutes using a glass syringe. The reaction mixture was maintained at 0 °C for 3 hours, filtered to remove the TEA salt, and then passed through a column of basic alumina. Approximately 1 mg of hydroquinone was added to the product solution before the solvent was evaporated in vacuo to afford 6.21 g polylactic acid ethyl ester methacrylate (PLA5EMA) with number average N = 5.3 in 77% yield.
:400 was prepared, then 2.05 g of this mixture (corresponding to 15.8 mmol HEMA) was added to the LA by syringe, and then allowed to react at 130 °C for 2 hours. The LA conversion was ≈96% for HEMA-LA5 with 2n = 5.3 (as determined by 1H NMR).
It should be noted that although HEMA-PLA5 and PLA5EMA have similar average MW, the former are predominantly even numbered oligomers (i.e., 2n = 2, 4, 6) while the latter are predominantly odd numbered oligomers (i.e., N = 3, 5, 7) because the ETL initiator fragment contributes a single PLA unit to each polyester chain.28
Pulsed laser polymerization
Low-conversion MMA, PLA1EMA, and PLA5EMA homopolymerizations were conducted using a pulsed laser setup consisting of a Coherent Xantos XS-500 laser operating at the XeF line of 351 nm and capable of producing laser energy of 1–6 mJ per pulse at repetition rates up to 500 Hz. Bulk (macro)monomer as well as (macro)monomer diluted with 75 wt% DMF, xylenes, and BuOH mixtures were prepared with 5 mmol L−1 DMPA photoinitiator. Approximately 1 mL of the monomer mixture (0.5 g for PLA1EMA and PLA5EMA bulk homopolymerizations) was added to a Quartz cuvette of 10 mm pathlength (CV10Q3500S, Thorlabs), heated to reaction temperature using an external circulating oil bath, and exposed to laser energy while the temperature was monitored and controlled to within ±0.5 °C. Experiments were conducted using laser repetition rates between 5 and 50 Hz (see Tables S5–S9 of the ESI† for the exact operating conditions employed for each sample).Following the PLP experiments, with pulsing time controlled to keep macromonomer conversion low, the residual solvent, MMA, or PLA1EMA was removed under constant air stream. At −20 °C, poly(MMA) and poly(PLA5EMA) samples were precipitated in methanol while poly(PLA1EMA) samples were precipitated in hexanes. Samples were centrifuged at 6000 rpm for 10 minutes followed by decantation of the supernatant to collect the homopolymer precipitate, which was then dried under air stream and used to determine sample conversion by gravimetry. Parameters relevant to kp determination are summarized by Table 1. The densities of MMA, PLA1EMA, and xylenes were measured at temperatures between 25 and 70 °C using a Paar DMA 48 Density Meter, while the density of highly viscous PLA5EMA was extrapolated from solution density measurements in xylenes, assuming volume additivity, as shown by Table S2 (and Table S3† for PLA7EMA).
| ρ (g mL−1) | [M] at 25 °C (mol L−1) | dn/dc (mL g−1) | Mark–Houwink parameters | ||
|---|---|---|---|---|---|
| K (10−5 dL g−1) | a | ||||
| a Pure species density extrapolated from xylenes solution assuming volume additivity. b Independent regression of literature data. | |||||
| MMA | 0.9671–0.001117 T/°C | 9.39 | 0.089 (ref. 38) | 9.44 | 0.719 (ref. 39) |
| PLA1EMA | 1.0478–0.001048 T/°C | 5.49 | 0.069 | 24.4 | 0.581 |
| PLA5EMA | 1.1892–0.000956 T/°Ca | 2.46 | 0.055 | 7.52 | 0.647 |
| Xylenes | 0.8833–0.000876 T/°C | — | — | — | — |
| DMF (ref. 40) | 0.9686–0.000958 T/°Cb | — | — | — | — |
| BuOH (ref. 41) | 0.8267–0.000809 T/°Cb | — | — | — | — |
The oligomeric distributions of macromonomers (Fig. S1†) as well as molar mass distributions (MMD) of all PLP samples were assessed by size exclusion chromatography (SEC). Since PLA5EMA is a distribution of macromonomers with an average of N = 5.3 PLA units per chain, 1H NMR analysis of several PLP-generated low-conversion poly(PLA5EMA) comb-polymers was employed to confirm preservation of graft density into the comb-polymer (average N = 5.0). The SEC setup consists of a Waters 2960 separation module instrument with a Waters 410 differential refractometer (DRI) and a Wyatt Instruments Dawn EOS 690 nm laser photometer multiangle light scattering (LS) detector. Four Styragel columns (HR 0.5, 1, 3, 4) were maintained at 35 °C with distilled THF as eluent at 0.3 mL min−1. The DRI detector was calibrated using 14 narrow poly(MMA) standards (302–853000 Da) and the LS detector was calibrated by toluene as recommended by the manufacturer. The LS output was interpreted using differential refractive indices (dn/dc) measured for poly(PLA1EMA) and poly(PLA5EMA) homopolymers in THF at 35 °C by a Wyatt Optilab DSP refractometer at 690 nm calibrated with sodium chloride. Six homopolymer samples of 1–20 mg mL−1 were prepared in THF and injected sequentially to construct a curve with slope dn/dc, as summarized by Table 1. In addition, the dn/dc of PLA5EMA macromonomer was measured as 0.040 mL g−1, while the dn/dc for comb-polymers with average graft lengths N = 7 and N = 9 plateaued at a value of 0.051 mL g−1, confirming that dn/dc becomes independent of the acrylic backbone length as well as the length of the polyester grafts.
