
Facile photo-flow synthesis of branched poly(butyl acrylate)s†
O. Eckardtab,
B. Wennc,
P. Biehlab,
T. Junkers *cd and
F. H. Schacher*ab
*cd and
F. H. Schacher*ab
aInstitute of Organic Chemistry and Macromolecular Chemistry (IOMC), Friedrich-Schiller-University Jena, Lessingstraße 8, D-07443 Jena, Germany. E-mail: felix.schacher@uni-jena.de
bJena Center for Soft Matter (JCSM), Friedrich-Schiller-University Jena, Philosophenweg 7, D-07743 Jena, Germany
cPolymer Reaction Design Group (PRD), Institute of Materials Research (IMO), Hasselt University, Martelarenlaan 42, BE-3500 Hasselt, Belgium. E-mail: tanja.junkers@uhasselt.be
dIMEC associated lab IMOMEC, Wetenschapspark 1, BE-3590 Diepenbeek, Belgium
First published on 13th April 2017
Abstract
We present the synthesis of branched poly(butyl acrylate)s using photo-induced free radical polymerization of (n/t)-butyl acrylate in the presence of tri(propylene glycol) diacrylate (TPGDA) as a crosslinker and varying amounts of dodecanethiol (DDT) as a chain transfer agent to prevent macroscopic gelation. Thereby, the monomer–thiol ratio strongly influences the molecular weight of the obtained materials from 5000 g mol−1 up to 100![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 g mol−1. Reactions were performed in a scalable photo-flow reactor to allow homogenous irradiation and prevention of local “hot spots”, to increase monomer conversion, and to improve control over reaction conditions (i.e. temperature and viscosity). The reactions were monitored by NMR spectroscopy and size exclusion chromatography (SEC) to determine the conversion and molecular weight, respectively. The robustness of this system is demonstrated by using different photo initiators, thiols and tube diameters. Due to their inherently low viscosity, branched poly(n-butyl acrylate)s could be synthesized in the bulk up to full conversion within 20 min and, hence, under solvent free conditions, which is very untypical of flow radical polymerization.
000 g mol−1. Reactions were performed in a scalable photo-flow reactor to allow homogenous irradiation and prevention of local “hot spots”, to increase monomer conversion, and to improve control over reaction conditions (i.e. temperature and viscosity). The reactions were monitored by NMR spectroscopy and size exclusion chromatography (SEC) to determine the conversion and molecular weight, respectively. The robustness of this system is demonstrated by using different photo initiators, thiols and tube diameters. Due to their inherently low viscosity, branched poly(n-butyl acrylate)s could be synthesized in the bulk up to full conversion within 20 min and, hence, under solvent free conditions, which is very untypical of flow radical polymerization.
Introduction
Although branched polymers have been theoretically described already by Flory in 1952,1 interest in both the synthesis and the characterization of such materials is still continuously growing.2–4 Depending on the synthetic method employed and on the overall degree of branching, these materials can exhibit lower solution viscosity, lowered melt rheology,5 a high number of functional groups,6 lower glass transition temperature,7 and an overall improved solubility, compared to their linear analogues.8 In addition, synthetic access to branched polymers is often relatively straightforward in contrast to synthetic access to perfectly defined and uniformly branched dendrimers.9 This, in combination with modern synthetic tools for post-polymerization modification, enables various possible applications of branched polymers in the fields of biomedicine10 and gene delivery (b-PEI Lupasol®),11 as viscosity modifiers,12 and within light emitting diodes.13One commonly used way to synthesize branched polymers is either polycondensation or polyaddition14 of AB and AB2 (ref. 15) or A2 and B3 monomers9 to produce, for example, polyesters (e.g. Boltorn™), polyethers, or polyurea. Unfortunately, most of these approaches require the presence of certain functional groups such as isocyanates and alcohols,14 cyclic precursor compounds,16 or specially designed catalysts. In 1995, Frechet et al. developed a new method to prepare branched vinyl-containing polymers, the so-called self-condensing vinyl polymerization (SCVP).17 This approach typically employs vinyl compounds featuring a second functionality to initiate controlled radical polymerization,18–20 group transfer polymerization (GTP),21 or cationic polymerization17 of other suitable vinyl compounds.
Recently, a facile and versatile procedure for the synthesis of branched (meth)acrylate-based polymers based on free radical copolymerization of a vinyl monomer and a bifunctional crosslinker has been described by Sherrington et al.22 To prevent the otherwise unavoidable gelation, a defined amount of a chain transfer agent, mostly alkane thiols, is added. This so-called “Strathclyde method” has already been used for the synthesis of branched polymers from a variety of monomers, e.g. styrene,23 methyl methacrylate (MMA),24 vinyl acetate (VAc),25 or methacrylic acid (MAA).26
Concerning scalability, Isaure et al. described a method to synthesize branched poly(dimethyl acrylamide)s (PDAM) in parallel reactors in 2006. The use of a Radley Carousel Reactor led to higher space-time yields of branched PDAM due to parallel synthesis being possible compared to the outcome from conventionally used round-bottom flasks.27 Another well-established technique for manufacturing large quantities of polymers is the use of continuous flow reactors. Quite often, flow chemistry can be regarded as more efficient than conventional batch reactors, e.g. due to a larger surface area to volume ratio and the concomitant highly efficient heat dissipation.28–30 Flow reactors are inherently simple to scale up and are regularly found to yield products with improved purity and definition (in the case of polymers) while keeping reaction times short. In recent years, different polymerization techniques have been adapted to flow reactors, including cationic polymerization of 2-oxazolines31 and anionic polymerization,32,33 as well as the RAFT34 and ATRP techniques.35 Significant interest has risen in the past few years on photo-controlled radical polymerizations,36 which show outstanding performance in flow reactors. Quite recently, Wenn et al. have developed a method to synthesize well-defined star-shaped block copolymers via photo-induced polymerization within such set-ups.37 A semi-quantitative assessment of important differences between photo-initiated batch and flow reactions has also been provided.38
In the present paper, we present the synthesis of branched poly(butyl acrylate)s via free radical polymerization of (n/t)-butyl acrylate in the presence of tri(propylene glycol) diacrylate (TPGDA) as a crosslinker and varying amounts of dodecanethiol (DDT) as a chain transfer agent. A photo-initiated continuous flow was thereby used and compared to conventional batch reactors. In the herein used flow reactors, polymerizations could be carried out to practically full conversion and high mass-average molecular weights in a solvent-free process. To the best of our knowledge, a solvent-free process is unique so far for flow polymerizations. Reactions conducted in a continuous flow reactor were further analyzed regarding their robustness towards various photo-initiators, aliphatic thiols, and different tube diameters.
