
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Efficient interfacial interaction for improving mechanical properties of polydimethylsiloxane nanocomposites filled with low content of graphene oxide nanoribbons†
Neng-Jian
Huang
a,
Jing
Zang
a,
Guo-Dong
Zhang
*a,
Li-Zhi
Guan
a,
Shi-Neng
Li
ab,
Li
Zhao
a and
Long-Cheng
Tang
 *a
*a
aKey Laboratory of Organosilicon Chemistry and Material Technology of Ministry of Education, Hangzhou Normal University, Hangzhou 310012, PR China. E-mail: zhangguodong@hznu.edu.cn; lctang@hznu.edu.cn
bInstitute for Advanced Ceramics, State Key Laboratory of Urban Water Resource and Environment, Harbin Institute of Technology, Harbin 150001, PR China
First published on 19th April 2017
Abstract
In this work, hydroxyl-terminated polydimethylsiloxane (H-t-PDMS) nanocomposites reinforced by different contents of graphene oxide nanoribbons (GONRs) were prepared via a facile solvent-free process, and the mechanical properties of the H-t-PDMS/GONR nanocomposites were investigated and compared with the corresponding nanocomposites containing pristine carbon nanotubes (CNTs) or functionalized CNTs (f-CNTs). It was found that the GONRs with abundant functional groups showed good compatibility with the H-t-PDMS matrix at appropriate content: both good dispersion levels of the GONR sheets and strong GONR/matrix interfacial interactions were achieved at low filler content (≤0.5 wt%), although the GONR sheets showed obvious clusters in the matrix at relatively high content. Mechanical testing indicated that the incorporation of a low content of GONRs into the H-t-PDMS polymer resulted in significant improvements in both the tensile and tear strengths, e.g. about 158 and 284% at 0.5 wt% GONRs, respectively; and such reinforcement efficiency of the GONRs in the PDMS nanocomposites was much better than those of the corresponding CNTs or f-CNTs, even superior to those of other carbon nanofillers in previous PDMS-based nanocomposite systems. Based on the morphology and fracture surface analysis, the possible reinforcing mechanisms were discussed and clarified to understand the discrepancies in the mechanical properties of the nanocomposite systems studied.
1. Introduction
Polydimethylsiloxane (PDMS) polymer is a well-known elastomer with versatile and desirable properties, showing wide application in various fields, e.g. aerospace, automobile, building, and electric industries.1 Its performance is superior to ordinary organic rubbers due to excellent chemical stability, high thermal resistance, outstanding insulating properties, low toxicity,2 and very low glass transition temperature (Tg), as well as unique linear elasticity over a broad range of temperatures (−40 to 400 °C) and strains.3 However, the intermolecular forces among PDMS polymer chains are relatively weak since Si–O bond exhibits larger bond length than C–C bond.4 Hence, PDMS polymer usually shows a low mechanical strength (0.1–0.4 MPa), which limits its mechanical component used in practical applications.To overcome the above shortages, numerous attempts such as mixing PDMS polymers with a second phase of superior nanofillers (e.g. nano-silica,5 carbon black,6 montmorillonite,7 polyhedral oligomeric silsesquioxanes (POSS),8 and their combination9), have been made to improve the stiffness and strength and even enhance the thermal properties. Typically, silica nanoparticles have been widely used to enhance the mechanical performance of PDMS-based nanocomposite materials;5 and high content of silica nanoparticles is normally required to be incorporated into PDMS polymer and thus obtain an ideal enhancement in tensile strength, e.g. from ∼0.48 MPa for pure PDMS to ∼1.10 MPa (∼129% increase) for PDMS nanocomposites containing 25 wt% fumed silica nanoparticles.10 While such a high content of nanofillers usually increases the viscosity of the PDMS nanocomposites and thus produces some drawbacks such as time- and energy-consumption and loss of control on the particle dispersion degree that results into serious filler clusters,11 thus inevitably inducing the difficultly in processing of the PDMS nanocomposites. Therefore, mechanical reinforcement in PDMS-based nanocomposites with low filler content is highly desired and required for developing lightweight and flexible materials in practical application.
Carbon nanofillers including carbon nanotubes (CNTs), graphite, graphene sheets and their derivatives have recently received considerable interest for improving the mechanical and functional properties of various polymers due to their unique structure and intriguing physical performance.12–23 Among these carbon nanofillers, graphene nanoribbons (GNRs) integrate the structures and properties of 1D CNTs and 2D graphene sheets,24,25 and exhibit excellent chemical/physical properties.26–28 Especially, graphene oxide nanoribbons (GONRs) are narrow strips of graphene oxide (GO) sheets with high length-to-width ratio (usually more than 10, similar to CNT) and high specific surface area (∼511 m2 g−1),29 which can be fabricated by longitudinal unzipping CNTs or chemical vapor deposition method.27,30–32 Moreover, many functional groups e.g. hydroxyl, carboxyl and epoxy groups can be created during the unzipping process,33 making them ideal fillers for polymer-based nanocomposites. Previous work has demonstrated that addition of the GONR sheets or their derivatives not only produced good reinforcement efficiency (enhancement% per filler content%) on epoxy polymer,33 but also enhanced the thermal and tribological performance of epoxy,34 even provided other functionalities such as electrical conductivity of polyimide fiber.35,36
During the past several years, carbon nanofillers were also reported as effective reinforcements for PDMS polymer,6,37–40 even endowing it with ideal functionalizes such as thermal conductivity.41 For example, the combined use of solution mixing (hexane as dispersing solvent) and ball-mill procedure provided produced improved tensile strength (∼113% at 3.0 wt% graphene) of methyl vinyl silicone rubber (MVSR) polymer,40 although the relatively good dispersion levels of graphene sheets were obtained in the matrix. Recently, GONRs unzipping the multi walled carbon nanotubes (MWCNTs) were synthesized to reinforce the MVSR using a tetrahydrofuran-based solution mixing approach; and the results revealed that addition of GONR sheets was found to significantly enhance thermal stability and mechanical properties, and at 2.0 wt% GONR content the tensile strength and elastic modulus of MVSR increased by 67% and 93%, respectively.39 However, in these above PDMS nanocomposites, the sheet/matrix interfacial interactions are still very poor due to their incompatible functional groups (e.g. methyl vinyl groups of MVSR vs. hydroxyl and carboxyl groups of GONRs), which greatly limit the capacity for the enhanced mechanical performance of the nanocomposite.42,43 Moreover, although the solution mixing method proves to be efficacious in dispersing the graphene and its derivatives, they still showed some deficiency and limitation for practical applications since they involve in liquid solvent phases are harmful to environment and health.
In this work, we synthesized GONR sheets by longitudinal unzipping MWCNTs to reinforce the PDMS polymer at low filler content via a facile and environmentally friendly solvent-free process. To obtain the good compatibility of GONRs in PDMS matrix, hydroxyl-terminated PDMS (H-t-PDMS) polymer was used as matrix since the hydroxyl end-groups of H-t-PDMS polymers are expected to react with the hydroxyl and carboxyl groups on the GONR surface during the cross-linking process. As expected, efficient GONR/matrix interfacial interactions in the H-t-PDMS nanocomposites were obtained at low filler content. The mechanical properties of the nanocomposites were measured and compared with those of the corresponding nanocomposites containing pristine or functionalized CNT (f-CNT). The results demonstrated that the reinforcement efficiency of GONR in the H-t-PDMS polymer is much higher than those of CNTs, f-CNTs and carbon nanofillers in the PDMS-based nanocomposites. Finally, the possible reinforcing mechanisms were discussed based on the microscopic structures and morphologies of the three carbon nanofillers and their nanocomposites studied.