SEC of low conversion polymers produced by PLP was also performed using a Viscotek 270 max separation module with a RI, viscosity (IV), and LS (low and right angle) triple detector setup. A set of two porous PolyAnalytik columns with an exclusion limit molecular weight of 20 × 106 g mol−1 was used in series at 40 °C with distilled THF as eluent at a flow rate of 1 mL min−1. The Mark-Houwink (MH) parameters for poly(PLA1EMA) and poly(PLA5EMA) homopolymers in Table 1 were estimated as an average of the output from the IV and LS detectors (using dn/dc summarized by Table 1) generated by several independent samples. The IV vs. MW data for poly(PLA1EMA) and poly(PLA5EMA) are included as Fig. S2 and S3† with global fits presented in Fig. S4.†
NP synthesis and degradation study
NP synthesis was carried out following a previously reported procedure with minor modification.17 In a 50 mL 3 neck round bottom flask equipped with condenser, 0.2 g SDS surfactant was dissolved in 18 mL deionized water, heated to 70 °C, and further purged with nitrogen for 10 min. About 16 mg APS was dissolved in 2 mL deionized water and added to the purged solution. One gram of macromonomer was dissolved in one gram of DCM and then the mixture was fed over one hour at constant injection rate using syringe pump (PLA1EMA was fed directly without DCM) under constant flow of nitrogen. The reaction was allowed to proceed for one additional hour (two additional hours for PLA5EMA macromonomer systems). No coagulum was formed during the emulsion polymerization of PLA1EMA; however, a coagulum less than 8 wt% (relative to macromonomer fed) was formed for the macromonomer systems, a result consistent with HEMA-PLA5 NP synthesis reported elsewhere.17 While feeding the macromonomer over a one hour period gives starved conditions for HEMA-PLA5, macromonomer, droplets were observed throughout the feeding period for the PLA5EMA homopolymerization and copolymerization systems, an observation indicative of the increased hydrophobicity of PLANEMA in comparison to HEMA-PLA5 macromonomers. The conversion of all systems was greater than 99% as estimated by concentrating 1 mL of latex under constant air stream overnight and dissolving the solids in DMSO-d6 for 1H NMR characterization.The particle size distributions (PSD) for all four NP suspensions are presented in Fig. S7–S10† where each PLANEMA (macro)monomer system exhibited a secondary peak near 4000 nm which continued to appear even after passing the latex multiple times through a 0.2 μm filter. Although less than 1% residual macromonomer was detected by 1H NMR analysis of the final dried PLA5EMA latex, longer reaction times as well as additional initiator and MMA shots at the end of the reaction were unsuccessful in removing the secondary peak. Despite its incomplete characterization or removal, this secondary peak accounts for less than 0.1 vol% of the PLANEMA NP systems, and therefore no further treatment was implemented. An accelerated degradation study was performed by maintaining NP latexes at 50 °C in an external water bath and periodically removing them for characterization, following procedures used previously.42 The solution pH was measured using a Mettler Toledo SevenExcellence pH meter, while NP size and polydispersity indices (PDI) were determined with a Malvern Zetasizer Nano ZS (size range 0.3 nm–10 μm) at 25 °C with backscattering optics (173°), using a 4 mW He–Ne (633 nm) laser. All samples were measured in DTS0012 disposable cuvettes. The reported sizes represent an intensity average of at least 30 scans.
Results and discussion
Bulk homopropagation kinetics
PLP experiments were performed for bulk homopolymerizations of MMA (equivalent to PLA0EMA), PLA1EMA, and PLA5EMA over a temperature range of 40–100 °C, with specific conditions summarized by Tables S5, S6, and S8.† As a highly viscous liquid, the PLA5EMA macromonomer is characterized by the low-termination limit of the PLP-SEC technique;43,44 therefore, to maximize the success of bulk PLA5EMA PLP experiments, relatively low pulse repetition rates (10–33 Hz) were employed43,44 and the total number of pulses was limited to 100.45 In conjunction with Tables S6 and S8,† the MMDs and corresponding first derivative plots in Fig. 1 demonstrate that bulk PLANEMA (both N = 1 and N = 5) homopolymerizations fulfil the PLP-SEC consistency criteria: at least two inflection points separated by a MW factor of 2 as well as good agreement between kp estimations made from a minimum of two different pulse repetition rates under otherwise identical conditions.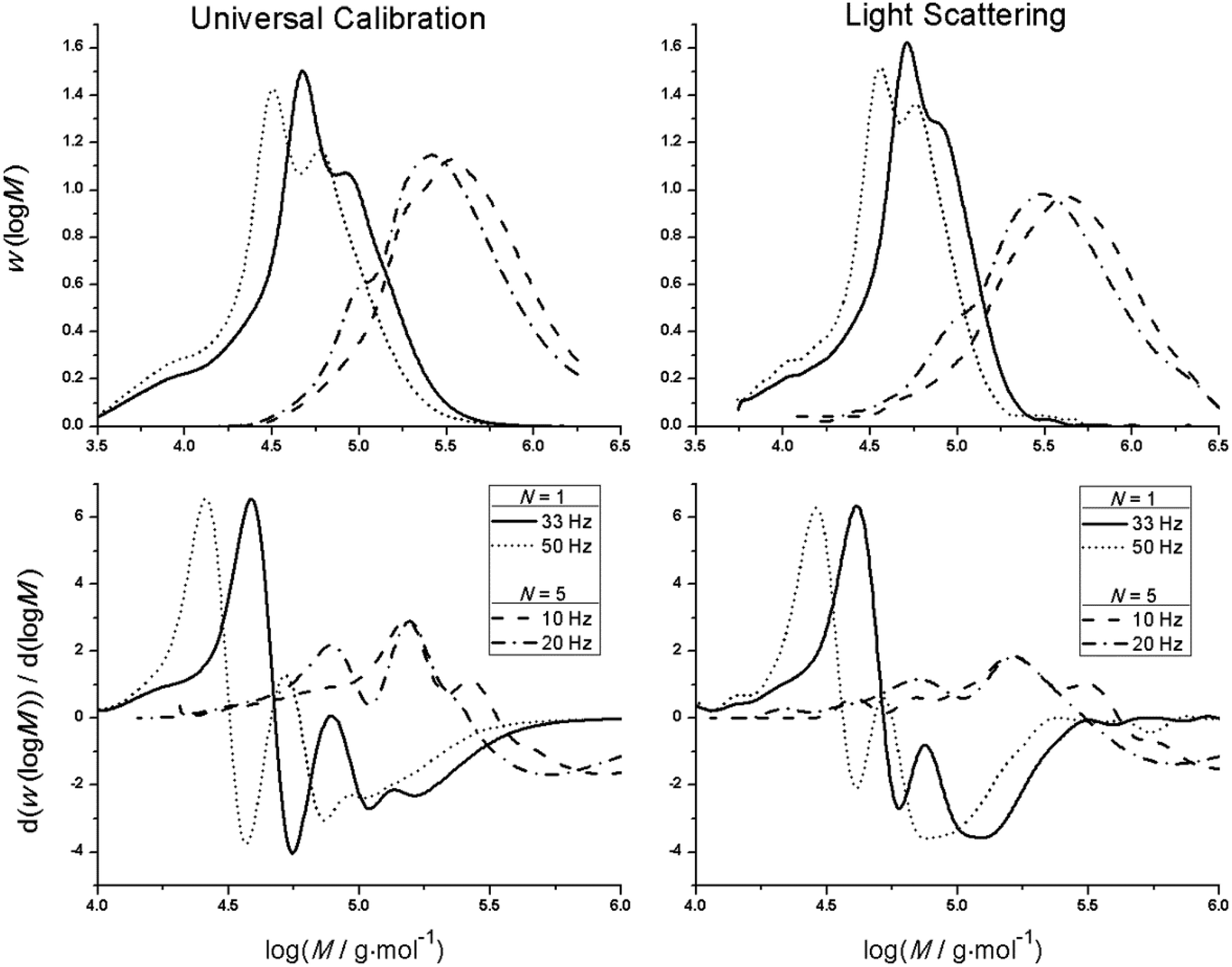 | ||
| Fig. 1 RI (left; interpreted by universal calibration) and LS (right) measures of MMDs (top) and corresponding first derivative plots (bottom) for polymer produced by PLP homopolymerization experiments of bulk PLA1EMA (N = 1) and PLA5EMA (N = 5) at 60 °C with 5 mmol L−1 DMPA, at pulse repetition rates as indicated in the legends. | ||
As shown by the poly(PLA5EMA) MMDs interpreted by universal calibration, the consequence of PLP experiments with lower pulse repetition rates is that higher MW polymer is produced which may exceed the calibration range of the SEC instrument; fortunately, the inflection point positions for all poly(PLA5EMA) samples were well within the polyMMA calibration range such that kp determination was not impeded. Universal calibration is applied throughout this work using MH parameters (Table 1) for poly(PLA1EMA) and poly(PLA5EMA) estimated by the Viscotek SEC (see Fig. S2 and S3,† respectively) over a MW range of approximately 25000–160000 g mol−1 and 126000–800000 g mol−1, respectively. The intrinsic viscosity of the comb-like polymers at identical MW decreases as the graft length is increased from N = 0 (PMMA), to N = 1, and to N = 5 (Fig. S4†), following the trends seen for calibrations established for polyMMA to polyDMA.46 Despite being estimated by calibrations established over a limited MW range, the kp values for poly(PLA1EMA) and poly(PLA5EMA) based on the determined MH parameters are corroborated by the reasonable agreement (within 15%; see Tables S6–S9†) with those estimated from light scattering.
