
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Degradable vinyl copolymers through thiocarbonyl addition–ring-opening (TARO) polymerization†
Nathaniel M.
Bingham
and
Peter J.
Roth
 *
*
Department of Chemistry, University of Surrey, Guildford, Surrey, GU2 7XH, UK. E-mail: p.roth@surrey.ac.uk
First published on 26th November 2018
Abstract
The radical copolymerization of the thionolactone dibenzo[c,e]oxepane-5-thione with acrylates, acrylonitrile, and N,N-dimethylacrylamide afforded copolymers containing a controllable amount of backbone thioesters which could be selectively cleaved. The process is compatible with RAFT polymerization and promising for the development of advanced degradable polymers.
Vinyl polymers constitute an important class of materials. The recent advent of reversible deactivation radical polymerization methods specifically has led to immense interdisciplinary research efforts in exploiting well-defined vinyl-based (nano)materials for biomedical applications including tissue engineering and drug delivery.1–3 Featuring C–C backbones, however, vinyl polymers are inherently non-degradable which limits their applicability.4–6 Radical ring-opening polymerization (RROP) enables the incorporation of labile linkages (typically esters) into vinyl polymer backbones. This method uses cyclic compounds that add onto a radical chain end and then ring-open to produce a secondary radical able to propagate the chain and install a modified section of the ring into the backbone.7,8 Since the development of RROP in the 1960s, two types of RROP monomers—cyclic ketene acetals9 and allyl sulfide lactones10–12—have emerged as the main candidates for bestowing degradability (Scheme 1A and B). However, these systems are not without drawbacks. Cyclic ketene acetals generally show low reactivity during radical copolymerizations with (meth)acrylic and styrenic monomers (meaning that only small quantities of the feed are incorporated into the product)8 although recent work has demonstrated high incorporation of cyclic ketene acetals during copolymerizations with vinyl ethers,13 vinyl esters,14N-vinyl pyrrolidone,15 and maleimides.16,17 The cyclic allyl sulfide monomer family, while allowing additional in-ring functionality,11,18 installs a methylene group into the product which remains reactive toward radicals and can lead to unwanted crosslinking.10 Further, both of these systems introduce esters into the backbone. Conditions required to cleave these backbone esters are likely to cleave also the esters of (meth)acrylic or vinyl ester repeat units and alter the chemical nature of the polymer.
 | ||
| Scheme 1 Synthesis of degradable vinyl copolymers through radical ring-opening polymerization (RROP): literature examples (A9 and B)10 that install esters into the backbone and proposed RROP of thionolactones (C) with monomer candidates prepared in this study (D). Of the shown structures the caprothionolactone derivative 6 underwent RROP. | ||
Herein, the RROP of thionolactones is presented. The process has been termed thiocarbonyl addition–ring-opening (TARO) polymerization and involves the radical addition onto a C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) S double bond, followed by ring-opening which produces an in-chain thioester (–C(O)S–) functionality, Scheme 1C. Radical addition to CS bonds and subsequent fragmentation through β-scission are well-documented in the literature; they form the basis of the Barton McCombie reaction and the mechanism of reversible addition–fragmentation chain transfer (RAFT).19 Meijs and co-workers investigated the non-cyclic analogue benzyl thionobenzoate as an irreversible chain transfer agent in radical polymerizations.20 Hannachi et al. calculated favourable radical addition and fragmentation reactions for two thionophthalides. Synthetic results were, however, limited to an example in which the open-ring radical was ill-suited for re-initiation.21 Cyclic trithiocarbonates have been used in an addition–ring-opening mechanism to provide (presumably degradable) in-chain trithiocarbonate functionality,22 which, however, remains reactive toward radicals. TARO polymerization, on the other hand, produces thioesters which are not susceptible to radical attack but offer chemistries distinct from those of oxoesters.23–25
S double bond, followed by ring-opening which produces an in-chain thioester (–C(O)S–) functionality, Scheme 1C. Radical addition to CS bonds and subsequent fragmentation through β-scission are well-documented in the literature; they form the basis of the Barton McCombie reaction and the mechanism of reversible addition–fragmentation chain transfer (RAFT).19 Meijs and co-workers investigated the non-cyclic analogue benzyl thionobenzoate as an irreversible chain transfer agent in radical polymerizations.20 Hannachi et al. calculated favourable radical addition and fragmentation reactions for two thionophthalides. Synthetic results were, however, limited to an example in which the open-ring radical was ill-suited for re-initiation.21 Cyclic trithiocarbonates have been used in an addition–ring-opening mechanism to provide (presumably degradable) in-chain trithiocarbonate functionality,22 which, however, remains reactive toward radicals. TARO polymerization, on the other hand, produces thioesters which are not susceptible to radical attack but offer chemistries distinct from those of oxoesters.23–25
Our initial attempts at TARO polymerization were based on γ-phenyl-γ-butyrothionolactone, 1, and four thionophthalides, 2–5, prepared through thionation with Lawesson's reagent of the commercially available lactones (see ESI†). Compounds 1–5 inhibited attempted radical polymerizations of vinyl acetate (presumably through H abstraction or CS addition in the absence of ring-opening and re-initiation) and were not incorporated during the radical polymerizations of styrene, methyl acrylate, and methyl methacrylate.