2. Experimental
2.1. Materials
Commercially available MWCNTs (50 μm in length and 20–30 nm in outer diameters, Timestub TM) were supplied by Chengdu Organic Chemicals Co., Ltd., China. H-t-PDMS polymer was purchased from Zhejiang Xin'an Chemicals Co., Ltd., China. Various acids including concentrated sulfuric acid, hydrochloric acid and concentrated nitric acid were provided from Beijing Chemical Factory. Potassium permanganate was supplied by Shanghai Lingfeng Chemical Reagent Co., Ltd., China. Hydrogen peroxide (H2O2, 30%) and tetraethylorthosilicate (TEOS) were supplied by Sinopharm Chemical Reagent Co., Ltd., China (SCRC). Dibutyltindilaurate (DBTDL) as catalyst was purchased from Alfa Aesar. All of the chemicals and solvents were of analytical grade and used as received.2.2. Preparation of f-CNT and GONR
The f-CNTs were prepared according to the previous work.44 Typically, 3 g MWCNTs were added into a mixture of concentrated H2SO4/HNO3 (675![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :225 mL) with stirring and sonication for 0.5 h. Then the solution was refluxed for 6 h at 80 °C, and kept over night after being added into a mass of ice-deionized water. The mixture was washed with deionized water (3–5 times) and dialyzed in deionized water for several days until it become neutral pH solution.
:225 mL) with stirring and sonication for 0.5 h. Then the solution was refluxed for 6 h at 80 °C, and kept over night after being added into a mass of ice-deionized water. The mixture was washed with deionized water (3–5 times) and dialyzed in deionized water for several days until it become neutral pH solution.
GONRs were synthesized by longitudinal unzipping of MWCNTs, as reported by James Tour and co-workers.27 Typically, 1 g MWCNTs were dispersed in 200 ml concentrated H2SO4 and sonicated for 0.5 h, then stirred at room temperature for 1.0 h. 5 g KMnO4 was slowly added to the dispersion and stirred at room temperature for 1.0 h. After sonicated for 0.5 h, the solution was stirred for another 1.0 h. The mixture was heated to 70 °C for 1.0 h with stirring and quenched into a mass of ice distilled water with excessive hydrogen peroxide (H2O2, 30%), and kept overnight. The dispersion was centrifuged and washed for several times with 5% aqueous hydrochloric acid (HCl) to remove metal ions, then washed with distilled water to remove acid and dialyzed in deionized water for several days to neutral or weak acidic state.
2.3. Fabrication of H-t-PDMS-based nanocomposites
For preparation of H-t-PDMS/GONR nanocomposite samples (Fig. S1†), GONR/water suspension from dialysis was firstly sonicated for 0.5 h and directly dispersed in H-t-PDMS polymer without using any solvents. The mixture was then stirred by mechanical mixing (800 rpm) at 80 °C for ∼6.0 h to evaporate the water. After adding TEOS and DBTDL, the blend was put in a vacuum oven for 0.5 h to degas, followed by curing in a mould at room temperature for 7 d. The filler content of GONR in H-t-PDMS nanocomposites varied from 0 to 1.0 wt%. For comparison, all the pure H-t-PDMS, H-t-PDMS/CNT and H-t-PDMS/f-CNT nanocomposites were also fabricated according to the same procedure for the H-t-PDMS/GONR nanocomposites.2.4. Characterizations
Fourier transform infrared spectroscopy (FTIR) was conducted using the KBr pellet technique (Bruker Alpha-T) to detect the various bond and groups in the fillers, recorded in the scan ranging from 4000 to 400 cm−1. The structures and morphologies of CNTs, f-CNTs and GONRs were observed by both the transmission electron microscopy (TEM, HITACHI H-7650) and scanning electron microscopy (SEM, Zeiss ultra plus; and Nova Nano SEM450, FEI). All the samples were collected on copper grids (200 mesh). And, the dispersion of different content of GONRs in H-t-PDMS was observed by transmission optical microscopy (TOM, Nikon Eclipse LV100 POL). X-ray diffraction (XRD) measurements were conducted using D/Max2550V X-ray diffractor (Rigaku, Japan) and scanned in the range 2θ from 5° to 40° at a scanning rate of 2° min−1. Raman spectra were recorded with SENTERRA Micro Raman Spectroscopy (Bruker Instrument, Germany). Thermo gravimetric analysis (TGA) of CNTs, f-CNTs and GONRs were conducted by using TA Q500 Instruments from room temperature to 800 °C with a heating rate of 10 °C min−1 in a nitrogen atmosphere.The mechanical properties of the nanocomposites were measured by a universal testing machine (Ametek Ls100plus) at room temperature. According to GB/T 528-2009, tensile tests used standard dumbbell-shaped specimens (115 mm long × 25 mm width × 2 mm thick) at a crosshead speed of 500 mm min−1. For tear tests, the un-nicked 90° angle specimens (100 mm × 19 mm width × 2 mm thick, according to GB/T 529-2008) were stretched at a crosshead speed of 500 mm min−1. At least five samples were measured to obtain average values. The fracture surface of the samples after tensile test was characterized by sputtering gold of 20 s and observed SEM.
