Three-dimensional co-cultures of human endothelial cells and embryonic stem cell-derived pericytes inside a microfluidic device†
Andries D.
van der Meer‡
*ab,
Valeria V.
Orlova‡
cd,
Peter
ten Dijke
c,
Albert
van den Berg
a and
Christine L.
Mummery
d
aBIOS/Lab on a Chip, MESA+ Institute for Nanotechnology, University of Twente, The Netherlands
bWyss Institute for Biologically Inspired Engineering, Harvard University, USA. E-mail: andries.vandermeer@wyss.harvard.edu
cDepartment of Molecular Cell Biology, Leiden University Medical Center, The Netherlands. E-mail: v.orlova@lumc.nl
dDepartment of Anatomy and Embryology, Leiden University Medical Center, The Netherlands
First published on 23rd May 2013
Abstract
Organs-on-chips are microengineered in vitro tissue structures that can be used as platforms for physiological and pathological research. They provide tissue-like microenvironments in which different cell types can be co-cultured in a controlled manner to create synthetic organ mimics. Blood vessels are an integral part of all tissues in the human body. Development of vascular structures is therefore an important research topic for advancing the field of organs-on-chips since generated tissues will require a blood or nutrient supply. Here, we have engineered three-dimensional constructs of vascular tissue inside microchannels by injecting a mixture of human umbilical vein endothelial cells, human embryonic stem cell-derived pericytes (the precursors of vascular smooth muscle cells) and rat tail collagen I into a polydimethylsiloxane microfluidic channel with dimensions 500 μm × 120 μm × 1 cm (w × h × l). Over the course of 12 h, the cells organized themselves into a single long tube resembling a blood vessel that followed the contours of the channel. Detailed examination of tube morphology by confocal microscopy revealed a mature endothelial monolayer with complete PECAM-1 staining at cell–cell contacts and pericytes incorporated inside the tubular structures. We also demonstrated that tube formation was disrupted in the presence of a neutralizing antibody against transforming growth factor-beta (TGF-β). The TGF-β signaling pathway is essential for normal vascular development; deletion of any of its components in mouse development results in defective vasculogenesis and angiogenesis and mutations in humans have been linked to multiple vascular genetic diseases. In the engineered microvessels, inhibition of TGF-β signaling resulted in tubes with smaller diameters and higher tortuosity, highly reminiscent of the abnormal vessels observed in patients with one particular vascular disease known as hereditary hemorrhagic telangiectasia (HHT). In summary, we have developed microengineered three-dimensional vascular structures that can be used as a model to test the effects of drugs and study the interaction between different human vascular cell types. In the future, the model may be integrated into larger tissue constructs to advance the development of organs-on-chips.
Introduction
Organs-on-chips are microengineered laboratory models of human organs that combine physiological realism with specific control over the cell culture microenvironment.1–5 The models usually feature multiple cell types in a three-dimensional organization that preserves tissue geometry. In addition, tissue morphology and function as well as specific pathologies can be recapitulated by biophysical and biochemical stimuli that can be controlled by the researcher. With their combination of realism, control and ease-of-use, organs-on-chips are increasingly being applied in studies of human physiology and disease, as well as in the development of new clinical drugs.Vascular structures are an integral part of all human tissues and are therefore of great interest for the development of more complex organs-on-chips. Not only do cells of the synthetic vasculature interact with cells of the organ of interest but they can also serve as a route to supplying nutrients and drugs. Moreover, the microfluidic blood vessel models can be applied in research on vascular function and dysfunction.6,7 Many studies of vascular structures in microengineered devices have already appeared in the literature.8,9 In all of these studies, one of three distinct strategies is employed. Firstly, vascular cells are cultured on the walls of a polymeric or glass microchannel, thus forming a cell-coated microstructure.10,11 Secondly, instead of constructing the microchannels from inorganic polymers or glass, they are made inside a polymeric network of extracellular matrix proteins, such as collagen. This can be accomplished by lithographic techniques or by viscous fingering.12,13 The advantage of this approach is that when vascular cells are seeded inside the device, they are exposed to a surface that more closely resembles the basement membrane present in the human body. The final approach to generate vascular structures in microfluidic devices is by filling the microchannels entirely or in part with a protein gel, followed by growth of the vascular cells inside or into these gel-laden microstructures.14–16 The advantage of using such an approach is that it allows for a more natural self-organization of vascular structures. Each of the three strategies has its advantages and disadvantages in terms of ease-of-use, realism and control over the cell culture microenvironment. Which of the strategies should be employed is mostly dependent on which physiological parameters are of interest. In this study, we were mostly interested in the organization and cellular interaction between endothelial cells and associated pericytes or smooth muscle cells in three-dimensional vascular structures and therefore employed the last of the three strategies.