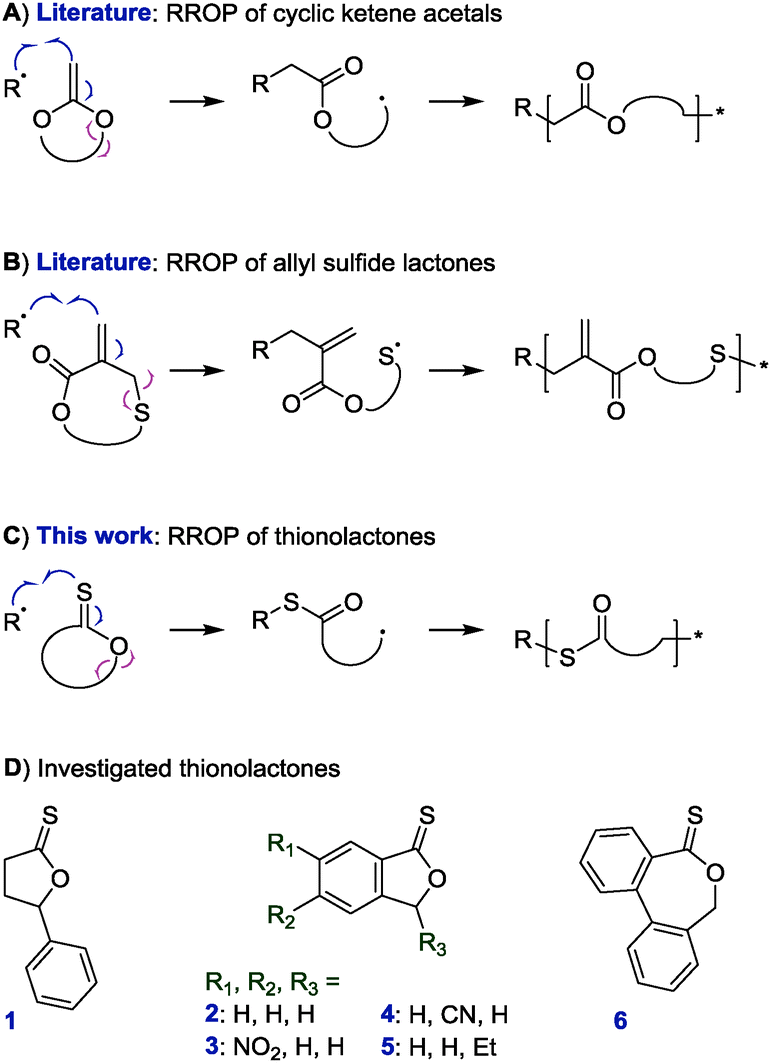
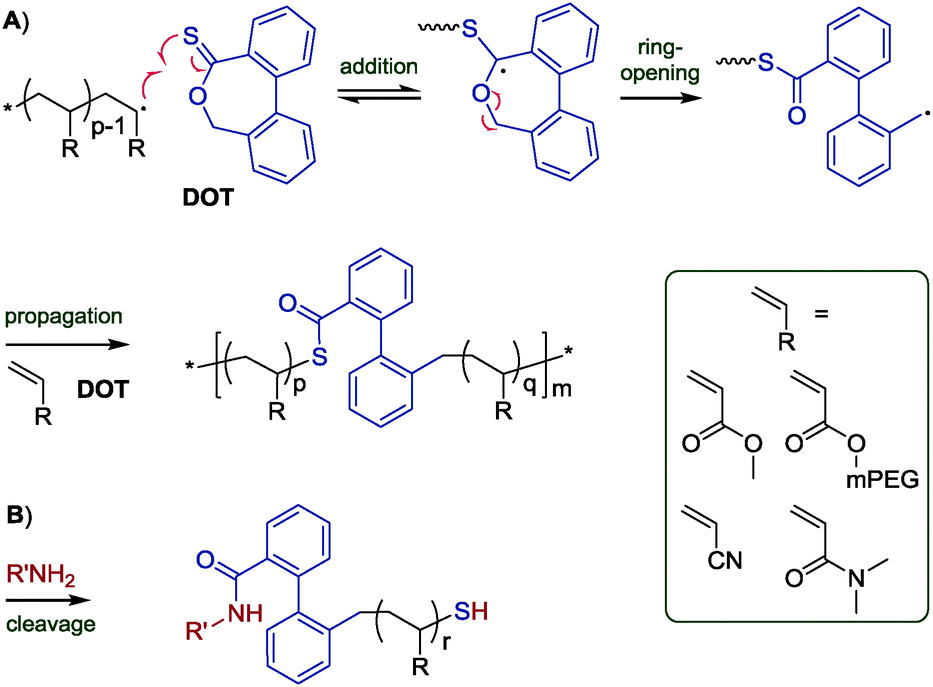
Thereupon, we designed and prepared dibenzo[c,e]oxepane-5-thione (DOT, 6) under the assumption that the biphenyl system would provide additional ring-strain and would, when ring-opened, rotate to move the benzylic radical into a position ideal for propagation (Scheme 2A). At the same time, the two benzene rings were expected to stabilise the adduct and open-ring radical, respectively. Benzylic radicals have been employed for the initiation of acrylic polymerizations.26 DOT was prepared in two steps from diphenic anhydride (see ESI†) and presented as yellow crystals (mp 134–135 °C). It could be stored for ten months in a fridge (darkness, 5 °C, air) or for 45 days on a bench (ambient light, RT, air) without observable signs of change. Heating DOT in DMSO to 150 °C, however, resulted in isomerization to the thioester (–C(O)S–) derivative. DOT was found to inhibit radical polymerizations of vinyl acetate and N-vinylcarbazole and to retard radical polymerizations of styrene without being incorporated. It was a bystander in radical polymerizations of methyl methacrylate, presumably due to the addition equilibrium (first reaction in Scheme 2A) being on the left side and favouring the tertiary methacrylic radicals. An attempted AIBN-initiated homopolymerization of DOT in acetonitrile-d3 reached <1% conversion after heating to 80 °C overnight. Conversely, DOT copolymerised with methyl acrylate, poly(ethylene glycol)methyl ether acrylate (PEGA), N,N-dimethyl acrylamide, and acrylonitrile. IR and NMR spectroscopic analysis of purified polymers confirmed the incorporation of aromatics into the polymers (see Fig. 1 and ESI†). The proposed TARO mechanism (Scheme 2A) is supported by observed IR vibrations at ν = 1680 cm−1 (assigned to the thioester CO stretching) and ν = 907 cm−1 (C–S stretching) which increased with an increasing DOT feed and the presence of 13C NMR signals at δ = 192.6 ppm and 191.1 ppm, characteristic of a thioester carbonyl (–C(O)S–) (see ESI†).