3. Results and discussion
3.1. Morphology and structural characterization
Fig. 1a shows the schematic fabrication process and representative SEM images of GONR and H-t-PDMS/GONR nanocomposites. GONR was synthesized by longitudinal unzipping MWCNTs and then filled into the H-t-PDMS polymer by the facile fabricating process (Fig. S1†). As expected, compared to the pristine MWCNTs with large aspect ratio and smooth surface along their axis (Fig. 1b), the GONR sheets show slightly rougher surface with wider ribbon structure (Fig. 1c), which is almost similar to the structure of GO sheet.45 After incorporation of GONRs into the H-t-PDMS polymer, these GONR sheets present well-embedded state and some of them are even curly in the H-t-PDMS matrix (see the white arrows in Fig. 1d), suggesting the efficient interfacial interactions obtained between the sheet and the matrix.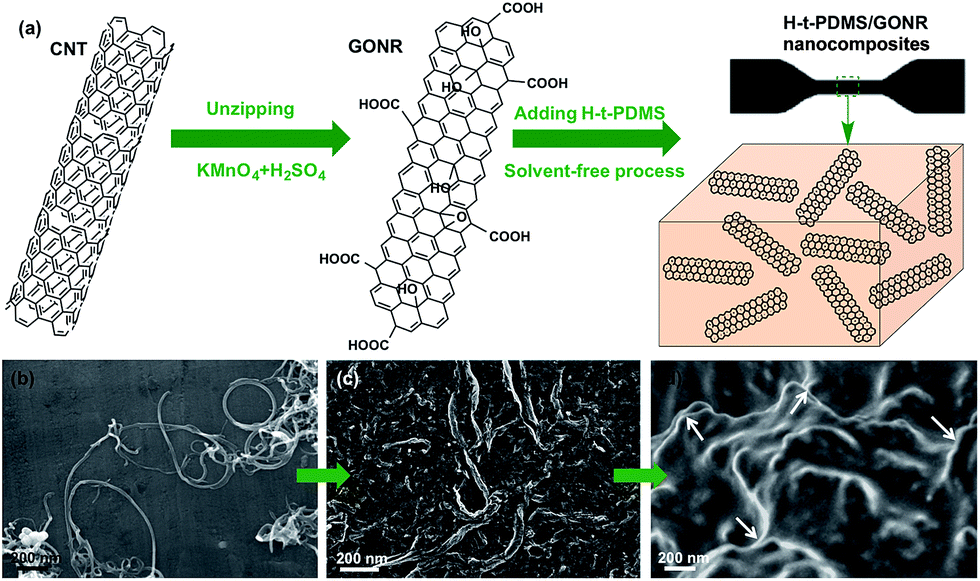 | ||
| Fig. 1 (a) Schematic of preparation of GONR sheets and H-t-PDMS/GONR nanocomposites and their high-magnification SEM images: (b) CNTs, (c) GONRs and (d) H-t-PDMS nanocomposites containing 0.25 wt% GONRs. | ||
Dispersion stability and TEM images of various fillers can offer more information about morphology and structural differences. Due to the chemical inactivity and nonpolarity, pristine CNTs are bundled and entangled and tend to aggregated (Fig. 2a and b), thus leading to the unstable dispersion in the aqueous solution (Fig. S2†). An enlarged image in Fig. 2c reveals that the diameter of pristine CNT is 30–50 nm with a hollow structure and many inner nanotube layers (Fig. 2d). Comparatively, the digital images in Fig. 2e and S2† show the GONR/water solution after sonication for 10 min to yield good dispersion and excellent stability, or even after 17 d of standing at ambient temperature, implying that CNTs would be unzipped to form GONRs with abundant hydrophilic groups. TEM analysis further shows that the GONRs remain long (∼5 μm in Fig. 2f) when not cut by the sonication; they display almost uniform width and rough structure, and the width of GONRs increased to 120–150 nm after oxidation (Fig. 2g). Notably, some of these GONRs can be well exfoliated after sonication, and some rough surfaces with a few wrinkled structures and defects can be seen in Fig. 2h, which would be attributed to the extremely thin thickness and the isolated oxygen reaction sites during the oxidation process. Moreover, the f-CNTs display similar structure and feature as the pristine CNTs (Fig. S3†), which are quite different from those of GONRs (Fig. 2g),46 although the dispersion stability of f-CNTs in water is also greatly improved after surface modification (Fig. S2†).
 | ||
| Fig. 2 Dispersion stability in water and typical TEM images of (a–d) pristine CNTs with smooth surface, and (e–h) as-prepared GONRs, showing the wrinkled structures. | ||
The surface chemical compositions and their changes of CNTs, f-CNTs and GONRs were further analyzed and verified by the FTIR, TGA, XRD and Raman spectra. Fig. 3a shows a comparison of FTIR spectra of various fillers. It can be observed that the GONRs show the appearance of the absorption bands assigned to the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O stretching vibrations at ∼1720 cm−1, and the intensity changes of CC and C–H peaks (at ∼1380 cm−1, ∼2970 and ∼2870 cm−1) imply a number increase of carboxyl and hydroxyl groups. TGA shows a significant enhancement in the total weight loss (6, 28 and 57% for CNTs, f-CNTs and GONRs, respectively) with increasing exposure of strong oxidants (Fig. 3b), suggesting a number increase of volatile side-wall functionalities, which confirms the higher degree of oxidation for GONRs.27
O stretching vibrations at ∼1720 cm−1, and the intensity changes of CC and C–H peaks (at ∼1380 cm−1, ∼2970 and ∼2870 cm−1) imply a number increase of carboxyl and hydroxyl groups. TGA shows a significant enhancement in the total weight loss (6, 28 and 57% for CNTs, f-CNTs and GONRs, respectively) with increasing exposure of strong oxidants (Fig. 3b), suggesting a number increase of volatile side-wall functionalities, which confirms the higher degree of oxidation for GONRs.27
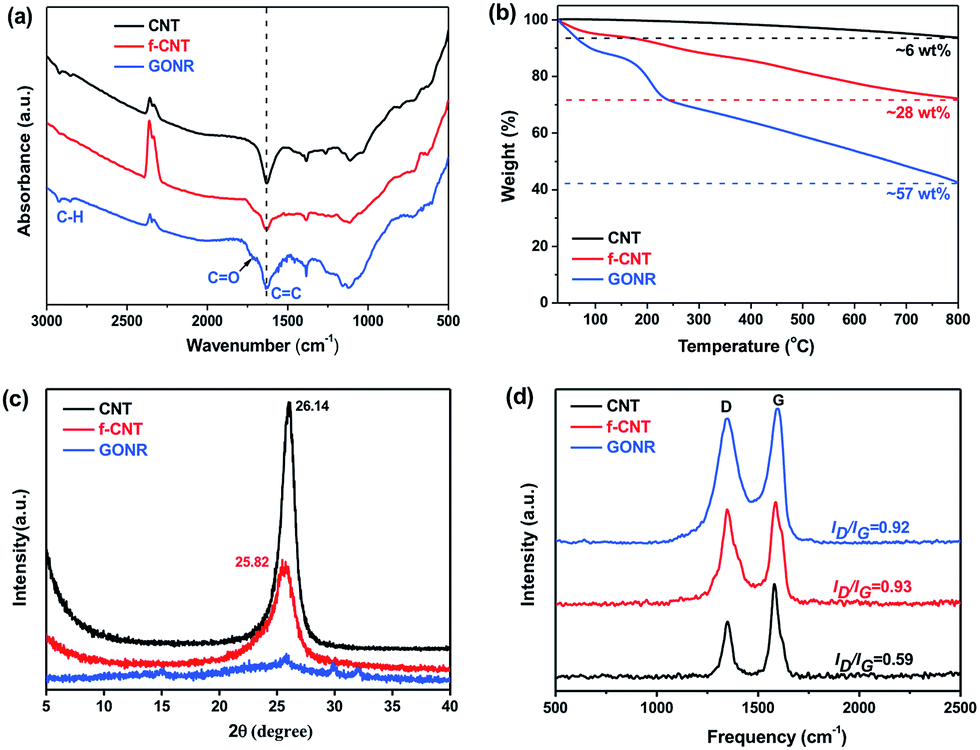 | ||
| Fig. 3 Structural characterizations of pristine CNTs, f-CNTs and GONRs: (a) FTIR spectra, (b) TGA curves, (c) XRD results and (d) Raman spectra. | ||
XRD analysis was also performed to further investigate the structure change of CNTs, f-CNTs and GONRs. Typically, both the pristine CNTs and f-CNTs have characteristic diffraction peaks at 2θ values of ∼26° (graphite (002) reflection peak,47Fig. 3c), corresponding to a d spacing of 0.34 nm. While the GONRs have no visible and obvious peak, suggesting that they stack together in a loose manner (see Fig. 2h). This indicates the larger d spacing than that typical bulk graphite structure of both CNTs and f-CNTs due to considerable functional groups on the GONR surfaces coming from the strong oxidation process. The sp2 or sp3 bonded crystalline domains would be greatly reduced during the oxidation process, thus producing the loose GONR structures, which should result in the broadening XRD peak, and similar phenomenon was also found in XRD spectra of GO sheets.48 Moreover, Raman spectroscopy shown in Fig. 3d exhibits an increased level of disorder (intensity of a D band at 1320–1330 cm−1) with increasing the degree of oxidation, and the increased intensity ratio of the D/G band (ID/IG, 0.59, 0.93 and 0.92 for CNTs, f-CNTs and GONRs, respectively) reveals the presence of abundant defects in both f-CNTs and GONRs, consistent with FTIR and TGA results.
