
Palladium-catalyzed oxidative borylation of conjugated enynones through carbene migratory insertion: synthesis of furyl-substituted alkenylboronates†
Yifan
Ping
a,
Taiwei
Chang
 a,
Kang
Wang
a,
Jingfeng
Huo
a and
Jianbo
Wang
*ab
a,
Kang
Wang
a,
Jingfeng
Huo
a and
Jianbo
Wang
*ab
aBeijing National Laboratory of Molecular Sciences (BNLMS), Key Laboratory of Bioorganic Chemistry and Molecular Engineering of Ministry of Education, College of Chemistry, Peking University, Beijing 100871, China. E-mail: wangjb@pku.edu.cn
bThe State Key Laboratory of Organometallic Chemistry, Shanghai Institute of Organic Chemistry, Chinese Academy of Sciences, Shanghai 200032, China
First published on 26th November 2018
Abstract
A palladium-catalyzed oxidative borylation reaction of conjugated enynones is developed. This reaction represents a new method for the synthesis of furyl-substituted alkenylboronates. The reaction works well with a series of conjugated enynones. Boryl migratory insertion of the palladium carbene intermediate is proposed as the key step in these transformations.
Transition-metal-catalyzed carbene-based cross-coupling reactions have emerged as powerful synthetic methods for C–C and C
![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C bond formation.1 In these transformations, the organometallic species, which can be generated from oxidative addition,2 transmetalation,3 C–H activation4 or other processes,5 reacts with a carbene precursor to form a metal carbene intermediate. Then, carbene migratory insertion occurs to generate a new organometallic species that undergoes further transformation to afford various coupling products. Diazo compounds are the most common carbene precursors, which can be decomposed by transition-metal catalysts to generate a metal carbene species with the release of nitrogen gas.6 Recently, we have explored other carbene precursors for this type of coupling reaction.7 In particular, we have previously focused on the carbene coupling with conjugated enynones, which have been used as furyl carbene precursors in traditional carbene transformations,8 such as X–H insertion,9 cyclopropanation,10 ylide formation11 and other transformations.12 We have developed a series of transition-metal-catalyzed cross-coupling reactions between conjugated enynones and various coupling partners, demonstrating that the migratory insertion can also be applied to these non-diazo carbene precursors.13 These reactions constitute efficient approaches for the synthesis of furan derivatives, which are important frameworks of many bioactive compounds and pharmaceuticals.14
C bond formation.1 In these transformations, the organometallic species, which can be generated from oxidative addition,2 transmetalation,3 C–H activation4 or other processes,5 reacts with a carbene precursor to form a metal carbene intermediate. Then, carbene migratory insertion occurs to generate a new organometallic species that undergoes further transformation to afford various coupling products. Diazo compounds are the most common carbene precursors, which can be decomposed by transition-metal catalysts to generate a metal carbene species with the release of nitrogen gas.6 Recently, we have explored other carbene precursors for this type of coupling reaction.7 In particular, we have previously focused on the carbene coupling with conjugated enynones, which have been used as furyl carbene precursors in traditional carbene transformations,8 such as X–H insertion,9 cyclopropanation,10 ylide formation11 and other transformations.12 We have developed a series of transition-metal-catalyzed cross-coupling reactions between conjugated enynones and various coupling partners, demonstrating that the migratory insertion can also be applied to these non-diazo carbene precursors.13 These reactions constitute efficient approaches for the synthesis of furan derivatives, which are important frameworks of many bioactive compounds and pharmaceuticals.14
As a continuation of our interest in carbene-based coupling reactions, we have envisioned that a boron–metal species generated from transmetalation between diboron compounds and transition-metal catalysts may also participate in carbene migratory insertion to form C–B bonds followed by β-H elimination, which may provide a new method for the synthesis of alkenylboronates (Scheme 1a).15,16 Alkenylboronates are versatile building blocks in transition-metal-catalyzed cross-coupling reactions and other functional group transformations.17 The established methods for their synthesis include alkyne borylation,18 alkene cross-metathesis,19 Miyaura borylation,20 alkene C–H borylation,21 Boryl–Heck reaction22 and Boron–Wittig reaction.23 Nevertheless, the methods for the synthesis of furyl-substituted alkenylboronates are still rather limited.24 Herein, we reported a palladium-catalyzed oxidative cross-coupling reaction between conjugated enynones and diboron compounds for the synthesis of furyl-substituted alkenylboronates (Scheme 1b). We have also demonstrated the transformations of the furyl-substituted alkenylboronates.