 | ||
| Scheme 2 Proposed mechanism of TARO copolymerization of DOT with structures of vinyl comonomers (A) and expected products of thioester cleavage (B). | ||
 | ||
| Fig. 1 1H NMR spectrum of poly(DOT26-co-PEGA141) with assignments. The integrals are normalised to 1 H (PEGA) = 1.00. For this composition, 1 H (DOT and direct neighbours) ≈ 0.20. | ||
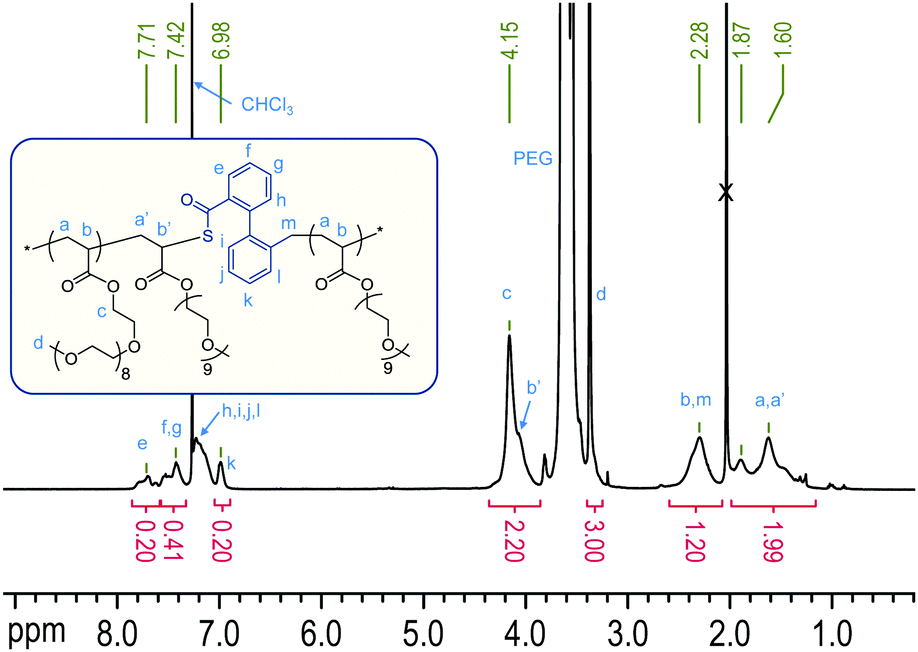
The relative reactivities of two monomers in a copolymerization can be expressed through reactivity ratios, r1, r2, assigned to the comonomers (1, 2) for a given combination. A reactivity ratio r1 > 1 indicates a preference of monomer 1 to homopolymerise; r1 < 1 (and the extreme r1 = 0) indicates that a growing chain terminated in monomer 1 prefers to cross-propagate and add monomer 2, while ideal behaviour (i.e. no apparent distinction between the monomers) is found for r1 = r2 = 1. Apparent reactivity ratios (thus called because RROP monomers involve two radical species)8 were estimated through non-linear regression for the AIBN-initiated free radical copolymerization of DOT and methyl acrylate as a model vinyl monomer to be rDOT = 0.003 and rMA = 0.424, see Fig. 2 and ESI.† Unlike cyclic ketene acetals, DOT copolymerizes rapidly with acrylates with a tendency of both monomers to form alternating sequences. Retardation (i.e. lower-than-expected monomer consumption at a given time) was found for increasing amounts of DOT in the feed, with DOT-rich formulations requiring 6 hours to reach global monomer conversions of 5–15%, see Table S1 (ESI†). The situation appears similar to that of the related dithiobenzoates (PhC(S)SR′) which can cause retardation in RAFT radical polymerizations, attributed to slow fragmentation (here: ring-opening) and termination reactions of the intermediate (adduct) radical.27–29
 | ||
| Fig. 2 Mole fraction of degradable units in the copolymer, F, versus mole fraction of cyclic monomer in the feed, f: data measured for DOT–methyl acrylate (MA) (black squares) and least-squares fit of measured data for rDOT = 0.003 and rMA = 0.424 (line a). The system has an azeotrope around fDOT = 0.37. For comparison, plots of two extreme (and rarely encountered) scenarios, r1 = r2 = 0 (alternating copolymer, horizontal line b) and r1 = r2 = 1 (ideal copolymerization, diagonal line c) and two literature examples of cyclic ketene acetals with acrylate comonomers, rBMDO = 0.08, rBuA = 3.7 (reported for 5,6-benzo-2-methylene-1,3-dioxepane and n-butyl acrylate,30 line d), and rMDO = 0.00235, rMA = 26.535 (reported for 2-methylene-1,3-dioxepane and methyl acrylate,31 line e) are shown. | ||
Next, a series of copolymers was prepared from DOT and PEGA with varying comonomer feed ratios and in the presence of S-benzyl-S′-propyl trithiocarbonate as a RAFT agent. Tuneable molar compositions (measured DOT mole fraction in copolymers 0–0.30) and narrow molecular weight distributions, Đ = 1.19–1.36, were achieved indicating that the two sulfur-based mechanisms, RAFT and TARO, can function side-by-side, Fig. 3.