3.2. Microstructure in the H-t-PDMS/GONR nanocomposites
Normally, nanofillers with high specific surface area tend to attract one another to form the clusters in polymer matrix due to the complicated van der Waals forces attractions.49,50 Our previous work demonstrated that graphene derivatives at relatively high content (>0.5 wt%) caused serious agglomerate phenomenon in epoxy resin.43,51 In order to evaluate the microstructures of GONRs in the H-t-PDMS matrix, both TOM and SEM images of the H-t-PDMS/GONR nanocomposites were carried out and shown in Fig. 4. Compared to pure transparent H-t-PDMS polymer, the H-t-PDMS/GONR nanocomposites turn into light grey at a low filler content of 0.1 wt%, and gradually transform into dark grey until black with further increasing the GONR content (Fig. 4a). Fig. 4b–g present the TOM images of the dispersion state of H-t-PDMS/GONR nanocomposites containing different filler contents of 0.1, 0.5 and 1.0 wt%. Noted that the liquid H-t-PDMS/GONR nanocomposite sample was put in a glass vessel with a fixed thickness of ∼0.2 mm. The GONRs can be highly dispersed in the H-t-PDMS polymer at 0.1 wt% (Fig. 4b and e); and with increasing the filler content the dispersion level of GONRs in the H-t-PDMS polymer shows almost unchanged at 0.5 wt% (Fig. 4c and f), although several GONR clusters can be observed in the matrix (see the white arrows). With further increasing the GONR content to ∼1.0 wt%, the dispersion level of GONRs shows obvious change: many black dots (GONR rich zone) can be seen and the cluster size indeed becomes larger (see Fig. 4d and g), which is likely due to the possibility of the complicated interactions between the adjacent GONR sheets; this phenomenon is very similar to those of CNTs, graphene sheets or their derivatives in other polymer nanocomposite systems.52–54 | ||
| Fig. 4 (a) Digital photograph of pure H-t-PDMS and H-t-PDMS/GONR nanocomposites at different filler content, and TOM images of the H-t-PDMS/GONR nanocomposites at different magnification: (b and e) 0.1 wt%, (c and f) 0.5 wt% and (d and g) 1.0 wt%. | ||
To further evaluate the microstructures in the H-t-PDMS/GONR nanocomposites, tensile fracture surfaces of the nanocomposite samples were observed with SEM. Normally, compared to the relatively smooth fracture surface of pure H-t-PDMS polymer with low crack resistance (Fig. S4†), the H-t-PDMS/GONR nanocomposites show much rougher fracture surfaces at different magnification (Fig. 5). As shown in Fig. 5a, the GONRs in H-t-PDMS polymer at 0.5 wt% content exhibit relatively good dispersion levels, although the high magnification of SEM image in Fig. 5b reveals the existence of small GONR clusters in the matrix, consistent with the TOM result (Fig. 4c). Many tortuous river-like structures with ribbons and hackles induced by the dispersed GONRs are dominant in the surface, which would restrict the micro-crack propagation in the nanocomposites (Fig. 1d). Moreover, the GONR/matrix interfacial quality may be relatively strong and the GONR sheets are well-embedded in the H-t-PDMS polymer in Fig. 5c. The strong interfacial bonding could be ascribed to the possible condensation between the hydroxyl and carboxyl groups on the GONRs and the hydroxyl groups of H-t-PDMS.54 However, at 1.0 wt% GONR content, the fracture surface of the nanocomposite exhibits different fracture morphologies and features. Some pseudo-spherical GONR aggregates with ∼5 μm can be visible on the surface, as indicated by the black arrows in Fig. 5d; and such GONR aggregate can debond from the matrix (see Fig. 5e), suggesting the weak interface between the matrix and the aggregate at high GONR content (see the obvious gap in Fig. 5f). Although it is difficult to well understand the reason why caused these GONR clusters in the H-t-PDMS matrix at this stage, the abundant functional groups on GONR surface that could tend to induce complicated sheet/sheet interactions such as hydrogen bonding,55 could be an important role in inducing the formation of the GONR clusters in the matrix. In fact, similar GO clusters and the GO/matrix debonding were also observed in the previous epoxy/GO nanocomposites.45 Hence, the complicated interactions between sheet/matrix and sheet/sheet in the H-t-PDMS matrix determine the above microstructures at different GONR contents, and these phenomena would result in different impacts on the mechanical performance of the H-t-PDMS nanocomposites, which will be discussed in the following section.