One of the important considerations in designing new organs-on-chips is the origin of the cells to be included. Many cell-based in vitro assays for drug discovery currently employ immortalized or cancerous mammalian cell lines, because they are readily available and easy to culture.17 However, these cells are not the most relevant cell source and may not recapitulate normal physiology. Primary human cells or human stem cell derivatives are considered increasingly as superior cell sources. Human pluripotent stem cells are of particular interest in the development of organs-on-chips since they can be differentiated into any cell type of the human body. More importantly, they can now be derived as human induced pluripotent stem cells and capture the genome of any individual of interest including those with genetic diseases.18 These cells are becoming increasingly easy to culture and differentiate so that there is growing interest in using them for human organs-on-chips applications. In order to explore this concept further, we used human embryonic stem cells as a source of pluripotent cells and derived pericytes from them to test their ability to interact with primary human endothelial cells in a microfluidic organ-on-chip device. Proper interaction between pericytes and endothelial cells should result in their differentiation to smooth muscle cells.
A number of requirements have to be met for an organ-on-a-chip to become a useful tool in physiological research. Briefly, the model needs to be realistic, yet controlled, meaning it should feature co-cultures of relevant human cell types in a controlled microenvironment. In addition, the model should be easy to set up and operate. Finally, it should also preferably be compatible with read-out methods for evaluating the effects of drugs.
In the present study, we describe a microfluidic device that meets these criteria. For the first time, we have cultured human endothelial cells and embryonic stem cell-derived pericytes in a controlled three-dimensional, microfluidic setting. We evaluated the effects of drugs on the morphology of the vascular structures and studied the three-dimensional interaction of endothelial cells and pericytes using confocal microscopy. The model we describe demonstrates the potential for human embryonic stem cell-derived cells (and by extrapolation, induced pluripotent stem cells) to be used in the engineering of organs-on-chips. Moreover, our study is part of a growing body of literature that highlights the great potential of organs-on-chips for studying human physiology in general and vascular biology in particular.
Results and discussion
Setting up a three-dimensional co-culture
The polydimethylsiloxane–glass microfluidic devices we used in this study were produced by standard soft lithography. The Y-shaped channels had dimensions of 120 μm × 500 μm × 1 cm (h × w × l), as is shown schematically in Fig. 1A. Human umbilical vein endothelial cells and human embryonic stem cell-derived pericytes were harvested, counted and mixed in a 10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 ratio. In order to distinguish different cell types during the time lapse imaging, pericytes were additionally labeled with a general cytoplasm stain (PKH-26) that resulted in a prominent bright fluorescent signal in the red channel. The cell suspension was mixed with the rat-tail collagen and injected into the channels, as is shown in Fig. 1B. A single cell suspension of endothelial cells and pericytes was equally distributed along the entire length of the channel (ESI† Fig. S4). Upon seeding, microchannels were left in a humidified atmosphere at 37 °C for complete tube formation.
:1 ratio. In order to distinguish different cell types during the time lapse imaging, pericytes were additionally labeled with a general cytoplasm stain (PKH-26) that resulted in a prominent bright fluorescent signal in the red channel. The cell suspension was mixed with the rat-tail collagen and injected into the channels, as is shown in Fig. 1B. A single cell suspension of endothelial cells and pericytes was equally distributed along the entire length of the channel (ESI† Fig. S4). Upon seeding, microchannels were left in a humidified atmosphere at 37 °C for complete tube formation.