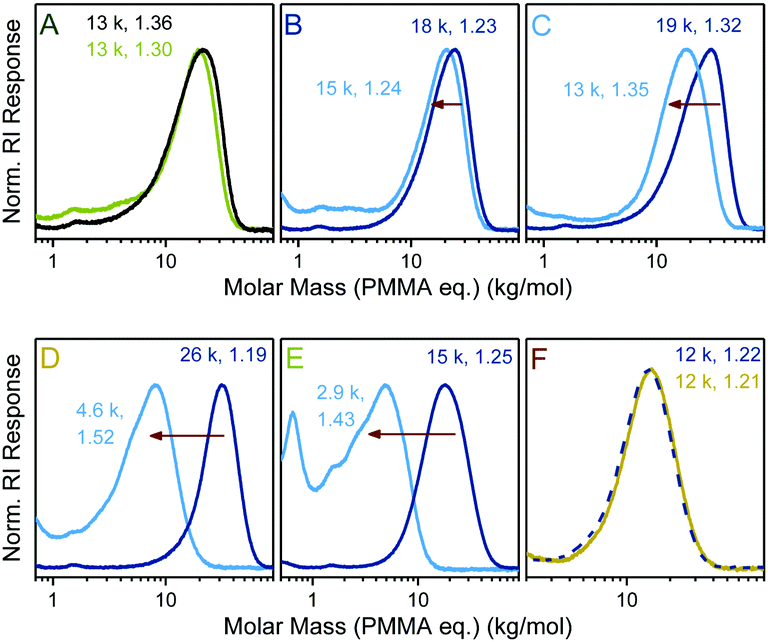 | ||
| Fig. 3 Size exclusion chromatograms of PEGA homopolymer (black line) and after stirring with isopropylamine in dichloromethane (green line) (A) and of DOT–PEGA copolymers (dark blue lines) and after identical amine treatment (light blue lines) for poly(DOT2-co-PEGA244) (B), poly(DOT5-co-PEGA235) (C), poly(DOT26-co-PEGA141) (D), poly(DOT20-co-PEGA46) (E), and of poly(DOT22-co-PEGA64) (dashed line) and after stirring in methanol at RT overnight (yellow line) (F) with measured PMMA-equivalent molar masses (first respective number, in g mol−1) and dispersities (second respective numbers, Đ = Mw/Mn) indicated. | ||
Thioester-functional polymers (that are accessible through other synthetic routes) have been attracting an increasing amount of interest in the smart materials arena.32 Herein, the degradation of TARO-made DOT–PEGA copolymers through aminolysis was investigated. Copolymers, together with a PEGA homopolymer control, were stirred in 5.8 M isopropylamine in dichloromethane overnight to ensure complete cleavage of thioesters. Products were isolated through evaporation and analysed by size exclusion chromatography, Fig. 3. For the PEGA control sample a slight shift (though only at the higher molar mass end) of the molar mass distribution curve was observed (Fig. 3A). 1H NMR spectroscopic analysis of the product showed that, within the error of integration, >95% of side group esters were still intact, indicating that no significant cleavage of side group esters had occurred. For DOT–PEGA copolymers, however, an observable shift of the molar mass distribution curve occurred, which increased with the molar DOT content and was attributed to a cleavage of the backbone thioesters, Fig. 3B–E. The measured molar masses of the degraded fragments were larger than expected from the molar copolymer compositions when assuming complete thioester cleavage. For example, sample poly(DOT2-co-PEGA244), with a measured MSECn = 18 kg mol−1 and featuring an average of two cleavable sites per chain, was degraded into a product with a measured MSECn = 15 kg mol−1 (Fig. 3B). Disparate reactivity ratios can cause a compositional drift along the copolymer chains. For the DOT–acrylate system it is presumed that low feeds of DOT are incorporated fully into copolymers in the early stages of polymerization, resulting in the formation of non-degradable homo-acrylate chains for the remainder of the polymerization.33 However, in this context it is also worth noting that SEC separates polymers by size (not mass) and that different (co-)polymers (including poly(DOT-co-PEGA), its PEGA-based fragments, and the PMMA calibration standards) do not usually show the same mass–size relationships. Nonetheless, the SEC data shown in Fig. 3 demonstrates that it was possible to create well-defined copolymers with varying molar compositions which degraded into fragments of tuneable sizes. More significantly, the TARO concept allowed for a selective degradation of the main chain without affecting the side chains by virtue of the established reactivity difference between esters and thioesters.25 Conversely, in a control experiment DOT–PEGA copolymers could be stirred (and dialysed) in methanol without observable signs of backbone degradation, suggesting that these materials can be stored and handled without unintended degradation, Fig. 3F.