In Fig. 2, the kp measured for bulk PLA1EMA, and PLA5EMA homopolymerizations over the temperature range of 40–100 °C are compared to values measured for bulk MMA, which are within 10% of the IUPAC benchmark values.30 Following family type behaviour,31 the kp for an alkyl ester methacrylate with linear ester side chain length of 5 atoms (e.g., PLA1EMA), is expected to be 20% greater than kp,MMA at 50 °C.32 However, the data in Fig. 2 show that the kp for PLA1EMA is augmented by 60% compared to kp,MMA at 50 °C, with the difference maintained over the complete range of temperatures studied. The structure of PLA1EMA is unique in that it comprises both branched methyl groups as well as polar ester functionalities in its ester side chain. A combination of these features is thought to contribute to the comparatively elevated kp estimate for PLA1EMA at 50 °C of 952 L mol−1 s−1, which is in a similar range as other diverse non-hydroxylated and non-linear heteroatom-containing methacrylates.47–51 In particular, similar values were established for four bulky tertiary amine substituted ethylmethacrylates,48 with the kp of 2-(N,N-dimethylamino)ethyl methacrylate (DMAEMA) slightly higher, presumably related to its less encumbered tertiary amine, and the kp for 3-(N,N-dimethylamino)propyl methacrylate (DMAPMAE) clearly below the proposed family behaviour due to the additional methylene of its propyl spacer. Thus, an interpretation of bulk PLA1EMA kp behaviour is likely rooted in the proximity of its polar and steric substituents to the methacrylic group.
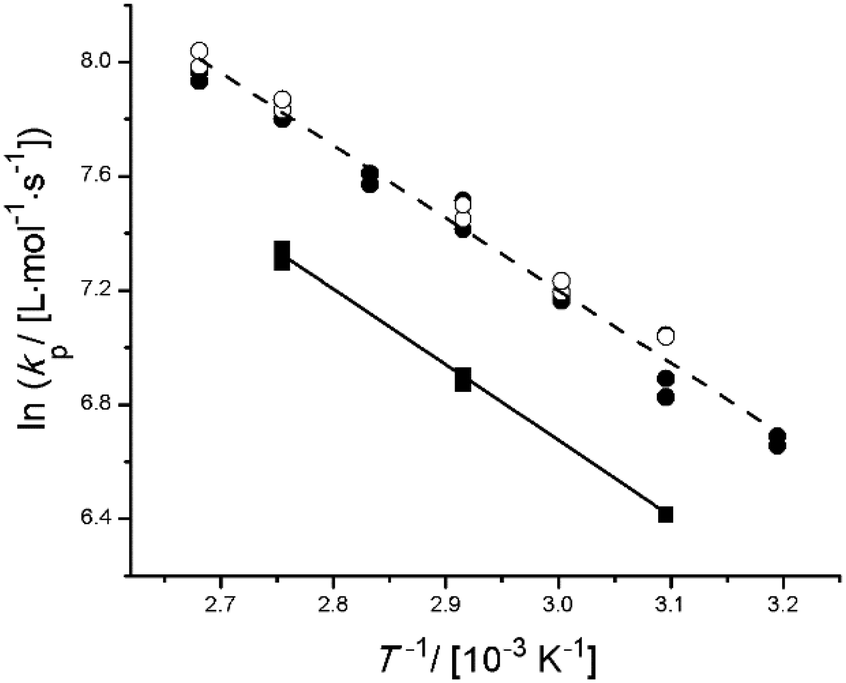 | ||
| Fig. 2 Arrhenius plot for kp determined from bulk PLP experiments of MMA (■), PLA1EMA (●), and PLA5EMA (○) using universal calibration to interpret SEC output. Best fit lines for MMA (solid) and the combined N = 1 and N = 5 data sets (dashed) are provided. | ||
In the case of the PLA5EMA macromonomer, inspection of Fig. 2 reveals that the addition of 4 more PLA units into the ester side chain does not significantly alter its bulk kp from that of PLA1EMA, certainly not to the extent that the bulk kp was increased from MMA (N = 0) to PLA1EMA. Since the bulk kp Arrhenius estimates for PLA1EMA and PLA5EMA in Table 2 encompass values typical of the alkyl ester methacrylate family,30–31 and the N = 1 and N = 5 bulk kp data are reasonably well represented by fitting a pre-exponential using the bulk MMA IUPAC benchmarked activation energy (EA) for kp of 22.36 kJ mol−1 (Fig. S5†), a combined Arrhenius fit is justified. Thus, the chemical features of the isobutyrate bridge adjacent to the methacrylic group must be solely responsible for the elevated PLA1EMA kp measurements in bulk. (As stated previously, 1H NMR analysis of the low-conversion PLP-generated polymers showed that the average N of the PLA5EMA macromonomer is preserved during the “grafting through” polymerization.) In other words, the presence of polar or steric groups further along the ester side chain does not decisively impact the bulk kp of PLANEMA systems. In support of this claim, we found that polyester type, length, and end-group functionality did not contribute to the relative reactivity of various methacrylate macromonomers and ST copolymerization systems,28 and other work showed that the influence of the N,N-dimethylamino substituent on bulk kp is diluted from DMAEMA to DMAPMAE.48 Furthermore, while Siegmann et al. measured a 50% increase in bulk kp for PEGEEMA (3 PEG units) compared to EEMA (1 PEG unit) at 25 °C,33 Smolne et al. found no further increase in the bulk kp of PEGMA (7–8 PEG units) compared to that reported for PEGEEMA.34 Interestingly, the values at which the PEGylated methacrylates appear to plateau (707 and 1954 L mol−1 s−1 at 40 and 80 °C, respectively) are close to the values measured for PLA1EMA (780 and 1978 L mol−1 s−1 at 40 and 80 °C, respectively). Finally, we recall that the pre-exponential for kp is largely governed by the degree to which the internal rotations of the transition state (TS) for propagation are hindered,52 and that Buback has explained the trend of increasing bulk kp from MMA to DMA in terms of the longer aliphatic ester side chain which can better shield the dipolar interactions between methacrylic esters, causing less friction in the TS.53 As the addition of 4 more “frictional” dipolar esters in the ester side chain did not cause a significant change in bulk kp for PLANEMA, it seems that the nature of the substituents several units beyond the methacrylic ester does not decisively influence kp.