Experimental
Materials
The monomers n-butyl acrylate (nBA, Acros, 99%), t-butyl acrylate (tBA, Acros, 99%) and tri(propylene glycol) diacrylate (TPGDA, VWR, mixture of isomers) contained monomethyl ether hydroquinone as an inhibitor, which was removed by column chromatography over aluminum oxide prior to use. Benzoin (Janssen Chimica, 98%), 2,2-dimethoxy-2-phenyl-acetophenone (DMPA, Acros, 99%), phenylbis(2,4,6-trimethyl-benzoyl)phosphine oxide (Irgacure 819, Tokyo Chemical Industry, 96%), 2-hydroxy-4′-(2-hydroxyethoxy)-2-methyl-propiophenone (Irgacure 2959, Sigma Aldrich, 98%), propane thiol (Sigma Aldrich, 99%), hexane thiol (Acros, 96%), octane thiol (Acros, 97%), decane thiol (Sigma-Aldrich, 96%) and dodecane thiol (Sigma Aldrich, 98%) were used as received. Deuterated chloroform and dimethylsulfoxide were purchased from Deutero.Instruments
040 000 Da). N,N-dimethylacetamide with 5 mmol lithium chloride was used as the eluent at 1 mL min−1 flow rate, and the column oven was set to 40 °C. The system was calibrated with low dispersity poly(methyl methacrylate) (PMMA) standards.Further size exclusion chromatography (SEC) measurements were performed using a Tosoh EcoSEC HLC-8320GPC, comprising an autosampler and a PSS guard column SDV (50 × 7.5 mm), followed by three PSS SDV analytical linear XL (5 μm, 300 × 7.5 mm) columns thermostated at 40 °C (column molecular weight range: 1 × 102–1 × 106 g mol−1), and a differential refractive index detector (Tosoh EcoSEC RI) using THF as the eluent at a flow rate of 1 mL min−1 with toluene as a flow marker. Calibration was performed using linear narrow polystyrene (PS) standards from PSS Laboratories in the range of 470–7.5 × 106 g mol−1. For the analysis, MHKS parameters for PnBA (α = 0.7, K = 12.2 × 10−5 dL g−1, THF 40 °C)39 and PtBA (α = 0.66, K = 19.7 × 10−5 dL g−1, THF 30 °C)40 were applied.
Synthesis
Results and discussion
Branched poly(n-butyl acrylate)s (PnBAs) and poly(tert-butyl acrylate) (PtBA) of different molecular weights were synthesized according to the “Strathclyde” methodology;41 therefore, monomers (nBA, tBA), tri(propylene glycol) diacrylate (TPGDA), and dodecanethiol (DDT) were mixed in different ratios with a suitable UV photo-initiator as shown in Scheme 1. Photo-initiation (Irgacure 819; Fig. S2†) was chosen due to easy handling, low energy requirements, low temperature treatment, and fast decomposition.42 Altogether, these effects lead to improved control over reaction time and temperature. Tri(propylene glycol) diacrylate as a difunctional co-monomer introduces branching points to the generated polyacrylates, and the addition of dodecanethiol as a chain transfer agent prevents macroscopic gelation. One further advantage of this method is that (at least in the case of PnBA) no solvent is necessary, and polymerizations can be carried out under bulk conditions.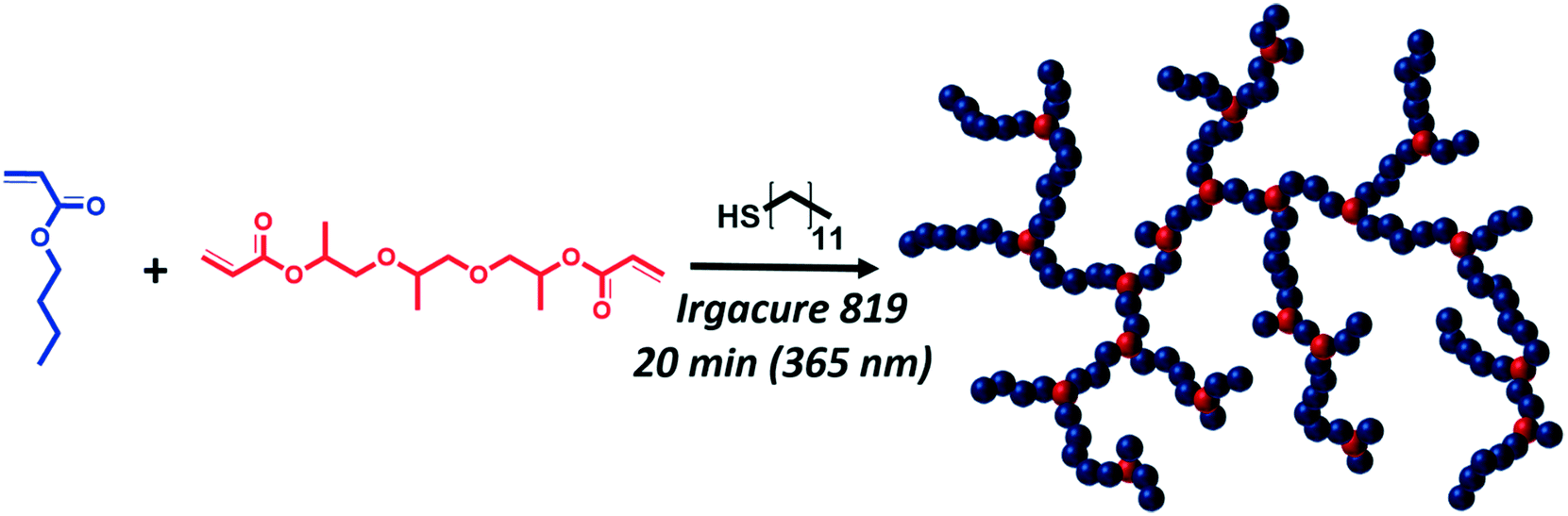 | ||
| Scheme 1 Synthesis route of branched poly(n-butyl acrylate) following the Strathclyde method. | ||
We start with the batch synthesis of branched PnBAs. Here, the molecular weight of the obtained polymer can be adjusted using the ratio of the monomer to thiol (nBA/DDT). With decreasing amount of DDT in the reaction mixture, higher molar masses are obtained, accompanied by broader dispersities, as can be seen in the SEC traces in Fig. 1A and Table 1. This can also be shown in a Mayo-plot, where the DDT/nBA ratio is plotted versus the reciprocal (apparent) degree of polymerization for different PnBAs (Fig. 1B). The linear relationship indicates a successful chain transfer mechanism with a calculated chain transfer constant (slope) of 0.44, which is similar to previous reports of Sherrington.43 Nevertheless, one has to be careful regarding interpretation, as only apparent molar masses are provided by the underlying SEC measurements.