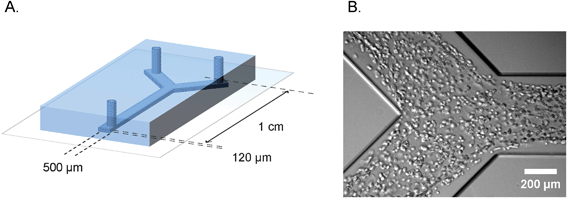 | ||
| Fig. 1 Microfluidic device for defined engineering of vascular structures. A. Schematic overview of the polydimethylsiloxane–glass device showing the dimensions and geometry of the microchannel. B. Bright field micrograph of the cell suspension inside the microchannel, one hour after seeding. | ||
The method used is very similar to previously reported methods for seeding vascular cells in micropatterned grooves.14 The advantage of our method is that the chips are closed systems and do not have to be centrifuged for efficient cell seeding. The simplicity of cell mixing and seeding can be further translated for drug screening applications. The compound of interest can be simply incorporated into the cell–collagen suspension and be seeded in parallel with the control channel. Moreover, due to the technical similarities of our system with other microfluidic tissue models,2 it can potentially be incorporated in more complex microfluidic organ-models. Finally, our system incorporates a branched channel geometry, which allows us to study vascular cell interaction in a controlled branching pattern.
Formation of vascular structures
After seeding, the cell pattern was studied with time-lapse microscopy. Over the course of the first 12 h, the cells organized themselves into a long, tubular structure that followed the contours of the microchannel, including the branched morphology, as is shown in Fig. 2A. In vivo, the formation of tubular structures is a complex process and involves multiple signaling pathways, including that of TGF-β which is important in inducing pericytes recruited to endothelial cells to differentiate to vascular smooth muscle cells.19 In order to test whether TGF-β signaling would affect formation of the tubular structure in vitro, we utilized a neutralizing antibody that has been shown to inactivate all three major TGF-β isoforms (TGF-β1, -β2 and -β3).20 Interestingly, the dynamics of the tube formation was significantly different upon inhibition of TGF-β signaling compared to the control channel. The TGF-β-depleted cells contracted faster and formed tubes with a smaller diameter, as is visible in the plot in Fig. 2B. Moreover, additional morphological examination after 24 h showed a dramatic alteration of tubular structures when all TGF-β isoforms were neutralized. Abnormal tubules with irregular and tortuous morphology were evident that were highly reminiscent of the phenotype observed upon defective TGF-β signaling in vivo both in patients with HHT21,22 and in mice with mutant TGF-β receptors known to cause HHT.23,24 In contrast, control conditions resulted in the formation of smooth and straight tubular structures (Fig. 3).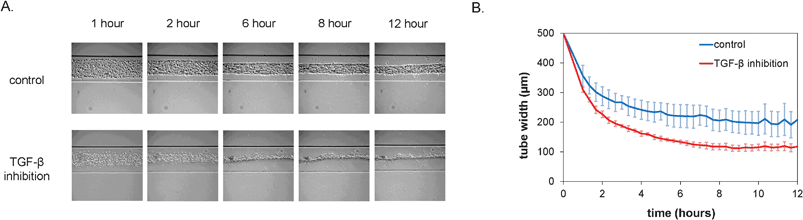 | ||
| Fig. 2 Formation of vascular structures within the first 12 h after seeding under control conditions and in the presence of inhibitory antibodies. A. Representative brightfield micrographs at different time-points after seeding. Channels are 500 μm wide. B. Quantification of tube width over time (n = 4, error bars denote standard deviation). | ||
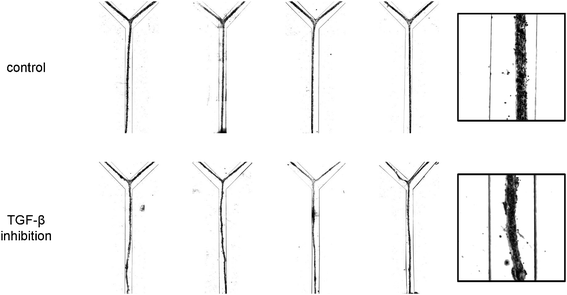 | ||
| Fig. 3 High-contrast bright field micrographs of the morphology of the vascular structures after 24 h of culturing under control conditions and in the presence of inhibitory antibodies. For each condition, four representative examples are given (left). The insets (right) provide a higher magnification view of the structures. Channels are 500 μm wide. | ||
The underlying mechanism of formation of the tubular structures in our model will be the object of future studies. Many parameters of the cell state and the culture microenvironment have an effect on endothelial structure formation in vitro.25 Initial observations of structure formation in our model seem to favor a mechanism in which all collagen I is actively recruited into the structures by cell-driven contraction (ESI† Fig. S3). Interestingly, this phenomenon may be affected by matrix material and concentration, degree of interaction of the matrix with the channel surfaces, degree of interaction of the cells with the matrix and the channel surfaces, and the state of cellular pathways involved in adhesion, contraction and migration. Our system offers the opportunity to study all these parameters in a systematic fashion.