| k 70°Cp (L mol−1 s−1) | E A (kJ mol−1) | ± | A (L mol−1 s−1) | Range | |
|---|---|---|---|---|---|
| MMAN = 0 | 979 | 22.1 | 2.2 | 2.3 × 106 | 4.9 × 106 1.1 × 106 |
| PLA1EMAN = 1 | 1744 | 21.6 | 1.6 | 3.1 × 106 | 5.4 × 106 1.8 × 106 |
| PLA5EMAN = 5 | 1764 | 19.9 | 1.2 | 1.8 × 106 | 2.8 × 106 1.2 × 106 |
| CombinedN = 1, 5 | — | 21.1 | 1.3 | 2.7 × 106 | 4.2 × 106 1.7 × 106 |
Solution homopropagation kinetics
In order to investigate the influence of hydrogen bonding and (macro)monomer molar volume on PLANEMA homopropagation kinetics, the PLP-SEC study was extended to MMA, PLA1EMA, and PLA5EMA solution homopolymerizations in 75 wt% BuOH, DMF, and xylenes with conditions summarized by Tables S5, S7, and S9;† additional experiments with MMA and PLA1EMA were conducted at an equimolar ratio of BuOH to monomer. The resulting kp estimates at 70 °C and 90 °C are presented in Fig. 3 and S6,† respectively, to compare specific solvent influences on each system (N = 0, 1, and 5). However, due to the marked decrease in monomer molar concentration from N = 0 to N = 5 at constant solvent weight fraction (see bulk monomer concentrations in Table 1), direct comparison of solution kp estimates from different N are not necessarily meaningful. In the case of MMA (N = 0), for which differences in monomer and solvent molar volumes (Vmon − Vsol) are not very large, there is no significant difference between the solution kp measured in xylenes compared to bulk, the kp is slightly elevated in DMF, and in BuOH the kp of MMA is clearly enhanced because of the well-documented hydrogen bond formation between hydroxyl and methacryloyl carbonyl which reduces the electron density at the double bond making MMA more reactive towards radical addition.35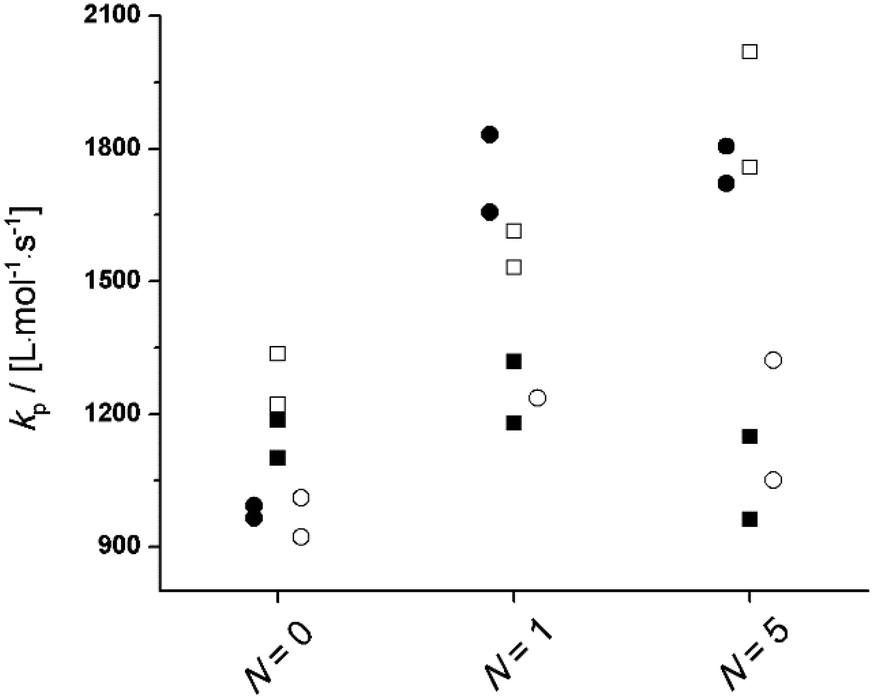 | ||
| Fig. 3 Plots for kp of MMA, PLA1EMA, and PLA5EMA determined by universal calibration in bulk (●), 75 wt% xylenes (○), 75 wt% DMF (■), and 75 wt% BuOH (□) solutions at 70 °C with 5 mmol L−1 DMPA. | ||
The situation is different for the PLA1EMA and PLA5EMA systems, for which a reduction in kp compared to bulk is measured (modest in xylenes and pronounced in DMF), even though no specific interaction between PLANEMA and either solvent is expected. This same trend in apparent kp was reported in the PLP-SEC study of PEGEEMA in toluene (Vmon − Vsol = 139 cm3 mol−1) and THF (Vmon − Vsol = 164 cm3 mol−1),33 solvents similar to xylenes and DMF (in terms of relative polarities and molar volumes), respectively, a result that was reconciled in terms of the previously established linear relationship between Vmon − Vsol and kp,∞/kp,bulk.37 Although this correlation formally predicts that systems with very large Vmon − Vsol (i.e., >226 cm3 mol−1) yield negative ratios of kp,∞/kp,bulk, the logic can still be applied to explain the PLANEMA results: at 70 °C the molar volumes of PLA1EMA (191 cm3 mol−1) and PLA5EMA (average of 422 cm3 mol−1) are larger than those of DMF (81 cm3 mol−1) and xylenes (129 cm3 mol−1) such that solvent molecules outcompete (macro)monomers for positions at the reaction site, leading to a lower than analytical local (macro)monomer concentration and corresponding reduction in apparent kp measured by PLP-SEC. The fact that there is only a small difference in the apparent kp (measured as 75 wt% solvent) for 0.50 mol L−1 PLA5EMA in DMF (Vmon − Vsol = 341 cm3 mol−1) and 1.23 mol L−1 PLA1EMA in DMF (Vmon − Vsol = 110 cm3 mol−1) at 70 and 90 °C (Fig. 3 and S6,† respectively), indicates that there should exist a minimum kp,∞/kp,bulk which cannot be exceeded by further increases in Vmon − Vsol of monomer/solvent pairings. The physical interpretation is that the volume around the chain-end radical which can be preferentially occupied by solvent molecules is finite. This reasoning is consistent with Buback's interpretation of linear alkyl acrylate homopropagation trends in toluene: a larger molar volume of the solvent (compared to monomer) allows for higher mobility of the TS structure and thus a higher kp results from the lower entropic penalty.53,54
Turning now to the PLANEMA homopolymerization experiments in 75 wt% BuOH, the kp values in Fig. 3 are increased compared to bulk PLA5EMA and slightly decreased compared to bulk for the PLA1EMA system. Since the effect of hydrogen bonding on the kp of a generic methacrylate (xMA) is known to depend on the relative concentrations of alcohol and xMA,36,55 additional experiments were performed so that the kp data estimated for MMA, PLA1EMA, and PLA5EMA in BuOH could be examined as a function of δ (molar ratio of alcohols to methacryloyl carbonyls; eqn (2)) for solution homopolymerizations, as summarized at 70 °C in Table 3 and at 90 °C in Table S4,† respectively.