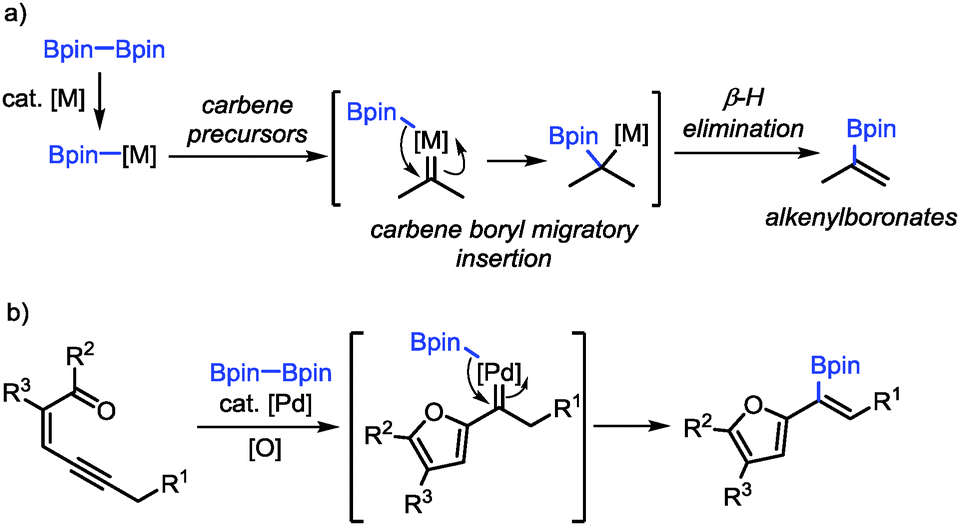 | ||
| Scheme 1 Carbene boryl migratory insertion for the synthesis of alkenylboronates. | ||
The investigation was initiated with conjugated enynone 1a and bis(pinacolato)diboron 2a as the model substrates. The reaction was carried out in methanol at 40 °C with Pd(OAc)2 as the catalyst, PPh3 as the ligand, KOAc as the base and benzoquinone (BQ) as the oxidant. Gratifyingly, we could observe the corresponding product 3a in 51% yield (Table 1, entry 1). Some other palladium catalysts such as Pd(PPh3)4, Pd(PPh3)2Cl2 and Pd(TFA)2 were then examined, however, the yields were slightly diminished with these palladium catalysts (entries 2–4). We also investigated the effect of substituted benzoquinones (entries 8 and 9). The yield could be improved to 65% with sterically-bulky 2,6-dimethylbenzoquinione (2,6-DMBQ) as the oxidant (entry 9). Inorganic bases such as K2CO3, K3PO4 and KOMe gave inferior results due to the side reactions (entries 5–7). Organic bases such as triethylamine and diisopropyl ethylamine were found to afford better results (entries 10 and 11). With diisopropyl ethylamine as the base, the catalyst loading was further reduced to 2 mol% (entry 12). Finally, increasing the loading of 2a and oxidant to 2 equiv., the reaction could give 80% yield with 10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 E/Z ratio (entry 13). The configuration of the double bond of 3a was confirmed by 1H NMR analysis of the protodeboronation product of 3a.25
:1 E/Z ratio (entry 13). The configuration of the double bond of 3a was confirmed by 1H NMR analysis of the protodeboronation product of 3a.25
|
|
|||||
|---|---|---|---|---|---|
| Entry | Pd catalyst | Ligand | Base | Oxidant | Yieldb (%) |
|
a Reaction conditions are the following if not otherwise noted: 1a (0.1 mmol), 2a (0.12 mmol), Pd catalyst (5 mol%), ligand (10 mol%), base (2 equiv.) and oxidant (1.2 equiv.) in methanol (2 mL) at 40 °C for 10 h.
b Determined by 1H NMR using nitromethane as the internal standard if not otherwise noted.
c Pd(OAc)2 (2 mol%), PPh3 (5 mol%).
d
2a (2 equiv.), 2,6-DMBQ (2 equiv.).
e
E/Z = 10:1. Isolated yield shown in brackets.
|
|||||
| 1 | Pd(OAc)2 | PPh3 | KOAc | BQ | 51 |
| 2 | Pd(PPh3)4 | — | KOAc | BQ | 35 |
| 3 | Pd(PPh3)2Cl2 | — | KOAc | BQ | 37 |
| 4 | Pd(TFA)2 | PPh3 | KOAc | BQ | 48 |
| 5 | Pd(OAc)2 | PPh3 | K2CO3 | BQ | 19 |