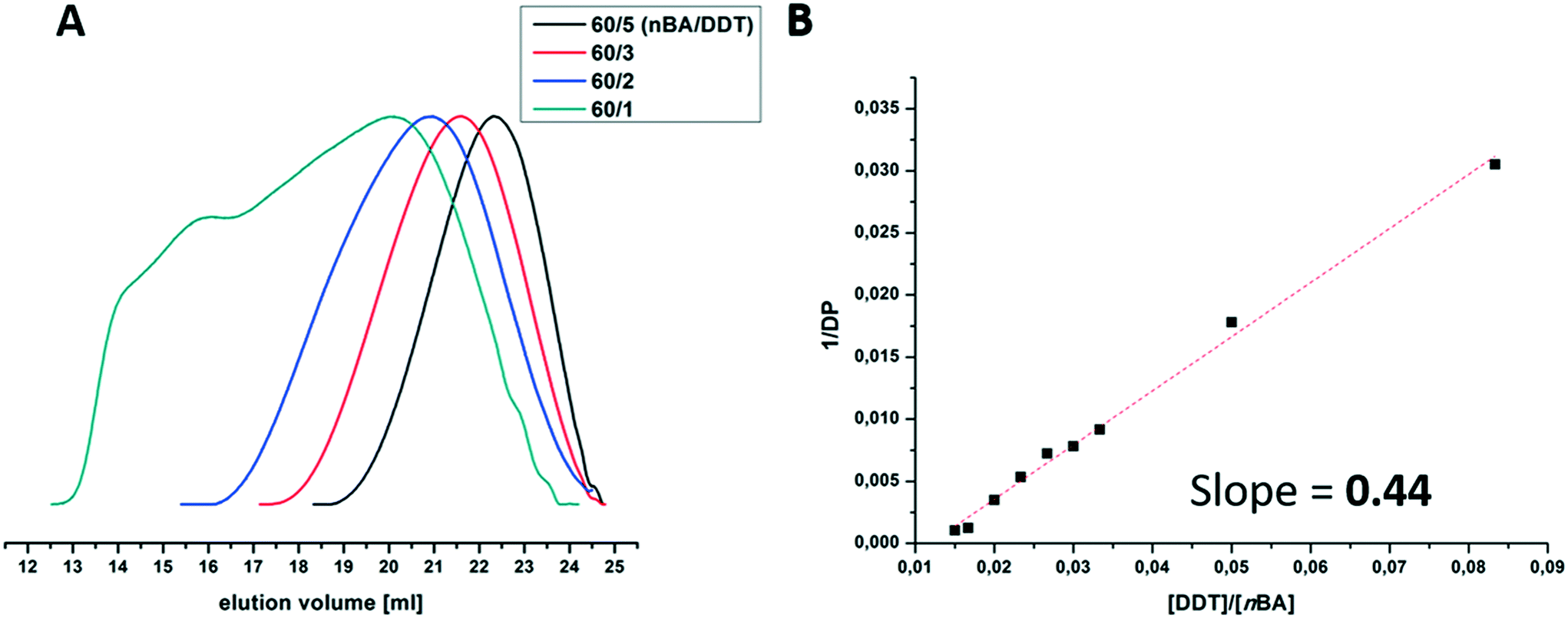 | ||
| Fig. 1 Selected SEC elution traces (A) and Mayo-plot with linear FIT (B) of branched PnBAs at different nBA to DDT molar feed ratios; the apparent degree of polymerization was determined by SEC: DMAc/LiCl (5 mmol L−1), calibrated against PMMA standards. | ||
| Feed ratio (mol) | Flow | Batch | |||||||
|---|---|---|---|---|---|---|---|---|---|
| M n [g mol−1] | M w [g mol−1] | Conv.c[%] | Viscosityg[mPa s] | M n [g mol−1] | M w [g mol−1] | Conv.c [%] | |||
| nBA | TPGDA | DDT | |||||||
| a SEC (THF) (PnBA-calib.). b SEC (DMAc/LiCl) (PMMA-calib.). c Conversion of the monomer as determined by 1H-NMR (300 MHz). d Viscosity at 20 °C. e Viscosity at 65 °C. f After 8 min irradiation. g Only viscosities for the solvent-free process in the flow reactor are described. | |||||||||
| 60 | 1 | 5 | 2100 | 5800 | 99 | 922;d 111e | 2300 | 5500 | 71 |
| 60 | 1 | 3 | 3400 | 14400 |
96 | 327e | 3900 | 12000 |
74 |
| 60 | 1 | 2 | 6200 | 78000 |
96 | >2500d,e | 6700 | 61500 |
76 |
| 60 | 1 | 1 | 12300f |
109000f |
73f | — | 14000 |
140000 |
78 |
| tBA | |||||||||
| 60 | 1 | 5 | 1340 | 2200 | 78 | — | 1600 | 2100 | 33 |
| 60 | 1 | 2 | 2600 | 5000 | 78 | — | 2900 | 4200 | 32 |
| 60 | 1 | 1 | 5400 | 9500 | 76 | — | 4700 | 7300 | 32 |
| 60 | 1 | 0.6 | 6500 | 11800 |
78 | — | 6600 | 10600 |
31 |
Although initial experiments were carried out in batch reactions with good results as shown above, we were interested to transfer this type of polymerization to photo-flow reactors, mainly due to its potential upscaling. Typically, upscaling of photoreactions is hampered by light gradients within the reaction vessel and varying light penetration depths, often leading to severe efficiency losses when carried out in larger volumes. Photoflow reactor set-ups remove these disadvantages and allow for fully scalable photopolymerizations.38,44 Therefore, a 2 mL photo-flow reactor consisting of a syringe pump or a Knauer Azura P2.1S HPLC pump, a 15 W Vilber–Lourmant UV-lamp with a maximum emission wavelength at 365 nm, and transparent PFA tubing (Ø = 0.75 mm; 2 mm or 4 mm) was employed for polymerization (see Scheme 2). The irradiation time of the polymerization reactions was adjusted by changing the flow rate from 2 mL min−1 (1 min of irradiation) to 0.1 mL min−1 (20 min of irradiation).
 | ||
| Scheme 2 Sketch (A) and photograph (B) of a photo-flow reactor with a HPLC pump, a UV lamp and a product collector. | ||
Photo-flow vs. photo-batch