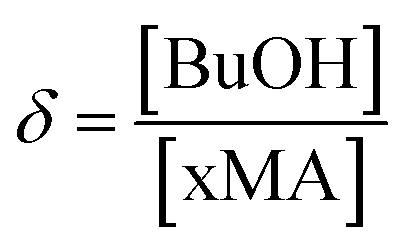 | (2) |
| Monomer | δ |
|
|
|---|---|---|---|
| MMA | 1.0 | 1.20 | — |
| 4.1 | 1.31 | 1.12 | |
| BMA (ref. 36) | 5.3 | 1.33 | — |
| 11.3 | 1.47 | — | |
| PLA1EMA | 1.0 | 0.81 | — |
| 7.5 | 0.90 | 1.26 | |
| PLA5EMA | 19.1 | 1.07 | 1.79 |
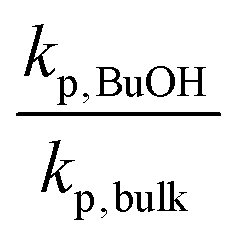
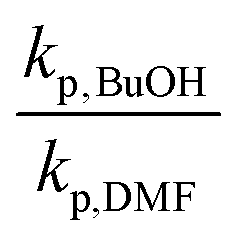
As δ increases from 1.0 to 4.1, the ratio of kp,BuOH/kp,bulk for MMA increases from 1.20 to 1.31 at 70 °C, in reasonable agreement with a 10% linear increase in kp per δ up to δ = 6.1 reported for MMA in benzyl alcohol at 30 °C.55 A similar increase in kp was observed at even greater dilutions (up to δ = 11.3) for BMA in BuOH at 70 °C,36 as also summarized by Table 3. In contrast, the ratio of kp,BuOH to kp,bulk decreases for PLA1EMA, as the comparison is confounded by the effect of large Vmon − Vsol on apparent kp. Thus, it is more meaningful to compare the kp in BuOH (96 cm3 mol−1) against the kp measured in DMF (81 cm3 mol−1) because the corresponding Vmon − Vsol for these solvents in PLANEMA systems are similar.
When the kp,BuOH/kp,DMF ratios are computed, it is clear that the hydrogen bonding provided by BuOH also leads to augmented kp for PLANEMA systems. The extent of the increase is quite significant, although the corresponding values of δ are also much higher (see Table 3), and the systems are complicated by the extra linkages in the methacrylate side chain: while for MMA and BMA, there is only a single methacryloyl carbonyl with which the alcohol's hydroxyl can interact, in PLANEMA systems there are N + 1 carbonyls which can accept hydrogen bonding. As the measured increases in kp,BuOH/kp,DMF for the limited N = 1 and N = 5 data sets can be reasonably accounted for by the proportionalities with δ estimated for BMA and MMA, it is unlikely that hydrogen bonding interactions with the N polyesters in the methacrylic ester side chain have any significant intrinsic kinetic effect on PLANEMA homopropagation, in agreement with our conclusion that the substituents several units beyond the methacrylic ester do not decisively influence the bulk kp measurements for PLANEMA. Nevertheless, the extent to which system specific enhanced kpvia hydrogen bonding between hydroxyl and methacryloyl carbonyl is diluted by the repeating esters in the PLANEMA side chain should be further investigated by controlling for δ as a function of the N + 1 carbonyls at various temperatures. Furthermore, since Vmon − Vsol is an important parameter for PLANEMA homopolymerizations, future work should include the influence of macromonomer molar volume on apparent reactivity ratios as well as kp,cop measurements for PLANEMA bulk copolymerizations with smaller molar volume comonomers such as MMA.
Nanoparticle degradation study
As previously mentioned, the ability to tune the degradation time of NPs produced from polyester macromonomers by specifying the average number and type of polyester units in the ROP step of the macromonomer synthesis has been demonstrated.15,18 However, whether the rate of NP degradation can be further controlled by end-group design needs to be evaluated, since the terminal hydroxyl of HEMA-PLA5 and the terminal ethyl ester of PLA5EMA distinguish macromonomers with the same average chain lengths by different hydrophobicities. To investigate the effect of PLANEMA's alkyl end-group on degradation, 5 wt% NP suspensions were prepared by semi-batch radical emulsion polymerizations at 70 °C with 1% SDS as surfactant using the following four (macro)monomer systems: PLA1EMA, PLA5EMA, HEMA-PLA5, and a comonomer mixture of 50 wt% PLA5EMA and 50 wt% HEMA-PLA5, with PSDs presented in Fig. S7–S10,† respectively.An accelerated degradation test for each NP suspension was performed over several weeks at 50 °C with the progress of the degradative swelling mechanism monitored by periodically measuring the increase in average particle size, as shown in Fig. 4; as degradation proceeds, the NPs become more hydrophilic leading to an increase in water absorption and apparent particle size at constant polydispersity index (PDI, pertaining to particle size), thus confirming that the increase in size is not due to coagulation.15,42 The corresponding PDI values from this study summarized by Fig. S11† are near constant with time, only increasing slightly at the final stages of degradation to suggest the formation of a small quantity of aggregates late in the process. The important feature of the data in Fig. 4 is the time at which the particle size begins to rapidly increase, as this indicates that the NPs have almost degraded to the final water-soluble material. For example, the rapidly increasing size of the HEMA-PLA5 latex from days 7–9 leads to the complete hydrolysis of the PLA grafts by day 10 to yield a poly(HEMA) backbone (which is observed as a swollen polymer at room temperature that can be dissolved upon dilution), in close agreement with an 8 day degradation time measured for very similar HEMA-PLA4 NPs.18 On the other hand, after 40 days the PLA5EMA homopolymer latex has finally degraded while the PLA1EMA latex is still observable.