| 6 | Pd(OAc)2 | PPh3 | K3PO4 | BQ | Trace |
| 7 | Pd(OAc)2 | PPh3 | KOMe | BQ | Trace |
| 8 | Pd(OAc)2 | PPh3 | KOAc | 2,5-DPhBQ | 63 |
| 9 | Pd(OAc)2 | PPh3 | KOAc | 2,6-DMBQ | 65 |
| 10 | Pd(OAc)2 | PPh3 | NEt3 | 2,6-DMBQ | 67 |
| 11 | Pd(OAc)2 | PPh3 | iPr2NEt | 2,6-DMBQ | 78 |
| 12c | Pd(OAc)2 | PPh3 | iPr2NEt | 2,6-DMBQ | 78 |
| 13d | Pd(OAc)2 | PPh3 | iPr2NEt | 2,6-DMBQ | 80(80)e |
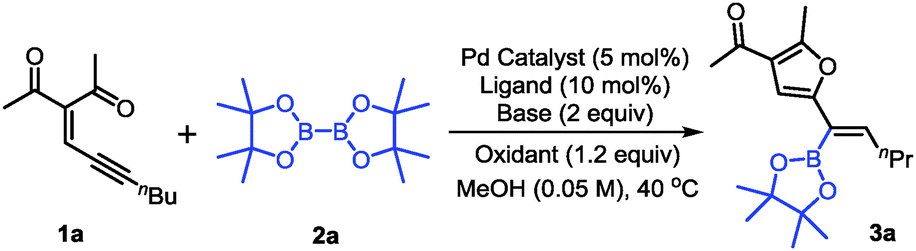
With the optimized conditions in hand, the scope of conjugated enynones was evaluated (Scheme 2). It should be mentioned that the hydrogens on the carbon adjacent to the alkyne moiety of the enynone substrates are required. Thus, a β-hydride elimination process can occur to complete the catalytic cycle. We first investigated the conjugated enynones bearing the primary alkyl group adjacent to the alkyne moiety (Scheme 2). The coupling reaction worked smoothly in all cases, affording a series of trisubstituted alkenylboronates in moderate to good yields and high stereoselectivities (3a–n). Notably, the enynone substrates 1f–l are all 1:1 mixtures of E/Z isomers. However, the reaction with the mixture of (E)- and (Z)-enynones afforded the corresponding products through cyclization of the ketone carbonyl oxygen in moderate to good yields (3f–l), which indicated that the isomerization of the double bonds occurred easily under the reaction conditions. Functional groups, such as phosphate and cyano groups, were tolerated in this reaction (3m–n). Furthermore, several conjugated enynones bearing secondary alkyl groups adjacent to the alkyne moiety were explored, leading to the formation of tetra-substituted alkenylboronates. The reaction proceeded smoothly and afforded the acyclic or exocyclic products in good yields (3o–s).
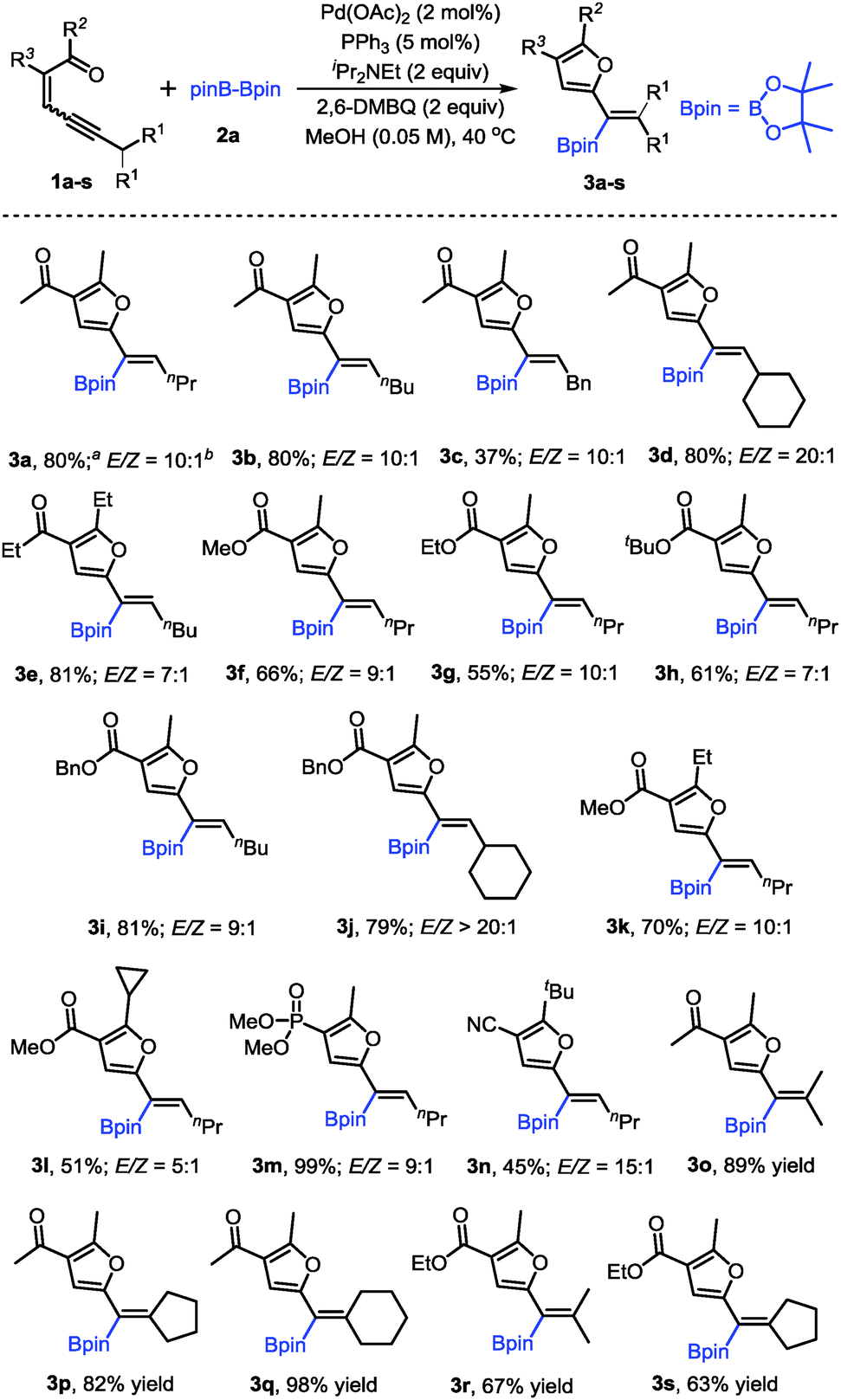 | ||
| Scheme 2 Scope of the enynones. Reaction conditions: 1a–g (0.2 mmol), 2a (0.4 mmol), Pd(OAc)2 (2 mol%), PPh3 (5 mol%), iPr2NEt (0.4 mmol), and 2,6-DMBQ (0.4 mmol) in MeOH (4 mL) at 40 °C for 10 h. aIsolated yields using column chromatography. bThe E/Z ratio was determined by 1H NMR. | ||
We also evaluated other diboron compounds, which are typically used in borylation reactions (Scheme 3). The use of B2mpd2 and B2dmpd2 gave the corresponding alkenylboronates in good yields and good stereoselectivities (4a–b). However, for the diboron compounds B2neo2 and B2cat2, no desired products were observed (4c, 4d).