Summarising, a novel type of RROP monomers is presented. The investigated thionolactone dibenzo[c,e]oxepane-5-thione showed a tendency to form alternating copolymers with acrylates. This behaviour is encouraging for the future development of fully alternating degradable vinyl copolymers that degrade exclusively into small molecule fragments. At the same time, thiocarbonyl addition–ring-opening is compatible with RAFT radical polymerization, suggesting that the concept can be combined with other advanced macromolecular design strategies in the preparation of materials with controllable degradability. Based on the established reactivity of thioesters toward thiolates,24 it is anticipated that TARO-made vinyl copolymers that degrade through intracellular glutathione may be beneficial for drug delivery applications.
The authors acknowledge the Department of Chemistry at the University of Surrey for funding, Nicolas Busatto for the synthesis of a PEGA control polymer, Max Attwood for DFT calculations, and Daniel Driscoll and Janella de Jesus for mass spectrometry measurements.
Conflicts of interest
There are no conflicts to declare.Notes and references
- S. Binauld and M. H. Stenzel, Chem. Commun., 2013, 49, 2082–2102 RSC.
- C. Boyer, V. Bulmus, T. P. Davis, V. Ladmiral, J. Liu and S. Perrier, Chem. Rev., 2009, 109, 5402–5436 CrossRef CAS PubMed.
- J. F. Quinn, M. R. Whittaker and T. P. Davis, Polym. Chem., 2017, 8, 97–126 RSC.
- R. P. Brannigan and A. P. Dove, Biomater. Sci., 2017, 5, 9–21 RSC.
- V. Delplace and J. Nicolas, Nat. Chem., 2015, 7, 771 CrossRef CAS PubMed.
- B. D. Ulery, L. S. Nair and C. T. Laurencin, J. Polym. Sci., Part B: Polym. Phys., 2011, 49, 832–864 CrossRef CAS.
- J. Bailey, J. L. Chou, P. Z. Feng, V. Kuruganti and L. L. Zhou, Acta Polym., 1988, 39, 335–341 CrossRef CAS.
- A. Tardy, J. Nicolas, D. Gigmes, C. Lefay and Y. Guillaneuf, Chem. Rev., 2017, 117, 1319 CrossRef CAS PubMed.
- S. Agarwal, Polym. Chem., 2010, 1, 953–964 RSC.
- R. A. Evans, G. Moad, E. Rizzardo and S. H. Thang, Macromolecules, 1994, 27, 7935 CrossRef CAS.
- J. M. J. Paulusse, R. J. Amir, R. A. Evans and C. J. Hawker, J. Am. Chem. Soc., 2009, 131, 9805 CrossRef CAS.
- M. Phelan, F. Aldabbagh, P. B. Zetterlund and B. Yamada, Polymer, 2005, 46, 12046–12056 CrossRef CAS.