To demonstrate the difference between photo-flow and photo-batch polymerization of branched polymers, poly(n-butyl acrylate)s of different molar masses were synthesized (Fig. S1†). Due to the branched nature of the polymers, all poly(n-butyl acrylate)s appeared as sticky and highly viscous oils in comparison with their solid linear analogue, which allows a solvent-free process. At room temperature, viscosity values from 900 mPa s up to several thousand mPa s were obtained (Table 1). By synthesizing branched PnBAs with MW above 100000 g mol−1 (green dashed line, Fig. S1†), the viscosity increased rapidly, which could potentially damage the photo-flow set-up by clogging and, eventually, rupture the tubing.
In a conventional 2 mL photo-batch reactor, no significant change in the conversion and molar mass between 1 min and 20 min of irradiation was observed. By using continuous photo-flow reactors, full conversion (>99% of the monomer) is reached after 20 min, as shown in Fig. 2A. The temperature inside the photo-flow reactor was permanently below 65 °C, presumably due to improved heat transfer, leading to milder reaction conditions compared to those of the earlier discussed photo-batch reactor (above 90 °C). This effect leads to higher molar masses in the batch reactor, as shown in Fig. 2B. Furthermore, all the data for the branched PnBAs synthesized in the flow and batch reactors are listed in Table 1. After 20 min of irradiation, branched PnBAs in flow reactors exhibit similar Mn-values at broader molecular weight distribution compared to batch synthesized analogues.
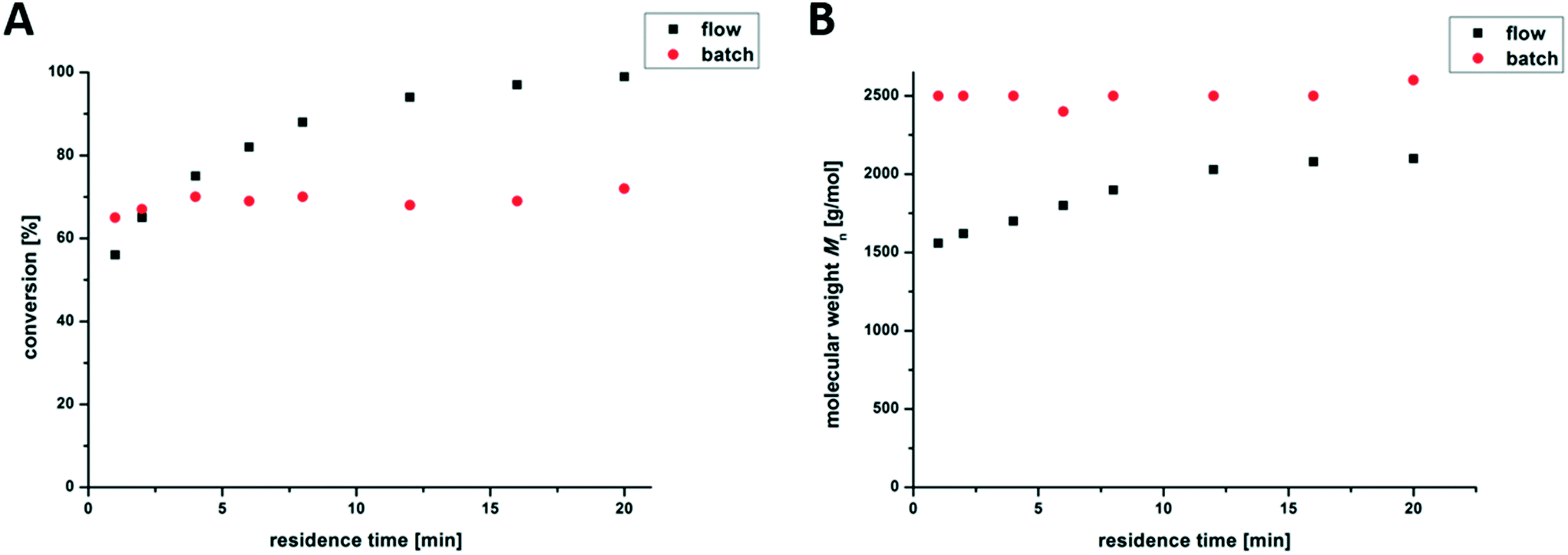 | ||
| Fig. 2 Time-dependent conversion of the monomer determined by NMR spectroscopy (A) and number weighted molecular weight (B) of branched PnBAs synthesized in a flow (flow rates between 2 mL min−1 and 0.1 mL min−1) (black) and a batch reactor (red). | ||
The second monomer used for polymerization was tert-butyl acrylate, which can be converted to poly(acrylic acid) for further investigation via simple hydrolysis. In order to reduce the overall viscosity, branched PtBA was synthesized in 25 wt% THF solution. In the flow reactor, a Back Pressure Regulator (BPR) with 40 psi was used to prevent evaporation of THF. The molecular weight of the produced branched PtBA in solution increases linearly with time (Fig. 3A) equally to the conversion. In comparison with the photo-batch reaction on a comparable scale, the synthesis of branched PtBA in the photo-flow set-up leads to equal Mn and higher conversion, respectively (Table 1). Higher conversion, on the other hand, presumably increases viscosity, which in turn results in broader molar mass distributions. In that regard, all branched polymers discussed here exhibited dispersities above 1.4, which is typical of randomly branched systems prepared under these conditions.45 These results indicate that photo-flow reactions are more efficient and economical than batch reactions considering the free radical polymerization of branched polyacrylates.