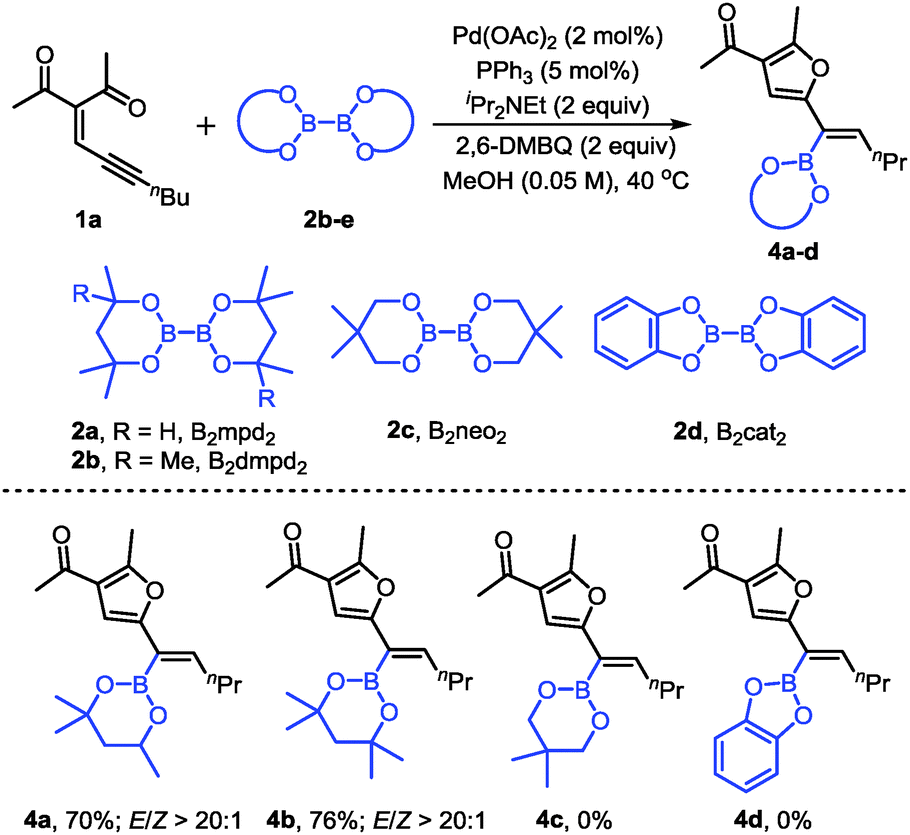 | ||
| Scheme 3 Scope of the diboron compounds. Reaction conditions: 1a (0.2 mmol), 2b–e (0.4 mmol), Pd(OAc)2 (2 mol%), PPh3 (5 mol%), iPr2NEt (0.4 mmol), and 2,6-DMBQ (0.4 mmol) in MeOH (4 mL) at 40 °C for 10 h. Isolated yields. The E/Z ratio was determined by 1H NMR. | ||
To demonstrate the synthetic utility of furyl-substituted alkenylboronates, further transformations of 3q were carried out (Scheme 4). First, protodeboronation could occur in the presence of a silver catalyst (5a). Alkenyl chloride or bromide could be formed with stoichiometric copper halides (5b–c). Besides, the alkenylboronate could be easily oxidized into a ketone with sodium perborate as a mild oxidant (5d). Finally, Suzuki–Miyaura coupling of 3q with 4-bromo-1,1′-biphenyl or (1-bromovinyl)benzene could afford the furyl-substituted alkene and 1,3-diene in good yields (5e–f).