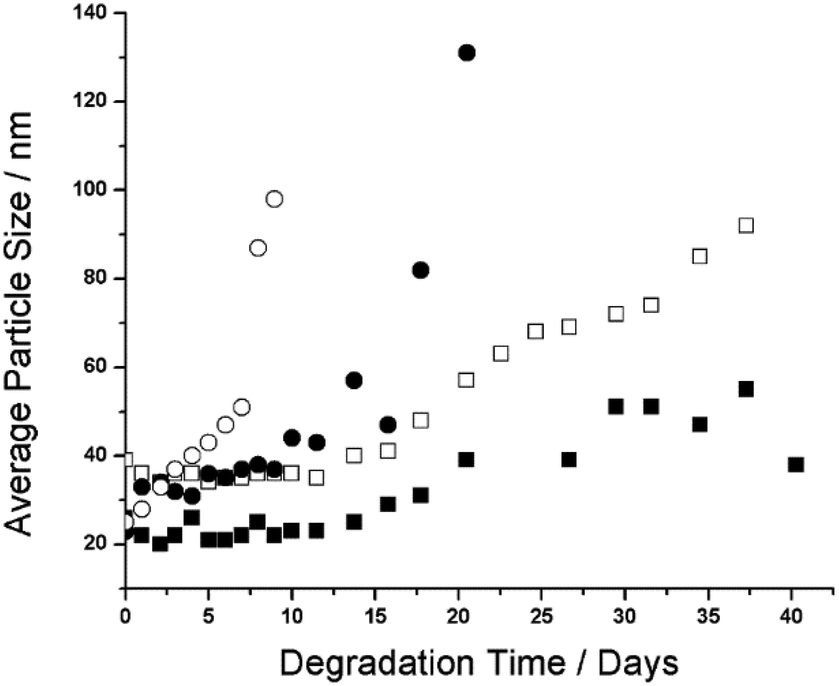 | ||
| Fig. 4 Intensity average size measurements at 25 °C for NPs produced from PLA1EMA (■), PLA5EMA (□), an equal mass copolymer of PLA5EMA and HEMA-PLA5 (●), and HEMA-PLA5 (○) throughout the accelerated degradation study performed at 50 °C. | ||
The continual increase in NP sizes over 40 days indicates that hydrolytic degradation of the NPs produced from the hydrophobic alkyl-terminated macromonomer occurs at a much slower rate because of the added hydrophobicity of PLANEMA's ethyl ester end-group. For comparison, NPs produced from the PCL (more hydrophobic than PLA) based HEMA-PCL3 macromonomer, with similar average MW as both PLA5EMA and HEMA-PLA5, were completely degraded after only 20 days under the same conditions.42 Furthermore, PLANEMA's lack of terminal hydroxyl precludes preferential chain-end scission hydrolysis, where protection of oligomeric PLA hydroxyl end-groups (through esterification or acetylation) has been shown to significantly reduce the rate of hydrolysis under both acidic and basic conditions.23,24 However, as illustrated by Scheme 2, any hydrolysis event in a PLANEMA chain yields a carboxyl-terminated graft. Since Codari et al. showed that preferential chain-end hydrolysis of bifunctional hydroxyl- and carboxyl- terminated oligomeric PLA can be ascribed to the increased end-group hydrophilicities,22 the slow hydrolysis of PLANEMA NPs must be due to the initial ethyl ester protection of PLA end-groups. Consideration must also be given to the pH of the NP environment because the terminal units of the hydrolysed PLANEMA grafts could exist as carboxylic acids or carboxylates which would certainly influence their degradability. Nevertheless, the delayed degradation (in comparison to HEMA-PLA5 NPs) afforded by the initial ethyl ester protection should remain effective regardless of the pH.
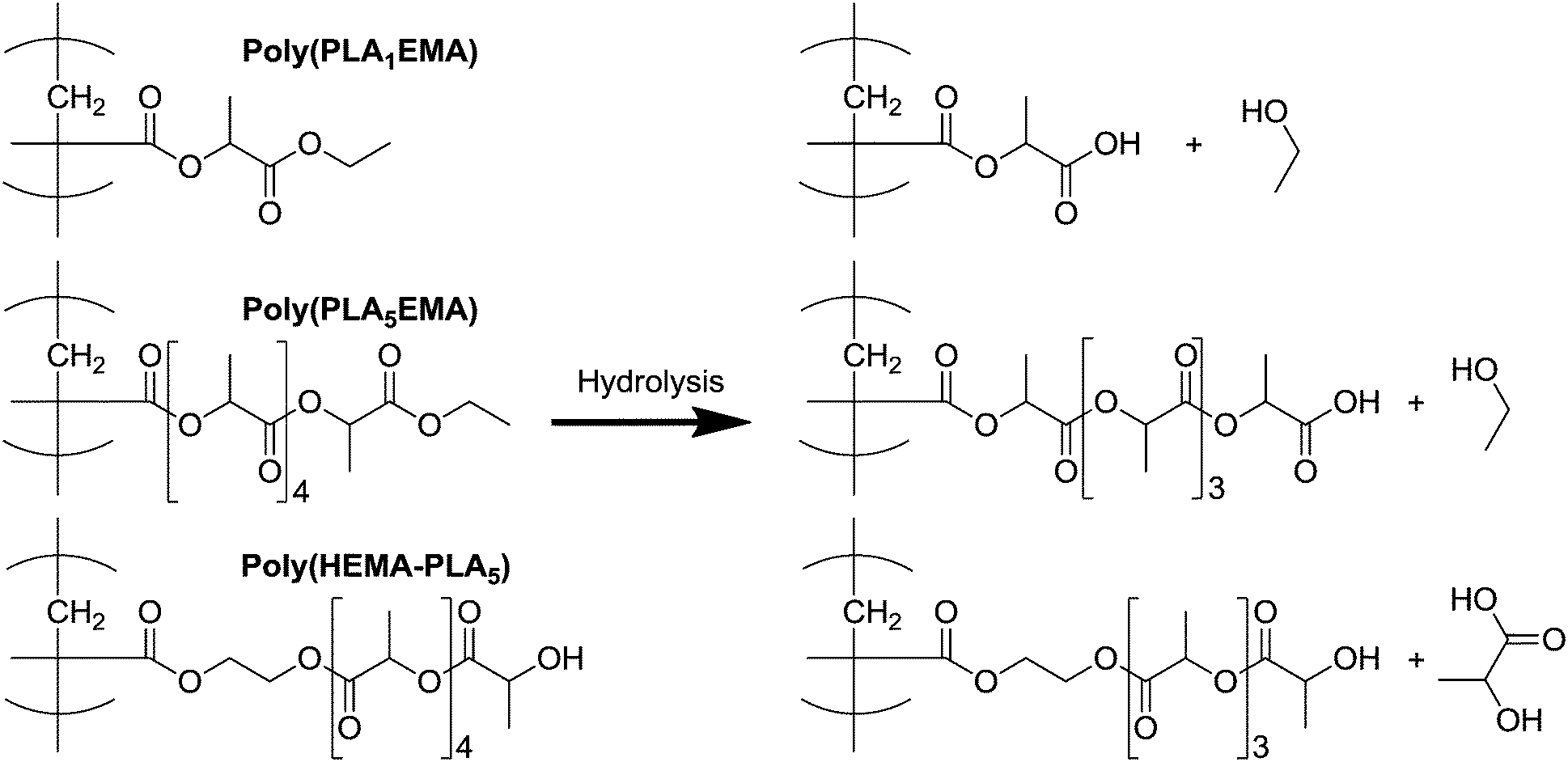 | ||
| Scheme 2 Proposed degradation products after one hydrolysis event at the terminal grafted unit of poly(PLA1EMA), poly(PLA5EMA), and poly(HEMA-PLA5) comb-polymers. | ||
In the case of PLA1EMA NPs, the reduction in pH after 35 days of accelerated degradation (Table S10†) indicates hydrolysis of the terminal ethyl ester to release ethanol; however, minimal increases in PLA1EMA NP size measurements to 40 days suggests that hydrolysis of the single grafted PLA unit to yield a poly(methacrylic acid) (PMAA) backbone is unfavourable such that complete degradation of PLA1EMA homopolymer will not occur over a timescale typically associated with oligomeric PLA based materials. As seen in Fig. 4, the PLA5EMA homopolymer latex degrades more quickly than the PLA1EMA latex; although it seems counterintuitive that complete degradation of the PLA5EMA homopolymer was observed before that of the PLA1EMA homopolymer, the latter has roughly 2.5 times more esters attached to the methacrylic backbone per unit mass due to the macromonomer's larger MW. In support of the notion that these units are more difficult to hydrolyse than typical ester linkages in the polyester backbone, the release of ethylene glycol was not detected at the end of the polyester graft degradation for previous HEMA-PLA3 NP degradation studies, indicating that the poly(HEMA) backbone did not degrade to PMAA.17
Under unbuffered degradation conditions (i.e., acidic), the choice of macromonomer end-group affords a considerable range of NP degradation times from 10 to 40 days for HEMA-PLA5 and PLA5EMA homopolymer latexes, respectively; when 50 wt% PLA5EMA is added to the HEMA-PLA5 macromonomer recipe, an intermediate NP degradation time of 23 days is achieved (Fig. 4). This finding verifies the successful copolymerization of the two macromonomers, despite their differences in water solubility, and also demonstrates that end-group choice can be used to tune NP degradation time in acidic environments. Depending on the intended application's pH and functional timescale, formulations could benefit by using PLANEMA as a comonomer to modify system hydrophobicity and degradation time while still maintaining the properties of a PLA based system. In addition, PLANEMA can be used to improve selectivity of post-polymerization esterification reactions through use as a comonomer spacer with hydroxyl or carboxyl functionalized monomers.