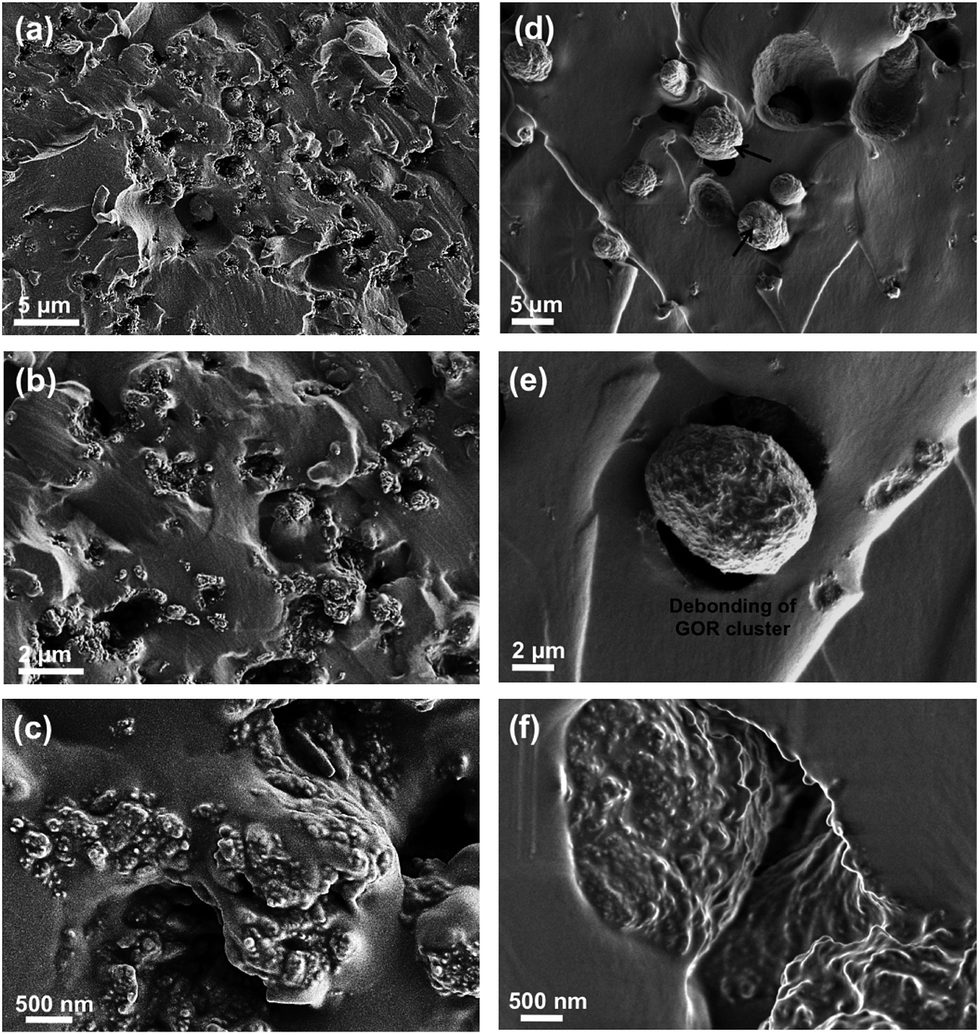 | ||
| Fig. 5 SEM images of tensile fracture surface at different magnification of H-t-PDMS/GONR nanocomposites with (a–c) 0.5 wt% and (d–f) 1.0 wt% GONRs. | ||
3.3. Mechanical properties
The mechanical properties of H-t-PDMS/GONR nanocomposites were measured and shown in Fig. 6 and Table S1.† As shown in Fig. 6a, the typical tensile stress–strain curves show the increased elongation at break after addition of GONRs, and obtains the maximum value at 0.5 wt%, and further decreases with the GONR content from 0.5 wt% to 1.0 wt%. The presence of GONR provides a significant enhancement of the area under the curves that indicates the enhanced ductility, thus producing H-t-PDMS with a flexible cross-linking network.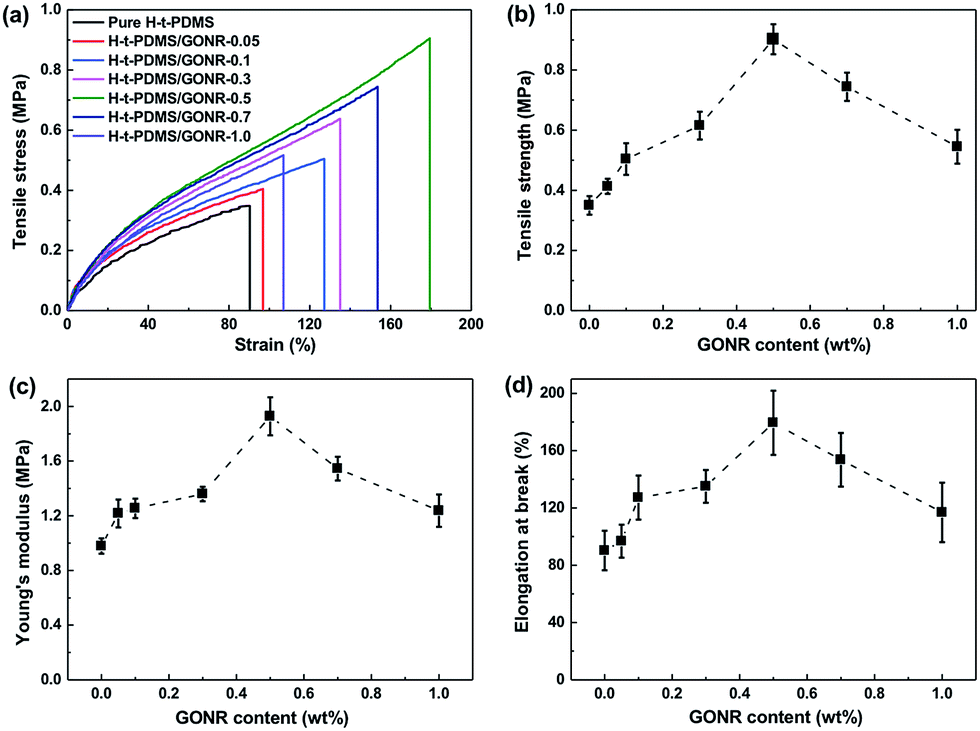 | ||
| Fig. 6 Mechanical properties of pure H-t-PDMS and H-t-PDMS/GONR composites as a function of filler content: (a) typical tensile stress–strain curves, (b) tensile strength, (c) Young's modulus and (d) elongation at break. | ||
Fig. 6b–d and Table S1† show the results of tensile testing of the nanocomposites, including ultimate tensile strength, Young's modulus, and elongation at break. For the pure H-t-PDMS, the tensile strength, Young's modulus and elongation at break are 0.350 MPa, 0.98 MPa and 90.28%. With the incorporation of GONRs, the mechanical properties are enhanced significantly, and saturated at appreciate filling content. The tensile strength of the H-t-PDMS composite is increased to 0.902 MPa at 0.5 wt% GONRs, ∼158% increase when compared to that of pure H-t-PDMS, but decreased to 0.545 MPa at 1.0 wt% GONRs. Similarly, incorporation of 0.5 wt% GONRs into H-t-PDMS polymer produced the highest improvements in both Young's modulus and elongation at break. As discussed in the above section, the good dispersion of GONRs in the matrix and the GONR/matrix strong interfacial interaction should improve the stress transferring from the H-t-PDMS matrix to the GONR sheets at low content, thus leading to the improved stiffness and strength. While the presence of GONR clusters at 1.0 wt% would induce the stress concentration, which would easily produce crack initiation and propagation to reduce the stress transferring efficiency between the matrix and the GONR, thus lowering the reinforcing effect.
We further performed and compared the mechanical properties of pure H-t-PDMS and H-t-PDMS nanocomposites with the three types of carbon nanofillers (CNTs, f-CNTs and GONRs) at optimum content of 0.5 wt%. Fig. S5† and 7 show the representative stress–strain curves, tensile strength and modulus as well as tear strength of these samples, and the results are summarized in Table 1. Clearly, compared with pure H-t-PDMS, the tensile strength and tear strength of the H-t-PDMS/CNT nanocomposites are enhanced by ∼53 and ∼96% respectively; and much higher improvements (∼85 and ∼156% increase in tensile and tear strength, respectively) are obtained for the H-t-PDMS/f-CNT nanocomposites. Notably, the reinforcing effects are more pronounced for the H-t-PDMS/GONR composites, corresponding to ∼158 and ∼284% enhancements in tensile strength and tear strength values. Furthermore, the GONRs also result in the higher improvement (∼97%) in the Young's moduli of the H-t-PDMS polymer than the other two carbon nanofillers (∼26 and ∼69% for CNTs and f-CNTs, respectively).