Although the observed phenotype of increased tubular contraction is somewhat contradictory with previous published reports implicating TGF-β as an inducer of the contractile phenotype, we cannot exclude the possibility of differences because of the heterotypic cell system. Indeed, most previous studies were performed on isolated vascular smooth muscle cell/pericytes in vitro. The situation in vivo is more complex, and the final effect of TGF-β signaling will be a cumulative outcome of the effects on both endothelial and perivascular cells. Therefore, development of systems that can address effects on both endothelial cells and pericytes is of great importance in creating disease models; the microchannel system described here could be well suited for this type of study.
Interaction of endothelial cells and pericytes in a three-dimensional setting
Time-lapse imaging and end-point bright field microscopy revealed tubular structures with pericytes incorporated inside the vessels wall, as shown in Fig. 4A. However, the resolution provided by the bright field microscopy was not sufficient to study cell–cell and cell–matrix interactions. Additionally, we could not determine whether the tubular structures contained a lumen. In order to examine this at higher resolution we developed microchannel devices that were permanently bound to thin coverslips that are compatible with the high-resolution imaging by confocal microscopy. In addition to the stably labeled pericytes, the structures were stained with a fluorescent dye for DNA and antibodies against the endothelial cell-specific adhesion marker, platelet-endothelial cell adhesion marker-1 (PECAM-1). Interestingly, examination of tubular channels with confocal microscopy confirmed formation of luminar hollow structures (Fig. 4B, right). In addition, endothelial cells formed well-organized cell–cell junctions, as demonstrated in Fig. 4B, left, and ESI† Fig. S1A. Visualization of pericytes revealed that they were localized inside the tubular structures, rather than on the outside, indicating that the tubules formed resembled ‘inside-out’ vessels with the core made of collagen and pericytes, surrounded by a monolayer of endothelial cells. A similar structural organization has been described before during the in vitro formation of endothelial-lined spheroids.25,26 The morphology of the ‘inside-out’ structures is interesting, because in vivo, the pericytes interact with endothelial cells from the outside of vascular tubes. Further studies will be needed to understand why this takes place and the difference between the structures described here and capillaries in vivo. Based on the presumed contraction-based mechanism of tubular structure formation in our model, the pericytes tend to localize to a microenvironment where they are in direct contact with high density collagen I and endothelial cells.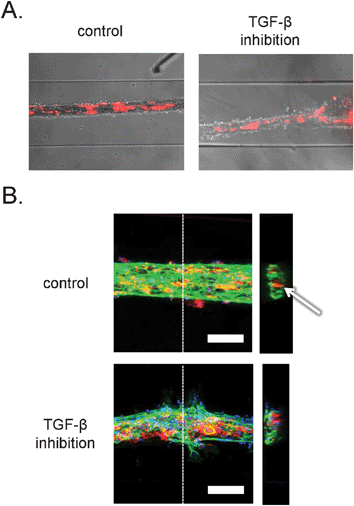 | ||
| Fig. 4 Organization of human embryonic stem cell-derived pericytes and endothelial cells in vascular structures. A. Combined bright field and epifluorescence images. Embryonic stem cell-derived pericytes are labeled with the general cell label PKH-26 (red). Channels are 500 μm wide. B. Confocal fluorescence microscopic analysis of three-dimensional structure. Vascular structures were labeled with antibodies against PECAM-1 (green) and the nuclear dye, DAPI (blue). The pericytes were pre-labeled with the general cell label PKH-26 (red). Left: projection of maximum intensity in the z-direction gives a general overview of the structures. Scale bar: 100 μm. Right: y–z projections of the vascular structures. Dashed lines denote the sections in the respective planes. Arrow denotes lumen in control structure. | ||
Interestingly, pericytes seemed to favour close interaction with the endothelial surface, because they were absent in the center of the branching regions (ESI† Fig. S2B).
Inhibition of TGF-β resulted in disorganization of endothelial cell–cell contacts and an increase in endothelial cell migration as observed by confocal microscopy. Strikingly, tortuous structures consisted of two cell types with a poorly organized monolayer of endothelial cells and pericytes that formed large agglomerates of cells.