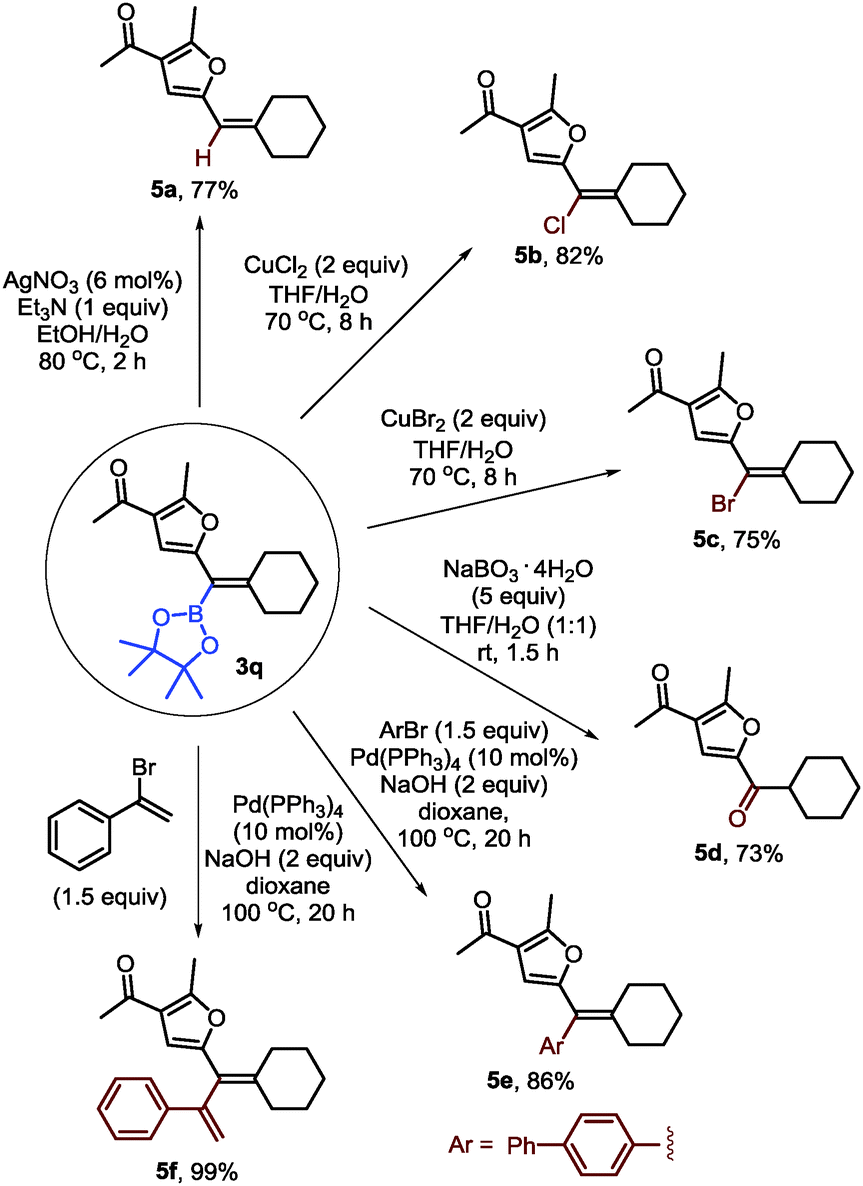 | ||
| Scheme 4 The transformations of furyl-substituted alkenylboronates. | ||
On the basis of our previous studies,13 a plausible mechanism has been proposed for the Pd-catalyzed oxidative borylation reaction (Scheme 5). First, palladium acetate reacts with diboron compound 2 through transmetalation to generate the palladium(II)–boron species B,20 which activates the alkyne moiety of conjugated enynones 1 to form palladium-carbene complexes C. Then boryl-migratory insertion occurs to generate intermediate D, followed by β-H elimination to produce the final product 3. The intermediate E undergoes reductive elimination in the presence of a base to produce Pd(0), which can be oxidized by 2,6-DMBQ to regenerate catalytically reactive Pd(II) species A to complete the catalytic cycle. The E selectivity is presumably attributed to the different steric hindrances of the furyl and boryl groups in the cis β-H elimination step.