Conclusions
Polylactic acid based methacrylate (macro)monomers (PLANEMA) with N = 1 and N = 5 average polyester units per chain were synthesized and their homopropagation kinetics studied in bulk and solution using the PLP-SEC technique. Reasonable agreement (within 15%) was obtained for kp estimates from low-conversion PLP samples analysed by SEC coupled with light scattering and universal calibration, validating the dn/dc and Mark-Houwink parameters measured in this work for both N = 1 and N = 5 comb-polymers. No significant difference between PLA1EMA and PLA5EMA bulk kp estimates was detected over the temperature range of 40–100 °C, indicating that polar and steric characteristics several units beyond the methacrylic group do not decisively influence kp. Furthermore, the apparent solution kp values measured for both PLA1EMA and PLA5EMA in DMF as well as in xylenes were markedly decreased compared to bulk due to differences in (macro)monomer and solvent molar volumes, while in BuOH the apparent kp of both systems increased relative to in DMF because of hydrogen bonding.PLANEMA (macro)monomers were used to produce NPs by semi batch radical emulsion polymerization. When an equal weight of PLA5EMA is used as comonomer, the time for accelerated degradation at 50 °C of hydroxyl terminated HEMA-PLA5 NPs is increased from 10 to 23 days, while the PLA5EMA homopolymer NPs took 40 days to degrade. An explanation for the slow degradation is proposed in terms of PLA graft orientation relative to the methacrylic backbone; ethyl ester protection of poly(PLANEMA) grafts' carboxyl end-groups delays the onset of preferential chain-end hydrolysis. The ability to affect degradation time by copolymerizing PLANEMA with HEMA-PLA2n provides further opportunities to tune the performance characteristics of this family of degradable NPs according to specific application requirements.
Acknowledgements
The authors thank the Natural Sciences and Engineering Research Council of Canada for financial support as well as Dr. Raffaele Ferrari and Dr. Claudio Colombo of ETH Zurich for technical discussions.References
- R. E. Drumright, P. R. Gruber and D. E. Henton, Adv. Mater., 2000, 12, 1841 CrossRef CAS.
- S. M. Davachi and B. Kaffashi, Polym.-Plast. Technol. Eng., 2015, 54, 944 CrossRef CAS.
- M. Jamshidian, E. A. Tehrany, M. Imran, M. Jacquot and S. Desobry, Compr. Rev. Food Sci. Food Saf., 2010, 9, 552 CrossRef CAS.
- F. Achmad, K. Yamane, S. Quan and T. Kokugan, Chem. Eng. J., 2009, 151, 342 CrossRef CAS.
- O. Dechy-Cabaret, B. Martin-Vaca and D. Bourissou, Chem. Rev., 2004, 104, 6147 CrossRef CAS PubMed.
- S. A. van den Berg, H. Zuilhof and T. Wennekes, Macromolecules, 2016, 49, 2054 CrossRef CAS.
- T. R. Rooney, S. P. Gumfekar, J. B. P. Soares and R. A. Hutchinson, Macromol. Mater. Eng., 2016, 301, 1248 CrossRef CAS.
- T. Okuda, K. Ishimoto, H. Ohara and S. Kobayashi, Macromolecules, 2012, 45, 4166 CrossRef CAS.
- J. Kiehl, C. Delaite, S. Bistac, A. S. Schuller and H. Farge, Polymer, 2012, 53, 694 CrossRef CAS.
- C. J. Hawker, D. Mecerreyes, E. Elce, J. Dao, J. L. Hedrick, I. Barakat, P. Dubois, R. Jérôme and W. Volksen, Macromol. Chem. Phys., 1997, 198, 155 CrossRef CAS.
- J. A. Wallach and S. J. Huang, Biomacromolecules, 2000, 1, 174 CrossRef CAS PubMed.
- K. Ishimoto, M. Arimoto, H. Ohara, S. Kobayashi, M. Ishii, K. Morita, H. Yamashita and N. Yabuuchi, Biomacromolecules, 2009, 10, 2719 CrossRef CAS PubMed.
- K. Ishimoto, M. Arimoto, T. Okuda, S. Yamaguchi, Y. Aso, H. Ohara, S. Kobayashi, M. Ishii, K. Morita, H. Yamashita and N. Yabuuchi, Biomacromolecules, 2012, 13, 3757 CrossRef CAS PubMed.
- D. W. Lim, S. H. Choi and T. G. Park, Macromol. Rapid Commun., 2000, 21, 464 CrossRef CAS.
- C. Colombo, L. Dragoni, S. Gatti, R. M. Pesce, T. R. Rooney, E. Mavroudakis, R. Ferrari and D. Moscatelli, Ind. Eng. Chem. Res., 2014, 53, 9128 CrossRef CAS.