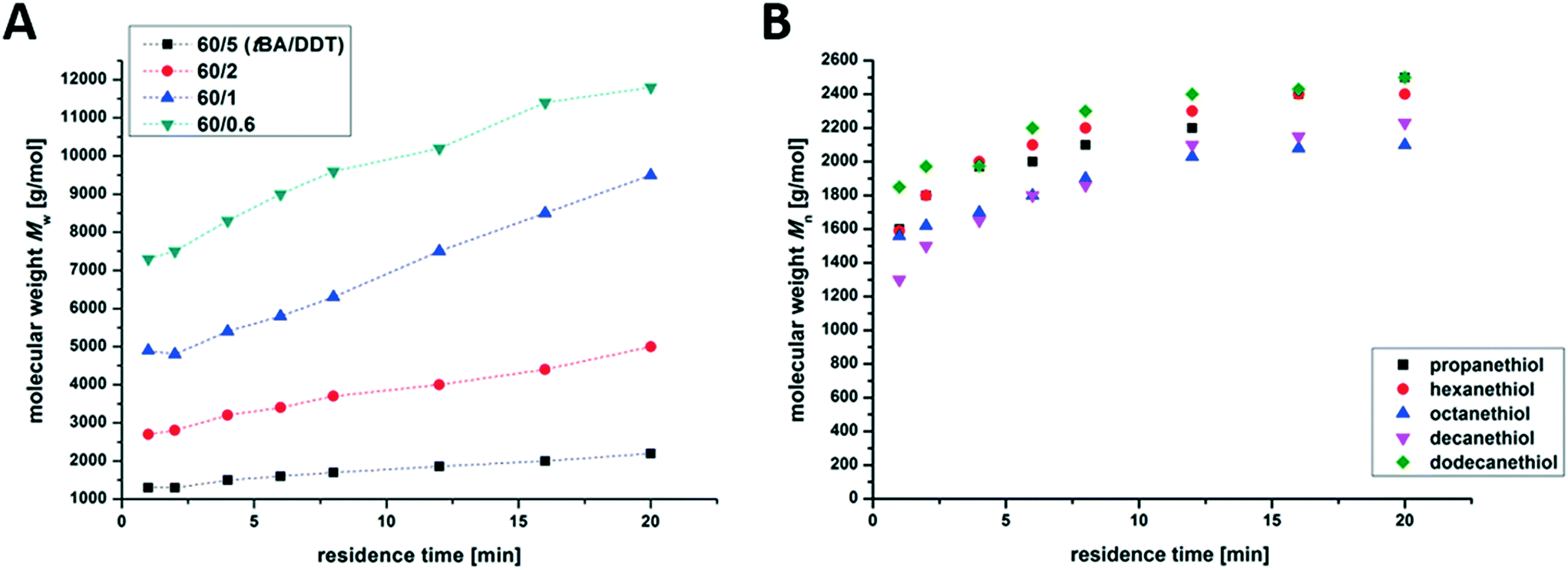 | ||
| Fig. 3 Time-dependent Mw-development of branched PtBA at different tBA to DDT molar feed ratios in a photo-flow reactor (A) and time-dependent Mn development of PnBAs with different thiols in a photo-flow reactor at flow rates between 2 mL min−1 and 0.1 mL min−1 (B). | ||
Robustness of the system
We also tested the influence of the thiol used. Herein, aliphatic thiols with different chain lengths (C3–C12) were added to the reaction mixture for the synthesis of branched PnBA with a monomer:thiol ratio of 60:5. Fig. 3B shows the evaluation of the molecular weight during the polymerization. As can be seen, both the molecular weight distribution and the overall conversion seem to be unaffected by the chosen thiol.
Further, the influence of the selected photo-initiator was analyzed. Different commercially available photo-initiators, Irgacure 819, DMPA, Irgacure 2959 and Benzoin (structural formulas are shown in Fig. S2†), were used to initiate the copolymerization of nBA and TPGDA in the presence of DDT (m/t-ratio = 60:5). Fig. 4 shows that there is no significant change in reaction kinetics, as can be seen via the comparable molecular weights and the evolution of the conversion during the reaction. All photo-initiators were fully decomposed within 5–10 min irradiation time.46 During irradiation, propagation is the dominating step, which should be the same for all the reactions discussed at this point (using the same m/t mixture).