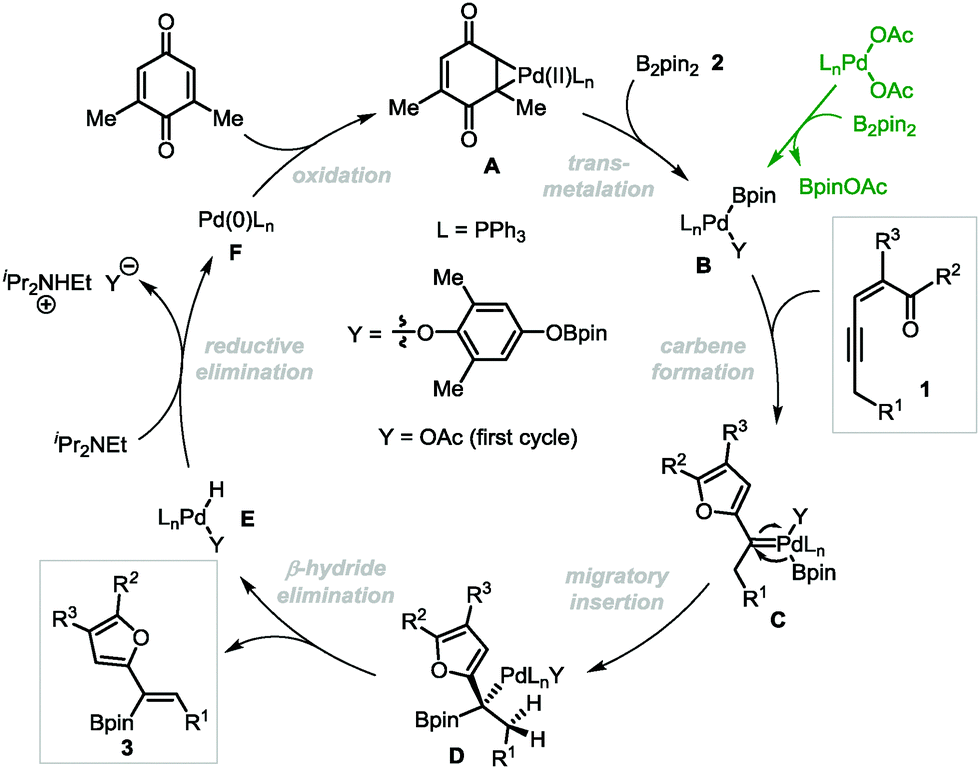 | ||
| Scheme 5 The proposed reaction mechanism. | ||
In summary, we have developed a Pd-catalyzed oxidative borylation reaction of conjugated enynones as the carbene precursors. A wide range of substrates were tolerated in this reaction, and various furyl-substituted alkenylboronates were obtained in good yields. The products could be converted into various furan derivatives, demonstrating the synthetic utility of this reaction. The strategy by using carbene boryl migratory insertion to form C–B bonds may open a new door for the synthesis of organoboron compounds.
This project is supported by the 973 Program (No. 2015CB856600) and NSFC (Grant 21871010).
Conflicts of interest
There are no conflicts to declare.Notes and references
- (a) For reviews, see: Y. Zhang and J. Wang, Eur. J. Org. Chem., 2011, 1015 CrossRef; (b) J. Barluenga and C. Valdés, Angew. Chem., Int. Ed., 2011, 50, 7486 CrossRef CAS; (c) Z. Shao and H. Zhang, Chem. Soc. Rev., 2012, 41, 560 RSC; (d) Q. Xiao, Y. Zhang and J. Wang, Acc. Chem. Res., 2013, 46, 236 CrossRef CAS PubMed; (e) Y. Xia, Y. Zhang and J. Wang, ACS Catal., 2013, 3, 2586 CrossRef CAS; (f) Z. Liu and J. Wang, J. Org. Chem., 2013, 78, 10024 CrossRef CAS; (g) Y. Xia, D. Qiu and J. Wang, Chem. Rev., 2017, 117, 13810 CrossRef CAS.
- For selected recent examples, see: (a) Z.-S. Chen, X.-H. Duan, P.-X. Zhou, S. Ali, J.-Y. Luo and Y.-M. Liang, Angew. Chem., Int. Ed., 2012, 51, 1370 CrossRef CAS; (b) S. Feng, F. Mo, Y. Xia, Z. Liu, Z. Liu, Y. Zhang and J. Wang, Angew. Chem., Int. Ed., 2016, 55, 15401 CrossRef CAS; (c) G. Qin, L. Li, J. Li and H. Huang, J. Am. Chem. Soc., 2015, 137, 12490 CrossRef CAS; (d) Z. Liu, H. Tan, T. Fu, Y. Xia, D. Qiu, Y. Zhang and J. Wang, J. Am. Chem. Soc., 2015, 137, 12800 CrossRef CAS; (e) Z. Liu, J. Huo, T. Fu, H. Tan, F. Ye, M. Hossain and J. Wang, Chem. Commun., 2018, 54, 11419 RSC.
- For selected examples, see: (a) C. Peng, Y. Wang and J. Wang, J. Am. Chem. Soc., 2008, 130, 1566 CrossRef CAS; (b) X. Zhao, J. Jing, K. Lu, Y. Zhang and J. Wang, Chem. Commun., 2010, 46, 1724 RSC; (c) Y.-T. Tsoi, Z. Zhou and W.-Y. Yu, Org. Lett., 2011, 13, 5370 CrossRef CAS PubMed; (d) Y. Xia, Z. Liu, S. Feng, F. Ye, Y. Zhang and J. Wang, Org. Lett., 2015, 17, 956 CrossRef CAS; (e) Z. Liu, Y. Xia, S. Feng, S. Wang, D. Qiu, Y. Zhang and J. Wang, Aust. J. Chem., 2015, 68, 1379 CrossRef CAS.
- For a review, see: F. Hu, Y. Xia, C. Ma, Y. Zhang and J. Wang, Chem. Commun., 2015, 51, 7986 RSC.
- (a) Y. Xia, Z. Liu, Z. Liu, R. Ge, F. Ye, M. Hossain, Y. Zhang and J. Wang, J. Am. Chem. Soc., 2014, 136, 3013 CrossRef CAS; (b) A. Yada, S. Fujita and M. Murakami, J. Am. Chem. Soc., 2014, 136, 7217 CrossRef CAS; (c) Y. Xia, S. Feng, Z. Liu, Y. Zhang and J. Wang, Angew. Chem., Int. Ed., 2015, 54, 7891 CrossRef CAS.