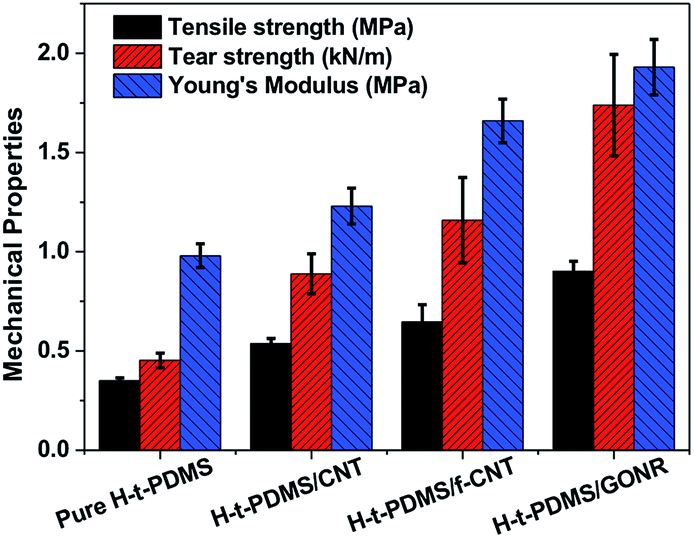 | ||
| Fig. 7 Comparison of tensile and tear strength of pure H-t-PDMS and its nanocomposites filled with 0.5 wt% various carbon nanofillers including CNTs, f-CNTs and GONRs. | ||
| Samples | E a [MPa] | σ b b [MPa] | ε c [%] | σ t d [MPa] |
|---|---|---|---|---|
| a E: elastic modulus. b σ b: tensile strength. c ε: elongation at break. d σ t: tear strength. | ||||
| Pure H-t-PDMS | 0.98 ± 0.06 | 0.350 ± 0.015 | 90.28 ± 13.77 | 0.453 ± 0.037 |
| H-t-PDMS/CNT | 1.23 ± 0.09 | 0.537 ± 0.026 | 161.31 ± 12.50 | 0.889 ± 0.101 |
| H-t-PDMS/f-CNT | 1.66 ± 0.11 | 0.646 ± 0.087 | 163.88 ± 17.47 | 1.159 ± 0.215 |
| H-t-PDMS/GONR | 1.93 ± 0.14 | 0.902 ± 0.050 | 179.42 ± 22.43 | 1.739 ± 0.255 |
3.4. Reinforcement efficiency and possible mechanism analysis
Based on the above mechanical properties of the H-t-PDMS/GONR nanocomposites, we compared the reinforcement efficiencies of PDMS-based nanocomposite systems filled with various carbon nano-fillers including graphite nanoplatelet (GNP),37 CNT,38 chemically reduced graphene oxide (CRGO),40 carbon nanofiber (CNF),6 carbon black (CB),6 and GNR,39 as shown in Fig. 8. The PDMS nanocomposite systems chosen in the figure have optimum improvement in the tensile strength. It can be found that our H-t-PDMS/GONR sample containing 0.5 wt% GONRs exhibits the highest reinforcement efficiency (∼300%) when compared to other carbon nanofillers. Especially, incorporation of GONRs into H-t-PDMS shows much more effective in enhancing the tensile strength compared to similar MVSR/GNR nanocomposites.39 Clearly, the increment of mechanical strength depends on not only the type of carbon nanofillers,33 but also other factors (such as aspect ratio, surface modification, dispersion of filler, matrix type and filler/matrix interfacial interaction);56 and the significance of the above results consists in the fact that the GONRs are indeed an ideal reinforcement to improve the mechanical performance of H-t-PDMS polymer. It is should be also noted such effect of GONRs that are highly dispersed in the H-t-PDMS polymer reinforces it at low content without adopting additional steps like surface functionalization.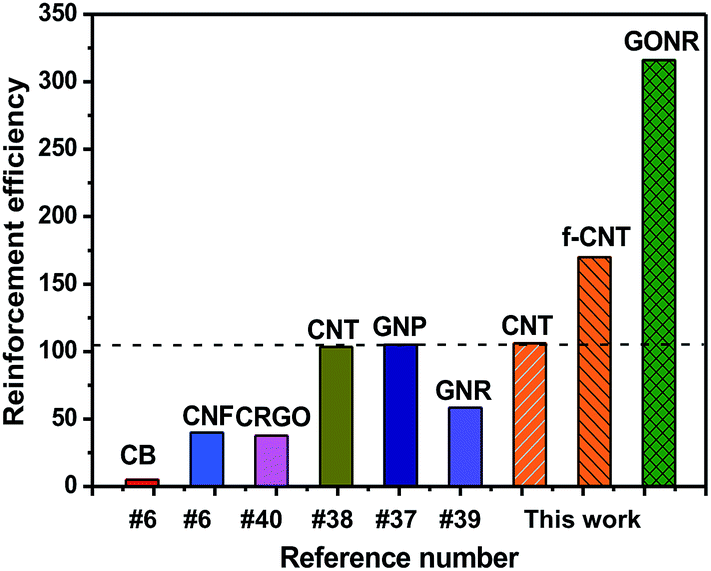 | ||
| Fig. 8 Comparison of reinforcement efficiency (strength enhancement%/filler content wt%) at optimal concentration among various PDMS/carbon nanofiller nanocomposite systems. | ||
Several reasons should be ascribed to the discrepancies in the mechanical properties of the three H-t-PDMS-based nanocomposite systems. First, the dispersion levels of carbon nanofillers are quite different. For pristine CNTs, simply physical mixing of them into H-t-PDMS polymer produces localized clusters, which leads to CNT-rich and CNT-poor regions and thus induces the micro-crack inside the clusters to initiate and propagate easily, leading to mild reinforcing effect in the polymer nanocomposites.57 In contrast, the f-CNTs and GONRs can be highly dispersed in the H-t-PDMS at appropriate filling content, which induces much denser and rougher surfaces with many tortuous surface structures (comparing Fig. S6a with S6c† and 5a), suggesting the effective stress transferring between the filler and the matrix.58 Second, the difference in the intrinsic structures of the CNTs, f-CNTs and GONRs is another important factor. Although no obvious clusters of f-CNTs were observed in the H-t-PDMS matrix (Fig. S6c†), the f-CNTs are curled up and thicker as pristine GONRs, leading to a reduced specific surface area in contact with the H-t-PDMS matrix and thus reducing their interfacial interaction to promote the stress transferring.51 In addition, careful comparison the Fig. 2h with the Fig. 2d and S2b† indicates that compared to the atomistically smooth surface of CNT and f-CNT fillers, both the wrinkled structure of GONRs likely induces a larger interphase zone around each sheet, which should remarkably constrains the molecular mobility of the H-t-PDMS polymer chains.51 This demonstrates that GONR sheets are ideal additives for reinforcing flexible H-t-PDMS polymers which are able to wrap around and interlock effectively with the rough surface.59
Finally, the interfacial interactions between nanofiller and the H-t-PDMS are also quite different. Due to their chemical inactivity, pristine CNTs are hardly reacted with the H-t-PDMS matrix and thus would be pulled out of the polymer matrix (see Fig. S6b†), although the relatively long CNTs may bridge the crack tip at the crack initiation to restrict the crack propagation.60 Comparatively, the surface functionalization of CNTs is speculated to improve the CNT/matrix interfacial interactions,57 which can be evidenced from most f-CNTs embedded tightly in the matrix and no obvious long CNT can be observed on the fracture surface (Fig. S6d†). In case of the H-t-PDMS/GONR nanocomposites at 0.5 wt% content, chemical reactions should occur between the GONRs and the H-t-PDMS molecular chains during the cross-linking process, and the highly dispersed GONR sheets would act as cross-linking sites to provide the strong sheet/matrix interfacial interactions in the H-t-PDMS nanocomposites, thus promoting their efficient stress transferring form the matrix to the GONR sheets. In fact, compared to f-CNTs, much more surface functionalities of GONRs (Fig. 3b) and their covalent bonding with the H-t-PDMS can produce efficient interfacial interactions with polymer chains (Fig. 1d),61 which can be well-illustrated by very short length of GONRs residue on the surface (less than 200 nm in Fig. 5c), suggesting the most GONRs would be broken instead of pullout during the failure process of the nanocomposites. Similar phenomenon could be also observed in the other edge-functionalized GNR-filled epoxy and epoxy/RGO nanocomposite.33,62 Therefore, the good dispersion levels of GONR sheets with the wrinkled morphology and high surface area and the strong GONR/matrix interfacial interactions provide an improved interphase zone in contact with the H-t-PDMS polymer chains, which is of great significance for the effective stress transferring from matrix to GONR,34 leading to an ideal reinforcing effect of H-t-PDMS nanocomposites.