- R. Ferrari, Y. Yu, M. Lattuada, G. Storti, M. Morbidelli and D. Moscatelli, Macromol. Chem. Phys., 2012, 213, 2012 CrossRef CAS.
- Y. Yu, R. Ferrari, M. Lattuada, G. Storti, M. Morbidelli and D. Moscatelli, J. Polym. Sci., Part A: Polym. Chem., 2012, 50, 5191 CrossRef CAS.
- R. Ferrari, C. Colombo, M. Dossi and D. Moscatelli, Macromol. Mater. Eng., 2013, 298, 730 CrossRef CAS.
- I. Grizzi, H. Garreau, S. Li and M. Vert, Biomaterials, 1995, 16, 305 CrossRef CAS PubMed.
- L. Xu, K. Crawford and C. B. Gorman, Macromolecules, 2011, 44, 4777 CrossRef CAS.
- S. Lazzari, F. Codari, G. Storti, M. Morbidelli and D. Moscatelli, Polym. Degrad. Stab., 2014, 110, 80 CrossRef CAS.
- F. Codari, S. Lazzari, M. Soos, G. Storti, M. Morbidelli and D. Moscatelli, Polym. Degrad. Stab., 2012, 97, 2460 CrossRef CAS.
- S. J. de Jong, E. R. Arias, D. T. S. Rijkers, C. F. van Nostrum, J. J. Kettenes-van den Bosch and W. E. Hennink, Polymer, 2001, 42, 2795 CrossRef CAS.
- C. F. van Nostrum, T. F. J. Veldhuis, G. W. Bos and W. E. Hennink, Polymer, 2004, 45, 6779 CrossRef CAS.
- H. Shinoda and K. Matyjaszewski, Macromolecules, 2001, 34, 6243 CrossRef CAS.
- J. Eguiburu, M. J. Fernandez-Berridi and J. S. Román, Polymer, 1996, 37, 3615 CrossRef CAS.
- R. Ferrari, T. R. Rooney, M. Lupi, P. Ubezio, R. A. Hutchinson and D. Moscatelli, Macromol. Biosci., 2013, 13, 1347 CrossRef CAS PubMed.
- T. R. Rooney, O. Monyatsi and R. A. Hutchinson, Macromolecules, 2017, 50, 784 CrossRef CAS.
- S. Beuermann and M. Buback, Prog. Polym. Sci., 2002, 27, 191 CrossRef CAS.
- S. Beuermann, M. Buback, T. P. Davis, R. G. Gilbert, R. A. Hutchinson, O. F. Olaj, G. T. Russell, J. Schweer and A. M. van Herk, Macromol. Chem. Phys., 1997, 198, 1545 CrossRef CAS.
- S. Beuermann, M. Buback, T. P. Davis, R. G. Gilbert, R. A. Hutchinson, A. Kajiwara, B. Klumperman and G. T. Russell, Macromol. Chem. Phys., 2000, 201, 1355 CrossRef CAS.
- A. P. Haehnel, M. Schneider-Baumann, K. U. Hiltebrandt, A. M. Misske and C. Barner-Kowollik, Macromolecules, 2013, 46, 15 CrossRef CAS.
- R. Siegmann, A. Jeličić and S. Beuermann, Macromol. Chem. Phys., 2010, 211, 546 CrossRef CAS.
- S. Smolne, S. Weber and M. Buback, Macromol. Chem. Phys., 2016, 217, 2391 CrossRef CAS.
- S. Beuermann, Macromol. Rapid Commun., 2009, 30, 1066 CrossRef CAS PubMed.
- S. Beuermann, Macromolecules, 2004, 37, 1037 CrossRef CAS.
- S. Beuermann and N. García, Macromolecules, 2004, 37, 3018 CrossRef CAS.
- M. Dossi, K. Liang, R. A. Hutchinson and D. Moscatelli, J. Phys. Chem. B, 2010, 114, 4213 CrossRef CAS PubMed.
- R. A. Hutchinson, J. H. McMinn, D. A. Paquet, S. Beuermann and C. Jackson, Ind. Eng. Chem. Res., 1997, 36, 1103 CrossRef CAS.
- J. M. Bernal-García, A. Guzmán-López, A. Cabrales-Torres, A. Estrada-Baltazar and G. A. Iglesias-Silva, J. Chem. Eng. Data, 2008, 53, 1024 CrossRef.
- M. G. Bravo-Sánchez, G. A. Iglesias-Silva, A. Estrada-Baltazar and K. R. Hall, J. Chem. Eng. Data, 2010, 55, 2310 CrossRef.
- R. Ferrari, Y. Yu, M. Morbidelli, R. A. Hutchinson and D. Moscatelli, Macromolecules, 2011, 44, 9205 CrossRef CAS.
- P. Drawe and M. Buback, Macromol. Theory Simul., 2016, 25, 74 CrossRef CAS.
- S. Beuermann, D. A. Paquet, J. H. McMinn and R. A. Hutchinson, Macromolecules, 1996, 29, 4206 CrossRef CAS.
- A. N. Nikitin, I. Lacík and R. A. Hutchinson, Macromolecules, 2016, 49, 9320 CrossRef CAS.
- R. A. Hutchinson, D. A. Paquet, J. H. McMinn, S. Beuermann, R. E. Fuller and C. Jackson, in 5th International Workshop on Polymer Reaction Engineering, DECHEMA Monographs Vol 131, ed. K.-H. Reichert and H.-U. Moritz, VCH Verlags, Weinheim Germany, 1995, p. 467 Search PubMed.
- K. B. Kockler, F. Fleischhaker and C. Barner-Kowollik, Polym. Chem., 2016, 7, 4342 RSC.
- K. B. Kockler, F. Fleischhaker and C. Barner-Kowollik, Macromolecules, 2016, 49, 8572 CrossRef CAS.
- A. Zoller, K. B. Kockler, M. Rollet, C. Lefay, D. Gigmes, C. Barner-Kowollik and Y. Guillaneuf, Polym. Chem., 2016, 7, 5518 RSC.
- A. P. Haehnel, M. Stach, A. Chovancová, J. M. Rueb, G. Delaittre, A. M. Misske, I. Lacik and C. Barner-Kowollik, Polym. Chem., 2014, 5, 862 RSC.
- M. Buback and C. H. Kurz, Macromol. Chem. Phys., 1998, 199, 2301 CrossRef CAS.
- J. P. A. Heuts, R. G. Gilbert and L. Radom, Macromolecules, 1995, 28, 8771 CrossRef CAS.
- M. Buback, Macromol. Symp., 2009, 90, 275 Search PubMed.
- M. Buback, Macromol. Rapid Commun., 2015, 36, 1979 CrossRef CAS PubMed.
- K. F. O'Driscoll, M. J. Monteiro and B. Klumperman, J. Polym. Sci., Part A: Polym. Chem., 1997, 35, 515 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c7re00019g |
| This journal is © The Royal Society of Chemistry 2017 |