These findings are in line with the studies by our group and others on the inhibitory effect of TGF-β on endothelial cell proliferation, migration and sprouting angiogenesis.27–29 The inhibitory effect can be reversed by addition of the Type I receptor inhibitor (SB-431542) or TGF-β neutralizing antibodies.29 In addition, TGF-β plays an important role in the stabilization of blood vessels via promoting recruitment and differentiation of pericytes.30,31 Therefore, the observed phenotype can also result from defective endothelial–pericyte interaction. In support of this explanation, we observed more dramatic homotypic clustering of pericytes under conditions of TGF-β inhibition (ESI† Fig. S2A).
As discussed previously, perturbation of the TGF-β signaling pathway has been linked to multiple genetic vascular diseases such as hereditary HHT, Marfan and Loeys–Dietz syndrome, where defective TGF-β signaling results in the formation of tortuous abnormal vessels.32 Our data indicates that inhibition of TGF-β signaling pathways in the microassay described here partly recapitulates the phenotype observed in vivo. Thus, the microfluidic system can potentially be applied to model pathological conditions with defective TGF-β signaling in vitro. In future studies, the assay may be used to mimic other vascular genetic diseases or conditions with pathological angiogenesis caused by deregulated behavior of endothelial cells and pericytes.
More importantly, we expect in the future to be able to combine endothelial cells or pericytes from patients with diseases like HHT with control equivalents to determine whether the disease phenotype is seen as here with neutralizing antibody and whether the effect derives from defects on the endothelial cells, pericytes or both.33
Conclusion
The formation of three-dimensional, tubular structures is a complex process. It involves multiple signaling pathways that coordinate endothelial cell–cell and endothelial–pericyte interactions, cell–matrix interactions, as well as matrix modification that all ultimately result in stabilization of the tubular structures.34 In the present study, we demonstrated that our ‘blood vessel-on-a-chip’ microfluidic assay recapitulates multiple aspects of in vivo angiogenesis, such as cell–matrix interaction, matrix remodeling, endothelial–mural cell interaction and stabilization of the vasculature via induction of the quiescent phenotype and cell survival. We additionally demonstrated that inhibition of TGF-β signaling results in aberrant tube formation, a finding that highly resembles pathological phenotypes observed in vivo. Finally, we showed that human pluripotent stem cell-derived cells can be used to set up microfluidic models of human tissues.The main shortcoming of our model is that the ‘inside-out’ morphology is not a direct representation of blood vessels in vivo. In our Discussion, we have outlined what we think are the main parameters that drive the formation of this specific morphology. Future work will be directed towards systematically identifying the parameters that affect structure morphology.
Development of in vitro assays that are close to the conditions present in vivo is of major importance for physiological studies and in drug discovery. Our current study is an integral part of the rapidly developing field of organs-on-a-chip. We demonstrated that tubulogenesis in microfluidic devices can be applied in mechanism-oriented studies and for drug screening applications. In the future, the model may be further optimized and integrated to recapitulate even more features of the in vivo microenvironment.
Materials and methods
Device fabrication
The design for the devices was drawn with CleWin software (WieWeb Software, The Netherlands). A chrome-fused silica photomask with the desired pattern was produced. Subsequently, a master mold was made by spincoating a 120 μm thick layer of SU-8 2-25 (Microchem, Germany) on a silicon wafer and using the photomask to produce the structures by UV-lithography. The master mold was used to prepare devices by standard polydimethylsiloxane-based soft lithography. Briefly, polydimethylsiloxane base and curing agent (Sylgard 184, Dow Corning, USA) were mixed in a 10:1 ratio, degassed and poured on top of the mold. After overnight crosslinking at 60 °C, the slab of polydimethylsiloxane was peeled off of the mold. The slab was cut into single chips and channel inlets of 1 mm diameter were punched with a needle. Finally, the surface of the chips was activated in a plasma sterilizer (Harrick PDC-001, USA) at 50 Pa vacuum and ‘Low’ setting for 40 s, after which the chips were permanently bonded to glass coverslips.