4. Conclusions
In the present work, the GONR sheets by longitudinal unzipping CNTs were synthesized to reinforce H-t-PDMS polymer by a facile fabrication processing. The morphology and structural analysis revealed that the strong oxidative process resulted in considerable functional groups and rough wrinkled structure on GONR surface, thus producing a good dispersion of GONRs in water. The GONR sheets were highly dispersed in the H-t-PDMS polymer matrix at low filling content (0.1–0.5 wt%), but formed serious agglomerate when the GONR content was more than 0.5 wt%. As a result, the H-t-PDMS/GONR nanocomposites showed the highest tensile properties with the 0.5 wt% GONRs, which shows better tear strength and reinforcement efficiency than the corresponding CNT and f-CNT fillers, providing the best reinforcing effect at 0.5 wt%. The good dispersion of GONRs with wrinkled structure and high specific surface area as well as the strong GONR/matrix interfacial interaction should produced the effective stress transferring between the filler and the matrix, which should be ascribed to the highest reinforcement efficiency obtained in the H-t-PDMS/GONR nanocomposites.Acknowledgements
We acknowledge the funding support from the Zhejiang Provincial Natural Science Foundation (LY15E030015), the National Natural Science Foundation of China (51403047), and the Science and Technology Project of Zhejiang Province (2016C31019).References
- S. C. Shit and P. Shah, Natl. Acad. Sci. Lett., 2013, 36, 355–365 CrossRef CAS
.
- W. Zhou, H. Yang, X. Guo and J. Lu, Polym. Degrad. Stab., 2006, 91, 1471–1475 CrossRef CAS
- S. Ansari, J. M. Varghese and K. R. Dayas, Polym. Adv. Technol., 2009, 20, 459–465 CrossRef CAS
- Q. Xu, M. Pang, L. Zhu, Y. Zhang and S. Feng, Mater. Des., 2010, 31, 4083–4087 CrossRef CAS
- D. Chen, F. Chen, H. Zhang, X. Yin, X. Liu and Y. Zhou, Polym. Int., 2015, 64, 1741–1746 CrossRef CAS
- K. Yoshimura, K. Nakano, T. Miyake, Y. Hishikawa, C. Kuzuya, T. Katsuno and S. Motojima, Carbon, 2007, 45, 1997–2003 CrossRef CAS
- M. L. Q. A. Kaneko, R. B. Romero, M. d. C. Gonçalves and I. V. P. Yoshida, Eur. Polym. J., 2010, 46, 881–890 CrossRef CAS
- L. Liu, M. Tian, W. Zhang, L. Zhang and J. E. Mark, Polymer, 2007, 48, 3201–3212 CrossRef CAS
- L. Bai, X. Wang, J. Tan, H. Li and J. Zheng, J. Mater. Sci., 2016, 51, 7130–7144 CrossRef CAS
- D. Chen, Y. Liu and C. Huang, Polym. Degrad. Stab., 2012, 97, 308–315 CrossRef CAS
- A. Camenzind, T. Schweizer, M. Sztucki and S. E. Pratsinis, Polymer, 2010, 51, 1796–1804 CrossRef CAS
- R. Wissert, P. Steurer, S. Schopp, R. Thomann and R. Mülhaupt, Macromol. Mater. Eng., 2010, 295, 1107–1115 CrossRef CAS
- A. R. Shafieizadegan Esfahani, A. A. Katbab, P. Dehkhoda, H. R. Karami, M. Barikani, S. H. H. Sadeghi and A. Ghorbani, Compos. Sci. Technol., 2012, 72, 382–389 CrossRef CAS
- J. Du and H. M. Cheng, Macromol. Chem. Phys., 2012, 213, 1060–1077 CrossRef CAS
- V. Mittal, Macromol. Mater. Eng., 2014, 299, 906–931 CrossRef CAS
- L.-Z. Guan, J.-F. Gao, Y.-B. Pei, L. Zhao, L.-X. Gong, Y.-J. Wan, H. Zhou, N. Zheng, X.-S. Du, L.-B. Wu, J.-X. Jiang, H.-Y. Liu, L.-C. Tang and Y.-W. Mai, Carbon, 2016, 107, 573–582 CrossRef CAS
- D. M. González, R. Quijada, M. Yazdani-Pedram, R. Benavente, G. B. Galland and M. A. Milani, Macromol. Mater. Eng., 2016, 301, 1503–1512 CrossRef
- H. Saleem, A. Edathil, T. Ncube, J. Pokhrel, S. Khoori, A. Abraham and V. Mittal, Macromol. Mater. Eng., 2016, 301, 231–259 CrossRef CAS
- Y. Zhan, J. Wu, H. Xia, N. Yan, G. Fei and G. Yuan, Macromol. Mater. Eng., 2011, 296, 590–602 CrossRef CAS
- K. Tschoppe, F. Beckert, M. Beckert and R. Mülhaupt, Macromol. Mater. Eng., 2015, 300, 140–152 CrossRef CAS
- D. Hofmann, M. Keinath, R. Thomann and R. Mülhaupt, Macromol. Mater. Eng., 2014, 299, 1329–1342 CrossRef CAS
- M. Beckert, F. J. Tölle, B. Bruchmann and R. Mülhaupt, Macromol. Mater. Eng., 2015, 300, 785–792 CrossRef CAS
- F. Beckert, S. Trenkle, R. Thomann and R. Mülhaupt, Macromol. Mater. Eng., 2014, 299, 1513–1520 CrossRef CAS
- Y. Ding, J. Zhu, C. Wang, B. Dai, Y. Li, Y. Qin, F. Xu, Q. Peng, Z. Yang, J. Bai, W. Cao, Y. Yuan and Y. Li, Carbon, 2016, 104, 133–140 CrossRef CAS
- Y. L. Liu, G. L. Xu and X. W. Zhang, J. Mater. Sci., 2015, 50, 3962–3969 CrossRef CAS
- L. Jiao, L. Zhang, X. Wang, G. Diankov and H. Dai, Nature, 2009, 458, 877–880 CrossRef CAS PubMed
- D. V. Kosynkin, A. L. Higginbotham, A. Sinitskii, J. R. Lomeda, A. Dimiev, B. K. Price and J. M. Tour, Nature, 2009, 458, 872–876 CrossRef CAS