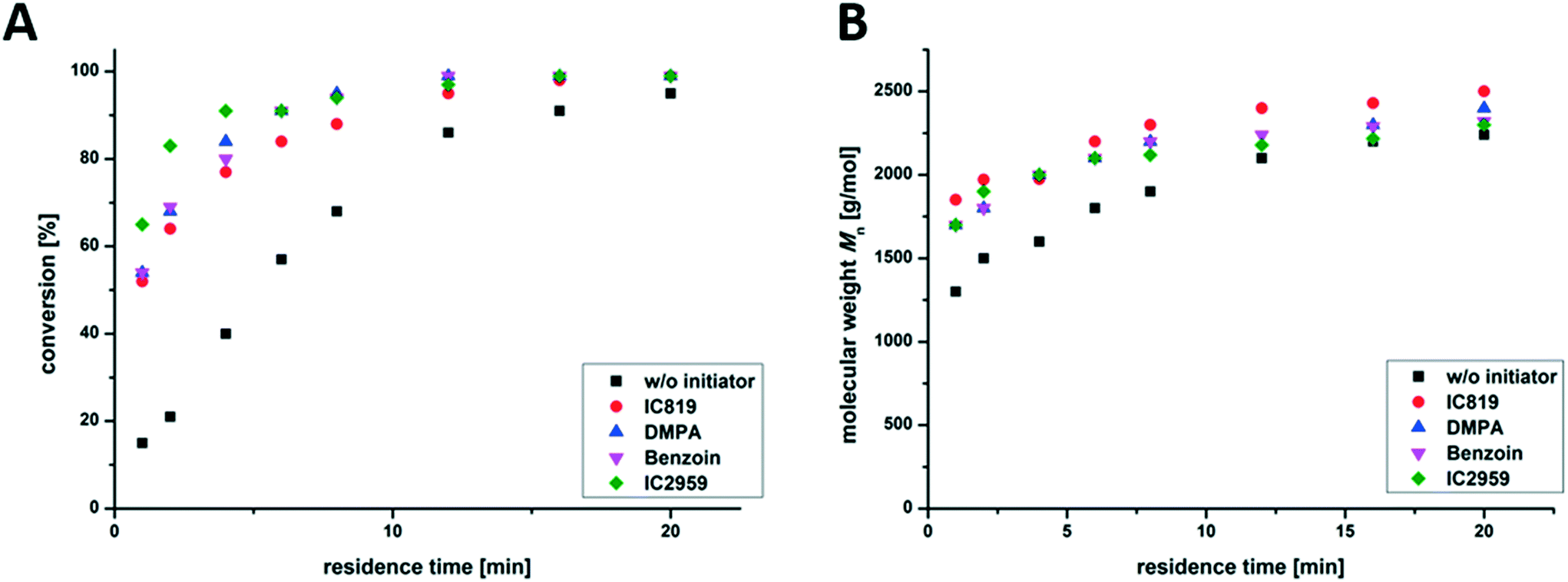 | ||
| Fig. 4 Time-dependent conversion of the monomer determined by NMR spectroscopy (A) and molecular weight (Mn) (B) of synthesized PnBAs with different initiators in a photo-flow reactor at flow rates between 2 mL min−1 and 0.1 mL min−1. | ||
An additional blank test without adding a photo-initiator also shows the formation of branched PnBAs, presumably by irradiation-induced autopolymerization, which leads to a conversion of 93% and comparable molecular weight and dispersity (Mn = 2200 g mol−1; Đ = 2.2) compared to samples containing a photo-initiator (e.g. Irgacure 819: Mn = 2500 g mol−1; Đ = 2.5) after 20 min of irradiation. This opens up the possibility of conducting initiator-free photo-induced synthesis of branched PnBAs in a flow reactor at the cost of slightly reduced reaction rates.
Upscaling
The main reason why flow reactors are often used in industry is their facile scalability. To increase the amount of synthesized material, longer tubes and/or larger tube diameters can be used. Thus, we further investigated the influence of different tube diameters on the formed branched PnBAs. An increase of tube diameter from 0.75 mm up to 2 mm leads to no significant difference in the molecular weight of the product (Fig. 5). We only observe a small increase of the reaction temperature by increasing the tube diameter (0.75 mm/65 °C; 2 mm/70 °C), which might explain the slightly higher conversion at the beginning of the reaction. At tube diameters above 2 mm, the laminar flow regime is left and turbulent flows occur. This has a substantial effect on the residence time distribution within the reactor and the irradiation time. Larger tube sizes strongly influence the amount of irradiated mixture, therefore resulting in higher initiator decomposition and increased reaction temperatures, e.g. 150 °C (4 mm) and 180 °C (6 mm). These effects also lead to broader molecular weight distributions.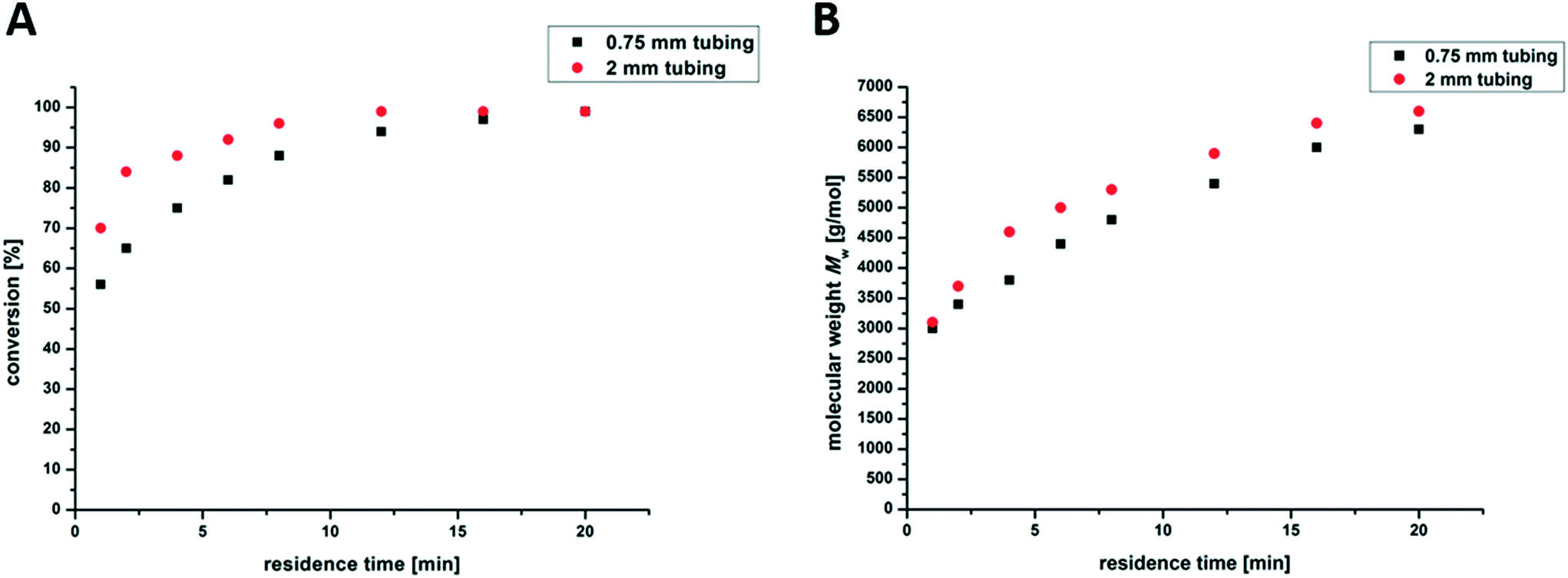 | ||
| Fig. 5 Kinetic studies of branched PnBAs synthesized in a flow reactor with two different tube diameters; conversion of the monomer determined by NMR spectroscopy (A) and molecular weight (B) at flow rates between 2 mL min−1 and 0.1 mL min−1. | ||
The most important property of a flow reactor set-up is the continuity of the obtained product. Therefore, 300 g of branched PnBA was synthesized during a 24 h continuous run test, in which the monomer mixture has a residence time of 20 min in the reactor. After different time intervals, samples were taken and analyzed by 1H-NMR spectroscopy and SEC to determine the conversion and the molecular weight distribution (Table 2). All the samples show identical parameters and therefore support the above drawn conclusions regarding the scalability of the herein described photo-flow polymerization set-up. Finally, 24 h of continuous run at lab-scale shows no considerable fouling, indicating the good stability of this system.