- For reviews, see: (a) T. Ye and M. A. McKervey, Chem. Rev., 1994, 94, 1091 CrossRef CAS; (b) M. P. Doyle, M. A. McKervey and T. Ye, Modern Catalytic Methods for Organic Synthesis with Diazo Compounds, Wiley, New York, 1998 Search PubMed; (c) H. M. L. Davies and R. E. J. Beckwith, Chem. Rev., 2003, 103, 2861 CrossRef CAS; (d) Z. Zhang and J. Wang, Tetrahedron, 2008, 64, 6577 CrossRef CAS; (e) A. Ford, H. Miel, A. Ring, C. N. Slattery, A. R. Maguire and M. A. McKervey, Chem. Rev., 2015, 115, 9981 CrossRef CAS.
- For reviews, see: (a) M. Jia and S. Ma, Angew. Chem., Int. Ed., 2016, 55, 9134 CrossRef CAS PubMed; (b) K. Wang and J. Wang, Synlett, 2018 DOI:10.1055/s-0037-1611020.
- For reviews on conjugated enynones as carbene precursors, see: (a) K. Miki, S. Uemura and K. Ohe, Chem. Lett., 2005, 34, 1068 CrossRef CAS; (b) A. Fürstner and P. W. Davies, Angew. Chem., Int. Ed., 2007, 46, 3410 CrossRef; (c) J. Ma, L. Zhang and S. Zhu, Curr. Org. Chem., 2016, 20, 102 CrossRef CAS; (d) L. Chen, K. Chen and S. Zhu, Chem, 2018, 4, 1208 CrossRef CAS.
- For selected examples, see: (a) J. González, J. González, C. Pérez-Calleja, L. A. López and R. Vicente, Angew. Chem., Int. Ed., 2013, 52, 5853 CrossRef; (b) S. Mata, L. A. López and R. Vicente, Chem. – Eur. J., 2015, 21, 8998 CrossRef CAS; (c) D. Zhu, J. Ma, K. Luo, H. Fu, L. Zhang and S. Zhu, Angew. Chem., Int. Ed., 2016, 55, 8452 CrossRef CAS; (d) J.-M. Yang, Z.-Q. Li, M.-L. Li, Q. He, S.-F. Zhu and Q.-L. Zhou, J. Am. Chem. Soc., 2017, 139, 3784 CrossRef CAS; (e) D. Zhu, L. Chen, H. Zhang, Z. Ma, H. Jiang and S. Zhu, Angew. Chem., Int. Ed., 2018, 57, 12405 CrossRef CAS.
- For selected examples, see: (a) K. Miki, Y. Washitake, K. Ohe and S. Uemura, Angew. Chem., Int. Ed., 2004, 43, 1857 CrossRef CAS; (b) C. H. Oh, S. J. Lee, J. H. Lee and Y. J. Na, Chem. Commun., 2008, 5794 RSC; (c) R. Vicente, J. González, L. Riesgo, J. González and L. A. López, Angew. Chem., Int. Ed., 2012, 51, 8063 CrossRef CAS; (d) J. Ma, H. Jiang and S. Zhu, Org. Lett., 2014, 16, 4472 CrossRef CAS; (e) K. Chen, D.-S. Ji, Y.-C. Luo, Z.-Y. Wang and P.-F. Xu, Org. Chem. Front., 2018, 5, 1768 RSC.
- Y. Kato, K. Miki, F. Nishino, K. Ohe and S. Uemura, Org. Lett., 2003, 5, 2619 CrossRef CAS.
- (a) M. J. González, L. A. López and R. Vicente, Org. Lett., 2014, 16, 5780 CrossRef; (b) M. J. González, E. López and R. Vicente, Chem. Commun., 2014, 50, 5379 RSC; (c) J. González, L. A. López and R. Vicente, Chem. Commun., 2014, 50, 8536 RSC; (d) P. Liu and J. Sun, Org. Lett., 2017, 19, 3482 CrossRef CAS; (e) H. Cao, H. Zhan, J. Cen, J. Lin, Y. Lin, Q. Zhu, M. Fu and H. Jiang, Org. Lett., 2013, 15, 1080 CrossRef CAS; (f) Y. Zheng, M. Bao, R. Yao, L. Qiu and X. Xu, Chem. Commun., 2018, 54, 350 RSC; (g) C. Pei, G.-W. Rong, Z.-X. Yu and X.-F. Xu, J. Org. Chem., 2018, 83, 13243 CrossRef CAS.