- A. L. Higginbotham, D. V. Kosynkin, A. Sinitskii, Z. Sun and J. M. Tour, ACS Nano, 2010, 4, 2059–2069 CrossRef CAS
- M. A. Rafiee, W. Lu, A. V. Thomas, A. Zandiatashbar, J. Rafiee, J. M. Tour and N. A. Koratkar, ACS Nano, 2010, 4, 7415–7420 CrossRef CAS
- W. Liu, B. L. Jackson, J. Zhu, C.-Q. Miao, C.-H. Chung, Y.-J. Park, K. Sun, J. Woo and Y.-H. Xie, ACS Nano, 2010, 4, 3927–3932 CrossRef CAS
- J. G. Son, M. Son, K.-J. Moon, B. H. Lee, J.-M. Myoung, M. S. Strano, M.-H. Ham and C. A. Ross, Adv. Mater., 2013, 25, 4723–4728 CrossRef CAS
- M. Liu, C. Zhang, W. W. Tjiu, Z. Yang, W. Wang and T. Liu, Polymer, 2013, 54, 3124–3130 CrossRef CAS
- R. Nadiv, M. Shtein, M. Buzaglo, S. Peretz-Damari, A. Kovalchuk, T. Wang, J. M. Tour and O. Regev, Carbon, 2016, 99, 444–450 CrossRef CAS
- P. Li, Y. Zheng, T. Shi, Y. Wang, M. Li, C. Chen and J. Zhang, Carbon, 2016, 96, 40–48 CrossRef CAS
- A. Joshi, A. Bajaj, R. Singh, A. Anand, P. S. Alegaonkar and S. Datar, Composites, Part B, 2015, 69, 472–477 CrossRef CAS
- M. Liu, Y. Du, Y.-E. Miao, Q. Ding, S. He, W. W. Tjiu, J. Pan and T. Liu, Nanoscale, 2015, 7, 1037–1046 RSC
- Y. Song, J. Yu, L. Yu, F. E. Alam, W. Dai, C. Li and N. Jiang, Mater. Des., 2015, 88, 950–957 CrossRef CAS
- S. Shang, L. Gan, M. C.-w. Yuen, S.-x. Jiang and N. Mei Luo, Composites, Part A, 2014, 66, 135–141 CrossRef CAS
- L. Gan, S. Shang, C. W. M. Yuen, S.-x. Jiang and N. M. Luo, Composites, Part B, 2015, 69, 237–242 CrossRef CAS
- H. Xu, L. X. Gong, X. Wang, L. Zhao, Y. B. Pei, G. Wang, Y. J. Liu, L. B. Wu, J. X. Jiang and L. C. Tang, Composites, Part A, 2016, 91, 53–64 CrossRef CAS
- W.-S. Ma, J. Li and X.-S. Zhao, J. Mater. Sci., 2013, 48, 5287–5294 CrossRef CAS
- I. Zaman, T. T. Phan, H. C. Kuan, Q. Meng, L. T. Bao La, L. Luong, O. Youssf and J. Ma, Polymer, 2011, 52, 1603–1611 CrossRef CAS
- Y. J. Wan, L. C. Tang, D. Yan, L. Zhao, Y. B. Li, L. B. Wu, J. X. Jiang and G. Q. Lai, Compos. Sci. Technol., 2013, 82, 60–68 CrossRef CAS
- K. Yang, M. Y. Gu, Y. P. Guo, X. F. Pan and G. H. Mu, Carbon, 2009, 47, 1723–1737 CrossRef CAS
- Y. J. Wan, L. C. Tang, L. X. Gong, D. Yan, Y. B. Li, L. B. Wu, J. X. Jiang and G. Q. Lai, Carbon, 2014, 69, 467–480 CrossRef CAS
- Z. Yang, M. Liu, C. Zhang, W. W. Tjiu, T. Liu and H. Peng, Angew. Chem., Int. Ed., 2013, 52, 3996–3999 CrossRef CAS
- Y. Wang, Z. Shi, J. Yu, L. Chen, J. Zhu and Z. Hu, Carbon, 2012, 50, 5525–5536 CrossRef CAS
- M. J. McAllister, J. L. Li, D. H. Adamson, H. C. Schniepp, A. A. Abdala, J. Liu, M. Herrera-Alonso, D. L. Milius, R. Car, R. K. Prud'homme and I. A. Aksay, Chem. Mater., 2007, 19, 4396–4404 CrossRef CAS
- P. C. Ma, S. Y. Mo, B. Z. Tang and J. K. Kim, Carbon, 2010, 48, 1824–1834 CrossRef CAS
- S. W. Kim, T. Kim, Y. S. Kim, H. S. Choi, H. J. Lim, S. J. Yang and C. R. Park, Carbon, 2012, 50, 3–33 CrossRef CAS
- L. C. Tang, Y. J. Wan, D. Yan, Y. B. Pei, L. Zhao, Y. B. Li, L. B. Wu, J. X. Jiang and G. Q. Lai, Carbon, 2013, 60, 16–27 CrossRef CAS
- J. F. Shen, W. S. Huang, L. P. Wu, Y. Z. Hu and M. X. Ye, Compos. Sci. Technol., 2007, 67, 3041–3050 CrossRef CAS
- S. Chandrasekaran, N. Sato, F. Tölle, R. Mülhaupt, B. Fiedler and K. Schulte, Compos. Sci. Technol., 2014, 97, 90–99 CrossRef CAS
- Y. J. Wan, L. X. Gong, L. C. Tang, L. B. Wu and J. X. Jiang, Composites, Part A, 2014, 64, 79–89 CrossRef CAS
- L. Z. Guan, Y. J. Wan, L. X. Gong, D. Yan, L. C. Tang, L. B. Wu, J. X. Jiang and G. Q. Lai, J. Mater. Chem. A, 2014, 2, 15058–15069 RSC
- T. V. Varghese, H. Ajith Kumar, S. Anitha, S. Ratheesh, R. S. Rajeev and V. Lakshmana Rao, Carbon, 2013, 61, 476–486 CrossRef CAS
- L. C. Tang, H. Zhang, J. H. Han, X. P. Wu and Z. Zhang, Compos. Sci. Technol., 2011, 72, 7–13 CrossRef CAS
- L.-X. Gong, Y.-B. Pei, Q.-Y. Han, L. Zhao, L.-B. Wu, J.-X. Jiang and L.-C. Tang, Compos. Sci. Technol., 2016, 134, 144–152 CrossRef CAS
- M. A. Rafiee, W. Lu, A. V. Thomas, A. Zandiatashbar, J. Rafiee, J. M. Tour and N. A. Koratkar, ACS Nano, 2010, 4, 7415–7420 CrossRef CAS PubMed
- L. C. Tang, H. Zhang, X. P. Wu and Z. Zhang, Polymer, 2011, 52, 2070–2074 CrossRef CAS
- M. Fang, K. Wang, H. Lu, Y. Yang and S. Nutt, J. Mater. Chem., 2009, 19, 7098–7105 RSC
- J. Zang, Y.-J. Wan, L. Zhao and L.-C. Tang, Macromol. Mater. Eng., 2015, 300, 737–749 CrossRef CAS
Footnote |
| † Electronic supplementary information (ESI) available: TEM of CNT and f-CNT and their dispersion stability in water, SEM images of pure H-t-PDMS and H-t-PDMS/CNT and H-t-PDMS/f-CNT composites. See DOI: 10.1039/c7ra02439h |
| This journal is © The Royal Society of Chemistry 2017 |