Conclusion
We successfully demonstrate the advantages of photo-flow chemistry by synthesizing branched poly(n-butyl acrylate)s in the bulk and poly(tert-butyl acrylate)s in THF solution with different molar masses up to 100000 g mol−1 and viscosities above 2500 mPa s. The use of a photo-flow reactor leads to higher conversions while operating under milder reaction conditions (low temperature evolution), and at the same time comparable molecular weight, compared to a batch reactor.
Further, the influence of the chain length (C3–C12) of the thiol added and the photo-initiators used was analyzed. Our results so far show no effect on the obtained branched polyacrylates regarding conversion, molar mass distribution, or overall molecular weight, confirming the robustness of the herein described system. The scalability of such a flow reactor set-up was also shown by increasing the used tube diameter from 0.75 mm up to 2 mm. Above 2 mm, the laminar flow turns into a turbulent flow, which changes the residence time of the monomer mixture inside the reactor, leading to inhomogeneity, broader dispersities, and high temperature evolution during the polymerization. Finally, the 24 h continuous run stability test produces more than 300 g b-PnBA with constant material quality.
Acknowledgements
O. E. acknowledges the Federal Ministry for Economic Affairs and Energy (BMWi) and the “Arbeitsgemeinschaft industrieller Forschungsvereinigung” (AiF) for financial support. T. J. is grateful for funding from the Fonds Wetenschappelijk Onderzoek (FWO) in the form of a project grant.References
- P. J. Flory, J. Am. Chem. Soc., 1952, 74, 2718–2723 CrossRef CAS.
- Y. H. Kim and O. W. Webster, J. Am. Chem. Soc., 1990, 112, 4592–4593 CrossRef CAS.
- Y. Zheng, S. Li, Z. Weng and C. Gao, Chem. Soc. Rev., 2015, 44, 4091–4130 RSC.
- S. Muthukrishnan, M. Nitschke, S. Gramm, Z. Ozyurek, B. Voit, C. Werner and A. H. Müller, Macromol. Biosci., 2006, 6, 658–666 CrossRef CAS PubMed.
- Y. H. Kim and O. W. Webster, Macromolecules, 1992, 25, 5561–5572 CrossRef CAS.
- B. I. Voit, Comp. Rev. Chim., 2003, 6, 821–832 CrossRef CAS.
- A. T. Slark, D. C. Sherrington, A. Titterton and I. K. Martin, J. Mater. Chem., 2003, 13, 2711–2720 RSC.
- B. Voit, J. Polym. Sci., Part A: Polym. Chem., 2000, 38, 2505–2525 CrossRef CAS.
- T. Emrick, H. Chang and J. M. J. Frechet, Macromolecules, 1999, 32, 6380–6382 CrossRef CAS.
- D. Wang, H. Chen, Y. Su, F. Qiu, L. Zhu, X. Huan, B. Zhu, D. Yan, F. Guo and X. Zhu, Polym. Chem., 2013, 4, 85–94 RSC.
- O. Boussif, F. Lezoualch, M. A. Zanta, M. D. Mergny, D. Scherman, B. Demeneix and J. Behr, Proc. Natl. Acad. Sci. U. S. A., 1995, 92, 7297–7301 CrossRef CAS.