- (a) Y. Xia, S. Qu, Q. Xiao, Z.-X. Wang, P. Qu, L. Chen, Z. Liu, L. Tian, Z. Huang, Y. Zhang and J. Wang, J. Am. Chem. Soc., 2013, 135, 13502 CrossRef CAS; (b) F. Hu, Y. Xia, C. Ma, Y. Zhang and J. Wang, Org. Lett., 2014, 16, 4082 CrossRef CAS; (c) Y. Xia, Z. Liu, R. Ge, Q. Xiao, Y. Zhang and J. Wang, Chem. Commun., 2015, 51, 11233 RSC; (d) Y. Xia, R. Ge, L. Chen, Z. Liu, Q. Xiao, Y. Zhang and J. Wang, J. Org. Chem., 2015, 80, 7856 CrossRef CAS; (e) F. Hu, Y. Xia, C. Ma, Y. Zhang and J. Wang, J. Org. Chem., 2016, 81, 3275 CrossRef CAS; (f) Y. Xia, L. Chen, P. Qu, G. Ji, S. Feng, Q. Xiao, Y. Zhang and J. Wang, J. Org. Chem., 2016, 81, 10484 CrossRef CAS.
- (a) P. A. Roethle and D. Trauner, Nat. Prod. Rep., 2008, 25, 298 RSC; (b) C. H. Woo, P. M. Beaujuge, T. W. Holcombe, O. P. Lee and J. M. J. Fréchet, J. Am. Chem. Soc., 2010, 132, 15547 CrossRef CAS; (c) X. Cui, X. Xu, L. Wojtas, M. M. Kim and X. P. Zhang, J. Am. Chem. Soc., 2012, 134, 19981 CrossRef CAS.
- For Pt-catalyzed cross-coupling of diazo compounds with diboron compounds, see: (a) H. Abu Ali, I. Goldberg and M. Srebnik, Organometallics, 2001, 20, 3962 CrossRef; (b) H. Abu Ali, I. Goldberg, D. Kaufmann, C. Burmeister and M. Srebnik, Organometallics, 2002, 21, 1870 CrossRef; (c) A. J. Wommack and J. S. Kingsbury, Tetrahedron Lett., 2014, 55, 3163 CrossRef CAS.
- For transition-metal-free cross-coupling of N-tosylhydrazones with diboron compounds, see: (a) H. Li, L. Wang, Y. Zhang and J. Wang, Angew. Chem., Int. Ed., 2012, 51, 2943 CrossRef CAS; (b) H. Li, X. Shangguan, Z. Zhang, S. Huang, Y. Zhang and J. Wang, Org. Lett., 2014, 16, 448 CrossRef CAS; (c) H. Li, Y. Zhang and J. Wang, Synthesis, 2013, 3090 CAS.
- D. G. Hall, Boronic Acids: Preparation, Applications in Organic Synthesis and Medicine, Wiley-VCH, Weinheim, 2011 Search PubMed.
- For reviews, see: (a) R. Barbeyron, E. Benedetti, J. Cossy, J.-J. Vasseur, S. Arseniyadis and M. Smietana, Tetrahedron, 2014, 70, 8431 CrossRef CAS; (b) H. Yoshida, ACS Catal., 2016, 6, 1799 CrossRef CAS.
- (a) C. Morrill and R. H. Grubbs, J. Org. Chem., 2003, 68, 6031 CrossRef CAS; (b) E. T. Kiesewetter, R. V. O’Brien, E. C. Yu, S. J. Meek, R. R. Schrock and A. H. Hoveyda, J. Am. Chem. Soc., 2013, 135, 6026 CrossRef CAS.
- J. Takagi, K. Takahashi, T. Ishiyama and N. Miyaura, J. Am. Chem. Soc., 2002, 124, 8001 CrossRef CAS.
- For reviews, see: (a) A. Ros, R. Fernandez and J. M. Lassaletta, Chem. Soc. Rev., 2014, 43, 3229 RSC; (b) L. Xu, G. Wang, S. Zhang, H. Wang, L. Wang, L. Liu, J. Jiao and P. Li, Tetrahedron, 2017, 73, 7123 CrossRef CAS.
- (a) W. B. Reid, J. J. Spillane, S. B. Krause and D. A. Watson, J. Am. Chem. Soc., 2016, 138, 5539 CrossRef CAS; (b) W. B. Reid and D. A. Watson, Org. Lett., 2018, 20, 6832 CrossRef CAS.
- For reviews, see: (a) N. Miralles, R. J. Maza and E. Fernández, Adv. Synth. Catal., 2018, 360, 1306 CrossRef CAS; (b) R. Nallagonda, K. Padala and A. Masarwa, Org. Biomol. Chem., 2018, 16, 1050 RSC; (c) C. Wu and J. Wang, Tetrahedron Lett., 2018, 59, 2128 CrossRef CAS.
- For a few examples on the synthesis of furan-substituted alkenylboronates, see: (a) C. Wang, C. Wu and S. Ge, ACS Catal., 2016, 6, 7585 CrossRef CAS; (b) E. K. Edelstein, S. Namirembe and J. P. Morken, J. Am. Chem. Soc., 2017, 139, 5027 CrossRef CAS; (c) K. Nagao, A. Yamazaki, H. Ohmiya and M. Sawamura, Org. Lett., 2018, 20, 1861 CrossRef CAS; (d) Z. Liu, W. Wei, L. Xiong, Q. Feng, Y. Shi, N. Wang and L. Yu, New J. Chem., 2017, 41, 3172 RSC.
- See the ESI,† for details.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental details, and characterization data. See DOI: 10.1039/c8cc09024f |
| This journal is © The Royal Society of Chemistry 2019 |