Cell culture and differentiation
Human umbilical vein endothelial cells (HUVECs, PromoCell, Heidelberg, Germany) were propagated on 0.1% gelatin-coated tissue culture plates in EGM-2 endothelial cell growth media (Lonza, USA). Human embryonic stem cell line HES3 was routinely maintained as a cut and paste culture on matrigel-coated plates in mTeSR1 defined media (Stem Cell Technologies, Vancouver, Canada). Pericytes were derived from human embryonic stem cells upon spontaneous differentiation as embryo bodies (EBs) (the detailed schematic overview of the protocol is presented in the ESI† Fig. S5). Human embryonic stem cell colonies were enzymatically dissociated with dispase (Invitrogen), and transferred into ultra-low attachment plates (Costar) in differentiation media with additional overnight supplementation with a Rho-associated protein kinase inhibitor (fasudil monohydrochloride (LC Laboratories), 5 μM) as described previously.35 Day 4 after induction of differentiation, EBs were transferred into gelatin-coated tissue culture plates for an additional 8 days and dissociated with TrypLE Select (Invitrogen). Pericytes were defined as a CD31−CD34−CD146+ population by immunostaining.36 Pericytes were isolated by immunomagnetic labeling with CD146-FITC and anti-FITC microbeads (Miltenyi) or flow activated cell sorting (FACS sort) (AriaIII). Isolated cells were further expanded on Collagen IV coated plates (BD Biosciences) for an additional 7 days in α-Minimal Essential Medium with additional TGF-β3 and platelet-derived growth factor-BB (Peprotech). For all experiments, differentiated pericytes were routinely maintained in gelatin-coated plates in EGM-2 media.Cell labeling and seeding
Pericytes and HUVECs were detached with TrypLE, and cells were counted. Pericytes were labeled with general cytoplasmic labeling agent PKH-26 (Sigma) according to the manufacturer's protocol. Prior to seeding HUVECs (60 × 106 cells mL−1) and pericytes (6 × 106 cells mL−1) were re-suspended in EGM-2, and mixed with an equal volume of rat tail collagen type I solution 5 mg mL−1 (BD Biosciences). The cell suspension was injected into the straight-section inlet of the microchannels, and uniform loading was confirmed by bright field microscopy and epifluorescence microscopy. The suspension was left to gelate for an additional 30 min at 37 °C. In some cases, TGF-β neutralizing antibody (40 μg mL−1 2G7, a kind gift from E. de Heer, LUMC, The Netherlands)20 was added to the collagen suspension before loading.Bright field microscopy
Time-lapse experiments were performed on a Leica AF6000 imaging system. Cells were imaged at 37 °C, 5% CO2 and humidified atmosphere. Images were acquired with the 10×/0.3 Ph1 dry objective. Differential interference contrast and fluorescence images were recorded continuously for 24 h with the time interval every 20 min. Static experiments in a standard humidified atmosphere were performed in parallel with the time-lapse imaging in order to ensure comparability of both conditions. End point static microchannels were imaged with a Nikon Eclipse Ti imaging system with the 10×/0.3 Ph1 dry objective.Confocal microscopy
Tubular structures in microchannels were fixed with 2% paraformaldehyde solution (Electron Microscopy Sciences) for 4 h at 4 °C. After fixation, microchannels were washed with physiological salt solution, and stored at 4 °C. For immunofluorescent staining tubular structures were permeabilized with 0.5% Triton X-100 in physiological salt solution for 30 min at room temperature, and washed additionally with physiological salt solution. Endothelial cells were visualized with rabbit anti-human-PECAM-1 antibody (1:200 in 2% (w/v) bovine serum albumin in physiological salt solution, Santa Cruz Biotechnology), and goat-anti-rabbit-Alexa488 (1:300 in 2% (w/v) bovine serum albumin in physiological salt solution). Nuclei were visualized with Hoechst 33342 (Molecular Probes). Droplets of fluorescent mounting media (Dako) were placed on inlets, and microchannels were stored at 4 °C.
A Leica SP5 confocal imaging system was used for acquisition of high-resolution images with the 20× (multi immersion, NA 0.7) or 40× (oil immersion, NA 1.25) objectives. ImageJ software37 was used for maximum projection images and for 3D reconstruction. Moreover, it was used to identify clusters of pericytes in the channels by automatically thresholding the red fluorescent images, automatically analysing particles in the thresholded images, and automatically color-coding the particles based on their area by using the ROI Color Coder plug-in by Tiago Ferreira.