- L. Cosimbescu, J. W. Robinson and P. Bhattacharya, US2016/0046885, 2016.
- J. Liu, L. Yu, C. Zhong, R. He, W. Yang, H. Wu and Y. Cao, RSC Adv., 2012, 2, 689–696 RSC.
- R. Spindler and J. M. J. Frechet, Macromolecules, 1993, 26, 4809–4813 CrossRef CAS.
- H. R. Kricheldorf, Q. Z. Zang and G. Schwarz, Polymer, 1982, 23, 1821–1829 CrossRef CAS.
- H. Magnusson, E. Malmström and A. Hult, Macromol. Rapid Commun., 1999, 20, 453–457 CrossRef CAS.
- J. M. J. Frechet, M. Henmi, I. Gitsov, S. Aoshima, M. R. Leduc and R. B. Grubbs, Science, 1995, 269, 1080–1083 CrossRef CAS PubMed.
- C. J. Hawker, J. M. J. Frechet, R. B. Grubbs and J. Dao, J. Am. Chem. Soc., 1995, 117, 10763–10764 CrossRef CAS.
- G. S. Gaynor, S. Edelman and K. Matyjaszewski, Macromolecules, 1996, 29, 1079–1081 CrossRef.
- K. Ishizu, Y. Ohta and S. Kawauchi, Macromolecules, 2002, 35, 3781–3784 CrossRef CAS.
- P. F. W. Simon, W. Radke and A. H. E. Müller, Macromol. Rapid Commun., 1997, 18, 865–873 CrossRef CAS.
- N. O'Brien, A. McKee, D. C. Sherrington, A. T. Slark and A. Titterton, Polymer, 2000, 41, 6027–6031 CrossRef.
- S. Durie, K. Jerabek, C. Mason and D. C. Sherrington, Macromolecules, 2002, 35, 9665–9672 CrossRef CAS.
- F. Isaure, P. A. G. Cormack and D. C. Sherrington, J. Mater. Chem., 2003, 13, 2701–2710 RSC.
- R. Baudry and D. C. Sherrington, Macromolecules, 2006, 39, 5230–5237 CrossRef CAS.
- S. Graham, P. A. G. Cormack and D. C. Sherrington, Macromolecules, 2005, 38, 86–90 CrossRef CAS.
- F. Isaure, P. A. G. Cormack and D. C. Sherrington, React. Funct. Polym., 2006, 66, 65–79 CrossRef CAS.
- L. D. Elliott, J. P. Knowles, P. J. Koovits, K. G. Maskill, M. J. Ralph, G. Lejeune, L. J. Edwards, R. I. Robinson, I. R. Clemens, B. Cox, D. D. Pascoe, G. Koch, M. Eberle, M. B. Berry and K. I. Booker-Milburn, Chemistry, 2014, 20, 15226–15232 CrossRef CAS PubMed.
- C. Tonhauser, A. Natalello, H. Löwe and H. Frey, Macromolecules, 2012, 45, 9551–9570 CrossRef CAS.
- J. I. Park, A. Saffari, S. Kumar, A. Günther and E. Kumacheva, Annu. Rev. Mater. Res., 2010, 40, 415–443 CrossRef CAS.
- E. Baeten, B. Verbraeken, R. Hoogenboom and T. Junkers, Chem. Commun., 2015, 51, 11701–11704 RSC.
- A. Nagaki, Y. Takahashi, K. Akahori and J.-i. Yoshida, Macromol. React. Eng., 2012, 6, 467–472 CrossRef CAS.
- E. Baeten, S. Vanslambrouck, C. Jerome, P. Lecomte and T. Junkers, Eur. Polym. J., 2016, 80, 208–218 CrossRef CAS.
- J. Vandenbergh, T. de Moraes Ogawa and T. Junkers, J. Polym. Sci., Part A: Polym. Chem., 2013, 51, 2366–2374 CrossRef CAS.
- T. Noda, A. J. Grice, M. E. Levere and D. M. Haddleton, Eur. Polym. J., 2007, 43, 2321–2330 CrossRef CAS.
- X. Pan, M. A. Tasdelen, J. Laun, T. Junkers, Y. Yagci and K. Matyjaszewski, Prog. Polym. Sci., 2016, 62, 73–125 CrossRef CAS.
- B. Wenn, A. C. Martens, Y. M. Chuang, J. Gruber and T. Junkers, Polym. Chem., 2016, 7, 2720–2727 RSC.
- T. Junkers and B. Wenn, React. Chem. Eng., 2016, 1, 60–64 CAS.
- E. Prenzel and N. Goetz, Angew. Makromol. Chem., 1990, 178, 191–200 CrossRef.
- B. Dervaux, T. Junkers, M. Schneider-Baumann, F. E. Du Prez and C. Barner-Kowollik, J. Polym. Sci., Part A: Polym. Chem., 2009, 47, 6641–6654 CrossRef CAS.
- P. A. Costello, I. K. Martin, A. T. Slark, D. C. Sherrington and A. Titterton, Polymer, 2002, 43, 245–254 CrossRef CAS.
- J. P. Fouassier, X. Allonas and D. Burget, Prog. Org. Coat., 2003, 47, 16–36 CrossRef CAS.
- P. Besenius, S. Slavin, F. Vilela and D. C. Sherrington, React. Funct. Polym., 2008, 68, 1524–1533 CrossRef CAS.
- S. Railian, B. Wenn and T. Junkers, J. Flow Chem., 2016, 6, 260–267 CrossRef.
- M. Chisholm, N. Hudson, N. Kirtley, F. Vilela and D. C. Sherrington, Macromolecules, 2009, 42, 7745–7752 CrossRef CAS.
- B. Wenn and T. Junkers, Macromolecules, 2016, 49, 6888–6895 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c7re00013h |
| This journal is © The Royal Society of Chemistry 2017 |