- A. Tardy, J.-C. Honoré, J. Tran, D. Siri, V. Delplace, I. Bataille, D. Letourneur, J. Perrier, C. Nicoletti, M. Maresca, C. Lefay, D. Gigmes, J. Nicolas and Y. Guillaneuf, Angew. Chem., Int. Ed., 2017, 56, 16515–16520 CrossRef CAS.
- G. G. Hedir, C. A. Bell, N. S. Ieong, E. Chapman, I. R. Collins, R. K. O’Reilly and A. P. Dove, Macromolecules, 2014, 47, 2847–2852 CrossRef CAS.
- G. G. Hedir, A. Pitto-Barry, A. P. Dove and R. K. O'Reilly, J. Polym. Sci., Part A: Polym. Chem., 2015, 53, 2699–2710 CrossRef CAS.
- M. R. Hill, E. Guégain, J. Tran, C. A. Figg, A. C. Turner, J. Nicolas and B. S. Sumerlin, ACS Macro Lett., 2017, 6, 1071–1077 CrossRef CAS.
- M. R. Hill, T. Kubo, S. L. Goodrich, C. A. Figg and B. S. Sumerlin, Macromolecules, 2018, 51, 5079–5084 CrossRef CAS.
- L. P. D. Ratcliffe, C. Couchon, S. P. Armes and J. M. J. Paulusse, Biomacromolecules, 2016, 17, 2277–2283 CrossRef CAS.
- G. Moad, E. Rizzardo and S. H. Thang, Aust. J. Chem., 2012, 65, 985–1076 CrossRef CAS.
- G. F. Meijs, E. Rizzardo, T. P. T. Le and Y.-C. Chen, Makromol. Chem., 1992, 193, 369–378 CrossRef CAS.
- D. Hannachi, N. Ouddai, M. Arotçaréna and H. Chermette, Mol. Phys., 2015, 113, 1541 CrossRef CAS.
- J. Hong, Q. Wang, Y. Lin and Z. Fan, Macromolecules, 2005, 38, 2691–2695 CrossRef CAS.
- E. A. Castro, J. Sulfur Chem., 2007, 28, 401 CrossRef CAS.
- P. J. Bracher, P. W. Snyder, B. R. Bohall and G. M. Whitesides, Origins Life Evol. Biospheres, 2011, 41, 399 CrossRef CAS.
- W. Yang and D. G. Drueckhammer, J. Am. Chem. Soc., 2001, 123, 11004–11009 CrossRef CAS.
- J. Y. Quek, P. J. Roth, R. A. Evans, T. P. Davis and A. B. Lowe, J. Polym. Sci., Part A: Polym. Chem., 2013, 51, 394–404 CrossRef CAS.
- C. Barner-Kowollik, M. Buback, B. Charleux, M. L. Coote, M. Drache, T. Fukuda, A. Goto, B. Klumperman, A. B. Lowe, J. B. McLeary, G. Moad, M. J. Monteiro, R. D. Sanderson, M. P. Tonge and P. Vana, J. Polym. Sci., Part A: Polym. Chem., 2006, 44, 5809–5831 CrossRef CAS.
- D. Konkolewicz, B. S. Hawkett, A. Gray-Weale and S. Perrier, Macromolecules, 2008, 41, 6400–6412 CrossRef CAS.
- G. Moad, Macromol. Chem. Phys., 2014, 215, 9–26 CrossRef CAS.
- J. Huang, R. Gil and K. Matyjaszewski, Polymer, 2005, 46, 11698–11706 CrossRef CAS.
- L. F. Sun, R. X. Zhuo and Z. L. Liu, J. Polym. Sci., Part A: Polym. Chem., 2003, 41, 2898 CrossRef CAS.
- S. Aksakal, R. Aksakal and C. R. Becer, Polym. Chem., 2018, 9, 4507–4516 RSC.
- D. Gigmes, P. H. M. Van Steenberge, D. Siri, D. R. D'Hooge, Y. Guillaneuf and C. Lefay, Macromol. Rapid Commun., 2018, 39, 1800193 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available: Instrumentation; synthesis of lactones, thionolactones, and (co-)polymers, determination of copolymerization parameters; degradation experiments. See DOI: 10.1039/c8cc08287a |
| This journal is © The Royal Society of Chemistry 2019 |