Acknowledgements
The authors would like to acknowledge Emile de Heer (Department of Pathology, LUMC) for providing the TGF-β neutralizing antibody (2G7), and Joop Wiegant and Annelies Boonzaier-van der Laan (Department of Molecular Cell Biology, LUMC) for help with confocal and time-lapse imaging microscopy.The collaborative study was facilitated by financial support from the Royal Netherlands Academy for Arts and Sciences (KNAW) Beyond Borders programme: Organs on Chips, Blood vessels-on-Chips Project (ADM, VO, AB, CLM), VO is sponsored by Gisela Thier fellowship from the LUMC and the SWORO foundation for research on HHT. Research of CLM is supported by the Netherlands Institute of Regenerative Medicine. ADM is supported by the Netherlands Organization for Scientific Research (NWO), Project ‘Nanotox chip’ (project 11521).
References
- D. Huh, G. A. Hamilton and D. E. Ingber, Trends Cell Biol., 2011, 21, 745–754 CrossRef CAS.
- D. Huh, Y. Torisawa, G. A. Hamilton, H. J. Kim and D. E. Ingber, Lab Chip, 2012, 12, 2156–2164 RSC.
- C. Moraes, G. Mehta, S. Lesher-Perez and S. Takayama, Ann. Biomed. Eng., 2012, 40, 1211–1227 CrossRef.
- J. H. Sung, M. Esch, J.-M. Prot, C. J. Long, A. Smith, J. Hickman and M. Shuler, Lab Chip, 2013, 13, 1201–1212 RSC.
- A. D. van der Meer and A. van den Berg, Integr. Biol., 2012, 4, 461–470 RSC.
- E. Westein, A. D. van der Meer, M. J. E. Kuijpers, J.-P. Frimat, A. van den Berg and J. W. M. Heemskerk, Proc. Natl. Acad. Sci. U. S. A., 2013, 110, 1357–1362 CrossRef CAS.
- L. M. Griep, F. Wolbers, B. de Wagenaar, P. M. ter Braak, B. B. Weksler, I. A. Romero, P. O. Couraud, I. Vermes, A. D. van der Meer and A. van den Berg, Biomed. Microdevices, 2013, 15, 145–150 CrossRef CAS.
- A. D. van der Meer, A. A. Poot, M. H. G. Duits, J. Feijen and I. Vermes, J. Biomed. Biotechnol., 2009, 2009, 823148 CrossRef CAS.
- K. H. K. Wong, J. M. Chan, R. D. Kamm and J. Tien, Annu. Rev. Biomed. Eng., 2012, 14, 205–230 CrossRef CAS.
- J. W. Song, W. Gu, N. Futai, K. A. Warner, J. E. Nor and S. Takayama, Anal. Chem., 2005, 77, 3993–3999 CrossRef CAS.
- M. Shin, K. Matsuda, O. Ishii, H. Terai, M. Kaazempur-Mofrad, J. Borenstein, M. Detmar and J. Vacanti, Biomed. Microdevices, 2004, 6, 269–278 CrossRef CAS.
- Y. Zheng, J. Chen, M. Craven, N. W. Choi, S. Totorica, A. Diaz-Santana, P. Kermani, B. Hempstead, C. Fischbach-Teschl, J. A. López and A. D. Stroock, Proc. Natl. Acad. Sci. U. S. A., 2012, 109, 9342–9347 CrossRef CAS.
- L. L. Bischel, E. W. K. Young, B. R. Mader and D. J. Beebe, Biomaterials, 2013, 34, 1471–1477 CrossRef CAS.
- S. Raghavan, C. M. Nelson, J. D. Baranski, E. Lim and C. S. Chen, Tissue Eng. A, 2010, 16, 2255–2263 CrossRef CAS.
- V. Vickerman, J. Blundo, S. Chung and R. Kamm, Lab Chip, 2008, 8, 1468–1477 RSC.
- J. H. Yeon, H. R. Ryu, M. Chung, Q. P. Hu and N. L. Jeon, Lab Chip, 2012, 12, 2815–2822 RSC.
- A. P. Li, Drug Discovery Today, 2001, 6, 357–366 CrossRef CAS.
- M. Bellin, M. C. Marchetto, F. H. Gage and C. L. Mummery, Nat. Rev. Mol. Cell Biol., 2012, 13, 713–726 CrossRef.
- R. L. C. Carvalho, L. Jonker, M.-J. Goumans, J. Larsson, P. Bouwman, S. Karlsson, P. t. Dijke, H. M. Arthur and C. L. Mummery, Development, 2004, 131, 6237–6247 CrossRef CAS.
- C. Lucas, L. N. Bald, B. M. Fendly, M. Mora-Worms, I. S. Figari, E. J. Patzer and M. A. Palladino, J. Immunol., 1990, 145, 1415–1422 CAS.
- T. Krings, A. Ozanne, S. M. Chng, H. Alvarez, G. Rodesch and P. L. Lasjaunias, Neuroradiology, 2005, 47, 711–720 CrossRef CAS.
- T. Haitjema, C. J. Westermann, T. T. Overtoom, R. Timmer, F. Disch, H. Mauser and J. W. Lammers, Arch. Intern. Med., 1996, 156, 714–719 CrossRef CAS.
- H. M. Arthur, J. Ure, A. J. H. Smith, G. Renforth, D. I. Wilson, E. Torsney, R. Charlton, D. V. Parums, T. Jowett, D. A. Marchuk, J. Burn and A. G. Diamond, Dev. Biol., 2000, 217, 42–53 CrossRef CAS.
- S. O. Park, M. Wankhede, Y. J. Lee, E.-J. Choi, N. Fliess, S.-W. Choe, S.-H. Oh, G. Walter, M. K. Raizada, B. S. Sorg and S. P. Oh, J. Clin. Invest., 2009, 119, 3487–3496 CAS.
- R. B. Vernon and E. H. Sage, Am. J. Pathol., 1995, 147, 873–883 CAS.
- T. Korff and H. G. Augustin, J. Cell Biol., 1998, 143, 1341–1352 CrossRef CAS.
- N. Rudini, A. Felici, C. Giampietro, M. Lampugnani, M. Corada, K. Swirsding, M. Garre, S. Liebner, M. Letarte, P. ten Dijke and E. Dejana, EMBO J., 2008, 27, 993–1004 CrossRef CAS.
- M.-J. Goumans, G. Valdimarsdottir, S. Itoh, A. Rosendahl, P. Sideras and P. ten Dijke, EMBO J., 2002, 21, 1743–1753 CrossRef CAS.
- Z. Liu, K. Kobayashi, M. van Dinther, S. H. van Heiningen, G. Valdimarsdottir, T. van Laar, M. Scharpfenecker, C. W. G. M. Löwik, M.-J. Goumans, P. t. Dijke and E. Pardali, J. Cell Sci., 2009, 122, 3294–3302 CrossRef CAS.
- D. C. Darland and P. A. D'Amore, Angiogenesis, 2001, 4, 11–20 CrossRef CAS.
- K. K. Hirschi, S. A. Rohovsky and P. A. D'Amore, J. Cell Biol., 1998, 141, 805–814 CrossRef CAS.
- V. V. Orlova, Z. Liu, M. J. Goumans and P. ten Dijke, Histol. Histopathol., 2011, 26, 1219–1230 CAS.
- C. Freund, R. P. Davis, K. Gkatzis, D. Ward-van Oostwaard and C. L. Mummery, Neth. Heart J., 2010, 18, 51–54 CAS.
- R. K. Jain, Nat. Med., 2003, 9, 685–693 CrossRef CAS.
- R. P. Davis, S. Casini, C. W. van den Berg, M. Hoekstra, C. A. Remme, C. Dambrot, D. Salvatori, D. W.-v. Oostwaard, A. A. M. Wilde, C. R. Bezzina, A. O. Verkerk, C. Freund and C. L. Mummery, Circulation, 2012, 125, 3079–3091 CrossRef.
- M. Crisan, S. Yap, L. Casteilla, C. Chen, M. Corselli, T. S. Park, G. Andriolo, B. Sun, B. Zheng, L. Zhang, C. Norotte, P. Teng, J. Traas, R. Schugar, B. M. Deasy, S. Badylak, H. Bűhring, J. Giacobino, L. Lazzari, J. Huard and B. Péault, Cell Stem Cell, 2008, 3, 301–313 CrossRef CAS.
- C. A. Schneider, W. S. Rasband and K. W. Eliceiri, Nature Methods, 2012, 9, 671–675 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c3lc50435b |
| ‡ Contributed equally. |
| This journal is © The Royal Society of Chemistry 2013 |